

Abstract
The synthesis and characterization of platinum(II) and palladium(II) complexes bearing two (dimers Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2), one (monomers Pt(Lpc)(Lref)Cl2 and Pd(Lpc)(Lref)Cl2), or no (reference compounds Pt(Lref)2Cl2 and Pd(Lref)2Cl2) pentacene-based pyridyl ligands are presented. Photophysical properties of the dimers are probed by means of steady-state and time-resolved transient absorption measurements in comparison to the monomer and model compounds. Our results document that despite enhanced spin–orbit coupling from the presence of heavy atoms, intramolecular singlet fission (iSF) is not challenged by intersystem crossing. iSF thus yields correlated triplet pairs and even uncorrelated triplet excited states upon decoherence. Importantly, significant separation of the two pentacenyl groups facilitates decoupling of the two chromophores. Furthermore, the mechanism of iSF is altered depending on the respective metal center, that is, Pt(II) versus Pd(II). The dimer based on Pt(II), Pt(Lpc)2Cl2, exhibits a direct pathway for the iSF and forms a correlated triplet pair with singlet–quintet spin-mixing within 10 ns in variable solvents. On the other hand, the dimer based on Pd(II), Pd(Lpc)2Cl2, leads to charge transfer mixing during the population of the correlated triplet pair that is dependent on solvent polarity. Moreover, Pd(Lpc)2Cl2 gives rise to a stable equilibrium between singlet and quintet correlated triplet pairs with lifetimes of up to 170 ns. Inherent differences in the size and polarizability, when contrasting platinum(II) with palladium(II), are the most likely rationale for the underlying trends.
Keywords: metal complexes, singlet fission, pentacene dimers, ultrafast spectroscopy, electronic coupling
Introduction
Singlet fission (SF) is a spin-allowed photophysical process, which allows the generation of two pairs of charges from absorption of a single photon, and it provides an avenue to break the detailed balance limit of single junction solar cells from ca. 32% to over 45%.1,2 When SF occurs, a chromophore in its singlet excited state (S1) interacts with a nearby chromophore in its ground state (S0), forming a spin-correlated triplet pair state 1(T1T1).3−6 Subsequently, the triplet pair can undergo decorrelation, resulting in two noninteracting, free triplet states (T1 + T1). These triplets are then available for completing the process of solar energy conversion.4,7−11
Over the years, many criteria have been identified for chromophores to perform effective SF. The most straightforward and basic criteria are, on one hand, the energetic relationship between the singlet (S1) and triplet (T1) excited states. For exothermic SF, the energy level of (S1) must be close to twice that of (T1), which also serves as a thermodynamic driving force for SF, namely E(S1) > 2 × E(T1). Alternatively, if E(S1) is equal or slightly below 2 × E(T1), endothermic SF is possible.1,12−14 Prominent representatives for exothermic and endothermic SF are pentacene-based15−19 and anthracene-based20−23 chromophores, respectively. E(S1) of pentacene is 1.83 eV, which is greater than 2 × E(T1) with a value of 1.72 eV, thus it is a typical sample capable of SF while the opposite pathway triplet–triplet annihilation up-conversion (TTA-UC) is undetectable. Tetracene, with similar E(S1) and 2 × E(T1) of 2.35 eV, exhibits the coexistence of SF and TTA-UC. Conversely, anthracene has E(S1) and 2 × E(T1) with values of 3.30 and 3.70 eV, respectively, which is an ideal sample for TTA-UC.2,24 The energy level of any higher triplet excited states, such as (T2), should exceed 2 × E(T1) to avoid its population via triplet–triplet annihilation (TTA) and avoid intersystem crossing (ISC) between (S1) and (T2), which is quite common in the case of strongly coupled chromophores.1,12,25 On the other hand, sufficient electronic interactions between the interacting chromophores need to be assured. Coupling is typically realized by either high concentrations in solution, or through control of the crystal packing in the solid state,15,26−31 and thus efficient intermolecular SF evolves. An alternative strategy relies on a spacer that provides molecular dimers and oligomers that drive intramolecular interactions for SF (iSF). The merit of oligomers is the control over, for instance, spatial overlap and degree of coupling, which offer the ability to fine-tune and analyze the impact of coupling via the spacer on the intricacies of the mechanism, kinetics, and efficiency of iSF as a whole.14,18,32−36 Despite the broad accomplishments toward efficient formation of 1(T1T1), the underlying mechanism of decorrelation toward formation of free triplet states (T1 + T1) remains to be tested and optimized.
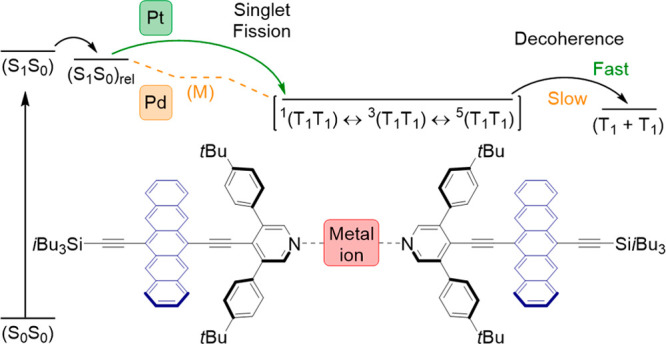
Conventionally, the design of SF materials excludes heavy atoms, since spin–orbit coupling (SOC) facilitates ISC. As well, the synthesis of heavy-atom chromophores can be more challenging when compared to small organic molecules.2,37,38 Triplets born from ISC are fundamentally different from triplets of SF and are clearly distinguishable.39−41 Studies have shown that SOC enhancement produced by a heavy atom, the so-called heavy-atom effect (HAE), can have profound effects on the decorrelation of the spin-correlated triplet pair (T1T1). Musser et al. have shown that the spin evolution within (T1T1) is faster through replacing sulfur in polythienylenevinylene with the heavy atoms selenium and tellurium. They suggest that the presence of heavy atoms may manipulate the dynamics of triplets formed by SF.42 Pt-bridged pentacene dimers have been synthesized, and the impact of the HAE has been investigated.40,43 It is reported that the presence of Pt has no impact on the formation of the singlet correlated triplet pair 1(T1T1) from (S1S0), but Pt did influence subsequent transitions (Figure 1). The enhanced SOC on 1(T1T1) caused by Pt leads to a spin-flip of 1(T1T1) into both the triplet correlated triplet pair 3(T1T1) and the quintet correlated triplet pair 5(T1T1). Ultimately, population of the triplet excited states (S0T1) and (T1 + T1) results from 3(T1T1) and 5(T1T1), respectively. Thus, enhanced SOC produced by a heavy atom may improve the yield of decorrelation. The presence of heavy atoms, however, is not always beneficial to SF. Recently, a tetramer has been synthesized through coordinating pyridyl pentacene-based ligands to Ru(II), and, in this case, the presence of the heavy atom gives rise to ultrafast ISC following photoexcitation rather than SF.44 Therefore, the influence of HAE on triplet decorrelation in SF remains unpredictable.
Figure 1.
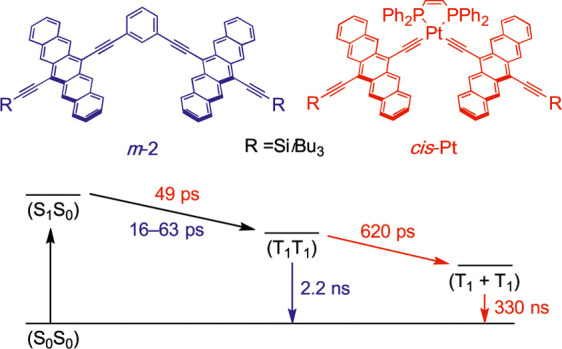
Comparison of iSF between cis-Pt with SOC and m-2 without SOC.40,45
Intrigued by these findings, we designed and synthesized two pentacene dimers that use heavy atoms Pt and Pd to link pyridyl pentacene ligands (Scheme 1). The pyridyl group is ubiquitous for bonding with metal ions by coordination and allows the versatile synthesis of pentacene oligomers featuring different metal ions.44,46 It is important to emphasize that by design, the two dimers, Pd(Lpc)2Cl2 and Pt(Lpc)2Cl2, are structurally identical except for the metal ion. As second- and third-row metals, Pd and Pt are congeners, and both dimers Pd(Lpc)2Cl2 and Pd(Lpc)2Cl2 feature metals in the M(II) oxidation state. Thus, while the valence electron structure is the same, the HAE is reduced in Pd compared to Pt as the effective nuclear charge of Pd is lower than Pt.47−49 In comparison to suitable model compounds that cannot undergo iSF, the electronic influence of the heavy atoms and their impact on the mechanism of SF can be carefully studied. In the presence of Pt in dimer Pt(Lpc)2Cl2, analyses show that 1(T1T1) forms directly from iSF. Conversely, the incorporation of Pd in dimer Pd(Lpc)2Cl2 leads to the formation of 1(T1T1) via a mediating species likely due to the lower spin–orbit coupling.
Scheme 1. Synthesis of Dimers M(Lpc)2Cl2, Monomers M(Lpc)(Lref)Cl2, and Model Compounds M(Lref)2Cl2.

Reagents and conditions: (a) M = Pt, Lpc, toluene, 80 °C; (b) M = Pd, Lpc, CH2Cl2, rt; (c) M = Pt, Lref, toluene, 110 °C then Lpc, toluene, 110 °C; (d) M = Pd, Lref, Lpc, CH2Cl2, rt; (e) M = Pt, Lref, toluene, 80 °C; (f) M = Pd, Lref, CH2Cl2, rt. Inset: X-ray crystallographic analysis of Pt(Lpc)(Lref)Cl2 (H atoms removed for clarity, ellipsoids at 30% level).
Results and Discussion
Synthesis
The pyridyl-end-capped pentacene ligand (Lpc) and its corresponding reference pyridyl ligand without pentacene (Lref) were synthesized following a published procedure.44 A ligand exchange reaction of Lpc with either Pt(PhCN)2Cl2 or Pd(PhCN)2Cl2 (1a or 1b) efficiently gives the dimers Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2 (Scheme 1). Unsymmetrical monomeric complexes Pt(Lpc)(Lref)Cl2 and Pd(Lpc)(Lref)Cl2 bearing a single pentacenyl unit were prepared by either a stepwise reaction for Pt(Lpc)(Lref)Cl2 (via the intermediate Pt(PhCN)(Lref)Cl2) or a one-pot reaction in the case of Pd(Lpc)(Lref)Cl2. While monomer Pt(Lpc)(Lref)Cl2 could be purified through a sequence of column chromatography and recrystallization, Pd(Lpc)(Lref)Cl2 was unstable to purification by column chromatography (using either SiO2 or Al2O3). Ultimately, preparative gel permeation chromatography gave pure product Pd(Lpc)(Lref)Cl2. Finally, ligand exchange of Lref with Pt(PhCN)2Cl2 or Pd(PhCN)2Cl2 gave the reference compounds Pt(Lref)2Cl2 or Pd(Lref)2Cl2. All products are highly soluble in common organic solvents such as CH2Cl2, CHCl3, tetrahydrofuran (THF), and toluene (≥20 mg/mL). The complexes are all stable as solids under normal laboratory conditions. However, all compounds containing a pentacene moiety decompose in solution upon exposure to air (O2) and light over days.
The successful formation of the desired metal complexes has been confirmed by mass spectrometry combined with 1H and 13C NMR spectroscopic analyses. 1H NMR spectra of M(Lref)2Cl2 and M(Lpc)2Cl2 (M = Pt, Pd) mirror the 1H NMR spectra of ligands Lref and Lpc, as the two ligands in the metal complexes are equivalent. It is noted, however, that the signals of pyridyl protons in M(Lref)2Cl2 (M = Pt, δ 8.78; and M = Pd, δ 8.70) and M(Lpc)2Cl2 (M = Pt, δ 9.03; and M = Pd, δ 8.95) are shifted downfield by 0.24–0.28 and 0.16–0.20 ppm for the Pt and Pd complexes (Figure S13),50 respectively, compared to the analogous protons of the free ligands Lref and Lpc (δ 8.54 and δ 8.75, respectively).44 The observed trends in 1H shifts for the pyridyl groups are consistent with literature reports, which indicate a decreased electron density upon metal coordination of a pyridyl ligand.51−55 The greater deshielding of these protons when bound to Pt(II) in comparison to Pd(II) corresponds to the lower spin–orbit coupling for Pd due to its nuclear charge.48,49
The molecular structures of monomer Pt(Lpc)(Lref)Cl2 and reference compounds Pt(Lref)2Cl2 and Pd(Lref)2Cl2 have been unambiguously established by X-ray crystallography (Scheme 1 and Figure S20). The structures confirm the trans orientation of the ligands as well as the expected square-planar geometry (full details are provided in the Supporting Information). In all cases, the opposing pyridyl ligands are coplanar, while the bulky tert-butyl substituents on the pyridyl ring prevent coplanarity between the pentacenyl and pyridyl moieties of Pt(Lpc)(Lref)Cl2 (with a torsion angle of ca. 16°).
Photophysics
Steady-state absorption measurements of compounds without pentacenyl groups (Lref, Pt(Lref)2Cl2, and Pd(Lref)2Cl2) show absorption bands in the high energy region of the UV spectrum between 300–400 nm (Figure S21). As such, they are transparent to time-resolved transient absorption (TAS) measurements, in which 610 nm serves as the excitation wavelength of the pentacene chromophores. Furthermore, emission is not detected for any of these reference compounds based on steady-state fluorescence measurements.
The steady-state absorption spectra of monomers Pt(Lpc)(Lref)Cl2 and Pd(Lpc)(Lref)Cl2 show well-resolved vibrational fine structures (Figure 2, Table 1), in contrast to Pt-acetylide complexes bearing acenes that show broadened features.40,43,56 Compared to the free ligand Lpc, the absorptions for monomers Pt(Lpc)(Lref)Cl2 and Pd(Lpc)(Lref)Cl2 are subtly red-shifted and slightly broadened. Extinction coefficients of both monomers and Lpc typically range between 30,000 and 40,000 M–1 cm–1. Increasing the solvent polarity from toluene to tetrahydrofuran (THF) and benzonitrile (BN) yields only minor differences, with only a slight broadening of the features in BN being the most visible change, especially for Pt(Lpc)(Lref)Cl2 (Figures S22–S24). Absorption spectra for pentacene dimers Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2 show similar features. The shift in the low-energy absorptions is minimal in spite of additional π-conjugated resulting from the second pentacenyl, pointing toward the presence of an effectively decoupled system in those dimers. The extinction coefficients of Pd(Lpc)2Cl2 and Pt(Lpc)2Cl2 are nearly twice that of the monomers, which further reinforced this fact (Figure 2, Table 1). Interestingly, the change of solvent to BN has a stronger effect on dimers than monomers, with a noticeable drop in extinction coefficient, suggesting possible changes in the electronic ground state of Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2 (Figures S22–S24).
Figure 2.
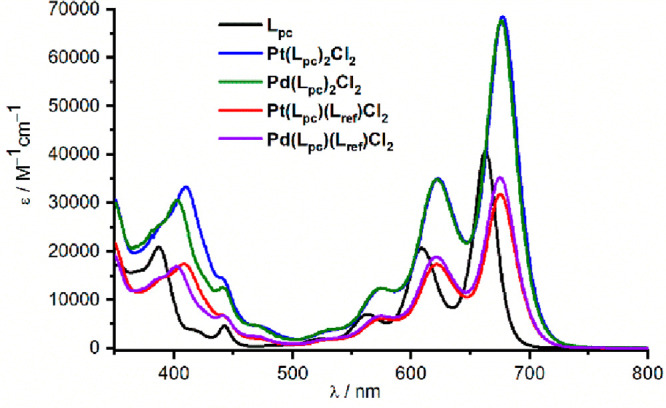
Steady-state absorption (toluene) of Lpc (black), Pt(Lpc)2Cl2 (blue), Pd(Lpc)2Cl2 (green), Pt(Lpc)(Lref)Cl2 (red), and Pd(Lpc)(Lref)Cl2 (purple).
Table 1. Long-Wavelength Extinction Coefficients (ε) and Fluorescence Quantum Yields (FQYs), Obtained from Steady-State Absorption and Fluorescence Measurements.
| Compound | Solvent | λmax/nm | ε/M–1 cm–1 | FQY/% |
|---|---|---|---|---|
| Lpc | toluene | 633 | 40,700 | 35.3 |
| THF | 660 | 34,700 | 23.4 | |
| BN | 670 | 37,000 | 9.1 | |
| Pt(Lpc)(Lref)Cl2 | toluene | 676 | 31,800 | 9.3 |
| THF | 672 | 31,900 | 3.0 | |
| BN | 685 | 28,800 | 0.7 | |
| Pd(Lpc)(Lref)Cl2 | toluene | 675 | 35,300 | 12.5 |
| THF | 671 | 35,800 | 7.4 | |
| BN | 684 | 27,400 | 3.3 | |
| Pt(Lpc)2Cl2 | toluene | 678 | 68,500 | 6.9 |
| THF | 674 | 71,500 | 2.6 | |
| BN | 687 | 51,400 | 0.6 | |
| Pd(Lpc)2Cl2 | toluene | 677 | 67,600 | 10.1 |
| THF | 671 | 61,700 | 6.0 | |
| BN | 681 | 46,400 | 2.5 |
Steady-state emission spectra provide initial insight into the excited-state deactivation of the metal complexes (Figures S25 and S26, Table 1). The fluorescence quantum yields in toluene drop consistently in the series Lpc (35.3%), Pt(Lpc)(Lref)Cl2 (9.3%), and Pt(Lpc)2Cl2 (6.9%). The fluorescence quantum yields of Pd(Lpc)(Lref)Cl2 (12.5%) and Pd(Lpc)2Cl2 (10.1%) show a similar trend. In polar solvents tetrahydrofuran and benzonitrile, fluorescence quantum yields trend to even lower (Table 1), and, for example, the values of dimers Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2 in benzonitrile drop to 0.6% and 2.5%, respectively.
Following the initial set of steady-state measurements, we turned to analysis by TAS (λex = 610 nm; E = 400 nJ), using fs-TAS to derive information related to the formation of the correlated triplet pair 1(T1T1) on the early time scales of SF, and ns-TAS to probe subsequent decoherence of 1(T1T1) and formation of (T1 + T1). The behavior of ligand Lpc was established first (Figures S28–S33). fs- and ns-TAS raw data for Lpc were best fit by global analysis with a kinetic model based on three species. In particular, the first and second species both have singlet excited state character and feature excited state absorptions (ESA) at 450 and 1360 nm, next to ground state bleaching (GSB) at ca. 670 nm. Considering that the initially formed excited state is likely more polar than the ground state, the first and second species are singlet excited states (S1) prior to and after ((S1)rel) vibrational and solvent relaxation. The corresponding lifetimes are 10.7 ps (S1) and 4.7 ns ((S1)rel) in benzonitrile. The third species shows a fundamentally different ESA at around 470 and 510 nm. This species evolves as a product of a slow and spin-forbidden intersystem crossing that is assigned as the triplet excited state (T1), with a lifetime of 20.2 μs.
Monomer Pt(Lpc)(Lref)Cl2 was then probed to establish the effect of metal coordination to Lpc (Figures S34–S39). The kinetic model established for Lpc fits the raw data of Pt(Lpc)(Lref)Cl2 well with the same three species, namely, (S1), (S1)rel, and (T1). In comparison to the Lpc, the lifetimes of (S1)rel and (T1) are, however, influenced by the HAE and reduced by a factor of about three with values of 12.4 and 27.1 μs for Lpc versus 4.9 ns and 7.9 μs for Pt(Lpc)(Lref)Cl2, respectively, in toluene. Hand-in-hand with a shorter lifetime for (S1)rel is an increased triplet quantum yield for Pt(Lpc)(Lref)Cl2 (5.2%), which is about 3.5 times greater than that of Lpc (1.5%) in toluene. The behavior in polar solvents, such as THF and benzonitrile, was similar.57
Finally, the photophysics of dimer Pt(Lpc)2Cl2 were explored. The development of a kinetic model based on four species was, however, needed to fit the fs- and ns-TAS raw data (Figure 3 and Figures S40–S45). The first and second species show ESA and GSB features consistent with singlet excited state. Conversely, the third and fourth species show ESA and GSB characteristics of triplet excited state. The first (6.3 ps) and second (0.5 ns) species of Pt(Lpc)2Cl2 in BN display similar deconvoluted spectra, which are analogous to that of (S1) and (S1)rel of Pt(Lpc)(Lref)Cl2. Furthermore, the first species exhibits a short lifetime of 6.3 ps, consistent with the initial formation of (S1S0) that transforms into (S1S0)rel. It is noted that the fourth species has a long-lived lifetime (7.0 μs) as well as triplet signatures that are characteristic of (T1) in Pt(Lpc)(Lref)Cl2. Thus, there is no doubt that the fourth species is a free triplet state. This leaves the third species as the only unidentified state. Considering this species is rapidly formed, and combined with triplet signatures in the ESA and GSB, we postulate a fast and spin-allowed triplet formation via iSF that results in a correlated triplet pair 1(T1T1). Subsequently, the final product of uncorrelated triplet excited state (T1 + T1), that is fourth species, is formed from decoherence of 1(T1T1) and then deactivates by means of recovery of the ground state. Importantly, this spin-allowed iSF for Pt(Lpc)2Cl2 (2.8 ns in toluene) is twice as fast as spin-forbidden ISC (4.9 ns in toluene), which results in an overall 1(T1T1) triplet quantum yield of around 140% in toluene and nearly 200% in benzonitrile.58 In Pt(Lpc)2Cl2, the final step of iSF is the decoherence of 1(T1T1), by which two separate triplet excited states, namely, (T1 + T1), are formed. The electronic interaction between the two triplets at short distances in singlet 1(T1T1) is stronger than that for either the triplet 3(T1T1) or quintet 5(T1T1). Moreover, this additional stabilizing energy has been related to the energy required to dissociate the two triplets.591(T1T1) can be stabilized by configurational interactions with other singlet configurations, while the 5(T1T1) preserves much larger pure diabatic character. The mixing of 5(T1T1) with other quintets is limited because these states are generally higher in energy.60 Hence, the binding energy of the two triplets can be approximated as Eb = E(5(T1T1)) – E(1(T1T1)).59,61 The role of the 5(T1T1) as an intermediate state in singlet fission has been hypothesized by means of the spin Hamiltonian of interacting triplets62,63 and has been established by time-resolved electron paramagnetic resonance (TR-EPR) in weakly coupled pentacene dimers.7 Along with the dissociation of 1(T1T1), the intermediate state is the quintet form of (T1T1), which is denoted as 5(T1T1). Moreover, the heavy atom effect enhances the spin–orbit coupling leading to the triplet correlated form of (T1T1), namely, 3(T1T1).40 In particular, the mixing of 3(T1T1) and 5(T1T1) determines the fate of SF. The involvement of 3(T1T1) strongly depends on the SF chromophore, the coupling between chromophores, as well as spin–orbit coupling.40,45,64 On one hand, the mixing of 3(T1T1) and 5(T1T1) is not experimentally observed in the case of large J values like those observed in heavy-atom free pentacene dimers.45 On the other hand, the small J value in weakly coupled terrylene diimides favors the formation of 3(T1T1) that triplet–triplet annihilates to produce a single rather than a pair of uncorrelated triplets.64 Here, spin mixing allows for annihilation through the triplet channel to compete with free triplet formation. The formation of 5(T1T1) is, nevertheless, essential as it shuts down annihilation from 1(T1T1) and 3(T1T1) channels.
Figure 3.

(a) fs-TAS (λex = 610 nm, 400 nJ) of Pt(Lpc)2Cl2 in BN with time delays between 0–5500 ps, together with the respective (b) deconvoluted spectra representing the singlet (S1S0) (black), solvent-relaxed singlet (S1S0)rel (red), and correlated triplet pair (T1T1) (blue) excited states as obtained by target analysis. (c) ns-TAS (λex = 610 nm, 400 nJ) of Pt(Lpc)2Cl2 in BN with time delays between 0 ns–400 μs, together with the respective (d) deconvoluted spectra representing the solvent-relaxed singlet (S1S0)rel (red), correlated triplet pair (T1T1) (blue), and uncorrelated triplet (T1 + T1) (green) excited states as obtained by target analysis.
Thus, we refer to the third species as the mixing of a singlet, triplet, and quintet (T1T1), namely 1/3/5(T1T1). Subsequently, decoherence dominates the transition to a free triplet pair (T1 + T1). Although the decoherence of 1/3/5(T1T1) is rather fast with 15.8, 14.2, and 12.9 ns in toluene, THF, and benzonitrile, respectively, the corresponding (T1 + T1) quantum yield is still reasonably high (52% in toluene, 49% in THF, and 46% in benzonitrile).
As a complement to the excited-state behavior of Pt(Lpc)(Lref)Cl2 and Pt(Lpc)2Cl2, the Pd(II) monomer Pd(Lpc)(Lref)Cl2 and the dimer Pd(Lpc)2Cl2 were examined. A kinetic model with three species, as used for Pt(Lpc)(Lref)Cl2, was also a good fit for the raw data of Pt(Lpc)(Lref)Cl2 (Table 2, Figures S46–S51). In particular, the first, second, and third species are assigned to (S1), (S1)rel, and (T1), respectively. In comparison to Pt(Lpc)(Lref)Cl2, the lifetimes of (S1)rel and (T1) are slightly longer, especially in BN with 4.2 ns and 13.6 μs, respectively, as a result of the weaker HAE from Pd(II). The longer (S1)rel lifetimes result in triplet quantum yields that are intermediate between those for Lpc and Pt(Lpc)(Lref)Cl2, namely, around 4%.
Table 2. Species and Lifetimes of Lpc, Pt(Lpc)(Lref)Cl2, Pd(Lpc)(Lref)Cl2, Pt(Lpc)2Cl2, and Pd(Lpc)2Cl2 Were Obtained from fs-TAS and ns-TAS Measurements.
|
Species |
||||||
|---|---|---|---|---|---|---|
| Compound | Solvent | τ(S1)/ps | τ(S1)rel/ns | τ(T1)/μs | ||
| Lpc | toluene | 8.0 | 12.4 | 27.1 | ||
| THF | 2.9 | 9.2 | 25.6 | |||
| BN | 10.7 | 4.7 | 20.2 | |||
| Pt(Lpc)(Lref)Cl2 | toluene | 6.7 | 4.9 | 7.9 | ||
| THF | 3.3 | 2.2 | 7.2 | |||
| BN | 11.3 | 0.7 | 7.4 | |||
| Pd(Lpc)(Lref)Cl2 | toluene | 6.7 | 4.2 | 18.5 | ||
| THF | 3.7 | 2.7 | 8.8 | |||
| BN | 8.9 | 4.2 | 13.6 | |||
| τ(S1S0)/ps | τ(S1S0)rel/ns | τ(T1T1)/ns | τ(T1+T1)/μs | |||
| Pt(Lpc)2Cl2 | toluene | 5.3 | 2.8 | 15.8 | 8.0 | |
| THF | 2.6 | 1.3 | 14.2 | 8.0 | ||
| BN | 6.3 | 0.5 | 12.9 | 7.0 | ||
| τ(S1S0)/ps | τ(S1S0)rel/ns | τ(M)/ns | τ(T1T1)/ns | τ(T1+T1)/μs | ||
| Pd(Lpc)2Cl2 | toluene | 4.0 | 3.1 | – | 138.1 | 25.7 |
| THF | 2.7 | 2.0 | 7.3 | 169.6 | 26.4 | |
| BN | 10.0 | 0.9 | 4.6 | 122.3 | 23.8 | |
For dimer Pd(Lpc)2Cl2 in toluene, the fs- and ns-TAS required the same kinetic model, which is based on four species as Pt(Lpc)2Cl2 to fit the raw data (Table 2, Figures 3 and 4, and Figures S52–S57). The first and second species feature ESA and GSB features of the singlet excited state, as established for Pt(Lpc)2Cl2. The lifetime (4.0 ps) of the first species is completely consistent with the unrelaxed state (S1S0), while that of the second species (3.1 ns) is assigned to the solvent-relaxed singlet (S1S0)rel. It is noteworthy that the weaker HAE of Pd(II) slightly retards the decay of (S1S0)rel in Pd(Lpc)2Cl2 (3.1 ns in toluene) relative to that of Pt(Lpc)2Cl2 (2.8 ns in toluene). ESA and GSB spectra confirm the triplet excited state character of the fourth and fifth species, which are thus assigned as 1(T1T1) and (T1 + T1). The changes of lifetimes for 1(T1T1) and (T1 + T1) are strongly metal dependent. For Pd(Lpc)2Cl2, the lifetime of 1(T1T1) is roughly an order of magnitude longer than Pt(Lpc)2Cl2, and it leads to decoherence times in the range of hundreds of nanoseconds. Due to the weak HAE of Pd, as well as the weak interpentacene coupling, we posit that the long-lived triplet pair is the result of an unresolvable equilibrium among the approximately isoenergetic states 1(T1T1), 3(T1T1), and 5(T1T1), namely, 1(T1T1) ⇔ 3(T1T1) ⇔ 5(T1T1). Similar relationships have been established for the behavior of other weakly coupled systems.7,65 The lifetime of (T1 + T1) for Pd(Lpc)2Cl2 (25.7 μs) is about three times that of Pt(Lpc)2Cl2 (8.0 μs) and, as a consequence, comparable to that of (T1) (Table 2).
Figure 4.

(a) fs-TAS (λex = 610 nm, 400 nJ) of Pd(Lpc)2Cl2 in BN with time delays between 0–5500 ps, together with the respective (b) deconvoluted spectra representing the singlet (S1S0) (black), solvent-relaxed excited state (S1S0)rel (red) excited states, and mixed singlet/triplet/charge transfer intermediate state (M) (brown) as obtained by target analysis. (c) ns-TAS (λex = 610 nm, 400 nJ) of Pd(Lpc)2Cl2 in BN with time delays between 0 ns–400 μs, together with the respective (d) deconvoluted spectra representing the solvent-relaxed singlet (S1S0)Sol (red), mixed intermediate (M) (brown), correlated triplet pair (T1T1) (orange), and uncorrelated triplet (T1 + T1) (green) excited states as obtained by target analysis.
For Pd(Lpc)2Cl2 in the polar solvents THF and benzonitrile, a different picture is observed. The fs- and ns-TAS of Pd(Lpc)2Cl2 in polar solvents required a kinetic model that included five species to fit the raw data. It is noted that 1(T1T1) is not formed directly from (S1S0)rel in the case of Pd(Lpc)2Cl2 in polar solvents. Rather, it is formed via a third species, which is best described as a spectroscopic superimposition of singlet and triplet excited state features that mix with those of a charge transfer state (M).17,66 Furthermore, (M) is observed only in the more polar solvents THF and benzonitrile, supporting its charge transfer character. Such an indirect mechanism, that is, the population of an SF intermediate, significantly impacts the quantum yield of 1(T1T1) relative to a direct mechanism. Thus, the quantum yield for Pd(Lpc)2Cl2 is around 40%, relative to 140–200% found for Pt(Lpc)2Cl2. Turning to the quantum yields for decoherence, comparable values are derived in toluene (58%), THF (54%), and benzonitrile (55%). The kinetic models used for the pentacene dimers Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2 are shown in Figure 5, which depicts the influence of different metal ions, namely, Pt(II) and Pd(II), on the formation of 1(T1T1).
Figure 5.

Kinetic models used to fit the fs-TAS and ns-TAS data for (a) Pt(Lpc)2Cl2 via relaxation, iSF, and decoherence, and (b) Pd(Lpc)2Cl2 via relaxation, iSF, and decoherence in toluene, or via relaxation, an intermediate (M), iSF, and decoherence in THF and benzonitrile.
Conclusion
A pyridyl group has been appended to the pentacenyl chromophore, providing a ligand for the formation of complexes with platinum(II) and palladium(II). The third row metal transition metal, Pt(II), facilitates interpentacene electronic communication that leads to iSF in the dimer Pt(Lpc)2Cl2 via a direct mechanism. In the polar solvent benzonitrile, iSF is very efficient and gives quantum yields of the correlated triplet (T1T1) as high as 200%. For the second row transition metal Pd(II), electronic communication is also present but weaker. Thus, iSF is observed for Pd(Lpc)2Cl2, but through a mechanism that requires a mediating species in polar solvents. In view of the fact that the dimers are structurally identical, aside from the metal atom, mechanistic changes are attributed to differences in the size and polarizability of the metal species. Furthermore, it is noteworthy that iSF is operative in both Pt(Lpc)2Cl2 and Pd(Lpc)2Cl2, despite a rather large interpentacene separation in both dimers as well as enhanced spin–orbit coupling from the presence of heavy atoms.
Methods
Hazards
No unexpected or unusually high safety hazards were encountered during the course of this research.
Synthesis of Pt(Lpc)2Cl2
A mixture of Lpc (36 mg, 0.042 mmol) and Pt(PhCN)2Cl2 (10 mg, 0.021 mmol) in dry toluene (5 mL) was stirred at 80 °C for 16 h under an atmosphere of argon. The flask was wrapped in aluminum foil during the reaction to limit the light exposure. The reaction mixture was cooled to room temperature, and the solvent was then removed under reduced pressure. The residue was purified by column chromatography (alumina, CH2Cl2). The eluent volume was reduced in vacuo to about 1 mL followed by the addition of hexanes (15 mL). The resulting suspension was filtered, and the residue was washed with hexanes (3 × 2 mL), affording Pt(Lpc)2Cl2 as a dark blue solid (32 mg, 76%). Mp 322 °C (decomp). Rf = 0.50 (CH2Cl2/hexanes 1:2). UV–vis (CH2Cl2) λmax (ε) 271 (128000), 306 (sh, 202000), 315 (544000), 347 (33200), 407 (36700), 441 (15600), 577 (sh, 13200), 626 (34700), 679 nm (63500). IR (CH2Cl2, cast) 3086 (w), 3050 (w), 2953 (s), 2927 (m), 2901 (m), 2867 (m), 2168 (m), 2122 (w), 1590 (m), 1461 (m) cm–1. 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 4H), 9.03 (s, 4H), 8.37 (s, 4H), 7.94–7.86 (m, 12H), 7.56–7.50 (m, 12H), 7.41–7.33 (m, 8H), 2.18 (nonet, J = 6.6 Hz, 6H), 1.20 (s, 36H), 1.19 (d, J = 6.6 Hz, 36H), 0.96 (d, J = 7.0 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ 152.5, 151.9, 140.2, 133.0, 132.4, 132.3, 130.5, 130.4, 129.5, 129.2, 128.4, 126.2, 126.1, 125.9, 125.7, 120.3, 116.4, 111.2, 104.6, 102.0, 100.4, 34.8, 31.2, 26.6, 25.5, 25.4 (two signals coincident or not observed). MALDI HRMS (DCTB) calcd for C126H13435Cl2N2195PtSi2 (M+) 1995.9105, found 1995.9082. DSC: Decomposition, 347 °C (onset), and 353 °C (peak).
Synthesis of Pd(Lpc)2Cl2
A mixture of Lpc (45 mg, 0.052 mmol) and Pd(PhCN)2Cl2 (10 mg, 0.026 mmol) in dry CH2Cl2 (5 mL) was stirred at room temperature for 15 h under an atmosphere of argon. The flask was wrapped in aluminum foil during the reaction to limit light exposure. The reaction mixture was plugged through a pad of alumina with CH2Cl2. The eluent volume was reduced in vacuo to ca. 1 mL followed by the addition of hexanes (15 mL). The resulting suspension was filtered, and the residue was washed with hexanes (3 × 2 mL), affording Pd(Lpc)2Cl2 as a dark blue-green solid (34 mg, 69%). Mp 309 °C (decomp). Rf = 0.75 (CH2Cl2/hexanes 1:1). UV–vis (CH2Cl2) λmax (ε) 271 (118000), 306 (sh, 183000), 315 (507000), 348 (28000), 400 (29600), 441 (12400), 573 (sh, 10700), 625 (31300), 677 nm (57100). IR (CH2Cl2, cast) 3080 (w), 3050 (w), 2953 (s), 2927 (m), 2902 (m), 2867 (m), 2166 (m), 2120 (w), 1588 (m), 1461 (m) cm–1. 1H NMR (500 MHz, CDCl3) δ 9.18 (s, 4H), 8.95 (s, 4H), 8.36 (s, 4H), 7.92–7.87 (m, 12H), 7.56–7.50 (m, 12H), 7.41–7.33 (m, 8H), 2.18 (nonet, J = 6.7 Hz, 6H), 1.20 (s, 36H), 1.18 (d, J = 6.7 Hz, 36H), 0.96 (d, J = 7.0 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ 152.5, 151.6, 140.0, 133.1, 132.4, 132.3, 130.8, 130.5, 130.4, 129.5, 129.2, 128.4, 126.2, 126.1, 125.9, 125.7, 120.4, 116.3, 111.2, 104.5, 101.9, 100.3, 34.8, 31.2, 26.6, 25.5, 25.4 (one signal coincident or not observed). MALDI HRMS (DCTB) calcd for C126H13435Cl2N2106PdSi2 (M+) 1906.8492, found 1906.8510. DSC: Decomposition, 321 °C (onset), and 330 °C (peak).
Spectroscopy
Steady-state absorption measurements were performed on a PerkinElmer Lambda2 UV/vis two-beam spectrophotometer with a slit width of 2 nm and a scan rate of 240 nm/min. Steady-state fluorescence measurements were performed on a Horiba Jobin Yvon FluoroMax-3 spectrometer with a slit width of 3 nm for excitation and emission and an integration time of 0.1 s within a wavelength range between 625–900 nm. Fluorescence quantum yields (FQYs) were determined from corrected emission spectra following the standard methods using zinc 2,9,16,23-tetra-tert-butyl-29H,31H-phthalocyanine (ZnPc – FQY = 0.3 in toluene)67 as standard for all the pentacene monomer references and pentacene dimers. Femto- (fs-TAS) and nanosecond (ns-TAS) transient absorption spectroscopy measurements were performed on an amplified CPA-2110 titanium:sapphire laser for excitation (1 kHz; 150 fs pulse width; 400 nJ laser energy) from Clark-MXR, Inc. In the EOS SYSTEM from Ultrafast Systems, a 1 kHz pump laser was used at an excitation wavelength of 610 nm. Probing was executed on a 2 kHz continuous white light fiber laser. Data evaluation of the fs-TAS and ns-TAS raw data has been conducted by means of multiwavelength and global/target analysis using the GloTarAn68 package. Target analysis was performed on the TAS raw data sets using the proposed kinetic models. The analytic solution to the coupled differential equations that describe the kinetic model is convoluted with a Gaussian instrument response function. After the least-squares fitting has converged, the raw data matrix is deconvoluted using the specific solution to the kinetic model and parameters from the fit to obtain the species-associated spectra and their populations as a function of time.
Acknowledgments
The authors thank the financial support from Natural Sciences and Engineering Research Council of Canada (NSERC), Canada Foundation for Innovation (CFI), and “Solar Energy goes Hybrid” Initiative of the Bavarian Ministry for Science, Culture and Education (SolTech). Y.B. acknowledges a fellowship from the Chinese Scholarship Council.
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2288684–2288686. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/prechem.3c00082.
General experimental methods, descriptions of synthetic route, experimental data, and HRMS and 1H/13C NMR spectra; figures and tables illustrating results from ground state and excited spectroscopy, time-resolved transient absorption spectroscopy, and kinetic models (PDF)
Author Present Address
∥ Department of Applied Chemistry, Graduate School of Engineering, Center for Molecular Systems (CMS), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
Author Contributions
§ Y.H. and I.P. contributed equally. R.R.T. and D.M.G. designed and oversaw the project. R.R.T and Y.H. designed the molecules. Y.H. synthesized and characterized the molecules. I.P., Y.B., A.-S.W., and L.A.M carried out photophysical characterization. M.J.F. conducted X-ray crystallographic characterization, refinement, and analysis. Y.H, I.P., Y.B., D.M.G., and R.R.T. wrote the paper with contributions from all authors. All authors analyzed the results and commented on the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Shockley W.; Queisser H. J. Detailed Balance Limit of Efficiency of p-n Junction Solar Cells. J. Appl. Phys. 1961, 32, 510–519. 10.1063/1.1736034. [DOI] [Google Scholar]
- Smith M. B.; Michl J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. 10.1021/cr1002613. [DOI] [PubMed] [Google Scholar]
- Xia J.; Sanders S. N.; Cheng W.; Low J. Z.; Liu J.; Campos L. M.; Sun T. Singlet Fission: Progress and Prospects in Solar Cells. Adv. Mater. 2017, 29, 1601652 10.1002/adma.201601652. [DOI] [PubMed] [Google Scholar]
- Rao A.; Friend R. H. Harnessing Singlet Exciton Fission to Break the Shockley-Queisser Limit. Nat. Rev. Mater. 2017, 2, 17063. 10.1038/natrevmats.2017.63. [DOI] [Google Scholar]
- Michl J. Unconventional Solar Energy: Singlet Fission. Mol. Front. J. 2019, 03, 84–91. 10.1142/S2529732519400042. [DOI] [Google Scholar]
- Gish M. K.; Pace N. A.; Rumbles G.; Johnson J. C. Emerging Design Principles for Enhanced Solar Energy Utilization with Singlet Fission. J. Phys. Chem. C 2019, 123, 3923–3934. 10.1021/acs.jpcc.8b10876. [DOI] [Google Scholar]
- Basel B. S.; Zirzlmeier J.; Hetzer C.; Phelan B. T.; Krzyaniak M. D.; Reddy S. R.; Coto P. B.; Horwitz N. E.; Young R. M.; White F. J.; Hampel F.; Clark T.; Thoss M.; Tykwinski R. R.; Wasielewski M. R.; Guldi D. M. Unified Model for Singlet Fission within a Non-conjugated Covalent Pentacene Dimer. Nat. Commun. 2017, 8, 15171. 10.1038/ncomms15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer C.; Guldi D. M.; Tykwinski R. R. Pentacene Dimers as a Critical Tool for the Investigation of Intramolecular Singlet Fission. Chem. Eur. J. 2018, 24, 8245–8257. 10.1002/chem.201705355. [DOI] [PubMed] [Google Scholar]
- Sakai H.; Inaya R.; Nagashima H.; Nakamura S.; Kobori Y.; Tkachenko N. V.; Hasobe T. Multiexciton Dynamics Depending on Intramolecular Orientations in Pentacene Dimers: Recombination and Dissociation of Correlated Triplet Pairs. J. Phys. Chem. Lett. 2018, 9, 3354–3360. 10.1021/acs.jpclett.8b01184. [DOI] [PubMed] [Google Scholar]
- Hudson R. J.; Stuart A. N.; Huang D. M.; Kee T. W. What Next for Singlet Fission in Photovoltaics? The Fate of Triplet and Triplet-Pair Excitons. J. Phys. Chem. C 2022, 126, 5369–5377. 10.1021/acs.jpcc.2c00273. [DOI] [Google Scholar]
- Wang T.; Liu H.; Wang X.; Tang L.; Zhou J.; Song X.; Lv L.; Chen W.; Chen Y.; Li X. Intramolecular Singlet Fission and Triplet Exciton Harvesting in Tetracene Oligomers for Solar Energy Conversion. J. Mater. Chem. A 2023, 11, 8515–8539. 10.1039/D3TA00193H. [DOI] [Google Scholar]
- Smith M. B.; Michl J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. 10.1146/annurev-physchem-040412-110130. [DOI] [PubMed] [Google Scholar]
- Ullrich T.; Munz D.; Guldi D. M. Unconventional Singlet Fission Materials. Chem. Soc. Rev. 2021, 50, 3485–3518. 10.1039/D0CS01433H. [DOI] [PubMed] [Google Scholar]
- Casillas R.; Papadopoulos I.; Ullrich T.; Thiel D.; Kunzmann A.; Guldi D. M. Molecular Insights and Concepts to Engineer Singlet Fission Energy Conversion Devices. Energy Environ. Sci. 2020, 13, 2741–2804. 10.1039/D0EE00495B. [DOI] [Google Scholar]
- Walker B. J.; Musser A. J.; Beljonne D.; Friend R. H. Singlet Exciton Fission in Solution. Nat. Chem. 2013, 5, 1019–1024. 10.1038/nchem.1801. [DOI] [PubMed] [Google Scholar]
- Yost S. R.; Lee J.; Wilson M. W. B.; Wu T.; McMahon D. P.; Parkhurst R. R.; Thompson N. J.; Congreve D. N.; Rao A.; Johnson K.; Sfeir M. Y.; Bawendi M. G.; Swager T. M.; Friend R. H.; Baldo M. A.; Van Voorhis T. A Transferable Model for Singlet-Fission Kinetics. Nat. Chem. 2014, 6, 492–497. 10.1038/nchem.1945. [DOI] [PubMed] [Google Scholar]
- Papadopoulos I.; Zirzlmeier J.; Hetzer C.; Bae Y. J.; Krzyaniak M. D.; Wasielewski M. R.; Clark T.; Tykwinski R. R.; Guldi D. M. Varying the Interpentacene Electronic Coupling to Tune Singlet Fission. J. Am. Chem. Soc. 2019, 141, 6191–6203. 10.1021/jacs.8b09510. [DOI] [PubMed] [Google Scholar]
- Papadopoulos I.; Reddy S. R.; Coto P. B.; Lehnherr D.; Thiel D.; Thoss M.; Tykwinski R. R.; Guldi D. M. Parallel versus Twisted Pentacenes: Conformational Impact on Singlet Fission. J. Phys. Chem. Lett. 2022, 13, 5094–5100. 10.1021/acs.jpclett.2c01395. [DOI] [PubMed] [Google Scholar]
- Yablon L. M.; Sanders S. N.; Miyazaki K.; Kumarasamy E.; He G.; Choi B.; Ananth N.; Sfeir M. Y.; Campos L. M. Singlet Fission and Triplet Pair Recombination in Bipentacenes with a Twist. Mater. Horizons 2022, 9, 462–470. 10.1039/D1MH01201K. [DOI] [PubMed] [Google Scholar]
- Korovina N. V.; Joy J.; Feng X.; Feltenberger C.; Krylov A. I.; Bradforth S. E.; Thompson M. E. Linker-Dependent Singlet Fission in Tetracene Dimers. J. Am. Chem. Soc. 2018, 140, 10179–10190. 10.1021/jacs.8b04401. [DOI] [PubMed] [Google Scholar]
- Mattos R. S.; Burghardt I.; Aquino A. J. A.; Cardozo T. M.; Lischka H. On the Cooperative Origin of Solvent-Enhanced Symmetry-Breaking Charge Transfer in a Covalently Bound Tetracene Dimer Leading to Singlet Fission. J. Am. Chem. Soc. 2022, 144, 23492–23504. 10.1021/jacs.2c10129. [DOI] [PubMed] [Google Scholar]
- Mardazad S.; Xu Y.; Yang X.; Grundner M.; Schollwöck U.; Ma H.; Paeckel S. Quantum Dynamics Simulation of Intramolecular Singlet Fission in Covalently Linked Tetracene Dimer. J. Chem. Phys. 2021, 155, 194101 10.1063/5.0068292. [DOI] [PubMed] [Google Scholar]
- Buchanan E. A.; Havlas Z.; Michl J. Optimal Arrangements of Tetracene Molecule Pairs for Fast Singlet Fission. Bull. Chem. Soc. Jpn. 2019, 92, 1960–1971. 10.1246/bcsj.20190229. [DOI] [PubMed] [Google Scholar]
- Qiao X.; Ma D. Nonlinear Optoelectronic Processes in Organic Optoelectronic Devices: Triplet-triplet Annihilation and Singlet Fission. Mater. Sci. Eng., R 2020, 139, 100519 10.1016/j.mser.2019.100519. [DOI] [Google Scholar]
- Minami T.; Ito S.; Nakano M. Theoretical Study of Singlet Fission in Oligorylenes. J. Phys. Chem. Lett. 2012, 3, 2719–2723. 10.1021/jz3011749. [DOI] [PubMed] [Google Scholar]
- Monahan N.; Zhu X.-Y. Charge Transfer–Mediated Singlet Fission. Annu. Rev. Phys. Chem. 2015, 66, 601–618. 10.1146/annurev-physchem-040214-121235. [DOI] [PubMed] [Google Scholar]
- Felter K. M.; Grozema F. C. Singlet Fission in Crystalline Organic Materials: Recent Insights and Future Directions. J. Phys. Chem. Lett. 2019, 10, 7208–7214. 10.1021/acs.jpclett.9b00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas R.; Adam M.; Coto P. B.; Waterloo A. R.; Zirzlmeier J.; Reddy S. R.; Hampel F.; McDonald R.; Tykwinski R. R.; Thoss M.; Guldi D. M. Intermolecular Singlet Fission in Unsymmetrical Derivatives of Pentacene in Solution. Adv. Energy Mater. 2019, 9, 1802221 10.1002/aenm.201802221. [DOI] [Google Scholar]
- Tayebjee M. J. Y.; Schwarz K. N.; MacQueen R. W.; Dvořák M.; Lam A. W. C.; Ghiggino K. P.; McCamey D. R.; Schmidt T. W.; Conibeer G. Morphological Evolution and Singlet Fission in Aqueous Suspensions of TIPS-Pentacene Nanoparticles. J. Phys. Chem. C 2016, 120, 157–165. 10.1021/acs.jpcc.5b11353. [DOI] [Google Scholar]
- Stuart A. N.; Tapping P. C.; Schrefl E.; Huang D. M.; Kee T. W. Controlling the Efficiency of Singlet Fission in TIPS-Pentacene/Polymer Composite Nanoparticles. J. Phys. Chem. C 2019, 123, 5813–5825. 10.1021/acs.jpcc.8b10163. [DOI] [Google Scholar]
- Gao C.; Prasad S. K. K.; Zhang B.; Dvořák M.; Tayebjee M. J. Y.; McCamey D. R.; Schmidt T. W.; Smith T. A.; Wong W. W. H. Intramolecular Versus Intermolecular Triplet Fusion in Multichromophoric Photochemical Upconversion. J. Phys. Chem. C 2019, 123, 20181–20187. 10.1021/acs.jpcc.9b07098. [DOI] [Google Scholar]
- Korovina N. V.; Pompetti N. F.; Johnson J. C. Lessons from Intramolecular Singlet Fission with Covalently Bound Chromophores. J. Chem. Phys. 2020, 152, 040904 10.1063/1.5135307. [DOI] [PubMed] [Google Scholar]
- Zeng T.; Goel P. Design of Small Intramolecular Singlet Fission Chromophore : An Azaborine Candidate and General Small Size Effects. J. Phys. Chem. Lett. 2016, 7, 1351–1358. 10.1021/acs.jpclett.6b00356. [DOI] [PubMed] [Google Scholar]
- Busby E.; Xia J.; Wu Q.; Low J. Z.; Song R.; Miller J. R.; Zhu X. Y.; Campos L. M.; Sfeir M. Y. A Design Strategy for Intramolecular Singlet Fission Mediated by Charge-Transfer States in Donor-Acceptor Organic Materials. Nat. Mater. 2015, 14, 426–433. 10.1038/nmat4175. [DOI] [PubMed] [Google Scholar]
- Hu J.; Xu K.; Shen L.; Wu Q.; He G.; Wang J.-Y.; Pei J.; Xia J.; Sfeir M. Y. New Insights into the Design of Conjugated Polymers for Intramolecular Singlet Fission. Nat. Commun. 2018, 9, 2999. 10.1038/s41467-018-05389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Nagami T.; Nakano M. Design Principles of Electronic Couplings for Intramolecular Singlet Fission in Covalently-Linked Systems. J. Phys. Chem. A 2016, 120, 6236–6241. 10.1021/acs.jpca.6b07153. [DOI] [PubMed] [Google Scholar]
- Miller J. C.; Meek J. S.; Strickler S. J. Heavy Atom Effects on the Triplet Lifetimes of Naphthalene and Phenanthrene. J. Am. Chem. Soc. 1977, 99, 8175–8179. 10.1021/ja00467a008. [DOI] [Google Scholar]
- Lin L.; Zhu J. Computational Predictions of Adaptive Aromaticity for the Design of Singlet Fission Materials. Inorg. Chem. Front. 2022, 9, 914–924. 10.1039/D1QI01442K. [DOI] [Google Scholar]
- Lubert-Perquel D.; Salvadori E.; Dyson M.; Stavrinou P. N.; Montis R.; Nagashima H.; Kobori Y.; Heutz S.; Kay C. W. M. Identifying Triplet Pathways in Dilute Pentacene Films. Nat. Commun. 2018, 9, 4222. 10.1038/s41467-018-06330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel B. S.; Young R. M.; Krzyaniak M. D.; Papadopoulos I.; Hetzer C.; Gao Y.; La Porte N. T.; Phelan B. T.; Clark T.; Tykwinski R. R.; Wasielewski M. R.; Guldi D. M. Influence of the Heavy-Atom Effect on Singlet Fission: A Study of Platinum-Bridged Pentacene Dimers. Chem. Sci. 2019, 10, 11130–11140. 10.1039/C9SC04410H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity N.; Kim W.; Panjwani N. A.; Kundu A.; Majumder K.; Kasetty P.; Mishra D.; Bittl R.; Nagesh J.; Dasgupta J.; Musser A. J.; Patil S. Parallel Triplet Formation Pathways in a Singlet Fission Material. Nat. Commun. 2022, 13, 5244. 10.1038/s41467-022-32844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser A. J.; Al-Hashimi M.; Heeney M.; Clark J. Heavy-atom Effects on Intramolecular Singlet Fission in a Conjugated Polymer. J. Chem. Phys. 2019, 151, 044902 10.1063/1.5110269. [DOI] [PubMed] [Google Scholar]
- Papadopoulos I.; Gao Y.; Hetzer C.; Tykwinski R. R.; Guldi D. M. Singlet Fission in Enantiomerically Pure Pentacene Dimers. ChemPhotoChem 2020, 4, 5168–5174. 10.1002/cptc.202000016. [DOI] [Google Scholar]
- Hou Y.; Papadopoulos I.; Ferguson M. J.; Jux N.; Tykwinski R. R.; Guldi D. M. Photophysical Characterization of a Ruthenium-based Tetrameric Pentacene Complex. J. Porphyrins Phthalocyanines 2023, 27, 686–693. 10.1142/S1088424623500645. [DOI] [Google Scholar]
- Zirzlmeier J.; Lehnherr D.; Coto P. B.; Chernick E. T.; Casillas R.; Basel B. S.; Thoss M.; Tykwinski R. R.; Guldi D. M. Singlet Fission in Pentacene Dimers. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 5325–5330. 10.1073/pnas.1422436112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribson R. D.; Choi G.; Hadt R. G.; Agapie T. Controlling Singlet Fission with Coordination Chemistry-Induced Assembly of Dipyridyl Pyrrole Bipentacenes. ACS Cent. Sci. 2020, 6, 2088–2096. 10.1021/acscentsci.0c01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E.; Raimondi D. L.; Reinhardt W. P. Atomic Screening Constants from SCF Functions. II. Atoms with 37 to 86 Electrons. J. Chem. Phys. 1967, 47, 1300–1307. 10.1063/1.1712084. [DOI] [Google Scholar]
- Strassert C. A.; Mauro M.; De Cola L. Photophysics of Soft and Hard Molecular Assemblies Based on Luminescent Complexes. Adv. Inorg. Chem. 2011, 63, 47–103. 10.1016/B978-0-12-385904-4.00009-3. [DOI] [Google Scholar]
- Theiss T.; Buss S.; Maisuls I.; López-Arteaga R.; Brünink D.; Kösters J.; Hepp A.; Doltsinis N. L.; Weiss E. A.; Strassert C. A. Room-Temperature Phosphorescence from Pd(II) and Pt(II) Complexes as Supramolecular Luminophores: The Role of Self-Assembly, Metal–Metal Interactions, Spin–Orbit Coupling, and Ligand-Field Splitting. J. Am. Chem. Soc. 2023, 145, 3937–3951. 10.1021/jacs.2c09775. [DOI] [PubMed] [Google Scholar]
- 1H–13C HSQC NMR spectra for Lpc, Pt(Lpc)2Cl2, and Pd(Lpc)2Cl2 are used to identify signals of the pyridyl protons (Figure S13).
- Tessier C.; Rochon F. D. Multinuclear NMR Study and Crystal Structures of Complexes of the Types cis- and trans-Pt(Ypy)2X2, where Ypy = pyridine derivative and X = Cl and I. Inorg. Chim. Acta 1999, 295, 25–38. 10.1016/S0020-1693(99)00303-5. [DOI] [Google Scholar]
- Pazderski L.; Toušek J.; Sitkowski J.; Maliňáková K.; Kozerski L.; Szłyk E. Experimental and Quantum-chemical Studies of 1H, 13C and 15N NMR Coordination Shifts in Au(III), Pd(II) and Pt(II) Chloride Complexes with Picolines. Magn. Reson. Chem. 2009, 47, 228–238. 10.1002/mrc.2378. [DOI] [PubMed] [Google Scholar]
- Pazderski L.; Pawlak T.; Sitkowski J.; Kozerski L.; Szłyk E. Structural Correlations for 1H, 13C and 15N NMR Coordination Shifts in Au(III), Pd(II) and Pt(II) Chloride Complexes with Lutidines and Collidine. Magn. Reson. Chem. 2010, 48, 417–426. 10.1002/mrc.2594. [DOI] [PubMed] [Google Scholar]
- Lewis N. A.; Pakhomova S.; Marzilli P. A.; Marzilli L. G. Synthesis and Characterization of Pt(II) Complexes with Pyridyl Ligands: Elongated Octahedral Ion Pairs and Other Factors Influencing 1H NMR Spectra. Inorg. Chem. 2017, 56, 9781–9793. 10.1021/acs.inorgchem.7b01294. [DOI] [PubMed] [Google Scholar]
- Kurpik G.; Walczak A.; Gołdyn M.; Harrowfield J.; Stefankiewicz A. R. Pd(II) Complexes with Pyridine Ligands: Substituent Effects on the NMR Data, Crystal Structures, and Catalytic Activity. Inorg. Chem. 2022, 61, 14019–14029. 10.1021/acs.inorgchem.2c01996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M.-H.; Yip J. H. K. Platinum-Conjugated Homo- and Heterobichromophoric Complexes of Tetracene and Pentacene. Organometallics 2012, 31, 7522–7531. 10.1021/om300815b. [DOI] [Google Scholar]
- TQY 4% versus 6.7% in THF and 1% versus 7.1% in benzonitrile for Lpc and Pt(Lpc)(Lref)Cl2, respectively.
- TQY determination via the traditional bleaching method is rather unprecise as GSB of (S1S0)rel nearly completely depletes and, in turn, cannot be used to normalize the SASs.
- Kolomeisky A. B.; Feng X.; Krylov A. I. A Simple Kinetic Model for Singlet Fission: A Role of Electronic and Entropic Contributions to Macroscopic Rates. J. Phys. Chem. C 2014, 118, 5188–5195. 10.1021/jp4128176. [DOI] [Google Scholar]
- Schrauben J. N.; Akdag A.; Wen J.; Havlas Z.; Ryerson J. L.; Smith M. B.; Michl J.; Johnson J. C. Excitation Localization/Delocalization Isomerism in a Strongly Coupled Covalent Dimer of 1,3-Diphenylisobenzofuran. J. Phys. Chem. A 2016, 120, 3473–3483. 10.1021/acs.jpca.6b00826. [DOI] [PubMed] [Google Scholar]
- Casanova D. Theoretical Modeling of Singlet Fission. Chem. Rev. 2018, 118, 7164–7207. 10.1021/acs.chemrev.7b00601. [DOI] [PubMed] [Google Scholar]
- Bencini A.; Gatteschi D.. Electron Paramagnetic Resonance of Exchange Coupled Systems; Springer-Verlag: 1990. [Google Scholar]
- Benk H.; Sixl H. Theory of Two Coupled Triplet States. Mol. Phys. 1981, 42, 779–801. 10.1080/00268978100100631. [DOI] [Google Scholar]
- Chen M.; Krzyaniak M. D.; Nelson J. N.; Bae Y. J.; Harvey S. M.; Schaller R. D.; Young R. M.; Wasielewski M. R. Quintet-triplet Mixing Determines the Fate of the Multiexciton State Produced by Singlet Fission in a Terrylenediimide Dimer at Room Temperature. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 8178–8183. 10.1073/pnas.1820932116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel B. S.; Zirzlmeier J.; Hetzer C.; Phelan B. T.; Krzyaniak M. D.; Reddy S. R.; Coto P. B.; Horwitz N. E.; Young R. M.; White F. J.; Hampel F.; Clark T.; Thoss M.; Tykwinski R. R.; Wasielewski M. R.; Guldi D. M. Unified model for singlet fission within a non-conjugated covalent pentacene dimer. Nat. Comm 2017, 8, 15171. 10.1038/ncomms15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos I.; Zirzlmeier J.; Hetzer C.; Bae Y. J.; Krzyaniak M. D.; Wasielewski M. R.; Clark T.; Tykwinski R. R.; Guldi D. M. Varying the Interpentacene Electronic Coupling to Tune Singlet Fission. J. Am. Chem. Soc. 2019, 141, 6191–6203. 10.1021/jacs.8b09510. [DOI] [PubMed] [Google Scholar]
- Vincett P. S.; Voigt E. M.; Rieckhoff K. E. Phosphorescence and Fluorescence of Phthalocyanines. J. Chem. Phys. 1971, 55, 4131–4140. 10.1063/1.1676714. [DOI] [Google Scholar]
- Snellenburg J. J.; Laptenok S.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. 10.18637/jss.v049.i03. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2288684–2288686. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.