

Abstract
The interplay between genetic alterations and metabolic dysregulation is increasingly recognized as a pivotal axis in cancer pathogenesis. Both elements are mutually reinforcing, thereby expediting the ontogeny and progression of malignant neoplasms. Intriguingly, recent findings have highlighted the translocation of metabolites and metabolic enzymes from the cytoplasm into the nuclear compartment, where they appear to be intimately associated with tumor cell proliferation. Despite these advancements, significant gaps persist in our understanding of their specific roles within the nuclear milieu, their modulatory effects on gene transcription and cellular proliferation, and the intricacies of their coordination with the genomic landscape. In this comprehensive review, we endeavor to elucidate the regulatory landscape of metabolic signaling within the nuclear domain, namely nuclear metabolic signaling involving metabolites and metabolic enzymes. We explore the roles and molecular mechanisms through which metabolic flux and enzymatic activity impact critical nuclear processes, including epigenetic modulation, DNA damage repair, and gene expression regulation. In conclusion, we underscore the paramount significance of nuclear metabolic signaling in cancer biology and enumerate potential therapeutic targets, associated pharmacological interventions, and implications for clinical applications. Importantly, these emergent findings not only augment our conceptual understanding of tumoral metabolism but also herald the potential for innovative therapeutic paradigms targeting the metabolism–genome transcriptional axis.
Keywords: cancer therapy, DNA methylation, histone modifications, nuclear metabolic enzymes, nuclear metabolic signaling, tumor metabolism
1. Cell metabolism and gene control are significantly integrated and regulated by one another.
2. There is a positive feedback loop of metabolism‐epigenetics‐gene regulation which control cancer progression.
3. The discovery of metabolic enzymes and their functions in the nucleus will provide more insights into cancer therapy.
1. INTRODUCTION
The reciprocal modulation of genetic information and metabolic dysregulation serves as a cornerstone in cancer pathogenesis. The crosstalk between these two domains is an imperative subject that holds significant implications for both our understanding of cancer progression and therapeutic innovation. In eukaryotic cells, genetic information is primarily housed within chromatin structures, whose integrity is governed by complex processes such as DNA methylation and an array of histone modifications. 1 , 2 These chromatin modifications exert a profound influence on a myriad of cellular and developmental events, functioning as quintessential regulators of both gene and genomic landscapes.
Chromatin architecture is orchestrated through the strategic assembly of nucleosomes, which are composed of histone octamers—formed by histone proteins H2A, H2B, H3, and H4—around which superhelical DNA is wrapped. 1 , 2 An extensive spectrum of covalent modifications, including but not limited to acetylation, methylation, phosphorylation, and O‐GlcNAcylation, have been documented on both histone amino‐terminal tails and globular histone cores. 3 , 4 Furthermore, variations in histone methylation—mono‐, di‐, and tri‐methylation—have been discerned to have distinct functional roles. 3 , 4 DNA is also susceptible to diverse modifications; in mammalian cells, the most prevalent include 5‐methylcytosine (5mC) and its oxidative derivatives—5 hydroxymethylcytosine (5hmC), 5 formylcytosine, and 5 carboxylcytosine—all of which contribute to the intricate regulatory network governing gene and genome function. 5 , 6 It is evident that these chromatin modifications are pivotal in modulating a plethora of biological activities, encompassing gene transcription and expression, DNA replication, DNA damage repair, and DNA recombination. Emerging evidence implicates that malignant transformation is associated with aberrant epigenetic landscapes and altered posttranslational modifications of histones. Importantly, these epigenetic changes are often fueled by metabolic intermediates, thereby establishing a feedback loop with cancer metabolism.
Metabolic reprogramming constitutes a hallmark of cancer pathobiology, significantly influencing a diverse range of abnormal metabolic pathways, including glycolysis, lipogenesis, and amino acid biosynthesis and catabolism, all of which have ramifications for cancer progression and treatment responsiveness. 7 , 8 , 9 , 10 , 11 , 12 , 13 Various metabolic enzymes catalyze these processes, generating a plethora of intracellular metabolites from extracellular nutrients such as glucose, lipids, amino acids, and vitamins. Notably, the regulatory circuitry of cellular metabolism can be tuned through the modulation of metabolic genes, which respond to both extrinsic and intrinsic nutrient cues. Additionally, the activities of chromatin can be altered via changes in metabolic enzymes or metabolites. Recent studies have illuminated that some metabolic enzymes undergo nuclear translocation in neoplastic tissues and serve dual functional roles: metabolic catalysis and the regulation of gene transcription pertinent to tumor cell proliferation. 13 , 14 , 15 , 16
Elucidating the nuclear functions of metabolic signaling—including both metabolites and metabolic enzymes, collectively termed as “nuclear metabolic signaling”—promises not only to unveil novel therapeutic targets but also to inspire innovative treatment strategies targeting the dual‐function metabolic enzymes or the intricate metabolism‐gene transcription axis. These potential targets are implicated not merely in cellular metabolism but also in an array of nuclear processes that influence tumoral proliferation, such as gene transcription, DNA damage repair, and chromatin modification. Hence, the identification and characterization of these nuclear roles are of paramount importance for advancing cancer research and therapeutic modalities.
In this review, we aim to provide a comprehensive analysis of metabolic signaling within the nuclear compartment. We will examine the multifaceted functions of these signaling components in the nuclear environment, their impact on other nuclear biological processes, and the ensuing therapeutic strategies and clinical applications.
2. NUCLEAR METABOLIC DYSREGULATION IN CANCER
Metabolic reprogramming is increasingly acknowledged as a salient hallmark of cancer pathobiology. Such reprogramming culminates in an overabundance of metabolites and metabolic enzymes, which serve not only as nutritional substrates but also infiltrate the nuclear compartment to modulate gene expression, thereby fueling cancer cell proliferation. Consequently, elucidating the intricacies of nuclear metabolic signaling and its regulatory mechanisms has become an imperative avenue of inquiry. We will introduce roles of nuclear metabolism in cancer growth and mechanisms of nuclear metabolic dysregulation in following discussion. It includes various modifications which affect the epigenetic characteristics and nuclear metabolic enzymes which usually present in the cytoplasm.
2.1. Role of nuclear metabolism in cancer cell growth and proliferation
The pathogenesis of cancer is an intricate tapestry woven from multiple contributing factors, among which metabolic anomalies stand as pivotal elements. Within this context, nuclear metabolic signaling represents an indispensable component of cellular metabolism, orchestrating a myriad of cellular functions essential for growth and proliferation. Metabolites that encroach upon the nuclear domain in tumor cells serve multifaceted roles—acting as cofactors, molecular donors for modifications, or agents that either potentiate or attenuate biochemical activities, thereby exerting a profound influence on the epigenetic framework.
For instance, the S‐adenosylmethionine (SAM) to S‐adenosylhomocysteine (SAH) ratio serves as a crucial regulatory axis, modulating the activities of an array of methyltransferases, and thereby overseeing methylation patterns across DNA, RNA, and histones. 17 , 18 , 19 Similarly, acetyl coenzyme A and nicotinamide adenine dinucleotide (NAD+) play regulatory roles in histone acetylation, thereby adding nuanced layers to an already complex epigenetic landscape. 20 , 21 Jumonji C domain‐containing histone demethylases (JHDMs) are activated by α‐ketoglutarate (α‐KG) and inhibited by 2‐hydroxyglutarate (2HG), succinate, and fumarate esters. α‐KG serves as a cosubstrate, facilitating the removal of methyl groups from histones, thereby generating succinate and formaldehyde as byproducts. 22 Furthermore, dysregulation in mRNA methylation profiles has been shown to wield influence over the fate of tumor cells by selectively modulating genes implicated in diverse malignancies. 23 Interestingly, recent report suggests that the translation process can also occur within the nucleus and regulate tumor growth. 24 Amino acids, mature transfer RNA (tRNA), messenger RNA (mRNA), and ribosome which are required for the translation process, are found to appear in the nucleus. Zou et al. 25 demonstrate numerous oncoproteins that are preferentially translated in the nucleus (e.g., TGFβ2 and NMP1), and high mRNA level in the nucleus increase protein pression and promote tumorigenesis.
Significantly, these events are metabolically driven, requiring substrates or cofactors such as SAM, acetyl coenzyme A, α‐KG, NAD+, ATP, and succinate. Alterations in the patterns of histone or DNA/RNA modification are frequently correlated with oncogenesis. Hence, nuclear metabolic signaling plays an instrumental role in the promotion of cancer cell growth and proliferation by modulating an array of oncogenes. Comprehensive insights into the role of nuclear metabolism could pave the way for the development of innovative and more efficacious therapeutic paradigms, thereby enhancing the landscape of cancer treatment.
2.2. Mechanisms underlying nuclear metabolic dysregulation in cancer
Nuclear metabolic signaling orchestrates gene expression by influencing a spectrum of molecular mechanisms, encompassing epigenetic modifications, RNA/protein posttranslational modifications, and the moonlighting functions of metabolic enzymes within the nuclear milieu. These represent the predominant mechanisms through which nuclear metabolic signaling exerts its regulatory influence on gene expression.
2.2.1. Metabolic control of epigenetic modification
Histone methylation
Histone methylation, a prevalent posttranslational modification regulated by histone methylases (HMTs), is ubiquitously identified across mammalian species and demonstrates a significant association with oncogenesis. This modification principally involves the methylation of lysine (K) or arginine (R) residues, predominantly localized at the amino termini of core histones H3 and H4, including but not limited to the loci H3K4, H3K9, H3K36, H3K79, and H4K20. 26 A spectrum of methylation states—mono‐, di‐, and trimethylation (notated as me1, me2, and me3, respectively)—can be engendered at the lysine residues by the enzymatic activity of histone lysine methyltransferases. 27 Conversely, the methylation of arginine residues is orchestrated by arginine methyltransferases, culminating in either mono‐ or dimethylated arginine derivatives. 28
SAM serves as the universal methyl donor, predominantly synthesized from methionine via the one‐carbon metabolism pathway, facilitated by methionine adenosyl transferases. 29 Methylation events are constrained in a cellular milieu characterized by low SAM concentrations. 5 The subsequent demethylation of SAM yields SAH, a potent inhibitor of all methyltransferase activity. 17 , 30 Consequently, the intracellular SAM:SAH ratio serves as a significant modulator of global methylation profiles. Importantly, cellular SAM and SAH concentrations are conspicuously attenuated under conditions of methionine scarcity. 17 Depletion of methionine in the methionine cycle reverberates through alterations in the SAM/SAH ratio, thereby influencing histone methylation profiles 17 (Figures 1 and 2).
FIGURE 1.
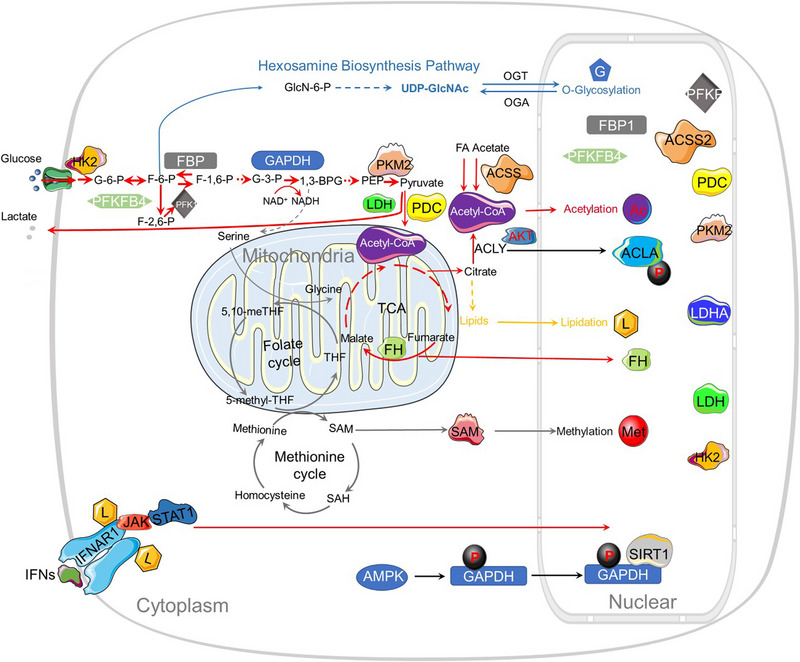
Metabolic reprogramming affects the abundance of key metabolites and metabolic enzymes in the nucleus. Within the tumorigenic microenvironment, aberrant metabolic activity amplifies metabolic flux, thereby accelerating the turnover rates of molecular entities and augmenting the concentration of intranuclear metabolites. Elevated levels of metabolic enzymes stand as a primary determinant in shaping the metabolic landscape of cancer cells. These enzymes translocate into the nuclear compartment, where they contribute to the augmented abundance of pivotal metabolites and enzymes. These, in turn, modulate gene expression through a diverse array of molecular modifications including methylation (denoted as Met), acetylation (Ac), O‐glycosylation (G), and lipidation (L). Nuclear metabolic enzymes are emphasized within the figure for clarity.
FIGURE 2.
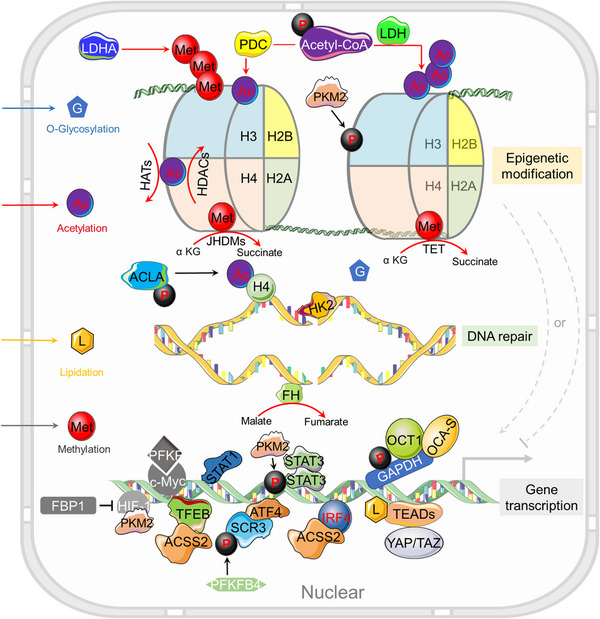
How metabolic alterations impact nuclear gene regulation in cancer. Abnormal metabolic activities in tumor cells facilitate alterations in chromatin or protein modifications such as methylation (Met), acetylation (Ac), O‐glycosylation (G), and lipidation (L), mediated through the control of key metabolites like SAM, acetyl‐CoA, UDP‐GlcNAc, and lipids. Nuclear metabolic enzymes, originating from specific metabolic pathways, exert their influence on chromatin architecture, DNA repair mechanisms, and gene transcription through diverse regulatory avenues. Perturbations in epigenomic stability or genomic fidelity can reciprocally affect the transcriptional profile of metabolic genes, thus establishing a dynamic metabolism‐epigenetics nexus that enables tumor cells to adapt and thrive in variable environments.
It is of considerable note that deletions involving methylthioadenosine phosphorylase (MTAP)—a pivotal enzyme in the methionine salvage pathway—have been implicated in multiple cancer phenotypes. 30 A compromised MTAP expression precipitates a reduction in intracellular methionine levels, engendering a heightened dependency of malignant cells on exogenous methionine. 30 Remarkably, in juxtaposition with normal cells, neoplastic cells manifest an elevated consumption of methionine, a metabolite of critical importance in the proliferation of certain cancer subtypes. 30 , 31 As early as 1958, a correlation between dietary methionine levels and tumorigenesis was reported. Specifically, the rate of tumor growth markedly decelerates in animal models subjected to methionine‐deficient diets. 32 Contemporary research further corroborates that dietary methionine restriction can not only attenuate tumor proliferation but also potentiate the efficacy of chemotherapeutics such as 5‐FU in patient‐derived xenograft models of colorectal cancer. 33 Additionally, methionine restriction has been shown to downregulate H3K4 methylation levels, thereby modulating the transcriptional activity of cancer‐associated genes like Myc, MAPK, and AKT, as evidenced through chromatin immunoprecipitation followed by sequencing (ChIP‐seq) analyses. 17 H3K4 methylation, a cardinal chromatin modification, is intricately associated with gene activation. The trimethylated form, H3K4me3, preferentially recruits transcriptional activators to gene promoters whilst precluding the association of transcriptional repressors, such as nucleosome remodeling and deacetylase complexes. 34 , 35 , 36 Within the specific context of pancreatic neoplasia, elevated levels of H3K4 trimethylation (H3K4me3) have been localized at the cd274 promoter region, thereby amplifying the transcription of PD‐L1 and facilitating immune evasion by the tumor. 37
Defects in serine metabolism exert a consequential impact on the levels of SAM in neoplastic cells. Serine serves as a principal donor of one‐carbon units to the folate cycle, an integral component of one‐carbon metabolism 38 (Figure 1). Within the folate cycle, serine undergoes a transformative process to yield glycine and one‐carbon moieties that subsequently conjugate with tetrahydrofolate (THF), culminating in the synthesis of 5,10‐methylene tetrahydrofolate (MTHF). This compound is subsequently metabolized into 5‐methyltetrahydrofolate (5‐MTHF) via the enzymatic activity of 5‐methylene THF reductase (MTHFR), thereby regenerating THF. 39 , 40 The methyl group liberated from 5‐MTHF engages in a binding interaction with homocysteine (HCys), resulting in the synthesis of methionine and instigating the methionine cycle. 29 , 41 Consequently, serine bioavailability critically influences cellular SAM concentrations and overarching methylation dynamics. Furthermore, intracellular serine concentrations can be augmented either via the biosynthetic conversion of glycolytic byproduct 3‐phosphoglycerate or through direct uptake. Notably, phosphoglycerate dehydrogenase, the cardinal enzyme in the serine biosynthetic pathway, is observed to be amplified in specific malignancies, such as breast cancer and melanoma. 42
Histone demethylases (HDMs), colloquially termed “erasers,” possess the capability to expunge methylation moieties from histones. Importantly, these enzymes leverage a variety of metabolites as cofactors in the aforementioned biological cascade (Figure 1). Two primary categories of HDMs have been identified: lysine‐specific protein demethylases 1 (LSD1) and JHDMs. JHDMs are allosterically activated by α‐KG and inhibited by 2HG, succinic acid, and fumarate. α‐KG, serving as a cosubstrate, facilitates the removal of methyl groups from histones through JHDM‐mediated reactions, subsequently yielding succinate and formaldehyde. 22 Typically, α‐KG is biosynthetically derived from isocitrate via isocitrate dehydrogenase (IDH) within the context of the tricarboxylic acid (TCA) cycle. Intriguingly, mutations in IDH have been implicated in a spectrum of malignancies, including acute myeloid leukemia (AML), glioblastoma, and T‐cell lymphoma. 39 Mutations in IDH isoforms (IDH1 and IDH2) abrogate the enzymatic synthesis of α‐KG, while concurrently engendering the accumulation of the oncometabolite 2HG. Subsequently, α‐KG‐dependent enzymes, such as HDMs, are competitively inhibited by the structurally analogous metabolite (R)−2‐hydroxyglutarate [(R)−2HG]. 2‐Oxoglutarate serves as an integral intermediate of the TCA cycle and constitutes a pivotal cofactor for a diverse array of enzymes. 40 , 42 , 43
DNA methylation
The mammalian genome is characterized by a high prevalence of CpG DNA methylation, with approximately 60−80% of CpG dinucleotides being methylated, excluding CpG islands—CpG‐rich domains that are predominantly unmethylated and encapsulate the promoters for up to 60% of genes. 44 , 45 , 46 It is noteworthy that genomic hypomethylation of CpG loci is typically associated with elevated gene expression, while hypermethylation, particularly within promoter or enhancer regions, often culminates in transcriptional silencing. 47 , 48 Distinct from normal cellular states, cancerous cells exhibit aberrant methylation profiles, characterized by hypomethylation in regions with low CpG density and selective hypermethylation of CpG islands. 42 This methylation landscape is perpetuated across generations through the enzymatic action of DNA methyltransferases (DNMTs) during DNA replication. All three isoforms of DNMTs—DNMT1, DNMT3A, and DNMT3B—are found to be overexpressed in various malignancies. 49 SAM, a one‐carbon donor derived from the methionine cycle, serves a dual role in both DNA and histone methylation. Consequently, perturbations in methionine metabolism can profoundly influence DNA methylation via modulating SAM availability. In colorectal cancer cells, for example, de novo ATP production, facilitated by serine metabolism, augments the conversion of methionine to SAM within the methionine cycle. Serine deficiency subsequently elevates the methionine‐to‐SAM ratio, attenuating the transfer of methyl groups to DNA. 50 Recent investigations have demonstrated that inhibited protein kinase C activity in both de novo and therapy‐induced neuroendocrine prostate cancer (NEPC) activates mTOR, thereby upregulating serine biosynthesis and increasing SAM levels to facilitate NEPC development. 38 Additionally, dysregulated tumor methionine metabolism has been implicated in T‐cell modulation. Integrative omics studies have posited that in hepatocellular carcinoma, T‐cell exhaustion is associated with alterations in methionine recycling, evidenced by elevated levels of 5‐methylthioadenosine and SAM, which contribute to T‐cell dysfunction in vitro. 49
DNA methylation represents a dynamic equilibrium between two antithetical processes: DNA methylation and DNA demethylation. The ten‐eleven translocation (TET) family of proteins and thymine DNA glycosylase function as catalytic facilitators of DNA demethylation. 51 , 52 Fe2+, O2, and α‐KG serve as pivotal cofactors in TET‐mediated reactions 53 , 54 (Figures 1 and 2). Neoplastic cells harboring mutations in metabolic enzymes, such as IDH, succinate dehydrogenase, and fumarate hydratase (FH), are prone to the intracellular accumulation of TCA cycle intermediates, including 2HG, succinate, and fumarate. These metabolites act as inhibitory agents against TET activity, thereby exacerbating tumorigenesis through increased DNA methylation. 55 Moreover, gene silencing through promoter hypermethylation plays a modulatory role in cancer metabolism. For instance, fructose‐1,6‐bisphosphatase (FBP1), the rate‐limiting enzyme in gluconeogenesis, catalyzes the conversion of fructose‐1,6‐diphosphate to fructose‐6‐phosphate (F6P). 56 The hypermethylation of the FBP1 promoter results in downregulation of its expression, a phenomenon observed in both lung and breast cancers. 56 , 57
Histone acetylation
Histone acetylation constitutes a ubiquitous, reversible posttranslational modification mediated by two distinct classes of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Figure 2). HATs orchestrate the covalent attachment of an acetyl moiety to a lysine residue, whereas HDACs facilitate the removal of the acetyl group. The dysregulation of these enzymes is intimately implicated in the pathogenesis of tumorigenesis. 58 Acetylation alters the electrostatic attributes and the local microenvironment of chromatin, thereby modulating its structure to a more accessible state, facilitating the recruitment of transcription factors, and expediting gene transcription. Conversely, deacetylation operates to restrain transcriptional activity. 59 Intriguingly, neoplastic cells typically manifest elevated levels of histone acetylation, aligning with their heightened transcriptional activities.
The rate of enzymatic acetylation and deacetylation is contingent upon the availability of acyl group donors. Acetyl‐coenzyme A (acetyl‐CoA) serves as a principal substrate for the acetylation catalyzed by HATs (Figure 1). The metabolic substrates—pyruvate, citrate, and acetate—are converted into acetyl‐CoA through the enzymatic activities of the pyruvate dehydrogenase complex (PDC), ATP citrate lyase (ACLY), and acetyl‐CoA synthetase short‐chain family member (ACSS), respectively. 60 Additionally, acetyl‐CoA can be synthesized through the β‐oxidation of fatty acids and other nutrient metabolism pathways, such as those involving amino acids and ketones. Hence, the global histone acetylation state is potentially influenced by the cellular abundance of acetyl‐CoA.
In a Ca2+‐responsive mechanism involving the NFAT1 transcription factor, acetyl‐CoA has been observed to promote glioblastoma cell migration and adhesion to the extracellular matrix. 61 Elevated intracellular concentrations of acetyl‐CoA lead to an increase in intracellular Ca2+ levels, thereby facilitating the nuclear translocation of Ca2+‐dependent NFAT1. The transfer of acetyl‐CoA from the mitochondrial matrix to the cytosol is mediated by citrate, which is subsequently cleaved back into oxaloacetate and acetyl‐CoA by ACLY. 62 Given the pivotal role of acetyl‐CoA in histone acetylation, the downregulation or loss of ACLY/ATPCL function precipitates a reduction in histone acetylation levels. 62 Therefore, cellular metabolism is intricately entwined with epigenetic modifications, specifically histone acetylation.
Notably, histone acetylation serves as a critical facilitator of proficient DNA repair mechanisms. It aids in the recruitment of specialized repair proteins and ensures unimpeded access to double‐strand break (DSB) loci for the DNA repair machinery. 63 Thus, the modulation of histone acetylation has far‐reaching implications for cellular integrity and function.
Recent investigations have elucidated those posttranslational modifications of specific nuclear genes, including but not limited to ACSS, ACLY or ACLA, and PDC, exhibit a nuanced interplay with HATs or nuclear transcription factors, thus potentially influencing chromatin regulation 64 (Figure 2). Oncogenic signaling pathways have been demonstrated to induce ACLY‐dependent synthesis and utilization of acetyl‐CoA in malignancies, and empirical evidence substantiates the existence of a positive correlation between global histone acetylation levels and phosphorylated AKT at Ser473 (pAKT–S473) in gliomas and prostate cancers. 20
ACLY serves as a unique substrate that is sensitive to mechanistic target of rapamycin complex 2 (mTORC2), which subsequently modulates ACLY activity to enhance ACSS2‐mediated synthesis of acetyl‐CoA from acetate, thereby augmenting histone acetylation. 65 Notably, AKT‐mediated phosphorylation of ACLY at the S455 site enhances its enzymatic activation. 66 This activated AKT is instrumental in escalating acetyl‐CoA production and promoting acetylation of histones H3 and H4 via the phosphorylation of ACLY. 20 Moreover, phosphorylated nuclear ACLY is responsive to DNA damage signals and fortifies homologous recombination DNA repair mechanisms through the orchestrated spatiotemporal synthesis of nuclear acetyl‐CoA, thereby modulating H4 acetylation at DSB sites. 64
Furthermore, ACSS2 plays a discernible role in furnishing acetyl‐CoA requisite for nuclear histone acetylation. 67 Under low‐glucose conditions, ACSS2, when phosphorylated by AMP‐activated protein kinase (AMPK), engages in interactions with transcription factor EB (TFEB) and repositions itself at gene promoters to initiate lysosomal biogenesis and autophagy as an adaptive response. 67 In myeloma cells sourced from obese patients, ACSS2 is regulated by adipocyte‐secreted angiotensin II, where it stabilizes interferon regulatory factor 4 (IRF4) through acetylation, thereby upregulating IRF4‐controlled gene expression. 67
Moreover, in the realm of prostate cancer, nuclear PDC has been shown to induce acetylation at histone 3 lysine 9 (H3K9) in the promoter regions of lipid‐associated genes, such as ACLY and squalene epoxidase, thereby facilitating lipogenesis and cellular proliferation. 68 In a parallel mechanism, mitochondrial PDC provides cytosolic citrate as a substrate for lipid synthesis to sustain cellular metabolism. 68 In HeLa cells, an association between nuclear PDC and the aryl hydrocarbon receptor has been reported. 69 Additionally, it has been corroborated that the growth of breast cancer cells exhibiting high ACSS2 expression can be mitigated by a small‐molecule inhibitor targeting ACSS2. 70
HDACs are zinc ion‐dependent enzymes and display subtype‐specific roles across a plethora of tumors. Elevated levels of HDAC1 have been reported in gastric and prostate cancers, while HDAC2 expression is abundant in colon, cervical, and gastric cancers. 71 Interestingly, fatty acid hydrolysis can directly inhibit HDAC1 and HDAC2a by generating butyrate and β‐hydroxybutyrate (β‐HB). 72 Another cadre of HDACs is constituted by the sirtuin family, notably SIRT1 and SIRT2. Elevated levels of NAD+ modulate and amplify the enzymatic activity of the sirtuin family, counteracting the inhibitory effect of nicotinamide (NAM), a precursor to NAD+. 73
Histone O‐glycosylation
Nucleotide sugars, which function as substrates for glycosylation processes, are generated through the hexosamine biosynthetic pathway (HBP). A salient substrate implicated in both N‐ and O‐linked glycosylation is uridine diphosphate N‐acetylglucosamine (UDP‐GlcNAc). 74 , 75 The O‐GlcNAcylation process specifically entails the singular O‐linked attachment of GlcNAc moieties to serine and threonine residues on proteins. This dynamic posttranslational modification is orchestrated by two pivotal enzymes: O‐GlcNAc transferase (OGT) and O‐GlcNAcase, which catalyze the addition and removal of O‐GlcNAc modifications, respectively. 76
Dysregulated O‐GlcNAcylation has been implicated in the modulation of signal transduction pathways, gene expression profiles, and metabolic processes, cumulatively contributing to oncogenic transformation and cancer progression. 77 , 78 , 79 Notably, an augmented level of O‐GlcNAcylation is frequently observed in various malignancies, and targeted suppression of OGT has been demonstrated to mitigate cancer growth. 80 , 81 , 82 Histone O‐GlcNAcylation is a ubiquitous phenomenon that engages in intricate crosstalk with other posttranslational modifications, such as methylation, acetylation, phosphorylation, and ubiquitination, to govern gene expression 83 (Figures 1 and 2).
In specific cancer types such as breast and colon cancer, the knockdown of OGT engenders a significant diminution in H3K27me3 levels and in the protein stability of Enhancer of Zeste Homolog 2 (EZH2), without influencing the concentrations of H3K27 demethylases, UTX, and JMJD3. 84 Elevated O‐GlcNAcylation in tumorigenic cells has also been shown to attenuate the intracellular levels of α‐KG. This in turn affects the α‐KG‐dependent prolyl hydroxylation of hypoxia inducible factor‐1 (HIF‐1), thereby stabilizing HIF‐1 and modulating target gene expression, including that of GLUT1. This cascade of events serves to accelerate glycolysis, suggesting that tumors employ a feed‐forward metabolic signaling loop mediated via the HBP to sustain their metabolic idiosyncrasies. 80
OGT itself is subject to direct phosphorylation at the Thr 444 site by AMPK. This phosphorylation disrupts OGT's chromatin interaction and attenuates histone H2B O‐GlcNAcylation, albeit without exerting a direct influence on OGT's enzymatic capabilities. 85 , 86 O‐ or N‐GlcNAcylation also contribute to other oncogenic mechanisms, such as the upregulation of lipid biosynthesis via modulation of SCAP/SREBP. 12 , 87
Beyond histones, a repertoire of epigenetic regulators are also susceptible to O‐GlcNAcylation. For instance, OGT forms a complex with the methylcytosine dioxygenases TET1, TET2, and TET3—each of which is highly O‐GlcNAcylated. In a reciprocal fashion, these TET proteins anchor OGT to chromatin, where they regulate the conversion of 5mC to 5hmC, a process intricately modulated by both OGT and O‐GlcNAcylated TET proteins. 79 , 83
Protein lipid modification
Lipid moieties, characterized by their hydrophobic groups, can be covalently affixed to proteins via an array of linkages, including amide, ester, thioester, or thioether bonds. Such lipid‐modified proteins fulfill multifaceted roles, encompassing the regulation of protein functionality, subcellular localization, and intricate interactions with other cellular entities, such as proteins, lipids, cofactors, and nucleic acids. 88 , 89 , 90 , 91 , 92 , 93 Protein lipid modifications are generally categorized into three predominant types: S‐ or N‐palmitoylation, N‐myristoylation, and S‐prenylation. Specifically, S‐palmitoylation and N‐myristoylation typically target cysteine thiols (S‐acylation) and the amine groups of glycine residues (N‐myristoylation), respectively. In contrast, S‐prenylation involves the formation of thioether bonds between thiols on cysteine residues and isoprenoid groups. These lipid modifications enhance the hydrophobic character of proteins, thereby facilitating their affinity for membrane interaction or nuclear translocation. For example, the estrogen receptor (ER) α undergoes a translocation from the plasma membrane to the nucleus subsequent to its de‐S‐palmitoylation. 94 Additionally, palmitoylation has been demonstrated to stabilize transcriptional enhanced associate domains and augment their interactions with the transcriptional coactivators Yes‐associated protein and Tafazzin 95 (Figure 2).
The cell surface receptor interferon alpha receptor 1 (IFNAR1) serves as a ligand for type I interferons (IFNs), thereby facilitating the activation of Janus kinase (JAK) and signal transducer and activator of transcription (STAT) to modulate the transcription of downstream genes via IFN‐stimulated response elements (Figures 1 and 2). However, the absence of palmitoylation in IFNAR1 leads to a compromised STAT2 activation, which consequentially hampers STAT1 activation and nuclear translocation, culminating in the inhibition of IFN‐α‐activated gene transcription. 96 Recent scholarly contributions have revealed that ZDHHC19 (Z19) functions as a palmitoyl acyltransferase that modulates STAT3 in lung cancer. Elevated expression levels of Z19 have been correlated with increased nuclear STAT3 expression in cancer patients. Furthermore, the SRC Homology 2 (SH2) domain of STAT3 undergoes posttranslational S‐palmitoylation, thereby stimulating STAT3 transcriptional activation and dimerization. Fatty acids have also been identified to potentiate STAT3 activation via palmitoylation, in synergy with cytokine activation. 97
Oct4 (POU5F1), a transcription factor, exists in variants including Oct4A, Oct4B, and Oct4B1. 98 Palmitoylation orchestrated by ZDHHC17 (Z17) is critically implicated in shielding Oct4A from lysosomal degradation, thereby preserving its protein stability. 99 This modification also facilitates the assembly of transcription factors, such as SOX4 and Oct4A, in the SOX2 enhancer region, thereby sustaining the stemness attributes of glioma stem cells. 99 Analogous to palmitoylation, myristoylation serves as a regulatory mechanism for membrane anchoring, protein–protein or protein–lipid interactions, while concurrently inhibiting protein degradation and gene transcription. 90 , 100 , 101
EZH2, a lysine methyltransferase, orchestrates DNA methylation to repress gene transcription. 102 , 103 Within the context of oncogenic signaling, STAT3 is identified as a bona fide substrate for EZH2. 104 The N‐terminal glycine of EZH2 undergoes myristoylation in lung cancer cells, thereby instigating liquid–liquid phase separation (LLPS). This process enables the compartmentalization of STAT3 by EZH2's LLPS and concurrently activates STAT3 signaling. 105 Myristoylation of EZH2 also augments its interaction with STAT3, thereby enhancing STAT3 Y705 phosphorylation and transcriptional activity, which ultimately propels lung cancer progression. 105
Wilms' tumor 1 protein undergoes a functional transformation from a transcriptional activator to a repressor via its interaction with the transcriptional corepressor BASP1. 106 Within the nuclear milieu, myristoylated BASP1 associates with phosphatidylinositol 4,5‐bisphosphate (PIP2), facilitating the recruitment of PIP2 to the promoter regions of target genes. 98 This intricate interaction between BASP1 and PIP2 catalyzes the recruitment of HDAC1 to gene promoters, thereby effectuating transcriptional repression. 98
RNA modification
The burgeoning advances in RNA research have culminated in the identification of an expansive repertoire of RNA modifications, enabled by the advent of highly specialized, quantitatively accurate, and sensitively precise investigative methodologies. To this point, an array of over 170 discrete modifications have been meticulously characterized within various RNA classes, including tRNAs, ribosomal RNAs (rRNAs), long noncoding RNAs (lncRNAs), mRNAs, microRNAs (miRNAs), among others. 23 Of particular prominence is the substantial body of research focusing on mRNA methylation, comprising modifications such as m6A, m5C, m1A, and m7G. RNA methylation serves as a burgeoning category of epigenetic modification with significant roles in modulating a myriad of biological processes, including but not limited to gene expression, RNA stability, translational control, and intricate RNA–protein interactions. 107 , 108 , 109 , 110 , 111 In the medical landscape, the study of m6A modifications has garnered considerable attention for its pertinence to a multitude of diseases, most notably various oncological conditions (Figure 3). This amplified focus emanates from observations that both m6A and its associated regulatory machinery exhibit dysregulation across different cancer types. 112
FIGURE 3.
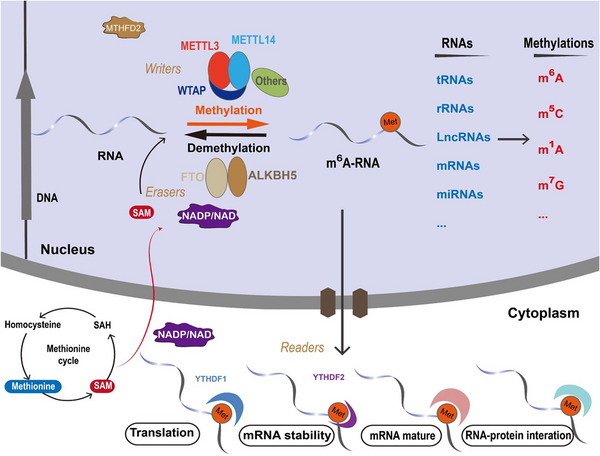
The interplay between nuclear metabolic signaling and RNA methylation. Nuclear metabolic signaling components, including SAM, methionine, NADP, and NAD, impact an array of RNA methylation modifications, such as m6A, m5C, m1A, and m7G, among others. These modifications can be enzymatically installed or removed by “writers” (methyltransferases) and “erasers” (demethylases), and are discerned by their respective “readers” (e.g., YTHDF1 and 2 for m6A modification). In the cytoplasmic milieu, m6A modifications and their regulatory elements partake in mRNA translation, stability, maturation, and RNA–protein interactions, collectively contributing to the advancement of oncogenesis.
As a pivotal regulatory modality, RNA methylation exerts influence over an assortment of metabolic pathways, thereby shaping the trajectory of tumor initiation and subsequent progression. Recent scholarly investigations suggest that nuclear metabolic signaling has the potential to fine‐tune RNA methylation levels through a variety of mechanistic avenues, offering profound implications for the dynamics of cancer development. Analogous to DNA or histone methylation, the methyl groups pertinent to RNA methylation are derived from SAM, implicating that both the methionine and folate cycles exert an intricate regulatory influence on RNA methylation levels. 18 , 19 , 113 Empirical evidence demonstrates that nutritional regimens characterized by restriction, high‐fat, or high‐sugar content can modulate m6A levels through their impact on FTO (a demethylase or “eraser”) and METTL3 (a methyltransferase or “writer”). 113 Furthermore, dietary restriction has been shown to precipitate an increase in hypothalamic FTO levels in rat models. 113 Concurrently, methionine and SAM have been documented to upregulate the expression of METTL3, thereby instigating augmented c‐Myc expression and Avpr2 mRNA m6A modification. Such alterations further activate the c‐Myc and cAMP signaling pathways. 19 An additional seminal study revealed that curtailing dietary methionine intake in murine models led to a marked reduction in levels of l‐cysteine hydropersulfide (LCYH), SAM, SAH, GSH, and l‐methionine in tumor tissues. This diminution subsequently resulted in a significant decrement in m6A methylation and expression of immune checkpoints, including PD‐L1 and V‐domain Ig suppressor of T cell activation (VISTA). 18 These collective findings advance the notion that targeting methionine metabolism as a modulatory tactic for m6A levels might present a novel therapeutic avenue in cancer treatment. Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is a mitochondrial enzyme intricately involved in the folate cycle; it modulates m6A methylation and impacts the clinical course of renal cell carcinoma. MTHFD2 enhances METTL3‐dependent methylation of HIF‐2α mRNA, subsequently promoting HIF‐2α expression. In a reciprocal manner, HIF‐2α binds to the promoter of the MTHFD2 gene, culminating in elevated MTHFD2 levels, thereby perpetuating a positive feedback loop contributing to metabolic reprogramming and tumoral progression. 114
Apart from the methionine and folate cycles, additional metabolic pathways and metabolites also hold the capacity to modulate m6A modifications. Under hypoxic conditions, HIF‐1α governs the transcription of YTHDF1, a recognized m6A reader, by directly engaging with its promoter region. Subsequently, YTHDF1 associates with m6A‐modified ATG2A and ATG14 mRNA, thereby facilitating the translational processes of autophagy‐related genes ATG2A and ATG14. This intricate cascade amplifies the autophagic process and contributes significantly to the pathogenesis of autophagy‐associated malignancies. 115
Recent investigations have illuminated that nicotinamide adenine dinucleotide phosphate (NADP) and NAD are also instrumental in modulating mRNA m6A modification. Specifically, NADP serves to potentiate the enzymatic activity of the RNA demethylase FTO, thereby fine‐tuning mRNA m6A methylation profiles, which subsequently wield an influence on adipocyte differentiation during the adipogenesis process. This insight delineates a direct nexus between cellular metabolic processes and the intricate regulatory mechanisms of RNA m6A methylation. 116
Moreover, the accretion of lactate within the tumor microenvironment has been shown to instigate histone H3K18 lactylation, engendering elevated expression of Mettl3 and m6A modification of Jak1 mRNA within tumor‐infiltrating myeloid cells. This sequential cascade amplifies the translation of Jak1 mRNA and fosters the phosphorylation of STAT3, thereby facilitating immune‐suppressive functionalities in these myeloid cells and mediating mechanisms for tumor immune evasion. 117 Furthermore, inhibiting glycolysis or lactate dehydrogenase (LDH) enzymes, specifically LDHA and LDHB, culminates in a lactate decrement, concomitantly attenuating H3K18 lactylation modifications that govern the transcription of the m6A reader protein YTHDF2. 118 Notably, YTHDF2 possesses the ability to recognize m6A modifications on the 3′ UTR of tumor‐suppressor genes PER1 and TP53 mRNA, thereby diminishing their mRNA stability, particularly in ocular melanoma contexts. 118
In addition to m6A modifications, other methylation variances are also subject to modulation by metabolic pathways or specific metabolites. For instance, upon the assimilation of fatty acids derived from omental adipocytes, gastric cancer cells experience an upregulation in the transcription factor E2F1. This elevation subsequently propels the expression of m5C methyltransferase NSUN2 via cis‐regulatory mechanisms. 119 NSUN2, acting through m5C modifications, modulates the stability of ORAI2 mRNA, thereby facilitating its expression and, subsequently, promoting peritoneal metastasis in gastric cancer. In this complex regulatory landscape, YBX1 operates as a “reader,” recognizing and binding to the m5C modification site on ORAI2 and enhancing its mRNA stability. 119
Currently, burgeoning empirical evidence is increasingly establishing a compelling correlation between dysregulated RNA methylation and perturbed tumor metabolism, thereby contributing to the complexity of tumor progression. The frontier of anticancer therapeutics is being reshaped by an emergent focus on developing drugs targeting RNA methylation (Table 1). Nonetheless, the intricate mechanisms that interlink RNA methylation dysregulation with tumor metabolism warrant more comprehensive, in‐depth investigation. Investigations aimed at drug development targeting RNA methylation‐modulating proteins are still nascent. Hence, the synthesis of more selective inhibitors or activators targeting these RNA methylation‐modulating entities holds the promise for furthering advancements in the burgeoning field of RNA‐based precision medicine.
TABLE 1.
RNA m6A modification, targets, and drugs in clinical trials.
| Target | Inhibitor | Mechanism | Cancer type | Clinical trial | Phase | Status | References |
|---|---|---|---|---|---|---|---|
| METTL3 | STC‐15 | Inhibiting the catalytic activity of METTL3 | Acute myeloid leukemia | NCT05584111 | I | Recruiting | N/A |
| STM2457 | Competitive binding to the binding site of the SAM | myeloid leukemia | N/A | N/A | N/A | 120 | |
| FTO | Entacapone | Regulating the FTO–FOXO1 signaling pathway | Gastrointestinal stromal tumor | NCT04006769 | I | Recruiting | 121 |
| R‐2HG | Activating expression of RARA | Leukemia | N/A | N/A | N/A | 122 | |
| FB23‐2 | Activating expression of ASB2 | Leukemia | N/A | N/A | N/A | 123 | |
| Saikosaponin‐D | Increasing global m6A RNA methylation | Leukemia | N/A | N/A | N/A | 124 | |
| CS1/CS2 | Inhibiting expression of immune checkpoint genes | Leukemia | N/A | N/A | N/A | 125 | |
| Dac51 | Blocking immune evasion | Lung cancer and melanoma | N/A | N/A | N/A | 126 | |
| ALKBH5 | ALK‐04 | Reducing m6A levels, abnormal splicing, Mct4/Slc16a3 expression, and lactate level | Melanoma | N/A | N/A | N/A | 127 |
| IGF2BP2/3 | CWI1‐2 | Inhibiting glutamine metabolism regulated by MYC, GPT2, and SLC1A5 | Acute myeloid leukemia | N/A | N/A | N/A | 128 |
Others
Phospholipids constitute a pivotal component of cellular membranes. While the primary understanding of phospholipids emanates from their salient biological functions within these membranes, it should be acknowledged that phospholipids and their derivatives, namely inositol phosphates (IPs), are ubiquitously present in the cellular nucleus and fulfill consequential biological roles therein. 129
Phosphatidylethanolamine (PE) methylation serves as a paramount consumer of SAM. This methylation event participates in catalyzing the transsulfuration pathways imperative for the biosynthesis of cysteine and glutathione (GSH). Diminished PE methylation culminates in the intracellular accretion of SAM, thereby engendering hypermethylation of both histones and the key phosphatase PP2A. In the absence of PE methylation, particular loci on histones function as methyl sinks, catalyzing the conversion of SAM to SAH. 30 Inositol pyrophosphates represent a distinct class of phospholipids characterized by a dynamically regulated metabolism orchestrated by inositol hexakisphosphate kinases (IP6Ks) and diphosphoinositol pentakisphosphate kinases (PPIP5K/VIP). Notably, Burton et al. 130 unveiled that IP6K1 localizes to the nucleus where it interacts with Jumonji domain‐containing 2C (JMJD2C) HDM and modulates H3K9me3. Their in vitro analyses divulged that IP6K1 knockout instigated a decrease in H3K9me3 levels, counterbalanced by a surge in H3K9 acetylation and H3S10 phosphorylation, thereby elucidating the regulatory role of IP6K1 in specific histone modifications through JMJD2C inhibition. 130
Phosphatidylinositol 4,5‐bisphosphate (PIP2), a derivative of inositol phosphoric acid, engages in complex interactions with UBF, RNA polymerase I, and prefibrillarin, thus modulating the transcriptional activity of RNA polymerase I and nucleolar RNA processing. 131 In human osteosarcoma cells, PIP2 facilitates transcription by binding to the Ser5‐phosphorylated RNA polymerase II complex. Concurrently, Myosin Phosphatase Rho‐Interacting Protein mediates the recruitment of Tyr1‐phosphorylated CTD (Tyr1P‐CTD) to PIP2‐enriched nuclear structures, thereby modulating transcriptional elongation. 132 Sphingosine 1‐phosphate (S1P), a vital component of bilayer membranes, exerts critical regulatory functions in cellular processes. Nuclear S1P interacts with HDAC1 and HDAC2, inhibiting their catalytic activity and thus modulating histone acetylation profiles. 133 Elevated levels of nuclear S1P, as a result of sphingosine kinase upregulation, attenuate HDAC enzyme activities and augment p53 acetylation. IPs have been implicated in an array of nuclear functions such as DNA damage response, chromatin remodeling, mRNA export, and gene expression. 134 IPs can potentiate HDAC activity by facilitating interactions between the catalytic domains and the SANT (Swi3, Ada2, nuclear receptor corepressor [N‐Cor], and TFIIIB) motif in almost all HDAC complexes, barring those that incorporate the Sin3 transcriptional corepressor factor. 135
Collectively, these studies elucidate that aberrant nuclear lipid metabolism is intrinsically entangled with epigenomic regulation and may serve as a contributory factor in tumorigenesis. Despite the valuable insights garnered, our comprehension of nuclear lipid metabolism remains conspicuously incomplete. Additional research is imperative for elucidating these multifaceted molecular mechanisms, and may proffer innovative avenues for targeted cancer therapeutics.
2.2.2. Nuclear function of metabolic enzymes
Emerging evidence posits that a multitude of metabolic enzymes, typically confined to the cytoplasm or mitochondria, can translocate into the nucleus and exercise regulatory influence over gene transcription by altering chromatin conformation. In addition to this epigenetic role, these enzymes execute additional nuclear functions that encompass gene transcription, DNA damage repair, and posttranslational modifications of proteins (Figure 2). These myriad functions collectively foster tumoral growth and pose impediments to effective cancer treatments. The “moonlighting” roles of these metabolic enzymes within the nuclear milieu are elaborated upon below.
Hexokinase 2
Recent investigations have elucidated that hexokinase 2 (HK2), an enzyme belonging to the transferase kinase category that phosphorylates glucose to glucose‐6‐phosphate, is translocated to the nucleus in direct association with the DNA‐binding transcriptional repressor Mig1. This translocation is modulated by glucose concentration and is under the regulatory influence of phosphorylation by Snf1 kinase and dephosphorylation by the Glc7‐Reg1 phosphatase complex. 136 , 137 , 138 Subsequent studies indicate that nuclear HK2 acts as an activator for nuclear factor‐erythroid 2‐associated factor 2 (NRF2), a transcription factor that mitigates oxidative damage under conditions of metabolic stress in glioma cells. 139 The AKT pathway is implicated in regulating nuclear accumulation of HK2, thereby augmenting its association with mitochondria and enhancing glucose uptake in cancer cells. 138 In HeLa cells, AKT inhibitor IV (Ai4) serves to intensify the nuclear presence of HK2; conversely, in MDA‐MB‐231 breast cancer cells, Ai4 merely redistributes HK2 throughout the cytoplasm without inducing its nuclear accumulation. 140 High‐glucose conditions retain glucokinase, a hexokinase isozyme, in the cytoplasm, whereas a glucose‐deficient environment instigates its nuclear translocation, a process reliant on its interaction with glucokinase regulatory protein. 141 , 142 Existing research posits that in both leukemic and standard hematopoietic stem cells, HK2 localization to the mitochondria and nucleus allows its interaction with nuclear proteins to regulate chromatin accessibility, thereby enhancing chromatin openness in leukemic stem cells 143 (Figures 1 and 2).
Moreover, HK2 contains highly conserved cysteine residues that are sensitive to redox modification in response to reactive oxygen species (ROS) stimulation. For example, dehydroascorbic acid (DHA) forms a covalent bond with the active cysteine site of HK1, leading to irreversible enzymatic inactivity. 144 , 145
Benitrobenrazide, an innovative selective HK2 inhibitor, demonstrates the capability to target the binding site and preclude glucose association with HK2, thus efficaciously inhibiting pancreatic cancer growth by disrupting glycolytic pathways. 146 Nevertheless, its potential impact on nuclear HK2 remains ambiguous. 3‐Bromopyruvic acid, a pyruvate analogue, is another plausible HK2 inhibitor, 147 which has been shown to induce apoptosis and inhibit proliferation in a range of cancer types. 147 Prior research has validated that lonidamine (LND), an indazole‐3‐carboxylic acid derivative, specifically inhibits the aerobic glycolysis and energy metabolism of cancer cells. 148 , 149 Additionally, LND fosters the generation of ROS and compromises melanoma cell viability by inhibiting the succinate panquinone reductase activity of respiratory complex II 150 (Table 2).
TABLE 2.
Nuclear metabolic enzymes, their roles, and drugs in cancer.
| Metabolic enzyme | Nuclear function | Mechanism | Cancer type | Drugs | Chemical structure | Clinical trial | References |
|---|---|---|---|---|---|---|---|
| HK2 | Provide protection against oxidative stress | Activate NRF 2 | Glioma | Benitrobenrazide |
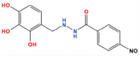
|
Preclinical | 146 |
| Regulate chromatin openness | Interact with nuclear proteins | Acute myeloid leukemia | 3‐Bromopyruvic acid |
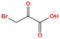
|
Preclinical | 147 | |
| Lonidamine |
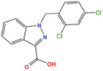
|
N/A | 149 , 150 | ||||
| PFK1 | Activate CXCR4 and T‐ALL cells invasion | Interacts with c‐Myc | Acute lymphoblastic leukemia | Citrate |
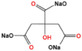
|
Preclinical | 155 , 156 |
| ML251 |
|
N/A | 157 | ||||
| GAPDH | Induce nuclear protein degradation and cell apoptosis | GAPDH S‐nitrosylation enhances its interaction with and stabilization of Siah1 | Neuroblastoma/ prostate cancer | Selegiline |
|
NCT04586543 (Phase I) | 158 , 165 , 166 |
| Increase expression of histone H2B | AMPK‐induced phosphorylation of GAPDH interacts with Oct‐1 and OCA‐S | Osteosarcoma/cervical cancer | Heptelidic acid |
|
Preclinical | 167 | |
| Protection and maintenance of telomeres | Interacts with telomere DNA | Lung cancer | 1,2,3,4,6‐penta‐O‐galloyl‐β‐d‐glucopyranose |
|
Preclinical | 168 | |
| PFKFB4 | Stimulate gene expression of metabolic enzymes and tumor growth | Phosphorylate SRC‐3 | Breast cancer | 5‐(N‐(8‐methoxy‐4‐quinolyl)amino)pentylnitrate |
|
Preclinical | 171 |
| PKM2 | Transcription | Phosphorylate STAT3 and H3 | Glioma/retinoblastoma | Shikonin |
|
Preclinical | 184 , 185 , 186 |
| Cell cycle progression/proliferation | Phosphorylate H3 | Retinoblastoma/glioma | Alkannin |
|
Preclinical | 186 | |
| Upregulation of glycolytic enzymes | Promote HIF‐binding activity to DNA | Cervical cancer | Scutellarin |
|
Preclinical | 187 | |
| LDH | Histone H3 hyper‐acetylation/damage response genes | Increase nuclear acetyl‐CoA and lactate | Hepatocellular carcinoma | Galloflavin |
|
Preclinical | 190 , 191 |
| NRF2 antioxidant genes and Wnt target genes/histone H3K79 hypermethylation | Gains a noncanonical enzyme activity to produce α‐hydroxybutyrate and triggers DOT1L | Cervical cancer | Oxamic acid sodium |
|
N/A | 192 | |
| FX11 |
|
Preclinical | 193 , 194 | ||||
| Fumarase | DNA damage and repair | Preserve histone H3 methylation at double‐strand break (DSB) regions | Hepatocellular carcinoma | Fumarate hydratase‐IN‐1 |
|
Preclinical | 198 |
| Transcription | Interact with AF2 at promoter region | Pancreatic cancer | |||||
| Inhibit cell growth | Inhibit lysine‐specific demethylase 2A (KDM2A) and promotion of histone H3K36me2 | Pancreatic cancer | |||||
| FBP | Inhibit HIF nuclear function | Interact with HIF inhibitory domain and restrain the nuclear function of HIF1α and HIF2α | Renal carcinoma | FBPase‐1 inhibitor‐1 |
|
Preclinical | 201 |
| Suppress mitochondrial respiration and the TCA cycle in a c‐Myc‐dependent manner | Inhibit NRF1 and TFAM‐dependent mitochondrial biogenesis gene expression | Sarcoma | FBPase‐IN‐1 |
|
Preclinical | 202 |
Phosphofructokinase 1
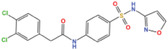
Within the glycolytic cascade, phosphofructokinase 1 (PFK1) functions as a rate‐limiting enzyme, facilitating the conversion of fructose 6‐phosphate to fructose 1,6‐bisphosphate. Various tetrameric isozymic variants of PFK1 coalesce to produce three distinct subunit types—namely, PFKM (muscle), PFKL (liver), and PFKP (platelet). 151 , 152 , 153 PFKP, the predominant isoform of PFK1, is discernible in T cell acute lymphoblastic leukemia (T‐ALL). D3/cyclin‐dependent kinase 6 (CDK6) phosphorylates PFKP at the Ser 679 site, thereby promoting an interaction with importin 9 and culminating in nuclear translocation of PFKP. 154 The chemokine receptor, C‐X‐C chemokine receptor type 4 (CXCR4), modulates the homing and infiltration of human leukemia cells, consequently facilitating T‐ALL cell invasion. Nuclear PFKP elevates CXCR4 expression via interaction with c‐Myc 154 (Figures 1 and 2). Citrate, an efficacious physiological inhibitor of PFK1, has been shown to significantly attenuate aerobic glycolysis and tumor proliferation in non‐small cell lung cancer. 155 , 156 ML251, another potent PFK inhibitor, exhibits promise, although its precise role in oncological therapy remains uncertain 157 (Table 2).
Glyceraldehyde‐3‐phosphate dehydrogenase
Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) plays an instrumental role in the glycolytic pathway, catalyzing the conversion of glyceraldehyde 3‐phosphate to 1,3‐bisphosphoglycerate. Remarkably, it also exhibits a proclivity for direct interaction with the substrates of the G1/S‐specific cyclin E‐CDK2 complex, specifically OCT1 and NPAT, thereby orchestrating the transcriptional regulation of histone genes (Figures 1 and 2).
The enzyme undergoes posttranslational modification via S‐nitrosylation at the Cys150 site, a process incited by the release of nitric oxide (NO) during the initiation of apoptosis. This modification facilitates GAPDH's binding to Siah1, an E3 ubiquitin ligase, and culminates in its nuclear translocation, ultimately inducing cellular apoptosis. Importantly, S‐nitrosylation accentuates GAPDH's interaction with Siah1, whose nuclear localization signal acts as a catalyst for GAPDH's nuclear translocation. 158 Within the nuclear domain, GAPDH stabilizes Siah1, leading to ubiquitination and subsequent degradation of specific nuclear proteins such as the N‐CoR, thereby triggering neuronal death. 158
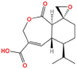
In glucose‐deprived conditions, cytoplasmic GAPDH is phosphorylated at the Ser122 site by activated AMPK. This phosphorylation serves as an impetus for GAPDH's nuclear translocation. Once localized to the nucleus, GAPDH engages in direct interactions with Sirt1, effectively displacing its repressor and activating the enzyme. 159 Furthermore, nuclear GAPDH enhances the expression levels of histone H2B during the S‐phase, achieved through the assembly of a complex with Oct‐1 and OCA‐S (Oct‐1 coactivator at the S phase). 160
Ceramide exerts a regulatory influence over GAPDH's nuclear localization in lung cancer cells through a cell cycle‐dependent mechanism, in addition to inhibiting its telomere‐binding function. Intriguingly, a rapid telomeric shortening is observed upon the inhibition of GAPDH expression, highlighting the critical role that nuclear GAPDH plays in telomere maintenance and protection. 161
Under conditions of oxidative stress, cysteine 152 (Cys152), a conserved active site within GAPDH, undergoes posttranslational modifications via sulfonation or glutathionylation. Such redox‐mediated modifications possess the capacity to attenuate GAPDH enzymatic activity, thereby rerouting metabolic flux away from glycolysis toward the pentose phosphate pathway (PPP). 162 This metabolic diversion serves to augment the viability and adaptive capabilities of neoplastic cells. Intriguingly, the oxidative alteration of Cys152 is not directly orchestrated by ROS; rather, it is facilitated through an intermediary cysteine at the 156 position (Cys156), which undergoes initial oxidation followed by a proton relay mechanism to Cys152. 162 , 163
High‐dose vitamin C therapy proves efficacious in mitigating the extensive depletion of GSH as well as the accumulation of ROS, thereby expediting the glutathionylation of GAPDH at the Cys152 site. Such modulation effectively inactivates GAPDH and exerts inhibitory effects on colon cancer cells harboring KRAS and BRAF mutations, primarily through the obstruction of glycolytic pathways. 164 Nonetheless, it is imperative to acknowledge that the ROS accumulation instigated by vitamin C is a nonspecific phenomenon; therefore, the potential for oxidative modifications to proteins other than GAPDH cannot be categorically dismissed, thereby implicating a more generalized tumor‐suppressive effect.
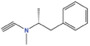
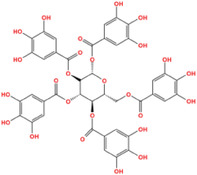
In a therapeutic context, selegiline—a pharmacological agent employed in Parkinson's disease treatment—can inhibit the S‐nitrosylation of GAPDH, thereby precluding its interaction with Siah1 and constraining the nuclear translocation of the GAPDH‐Siah complex, thus limiting cellular apoptosis. 158 , 165 , 166 Heptelidic acid, also known as Koningic acid, serves as a specialized GAPDH inhibitor, attenuating Etoposide‐induced apoptosis through the downregulation of caspases. 167 1,2,3,4,6‐penta‐O‐galloyl‐β‐d‐glucopyranose has been reported to compete with NAD+ and inorganic phosphate (Pi) to impede GAPDH activity; however, its inhibitory efficacy in tumor models remains to be empirically validated 168 (Table 2). In summation, GAPDH inhibitors hold promising therapeutic potential for cancer treatment applications.
N6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphos‐phatase 4
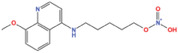
N6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphos‐phatase 4 (PFKFB4) serves as an indispensable catalyst in the glycolytic pathway and is manifestly overexpressed in neoplastic cells 169 (Figures 1 and 2). Emerging evidence delineates the modulatory impact of PFKFB4 on the biological transcriptional machinery, specifically through its regulatory interaction with the oncogenic steroid receptor coactivator‐3 (SRC‐3). SRC‐3, in turn, liaises with a multitude of nuclear receptors. A seminal study by Dasgupta et al. 170 illuminates that tumors positive for ERs exhibit concomitantly elevated levels of PFKFB4 and phosphorylated SRC‐3, thereby indicating a correlative association between the two entities. 171
SRC‐3 undergoes specific phosphorylation at the serine 857 residue, mediated by the protein kinase activity of PFKFB4. Once phosphorylated, SRC‐3 engages in intricate interactions with ATF4 at the gene promoter locales, thereby engendering an upsurge in the expression of an array of metabolic enzymes. Noteworthy among these are transketolase, xanthine dehydrogenase, and adenosine monophosphate deaminase‐1. 170 Importantly, targeted inhibition of either SRC‐3 or PFKFB4 culminates in the attenuation of breast tumor development in murine models and curtails metastatic propagation to pulmonary tissues when assessed in an orthotopic milieu. These salient findings accentuate the pivotal role of PFKFB4, not merely as a glycolytic enzyme but as a protein kinase, in orchestrating the transcriptional landscape through the direct phosphorylation of the coactivator SRC‐3. 170 , 171
In the realm of therapeutic interventions, 5‐(N‐(8‐methoxy‐4‐quinolyl)amino)pentyl nitrate (5MPN) has been unequivocally identified as a potent, orally bioavailable, and selective inhibitor of PFKFB4. 5MPN operates as a competitive antagonist at the F6P binding site, consequently exerting a targeted inhibition on glucose metabolism. This inhibition effectually constrains the proliferative trajectory of a diverse range of malignancies 171 (Table 2).
Pyruvate kinase
Pyruvate kinase (PK) serves as a pivotal enzyme in the glycolytic pathway, facilitating the conversion of phosphoenolpyruvate (PEP) and adenosine diphosphate into pyruvate and ATP. 172 Notably, two isoforms of PK, namely PKM1 and PKM2, have been delineated, with the latter uniquely localized in the nuclear compartment. 173 Within the nucleus, PKM2 predominantly exists in either dimeric or monomeric configurations, whereas its cytosolic analogue principally manifests as a tetramer. 174
In addition to its canonical role in glycolysis, PKM2 exhibits multifunctional capabilities as a protein kinase. In the nuclear milieu, PKM2 dimers function as active protein kinases, whereas the tetrameric form operates as an efficacious PK. 175 Employing PEP as the phosphoryl donor, nuclear PKM2 is implicated in the phosphorylation of STAT3 and histone H3, thereby modulating gene transcription 176 , 177 , 178 (Figures 1 and 2). In oncogenic contexts, nuclear PKM2 specifically phosphorylates STAT3 at the 705th amino acid residue, thereby functioning as a transcriptional coactivator to upregulate MEK5 (also known as MAP2K5) expression. 176 Furthermore, nuclear PKM2 and STAT3 synergize, enabling the facilitation of STAT3 phosphorylation and concomitantly promoting Th17 cell differentiation; however, PKM2 is dispensable for metabolic reprogramming or proliferation in Th17 cells. 177
In response to epidermal growth factor (EGF) stimuli, PKM2 directly phosphorylates and associates with histone H3. This interaction is requisite for K9 histone H3 acetylation, and such PKM2‐dependent chromatin modifications bear significant implications for brain carcinogenesis, tumor cell proliferation, cell cycle dynamics, and EGF‐mediated expression of cyclin D1 and c‐Myc. 176 In retinoblastoma, hepatocyte growth factor‐induced CMET‐dependent signaling leads to the phosphorylation of ERK 1/2, subsequently enhancing the nuclear translocation of PKM2. Therein, PKM2 collaborates with histone H3, thereby stimulating tumor cell growth and facilitating C‐MET‐dependent synthesis of cyclin D1 and c‐Myc. 178
Furthermore, PKM2 modulates the G1/S phase transition by orchestrating the expression of cyclin D1 and effecting phosphorylation of the Bub3 constituent of the spindle assembly checkpoint complex (SAC). Intriguingly, Jiang et al. reported that glioblastoma prognosis correlates with the phosphorylation levels of Bub3 at the Y207 residue. 179 PKM2 also functions as a cotranscriptional modulator, independent of its kinase activity, thereby bolstering HIF binding to DNA. This interaction initiates a positive feedback loop with HIF‐1, culminating in the upregulation of multiple glycolytic enzymes, inclusive of PKM2 itself. 180 , 181
The functionality of nuclear PKM2 extends beyond enzymatic catalysis to the regulation of gene expression, chromatin modification, and cell cycle progression, thereby implicating it in oncogenesis. Nevertheless, comprehensive investigations remain imperative for elucidating the intricate roles of PKM2 as a protein kinase.
Emerging research posits that ROS exert a direct modulatory impact on PKM2, subsequently altering tumor metabolic dynamics. Under conditions of oxidative stress, the acetylation state of PKM2 diminishes, thereby resisting lysosomal degradation and contributing to pharmacological resistance in renal cell carcinoma. 182 Additional studies have reported that PKM2 undergoes oxidation and consequent inactivation at the cysteine 358 residue (Cys358). This modification impedes glycolytic flux and redirects cellular metabolism toward the PPP, thereby generating NADPH essential for ROS detoxification. This metabolic shift confers a survival advantage to lung cancer cells under oxidative stress. 183
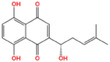
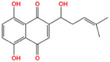
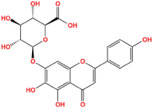
Phytochemicals such as Shikonin and alkannin, derived from traditional Chinese herbal medicine, have been identified as specific PKM2 inhibitors. 184 , 185 , 186 Historically employed for the treatment of dermatitis, burns, and traumatic injuries, 182 recent investigations have unveiled Shikonin's antitumorigenic properties via PKM2 inhibition, resulting in the suppression of glycolytic activity. 184 , 185 , 186 Another PKM2 antagonist, Scutellarin, exhibits the ability to directly bind to PKM2 and attenuate its enzymatic function. Subsequently, this interaction diminishes glycolytic metabolism and heightens nuclear translocation of PKM2, thereby mitigating apoptotic pathways in tumor cells through the modulation of the MEK/ERK/PIN1 signaling axis 187 (Table 2).
Lactate dehydrogenase
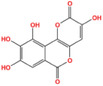
LDH is a pivotal enzyme that mediates the interconversion between pyruvate and lactate, while concurrently catalyzing the redox reaction involving NADH and NAD+ 188 (Figures 1 and 2). As hepatic function deteriorates, LDH accrues within the nuclear compartment, leading to elevated levels of acetyl‐CoA and lactate, as well as the induction of histone H3 hyper‐acetylation and damage response genes. 188 Interestingly, an aggregation of intracellular ROS can disassemble the LDHA tetramer, concurrently augmenting nuclear LDHA concentration in Human papilloma virus (HPV) 16 E7 oncoprotein‐positive cervical cancer cells. 189 In a noncanonical enzymatic function, nuclear LDHA catalyzes the production of α‐hydroxybutyrate, which facilitates histone H3K79 hypermethylation via methyltransferase disruptor of telomeric silencing 1‐like (DOT1L). This modification upregulates target genes in the Wnt signaling pathway and NRF2 antioxidant genes, thereby implicating it in oncogenesis. 189
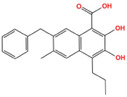
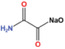
Therapeutically, Galloflavin has been identified as a putative LDH inhibitor that induces cancer cell apoptosis by obstructing glycolysis and ATP synthesis. 190 , 191 Oxamate, a classic LDHA inhibitor, operates by competitively inhibiting the enzyme's affinity for its substrate, pyruvate. 192 Prior studies substantiate Oxamate's potential to abrogate proliferation, invasion, and migration across a plethora of cancers, including but not limited to breast, liver, and non‐small cell lung cancer, whilst simultaneously enhancing the efficacy of other antineoplastic agents and radiotherapy. 192 Notwithstanding, the inhibitory effect of oxalate on LDHA is characterized as nonspecific and relatively weak. Conversely, FX‐11 has garnered attention as a potent, selective, and competitive inhibitor specific to LDHA, significantly reducing ATP levels while inducing ROS generation and consequent tumor cell death 193 , 194 (Table 2).
Fumarase
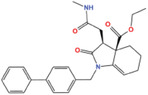
Within the TCA (tricarboxylic acid) cycle, fumarase, also known as FH, serves as an enzymatic catalyst facilitating the reversible hydration and dehydration of fumarate to malate, thereby promoting energy production via the generation of reduced NAD (NADH) (Figures 1 and 2). Remarkably, in response to genotoxic stress, fumarase catalyzes the transformation of fumarate into malate, a metabolite that can be translocated to the nuclear compartment and participate in DNA repair mechanisms. 195 Deficiency in fumarase functionality can drive oncogenesis, primarily by compromising DNA repair processes and consequently facilitating the accrual of genomic mutations. Intriguingly, localized fumarate production within subnuclear domains preserves histone H3 methylation at sites of DSBs, representing a critical component of the cellular DNA damage response architecture. 195 , 196 During glucose scarcity, AMPK phosphorylates fumarase, which then interacts with the transcription factor ATF2. This complex translocates to the promoters of ATF2‐regulated genes, where locally generated fumarate inhibits the activity of KG‐dependent demethylase (KDM2A), culminating in dimethylation of H3K36 and ensuing cell growth arrest. 197 Notably, FH‐IN‐1, a cell‐permeable fumarase inhibitor, has displayed antineoplastic activity across a diverse array of malignancies 198 (Table 2).
Fructose‐1,6‐bisphosphatase
FBP acts as a rate‐limiting enzyme, orchestrating the hydrolysis of fructose 1,6‐bisphosphate to fructose 6‐phosphate, a pivotal step in gluconeogenesis — the biochemical antithesis of glycolysis. Vertebrates express two isozymic forms of FBP: FBP1, predominantly expressed in hepatic and renal tissues, and FBP2, which is primarily localized in skeletal muscle and mesenchymal tissues. 199 Recent investigations have elucidated that FBP1 can translocate to the nucleus, where it directly engages with the HIF “inhibitory domain,” thus attenuating the activities of HIF1α and HIF2α (also referred to as EPAS1) (Figures 1 and 2). Such downregulation of FBP1 in renal carcinoma cells abolishes its HIF inhibitory function, thereby exacerbating tumorigenic processes. 200 Complementary research in sarcoma has ascertained that FBP2 is epigenetically silenced across various sarcoma subtypes and its presence inhibits cellular proliferation. Notably, nuclear FBP2 suppresses the expression of mitochondrial biogenesis genes regulated by nuclear respiratory factor 1 (NRF1) and transcription factor A (TFAM). This results in diminished mitochondrial respiration and TCA cycle activity in a c‐Myc‐dependent manner. Specifically, c‐Myc colocalizes with nuclear FBP2 at the TFAM binding site, leading to the inhibition of TFAM expression and, consequently, sarcoma tumorigenesis. 199 With regard to therapeutic avenues, FBPase‐1 inhibitor‐1 functions as an allosteric inhibitor of FBP, 201 while FBPase‐IN‐1 has been identified as another FBP inhibitor with the capability to ameliorate hyperglycemia and glucose intolerance in Type 2 diabetes. 202 However, the efficacy of these inhibitors in the context of cancer remains unexplored (Table 2).
3. THERAPEUTIC STRATEGIES AND CLINICAL APPLICATIONS TO TARGET NUCLEAR METABOLIC REGULATION
Since the seminal discovery of metabolic aberrations in neoplastic tissues, the field of cancer metabolism has witnessed significant advancements over the past several decades. Emerging therapeutic paradigms grounded in metabolic regulation have been developed and integrated into the clinical management of malignancies. These include the employment of small‐molecule inhibitors targeting pivotal metabolic pathways, the combination of metabolic interventions with immune checkpoint inhibitors, and multimodality treatment strategies that incorporate metabolic drugs into conventional oncological regimens such as surgical resection, radiation, and chemotherapy. 8 , 9 , 11 , 203 Nonetheless, the therapeutic landscape focusing explicitly on nuclear metabolic signaling remains comparatively nascent, despite its critical implications in cancer metabolism. In the subsequent sections, we delineate the extant nuclear metabolism‐centric therapeutic strategies and their clinical ramifications, and provide insights for the potential translation or alternative approach of tumor metabolism‐based preclinical and clinical study.
3.1. Targeting nucleotide metabolism, RNA, and DNA synthesis
In the clinical armamentarium against cancer, metabolic chemotherapy agents are predominantly classified into two categories: folate antagonists and nucleotide base analogs. These compounds primarily exert their antineoplastic effects by obstructing nucleotide synthesis and thereby impeding cellular replication. 204 Folate antagonists, represented by drugs such as methotrexate and pemetrexed, inhibit dihydrofolate reductase, effectively thwarting the conversion of dihydrofolate into its physiologically active form, THF. This enzymatic blockade disrupts the one‐carbon unit transfer essential for purine and pyrimidine nucleotide biosynthesis, thereby stalling DNA synthesis. 205 , 206 , 207 Base analogs like 5‐fluorouracil, gemcitabine, thioguanine, and fludarabine operate by mimicking nucleotide bases, thereby disrupting the integrity of DNA synthesis. 204 , 207 , 208 As such, the therapeutic modulation of nucleotide metabolism presents an efficacious avenue in oncological treatment.
3.2. Targeting nuclear metabolic products via inhibiting metabolic pathway
As elucidated in preceding discussions, key metabolic pathways such as the TCA cycle, one‐carbon metabolism, and glucose metabolism are instrumental in regulating nuclear processes including RNA modifications, DNA methylation, and histone modifications. These pathways supply essential metabolites that function as cofactors in these nuclear modifications. Additionally, the intermediates generated from these metabolic cycles contribute to nucleotide synthesis, ribose generation, and the production of purine and pyrimidine nucleotides. 202 The strategic attenuation or abrogation of these substrate supplies by targeting key metabolic pathways could unlock novel therapeutic avenues in the battle against cancer. However, the realization of these promising therapies is contingent upon the development of highly specific and effective inhibitors.
3.3. Targeting nuclear metabolic enzymes
In addition to metabolites that wield significant epigenetic influence on oncogenesis, metabolic enzymes serve as pivotal regulatory entities in the trajectory of tumor development. Evidence suggests that these enzymes undergo nuclear translocation, where they manifest kinase or nonkinase functions that modulate gene transcription, DNA damage repair, and posttranslational modifications, thereby influencing oncogene expression (Table 2). Although pharmacological inhibitors targeting these nuclear metabolic enzymes have been synthesized, the extent to which these inhibitors modulate their intranuclear biological activities warrants further rigorous investigation. Continued research could elucidate the complex interrelationships between metabolic enzymes and nuclear processes, thus unveiling innovative paradigms for therapeutic interventions specifically aimed at nuclear metabolic regulation in neoplastic conditions.
3.4. Targeting metabolism‐related epigenetic modifications
The advancement of scientific research has laid bare a myriad of nuclear protein, DNA, and RNA modifications that serve as cardinal regulators of nuclear function and the transmission of epigenetic information. HDAC inhibitors, including compounds such as Vorinostat (Zolinza), Belinostat, Panobinostat (Faridak), and Romidepsin (Istodax), have received approval from the United States Food and Drug Administration (US FDA) and have demonstrated clinical efficacy in oncological applications 209 , 210 (Table 3). More recently, the histone methyltransferase inhibitor Tazemetostat achieved US FDA endorsement, thereby augmenting the available repertoire of therapeutic agents. Moreover, two DNMT inhibitors, 5‐azacytidine (Vidaza) and SGI‐110 (guadecitabine), have been recognized by both the US FDA and the European Medicines Agency (EMA), offering innovative therapeutic modalities for malignancy management. Concurrently, several prospective inhibitors targeting these epigenetic modifications are at various stages of clinical evaluation, portending future therapeutic advancements (Table 3). This burgeoning domain of research has catalyzed significant innovation within the realm of pharmacotherapy, fostering the development of agents with a high degree of specificity for these critical epigenetic modifications.
TABLE 3.
Epigenetic modification, targets, and drugs in cancer.
| Targets | Drugs | Mechanism | Cancer type | Clinical trial | Phase | Status | References |
|---|---|---|---|---|---|---|---|
| DNMT | 5‐Azacytidine (Vidaza) | Reversing hypermethylation to restore transcription of tumor suppressor genes | Squamous cell carcinoma, head and neck cancer | NCT05317000 | I | Recruiting | 221 |
| SGI‐110 (guadecitabine) | The precursor drug of decitabine/ extending drug exposure | Leukemia | NCT02907359 | III | Completed | 210 | |
| Decitabine (Dacogen) | Inhibit DNA methylation of tumor suppressor genes | Pancreatic cancer | NCT05360264 | II | Recruiting | 222 | |
| HDAC | Vorinostat (Zolinza) | Modulating chromatin conformation, inducing cellular differentiation, and blocking cell cycle | Rhabdomyosarcom | NCT04308330 | I | Recruiting | 223 |
| Belinostat | Inducing accumulation of acetylated histones and apoptosis‐related proteins | Refractory peripheral T‐cell lymphoma | NCT05170334 | II | Recruiting | 224 | |
| Citarinostat | Decreasing Th2 cytokine production (i.e., IL‐4, IL‐5, IL‐6, IL‐10, and IL‐13) | Smoldering multiple myeloma | NCT02886065 | I | Recruiting | 225 | |
| Entinostat | Promoting immune editing of tumor neoantigens and remodeling the tumor immune microenvironment | Breast cancer | NCT02115282 | III | Recruiting | 226 | |
| Valproic acid | Inducing proteasomal degradation of HDAC2 | Non‐small‐lung cancer | NCT01203735 | II | N/A | 227 | |
| Trichostatin A | Inhibiting dendritic cell maturation andadipogenesis | Hematologic malignancies | NCT03838926 | I | N/A | 228 , 229 | |
| Tacedinaline (CI‐994) | Suppressing NF‐κB pathway | Lung cancer | NCT00005093 | III | Completed | 230 | |
| Panobinostat (Faridak) | Modulating chromatin conformation, facilitating transcription, and inhibiting protein metabolism | Multiple myeloma | N/A | N/A | Approved (US FDA) | 231 | |
| Romidepsin (Istodax) | Inducing cell death | Cutaneous T‐cell lymphoma | N/A | N/A | Approved (US FDA) | 232 | |
| Chidamide | Inducing growth arrest and apoptosis and enhancing antitumor immunity | Peripheral T‐cell lymphoma | N/A | N/A | N/A | 233 | |
| HMTs | Tazemetostat | Competitively inhibiting the methylation of histone lysine | Relapsed or refractory follicular lymphoma | NCT01897571 | II | Completed | 234 |
| Pinometostat | Reducing H3K79 methylation | Acute myeloid leukemia | NCT03701295 | II | Completed | 235 | |
| GSK126 | Suppressing antitumor immunity by driving production of myeloid‐derived suppressor cells | Leukemia | N/A | N/A | N/A | 236 | |
| AZ505 | Inhibiting the binding of SMYD2 to miR‐125b promoter | Renal cell carcinoma | N/A | N/A | N/A | 237 | |
| EPZ015666 | Inhibiting EMT by promoting expression of TET1 and increasing 5hmC | Cervical cancer | N/A | N/A | N/A | 238 | |
| MS023 | Activation of interferon responses | Breast cancer | N/A | N/A | N/A | 239 | |
| HDMs | GSK2879552 | Inducing the expression of KDM1A‐targeted genes to counteract cell proliferation | Leukemia | NCT02177812 | I | Terminated | 240 |
| Seclidemstat (SP‐2577) | Suppressing the viability and metabolism of NK cells | Chronic myelomonocytic leukemia | NCT04734990 | II | Recruiting | 241 | |
| JIB‐04 | Inducing cell apoptosis | Liver cancer | N/A | N/A | N/A | 242 | |
| GSK‐J4 | Inhibiting the JMJD3/UTX enzyme to upregulate H3K27me3 level | Acute myeloid leukemia | N/A | N/A | N/A | 243 | |
| ZY0511 | Inducing H3K4 methylation at the promoter of GADD45B | Hepatocellular carcinoma | N/A | N/A | N/A | 244 |
Abbreviations: DNMT, DNA methyltransferases; HDAC, histone deacetylases; HMTs, histone methylases; HDMs, histone demethylases.
3.5. Targeting RNA modifications
In contrast to the more mature understanding, we possess concerning protein and DNA modifications, the scientific comprehension of RNA modifications remains in its formative stage. This nascent understanding has led to ongoing research endeavors that have yet to fully elucidate the complexities inherent in RNA modifications. As a result, pharmacological inhibitors targeting these modifications are still in their incipient phases and have generally not progressed to the stage of clinical trials (Table 1). Nevertheless, the continuous, incremental advancements in this burgeoning field are propitious for the eventual identification and development of novel inhibitors capable of modulating RNA modifications, thereby enriching the compendium of therapeutic avenues available for future clinical applications.
3.6. Others
Beyond the aforementioned therapeutic strategies, alternative approaches are under exploration in both preclinical and clinical settings. These include: (1) manipulating transcription factor regulation as a means of controlling or responding to metabolic aberrations, such as the role played by Sterol Regulatory Element‐Binding Proteins (SREBPs) in lipid metabolism, and transcription factors like p53 and c‐Myc. 11 (2) The synergistic application of nuclear metabolic inhibitors in conjunction with traditional therapeutic modalities, including surgical interventions, 211 , 212 radiotherapy, 213 , 214 chemotherapy, 215 , 216 and immunotherapy. 217 , 218 Such metabolic perturbations within cancer cells have been demonstrated to enhance their sensitivity to these conventional treatments. For instance, in reaction to radiation or chemotherapy, cancer cells invoke adaptive metabolic shifts that fortify mechanisms of radioresistance and chemoresistance. 219 Interventions timed to disrupt this metabolic homeostasis may potentiate enhanced clinical responses. (3) The multifaceted targeting of multiple metabolic pathways. Given the metabolic heterogeneity or metabolic plasticity observed within tumors, monotherapeutic approaches targeting a single pathway often prove inadequate. Hence, polypharmacological strategies employing multiple inhibitors are being pursued for comprehensive cancer treatment. 220 Despite these efforts, methodologies to specifically target nuclear metabolic signaling require further developmental refinement.
4. CONCLUSION AND PERSPECTIVES
Cellular metabolism and genomic regulation are inextricably intertwined, operating in a highly coordinated manner to facilitate rapid cellular adaptation to fluctuating extracellular nutrient conditions (Figure 4). A failure in this intricate regulatory balance can precipitate a cascade of adverse outcomes, including developmental aberrations, cellular apoptosis, and the onset of multifaceted pathologies. In navigating the complexities of a dynamic milieu—replete with fluctuating nutrient levels, growth factors, cytokines, and varying oxygen concentrations—metabolic enzymes undergo intricate regulatory modulations to satisfy the metabolic imperatives of tumorigenic growth. These modulations manifest through alterations in enzymatic activity, expression levels, and subcellular compartmentalization.
FIGURE 4.
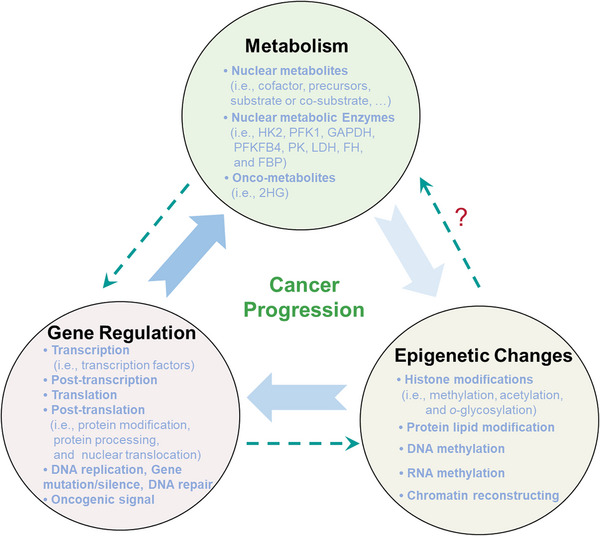
The positive feedback loop of metabolism–epigenetics–gene regulation. As delineated throughout this review, a positive feedback loop underpins the progression of malignant growth. Nuclear metabolites, which may be derived from extracellular sources, along with intranuclear metabolic enzymes, serve as cosubstrates, cofactors, or regulators in instigating epigenetic changes. These changes either promote or inhibit the expression of a myriad of genes, subsequently amplifying metabolic activity within the cell. Metabolism, in turn, exerts a direct influence on gene functionality via posttranslational modifications, protein processing, and nuclear translocation, among other mechanisms. Research exploring the direct ramifications of epigenetic changes on metabolic activity—specifically, how chromatin modifications may influence cellular metabolism via nongenomic routes—is relatively scant, underscoring the complexity of the interactions shaping malignant tumorigenesis.
As elucidated in this review, a salient feedback loop involving metabolism, epigenetics, and gene regulation serves as a linchpin in the progression of malignancies (Figure 4). Within the tumor cell milieu, nuclear metabolites and metabolic enzymes—primarily of cytoplasmic origin—act as cosubstrates, cofactors, or regulators that influence epigenetic modifications of DNA and histones. These epigenetic alterations, in turn, modulate the expression profiles of relevant genes, thereby facilitating or amplifying the neoplastic metabolic landscape. Within the nuclear compartment, these entities execute their regulatory functions through several intricate mechanisms. Firstly, they modulate carcinoma‐associated gene expression primarily by influencing DNA and histone modifications as well as gene transcription. Metabolic byproducts, such as acyl‐CoA, SAM, fumarate, amino acids, α‐KG, 2‐HG, and NAD+, serve as requisite precursors for these modifications while simultaneously affecting the activity of the enzymes that govern them.
Secondly, nuclear metabolic enzymes, exemplified by but not limited to PKM2, orchestrate gene transcription via distinct pathways, extending beyond their conventional metabolic roles. For instance, PKM2 is capable of phosphorylating histone H3 to directly modify chromatin architecture. Beyond these traditionally understood metabolic functions, metabolic enzymes can engage in direct interactions with epigenetic regulatory proteins (e.g., PFKFB4‐regulated SRC‐3, LDHA‐regulated DOT1L, FBP1‐regulated EZH2, GAPDH‐regulated Sirt1, and HDAC2. Furthermore, metabolic enzymes can enter into direct interactions with transcription factors or coregulators, as evidenced by PKM2‐phosphorylated STAT3 and PKM2‐regulated HIF.
To illuminate the intricate mechanisms governing the translocation of key metabolic enzymes or metabolites to the nuclear compartment, and to understand their complex functionalities therein, is to open new vistas in cancer therapeutics. Given the pleiotropic roles of these entities, targeted inhibition could offer a particularly efficacious approach in cancer treatment. These strategies encompass: (1) targeting nuclear metabolic enzymes; (2) modulating nuclear‐specific modifications; (3) interfering with the trafficking of metabolites or metabolic enzymes; and (4) targeting modulators of these metabolic enzymes. However, the ultimate efficacy of these novel therapeutic modalities is contingent upon an enhanced understanding of the specific metabolic requirements of tumor cells and the concomitant development of innovative antimetabolic strategies.
The metabolic underpinnings of cancer are likely more expansive than currently anticipated, potentially influencing every facet of neoplastic progression. Advancements in research focusing on nuclear metabolic signaling—still in its nascent stage—will undoubtedly yield groundbreaking insights and perhaps paradigm shifts in our understanding of tumorigenesis. The identification of novel metabolic enzymes within the nuclear compartment, accompanied by a comprehensive elucidation of their functional roles and regulatory mechanisms, will enrich our understanding of tumor metabolism and highlight hitherto undiscovered targets for future pharmacological interventions.
AUTHOR CONTRIBUTIONS
P. Y. and C. C. conceived the manuscript; Y. C., J. X., and X. L. wrote the initial draft; L. G., J. X., C. C., and Y. C. prepared the figure; C. C. revised the manuscript. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
The figure is created by the authors and some components in the figure are from the “Servier Medical Art” (https://smart.servier.com/). This research was funded by an NSFC grant (No. 82072886), Natural Science Foundation of ChongqingChina (No. cstc2020jcyj‐msxmX0344), CQMU Program for Youth Innovation in Future Medicine, and the Science and Technology Innovation Project (2020) of Yubei District, Chongqing to Dr. Ping Yi and Pelotonia Postdoc Fellowship and an OSU Department of Radiation‐Oncology Basic Research seed Grant to Dr. Chunming Cheng.
Chen Y, Xu J, Liu X, Guo L, Yi P, Cheng C. Potential therapies targeting nuclear metabolic regulation in cancer. MedComm. 2023;4:e421. 10.1002/mco2.421
Contributor Information
Ping Yi, Email: yiping@cqmu.edu.cn.
Chunming Cheng, Email: chunming.cheng@gmail.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERRENCES
- 1. Zhang Y, Sun Z, Jia J, et al. Overview of Histone Modification. Adv Exp Med Biol. 2021;1283:1‐16. [DOI] [PubMed] [Google Scholar]
- 2. Kinnaird A, Zhao S, Wellen K, Michelakis E. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16(11):694‐707. [DOI] [PubMed] [Google Scholar]
- 3. Zhao S, Allis C, Wang G. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21(7):413‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15(11):703‐708. [DOI] [PubMed] [Google Scholar]
- 5. Wu X, Zhang Y. TET‐mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517‐534. [DOI] [PubMed] [Google Scholar]
- 6. Ren R, Horton J, Zhang X, Blumenthal R, Cheng X. Detecting and interpreting DNA methylation marks. Curr Opin Struct Biol. 2018;53:88‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng C, Geng F, Li Z, et al. Ammonia stimulates SCAP/Insig dissociation and SREBP‐1 activation to promote lipogenesis and tumour growth. Nat Metab. 2022;4(5):575‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Wang Y, Ren Y, Zhang Q, Yi P, Cheng C. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol. 2022;86:542‐565. [DOI] [PubMed] [Google Scholar]
- 9. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of Amino Acids in Cancer. Front Cell Dev Biol. 2020;8:603837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu J, Liu X, Chen Y, Wang Y, Liu T, Yi P. RNA 5‐Methylcytosine Regulators Contribute to Metabolism Heterogeneity and Predict Prognosis in Ovarian Cancer. Front Cell Dev Biol. 2022;10:807786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng C, Ru P, Geng F, et al. Glucose‐Mediated N‐glycosylation of SCAP Is Essential for SREBP‐1 Activation and Tumor Growth. Cancer Cell. 2015;28(5):569‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng C, Guo J, Geng F, et al. Analysis of SCAP N‐glycosylation and Trafficking in Human Cells. J Vis Exp. 2016;(117):54709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang H, Lin Q, Ma L, et al. Fructose and fructose kinase in cancer and other pathologies. J Genet Genomics. 2021;48(7):531‐539. [DOI] [PubMed] [Google Scholar]
- 15. Xu D, Shao F, Bian X, Meng Y, Liang T, Lu Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell Metab. 2021;33(1):33‐50. [DOI] [PubMed] [Google Scholar]
- 16. Li X, Egervari G, Wang Y, Berger S, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. 2018;19(9):563‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mentch S, Mehrmohamadi M, Huang L, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One‐Carbon Metabolism. Cell Metab. 2015;22(5):861‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li T, Tan Y, Chen Y, et al. Methionine deficiency facilitates antitumour immunity by altering mA methylation of immune checkpoint transcripts. Gut. 2023;72(3):501‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramalingam H, Kashyap S, Cobo‐Stark P, et al. A methionine‐Mettl3‐N‐methyladenosine axis promotes polycystic kidney disease. Cell Metab. 2021;33(6):1234‐1247.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee J, Carrer A, Shah S, et al. Akt‐dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20(2):306‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li X, Kazgan N. Mammalian sirtuins and energy metabolism. Int J Biol Sci. 2011;7(5):575‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Y, Tsukada Y. The discovery of histone demethylases. Cold Spring Harb Perspect Biol. 2013;5(9):a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y, Yang D, Liu T, Chen J, Yu J, Yi P. N6‐methyladenosine‐mediated gene regulation and therapeutic implications. Trends Mol Med. 2023;29(6):454‐467. [DOI] [PubMed] [Google Scholar]
- 24. Iborra F, Jackson D, Cook P. Coupled transcription and translation within nuclei of mammalian cells. Science. 2001;293(5532):1139‐1142. [DOI] [PubMed] [Google Scholar]
- 25. Zou S, Kim B, Tian Y, et al. Enhanced nuclear translation is associated with proliferation and progression across multiple cancers. MedComm. 2023;4(3):e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bannister A, Kouzarides T. Reversing histone methylation. Nature. 2005;436(7054):1103‐1106. [DOI] [PubMed] [Google Scholar]
- 27. Li Y, Chen X, Lu C. The interplay between DNA and histone methylation: molecular mechanisms and disease implications. EMBO Rep. 2021;22(5):e51803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wesche J, Kühn S, Kessler B, Salton M, Wolf A. Protein arginine methylation: a prominent modification and its demethylation. Cell Mol Life Sci. 2017;74(18):3305‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Serine Locasale J., glycine and one‐carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye C, Sutter B, Wang Y, Kuang Z, Tu B. A Metabolic Function for Phospholipid and Histone Methylation. Mol Cell. 2017;66(2):180‐193.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei F, Locasale J. Methionine restriction and antitumor immunity. Trends Cancer. 2023;9(9):705‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. SUGIMURA T, BIRNBAUM S, WINITZ M, GREENSTEIN J. Quantitative nutritional studies with water‐soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Arch Biochem Biophys. 1959;81(2):448‐455. [DOI] [PubMed] [Google Scholar]
- 33. Gao X, Sanderson S, Dai Z, et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature. 2019;572(7769):397‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deb M, Kar S, Sengupta D, et al. Chromatin dynamics: H3K4 methylation and H3 variant replacement during development and in cancer. Cell Mol Life Sci. 2014;71(18):3439‐3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bannister A, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin B, McBurney K, Maltby V, Jensen K, Brind'Amour J, Howe L. Histone H3K4 and H3K36 methylation independently recruit the NuA3 histone acetyltransferase in Saccharomyces cerevisiae. Genetics. 2017;205(3):1113‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Shay J. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J Natl Cancer Inst. 2017;109(8):djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reina‐Campos M, Linares J, Duran A, et al. Increased Serine and One‐Carbon Pathway Metabolism by PKCλ/ι Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell. 2019;35(3):385‐400.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Losman J, Kaelin W. What a difference a hydroxyl makes: mutant IDH, (R)‐2‐hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu W, Yang H, Liu Y, et al. Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of α‐ketoglutarate‐dependent dioxygenases. Cancer Cell. 2011;19(1):17‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geeraerts S, Heylen E, De Keersmaecker K, Kampen K. The ins and outs of serine and glycine metabolism in cancer. Nat Metab. 2021;3(2):131‐141. [DOI] [PubMed] [Google Scholar]
- 42. Chowdhury R, Yeoh K, Tian Y, et al. The oncometabolite 2‐hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiang X, Liu Z, Zhang C, et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv Sci (Weinh). 2021;8(17):e2101230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37(11):1012‐1027. [DOI] [PubMed] [Google Scholar]
- 45. Ehrlich M, Gama‐Sosa M, Huang L, et al. Amount and distribution of 5‐methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10(8):2709‐2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bird A. CpG‐rich islands and the function of DNA methylation. Nature. 1986;321(6067):209‐213. [DOI] [PubMed] [Google Scholar]
- 47. Browne J, Yang R, Leir S, Eggener S, Harris A. Expression profiles of human epididymis epithelial cells reveal the functional diversity of caput, corpus and cauda regions. Mol Hum Reprod. 2016;22(2):69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He L, Huang H, Bradai M, et al. DNA methylation‐free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat Comm. 2022;13(1):1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81‐92. [DOI] [PubMed] [Google Scholar]
- 50. Maddocks O, Labuschagne C, Adams P, Vousden K. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell. 2016;61(2):210‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ross S, Bogdanovic O. TET enzymes, DNA demethylation and pluripotency. Biochem Soc Trans. 2019;47(3):875‐885. [DOI] [PubMed] [Google Scholar]
- 52. Wang D, Wu W, Callen E, et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science. 2022;378(6623):983‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kohli R, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thienpont B, Steinbacher J, Zhao H, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537(7618):63‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xiao M, Yang H, Xu W, et al. Inhibition of α‐KG‐dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li L, Yang L, Fan Z, et al. Hypoxia‐induced GBE1 expression promotes tumor progression through metabolic reprogramming in lung adenocarcinoma. Signal Transduct Target Ther. 2020;5(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dong C, Yuan T, Wu Y, et al. Loss of FBP1 by Snail‐mediated repression provides metabolic advantages in basal‐like breast cancer. Cancer Cell. 2013;23(3):316‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hansen B, Gupta R, Baldus L, et al. Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat Comm. 2019;10(1):1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. He W, Li Q, Li X. Acetyl‐CoA regulates lipid metabolism and histone acetylation modification in cancer. Biochim Biophys Acta Rev Cancer. 2023;1878(1):188837. [DOI] [PubMed] [Google Scholar]
- 60. Sivanand S, Viney I, Wellen K. Spatiotemporal Control of Acetyl‐CoA Metabolism in Chromatin Regulation. Trends Biochem Sci. 2018;43(1):61‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee J, Berry C, Kim K, et al. Acetyl‐CoA promotes glioblastoma cell adhesion and migration through Ca‐NFAT signaling. Genes Dev. 2018;32:497‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wellen K, Hatzivassiliou G, Sachdeva U, Bui T, Cross J, Thompson C. ATP‐citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9(1):40‐48. [DOI] [PubMed] [Google Scholar]
- 64. Sivanand S, Rhoades S, Jiang Q, et al. Nuclear Acetyl‐CoA Production by ACLY Promotes Homologous Recombination. Mol Cell. 2017;67(2):252‐265.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martinez Calejman C, Trefely S, Entwisle S, et al. mTORC2‐AKT signaling to ATP‐citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Comm. 2020;11(1):575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Berwick D, Hers I, Heesom K, Moule S, Tavare J. The identification of ATP‐citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277(37):33895‐33900. [DOI] [PubMed] [Google Scholar]
- 67. Li Z, Liu H, He J, et al. Acetyl‐CoA Synthetase 2: A Critical Linkage in Obesity‐Induced Tumorigenesis in Myeloma.Cell Metab. 2021;33(1):78‐93.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen J, Guccini I, Di Mitri D, et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nature Genet. 2018;50(2):219‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Matsuda S, Adachi J, Ihara M, et al. Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res. 2016;44(2):636‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller K, Pniewski K, Perry C, et al. Targeting ACSS2 with a Transition‐State Mimetic Inhibits Triple‐Negative Breast Cancer Growth. Cancer Res. 2021;81(5):1252‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Segré C, Chiocca S. Regulating the regulators: the post‐translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol. 2011;2011:690848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shimazu T, Hirschey M, Newman J, et al. Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cantó C, Menzies K, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus.Cell Metab. 2015;22(1):31‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Denzel M, Antebi A. Hexosamine pathway and (ER) protein quality control. Curr Opin Cell Biol. 2015;33:14‐18. [DOI] [PubMed] [Google Scholar]
- 75. Ferrer C, Sodi V, Reginato M. O‐GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol. 2016;428(16):3282‐3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang X, Qian K. Protein O‐GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18(7):452‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Makwana V, Ryan P, Patel B, Dukie S, Rudrawar S. Essential role of O‐GlcNAcylation in stabilization of oncogenic factors. Biochim Biophys Acta. 2019;1863(8):1302‐1317. [DOI] [PubMed] [Google Scholar]
- 78. Hanover J, Chen W, Bond M. O‐GlcNAc in cancer: An Oncometabolism‐fueled vicious cycle. J Bioenerg Biomembr. 2018;50(3):155‐173. [DOI] [PubMed] [Google Scholar]
- 79. Hart G, Slawson C, Ramirez‐Correa G, Lagerlof O. Cross talk between O‐GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ferrer C, Lynch T, Sodi V, et al. O‐GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF‐1 pathway. Mol Cell. 2014;54(5):820‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Singh J, Zhang K, Wu J, Yang X. O‐GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 2015;356:244‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Caldwell S, Jackson S, Shahriari K, et al. Nutrient sensor O‐GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29(19):2831‐2842. [DOI] [PubMed] [Google Scholar]
- 83. Hardivillé S, Hart G. Nutrient regulation of gene expression by O‐GlcNAcylation of chromatin. Curr Opin Chem Biol. 2016;33:88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chu C, Lo P, Yeh Y, et al. O‐GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci USA. 2014;111(4):1355‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xu Q, Yang C, Du Y, et al. AMPK regulates histone H2B O‐GlcNAcylation. Nucleic Acids Res. 2014;42(9):5594‐5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bullen J, Balsbaugh J, Chanda D, et al. Cross‐talk between two essential nutrient‐sensitive enzymes: O‐GlcNAc transferase (OGT) and AMP‐activated protein kinase (AMPK). J Biol Chem. 2014;289(15):10592‐10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sodi V, Bacigalupa Z, Ferrer C, et al. Nutrient sensor O‐GlcNAc transferase controls cancer lipid metabolism via SREBP‐1 regulation. Oncogene. 2018;37(7):924‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z, Lin H. Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem Rev. 2018;118(3):919‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Running M. The role of lipid post‐translational modification in plant developmental processes. Front Plant Sci. 2014;5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Resh M. Fatty acylation of proteins: The long and the short of it. Prog Lipid Res. 2016;63:120‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Suazo K, Park K, Distefano M. A Not‐So‐Ancient Grease History: Click Chemistry and Protein Lipid Modifications. Chem Rev. 2021;121(12):7178‐7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Venne A, Solari F, Faden F, Paretti T, Dissmeyer N, Zahedi R. An improved workflow for quantitative N‐terminal charge‐based fractional diagonal chromatography (ChaFRADIC) to study proteolytic events in Arabidopsis thaliana. Proteomics. 2015;15(14):2458‐2469. [DOI] [PubMed] [Google Scholar]
- 93. Pedram A, Razandi M, Sainson R, Kim J, Hughes C, Levin E. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282(31):22278‐22288. [DOI] [PubMed] [Google Scholar]
- 94. Noland C, Gierke S, Schnier P, et al. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure. 2016;24(1):179‐186. [DOI] [PubMed] [Google Scholar]
- 95. Linder M, Deschenes R. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8(1):74‐84. [DOI] [PubMed] [Google Scholar]
- 96. Claudinon J, Gonnord P, Beslard E, et al. Palmitoylation of interferon‐alpha (IFN‐alpha) receptor subunit IFNAR1 is required for the activation of Stat1 and Stat2 by IFN‐alpha. J Biol Chem. 2009;284(36):24328‐24340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Niu J, Sun Y, Chen B, et al. Fatty acids and cancer‐amplified ZDHHC19 promote STAT3 activation through S‐palmitoylation. Nature. 2019;573(7772):139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98. Toska E, Campbell H, Shandilya J, et al. Repression of transcription by WT1‐BASP1 requires the myristoylation of BASP1 and the PIP2‐dependent recruitment of histone deacetylase. Cell Rep. 2012;2(3):462‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen X, Niu W, Fan X, et al. Oct4A palmitoylation modulates tumorigenicity and stemness in human glioblastoma cells.Neuro Oncol. 2023;25(1):82‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Timms R, Zhang Z, Rhee D, Harper J, Koren I, Elledge S. A glycine‐specific N‐degron pathway mediates the quality control of protein ‐myristoylation. Science. 2019;365(6448):eaaw4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhu X, Nicholson Puthenveedu S, Shen Y, et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol Cell. 2019;74(1):45‐58.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Xu K, Wu Z, Groner A, et al. EZH2 oncogenic activity in castration‐resistant prostate cancer cells is Polycomb‐independent. Science. 2012;338(6113):1465‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kim E, Kim M, Woo D, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem‐like cells. Cancer Cell. 2013;23(6):839‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rothbart S, Baylin S. Epigenetic Therapy for Epithelioid Sarcoma. Cell. 2020;181(2):211. [DOI] [PubMed] [Google Scholar]
- 105. Zhang J, Zeng Y, Xing Y, et al. Myristoylation‐mediated phase separation of EZH2 compartmentalizes STAT3 to promote lung cancer growth. Cancer Lett. 2021;516:84‐98. [DOI] [PubMed] [Google Scholar]
- 106. Green L, Wagner K, Campbell H, Addison K, Roberts S. Dynamic interaction between WT1 and BASP1 in transcriptional regulation during differentiation. Nucleic Acids Res. 2009;37(2):431‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Squires J, Patel H, Nousch M, et al. Widespread occurrence of 5‐methylcytosine in human coding and non‐coding RNA. Nucleic Acids Res. 2012;40(11):5023‐5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen X, Li A, Sun B, et al. 5‐methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs.Nat Cell Biol. 2019;21(8):978‐990. [DOI] [PubMed] [Google Scholar]
- 109. Su J, Wu G, Ye Y, et al. NSUN2‐mediated RNA 5‐methylcytosine promotes esophageal squamous cell carcinoma progression via LIN28B‐dependent GRB2 mRNA stabilization. Oncogene. 2021;40(39):5814‐5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu Y, Zhao Y, Wu R, et al. mRNA m5C controls adipogenesis by promoting CDKN1A mRNA export and translation. RNA Biol. 2021;18:711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen Y, Zuo X, Wei Q, et al. Upregulation of LRRC8A by mC modification‐mediated mRNA stability suppresses apoptosis and facilitates tumorigenesis in cervical cancer. Int J Biol Sci. 2023;19(2):691‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N‐methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vujovic P, Stamenkovic S, Jasnic N, et al. Fasting induced cytoplasmic Fto expression in some neurons of rat hypothalamus. PLoS One. 2013;8(5):e63694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Green N, Galvan D, Badal S, et al. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene. 2019;38(34):6211‐6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li Q, Ni Y, Zhang L, et al. HIF‐1α‐induced expression of m6A reader YTHDF1 drives hypoxia‐induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang L, Song C, Wang N, et al. NADP modulates RNA mA methylation and adipogenesis via enhancing FTO activity. Nat Chem Biol. 2020;16(12):1394‐1402. [DOI] [PubMed] [Google Scholar]
- 117. Xiong J, He J, Zhu J, et al. Lactylation‐driven METTL3‐mediated RNA mA modification promotes immunosuppression of tumor‐infiltrating myeloid cells. Mol Cell. 2022;82(9):1660‐1677.e10. [DOI] [PubMed] [Google Scholar]
- 118. Yu J, Chai P, Xie M, et al. Histone lactylation drives oncogenesis by facilitating mA reader protein YTHDF2 expression in ocular melanoma. Genome Biol 2021;22(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu K, Xu P, Lv J, et al. Peritoneal high‐fat environment promotes peritoneal metastasis of gastric cancer cells through activation of NSUN2‐mediated ORAI2 m5C modification. Oncogene. 2023;42(24):1980‐1993. [DOI] [PubMed] [Google Scholar]
- 120. Yankova E, Blackaby W, Albertella M, et al. Small‐molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Peng S, Xiao W, Ju D, et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Transl Med. 2019;11(488):eaau7116. [DOI] [PubMed] [Google Scholar]
- 122. Su R, Dong L, Li C, et al. R‐2HG Exhibits Anti‐tumor Activity by Targeting FTO/mA/MYC/CEBPA Signaling. Cell. 2018;172:90‐105.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Huang Y, Su R, Sheng Y, et al. Small‐Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35(4):677‐691.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sun K, Du Y, Hou Y, et al. Saikosaponin D exhibits anti‐leukemic activity by targeting FTO/mA signaling. Theranostics. 2021;11(12):5831‐5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Su R, Dong L, Li Y, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell. 2020;38(1):79‐96.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Liu Y, Liang G, Xu H, et al. Tumors exploit FTO‐mediated regulation of glycolytic metabolism to evade immune surveillance.Cell Metab. 2021;33(6):1221‐1233.e11. [DOI] [PubMed] [Google Scholar]
- 127. Li N, Kang Y, Wang L, et al. ALKBH5 regulates anti‐PD‐1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci USA. 2020;117(33):20159‐20170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Weng H, Huang F, Yu Z, et al. The mA reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40(12):1566‐1582.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Uličná L, Paprčková D, Fáberová V, Hozák P. Phospholipids and inositol phosphates linked to the epigenome. Histochem Cell Biol. 2018;150(3):245‐253. [DOI] [PubMed] [Google Scholar]
- 130. Burton A, Azevedo C, Andreassi C, Riccio A, Saiardi A. Inositol pyrophosphates regulate JMJD2C‐dependent histone demethylation. Proc Natl Acad Sci USA. 2013;110(47):18970‐18975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yildirim S, Castano E, Sobol M, et al. Involvement of phosphatidylinositol 4,5‐bisphosphate in RNA polymerase I transcription. J Cell Sci. 2013;126:2730‐2739. [DOI] [PubMed] [Google Scholar]
- 132. Balaban C, Sztacho M, Antiga L, Miladinović A, Harata M, Hozák P. PIP2‐Effector Protein MPRIP Regulates RNA Polymerase II Condensation and Transcription. Biomolecules. 2023;13(3):426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hait N, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine‐1‐phosphate. Science. 2009;325(5945):1254‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gong L, Shen Y, Wang S, et al. Nuclear SPHK2/S1P induces oxidative stress and NLRP3 inflammasome activation via promoting p53 acetylation in lipopolysaccharide‐induced acute lung injury. Cell Death Discov. 2023;9(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Marcum R, Radhakrishnan I. Inositol phosphates and core subunits of the Sin3L/Rpd3L histone deacetylase (HDAC) complex up‐regulate deacetylase activity. J Biol Chem. 2019;294(38):13928‐13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Neary C, Pastorino J. Nucleocytoplasmic shuttling of hexokinase II in a cancer cell. Biochem Biophys Res Commun. 2010;394(4):1075‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kayikci Ö, Nielsen J. Glucose repression in Saccharomyces cerevisiae. FEMS Yeast Res. 2015;15(6):fov068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Moreno F, Ahuatzi D, Riera A, Palomino C, Herrero P. Glucose sensing through the Hxk2‐dependent signalling pathway. Biochem Soc Trans. 2005;33:265‐268. [DOI] [PubMed] [Google Scholar]
- 139. Sheikh T, Gupta P, Gowda P, Patrick S, Sen E. Hexokinase 2 and nuclear factor erythroid 2‐related factor 2 transcriptionally coactivate xanthine oxidoreductase expression in stressed glioma cells. J Biol Chem. 2018;293(13):4767‐4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Neary C, Pastorino J. Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J Cell Physiol. 2013;228(9):1943‐1948. [DOI] [PubMed] [Google Scholar]
- 141. Massa M, Gagliardino J, Francini F. Liver glucokinase: An overview on the regulatory mechanisms of its activity. IUBMB Life. 2011;63(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 142. de la Iglesia N, Veiga‐da‐Cunha M, Van Schaftingen E, Guinovart J, Ferrer J. Glucokinase regulatory protein is essential for the proper subcellular localisation of liver glucokinase. FEBS Lett. 1999;456(2):332‐338. [DOI] [PubMed] [Google Scholar]
- 143. Thomas G, Egan G, García‐Prat L, et al. The metabolic enzyme hexokinase 2 localizes to the nucleus in AML and normal haematopoietic stem and progenitor cells to maintain stemness.Nat Cell Biol. 2022;24(6):872‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fiorani M, De Sanctis R, Scarlatti F, et al. Dehydroascorbic acid irreversibly inhibits hexokinase activity. Mol Cell Biochem. 2000;209:145‐153. [DOI] [PubMed] [Google Scholar]
- 145. Heneberg P. Redox Regulation of Hexokinases. Antioxid Redox Signal. 2019;30(3):415‐442. [DOI] [PubMed] [Google Scholar]
- 146. Zheng M, Wu C, Yang K, et al. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol Res. 2021;164:105367. [DOI] [PubMed] [Google Scholar]
- 147. Yadav S, Pandey S, Kumar A, Kujur P, Singh R, Singh S. Antitumor and chemosensitizing action of 3‐bromopyruvate: Implication of deregulated metabolism. Chem Biol Interact. 2017;270:73‐89. [DOI] [PubMed] [Google Scholar]
- 148. Abdel‐Wahab A, Mahmoud W, Al‐Harizy R. Targeting glucose metabolism to suppress cancer progression: prospective of anti‐glycolytic cancer therapy. Pharmacol Res. 2019;150:104511. [DOI] [PubMed] [Google Scholar]
- 149. Cheng G, Zhang Q, Pan J, et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat Comm. 2019;10(1):2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Guo L, Shestov A, Worth A, et al. Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine. J Biol Chem. 2016;291(1):42‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lee J, Liu R, Li J, et al. EGFR‐Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol Cell. 2018;70(2):197‐210.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lee J, Liu R, Li J, et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Comm. 2017;8(1):949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Amara N, Cooper M, Voronkova M, et al. Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell. 2021;184(17):4480‐4494.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Gao X, Qin S, Wu Y, et al. Nuclear PFKP promotes CXCR4‐dependent infiltration by T cell acute lymphoblastic leukemia. J Clin Invest. 2021;131(16):e143119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Icard P, Simula L, Fournel L, et al. The strategic roles of four enzymes in the interconnection between metabolism and oncogene activation in non‐small cell lung cancer: Therapeutic implications. Drug Resist Updat. 2022;63:100852. [DOI] [PubMed] [Google Scholar]
- 156. Davidson S, Papagiannakopoulos T, Olenchock B, et al. Environment Impacts the Metabolic Dependencies of Ras‐Driven Non‐Small Cell Lung Cancer. Cell Metab. 2016;23(3):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Brimacombe K, Walsh M, Liu L, et al. Identification of ML251, a Potent Inhibitor of T. brucei and T. cruzi Phosphofructokinase. ACS Med Chem Lett. 2014;5(1):12‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hara M, Agrawal N, Kim S, et al. S‐nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding.Nat Cell Biol. 2005;7(7):665‐674. [DOI] [PubMed] [Google Scholar]
- 159. Chang C, Su H, Zhang D, et al. AMPK‐Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol Cell. 2015;60(6):930‐940. [DOI] [PubMed] [Google Scholar]
- 160. Zheng L, Roeder R, Luo Y. S phase activation of the histone H2B promoter by OCA‐S, a coactivator complex that contains GAPDH as a key component. Cell. 2003;114(2):255‐266. [DOI] [PubMed] [Google Scholar]
- 161. Sundararaj K, Wood R, Ponnusamy S, et al. Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde‐3‐phosphate dehydrogenase. J Biol Chem. 2004;279(7):6152‐6162. [DOI] [PubMed] [Google Scholar]
- 162. Peralta D, Bronowska A, Morgan B, et al. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat Chem Biol. 2015;11(2):156‐163. [DOI] [PubMed] [Google Scholar]
- 163. Schneider M, Knuesting J, Birkholz O, Heinisch J, Scheibe R. Cytosolic GAPDH as a redox‐dependent regulator of energy metabolism. BMC Plant Biol. 2018;18(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Hara M, Thomas B, Cascio M, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci USA. 2006;103(10):3887‐3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ventura M, Mateo F, Serratosa J, et al. Nuclear translocation of glyceraldehyde‐3‐phosphate dehydrogenase is regulated by acetylation. Int J Biochem Cell Biol. 2010;42(10):1672‐1680. [DOI] [PubMed] [Google Scholar]
- 167. Liberti M, Dai Z, Wardell S, et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product.Cell Metab. 2017;26(4):648‐659.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li W, Liao L, Song N, et al. Natural product 1,2,3,4,6‐penta‐O‐galloyl‐β‐D‐glucopyranose is a reversible inhibitor of glyceraldehyde 3‐phosphate dehydrogenase. Acta Pharmacol Sin. 2022;43(2):470‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kotowski K, Rosik J, Machaj F, et al. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers. 2021;13(4):909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Dasgupta S, Rajapakshe K, Zhu B, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC‐3 to drive breast cancer. Nature. 2018;556(7700):249‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chesney J, Clark J, Lanceta L, Trent J, Telang S. Targeting the sugar metabolism of tumors with a first‐in‐class 6‐phosphofructo‐2‐kinase (PFKFB4) inhibitor. Oncotarget. 2015;6(20):18001‐18011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Witney T, James M, Shen B, et al. PET imaging of tumor glycolysis downstream of hexokinase through noninvasive measurement of pyruvate kinase M2. Sci Transl Med. 2015;7(310):310ra169. [DOI] [PubMed] [Google Scholar]
- 173. Noguchi T, Inoue H, Tanaka T. The M1‐ and M2‐type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261(29):13807‐13812. [PubMed] [Google Scholar]
- 174. Gao X, Wang H, Yang J, Liu X, Liu Z. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45(5):598‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol.. 2011;43(7):969‐980. [DOI] [PubMed] [Google Scholar]
- 176. Yang W, Xia Y, Hawke D, et al. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell. 2014;158(5):1210. [DOI] [PubMed] [Google Scholar]
- 177. Damasceno L, Prado D, Veras F, et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine‐tuning STAT3 activation. J Exp Med. 2020;217(10):e20190613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Dai H, Zeng W, Luo H. C‐MET‐dependent signal transduction mediates retinoblastoma growth by regulating PKM2 nuclear translocation. Cell Biochem Funct. 2020;38(2):204‐212. [DOI] [PubMed] [Google Scholar]
- 179. Jiang Y, Li X, Yang W, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014;53(1):75‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3‐stimulated coactivator for hypoxia‐inducible factor 1. Cell. 2011;145(5):732‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Wang H, Hsieh Y, Cheng W, et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF‐1α‐mediated glucose metabolism. Proc Natl Acad Sci USA. 2014;111(1):279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Shanmugasundaram K, Nayak B, Friedrichs W, Kaushik D, Rodriguez R, Block K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat Comm. 2017;8(1):997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Anastasiou D, Poulogiannis G, Asara J, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Boulos J, Rahama M, Hegazy M, Efferth T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019;459:248‐267. [DOI] [PubMed] [Google Scholar]
- 185. Li W, Zhang C, Ren A, et al. Shikonin Suppresses Skin Carcinogenesis via Inhibiting Cell Proliferation. PLoS One. 2015;10(5):e0126459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase‐M2. Oncogene. 2011;30(42):4297‐4306. [DOI] [PubMed] [Google Scholar]
- 187. You L, Zhu H, Wang C, et al. Scutellarin inhibits Hela cell growth and glycolysis by inhibiting the activity of pyruvate kinase M2. Bioorg Med Chem Lett. 2017;27(24):5404‐5408. [DOI] [PubMed] [Google Scholar]
- 188. Ferriero R, Nusco E, De Cegli R, Carissimo A, Manco G, Brunetti‐Pierri N. Pyruvate dehydrogenase complex and lactate dehydrogenase are targets for therapy of acute liver failure. J Hepatol. 2018;69(2):325‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Liu Y, Guo J, Liu Y, et al. Nuclear lactate dehydrogenase A senses ROS to produce α‐hydroxybutyrate for HPV‐induced cervical tumor growth. Nat Comm. 2018;9(1):4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Manerba M, Vettraino M, Fiume L, et al. Galloflavin (CAS 568‐80‐9): a novel inhibitor of lactate dehydrogenase. ChemMedChem. 2012;7(2):311‐317. [DOI] [PubMed] [Google Scholar]
- 191. Farabegoli F, Vettraino M, Manerba M, Fiume L, Roberti M, Di Stefano G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur J Pharm Sci. 2012;47(4):729‐738. [DOI] [PubMed] [Google Scholar]
- 192. Koukourakis M, Tsolou A, Pouliliou S, et al. Blocking LDHA glycolytic pathway sensitizes glioblastoma cells to radiation and temozolomide. Biochem Biophys Res Commun. 2017;491(4):932‐938. [DOI] [PubMed] [Google Scholar]
- 193. Le A, Cooper C, Gouw A, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107(5):2037‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wu H, Wang Y, Ying M, Jin C, Li J, Hu X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct Target Ther. 2021;6(1):242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Yogev O, Yogev O, Singer E, et al. Fumarase: a mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol 2010;8(3):e1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Jiang Y, Qian X, Shen J, et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol. 2015;17(9):1158‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Wang T, Yu Q, Li J, et al. O‐GlcNAcylation of fumarase maintains tumour growth under glucose deficiency.Nat Cell Biol. 2017;19(7):833‐843. [DOI] [PubMed] [Google Scholar]
- 198. Takeuchi T, Schumacker P, Kozmin S. Identification of fumarate hydratase inhibitors with nutrient‐dependent cytotoxicity. J Am Chem Soc. 2015;137(2):564‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Huangyang P, Li F, Lee P, et al. Fructose‐1,6‐Bisphosphatase 2 Inhibits Sarcoma Progression by Restraining Mitochondrial Biogenesis.Cell Metab. 2020;31(5):1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Li B, Qiu B, Lee D, et al. Fructose‐1,6‐bisphosphatase opposes renal carcinoma progression. Nature. 2014;513(7517):251‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. von Geldern T, Lai C, Gum R, et al. Benzoxazole benzenesulfonamides are novel allosteric inhibitors of fructose‐1,6‐bisphosphatase with a distinct binding mode. Bioorg Med Chem Lett. 2006;16(7):1811‐1815. [DOI] [PubMed] [Google Scholar]
- 202. Xu Y, Huang Y, Song R, et al. Development of disulfide‐derived fructose‐1,6‐bisphosphatase (FBPase) covalent inhibitors for the treatment of type 2 diabetes. Eur J Med Chem. 2020;203:112500. [DOI] [PubMed] [Google Scholar]
- 203. Cheng C, Zhang S, Guan D, Wang Y, Jyotsana N. Editorial: Tumor metabolism: From molecular mechanisms to clinical application. Front Cell Dev Biol. 2023;11:1158467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Stine Z, Schug Z, Salvino J, Dang C. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2):141‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Lu C, Lin C, Chang Y, et al. Antimetabolite pemetrexed primes a favorable tumor microenvironment for immune checkpoint blockade therapy. J Immunother Cancer. 2020;8(2):e001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Yang C, Zhang J, Liao M, et al. Folate‐mediated one‐carbon metabolism: a targeting strategy in cancer therapy. Drug Discov Today. 2021;26(3):817‐825. [DOI] [PubMed] [Google Scholar]
- 207. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5‐fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol Ther. 2020;206:107447. [DOI] [PubMed] [Google Scholar]
- 208. Pandit B, Royzen M. Recent Development of Prodrugs of Gemcitabine. Genes. 2022;13(3):466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Gore S, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361‐6369. [DOI] [PubMed] [Google Scholar]
- 210. Adès L, Sebert M, Fenaux P. Guadecitabine in myelodysplastic syndromes: promising but there is still progress to be made. Lancet Haematol. 2019;6(6):e290‐e291. [DOI] [PubMed] [Google Scholar]
- 211. Tachihara M, Dokuni R, Okuno K, et al. Phase II study of adjuvant chemotherapy with pemetrexed and cisplatin with a short hydration method for completely resected nonsquamous non‐small cell lung cancer. Thorac Cancer. 2020;11(9):2536‐2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zhai W, Duan F, Li D, et al. Risk stratification and adjuvant chemotherapy after radical resection based on the clinical risk scores of patients with stage IB‐IIA non‐small cell lung cancer. Eur J Surg Oncol. 2022;48(4):752‐760. [DOI] [PubMed] [Google Scholar]
- 213. Zhu X, Cao Y, Liu W, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open‐label, randomised, controlled, phase 2 trial. Lancet Oncol. 2021;22(8):1093‐1102. [DOI] [PubMed] [Google Scholar]
- 214. Coen J, Zhang P, Saylor P, et al. Bladder Preservation With Twice‐a‐Day Radiation Plus Fluorouracil/Cisplatin or Once Daily Radiation Plus Gemcitabine for Muscle‐Invasive Bladder Cancer: NRG/RTOG 0712—A Randomized Phase II Trial. J Clin Oncol. 2019;37(1):44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Coleman J, Yip W, Wong N, et al. Multicenter Phase II Clinical Trial of Gemcitabine and Cisplatin as Neoadjuvant Chemotherapy for Patients With High‐Grade Upper Tract Urothelial Carcinoma. J Clin Oncol 2023;41(8):1618‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Garassino M, Gadgeel S, Speranza G, et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non‐Small‐Cell Lung Cancer: 5‐Year Outcomes From the Phase 3 KEYNOTE‐189 Study. J Clin Oncol. 2023;41(11):1992‐1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Qiu J, Feng M, Yang G, et al. mTOR inhibitor, gemcitabine and PD‐L1 antibody blockade combination therapy suppresses pancreatic cancer progression via metabolic reprogramming and immune microenvironment remodeling in Trp53LSL‐KrasPdx‐1‐Cre murine models. Cancer Lett. 2023;554:216020. [DOI] [PubMed] [Google Scholar]
- 218. Lam T, Tsang K, Choi H, et al. Combination atezolizumab, bevacizumab, pemetrexed and carboplatin for metastatic EGFR mutated NSCLC after TKI failure. Lung Cancer. 2021;159:18‐26. [DOI] [PubMed] [Google Scholar]
- 219. Tang L, Wei F, Wu Y, et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Zhao H, Li Y. Cancer metabolism and intervention therapy. Mol Biomed. 2021;2(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361‐6369. [DOI] [PubMed] [Google Scholar]
- 222. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794‐1803. [DOI] [PubMed] [Google Scholar]
- 223. Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol. 2005;68(4):917‐932. [DOI] [PubMed] [Google Scholar]
- 224. Jensen MM, Erichsen KD, Johnbeck CB, et al. [18F]FDG and [18F]FLT positron emission tomography imaging following treatment with belinostat in human ovary cancer xenografts in mice. BMC Cancer. 2013;13:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Laino AS, Betts BC, Veerapathran A, et al. HDAC6 selective inhibition of melanoma patient T‐cells augments anti‐tumor characteristics. J Immunother Cancer. 2019;7(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Truong AS, Zhou M, Krishnan B, et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J Clin Invest. 2021;131(16):e138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Heers H, Stanislaw J, Harrelson J, Lee MW. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur J Pharmacol. 2018;835:61‐74. [DOI] [PubMed] [Google Scholar]
- 228. Lv X, Qiu J, Hao T, Zhang H, Jiang H, Tan Y. HDAC inhibitor Trichostatin A suppresses adipogenesis in 3T3‐L1 preadipocytes. Aging. 2021;13(13):17489‐17498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Yu Y, Liu B, Chen S, et al. Trichostatin A inhibits dendritic cell maturation through down‐regulating NF‐κ B (p65) pathway. Mol Biol Rep. 2022;49(4):2619‐2627. [DOI] [PubMed] [Google Scholar]
- 230. Hubeek I, Comijn EM, Van der Wilt CL, et al. CI‐994 (N‐acetyl‐dinaline) in combination with conventional anti‐cancer agents is effective against acute myeloid leukemia in vitro and in vivo. Oncol Rep. 2008;19(6):1517‐1523. [DOI] [PubMed] [Google Scholar]
- 231. Laubach JP, Moreau P, San‐Miguel JF, Richardson PG. Panobinostat for the Treatment of Multiple Myeloma. Clin Cancer Res. 2015;21(21):4767‐4773. [DOI] [PubMed] [Google Scholar]
- 232. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769‐784. [DOI] [PubMed] [Google Scholar]
- 233. Lu X, Ning Z, Li Z, Cao H, Wang X. Development of chidamide for peripheral T‐cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5(3):185‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open‐label, single‐arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21(11):1433‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Stein EM, Garcia‐Manero G, Rizzieri DA, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131(24):2661‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Huang S, Wang Z, Zhou J, et al. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid‐Derived Suppressor Cells. Cancer Res. 2019;79(8):2009‐2020. [DOI] [PubMed] [Google Scholar]
- 237. Yan L, Ding B, Liu H, et al. Inhibition of SMYD2 suppresses tumor progression by down‐regulating microRNA‐125b and attenuates multi‐drug resistance in renal cell carcinoma. Theranostics. 2019;9(26):8377‐8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Gao J, Liu R, Feng D, et al. Snail/PRMT5/NuRD complex contributes to DNA hypermethylation in cervical cancer by TET1 inhibition. Cell Death Differ. 2021;28(9):2818‐2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Eram M, Shen Y, Szewczyk M, et al. A Potent, Selective, and Cell‐Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem Biol. 2016;11(3):772‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Bauer TM, Besse B, Martinez‐Marti A, et al. Phase I, Open‐Label, Dose‐Escalation Study of the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of GSK2879552 in Relapsed/Refractory SCLC. J Thorac Oncol. 2019;14(10):1828‐1838. [DOI] [PubMed] [Google Scholar]
- 241. Noce B, Di Bello E, Fioravanti R, Mai A. LSD1 inhibitors for cancer treatment: Focus on multi‐target agents and compounds in clinical trials. Front Pharmacol. 2023;14:1120911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Liao W, Liu J, Liu B, et al. JIB‑04 induces cell apoptosis via activation of the p53/Bcl‑2/caspase pathway in MHCC97H and HepG2 cells. Oncol Rep. 2018;40(6):3812‐3820. [DOI] [PubMed] [Google Scholar]
- 243. Dalpatraj N, Naik A, Thakur N. GSK‐J4: An H3K27 histone demethylase inhibitor, as a potential anti‐cancer agent. Int J Cancer. 2023;153(6):1130‐1138. [DOI] [PubMed] [Google Scholar]
- 244. Sang N, Zhong X, Gou K, et al. Pharmacological inhibition of LSD1 suppresses growth of hepatocellular carcinoma by inducing GADD45B. MedComm. 2023;4(3):e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.