

Key Points
-
•
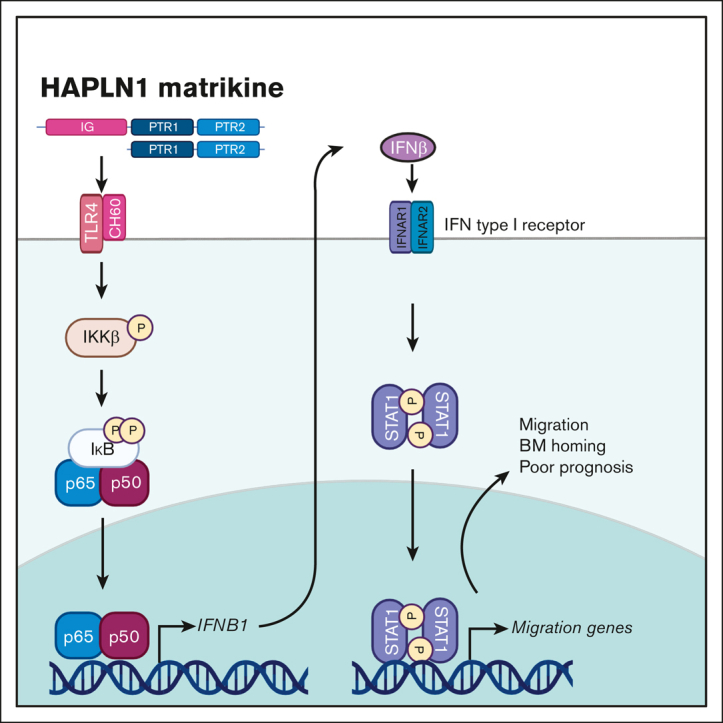
HAPLN1 matrikine induces adhesion, migration, and BM homing of myeloma cells by STAT1 activation via NF-κB–induced IFN-β.
-
•
Higher HAPLN1 levels in BM samples correlate with shorter progression-free survival of patients with newly diagnosed MM.
Visual Abstract
Abstract
The bone marrow (BM) microenvironment is critical for dissemination, growth, and survival of multiple myeloma (MM) cells. Homing of myeloma cells to the BM niche is a crucial step in MM dissemination, but the mechanisms involved are incompletely understood. In particular, any role of matrikines, neofunctional peptides derived from extracellular matrix proteins, remains unknown. Here, we report that a matrikine derived from hyaluronan and proteoglycan link protein 1 (HAPLN1) induces MM cell adhesion to the BM stromal components, such as fibronectin, endothelial cells, and stromal cells and, furthermore, induces their chemotactic and chemokinetic migration. In a mouse xenograft model, we show that MM cells preferentially home to HAPLN1 matrikine–conditioned BM. The transcription factor STAT1 is activated by HAPLN1 matrikine and is necessary to induce MM cell adhesion, migration, migration-related genes, and BM homing. STAT1 activation is mediated by interferon beta (IFN-β), which is induced by NF-κB after stimulation by HAPLN1 matrikine. Finally, we also provide evidence that higher levels of HAPLN1 in BM samples correlate with poorer progression-free survival of patients with newly diagnosed MM. These data reveal that a matrikine present in the BM microenvironment acts as a chemoattractant, plays an important role in BM homing of MM cells via NF-κB–IFN-β–STAT1 signaling, and may help identify patients with poor outcomes. This study also provides a mechanistic rationale for targeting HAPLN1 matrikine in MM therapy.
Introduction
Multiple myeloma (MM) is a plasma cell malignancy frequently detected in multiple bone marrow (BM) sites where cancer cells are dependent on the BM microenvironment for their survival and proliferation.1 As the disease progresses, MM cells further disseminate to distant BM sites via a BM homing process involving adhesion of circulating MM cells to vascular endothelium, transendothelial migration, and localization into the BM niche.2,3 BM homing of MM cells is dependent on chemoattractant gradients and the best studied is stromal cell–derived factor-1 (SDF-1)/CXCL12.4 SDF-1 is mainly secreted by BM stromal cells (BMSCs) and enriched in the BM, and its cognate receptor CXCR4 is highly expressed on MM cells. Nonetheless, blocking the SDF-1/CXCR4 axis is not sufficient to abrogate MM BM homing, indicating that SDF-1 is critical but not the sole BM homing factor.4,5
Cancer cell behaviors may be modulated by soluble factors not only secreted by other cell types but also produced by partial proteolysis of extracellular matrix (ECM) proteins, known as matrikines.6 Matrikines can control migration of different normal or malignant cell types, including neutrophils via collagen matrikine,7 monocytes and melanoma cells via elastin matrikine,8,9 and various other solid cancer cell types via laminin matrikine.10 In addition, in the context of MM, to date, 1 matrikine, versikine derived from versican, has been studied for the effect of tumor-infiltrating immune cells and immunosurveillance. Versikine acts as a damage-associated molecular pattern, promotes antitumor immunogenicity, and antagonizes the tolerogenic actions of intact versican.11 Finally, proteolytic processing of hyaluronan and proteoglycan link protein 1 (HAPLN1) by matrix metalloproteinase 2, both secreted by BMSCs, produces HAPLN1 matrikine, which can cause NF-κB signaling and resistance of MM cells to several classes of anti-MM therapeutic agents.12, 13, 14 In BM samples from patients with MM, variable levels of HAPLN1 matrikine forms could be detected.12 Despite known roles in the regulation of both normal and cancer cell behaviors, a role of any matrikine in MM cell BM homing and ability in the prediction of poor patient outcomes remain unknown.
In this study, we investigated the potential role of HAPLN1 matrikine in MM cell adhesion and migration and BM homing. HAPLN1 matrikine was found to function as a chemotactic factor that could induce MM cell migration and adhesion, and BM homing. In addition, further analyses have identified the transcription factor STAT1 as a critical mediator of HAPLN1 matrikine–induced MM cell migration and BM homing. We also provide evidence that STAT1 activation is mediated by interferon beta (IFN-β), which is induced by NF-κB after the stimulation of MM cells by HAPLN1 matrikine. Finally, we measured HAPLN1 levels in BM samples of patients with newly diagnosed MM (NDMM) and found their high levels of HAPLN1 to correlate with poor patient outcomes. This study reveals HAPLN1 matrikine to be a novel regulator of BM homing via STAT1 signaling in MM disease progression and its potential role as a marker for poor prognosis for certain patients with MM.
Methods
Primary CD138+ myeloma cell isolation and BM plasma collection
BM aspirates from patients with MM were obtained in accordance with the University of Wisconsin-Madison Institutional Review Board requirements (HO07403). CD138+ myeloma cells were positively sorted with CD138+ magnetic MACS beads (Miltenyi Biotec, Bergisch Gladbach, Germany) as described previously.12,17 The BM plasma fraction was collected by centrifuging the BM aspirate and stored at −80°C in multiple microcentrifuge tubes until analysis.
In vivo BM homing assay
NOD/SCID interleukin-2Rγ (IL-2Rγ)–null (NSG) mice were injected with HS-5 cells (105 cells per 10 μL phosphate-buffered saline per tibia) by intratibial injection at 6 to 8 weeks of age. Two weeks after the intratibial injection, a MM homing assay was conducted as described previously.18,19 In brief, MM.1S cells labeled with PKH26 fluorescent cell linkers (Sigma-Aldrich, St Louis, MO) were inoculated by intracardiac (IC) injection (3 × 106 cells per 100 μL phosphate-buffered saline per mouse). To facilitate the MM engraftment, 1 day before IC injection, mice were irradiated with a sublethal dose (3 Gy). Three days after MM engraftment, hind leg bones were harvested, and BM samples were collected after centrifugation.20 BM samples were incubated with red blood cell lysis buffer containing ammonium chloride and potassium bicarbonate. Subsequently, mononuclear cells were isolated by density gradient centrifugation using lymphocyte separation medium (Corning, Tewksbury, MA), following the manufacturer’s instructions, and fixed with 4% paraformaldehyde. Mouse femur and tibia BM samples were analyzed by flow cytometry. The injected PKH26-labeled MM cells were cultured for 3 days and harvested at the same time when the mice were euthanized. The cultured PKH26+ MM cells were analyzed together as a positive control to gate for PKH26+ cells.
HAPLN1 AlphaLISA on patient BM plasma fractions
BM aspirates from patients with MM were obtained in accordance with the University of Wisconsin-Madison Institutional Review Board requirements (HO07403). Cytogenetic risk was assessed using fluorescence in situ hybridization. The plasma layer of patient BM aspirates, which were previously kept frozen, was subjected to an amplified luminescent proximity homogenous assay–linked immunosorbent assay (AlphaLISA).
Additional detailed materials and methods can be found in the supplemental Methods.
Results
HAPLN1 matrikine stimulates MM cell adhesion and migration
HAPLN1 is composed of an N-terminal signal peptide, followed by 3 structural domains, immunoglobulin-like and proteoglycan tandem repeat 1 and 2 (PTR1 and PTR2).22 We previously reported that along with a fragment containing all 3 domains, smaller fragments of HAPLN1 containing PTR1 and PTR2 domains are detectable in patient BM samples,12 and similar fragments are produced by matrix metalloproteinase 2 proteolysis of recombinant full-length HAPLN1.13 Although both recombinant PTR1 and PTR2 domains can induce NF-κB signaling separately or together, the PTR1 domain was found to possess the stronger signaling activity.12 Thus, we used recombinant PTR1 tagged with maltose-binding protein (MBP-PTR1)23 and MBP alone as a negative control to assess whether HAPLN1 matrikine could modulate MM cell adhesion and migration. We first assessed MM cell adhesion to fibronectin, prevalent in the BM,24 using fluorescently labeled RPMI8226 and MM.1S human MM cell lines. Pretreatment with MBP-PTR1 induced an increase in adhesion of MM cells after 2 hours of incubation (supplemental Figure 1A-B). MBP-PTR1 also increased MM cell adhesion to human microvascular endothelial cell line, HMEC-1 (supplemental Figure 1C). Next, we assessed whether MBP-PTR1 could function as a chemoattractant for MM cells. Fluorescently labeled MM cells were placed in the upper transwell chamber in serum-free media, and MBP-PTR1, MBP, or SDF1, a well-established chemoattractant for MM cells, was added in the bottom chamber; cells that migrated through the pore and adhered to the bottom of the insert were imaged and counted 16 hours later. MBP-PTR1 induced a threefold and 1.5-fold increase in the migration of RPMI8226 and MM.1S cell lines, respectively, whose magnitudes were comparable with those induced by SDF-1 (Figure 1A-B). MBP-PTR1 could also induce transwell migration of primary cells of patients with MM (Figure 1C). Thus, HAPLN1 matrikine is capable of stimulating MM cell adhesion and migration.
Figure 1.

HAPLN1-PTR1 induces chemotactic and chemokinetic migration in MM cells. (A) Microscopic images of RPMI8226 cells on transwell membranes that had migrated from the upper chamber to the lower chamber containing MBP or MBP-PTR1 at 100 nM or SDF-1 at 30 nM after 16 hours. (B) Graphs depicting percentage migration of RPMI8226 and MM.1S MM cells treated as in panel A with MBP control being set as 100%. (C) Graphs depicting percentage migration of CD138+ primary MM cells isolated from 6 patients in response to MBP, MBP-PTR1, or SDF-1 with MBP control being set as 100%. (D) Migration of RPMI8226 cells in response to indicated MBP-PTR1 concentrations in the upper and lower chambers were quantified with the control (0 nM) set at 100%. (E) Individual cell track trajectories of 3 groups in time-lapse μ-slide migration assay using RPMI8226 cells recorded for 16 hours are shown: positive gradient (−/+; 30 nM MBP/MBP-PTR1), negative control (−/−; 30 nM MBP/MBP), and no gradient (+/+; 30 nM MBP-PTR1/MBP-PTR1). The y-axis is parallel to the chemotactic gradient in which cell trajectory going up along the y-axis is the migration toward the MBP-PTR1 in the positive-gradient group. (F) Graphs showing the comparison of averaged FMIII and FMI⊥ for each group from panel E. (G) Graph showing the comparison of the cell speed from panel E. All experiments were independently repeated 3 times for panel D and 4 times for panels B,E-G. Data are expressed as means ± standard error of the mean (SEM). ∗P < .05; ∗∗P < .01; ns, not significant.
The 2 most common cellular migratory behaviors are chemotaxis (a directional motility toward a chemical gradient) and chemokinesis (an increase in the motility speed without any directionality).25,26 Dual increases in chemotactic and chemokinetic motility are associated with metastasis of certain cancer types.27,28 SDF-1 is known to induce MM cell chemotaxis but not chemokinesis.29 To determine whether HAPLN1 matrikine could induce chemotactic and/or chemokinetic migrations in MM cells, the Zigmond-Hirsch migration assay30 was performed using the linear migration dose range of MBP-PTR1 (0-30 nM; supplemental Figure 1D). The largest positive gradient (0 nM in upper and 30 nM in lower chambers) caused the greatest MM cell migration (Figure 1D, bottom-left), demonstrating chemotactic migration toward MBP-PTR1. Increasing concentrations at both sides of the chamber without any gradient (diagonal grid) caused correspondingly increased MM cell migration, thus indicative of chemokinetic migration.
To further define the promigratory activities of HAPLN1 matrikine on MM cells, a time-lapse μ-slide migration assay15,16 was implemented for 16 hours, with each recording at 10-minute intervals. This assay tracks each MM cell to enable computation of migration parameters, such as directionality, traveled distance, and speed.16 We performed this assay using 3 groups, a positive-gradient group (−/+; MBP/MBP-PTR1), a no-gradient group (+/+; MBP-PTR1/MBP-PTR1), and a negative control group (−/−; MBP/MBP), and the ImageJ plugin was used to track cells in the time-lapse sequences (Figure 1E). The directionality of migration was assessed from the trajectory data using the forward migration index (FMI), which was calculated by averaging the end point of migrated cells divided by the accumulated distance, representing the efficiency of migration in a direction parallel (FMIII) or perpendicular (FMI⊥) to the chemotactic gradient.15,16 In the positive-gradient group, the FMIII was significantly greater, thus, more chemotactic than the other 2 groups (Figure 1F). Moreover, the FMIII of the positive-gradient group was significantly higher than its FMI⊥ counterpart, indicating the induction of directional migration toward MBP-PTR1. Finally, the positive-gradient group displayed the highest speed, and the cell speed in no-gradient group was higher than that in the negative control group (Figure 1G), indicative of chemokinetic migration. Taken together, both the Zigmond-Hirsch and time-lapse μ-slide migration assays demonstrated that HAPLN1 matrikine has dual chemotactic and chemokinetic effects on MM cells, thus distinguishing itself from the well-established, chemotactic-only factor SDF-1.
HAPLN1 matrikine promotes BM homing in vivo
Next, we tested the ability of HAPLN1 matrikine to induce MM cell BM homing in vivo. First, we established BMSCs capable of secreting biologically active HAPLN1 matrikine, which could be injected into mouse tibia to locally produce the matrikine. To this end, we initially attempted to generate a HS-5 human BMSC line secreting only the PTR1 domain of HAPLN1 but were unsuccessful. Instead, we were able to generate a HS-5 cell line (HS-5/H1) secreting HAPLN1 fragment (32-354 amino acids; supplemental Figure 2A), which represents a product of MMP cleavage31 and is capable of activating NF-κB, whereas the full-length HAPLN1 lacked such activity.12 An enzyme-linked immunosorbent assay of conditioned media demonstrated that HS-5/H1 cells secreted 69.5 nM HAPLN1 fragment (32-354 amino acids) at steady state, which was in the range of MBP-PTR1 concentrations used in the migration assays (Figure 1A-C; supplemental Figure 1D). Moreover, when MM cells were incubated above a confluent monolayer of HS-5/H1 or empty vector control (HS-5/EV) cells for 2 hours, significantly increased adhesion was observed for HS-5/H1 cells relative to that for HS-5/EV cells (supplemental Figure 2B). Similarly, transwell migration assays demonstrated that MM cells migrated significantly more toward HS-5/H1 in the bottom chamber than toward HS-5/EV (supplemental Figure 2C). To verify whether the overexpressed HAPLN1 fragment and not the endogenous HAPLN1 acts as a migratory factor, we generated HAPLN1 knockdown HS-5 cells. Endogenous HAPLN1 was not detected in the conditioned media (supplemental Figure 2D). The migratory potential induced by the HAPLN1 fragment was lost when HAPLN1 was knocked down in HS-5/H1 cells (supplemental Figure 2E). Thus, exogenous HAPLN1 matrikine produced by HS-5/H1 cells was responsible for the stimulation of MM cell migration.
We assessed whether HS-5 cells would remain at the injected mouse BM site without forming a tumor. HS-5 cells stably expressing the luciferase gene were injected into NSG mouse tibiae and monitored for their presence by bioluminescence imaging. The human HS-5 cells were maintained in the injected sites for at least 2 weeks without any obvious growth or migration to other BM sites (supplemental Figure 2F). To test the impact of HAPLN1 matrikine secreted by HS-5/H1 in vivo, both tibiae in individual NSG mice were injected with either HS-5/H1 or HS-5/EV cells (supplemental Figure 2G). Two weeks later a sublethal dose of radiation (3 Gy) was administered to facilitate MM cell BM engraftment and PKH26-labeled MM.1S cells were then injected via the IC route a day later to induce MM cell homing to the BM within 48 to 72 hours.18,19 The BM of 2 tibiae was collected 72 hours after the MM engraftment and subjected to flow cytometry to quantify percentages of the PKH26-labeled MM cells among BM mononuclear cells (Figure 2A). Because individual mice had different engraftment efficiencies, the MM cells that homed to injection-naive femur were used as an internal control to calculate the tibia-to-femur ratio for each mouse. Significantly, the tibia-to-femur ratio of MM cells was ∼2.5-fold greater for HS-5/H1 than for HS-5/EV–injected mice (Figure 2B).
Figure 2.

HAPLN1 matrikine induces MM cell BM homing in vivo. (A) Representative flow plots and gating strategy of PKH26+ MM.1S cells after cell culture (left) or from BM of mouse. (B) The percentage of PKH26+ MM cells in the tibia and femur of mice was obtained by flow cytometry, as in panel A, and the tibia/femur ratio of PKH26+ MM percentage was plotted. (C) Immunohistochemistry (IHC) images of tibia sections stained for MM cells (CD138, green color), HS-5 cells (HLA-I, red color) or nuclei (4′,6-diamidino-2-phenylindole [DAPI], blue color). The white dotted line indicates the outline of the cortical bone. The white arrowhead indicates CD138+ MM cells. (D) MM or HS-5 cells were counted in the imaged sections; their numbers were then normalized to the BM area from 2 to 4 sections of each tibia, averaged, and repeated for 5 mice. (E) Fifty ng of total genomic DNA collected from BM of individual mouse tibia and femur (n = 5) was used to quantify the luciferase copy number using quantitative polymerase chain reaction (qPCR) and luciferase gene standard, and the total copy number per tibia was calculated. The tibia-to-femur ratio of the luciferase copy number was plotted. (F) Total RNA was collected from above mouse tibia (n = 5) and human HAPLN1 mRNA was quantified using quantitative reverse transcription (qRT)-PCR and normalized to human SMA, and then fold changes were plotted using HS-5/EV–injected tibia set as 1. Data are expressed as means ± SEM. ∗P < .05; ∗∗P < .01; FSC, forward scatter.
To rule out the potential for any unintended systemic effect induced by the injection of HS-5/H1 cells, MM cell homing within individual mice was assessed by injecting HS-5/H1 cells in one tibia and HS-5/EV cells in the opposite tibia before systemic MM cell introduction (supplemental Figure 2H). Because initial analysis with the flow cytometry assay became unreliable because of low MM cell yields from a tibia, each tibia was subjected to immunohistochemistry analysis to quantify injected MM cells by anti-CD138 staining and HS-5 cells by anti-HLA class I (HLA-I) staining. CD138+ MM cells were not costained with the HLA-I antibody (Figure 2C), similar to many cancer cell types that display lower levels of HLA-I to evade immunity.32 The numbers of MM cells and HS-5 cells were normalized to the areas of BM sections imaged. The normalized CD138+ MM cell number in HS-5/H1-injected tibia was ∼2.5-fold greater than that in the HS-5/EV counterpart (Figure 2D). In contrast, the normalized HLA-I+ HS-5 staining was similar in both cases (Figure 2D); thus, the observed difference in MM cell homing was not due to different numbers of HS-5 cells retained in the BM.
To further validate the differences in MM cell homing to HS-5/H1- vs HS-5/EV–injected tibia, we quantified the number of MM cells in each tibia by means of MM cell–specific genomic DNA. An MM.1S cell clone with the luciferase gene stably integrated into the genome was used in BM homing experiments as in supplemental Figure 2H, and the luciferase copy number was quantified to measure the MM cells homed to each BM. The BM of femur and tibia were collected 72 hours after the MM cell injection and subjected to genomic DNA and total RNA extraction. The tibia-to-femur ratio of the luciferase copy number was calculated for each leg in each mouse. Comparable with the aforementioned 2 distinct BM homing experiments (Figure 2A-B vs Figure 2C-F), the number of luciferase-labeled MM cells was a ∼1.9-fold greater in the HS-5/H1-injected tibia than in the HS-5/EV–injected tibia (Figure 2E). When human HAPLN1 messenger RNA (mRNA) was quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using human SMA stromal gene as a normalization control, significantly higher HAPLN1 mRNA was detected in the HS-5/H1–injected tibia relative to that in its counterpart (Figure 2F), indicating that the injected HS-5/H1 cells were expressing the HAPLN1 gene. These 3 independent assays collectively demonstrated that HAPLN1 matrikine promoted BM homing of MM cells in vivo.
HAPLN1 matrikine activates STAT1 in MM cells
We previously performed RNA sequencing (RNA-seq) analysis of RPMI8226 MM cells exposed to recombinant PTR1 (GSE202672) and identified NF-κB and JAK/STAT as potentially enriched signaling pathways.14 We previously confirmed NF-κB activation,12 but, to our knowledge, STAT activation has not been demonstrated by stimulation of MM cells with HAPLN1 matrikine. To further assess the potential involvement of STAT factors, we also evaluated the RNA-seq data using a novel bioinformatics tool, Mining Algorithm for GenetIc Controllers21 and, again, identified NF-κB and STAT transcription factors as being among the top potential drivers of transcriptional changes in the HAPLN1 matrikine–induced genes (Figure 3A; supplemental Table 1). The RNA-seq data also showed that transcripts encoding NF-κB family members (RelA, RelB, c-Rel, NFKB1, and NFKB2) and STAT family members (STAT1, STAT3, STAT4, and STAT5A) were significantly induced in MM cells stimulated with HAPLN1 matrikine relative to that in control cells (Figure 3B). Interestingly, STAT1 had the highest reads per kilobase million among these potential master regulators. To test the activation of STAT transcription factors, we then performed a luciferase reporter assay using a construct containing the STAT1/3 responsive M67 promoter.33 Consistent with the bioinformatics predictions, MBP-PTR1 significantly increased the reporter activity in MM cells at a level similar to the positive control IL-6 (Figure 3C). Next, electrophoretic mobility shift and anti-STAT1/3 supershift assays were performed using an M67 probe, which demonstrated the presence of STAT1 but not STAT3 (Figure 3D). Consistently, western blot analysis demonstrated that STAT1 phosphorylation at tyrosine-701 was induced in a dose-dependent manner within 3 hours after MBP-PTR1 treatment, which lasted 24 hours (Figure 3E-F). Total STAT1 level was also increased after MBP-PTR1 treatment (Figure 3F), consistent with the induction of STAT1 gene expression (Figure 3B). In contrast, STAT3 tyrosine-705 phosphorylation and its total level were only minimally induced (Figure 3E-F). These results revealed that HAPLN1 matrikine primarily activates STAT1, not STAT3, in MM cells.
Figure 3.

HAPLN1 matrikine activates STAT1 in MM cells. (A) The graph depicts Mining Algorithm for GenetIc Controllers–identified enrichment scores of transcription factors and cofactors in response to PTR1 treatment in RPMI8226 cells. (B) Reads per kilobase million values of indicated genes from the RNA-seq results are plotted. (C) STAT-dependent luciferase reporter activities on RPMI8226 cells with the indicated stimuli are shown. The graph depicts the mean ± SEM of the quantification of 3 independent replicates. (D) Representative STAT1 and STAT3 supershift analysis of RPMI8226 cells incubated with MBP or MBP-PTR1 (100 nM; 6 hours) or IL-6 (50 ng/mL; 15 minutes). (E-F) Representative western blot analysis on RPMI8226 cells treated with the indicated dose of MBP or MBP-PTR1 for 6 hours (E) or 100 nM MBP-PTR1 for the indicated time (F). ∗P < .05.
STAT1 is required for HAPLN1 matrikine–induced MM cell migration and BM homing
To test whether STAT1 activation is critical for HAPLN1 matrikine functions, we generated STAT1 (short hairpin [shSTAT1]) or control (shControl) knockdown MM.1S cells (supplemental Figure 3A) and assayed for in vitro migration. Although basal migration (MBP only) was not affected, the increased migration observed in MBP-PTR1–treated shControl cells was absent in shSTAT1cells (Figure 4A). Similarly, in vivo BM homing of MM cells to control HS-5/EV–injected tibia was similar to but was reduced compared with the control level in HS-5/H1-injected tibia (Figure 4B; supplemental Figure 3B). To confirm that STAT3 does not play a role in HAPLN1-induced migration, we generated STAT3 knockout (KO) cells and conducted a migration assay. The STAT3 KO induced both STAT1 activation and HAPLN1-induced migratory potential comparable with those of the wild-type cells (supplemental Figure 3C-D). These results confirmed that STAT3 does not participate in HAPLN1-induced migration. Together, these data indicate that STAT1, not STAT3, is necessary for HAPLN1 matrikine–induced MM cell migration and BM homing.
Figure 4.

STAT1 is required for HAPLN1 matrikine–induced MM cell migration and BM homing. (A) Graphs depicting the percentage migration of MM.1S shControl or shSTAT1 clones in response to MBP or MBP-PTR1, with MBP-treated shControl being set as 100%. (B) The tibia-to-femur ratio of PKH26+ MM percentage was plotted as in Figure 2B. (C) Graph depicting the normalized expression of genes in the red cluster. Data are expressed as mean ± SD. (D) The dot plot represents the enrichment scores of the top 30 pathways in which the size of dot indicates the number of genes enriched for each pathway, and the color intensity correlates with the adjusted P. See “Methods” for enrichment score calculations. (E-F) mRNA levels of indicated genes was quantified using qRT-PCR and normalized to glyceraldehyde-3-phosphatedehydrogenase (GAPDH) and fold change relative to control (MBP in wild-type [WT] cells) were plotted. The graph represents the means ± SEM of 5 biological replicates for panel E and 3 biological replicates for panel F, each performed in duplicates. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
To identify STAT1-dependent, migration-related genes whose expression was induced by HAPLN1 treatment, a similar RNA-seq analysis as previously done on RPMI8226 cells was performed,14 except for using control and STAT1 KO cells (supplemental Figure 3E; GSE237216), the latter of which also showed defective MBP-PTR1–induced migration (supplemental Figure 3F). Cluster analysis identified 11 clusters in the differentially expressed genes (supplemental Figure 3G-H). We were particularly interested in the one in which the lack of STAT1 did not affect the basal expression but abrogated MBP-PTR1–induced gene expression (red cluster, Figure 4C; supplemental Table 1). Enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database showed that the genes in this cluster were enriched in JAK/STAT pathway and those previously identified to be involved in adhesion, migration, and BM homing of MM cells (eg, CD44, CXCR5, ICAM-1, VAV1, and DOCK10), among others. (Figure 4D; supplemental Table 2). We confirmed the MBP-PTR1–dependent induction of the well-established MM BM homing–related genes, CD44, SDF-1, and ICAM-1, using qRT-PCR (Figure 4E). These homing-related genes were not reduced in STAT3 KO cells, but some (ICAM-1 and CD44) were increased instead (Figure 4F). Taken together, these results demonstrated that HAPLN1 matrikine activates STAT1 to induce migration- and BM homing–related genes in MM cells.
HAPLN1 matrikine activates STAT1 via NF-κB–induced IFN-β in MM cells
We previously demonstrated that HAPLN1 matrikine can activate NF-κB signaling and induce NF-κB regulated drug resistance genes in MM cells.12,14 This study showed peak NF-κB activation occurring within 2 hours after HAPLN1-PTR1 treatment in RPMI8226 cells, whereas peak STAT1 phosphorylation was delayed and observed 3 to 6 hours after stimulation. This temporal sequence suggests that STAT1 phosphorylation takes place after NF-κB activation. Thus, we investigated whether NF-κB signaling was involved in HAPLN1-induced STAT1 activation. To test this possibility, we cotreated cells with MBP-PTR1 and an IκB kinase (IKK) inhibitor, IKK16 that was previously demonstrated to block MBP-PTR1–induced NF-κB signaling.12 Importantly, this cotreatment completely inhibited STAT1 phosphorylation (Figure 5A). Additionally, to determine whether any secreted factor downstream of NF-κB activates STAT1, we treated RPMI8226 cells with cycloheximide to block new protein synthesis or brefeldin A to block secretion. Both reagents effectively abrogated HAPLN1-induced STAT1 phosphorylation (Figure 5B). Furthermore, conditioned medium from HAPLN1-treated cells was capable of inducing STAT1 activity within 15 minutes of treatment (Figure 5C), indicating that newly synthesized and secreted factor(s), downstream of NF-κB signaling, mediate STAT1 activation in an autocrine or paracrine manner. To determine which factor activates STAT1 after HAPLN1 induces NF-κB activation, we identified candidate cytokines, for example, IL-6 and IFN-β, which are encoded by NF-κB target genes and are well-established activators of STAT1 signaling, in the red cluster genes from the RNA-seq analysis (Figure 5D). A previous study demonstrated that lipopolysaccharide stimulation of macrophages induces toll-like receptor 4 (TLR4)– and NF-κB–dependent STAT1 signaling via autocrine or paracrine production of IFN-β.34 Because HAPLN1 activates NF-κB through the TLR4-chaperonin 60 (CH60) cell surface receptor complex,23 we tested whether IFN-β is involved in the HAPLN1-activated STAT1 process. The cotreatment of IKK16 significantly reduced the HAPLN1-induced IFNB1 mRNA level (Figure 5E). In addition, the cotreatment of the Food and Drug Administration–approved type 1 IFN receptor blocking antibody, anifrolumab, completely inhibited HAPLN1-induced STAT1 phosphorylation (Figure 5F). In contrast, a neutralizing antibody against IL-6 had no effect (not shown). Taken together, these results support the notion that autocrine or paracrine production of IFN-β by NF-κB signaling is required for HAPLN1-induced STAT1 signaling.
Figure 5.

Autocrine/paracrine production of IFN-β by NF-κB contributes to HAPLN1 matrikine–induced STAT1 activation. (A) Representative western blot analysis of indicated proteins in RPMI8226 cells pretreated with IKK16 (10 μM) or dimethyl sulfoxide (DMSO) for 10 minutes and stimulated with 100 nM of MBP or MBP-PTR1 for 6 hours. (B) Representative western blot analysis of indicated proteins in RPMI8226 cells pretreated for 10 minutes with cycloheximide (CHX; 20 μg/mL) or brefeldin A (BFA; 3 μg/mL) and stimulated with 100 nM of MBP or MBP-PTR1 for 6 hours. (C) RPMI8226 cells were stimulated with 100 nM of MBP or MBP-PTR1 for 6 hours (lane 1 and 2). RPMI8226 cells were stimulated with 100 nM of MBP or MBP-PTR1 for 5 hours and subsequently washed with fresh media. Conditioned medium (CM) was harvested after a 1-hour incubation of fresh media without MBP or MBP-PTR1. CM was added to fresh RPMI8226 cells for 15 minutes (lane 3 and 4) and analyzed using western blotting for the indicated proteins. (D) RPMI8226 (WT and STAT1 KO) cells were stimulated with 100 nM of MBP or MBP-PTR1 for 6 hours. The IFNB1 mRNA level was quantified using qRT-PCR and normalized to GAPDH and fold change relative to control (MBP in WT cells) was plotted. (E) RPMI8226 cells were treated as in panel A. The mRNA level of IFNB1 was quantified using qRT-PCR and normalized to GAPDH and fold change relative to control (MBP and DMSO treated cells) was plotted. (F) Representative western blot analysis of indicated proteins in RPMI8226 cells pretreated with 10 μg/mL of anifrolumab or human immunoglobulin G (IgG) for 10 minutes and stimulated with 100 nM of MBP or MBP-PTR1 for 6 hours or IFN-β (50 pg/mL) for 15 minutes. The graph represents the means ± SEM of 3 biological replicates for panels D-E, each performed in duplicates. ∗P < .05, ∗∗P < .01.
High HAPLN1 levels in BM aspirates correlate with poor outcomes in patients
We previously reported that HAPLN1 matrikine is capable of inducing resistance to multiple classes of therapeutic agents in MM cells.14 We also showed that soluble HAPLN1 fragments are detectable in BM plasma but are highly variable from patient to patient, similar to HAPLN1 mRNA levels in patient BMSCs.12,13 Therefore, given the dual role of HAPLN1 in inducing drug resistance and BM homing, we hypothesized that soluble HAPLN1 in the BM may contribute to poor outcomes in patients with MM. To quantify soluble HAPLN1 levels in the plasma of BM aspirates, we developed an AlphaLISA (“Methods”), which detected HAPLN1 with minimal cross-reactivity with highly homologous HAPLN3 protein (supplemental Figure 4A). HAPLN1 levels in BM plasma samples from 26 patients with NDMM were measured using the AlphaLISA, and the patients were grouped into high and low groups based on the median cut-off value (Table 1). Both the sex and age of the patients were not different between these groups (supplemental Figure 4B). Significantly, the high-HAPLN1 group exhibited worse progression-free survival (PFS) than the low-HAPLN1 group (Figure 6A). The median PFS rates for the low- and high-HAPLN1 groups were 57 months and 28 months, respectively (hazard ratio [HR], 3.027). Furthermore, patients in the high-HAPLN1 group underwent a significantly higher number of therapies (Figure 6B). We further stratified the patients based on cytogenetic risk assessed using fluorescence in situ hybridization based on the Revised International Staging System and the second revision of the International Staging System.35,36 Interestingly, patients with standard cytogenetic risk showed a stark difference in PFS (HR, 5.861; Figure 6C). Although the sample size was too small to draw conclusive results, patients with high/ultrahigh cytogenetic risk did not exhibit a significant difference in PFS (supplemental Figure 4C). These results suggest that the level of HAPLN1, a MM cell-extrinsic factor derived from the ECM protein in the tumor microenvironment (TME), can serve as a prognostic marker in MM. Furthermore, patients with higher STAT1 mRNA level (n = 47) exhibited significantly worse PFS than the STAT1-low group (n = 48; HR, 1.7539; Figure 6D) in the CoMMpass trial RNA-seq data set from the Multiple Myeloma Research Foundation Researcher Gateway. Altogether, the results suggest that the HAPLN1 level in the BM and the STAT1 mRNA level in MM cells can be predictive markers of prognosis in patients with MM.
Table 1.
Characteristics of patients with NDMM
| All | Low | High | |
|---|---|---|---|
| Patient characteristics | |||
| Median age | 65 | 62 | 65 |
| Sex, no. (%) | |||
| Male | 17 (65.4) | 8 (61.5) | 9 (69.2) |
| Female | 9 (34.6) | 5 (38.5) | 4 (30.8) |
| Cytogenetic risk (no. of patients) | |||
| Standard | 14 | 7 | 7 |
| High | 7 | 2 | 5 |
| Ultrahigh | 3 | 2 | 1 |
| Not assessed | 2 | 2 | 0 |
| Medianfollow-uptime (mo) | 51 | 59 | 45 |
| HAPLN1 concentration (μg/mL) | |||
| Mean ± SD | 0.71 ± 0.80 | 0.36 ± 0.05 | 1.06 ± 1.03 |
| Median PFS (mo) | 42 | 57 | 28 |
| HR (95% CI) | 3.027 (1.135-8.072) | ||
| P value (log-rank) | .0164 | ||
CI, confidence interval; SD, standard deviation.
Figure 6.

High HAPLN1 levels in BM plasma fractions and high STAT1 mRNA levels in MM cells correlate with poor prognosis of patients with NDMM. (A) Kaplan-Meier PFS curve of the HAPLN1 high and low groups (n = 26). (B) Graph depicting the number of therapies that patients with NDMM received during the follow-up. (C) Kaplan-Meier PFS curve of patients at standard cytogenetic risk (n = 14). (D) Kaplan-Meier PFS curve of STAT1 high and low groups from the CoMMpass study. ∗P < .05; ∗∗P < .01.
Discussion
In this study, we uncovered for the first time, to our knowledge, that an ECM-derived matrikine could stimulate migration and BM homing of human MM cells. Specifically, the PTR1 domain of HAPLN1 or a larger fragment containing all 3 domains of HAPLN1 (immunoglobulin, PTR1, and PTR2 domains), representing HAPLN1 matrikine, could promote such activities. Similarly, larger and smaller fragments of HAPLN1 containing the PTR1 domain were detected in the BM aspirates of some patients with relapsed/refractory MM with progressive disease, an unfavorable category of treatment response.12 We also previously found that HAPLN1 matrikine could promote the resistance of MM cells to a variety of therapeutic agents.14 Thus, HAPLN1 matrikine may be a potentially important MM progression factor via 2 unique and distinct mechanisms: enhancing migration and homing to multiple BM sites and increasing multidrug resistance. Accordingly, higher HAPLN1 levels in the BM plasma correlated with a shorter PFS of patients with NDMM than that of the low-HAPLN1 group.
Although we previously found NF-κB to be a drug resistance factor,14 the discovery of STAT1 as the main MM cell migration and BM homing mechanism induced by HAPLN1 matrikine was unanticipated. Previous studies have demonstrated coactivation of NF-κB and STAT3 in a variety of human malignancies37,38 and the role for STAT3, not STAT1, as an oncogenic driver in MM.39,40 However, biochemical and genetic studies clearly revealed the activation of STAT1, not STAT3, and its requirement for HAPLN1 matrikine–induced MM cell migration and BM homing. Moreover, our study demonstrated that NF-κB is upstream of STAT1 activation and likely involves the NF-κB–dependent synthesis and secretion of IFN-β to induce subsequent STAT1 activation. Thus, HAPLN1 matrikine as a sequential activator of NF-κB and STAT1 adds to the emerging evidence supporting a tumor promoter role for STAT1 observed in other cancer types. For example, STAT1 has been implicated in the migration of colon cancer cells41 and cell adhesion, migration, and progression of serous papillary endometrial cancer cells.42 STAT1 has also been implicated in chemoresistance of head and neck cancer,43 epithelial-to-mesenchymal transition of lung cancer,44 and metastasis of melanoma.45 In addition, Boiarsky et al reported their single-cell RNA-seq analysis on CD138+ cells from patients with MM and premalignant cancer in whom IFN-inducible signatures, including the STAT1 level, were significantly upregulated in MM but not in precursor conditions.46 This report is consistent with our finding that HAPLN1 activates STAT1 in MM cells and that HAPLN1-induced IFN-β acts in an autocrine or paracrine manner. Moreover, our analysis of a larger scale clinical trial (CoMMpass) database also demonstrated that higher STAT1 mRNA level correlated with poor PFS of patients with MM. Thus, the new function of STAT1 in HAPLN1 matrikine–induced MM cell migration and BM homing expands its emerging context-dependent tumor promoter roles.
Our study also revealed that HAPLN1 matrikine may be a novel dual chemotactic and chemokinetic factor for MM cells. To date, the crucial role of SDF-1/CXCR4 axis in MM cell adhesion, migration, and BM homing is well established.47, 48, 49 SDF-1 induces MM cell chemotaxis at low doses but inhibits MM cell migration at high doses, thus promoting MM cell homing in circulation (ie, low concentrations) but retaining MM cells in the BM niche, where SDF-1 is enriched.4 In contrast, HAPLN1 matrikine increased MM cell migration not only when there was a positive concentration gradient acting as a chemotactic factor but also when concentrations were high without any gradient as a chemokinetic factor. It is noteworthy that a combination of increased chemotactic and chemokinetic motilities correlates with invasiveness and metastasis of other cancer types, including non–small cell lung cancer cells and others.27,28 These prior studies support the possibility that dual chemotactic and chemokinetic action of HAPLN1 matrikine contributes to not only BM homing through chemotaxis-mediated migration into the BM niche but also to egress from the BM into the circulation because of high motility when exposed to even higher concentrations of the matrikine in the BM niche, which can result in a vicious cycle of disease dissemination.
We demonstrated that high HAPLN1 levels in BM samples are associated with poor prognosis of patients with NDMM, supporting its potential use as a prognostic marker and therapeutic target. The current prognostic staging system for MM uses the Revised International Staging System, which can stratify the risk for patients based on protein biomarkers (β2-microglobulin, albumin, and lactate dehydrogenase) and cytogenetic aberrations.35 Although it is informative on tumor burden and risk assessment, it is not used for therapeutic decision-making.50 Given that the ECM is a major component of the TME, the role of ECM proteins per se as biomarkers remains incompletely understood. This study discovered that a TME factor could be a risk factor in MM pathogenesis. Our previous study also revealed that although HAPLN1 matrikine may induce resistance to multiple therapeutic agents, such as bortezomib, lenalidomide, dexamethasone, and others, it failed to induce resistance to the second generation proteasome inhibitor, carfilzomib and the nuclear export inhibitor, selinexor.14 These findings combined suggest the possibility that patients with NDMM with a high HAPLN1 status may benefit from therapies that contain either carfilzomib or selinexor or both. These findings also suggest a potential clinical benefit of ablating HAPLN1 matrikine function in MM therapy. However, a caveat is the relatively low number of samples of patients with NDMM analyzed (n = 26) in this study, especially for the patient groups who are at high or ultrahigh risk. Another weakness may be the generally low sensitivity of HAPLN1 AlphaLISA. Finally, the roles of full-length HAPLN1 and its matrikine in normal physiological processes in adult humans are currently lacking. Thus, development of a highly sensitive and specific assay to quantify HAPLN1 levels and an assessment of larger cohorts of patients with MM are necessary to establish this ECM-derived matrikine as a potential biomarker for predicting poor outcomes in patients. Once established, clinical studies could be conducted to test the potential benefits of stratifying patients with NDMM to carfilzomib and/or selinexor therapies. Finally, improved knowledge of the role of HAPLN1 and its matrikine in human physiology may shed light on the safety of HAPLN1 matrikine–targeting therapeutics against MM, which is currently considered an incurable cancer type.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Deane F. Mosher (University of Wisconsin-Madison) for the useful discussion on chemotaxis and chemokinesis and Lixin Rui for advice on STAT analyses. The authors also thank Miyamoto laboratory members for the helpful discussion and Debayan De Bakshi, in particular, for advice on MBP-PTR1 protein purification; the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory, the Experimental Animal Pathology Laboratory, and the Small Animal Imaging & Radiotherapy Facility for use of their facilities and services (University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520).
This work was supported by the National Institutes of Health R01 CA251595 and CA155192 (S.M.); and National Institutes of Health T32 CA009135 and SciMed Graduate Research Scholars Fellowship at UW-Madison (M.H.). The visual abstract was created with Biorender.com.
Authorship
Contribution: H.Y.C. performed and analyzed the experiments; M.H. performed initial RNA-seq analysis and A.R. performed Mining Algorithm for GenetIc Controllers and cluster analyses of all RNA-seq data; N.S.C. consented and obtained primary samples from patients; H.Y.C. and S.M. were responsible for overall study design; H.Y.C., M.H., and S.M. wrote the manuscript; and all authors reviewed, edited, and approved the manuscript.
Footnotes
The RNA sequencing data are deposited in the Gene Expression Omnibus database (accession number GSE237216).
The CoMMpass study data were generated as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiatives using the Genospace Population Analytics platforms (https://research.themmrf.org and www.themmrf.org).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Shain K. Metastatic myeloma? Blood. 2012;119(24):5612–5613. doi: 10.1182/blood-2012-04-417337. [DOI] [PubMed] [Google Scholar]
- 2.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 3.Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol. 2012;2012(6):157496. doi: 10.1155/2012/157496. 157495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alsayed Y, Ngo H, Runnels J, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109(7):2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roccaro AM, Sacco A, Purschke WG, et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014;9(1):118–128. doi: 10.1016/j.celrep.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maquart F-X, Pasco S, Ramont L, Hornebeck W, Monboisse J-C. An introduction to matrikines: extracellular matrix-derived peptides which regulate cell activity. Implication in tumor invasion. Crit Rev Oncol Hematol. 2004;49(3):199–202. doi: 10.1016/j.critrevonc.2003.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Weathington NM, van Houwelingen AH, Noerager BD, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12(3):317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 8.Hunninghake GW, Davidson JM, Rennard S, Szapiel S, Gadek JE, Crystal RG. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science. 1981;212(4497):925–927. doi: 10.1126/science.7233186. [DOI] [PubMed] [Google Scholar]
- 9.Pocza P, Süli-Vargha H, Darvas Z, Falus A. Locally generated VGVAPG and VAPG elastin-derived peptides amplify melanoma invasion via the galectin-3 receptor. Int J Cancer. 2008;122(9):1972–1980. doi: 10.1002/ijc.23296. [DOI] [PubMed] [Google Scholar]
- 10.Grahovac J, Wells A. Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Lab Invest. 2014;94(1):31–40. doi: 10.1038/labinvest.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hope C, Foulcer S, Jagodinsky J, et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood. 2016;128(5):680–685. doi: 10.1182/blood-2016-03-705780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huynh M, Pak C, Markovina S, et al. Hyaluronan and proteoglycan link protein 1 (HAPLN1) activates bortezomib-resistant NF-κB activity and increases drug resistance in multiple myeloma. J Biol Chem. 2018;293(7):2452–2465. doi: 10.1074/jbc.RA117.000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mark C, Warrick J, Callander NS, Hematti P, Miyamoto S. A hyaluronan and proteoglycan link protein 1 matrikine: role of matrix metalloproteinase 2 in multiple myeloma NF-κB activation and drug resistance. Mol Cancer Res. 2022;20(9):1456–1466. doi: 10.1158/1541-7786.MCR-21-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh M, Chang HY, Lisiero DN, et al. HAPLN1 confers multiple myeloma cell resistance to several classes of therapeutic drugs. PLoS One. 2022;17(12) doi: 10.1371/journal.pone.0274704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zengel P, Nguyen-Hoang A, Schildhammer C, Zantl R, Kahl V, Horn E. μ-Slide chemotaxis: a new chamber for long-term chemotaxis studies. BMC Cell Biol. 2011;12(1):21. doi: 10.1186/1471-2121-12-21. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomasova L, Guttenberg Z, Hoffmann B, Merkel R. Advanced 2D/3D cell migration assay for faster evaluation of chemotaxis of slow-moving cells. PLoS One. 2019;14(7) doi: 10.1371/journal.pone.0219708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markovina S, Callander NS, O'Connor SL, et al. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res. 2008;6(8):1356–1364. doi: 10.1158/1541-7786.MCR-08-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Runnels JM, Carlson AL, Pitsillides C, et al. Optical techniques for tracking multiple myeloma engraftment, growth, and response to therapy. J Biomed Opt. 2011;16(1) doi: 10.1117/1.3520571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood. 2014;123(14):2204–2208. doi: 10.1182/blood-2013-07-517136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lwin ST, Edwards CM, Silbermann R. Preclinical animal models of multiple myeloma. Bonekey Rep. 2016;5:772. doi: 10.1038/bonekey.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roopra A. MAGIC: a tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput Biol. 2020;16(4) doi: 10.1371/journal.pcbi.1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spicer AP, Joo A, Bowling RA. A hyaluronan binding link protein gene family whose members are physically linked adjacent to chondroitin sulfate proteoglycan core protein genes: the missing links. J Biol Chem. 2003;278(23):21083–21091. doi: 10.1074/jbc.M213100200. [DOI] [PubMed] [Google Scholar]
- 23.De Bakshi D, Chen Y-C, Wuerzberger-Davis SM, et al. Ectopic CH60 mediates HAPLN1-induced cell survival signaling in multiple myeloma. Life Sci Alliance. 2023;6(3) doi: 10.26508/lsa.202201636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diamond MS, Springer TA. The dynamic regulation of integrin adhesiveness. Curr Biol. 1994;4(6):506–517. doi: 10.1016/s0960-9822(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 25.Wilkinson PC. Assays of leukocyte locomotion and chemotaxis. J Immunol Methods. 1998;216(1-2):139–153. doi: 10.1016/s0022-1759(98)00075-1. [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson PC. How do leucocytes perceive chemical gradients? FEMS Microbiol Immunol. 1990;2(5-6):303–311. doi: 10.1111/j.1574-6968.1990.tb03533.x. [DOI] [PubMed] [Google Scholar]
- 27.Tchou-Wong K-M, Fok SYY, Rubin JS, et al. Rapid chemokinetic movement and the invasive potential of lung cancer cells; a functional molecular study. BMC Cancer. 2006;6(1):151. doi: 10.1186/1471-2407-6-151. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, Klominek J. Chemotaxis and chemokinesis of malignant mesothelioma cells to multiple growth factors. Anticancer Res. 2004;24(3a):1625–1630. [PubMed] [Google Scholar]
- 29.de Gorter DJJ, Reijmers RM, Beuling EA, et al. The small GTPase Ral mediates SDF-1-induced migration of B cells and multiple myeloma cells. Blood. 2008;111(7):3364–3372. doi: 10.1182/blood-2007-08-106583. [DOI] [PubMed] [Google Scholar]
- 30.Zigmond SH, Hirsch JG. Leukocyte locomotion and chemotaxis. New methods for evaluation, and demonstration of a cell-derived chemotactic factor. J Exp Med. 1973;137(2):387–410. doi: 10.1084/jem.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen Q, Murphy G, Hughes CE, Mort JS, Roughley PJ. Matrix metalloproteinases cleave at two distinct sites on human cartilage link protein. Biochem J. 1993;295(pt 2):595–598. doi: 10.1042/bj2950595. (pt 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fangazio M, Ladewig E, Gomez K, et al. Genetic mechanisms of HLA-I loss and immune escape in diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2021;118(22) doi: 10.1073/pnas.2104504118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvath CM, Wen Z, Darnell JE. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;9(8):984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 34.Toshchakov V, Jones BW, Perera PY, et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3(4):392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 35.Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863–2869. doi: 10.1200/JCO.2015.61.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D'Agostino M, Cairns DA, Lahuerta JJ, et al. Second Revision of the International Staging System (R2-ISS) for overall survival in multiple myeloma: a European Myeloma Network (EMN) report within the HARMONY Project. J Clin Oncol. 2022;40(29):3406–3418. doi: 10.1200/JCO.21.02614. [DOI] [PubMed] [Google Scholar]
- 37.Bharti AC, Shishodia S, Reuben JM, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103(8):3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 38.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10(12):1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 40.Chong PSY, Chng WJ, de Mel S. STAT3: a promising therapeutic target in multiple myeloma. Cancers (Basel) 2019;11(5) doi: 10.3390/cancers11050731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malilas W, Koh SS, Kim S, et al. Cancer upregulated gene 2, a novel oncogene, enhances migration and drug resistance of colon cancer cells via STAT1 activation. Int J Oncol. 2013;43(4):1111–1116. doi: 10.3892/ijo.2013.2049. [DOI] [PubMed] [Google Scholar]
- 42.Kharma B, Baba T, Matsumura N, et al. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res. 2014;74(22):6519–6530. doi: 10.1158/0008-5472.CAN-14-0847. [DOI] [PubMed] [Google Scholar]
- 43.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101(6):1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaowinn S, Kaewpiboon C, Koh SS, Krämer OH, Chung Y-H. STAT1-HDAC4 signaling induces epithelial-mesenchymal transition and sphere formation of cancer cells overexpressing the oncogene. Oncol Rep. 2018;40(5):2619–2627. doi: 10.3892/or.2018.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khodarev NN, Roach P, Pitroda SP, et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One. 2009;4(6) doi: 10.1371/journal.pone.0005821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boiarsky R, Haradhvala NJ, Alberge JB, et al. Single cell characterization of myeloma and its precursor conditions reveals transcriptional signatures of early tumorigenesis. Nat Commun. 2022;13(1):7040. doi: 10.1038/s41467-022-33944-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vila-Coro AJ, Rodríguez-Frade JM, Martín De Ana A, Moreno-Ortíz MC, Martínez-A C, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13(13):1699–1710. [PubMed] [Google Scholar]
- 48.Sanz-Rodríguez F, Hidalgo A, Teixidó J. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and VCAM-1. Blood. 2001;97(2):346–351. doi: 10.1182/blood.v97.2.346. [DOI] [PubMed] [Google Scholar]
- 49.Azab AK, Azab F, Blotta S, et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009;114(3):619–629. doi: 10.1182/blood-2009-01-199281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallington-Beddoe CT, Mynott RL. Prognostic and predictive biomarker developments in multiple myeloma. J Hematol Oncol. 2021;14(1):151. doi: 10.1186/s13045-021-01162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.