

Abstract
The first examples of enantioselective doubly decarboxylative cross coupling are disclosed. Malonate half amides are smoothly coupled to a variety of primary carboxylic acids after formation of the corresponding redox-active esters under Ni-electrocatalytic conditions using a new chiral ligand based on PyBox resulting in amides with a-alkylated stereocenters. The scope of the reaction is broad tolerating numerous functional groups and uniformly proceeds with high ee. Finally, the potential utility of this enantioselective radical-radical reductive cross coupling to simplify synthesis is demonstrated with numerous case studies.
Graphical Abstract

The enantioselective a-alkylation of carbonyl compounds is a staple transformation in organic synthesis that has been widely studied.1 With regards to ester and amide alkylation, the use of chiral auxiliaries is still commonplace due to the predictable outcomes and practical ease with which diastereomers can be separated.2 Such an approach is also intuitive as it takes advantage of polar-bond analysis resulting in an enolate nucleophile reacting with an alkyl halide electrophile. In 1983, Frejd reported a different approach wherein an electrophilic α-halocarbonyl compound could be cross coupled with an aryl zinc nucleophile via Ni catalysis.3 In 2005, the Fu group reported an enantioselective variant of such a reaction setting the stage for a number of advances in catalytic enantioselective access to a-alkylated ester and amide derivatives (Figure 1A).4 Indeed, numerous studies built off of those seminal findings to combine electrophilic ester and amide derivatives with olefins under Ni-catalysis (Figure 1A).5–7 In 2022, the Fu group further demonstrated that in situ generated Reformansky-type reagents could be coupled to alkyl halides in enantioselective fashion (Figure 1B).8 Recent findings from the Xu group have subsequently demonstrated that two electrophiles such as ahalo boronic esters and alkyl halides could also be coupled with high enantiocontrol under photoreductive conditions (Figure 1C).9 Meanwhile, doubly decarboxylative cross coupling (dDCC) has emerged as a powerful method to construct Csp3-Csp3 bonds under electrochemical conditions (with and without Ni, Figure 1D).10–12 In this Communication, the first examples of enantio- and diasteroselective dDCC between redox-active esters (RAEs) derived from readily available alkyl carboxylic acids and malonate derivatives are disclosed.13 This Ni-electrocatalytic reaction is simple to conduct, uses inexpensive components, and demonstrates a wide substrate scope. Its tactical application results in a significant reduction in step count in a variety of different contexts.14
Figure 1.
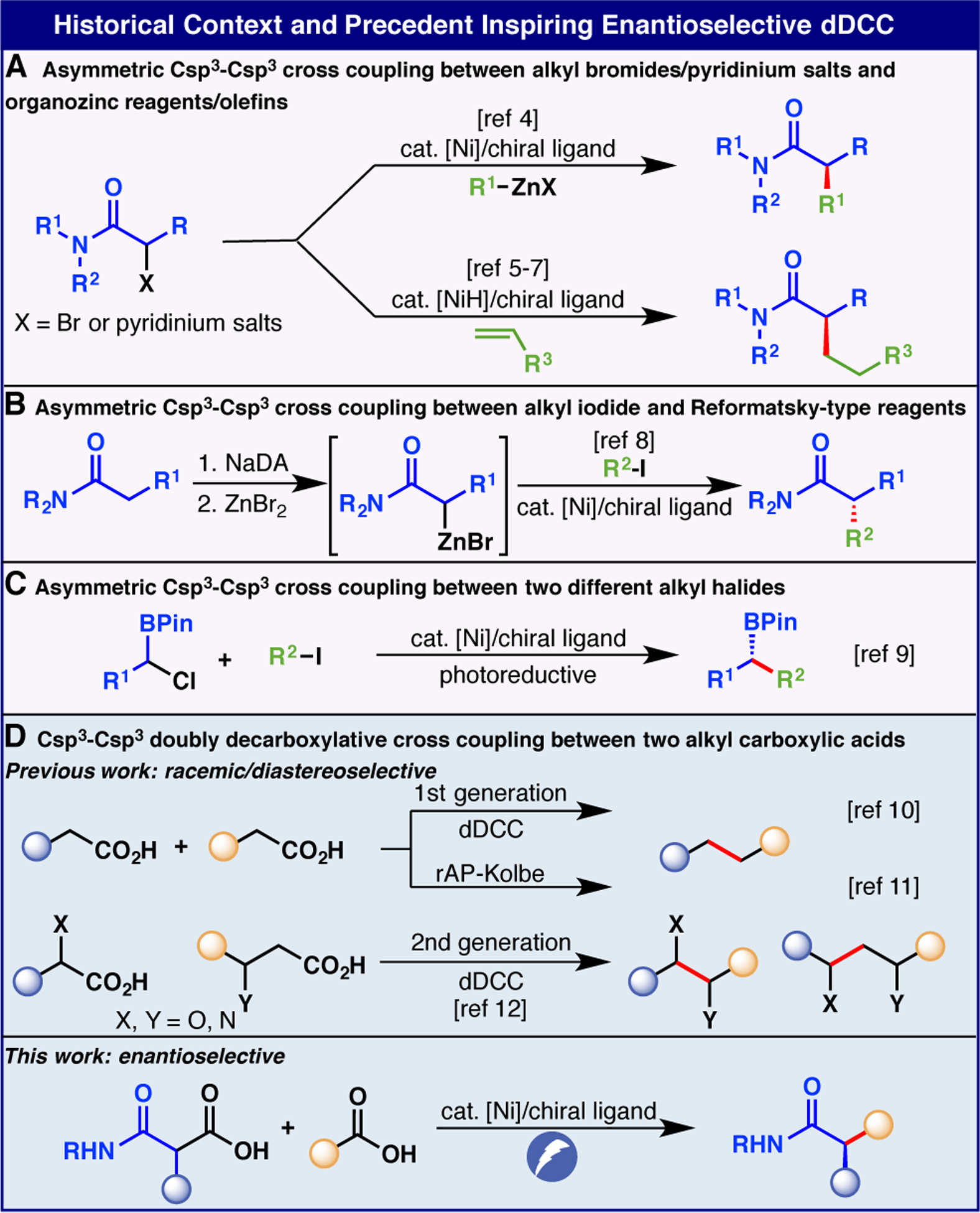
Historical Context and Precedent Inspiring Enantioselective dDCC
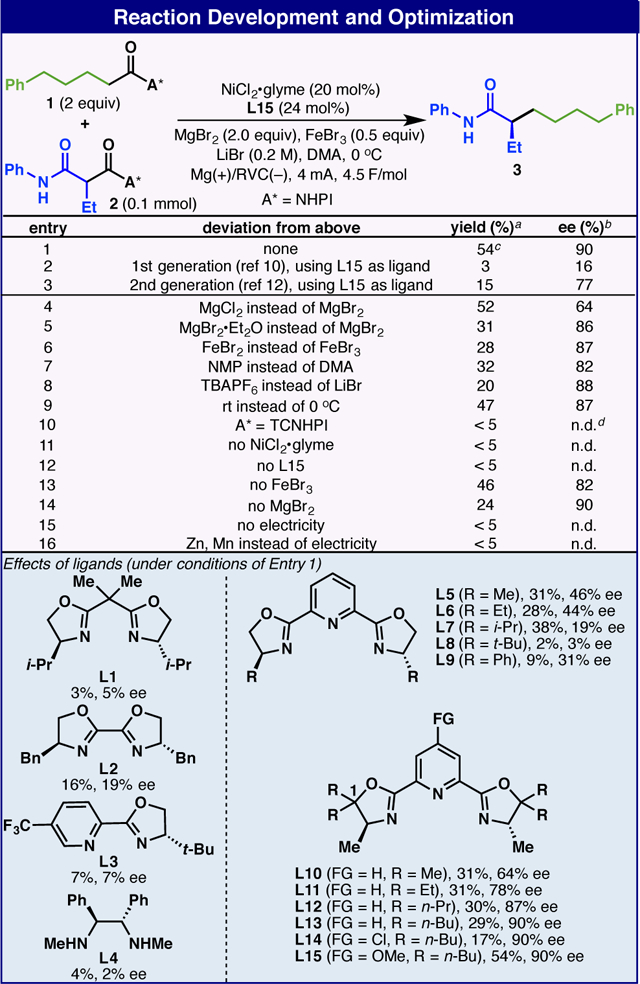
The development of enantioselective dDCC required extensive screening of conditions and ligands, some of which is summarized in Table 1 using RAEs 1 and 2. The final optimized conditions utilized NiCl2•glyme (20 mol%), chiral ligand L15 (24 mol%), MgBr2 (2.0 equiv.), FeBr3 (0.5 equiv.), and LiBr (0.2 M) as the electrolyte in DMA (0.04 M) at 0 °C, affording 3 in 54% isolated yield and 90% ee after 3 hours of electrolysis (0.1 mmol scale, Mg anode and RVC cathode). In contrast, either 1st 10 or 2nd 12 generation dDCC condition proved to be much less effective (Table 1, entries 2 and 3). In terms of additives, MgBr2 appeared to be crucial for this reaction whereas MgCl2 gave similar yield and much lower ee, and MgBr2•Et2O only provided 31% 3 (Table 1, entries 4 and 5). Replacing FeBr3 with FeBr2 also led to inferior results (Table 1, entry 6). Changing other parameters such as solvent, electrolyte and temperature resulted in unsatisfactory outcomes (Table 1, entries 7–9). In addition, TCNHPI-based RAEs proved to be too reactive under these conditions, undergoing unmediated cathodic reduction without affording desired cross-coupled product (Table 1, entry 10). A series of control experiments indicated that nickel catalyst, ligand and electricity were crucial to promote this reaction, while MgBr2 and FeBr3 were indispensable components to ensure its efficiency (Table 1, entries 11–16).
Table 1.
|
Yields determined by GC-MS analysis.
ee values determined by chiral SFC analysis.
Isolated yield.
Not determined.
The ligands had a substantial effect on the outcome of this reaction. As observed in our previous report,10 tridentate ligands proved to be superior to bidentate analogues in the Ni-catalyzed Csp3–Csp3 dDCC reaction. For example, bidentate ligands with different backbones (L1–L4) all gave very poor yield and ee. To our delight, preliminary screening of PyBox ligands provided promising results, and the use of L5 gave 3 in 31% yield and 46% ee. Further modification of L5 revealed that alkyl chain substituents at the C-1 position dramatically improved enantioselectivity (L10–L13). Final adjustment of the C-4 substituent of the pyridine ring increased the yield without loss of enantiopurity.
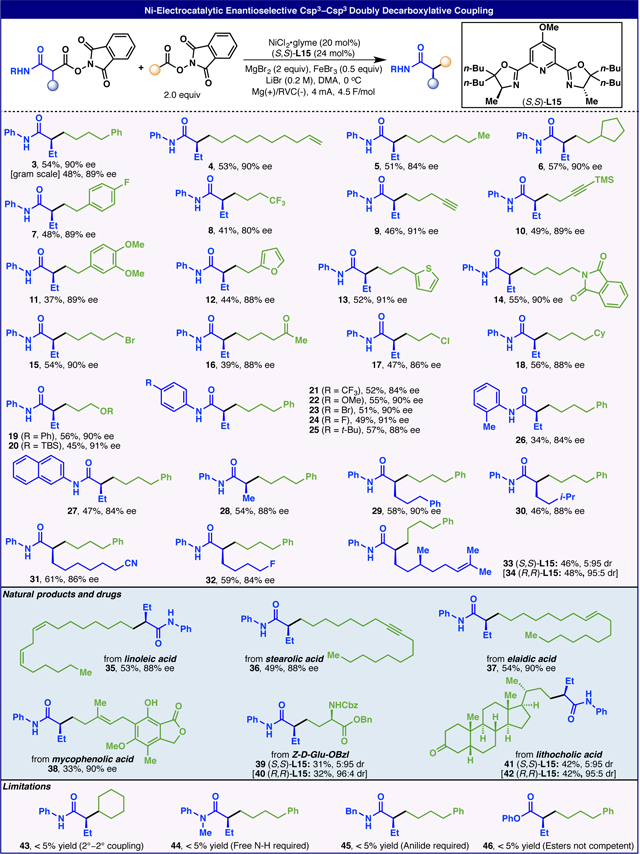
With the optimal conditions in hand, the scope of enantioselective doubly decarboxylative Csp3-Csp3 cross coupling was investigated, as shown in Table 2. A vast array of RAEs derived from readily available alkyl carboxylic acids were tested. Aside from simple alkyl chains (3, 5, 6, 18), a broad range of functional groups could be tolerated, such as terminal alkenes (4), internal alkenes (35, 37, 38), trifluoromethyl group (8), terminal alkynes (9), internal alkynes (10, 36), alkyl halides (15, 17), aryl halides (7), ketones (16, 41, 42), silyl ethers (20), ethers (11, 19), imides (14), heterocycles (12, 13), lactones (38), carba-mates (39, 40), and esters (39, 40). Several RAEs derived from malonate derivatives were also explored ranging from various substitutions on the phenyl ring (21–27) to substrates containing alkyl fluorides (32), nitriles (31) and internal alkenes (33, 34). Even with pre-existing stereocenters, the reaction can be programmed to access diastereomers with high control (33, 34, 39, 40, 41 and 42). With the exception of compounds 27 and 28, none of these structures have been prepared before. However, several related derivatives of some of these molecules (3, 6, 7, 14, 16, 17, 19 and 20) have been synthesized in racemic form, often through laborious routes (see SI for graphical comparison).
Table 2.
Scope of Ni-Electrocatalytic Enantioselective Csp3–Csp3 Doubly Decarboxylative Coupling (dDCC)a
|
Yields of isolated products are indicated in each case unless otherwise specified.
It is worth noting that compound 3 has been prepared on a gram scale with no significant reduction in both yield and enantiopurity. With regard to limitations, coupling of 2 and secondary alkyl carboxylic acids was unsuccessful (43). As for the malonate-derived RAEs, the secondary aryl amide group proved to be critical as evidenced by the fact that the corresponding analogs containg tertiary amides (44), secondary aliphatic amides (45) and esters (46) all gave unproductive results.
The enantioselective dDCC outlined herein can be applied to simplify the synthesis of both medicinally important structures and intermediates employed in natural product total synthesis (Figure 2). For instance, the bile-acid derivative 49 (Figure 2A) was previously prepared from commercial hyodeoxycholic acid 47 as an inseparable mixture of diasteromers at C-24 in 15 steps with only one of those steps making a key C–C bond.15 In contrast, the same starting material could be enlisted to afford the desired product in only four steps as the major isomer (96:4 dr). The key dDCC proceeded in 34% yield with high diastereoselectivity (96:4 dr) thereby enabling this rapid route. Similarly, indole building block 54 (Figure 2B) required a 10-step sequence featuring an Evans alkylation.16 In contrast, the dDCC approach was far more direct requiring only 3 steps from commercial 53 (42% yield and 88% ee for the coupling step). In the next case study, the total synthesis of penicitide A employed a simple chiral alcohol 57 that was prepared from D-aspartic acid 55 in 12 steps.17 In contrast, the same structure could be accessed in only five steps from carboxylic acid 56 followed by a diasteroselective dDCC (51% yield, 96:4 dr) and amide reduction. Finally, carboxylic acid 61 was recently employed to complete the total synthesis of fluvirucinin B1.18 Its preparation involved a circuitous 12-step route that could be completely circumvented by employing an enantioselective dDCC on carboxylic acid 60 (44% yield, 90% ee) followed by mild amide hydrolysis to enable 3-step access. In all four of these cases, the use of pyrophoric and/or toxic reagents and expensive transition metals was eliminated as well as numerous functional group inter-conversions, protecting group manipulations, and redox fluctuations. This is yet another example of how radical retrosynthetic logic14 can often lead to more direct and ideal synthetic routes.19
Figure 2.
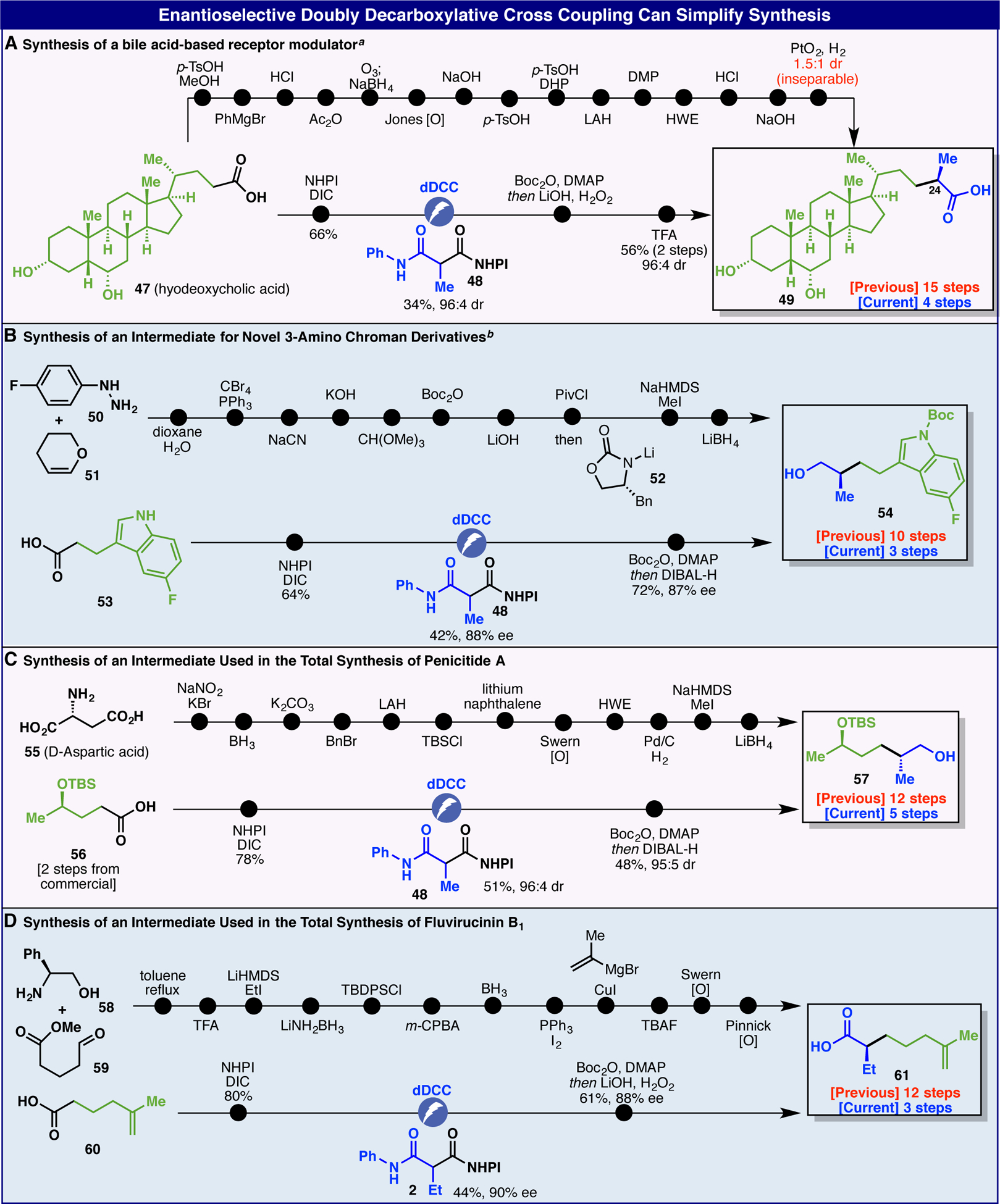
Enantioselective Doubly Decarboxylative Cross Coupling Can Simplify Synthesis. a47 (0.1 mmol), 48 (0.3 mmol) and 6.0 F/mol were used. bReaction was conducted at −5°C.
A mechanistic analysis of this reaction system will be the subject of a future study. Currently, the elementary steps are understood by analogy to prior art (See SI for proposed mechanism). The precise role of the enabling Mg- and Fe-based additives is unclear at the present moment. To elucidate the active catalytic species a non-linear effect study was performed (see SI) suggesting that a monomeric Ni-complex bearing a single chiral ligand is operative.20 A CV study (see SI) was also performed suggesting that the most reducible species in solution is the Ni-ligand complex. Two reduction peaks are observed, which may be attributed to the reduction potential of Ni(II)/Ni(I) and Ni(I)/Ni(0), respectively.21 The moderate yields observed in several cases might originate from the different rates of generating the alkyl radicals from the electronically differing RAEs. Based on this assumption, a fine-tuning the electronic properties of the NHPI moiety of the two different RAEs (e.g. install different substitution groups on the benzene ring) might be a potential strategy to enhance the yields.
This work discloses a unique example of forging C–C bonds adjacent to a carbonyl group using dDCC with high stereocontrol. As an addition to the growing body of literature wherein the stereochemical course of radical cross couplings can be controlled with judicious ligand choice it expands the scope of this newly emerging reaction class.22 It also represents a useful precedent for the development of electrocatalytic asymmetric transformations.23 The simple reaction setup, readily available reagents, and high enantiocontrol combined with illustrative examples that simplify synthesis through radical retrosynthetic logic are suggestive of broad applicability.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NSF Center for Synthetic Organic Electrochemistry, CHE-2002158NSF (the discovery and optimization effort). NIGMS (GM-118176) supported the scope and application study. We also acknowledge SIOC Fellowship for J.H. during this study. Authors are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy, to Dr. Jason Chen, Brittany Sanchez and Quynh Nguyen Wong (Scripps Automated Synthesis Facility) for assistance with chiral SFC analysis and HRMS.
REFERENCES
- (1).Kohler MC; Wengryniuk SE; Coltart DM Asymmetric α-Alkylation of Aldehydes, Ketones, and Carboxylic Acids. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko V, Andrushko N, Eds.; John Wiley & Sons, 2014; Vol. 1, pp 183–213. [Google Scholar]
- (2).(a) Marco JA; Carda M; Murga J; Falomir E Selected Diastereoselective Reactions: Enolate Alkylation. In Comprehensive Chirality; Carreira EM, Yamamoto H, Eds.; Academic, 2012; Vol. 2, Chapter 2.14. [Google Scholar]; (b) Evans DA Stereoselective Alkylation Reactions of Chiral Metal Enolates. In Asymmetric Synthesis; Morrison JD, Ed.; Academic, 1984; Vol. 3, pp 1–110. [Google Scholar]; (c) Myers AG; Yang BH; Chen H; Gleason JL Use of Pseudoephedrine as a Practical Chiral Auxiliary for Asymmetric Synthesis. J. Am. Chem. Soc 1994, 116, 9361–9362. [Google Scholar]; (d) Heravi MM; Zadsirjan V; Farajpour B Applications of Oxazolidinones as Chiral Auxiliaries in the Asymmetric Alkylation Reaction Applied to Total Synthesis. RSC Adv 2016, 6, 30498–30551. [Google Scholar]
- (3).Klingstedt T; Frejd T Nickel-Catalyzed Synthesis of Arylacetic Esters from Arylzinc Chlorides and Ethyl Bromoacetate. Organometallics 1983, 2, 598–600. [Google Scholar]
- (4).Fischer C; Fu GC Asymmetric Nickel-Catalyzed Negishi Cross-Couplings of Secondary α-Bromo Amides with Organozinc Reagents. J. Am. Chem. Soc 2005, 127, 4594–4595. [DOI] [PubMed] [Google Scholar]
- (5).Wang Z; Yin H; Fu GC Catalytic Enantioconvergent Coupling of Secondary and Tertiary Electrophiles with Olefins. Nature 2018, 563, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhou F; Zhang Y; Xu X; Zhu S NiH-Catalyzed Remote Asymmetric Hydroalkylation of Alkenes with Racemic α-Bromo Amides. Angew. Chem., Int. Ed 2019, 58, 1754–1758. [DOI] [PubMed] [Google Scholar]
- (7).Sun SZ; Cai YM; Zhang DL; Wang JB; Yao HQ; Rui XY; Martin R; Shang M Enantioselective Deaminative Alkylation of Amino Acid Derivatives with Unactivated Olefins. J. Am. Chem. Soc 2022, 144, 1130–1137. [DOI] [PubMed] [Google Scholar]
- (8).Tong X; Schneck F; Fu GC Catalytic Enantioselective α-Alkylation of Amides by Unactivated Alkyl Electrophiles. J. Am. Chem. Soc 2022, 144, 14856–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhou J; Wang D; Xu W; Hu Z; Xu T Enantioselective C(sp3)–C(sp3) Reductive Cross-Electrophile Coupling of Unactivated Alkyl Halides with α-Chloroboronates via Dual Nickel/Photoredox Catalysis. J. Am. Chem. Soc 2023, 145, 2081–2087. [DOI] [PubMed] [Google Scholar]
- (10).Zhang B; Gao Y; Hioki Y; Oderinde MS; Qiao JX; Rodriguez KX; Zhang H-J; Kawamata Y; Baran PS Ni-Electrocatalytic Csp3–Csp3 Doubly Decarboxylative Coupling. Nature 2022, 606, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hioki Y; Costantini M; Griffin J; Harper KC; Merini MP; Nissl B; Kawamata Y; Baran PS Overcoming the Limitations of Kolbe Coupling with Waveform-Controlled Electrosynthesis. Science 2023, 380, 81–87. [DOI] [PubMed] [Google Scholar]
- (12).Zhang B; He J; Gao Y; Levy L; Oderinde MS; Palkowitz MD; Murali Dhar TG; Mandler MD; Collins MR; Schmitt DC; Bolduc PN; Chen T; Clementson S; Petersen NN; Laudadio G; Kawamata Y; Baran PS Radical Simplification of Complex Molecule Retrosynthesis Enabled by Electrocatalytic Cross-Coupling of a-Substituted Carboxylic Acids. ChemRxiv 2023. DOI: 10.26434/chemrxiv-2023-0ndm7. [DOI] [Google Scholar]
- (13).For Ni-catalyzed reductive arylation of malonate-derived RAEs, see:; Gabbey AL; Michel NWM; Hughes JME; Campeau L-C; Rousseaux SAL Synthesis of α-Aryl Secondary Amides via Nickel-Catalyzed Reductive Coupling of Redox-Active Esters. Org. Lett 2022, 24, 3173–3178. [DOI] [PubMed] [Google Scholar]
- (14).Smith JM; Harwood SJ; Baran PS Radical Retrosynthesis. Acc. Chem. Res 2018, 51, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sabbatini P; Filipponi P; Sardella R; Natalini B; Nuti R; Macchiarulo A; Pellicciari R; Gioiello A Synthesis and Quantitative Structure-Property Relationships of Side Chain-Modified Hyodeoxycholic Acid Derivatives. Molecules 2013, 18, 10497–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hatzenbuhler NT; Evrard DA; Mewshaw RE; Zhou D-H; Shah US; Inghrim JA; Lenicek SE; Baudy RB; Butera JA; Sabb AL; Failli AA; Ramamoorthy PS 3-Amino Choman and 2-Amino Tetralin Derivatives WO2005012291, 2005. [Google Scholar]
- (17).Saha D; Guchhait S; Goswami RK Total Synthesis and Stereochemical Assignment of Penicitide A. Org. Lett 2020, 22, 745–749. [DOI] [PubMed] [Google Scholar]
- (18).Guignard G; Llor N; Molins E; Bosch J; Amat M Enantioselective Total Synthesis of Fluvirucinin B1. Org. Lett 2016, 18, 1788–1791. [DOI] [PubMed] [Google Scholar]
- (19).Gaich T; Baran PS Aiming for the Ideal Synthesis. J. Org. Chem 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- (20).Satyanarayana T; Abraham S; Kagan HB Nonlinear Effects in Asymmetric Catalysis. Angew. Chem., Int. Ed 2009, 48, 456–494. [DOI] [PubMed] [Google Scholar]
- (21).Tang T; Hazra A; Min DS; Williams WL; Jones E; Doyle AG; Sigman MS J. Am. Chem. Soc 2023, 145, 8689–8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For reviews, see:; (a) Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds. Chem. Rev 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu GC Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci 2017, 3, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Choi J; Fu GC Transition Metal–Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang F; Chen P; Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 2018, 51, 2036–2046. [DOI] [PubMed] [Google Scholar]
- (23).For reviews, see:; (a) Lin Q; Li L; Luo S Asymmetric Electrochemical Catalysis. Chem. Eur. J 2019, 25, 10033–10044. [DOI] [PubMed] [Google Scholar]; (b) Ghosh M; Shinde VS; Rueping M A Review of Asymmetric Synthetic Organic Electrochemistry and Electrocatalysis: Concepts, Applications, Recent Developments and Future Directions. Beilstein J. Org. Chem 2019, 15, 2710–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chang X; Zhang Q; Guo C Asymmetric Electrochemical Transformations. Angew. Chem., Int. Ed 2020, 59, 12612–12622. [DOI] [PubMed] [Google Scholar]; (d) Wang X; Xu X; Wang Z; Fang P; Mei T Advances in Asymmetric Organotransition Metal-Catalyzed Electrochemistry. Chin. J. Org. Chem 2020, 40, 3738–3747. [Google Scholar]; (e) Jiao K; Wang Z; Ma C; Liu H; Cheng B; Mei T The Applications of Electrochemical Synthesis in Asymmetric Catalysis. Chem Catal 2022, 2, 3019–3047. [Google Scholar]; For recent examples, see:; (f) Von Münchow T; Dana S; Xu Y; Yuan B; Ackermann L Enantioselective Electrochemical Cobalt-Catalyzed Aryl C–H Activation Reactions. Science 2023, 379, 1036–1042. [DOI] [PubMed] [Google Scholar]; (g) Hu X; Cheng-Sánchez I; Cuesta-Galisteo S; Nevado C Nickel-Catalyzed Enantioselective Electrochemical Reductive Cross-Coupling of Aryl Aziridines with Alkenyl Bromides. J. Am. Chem. Soc DOI: 10.1021/jacs.2c12869. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) DeLano TJ; Reisman SE Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal 2019, 9, 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Song L; Fu N; Ernst BG; Lee WH; Frederick MO; DiS-tasio RA Jr.; Lin S Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem 2020, 12, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Liu D; Liu Z; Wang Z; Ma C; Herbert S; Schirok H; Mei T Paired Electrolysis-Enabled Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Between α-Chloroesters and Aryl Bromides. Nat. Commun 2022, 13, 7318–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Gao Y; Hill DE; Hao W; McNicolas BJ; Vantourout JC; Hadt RG; Reisman SE; Blackmond D; Baran PS Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism. J. Am. Chem. Soc 2021, 143, 9478–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Zhang Q; Liang K; Guo C Enantioselective Nickel-Catalyzed Electrochemical Radical Allylation. Angew. Chem., Int. Ed 2022, 61, e202210632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.