

Abstract
Nonsense-mediated RNA decay (NMD) is an evolutionarily conserved RNA quality control process that serves both as a mechanism to eliminate aberrant transcripts carrying premature stop codons, and to regulate expression of some normal transcripts. For a quality control process, NMD exhibits surprising variability in its efficiency across transcripts, cells, tissues, and individuals in both physiological and pathological contexts. Whether an aberrant RNA is spared or degraded, and by what mechanism, could determine the phenotypic outcome of a disease-causing mutation. Hence, understanding the variability in NMD is not only important for clinical interpretation of genetic variants but also may provide clues to identify novel therapeutic approaches to counter genetic disorders caused by nonsense mutations. Here, we discuss the current knowledge of NMD variability and the mechanisms that allow certain transcripts to escape NMD despite the presence of NMD-inducing features.
Graphical Abstract

Variation in RNA quality control by NMD can lead to different gene expression outcomes of nonsense-containing transcripts.
Introduction
Nonsense-mediated RNA decay (NMD) safeguards cells against aberrant mRNAs and their translation products (Brogna et al., 2016; Celik et al., 2017). In mammalian cells, a ribosome translating a normal transcript evicts exon junction complexes (EJCs), which are protein marks left behind by the splicing machinery, and other RNA binding proteins (RBPs) as it makes its way through the transcript to the normal stop codon (Figure 1A). However, in the case of an aberrant transcript that contains a premature termination codon (PTC), the PTC causes the ribosome to terminate before it reaches the normal stop codon, thus failing to evict downstream EJCs. The continued presence of an EJC or other RBPs downstream of the terminating ribosome acts as a signal to recruit NMD machinery and initiate destruction of the aberrant mRNA (Figure 1B; (Fatscher et al., 2015; Hug et al., 2016)). As a result, nonsense mutations often inactivate genes and cause loss of function (Kurosaki and Maquat, 2016; Miller and Pearce, 2014).
Figure 1. Mammalian model of NMD activation.
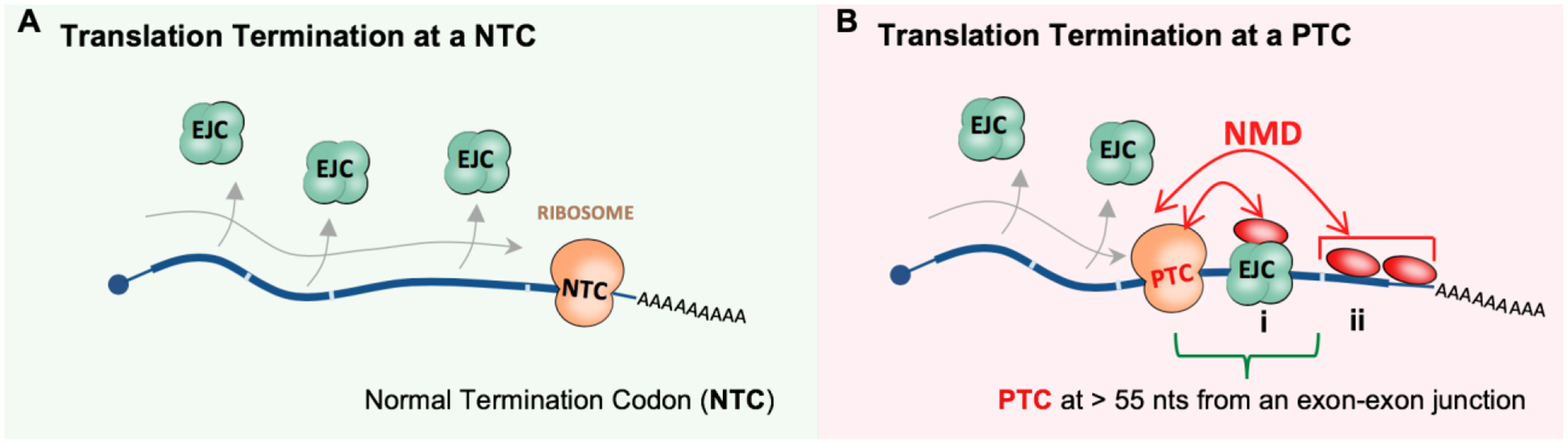
(A) During translation of a normal mRNA the ribosome dislodges EJCs as it traverses the RNA molecule up until the NTC. (B) Activation of NMD on a PTC-containing mRNA can occur by at least two mechanisms: i) During EJC-dependent NMD, a PTC found more than 50–55 nucleotides upstream of a downstream exon-exon junction triggers NMD. ii) EJC-independent NMD relies on proteins other than the EJC found downstream of the PTC, including UPF1. Red ovals represent NMD-associated factors.
One-third of all inherited human diseases are caused by nonsense or frameshift mutations that introduce a premature stop codon in a transcript (Mendell and Dietz, 2001; Stenson et al., 2003). Prominent examples include β-thalassemia, Duchenne muscular dystrophy, and cystic fibrosis (Miller and Pearce, 2014). Studying such diseases has offered a wealth of information on how NMD works and how aberrant transcripts are recognized by NMD. For example, nonsense mutations in early exons of β-globin (HBB) cause a recessive form of β-thalassemia. Here, mutant HBB mRNA is to degraded via NMD, which leads to insufficient levels of β-globin protein and disease (Neu-Yilik et al., 2011). Conversely, mutations in part of the penultimate exon or the last coding exon cause a dominant form of the disease because the mutant HBB mRNA escapes NMD and results in production of a C-terminally truncated β-globin protein (Neu-Yilik et al., 2011). Studies that identified mutations where the resulting transcript either underwent or escaped led to the proposal of the “50–55 nt rule” (Hentze and Kulozik, 1999; Li and Wilkinson, 1998; Maquat, 1995), where only those nonsense mutations that are at least 50–55 nucleotides upstream of the final exon-exon trigger efficient NMD (Figure 1B).
Several exceptions to the 50–55 nt rule have been observed. For example, nonsense mutations found close to the start codon often escape NMD because translation can reinitiate downstream of the PTC and EJCs that might otherwise trigger NMD are evicted (Inacio et al., 2004; Pereira et al., 2015; Zhang and Maquat, 1997). Stop codon readthrough also contributes to the escape of some nonsense variants from NMD for the same reason (Hogg and Goff, 2010; Keeling et al., 2004). Finally, NMD efficiency varies across cell types (Linde et al., 2007), tissues (Bateman et al., 2003; Zetoune et al., 2008), and individuals (Kerr et al., 2001; Nguyen et al., 2014), offering potential explanations for tissue tropism and varying penetrance across the population of disease-causing nonsense variants. Thus, NMD variability can compromise the accuracy of variant interpretation in clinical genetic diagnostics settings and, for this reason, is important to understand.
NMD machinery can also target normal transcripts that contain upstream open reading frames (uORFs), spliced introns in 3’ untranslated regions (UTRs), or long 3’ UTRs (Lykke-Andersen and Jensen, 2015; Mendell et al., 2004; Nasif et al., 2018; Nickless et al., 2017a). These features cause a transcript to engage the NMD machinery due to the continued presence of EJCs or other RBPs downstream of a terminating ribosome. The expression level of such transcripts can be dialled up or down by varying the efficiency with which they undergo NMD (Nasif et al., 2018). Moreover, depending on the context and mode of NMD inhibition, different subsets of physiological NMD substrates are stabilized, adding another layer of complexity to this regulatory mechanism. How and why are some NMD targets degraded and some spared? How does cellular context influence this choice? How do cells cope with regulated NMD inhibition given its role as a quality control process? These are open questions that can only be answered through better understanding of both the rules and exceptions of the NMD process. In this review, we will discuss the current state of knowledge of NMD variability and identify gaps that need to be filled through further investigations.
NMD MECHANISMS
Over the years, several cues that allow NMD to distinguish normal and aberrant transcripts have been discovered (Carter et al., 1996; Hentze and Kulozik, 1999; Thermann et al., 1998; Zhang et al., 1998). The presence of a premature stop codon >50–55 nucleotides upstream of an exon-exon junction is a strong predictor of NMD (Hsu et al., 2017), and induces a phenomenon called the EJC-dependent NMD (Ivanov et al., 2008; Kashima et al., 2006). The length of the 3’ UTR is another factor that also influences NMD, with longer 3’ UTRs inducing an EJC-independent mode of RNA surveillance termed long UTR-mediated NMD (Buhler et al., 2006; Eberle et al., 2008; Hogg and Goff, 2010; Hurt et al., 2013; Singh et al., 2008). However, it is worth noting that not all long 3’ UTRs induce NMD and additional factors exist that may modulate the efficiency with which a long UTR elicits NMD (Boehm et al., 2014; Toma et al., 2015). Yeast, which lacks introns in the majority of its genes, is thought to primarily utilize an EJC-independent NMD mechanism termed the “faux 3’ UTR model”, wherein premature termination causes the downstream RNA segment to function as a “faux” long UTR and trigger NMD via the long-UTR model (Amrani and Jacobson, 2013).
At the core of NMD substrate recognition via all mechanisms is the non-specific binding of UPF1, an RNA helicase, to RNA molecules (Hogg and Goff, 2010; Kurosaki and Maquat, 2013; Singh et al., 2008; Zund et al., 2013). When translation termination occurs close to the natural end of transcripts, the presence of the polyA binding protein, PABPC1, is thought to prevent the interaction between UPF1 and the eukaryotic release factors, eRF1 and eRF3, as well as increase the efficiency of translation termination (Amrani et al., 2004; Behm-Ansmant et al., 2007; Eberle et al., 2008; Singh et al., 2008). In the case of the EJC-dependent mode of NMD, a ribosome terminating >50–55 nucleotides upstream of an exon-exon junction fails to evict the downstream EJC. The combined presence of a terminating ribosome and the downstream EJC, as well as the efficient interaction of UPF1 with eRF1/3, is thought to induce the sequential recruitment and assembly of UPF2/3 and SMG1/5/6/7 factors on to the aberrant transcript, ultimately causing degradation of the RNA (Ivanov et al., 2008; Kashima et al., 2006). In long UTR-mediated NMD, UPF1 again interacts efficiently with eRF1/3, though UPF2/3 recruitment occurs unassisted by the EJC, with further assembly of SMGs and other degradation factors following similar mechanism as the EJC-dependent NMD (Eberle et al., 2008; Hogg and Goff, 2010; Singh et al., 2008). Finally, the combined action of the endonuclease SMG6, exonuclease XRN1, and the exosome complex, together with decapping and deadenylation factors, ensures efficient clearance of the aberrant RNA. In both of these models of NMD, the kinetics of premature termination appear distinct from that of normal termination in a way that facilitates the formation of various protein complexes in the context of the terminating ribosome to then recruit the NMD machinery (Celik et al., 2015; Ghosh et al., 2010).
For a more detailed discussion of the molecular mechanisms of NMD, we refer the reader to several excellent reviews on the subject (Brogna et al., 2016; Celik et al., 2017; Fatscher et al., 2015; Hug et al., 2016; Karousis et al., 2016; Kervestin and Jacobson, 2012; Kurosaki and Maquat, 2016; Lykke-Andersen and Jensen, 2015; Miller and Pearce, 2014; Nasif et al., 2018; Nickless et al., 2017a).
MECHANISMS OF ESCAPE FROM NMD
Nonsense variants can escape NMD via many routes (Buisson et al., 2006; Hamid et al., 2010; Harries et al., 2005; Hulsebos et al., 2014; Kerr et al., 2001; Krempely and Karam, 2018; Loughran et al., 2018; Poli et al., 2018; Sciezynska et al., 2017; Stump et al., 2012; Stump et al., 2013; Yamaguchi and Baba, 2018; Yamaguchi et al., 2012). Cis elements in the mRNA that modify splicing or translation as well as trans factors that affect these processes can influence the fate of a predicted NMD target. For example, if an exon containing the stop codon is excluded from the transcript via alternative splicing, the presumed target will no longer undergo NMD. Similarly, if a sole intron downstream of a PTC is inefficiently spliced, there will be no EJCs downstream of the PTC to signal NMD. Any variability in EJC deposition can also influence NMD efficiency. An unbiased, transcriptome-wide analysis of the RNA binding sites of four EJC components—eIF4A3, BTZ, UPF3B, and RNPS1— identified high-confidence EJC binding sites and revealed that EJC composition and deposition are largely homogenous with the notable exception of transcripts that encode ribosomal proteins (Hauer et al., 2016). While it is unknown whether the lack of EJC deposition on ribosomal transcripts is associated with a higher degree of escape from NMD, it is possible that they do. In fact, an anti-sense oligonucleotide (ASO) targeting the downstream EJC deposition site of a PTC-containing transcript was shown to block EJC assembly and thus inhibit NMD of the corresponding transcript (Nomakuchi et al., 2016), demonstrating that lack of EJC deposition on specific transcripts does allow such mRNAs to escape decay by NMD (reviewed here (Nasif et al., 2018)). Though Hauer et al only evaluated EJC deposition in healthy cells, it is possible that other transcripts might exhibit variability in EJC deposition during disease or cell stress, thus potentially escaping NMD in particular cellular contexts.
Other factors that can modify NMD efficiency include the sequence and architecture of the 3’ UTR, as well as the associated protein complexes and their relative abundance in different contexts (Singh et al., 2008). For example, some long 3’ UTRs contain cis elements that contribute to NMD evasion (Toma et al., 2015). It has also been shown in yeast that longer open reading frames desensitize transcripts with long 3’ UTRs to NMD (Decourty et al., 2014). As NMD is a translation-dependent process, cis and trans factors that influence translation can modulate NMD efficiency. Several stress response pathways culminate in phosphorylation of eIF2α that leads to inefficient translation and inhibition of NMD (Goetz and Wilkinson, 2017; Karousis et al., 2016; Li et al., 2017; Wang et al., 2011). At the mRNA level, inefficient translation termination at the PTC (a process termed stop codon readthrough or nonsense suppression), or reinitiation of translation downstream of the PTC, evicts downstream EJCs and causes NMD evasion by the PTC-containing transcript. Nonsense suppression via readthrough inducing small molecules is being investigated as a therapeutic strategy in several genetic disorders (Asiful Islam et al., 2017; Keeling et al., 2014; Lee and Dougherty, 2012; Mah, 2016; Wang and Gregory-Evans, 2015), highlighting the role of the ribosome and its interpretation of a stop codon in eliciting efficient NMD. In the following sections, we will separately consider gene-specific and context-specific modifiers of NMD, their mechanisms of action, and their molecular and phenotypic consequences.
GENE-SPECIFIC MODIFIERS OF NMD EFFICIENCY
Molecular studies of genetic disorders have led to the identification of hundreds of isolated instances of nonsense variants that escape NMD. However, we have only recently begun systematically assessing the fraction of nonsense variants that escape NMD, the mechanisms responsible for NMD evasion, and the quantity of protein produced from a nonsense allele (Jagannathan and Bradley, 2016; Lindeboom et al., 2016). Analyses of publicly available functional genomics datasets to assess the NMD efficiency of a subset of highly common nonsense mutations (i.e. those that have not been purged by negative selection during evolution) found that 52.3% of these common nonsense variants escape NMD (Jagannathan and Bradley, 2016). Reporter-based molecular validation further showed that translation reinitiation and stop codon readthrough, among other post-transcriptional mechanisms, contribute to NMD escape, enabling protein production from the nonsense alleles (Jagannathan and Bradley, 2016). Another study used 2,840 high-confidence somatic nonsense mutations found in the Cancer Genome Atlas (TCGA) and asked how well the known rules for NMD explain the observed NMD efficiency (Lindeboom et al., 2016). Surprisingly, only ~50% of the variance in NMD efficiency could be explained by the 50–55 nt rule (Lindeboom et al., 2016). These results highlight our incomplete understanding of the range of molecular mechanisms that could rescue or modify gene function in the presence of a predicted loss-of-function mutation (Figure 2). While the proteins produced from nonsense alleles that escape NMD may not be fully functional, plasticity in RNA processing and translation has the potential to convert loss-of-function variants from genetic nulls into hypomorphic, silent, or even neomorphic alleles (Jagannathan and Bradley, 2016).
Figure 2. Graphical illustration of the different gene-specific molecular events that promote escape from NMD.
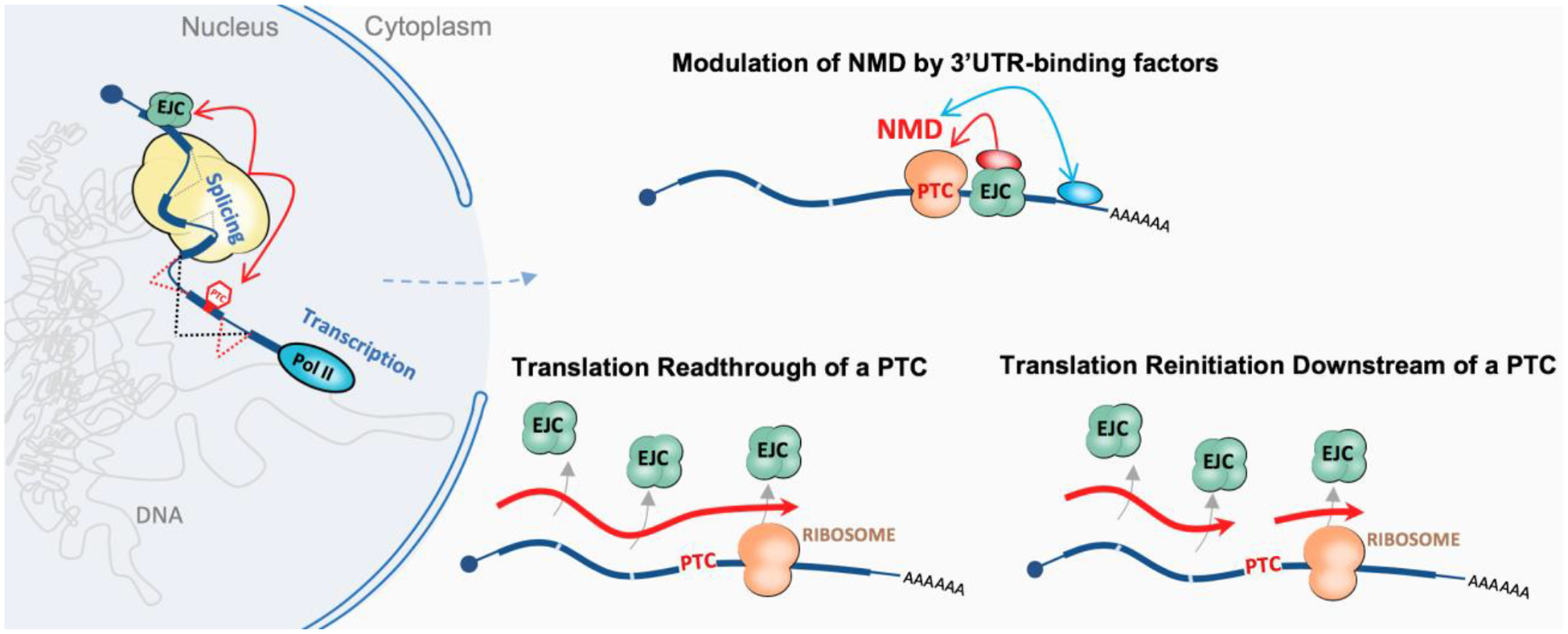
PTC-containing mRNAs can be generated via multiple mechanisms. Among co-transcriptional mechanisms that prevent the production of an NMD isoform, an alternative splicing decision that leads to the exclusion of an exon containing an in-frame PTC is shown. Not shown are alternative transcription initiation and alternative polyadenylation, both of which can allow exclusion of a PTC-containing. Post-transcriptional escape from NMD can be mediated by gene-specific elements such as nucleotide sequences or mRNA structure and may facilitate mRNA escape from NMD by two potential mechanisms: 1) facilitating recruitment of RNA-binding factors that can modulate NMD, or 2) by stimulating non-canonical translation at the PTC which causes removal of downstream EJCs and inhibition of NMD, including translation readthrough and translation reinitiation. Red oval represents NMD-associated factors; Blue oval represents NMD-modifying factors.
Co-transcriptional mechanisms that prevent the production of NMD isoforms
Co-transcriptional events, including alternative promoter usage, alternative splicing, and alternative polyadenylation, can all result in the exclusion of an exon containing a premature stop codon, and hence introduce variability of expression of a predicted NMD target in a gene-specific manner. Alternative splicing is perhaps the best understood mechanism that allows gene expression despite the presence of a nonsense mutation or genetic variant with numerous examples in the literature (da Costa et al., 2017; Lewis et al., 2003). For example, alternative-splicing coupled NMD (AS-NMD) serves as a homeostatic regulatory mechanism to regulate the expression of different mRNA isoforms produced from a gene (Lareau et al., 2007; Ni et al., 2007). In one prominent example, members of the SR-family splicing factors autoregulate their levels by modulating inclusion or exclusion of a PTC-containing exon (referred to as “poison exon”) by binding to an exonic splicing enhancer (ESE) within the poison exon. In the presence of high levels of an SR protein, its binding to the ESE promotes inclusion of the PTC-containing exon and leads to NMD of the transcript, and thus lowers the level of the protein (Lareau et al., 2007; Ni et al., 2007).
Several nonsense variants found in cassette exons (i.e. those that can be included or excluded in a regulated manner) appear to induce alternative splicing, particularly when the exon skipping preserves the translation reading frame (da Costa et al., 2017). With increasing use of CRISPR/Cas9 genome editing, there is accumulating evidence for nonsense mutations resulting in observed changes in in the abundance of mRNA isoforms generated by alternative splicing (Lalonde et al., 2017; Mou et al., 2017; Sui et al., 2018), though it is unclear if the splicing decision itself is affected in these cases or if it is simply a case of differential isoform targeting by NMD (i.e. preferential degradation of PTC-inclusion versus -exclusion isoform). Finally, tissue-specific and developmentally-regulated alternative splicing introduces yet another layer of complexity to NMD wherein a transcript containing a PTC is only produced in some tissues, thus allowing the PTC-containing gene to escape NMD-mediated loss of function in other tissues (Barberan-Soler et al., 2009; Jagannathan and Bradley, 2016; Yeo et al., 2004).
Post-transcriptional mechanisms that allow an NMD isoform to evade degradation
Sequence-specific RBP binding
Several RNA binding proteins are capable of coordinating with NMD factors to protect specific transcripts from NMD. For example, binding of cytoplasmic poly(A)-binding protein, PABPC1, to long 3’ UTRs effectively suppresses NMD activation (Fatscher et al., 2014). More broadly, short peptides consisting of PABP-interacting motif 2 (PAM2) found in a variety of proteins bound to long 3’ UTRs also suppress NMD activity (Fatscher and Gehring, 2016). Some long 3’ UTRs contain an RNA stability Element (RSE) that recruits polypyrimidine-tract-binding protein 1 (PTBP1) to form an mRNP structure that inhibits NMD (Ge et al., 2016). A recent study also found that some normal mRNAs with long 3’ UTRs and multiple 3’ UTR introns that contain features bound by the protein hnRNPL can evade NMD-mediated degradation (Kishor et al., 2019). Importantly, in many B cell lymphomas with BCL2:IGH translocations, hnRNPL recruitment protects and stabilizes aberrant BCL2 fusion mRNAs, which contributes to increased BCL2 expression (Kishor et al., 2019). Thus, 3’ UTR sequence elements regulate the extent of NMD-mediated degradation and gene expression.
Translation reinitiation downstream of a PTC
Reinitiation occurs when a post-termination ribosome is not recycled but continues to scan and then initiate at downstream start codons (Gunisova et al., 2018). Despite being well accepted as a cause for escape from NMD (Buisson et al., 2006; Hulsebos et al., 2014; Neu-Yilik et al., 2011; Paulsen et al., 2006; Stump et al., 2012; Stump et al., 2013; Zhang and Maquat, 1997), the mechanism that enables efficient reinitiation of a subset of nonsense variants is poorly understood. Transcripts with uORFs are often destabilized via NMD due to the presence of EJCs that are not evicted due to the uORF reducing translation of the downstream ORF (Colombo et al., 2017). However, there are several examples of transcripts with start-proximal nonsense codons that are not subject to NMD due to efficient reinitiation of translation at downstream start codons (Buisson et al., 2006; Hulsebos et al., 2014; Paulsen et al., 2006; Pereira et al., 2015; Stump et al., 2012; Stump et al., 2013; Zhang and Maquat, 1997), leading to the proposal of a “boundary” within each transcript where reinitiation is efficient (Pereira et al., 2015). In support of this model, by analyzing common nonsense variants in the human population, we observed a statistically significant enrichment of downstream in-frame methionine within 50 codons of the nonsense variants (Jagannathan and Bradley, 2016). Specifically, we found that 57% of rare and 85% of common nonsense variants contained a downstream in-frame methionine within 50 codons of the variant, compared to only 52% of synonymous variants (Jagannathan and Bradley, 2016). This observation only held, however, in the first 10% of a coding sequence, consistent with studies that have found that reinitiation is most efficient when a PTC is close to the start codon (Inacio et al., 2004; Kozak, 2001; Stump et al., 2012). These observations suggest that the post-termination ribosome may need to be close enough to the canonical start codon in order to be reinitiation-competent, as well as encounter a downstream start relatively quickly to reinitiate efficiently.
Nonetheless, this reinitiation boundary varies even across closely related genes such as alpha and beta globin (Pereira et al., 2015), which suggests transcript-specific features modulate reinitiation. Prior studies have shown that the eukaryotic Initiation Factor complex 3 (eIF3), but not other initiation factors, remains transiently associated with the ribosome through elongation and termination (Hronova et al., 2017; Mohammad et al., 2017; Munzarova et al., 2011). Those ribosomes that retain eIF3 are then able to resume scanning for downstream start codons and reinitiate post-termination. In some cases of reinitiation downstream of short uORFs, the association of eIF3 with the post-termination ribosome is facilitated by specific cis regulatory sequences (Hronova et al., 2017; Mohammad et al., 2017; Munzarova et al., 2011). Thus, the distance between the start codon and the premature stop, as well as the presence of specific sequence elements or RNA modifications, may influence whether or not the ribosome is able to reinitiate and define the “window of opportunity” for reinitiation.
Stop codon readthrough
A key step translation termination the competition between the release factor and a non-cognate tRNA, to bind to the A site in the ribosome. The outcome of this competition determines whether translation terminates prematurely to trigger NMD or continues to proceed via readthrough and incorporation of a non-cognate amino acid in the place of the PTC (Celik et al., 2015). Nonsense suppression therapies for genetic diseases target this step of NMD using suppressor tRNAs, aminoglycosides, and non-aminoglycosides such as ataluren—all of which induce stop codon readthrough (Asiful Islam et al., 2017; Keeling and Bedwell, 2011). However, the efficiency and specificity of nonsense suppression drugs for premature versus canonical stop codons has been debated and the drugs themselves are known to have significant side effects (Namgoong and Bertoni, 2016).
Decades of work on termination efficiency at “normal” stop codons has shown that readthrough is a rare and inefficient phenomenon in mammalian cells (Cassan and Rousset, 2001; Loughran et al., 2014; Loughran et al., 2018). Previous work on canonical stop codon readthrough in viral, yeast, Drosophila, and mammalian genes has identified several factors that influence the efficiency of readthrough, including stop codon identity and context, and RNA secondary structures such as pseudoknots (Brown et al., 1996; Cassan and Rousset, 2001; Green and Goff, 2015; Liu and Xue, 2004; Loughran et al., 2014; Loughran et al., 2018; Napthine et al., 2012; Stiebler et al., 2014; Xu et al., 2018). Moreover, termination at PTCs has been shown to be kinetically distinct from termination at canonical stop codons in yeast even though the underlying mechanism is unclear (Amrani et al., 2004; Amrani and Jacobson, 2013), and a recent study in mammalian cells found that PTC readthrough occurs in specialized cytoplasmic foci upon NMD inhibition (Jia et al., 2017). With the discovery of a subset of mRNAs with PTCs that are naturally read through with high efficiency (as indicated by the uninterrupted ribosome footprint density across a PTC and/or near-normal levels of full-length protein product from a homozygous NMD allele; (Jagannathan and Bradley, 2016), it is clear that there may be sequence contexts or other factors that facilitate natural PTC readthrough – especially since canonical stop codons are readthrough very poorly (Cassan and Rousset, 2001; Loughran et al., 2014; Loughran et al., 2018). For instance, RNA modifications can also influence stop codon readthrough. A recent study found that pseudouridylation of the ‘U’ in stop codons by sequence-specific pseudouridine synthases promotes readthrough (Karijolich and Yu, 2011). Therefore, the mechanism(s) that allow readthrough of a normal stop codon may also be different from that of readthrough of a premature stop codon, and understanding how to coax ribosomes into reading through PTCs can have an immense practical benefit in targeting genetic diseases caused by nonsense mutations.
While both co-transcriptional and post-transcriptional events contribute to NMD variability, the relative fractional contribution of these mechanisms to escape from NMD is yet to be determined. As we accumulate more ribosome footprinting and proteomics datasets to complement RNA-seq data on nonsense variants, quantitative estimates of the contribution of translational recoding to NMD escape may be derived.
CONTEXT-SPECIFIC MODIFIERS OF NMD EFFICIENCY
Because NMD functions in part to regulate normal gene expression, overall NMD efficiency and specific mRNAs targeted for degradation can vary based on genetic background, cell type, and developmental stage, among other contexts (Lykke-Andersen and Jensen, 2015; Nasif et al., 2018). As an example, studies in yeast have found that the background strain can influence levels of accumulated NMD substrate mRNAs, which suggests that many genes influence NMD efficiency (Kebaara et al., 2003). In addition, the recognition of cytoplasmic downregulation of normal mRNAs with PTCs as part of normal physiological processes (Li and Wilkinson, 1998) led to several investigations into the role of NMD in diverse cell types and during development.
Physiological variation of NMD during differentiation and development
Several cellular contexts exist in which NMD efficiency is modulated to affect specific physiological consequences (Figure 3). NMD is key for myogenic differentiation and yet plays seemingly contradictory roles. On the one hand, NMD is essential for downregulating c-Myc mRNA, which in turns drives myogenic differentiation (Yeilding et al., 1998). Importantly, initial studies showed that c-myc mRNA downregulation occurs in the cytoplasm, is translation-dependent, and is mediated by exons 2 and 3 (Yeilding and Lee, 1997; Yeilding et al., 1998). Recent reports in embryonic stem cells suggest the NMD pathway is responsible for forced down-regulation of c-Myc mRNA, which promotes cellular differentiation (Li et al., 2015; Lou et al., 2015). Here, genetic deletion of Smg6, as well as transient knockdown of other NMD factors, leads to increased levels of c-Myc mRNA and a differentiation block. Restoring NMD function by expressing NMD-proficient Smg6 constructs rescues differentiation. On the other hand, during differentiation of mouse C2C12 myoblasts to myotubes, the efficiency of NMD decreases due to preferential recruitment of UPF1 to a competing pathway called Staufen 1-mediated mRNA decay (SMD) (Gong et al., 2009). In this context, reduced NMD efficiency permits increased expression of myogenin—a muscle-specific transcription factor involved in muscle differentiation—and consequently, myogenesis (Gong et al., 2009). Furthermore, consistent with the idea that reduced NMD, or NMD factors, in this case, is critical for myogenesis, it was recently demonstrated that the E3 ligase function of the UPF1 protein (Kuroha et al., 2013; Kuroha et al., 2009) targets MYOD, a protein that promotes myogenesis, for ubiquitination and degradation (Feng et al., 2017; Takahashi et al., 2008; TD and Wilkinson, 2018). Knockdown of UPF1 mRNA leads to increased MYOD protein and increased myogenesis.
Figure 3. Cellular contexts that induce variability in NMD.
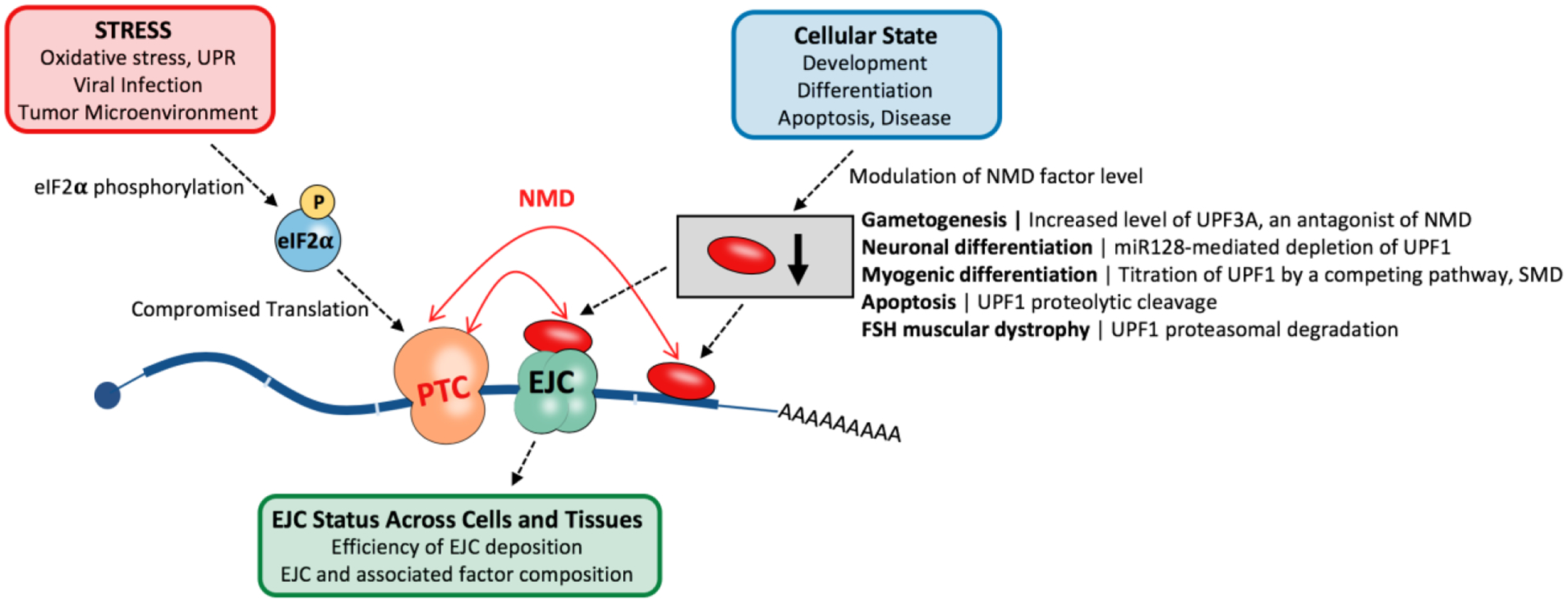
A) Cell stresses including oxidative stress, unfolded protein response (UPR), viral infection and the tumor microenvironment can induce phosphorylation of the translation initiation factor, eIF2α, and lead to translation attenuation and consequently, NMD inhibition. B) Variability in the level of EJC and associated factors as well as the efficiency of EJC deposition can vary NMD. C) Cellular states such precursor versus differentiated cells, cells undergoing apoptosis, and cells derived from disease states (e.g. FSHD) show characteristic modulation of NMD factor levels that allows variation in NMD efficiency. Red oval represents NMD-associated factors
NMD also plays a prominent role in neural development and in stem cell biology. These topics have been reviewed extensively elsewhere (Han et al., 2018; Jaffrey and Wilkinson, 2018; Lykke-Andersen and Jensen, 2015). Briefly, UPF1 is downregulated via a neuronally-expressed microRNA, miR-128, causing a decrease in NMD activity during mouse embryonic development and when neural progenitor cells undergo differentiation (Karam and Wilkinson, 2012; Lou et al., 2014). Here, the authors demonstrate that in mouse neuronal stem cells, UPF1 negatively regulates components of the TGF-b family of proteins, and that transient knockdown of UPF1 leads to increased levels of factors that promote differentiation. Further evidence that the NMD pathway is important for neural development is the finding that human genetic mutations in UPF3B are associated with intellectual disability (Nguyen et al., 2012; Tarpey et al., 2007): Upf3b-null mice exhibit a learning deficit (Huang et al., 2018), and cultured stem cells from Upf3b-null mice have an impaired ability to differentiate (Jolly et al., 2013).
Recent data suggest the composition of the assembled NMD protein complex also varies in certain developmental scenarios and helps determine the NMD susceptibility of specific substrates. In one example, researchers investigated the expression level of UPF3A mRNA in adult mouse tissues and found that it is highly expressed in spermatocytes and is developmentally regulated across different germ cell subsets. UPF3A is a paralog of UPF3B, and has a unique, antagonistic role in NMD (Chan et al., 2009; Shum et al., 2016). When bound by UPF3B, NMD function is maintained and the target transcript is degraded; when bound by UPF3A, the NMD complex is repressed and the target transcript remains stable (Shum et al., 2016). In addition, UPF3A exhibits variable expression in many adult mouse tissues (Shum et al., 2016) and is increased in cell lines derived from human patients with loss-of-function mutations in UPF3B (Nguyen et al., 2012). Taken together, these examples show the importance of NMD for development of many different tissues and that regulated modulation of NMD efficiency is also critical.
Physiological variation of NMD during cell stress
Accumulation of misfolded proteins in the lumen of the endoplasmic reticulum activates a signaling cascade called the unfolded protein response (UPR). Several groups have shown that the UPR is shaped by mRNA surveillance as well (Goetz and Wilkinson, 2017; Nasif et al., 2018). Early studies investigating the link between ER stress, UPR activation, and NMD activity found that the loss or deletion of many NMD factors cause ER stress (Sakaki et al., 2012). Notably, NMD factors are also known to colocalize with the ER, though whether their localization has any significance to NMD is poorly understood (Sakaki et al., 2012). These data support a model in which NMD functions to eliminate aberrant transcripts that could encode polypeptides capable of inducing ER stress. Later studies discovered that NMD functions to fine tune regulation of the ER stress response and UPR activation. Under mild ER stress conditions, NMD activity prevents full-scale UPR activation by raising the activation threshold and quickly resolving mild stress (Karam et al., 2015). Conversely, NMD is inhibited during the plateau phase of the UPR and during chronic activation of ER stress (Karam et al., 2015; Li et al., 2017), which allows for strong activation of the UPR. Furthermore, some transcripts encoding key mediators of the UPR—such as the transcription factors ATF3, ATF4, and CHOP—are NMD substrates and increase as a consequence of NMD inhibition (Oren et al., 2014). Following a strong ER stress response, reviving ER homeostasis and translation depends on restored NMD activity, which is evidenced by findings that chronic depletion of NMD reduces mammalian cell proliferation and survival (Karam et al., 2015; Sakaki et al., 2012).
During ER stress, the levels of mRNAs encoding key NMD factors do not change (Karam et al., 2015). However, global translation is inhibited by a mechanism that involves phosphorylation of eIF2α. As NMD is a translation-dependent process, reduced global translation results in increased stability of NMD substrates. UPR activation has also been shown to attenuate NMD by enhancing ribosome readthrough of PTCs (Oren et al., 2014). It is worth noting that most stress response pathways, including oxidative stress, anti-viral response, and heat shock response, lead to eIF2α phosphorylation and thus, can inhibit NMD. In fact, NMD inhibition was observed in the tumor microenvironment and found to promote tumorigenesis by stabilizing mRNAs that are involved in stress response and tumor-promoting pathways (Wang et al., 2011). Given the chronic activation of cell stress in many human diseases, pharmacologic therapies to restore NMD activity may provide clinical benefit and address unmet medical needs (Goetz and Wilkinson, 2017).
Pathological variability of NMD in disease
Aside from the regulated inhibition of NMD in specific physiological contexts, en masse inhibition of NMD is quite rare in pathological contexts, perhaps due to the critical role that many NMD factors play in mammalian embryogenesis (Han et al., 2018; Li et al., 2015; Lou et al., 2014; Nasif et al., 2018). Therefore, germline mutations that cause loss of function of an NMD factor do not survive past early embryonic stages (Han et al., 2018). One exception to this general trend is UPF3B, in which loss of function mutations are viable, but cause severe neurodevelopmental defects and intellectual disability (Jaffrey and Wilkinson, 2018). Recent studies have reported somatically acquired point mutations in UPF1 that are frequently found in pancreatic adenosquamous carcinoma (Liu et al., 2014) and in inflammatory myofibroblastic tumors (Lu et al., 2016). In most of these cases, inactivation of an NMD factor has been shown to affect a specific subset of endogenous NMD targets that are consistent with the observed pathology, rather than a general inhibition of NMD with a strong effect on all endogenous targets (Lykke-Andersen and Jensen, 2015). While robust knockdown of individual NMD factors does affect cell growth and viability, partial knockdowns often have modest effects on cell physiology, consistent with the idea that NMD factors are under an autoregulatory feedback loop that protects the NMD pathway from perturbations (Chan et al., 2009; Huang et al., 2011; Yepiskoposyan et al., 2011).
The only known example of en masse NMD inhibition playing a key role in a disease process was recently discovered in a genetically inherited muscular dystrophy called facioscapulohumeral muscular dystrophy (FSHD). FSHD is caused by epigenetic de-repression of a macrosatellite repeat array in chromosome 4q and the resulting activation of an early embryonic transcription factor, DUX4, in skeletal muscle cells (Lemmers et al., 2010). In addition to turning on expression of direct target genes (Geng et al., 2012), skeletal muscle DUX4 also leads to an unprecedented degree of inhibition of NMD (Feng et al., 2015). Interestingly, the DUX4 transcript is an endogenous target of NMD due to the presence of two introns in the 3’ UTR (Feng et al., 2015). Consequently, when DUX4 expression rises slightly, a feed-forward loop initiates because DUX4 expression decreases NMD function, which then relieves NMD-mediated inhibition of DUX4 and causes a characteristic burst pattern of DUX4 expression. This burst of DUX4 expression is observable in FSHD muscle cells and causes cell death and muscle deterioration in FSHD (Rickard et al., 2015). In addition to stabilizing DUX4 mRNA, the systemic inhibition of NMD also causes accumulation of hundreds of NMD-substrate mRNAs in DUX4-expressing cells and in the FSHD muscle (Feng et al., 2015; Jagannathan et al., 2019). Dramatically reduced NMD in DUX4-expressing skeletal muscle cells raises the possibility that the proteins translated from NMD-substrate mRNAs contribute to skeletal muscle deterioration in FSHD. It also raises the possibility that reduced NMD efficiency may play a role in other skeletal muscle diseases.
Mechanisms of NMD inhibition and their contribution to muscle cell death in FSHD are beginning to be explored. Interestingly, global inhibition of NMD in FSHD skeletal muscle cells is caused by post-translation degradation of core NMD factors. The first support of such a post-translational mechanism came from the finding that increased DUX4 expression causes decreased levels of UPF1 and SMG7 protein, but UPF1 and SMG7 mRNAs were either unchanged or increased (Feng et al., 2015). A recent unbiased proteomics study from FSHD myoblasts revealed a discordance between the protein and mRNA levels for many core NMD factors, including UPF1, UPF3B, SMG6, and the exonuclease XRN1 (Jagannathan et al., 2019). Despite recent progress, the upstream events that are responsible for targeted, post-translational depletion of core NMD factors are not known. Additional studies focused on identifying these upstream triggering events will lead to a better understanding of the underlying molecular pathology of FSHD and may lead to new therapeutic strategies.
NMD also varies between individuals (Nguyen et al., 2014), a finding especially important to consider in the context of human diseases and interpreting potential disease-causing mutations. For instance, a recent study showed that an identical nonsense mutation known to cause Choroideremia exhibited up to 40% variability in the RNA level (Sarkar et al., 2019). It is thus possible that genetic variation in NMD factors that affect their level or activity could modify the penetrance and the severity of diseases even if they do not cause diseases by themselves.
In summary, NMD activity is modulated in a number of different ways to affect specific physiological or pathological outcomes (Figure 3). Some of the mechanisms that lead to NMD inhibition include downregulation of NMD factors by microRNAs (Jin et al., 2016; Karam and Wilkinson, 2012; Wang et al., 2013) or proteasomal degradation (Feng et al., 2015; Jagannathan et al., 2019), cleavage of specific NMD factors to produce dominant negative products (Popp and Maquat, 2015), competition between alternative decay pathways (Gong et al., 2009), translation inhibition (Li et al., 2017), and activation of signaling pathways such as p38 MAPK (Nickless et al., 2017b). In physiological scenarios, NMD inhibition leads to the increased abundance of one or more physiological NMD targets that confer a benefit to the cell. In pathology, NMD inhibition often promotes or exacerbates the disease, again by stabilizing a subset of substrates. How the regulatory arm of NMD is balanced with the quality control arm, and how different subsets of NMD targets are stabilized under different scenarios are some of the biggest questions facing NMD researchers.
Future Directions
Understanding the rules and exceptions that govern NMD is essential for accurate clinical interpretation of genetic data (Popp and Maquat, 2016). For example, an NMD target may degrade and generate no protein or escape degradation and result in the production of a full-length or truncated protein (Anczukow et al., 2008), each scenario associated with a potentially different disease outcome (Figure 4). We have discussed numerous examples—both in normal physiology and in disease—of NMD targets escaping degradation and having a functional consequence. Further studies are needed to quantify the cumulative impact of gene-specific and context-specific NMD modifiers on NMD efficiency of a given transcript containing a PTC in a given scenario. Given the numerous factors that can alter the fate of a transcript containing a PTC, clinical genetic pipelines need to account for factors other than the 50–55 nt rule in order to make an accurate prediction. For example, if a gene carrying a nonsense variant also carries a sequence element known to induce escape from NMD, a pipeline could be trained to identify and report the variant accordingly. Thus, understanding gene-specific NMD escape mechanisms has immense practical value.
Figure 4.
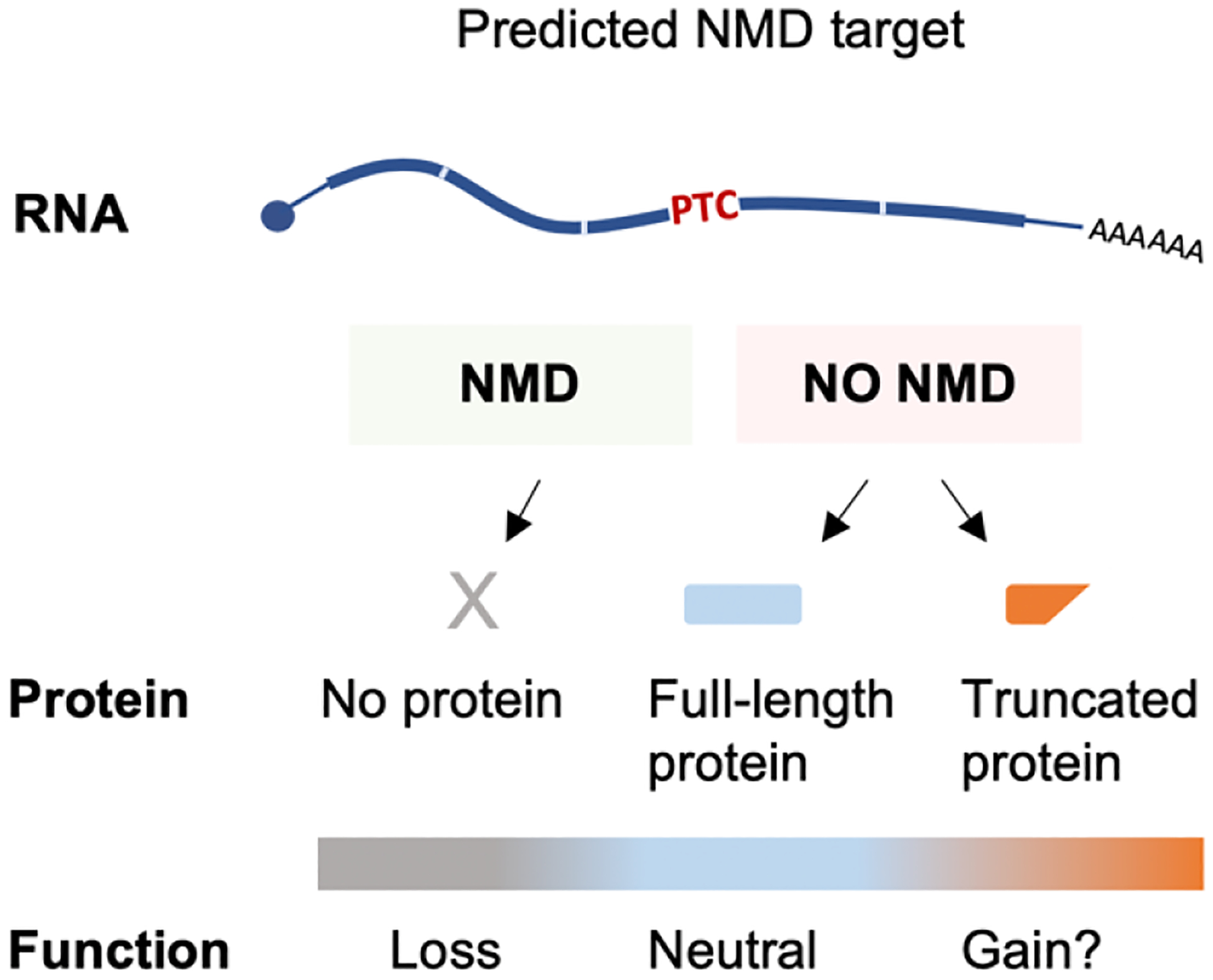
Varying RNA quality control efficiency modulates expression of protein-truncating variants.
With sufficient data, we can build better predictive models of NMD that consider the location of a PTC within a gene, the sequence context of a PTC, distal elements in the transcript that may modify NMD efficiency, and the abundance of various NMD factors in a relevant tissue and even individual, in order to accurately predict the fate of the transcript. If a transcript is predicted to escape NMD, the mechanism by which it does so can be used to predict the protein the transcript is most likely to make, which would be most useful for clinical interpretation of a disease-causing mutation. Finally, better understanding of the many routes to NMD escape may lead to novel therapeutics approaches that leverage these mechanisms to restore full function of gene despite the presence of a nonsense mutation.
Acknowledgments
The authors wish to acknowledge members of the Jagannathan laboratory for helpful discussions. We also thank current and past members of the Bradley and Tapscott laboratories for discussions that have contributed to this body of work over time. We thank Olivia Rissland for critical reading of the manuscript. This research was supported by start-up funds from the RBI (SJ), and the FSH Society FSHS-22014-01 (MCD).
Contributor Information
Michael C. Dyle, Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; RNA Bioscience Initiative, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Divya Kolakada, Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; RNA Bioscience Initiative, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; Molecular Biology Graduate Program, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Michael A. Cortazar, Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; RNA Bioscience Initiative, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Sujatha Jagannathan, Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; RNA Bioscience Initiative, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
References
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, and Jacobson A. 2004. A faux 3’-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 432:112–118. [DOI] [PubMed] [Google Scholar]
- Amrani N, and Jacobson A. 2013. All termination events are not equal: premature termination in yeast is aberrant and triggers NMD. In Madame Curie Bioscience Database. Landes Bioscience, Austin, TX. [Google Scholar]
- Anczukow O, Ware MD, Buisson M, Zetoune AB, Stoppa-Lyonnet D, Sinilnikova OM, and Mazoyer S. 2008. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum Mutat. 29:65–73. [DOI] [PubMed] [Google Scholar]
- Asiful Islam M, Alam F, Kamal MA, Gan SH, Wong KK, and Sasongko TH. 2017. Therapeutic Suppression of Nonsense Mutation: An Emerging Target in Multiple Diseases and Thrombotic Disorders. Curr Pharm Des. 23:1598–1609. [DOI] [PubMed] [Google Scholar]
- Barberan-Soler S, Lambert NJ, and Zahler AM. 2009. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA. 15:1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JF, Freddi S, Nattrass G, and Savarirayan R. 2003. Tissue-specific RNA surveillance? Nonsense-mediated mRNA decay causes collagen X haploinsufficiency in Schmid metaphyseal chondrodysplasia cartilage. Hum Mol Genet. 12:217–225. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Gatfield D, Rehwinkel J, Hilgers V, and Izaurralde E. 2007. A conserved role for cytoplasmic poly(A)-binding protein 1 (PABPC1) in nonsense-mediated mRNA decay. EMBO J. 26:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm V, Haberman N, Ottens F, Ule J, and Gehring NH. 2014. 3’ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep. 9:555–568. [DOI] [PubMed] [Google Scholar]
- Brogna S, McLeod T, and Petric M. 2016. The Meaning of NMD: Translate or Perish. Trends Genet. 32:395–407. [DOI] [PubMed] [Google Scholar]
- Brown CM, Dinesh-Kumar SP, and Miller WA. 1996. Local and distant sequences are required for efficient readthrough of the barley yellow dwarf virus PAV coat protein gene stop codon. J Virol. 70:5884–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Steiner S, Mohn F, Paillusson A, and Muhlemann O. 2006. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3’ UTR length. Nat Struct Mol Biol. 13:462–464. [DOI] [PubMed] [Google Scholar]
- Buisson M, Anczukow O, Zetoune AB, Ware MD, and Mazoyer S. 2006. The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mutat. 27:1024–1029. [DOI] [PubMed] [Google Scholar]
- Carter MS, Li S, and Wilkinson MF. 1996. A splicing-dependent regulatory mechanism that detects translation signals. EMBO J. 15:5965–5975. [PMC free article] [PubMed] [Google Scholar]
- Cassan M, and Rousset JP. 2001. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol. 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik A, He F, and Jacobson A. 2017. NMD monitors translational fidelity 24/7. Curr Genet. 63:1007–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik A, Kervestin S, and Jacobson A. 2015. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie. 114:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, and Wilkinson MF. 2009. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol. 16:747–753. [DOI] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, and Muhlemann O. 2017. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. 23:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa PJ, Menezes J, and Romao L. 2017. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int J Biochem Cell Biol. 91:168–175. [DOI] [PubMed] [Google Scholar]
- Decourty L, Doyen A, Malabat C, Frachon E, Rispal D, Seraphin B, Feuerbach F, Jacquier A, and Saveanu C. 2014. Long open reading frame transcripts escape nonsense-mediated mRNA decay in yeast. Cell Rep. 6:593–598. [DOI] [PubMed] [Google Scholar]
- Eberle AB, Stalder L, Mathys H, Orozco RZ, and Muhlemann O. 2008. Posttranscriptional gene regulation by spatial rearrangement of the 3’ untranslated region. PLoS Biol. 6:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatscher T, Boehm V, and Gehring NH. 2015. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell Mol Life Sci. 72:4523–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatscher T, Boehm V, Weiche B, and Gehring NH. 2014. The interaction of cytoplasmic poly(A)-binding protein with eukaryotic initiation factor 4G suppresses nonsense-mediated mRNA decay. RNA. 20:1579–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatscher T, and Gehring NH. 2016. Harnessing short poly(A)-binding protein-interacting peptides for the suppression of nonsense-mediated mRNA decay. Sci Rep. 6:37311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Jagannathan S, and Bradley RK. 2017. The RNA Surveillance Factor UPF1 Represses Myogenesis via Its E3 Ubiquitin Ligase Activity. Mol Cell. 67:239–251 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, and Bradley RK. 2015. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Quek BL, Beemon KL, and Hogg JR. 2016. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, and Tapscott SJ. 2012. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 22:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ganesan R, Amrani N, and Jacobson A. 2010. Translational competence of ribosomes released from a premature termination codon is modulated by NMD factors. RNA. 16:1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz AE, and Wilkinson M. 2017. Stress and the nonsense-mediated RNA decay pathway. Cell Mol Life Sci. 74:3509–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Kim YK, Woeller CF, Tang Y, and Maquat LE. 2009. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 23:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green L, and Goff SP. 2015. Translational readthrough-promoting drugs enhance pseudoknot-mediated suppression of the stop codon at the Moloney murine leukemia virus gag-pol junction. J Gen Virol. 96:3411–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunisova S, Hronova V, Mohammad MP, Hinnebusch AG, and Valasek LS. 2018. Please do not recycle! Translation reinitiation in microbes and higher eukaryotes. FEMS Microbiol Rev. 42:165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid R, Hedges LK, Austin E, Phillips JA 3rd, Loyd JE, and Cogan JD. 2010. Transcripts from a novel BMPR2 termination mutation escape nonsense mediated decay by downstream translation re-initiation: implications for treating pulmonary hypertension. Clin Genet. 77:280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wei Y, Wang H, Wang F, Ju Z, and Li T. 2018. Nonsense-mediated mRNA decay: a ‘nonsense’ pathway makes sense in stem cell biology. Nucleic Acids Res. 46:1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harries LW, Bingham C, Bellanne-Chantelot C, Hattersley AT, and Ellard S. 2005. The position of premature termination codons in the hepatocyte nuclear factor −1 beta gene determines susceptibility to nonsense-mediated decay. Hum Genet. 118:214–224. [DOI] [PubMed] [Google Scholar]
- Hauer C, Sieber J, Schwarzl T, Hollerer I, Curk T, Alleaume AM, Hentze MW, and Kulozik AE. 2016. Exon Junction Complexes Show a Distributional Bias toward Alternatively Spliced mRNAs and against mRNAs Coding for Ribosomal Proteins. Cell Rep. 16:1588–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, and Kulozik AE. 1999. A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 96:307–310. [DOI] [PubMed] [Google Scholar]
- Hogg JR, and Goff SP. 2010. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell. 143:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hronova V, Mohammad MP, Wagner S, Panek J, Gunisova S, Zeman J, Poncova K, and Valasek LS. 2017. Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells? RNA Biol. 14:1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MK, Lin HY, and Chen FC. 2017. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS One. 12:e0174798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, Karam R, Song HW, and Wilkinson MF. 2011. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 43:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Shum EY, Jones SH, Lou CH, Dumdie J, Kim H, Roberts AJ, Jolly LA, Espinoza JL, Skarbrevik DM, Phan MH, Cook-Andersen H, Swerdlow NR, Gecz J, and Wilkinson MF. 2018. A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol Psychiatry. 23:1773–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, and Caceres JF. 2016. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebos TJ, Kenter S, Verhagen WI, Baas F, Flucke U, and Wesseling P. 2014. Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis-associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors. Acta Neuropathol. 128:439–448. [DOI] [PubMed] [Google Scholar]
- Hurt JA, Robertson AD, and Burge CB. 2013. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 23:1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio A, Silva AL, Pinto J, Ji X, Morgado A, Almeida F, Faustino P, Lavinha J, Liebhaber SA, and Romao L. 2004. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem. 279:32170–32180. [DOI] [PubMed] [Google Scholar]
- Ivanov PV, Gehring NH, Kunz JB, Hentze MW, and Kulozik AE. 2008. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 27:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, and Wilkinson MF. 2018. Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat Rev Neurosci. 19:715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S, and Bradley RK. 2016. Translational plasticity facilitates the accumulation of nonsense genetic variants in the human population. Genome Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S, Ogata Y, Gafken PR, Tapscott SJ, and Bradley RK. 2019. Quantitative proteomics reveals key roles for post-transcriptional gene regulation in the molecular pathology of facioscapulohumeral muscular dystrophy. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Werkmeister E, Gonzalez-Hilarion S, Leroy C, Gruenert DC, Lafont F, Tulasne D, and Lejeune F. 2017. Premature termination codon readthrough in human cells occurs in novel cytoplasmic foci and requires UPF proteins. J Cell Sci. 130:3009–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhang F, Ma Z, and Ren Z. 2016. MicroRNA 433 regulates nonsense-mediated mRNA decay by targeting SMG5 mRNA. BMC Mol Biol. 17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly LA, Homan CC, Jacob R, Barry S, and Gecz J. 2013. The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet. 22:4673–4687. [DOI] [PubMed] [Google Scholar]
- Karam R, Lou CH, Kroeger H, Huang L, Lin JH, and Wilkinson MF. 2015. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 16:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, and Wilkinson M. 2012. A conserved microRNA/NMD regulatory circuit controls gene expression. RNA Biol. 9:22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karijolich J, and Yu YT. 2011. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature. 474:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis ED, Nasif S, and Muhlemann O. 2016. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA. 7:661–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, and Ohno S. 2006. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 20:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebaara B, Nazarenus T, Taylor R, and Atkin AL. 2003. Genetic background affects relative nonsense mRNA accumulation in wild-type and upf mutant yeast strains. Curr Genet. 43:171–177. [DOI] [PubMed] [Google Scholar]
- Keeling KM, and Bedwell DM. 2011. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip Rev RNA. 2:837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Lanier J, Du M, Salas-Marco J, Gao L, Kaenjak-Angeletti A, and Bedwell DM. 2004. Leaky termination at premature stop codons antagonizes nonsense-mediated mRNA decay in S. cerevisiae. RNA. 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Xue X, Gunn G, and Bedwell DM. 2014. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 15:371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr TP, Sewry CA, Robb SA, and Roberts RG. 2001. Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum Genet. 109:402–407. [DOI] [PubMed] [Google Scholar]
- Kervestin S, and Jacobson A. 2012. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 13:700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishor A, Ge Z, and Hogg JR. 2019. hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M 2001. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 29:5226–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempely K, and Karam R. 2018. A novel de novo CDH1 germline variant aids in the classification of carboxy-terminal E-cadherin alterations predicted to escape nonsense-mediated mRNA decay. Cold Spring Harb Mol Case Stud. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Ando K, Nakagawa R, and Inada T. 2013. The Upf factor complex interacts with aberrant products derived from mRNAs containing a premature termination codon and facilitates their proteasomal degradation. J Biol Chem. 288:28630–28640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Tatematsu T, and Inada T. 2009. Upf1 stimulates degradation of the product derived from aberrant messenger RNA containing a specific nonsense mutation by the proteasome. EMBO Rep. 10:1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, and Maquat LE. 2013. Rules that govern UPF1 binding to mRNA 3’ UTRs. Proc Natl Acad Sci U S A. 110:3357–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, and Maquat LE. 2016. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 129:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde S, Stone OA, Lessard S, Lavertu A, Desjardins J, Beaudoin M, Rivas M, Stainier DYR, and Lettre G. 2017. Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS One. 12:e0178700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, and Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 446:926–929. [DOI] [PubMed] [Google Scholar]
- Lee HL, and Dougherty JP. 2012. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 136:227–266. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, and van der Maarel SM. 2010. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 329:1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Green RE, and Brenner SE. 2003. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A. 100:189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, and Wilkinson MF. 1998. Nonsense surveillance in lymphocytes? Immunity. 8:135–141. [DOI] [PubMed] [Google Scholar]
- Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen YS, Groth M, Krueger A, Platzer M, Yang YG, Rudolph KL, and Wang ZQ. 2015. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 34:1630–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vuong JK, Zhang M, Stork C, and Zheng S. 2017. Inhibition of nonsense-mediated RNA decay by ER stress. RNA. 23:378–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde L, Boelz S, Neu-Yilik G, Kulozik AE, and Kerem B. 2007. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur J Hum Genet. 15:1156–1162. [DOI] [PubMed] [Google Scholar]
- Lindeboom RG, Supek F, and Lehner B. 2016. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet. 48:1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, Zhu J, Wang Y, Zhao Y, Foo WC, Zuo M, Valasek MA, Javle M, Wilkinson MF, and Lu Y. 2014. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med. 20:596–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, and Xue Q. 2004. Computational identification and sequence analysis of stop codon readthrough genes in Oryza sativa. Biosystems. 77:33–39. [DOI] [PubMed] [Google Scholar]
- Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, and Wilkinson MF. 2014. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 6:748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou CH, Shum EY, and Wilkinson MF. 2015. RNA degradation drives stem cell differentiation. EMBO J. 34:1606–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Chou MY, Ivanov IP, Jungreis I, Kellis M, Kiran AM, Baranov PV, and Atkins JF. 2014. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 42:8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Jungreis I, Tzani I, Power M, Dmitriev RI, Ivanov IP, Kellis M, and Atkins JF. 2018. Stop codon readthrough generates a C-terminally extended variant of the human vitamin D receptor with reduced calcitriol response. J Biol Chem. 293:4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Plank TD, Su F, Shi X, Liu C, Ji Y, Li S, Huynh A, Shi C, Zhu B, Yang G, Wu Y, Wilkinson MF, and Lu Y. 2016. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J Clin Invest. 126:3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, and Jensen TH. 2015. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 16:665–677. [DOI] [PubMed] [Google Scholar]
- Mah JK 2016. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 12:1795–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE 1995. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. 1:453–465. [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, and Dietz HC. 2001. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 107:411–414. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, and Dietz HC. 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 36:1073–1078. [DOI] [PubMed] [Google Scholar]
- Miller JN, and Pearce DA. 2014. Nonsense-mediated decay in genetic disease: friend or foe? Mutat Res Rev Mutat Res. 762:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad MP, Munzarova Pondelickova V, Zeman J, Gunisova S, and Valasek LS. 2017. In vivo evidence that eIF3 stays bound to ribosomes elongating and terminating on short upstream ORFs to promote reinitiation. Nucleic Acids Res. 45:2658–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou H, Smith JL, Peng L, Yin H, Moore J, Zhang XO, Song CQ, Sheel A, Wu Q, Ozata DM, Li Y, Anderson DG, Emerson CP, Sontheimer EJ, Moore MJ, Weng Z, and Xue W. 2017. CRISPR/Cas9-mediated genome editing induces exon skipping by alternative splicing or exon deletion. Genome Biol. 18:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzarova V, Panek J, Gunisova S, Danyi I, Szamecz B, and Valasek LS. 2011. Translation reinitiation relies on the interaction between eIF3a/TIF32 and progressively folded cis-acting mRNA elements preceding short uORFs. PLoS Genet. 7:e1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgoong JH, and Bertoni C. 2016. Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy. Degener Neurol Neuromuscul Dis. 6:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napthine S, Yek C, Powell ML, Brown TD, and Brierley I. 2012. Characterization of the stop codon readthrough signal of Colorado tick fever virus segment 9 RNA. RNA. 18:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif S, Contu L, and Muhlemann O. 2018. Beyond quality control: The role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin Cell Dev Biol. 75:78–87. [DOI] [PubMed] [Google Scholar]
- Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, and Kulozik AE. 2011. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA. 17:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LS, Jolly L, Shoubridge C, Chan WK, Huang L, Laumonnier F, Raynaud M, Hackett A, Field M, Rodriguez J, Srivastava AK, Lee Y, Long R, Addington AM, Rapoport JL, Suren S, Hahn CN, Gamble J, Wilkinson MF, Corbett MA, and Gecz J. 2012. Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol Psychiatry. 17:1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LS, Wilkinson MF, and Gecz J. 2014. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev. 46 Pt 2:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, and Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 21:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickless A, Bailis JM, and You Z. 2017a. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickless A, Cheruiyot A, Flanagan KC, Piwnica-Worms D, Stewart SA, and You Z. 2017b. p38 MAPK inhibits nonsense-mediated RNA decay in response to persistent DNA damage in noncycling cells. J Biol Chem. 292:15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomakuchi TT, Rigo F, Aznarez I, and Krainer AR. 2016. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol. 34:164–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren YS, McClure ML, Rowe SM, Sorscher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, Geiger T, and Kerem B. 2014. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med. 6:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen M, Lund C, Akram Z, Winther JR, Horn N, and Moller LB. 2006. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am J Hum Genet. 79:214–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FJ, Teixeira A, Kong J, Barbosa C, Silva AL, Marques-Ramos A, Liebhaber SA, and Romao L. 2015. Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic Acids Res. 43:6528–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, Mace EM, Vogel TP, Carisey AF, Benavides F, Coban-Akdemir ZH, Gibbs RA, Jhangiani SN, Muzny DM, Carvalho CMB, Schady DA, Jain M, Rosenfeld JA, Emrick L, Lewis RA, Lee B, m. Undiagnosed Diseases Network, Zieba BA, Kury S, Kruger E, Lupski JR, Bostwick BL, and Orange JS. 2018. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am J Hum Genet. 102:1126–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, and Maquat LE. 2015. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat Commun. 6:6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, and Maquat LE. 2016. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell. 165:1319–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard AM, Petek LM, and Miller DG. 2015. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. 24:5901–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K, Yoshina S, Shen X, Han J, DeSantis MR, Xiong M, Mitani S, and Kaufman RJ. 2012. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc Natl Acad Sci U S A. 109:8079–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar H, Mitsios A, Smart M, Skinner J, Welch A, Kalatzis V, Coffey P, Dubis AM, Webster A, and Moosajee M. 2019. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciezynska A, Ruszkowska E, Szulborski K, Rydz K, Wierzbowska J, Kosinska J, Rekas M, Ploski R, Szaflik JP, and Oldak M. 2017. Processing of OPA1 with a novel N-terminal mutation in patients with autosomal dominant optic atrophy: Escape from nonsense-mediated decay. PLoS One. 12:e0183866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH, Ramaiah M, Huang L, McCarrey JR, Peterson KJ, De Rooij DG, Cook-Andersen H, and Wilkinson MF. 2016. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell. 165:382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, and Lykke-Andersen J. 2008. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 6:e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, and Cooper DN. 2003. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 21:577–581. [DOI] [PubMed] [Google Scholar]
- Stiebler AC, Freitag J, Schink KO, Stehlik T, Tillmann BA, Ast J, and Bolker M. 2014. Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in Fungi and animals. PLoS Genet. 10:e1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump MR, Gong Q, Packer JD, and Zhou Z. 2012. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J Mol Cell Cardiol. 53:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump MR, Gong Q, and Zhou Z. 2013. LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation. Am J Physiol Heart Circ Physiol. 305:H1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui T, Song Y, Liu Z, Chen M, Deng J, Xu Y, Lai L, and Li Z. 2018. CRISPR-induced exon skipping is dependent on premature termination codon mutations. Genome Biol. 19:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Araki Y, Ohya Y, Sakuno T, Hoshino S, Kontani K, Nishina H, and Katada T. 2008. Upf1 potentially serves as a RING-related E3 ubiquitin ligase via its association with Upf3 in yeast. RNA. 14:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, O’Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Hills K, Jones D, Mironenko T, Perry J, Varian J, West S, Widaa S, Teague J, Dicks E, Butler A, Menzies A, Richardson D, Jenkinson A, Shepherd R, Raine K, Moon J, Luo Y, Parnau J, Bhat SS, Gardner A, Corbett M, Brooks D, Thomas P, Parkinson-Lawrence E, Porteous ME, Warner JP, Sanderson T, Pearson P, Simensen RJ, Skinner C, Hoganson G, Superneau D, Wooster R, Bobrow M, Turner G, Stevenson RE, Schwartz CE, Futreal PA, Srivastava AK, Stratton MR, and Gecz J. 2007. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 39:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TD MP, and Wilkinson MF. 2018. RNA Decay Factor UPF1 Promotes Protein Decay: A Hidden Talent. Bioessays. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, Hentze MW, and Kulozik AE. 1998. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 17:3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma KG, Rebbapragada I, Durand S, and Lykke-Andersen J. 2015. Identification of elements in human long 3’ UTRs that inhibit nonsense-mediated decay. RNA. 21:887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, and Gardner LB. 2011. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 31:3670–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Jiang B, Jia C, Chai B, and Liang A. 2013. MicroRNA 125 represses nonsense-mediated mRNA decay by regulating SMG1 expression. Biochem Biophys Res Commun. 435:16–20. [DOI] [PubMed] [Google Scholar]
- Wang X, and Gregory-Evans CY. 2015. Nonsense suppression therapies in ocular genetic diseases. Cell Mol Life Sci. 72:1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ju HJ, DeBlasio S, Carino EJ, Johnson R, MacCoss MJ, Heck M, Miller WA, and Gray SM. 2018. A Stem-Loop Structure in Potato Leafroll Virus Open Reading Frame 5 (ORF5) Is Essential for Readthrough Translation of the Coat Protein ORF Stop Codon 700 Bases Upstream. J Virol. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, and Baba H. 2018. Phylogenetically Conserved Sequences Around Myelin P0 Stop Codon are Essential for Translational Readthrough to Produce L-MPZ. Neurochem Res. 43:227–237. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Hayashi A, Campagnoni CW, Kimura A, Inuzuka T, and Baba H. 2012. L-MPZ, a novel isoform of myelin P0, is produced by stop codon readthrough. J Biol Chem. 287:17765–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeilding NM, and Lee WM. 1997. Coding elements in exons 2 and 3 target c-myc mRNA downregulation during myogenic differentiation. Mol Cell Biol. 17:2698–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeilding NM, Procopio WN, Rehman MT, and Lee WM. 1998. c-myc mRNA is down-regulated during myogenic differentiation by accelerated decay that depends on translation of regulatory coding elements. J Biol Chem. 273:15749–15757. [DOI] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, and Burge CB. 2004. Variation in alternative splicing across human tissues. Genome Biol. 5:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, and Muhlemann O. 2011. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA. 17:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetoune AB, Fontaniere S, Magnin D, Anczukow O, Buisson M, Zhang CX, and Mazoyer S. 2008. Comparison of nonsense-mediated mRNA decay efficiency in various murine tissues. BMC Genet. 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Maquat LE. 1997. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 16:826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sun X, Qian Y, and Maquat LE. 1998. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA. 4:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zund D, Gruber AR, Zavolan M, and Muhlemann O. 2013. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3’ UTRs. Nat Struct Mol Biol. 20:936–943. [DOI] [PubMed] [Google Scholar]