

Summary.
Lipid droplets (LDs) store lipids that can be utilized during times of scarcity via autophagic and lysosomal pathways, but how LDs and autophagosomes interact remained unclear. Here, we discovered that the E2 autophagic enzyme, ATG3, localizes to the surface of certain ultra-large LDs in differentiated murine 3T3-L1 adipocytes or Huh7 human liver cells undergoing prolonged starvation. Subsequently, ATG3 lipidates Microtubule-associated protein 1 light chain 3B (LC3B) to these LDs. In vitro, ATG3 could bind alone to purified and artificial LDs to mediate this lipidation reaction. We observed that LC3B-lipidated LDs were consistently in close proximity to collections of LC3B-membranes and were lacking Plin1. This phenotype is distinct from macrolipophagy but required autophagy, as it disappeared following ATG5 or Beclin1 knockout. Our data suggest that extended starvation triggers a noncanonical autophagy mechanism, similar to LC3B-associated phagocytosis, where the surface of large LDs serves as an LC3B lipidation platform for autophagic processes.
Introduction
Autophagy is a catabolic process that breaks down cellular components in response to various stimuli such as energy deprivation or cellular stresses 1. A major autophagic pathway is macroautophagy, wherein an autophagosome, a double-vesicle membrane phagophore, isolates compounds from the cytosol and delivers them to lysosomes for degradation 2,3. The formation of the autophagosome proceeds in multiple steps 2,4,5: initiation and nucleation of a pre-autophagosomal structure (~ 100 nm)6, elongation of the membrane and encapsulation of cargos, closure of the double-vesicle (~ 0.5 - 1.5 μm in size), i.e., the completed autophagosome 7,8. These steps involve several autophagy (ATG) related enzymes and take place at specific endoplasmic reticulum (ER) sub-regions 9,10.
The autophagosome maturation and the recognition of cargos require the lipidation of a ubiquitin-like protein ATG8 to phosphatidylethanolamine (PE) onto the double-membrane 11,12. In mammals, there are two ATG8 subfamilies, the microtubule-associated protein 1 light chain 3 A (also known as MAP1LC3)(LC3A), LC3B (the most ubiquitous) and LC3C, and the GABA type A receptor-associated protein (GABARAP) and GABARAP-like proteins, GABARAPL1 and GABARAPL2 13. The lipidation reaction is assisted by the ATG16L1/ATG5-ATG12 complex 14–16. ATG16 binds to the pre-autophagosomal structure through interactions with FIP200, WIPI2, and PI3P on the membrane. Membrane-bound ATG16 recruits ATG5-ATG12, which then recruits ATG3 onto the membrane, the E2 enzyme catalyzing ATG8 lipidation 17. Finally, ATG7, an E1 protein, activates and delivers ATG8 to ATG3, which lipidates it to PE onto the phagophore 18.
Apart from its role in autophagosomes, LC3 proteins can also be attached to PE on single bilayer membranes in a noncanonical autophagic pathway termed LC3-associated phagocytosis 19. This pathway breaks down large components or pathogens that are internalized through phagocytosis and involves the recruitment of lysosomes through the phagosome-lipidated LC3 20. Unlike conventional autophagy, this noncanonical pathway does not generate autophagosomes and does not involve crucial autophagic enzymes like Atg9 or the ULK complex 20. However, it still requires all the upstream lipidation machinery, including ATG16L1/ATG5-ATG12 21. The existence of this pathway suggests that autophagic proteins may serve other purposes beyond autophagosome biology 21. Interestingly, several autophagy proteins have been identified around lipid droplets (LDs), including Atg2, DFCP1, Atg14L, and possibly LC3 22–27, implying functional crosstalk between autophagy and LDs that is yet to be fully understood 28.
LDs are unique cellular organelles that have a neutral lipid core consisting mainly of triglycerides. They are surrounded by a phospholipid monolayer, which makes them structurally distinct from bilayer-bounded organelles 29. While LDs primarily function in maintaining cellular energy balance, they also have non-metabolic functions such as protein quality control, gene expression, and development 30. These functions are closely related to the proteins bound to their surface 31. LDs are targeted by two major classes of proteins: soluble proteins that often have amphipathic helix motifs or lipid anchors, and monotopic membrane proteins that typically move from the ER to LDs via physical contiguities 31,32,36,55,57. Most autophagic proteins that target LDs are from the cytosol, with the exception of DFCP1, which comes from the ER 23.
Autophagic proteins are involved in regulating both the biogenesis and catabolism of LDs 25,33. LDs also regulate autophagy, by providing lipids for autophagosome biogenesis 34,35,37–39. Furthermore, the adipose triglyceride lipase (ATGL), which acts on the LD surface, has an LC3 interacting motif (LIR) and its activity can modulate Sirtuin 1, an autophagy regulator 40,41. During energy scarcities, LDs can undergo lipophagy 42, a process where an LD is engulfed by an autophagosome that subsequently fuses with lysosomes. In the liver, ATGL hydrolyses triglycerides to reduce LD size to the point where they can fit into autophagosomes during energy depletion 43. Smaller LDs, typically less than 1μm, are majorly found in autophagosomes or autolysosomes 43,44 and targeted by lipophagy. These findings suggest that depending on metabolic cues, autophagy, and LDs communicate through distinct channels.
In the present study, we report a noncanonical autophagy pathway that is activated in response to prolonged starvation. Specifically, we found that LC3B is ligated to LD phospholipids and localizes to a few large LDs, which can reach up to 25 μm in size in 3T3-L1 adipocytes, through the involvement of the minimal ATG3,7 machinery. Following this process, the lapidated LDs were always found in close apposition with LC3-positive autophagosomes/autolysosomes-like membranes, serving for degradation.
Results.
Lipidated LC3B localizes to large LDs during long-term nutrient deprivation
Adipocytes serve as the primary cells for storing lipids, and they contain micrometric LDs that are considerably larger than LDs degraded by lipophagy. We asked whether autophagy could degrade such large LDs and sought to study their interaction with autophagosomes during nutrient deprivation. LC3B is the most ubiquitous ATG8 protein in mammals 45 that marks autophagosomes, although it does not exclusively localize there. Differentiated murine 3T3-L1 adipocytes were exposed to an eGFP-LC3B adenovirus and then cultured in Earle’s balanced salts (EBSS) devoid of bovine serum albumin (BSA), a nutrient-deprived media, for a period of up to 72hs.
We found that LC3B was present around ultra-large LDs, which were tens of μm in size at 24, 48, and 72hs (Figures 1A–1D and S1A). This phenomenon persisted even when BSA was added to the culture medium and rinsed, to remove released fatty acids during the starvation time (Figure S1B), suggesting that the localization of LC3B to the large LDs was not linked to de novo re-esterification of liberated fatty acids. When the cells were grown in DMEM or starved for two hours only, eGFP-LC3B localization to ultra-large LDs was rare. We found that in a few cases, the LC3B signal on the LDs could be colocalized with Plin1, a standard adipocyte LD surface marker (Figure S1C). However, the eGFP-LC3B signal on the LDs was often inhomogeneous, with both a uniform LC3B signal on the LD surface and another more intense accumulation of LC3B signal from structures adjacent or adhering to the LDs (Figures 1A and S1A).
Figure 1. LC3B is recruited to LDs during long-term nutrient deprivation.
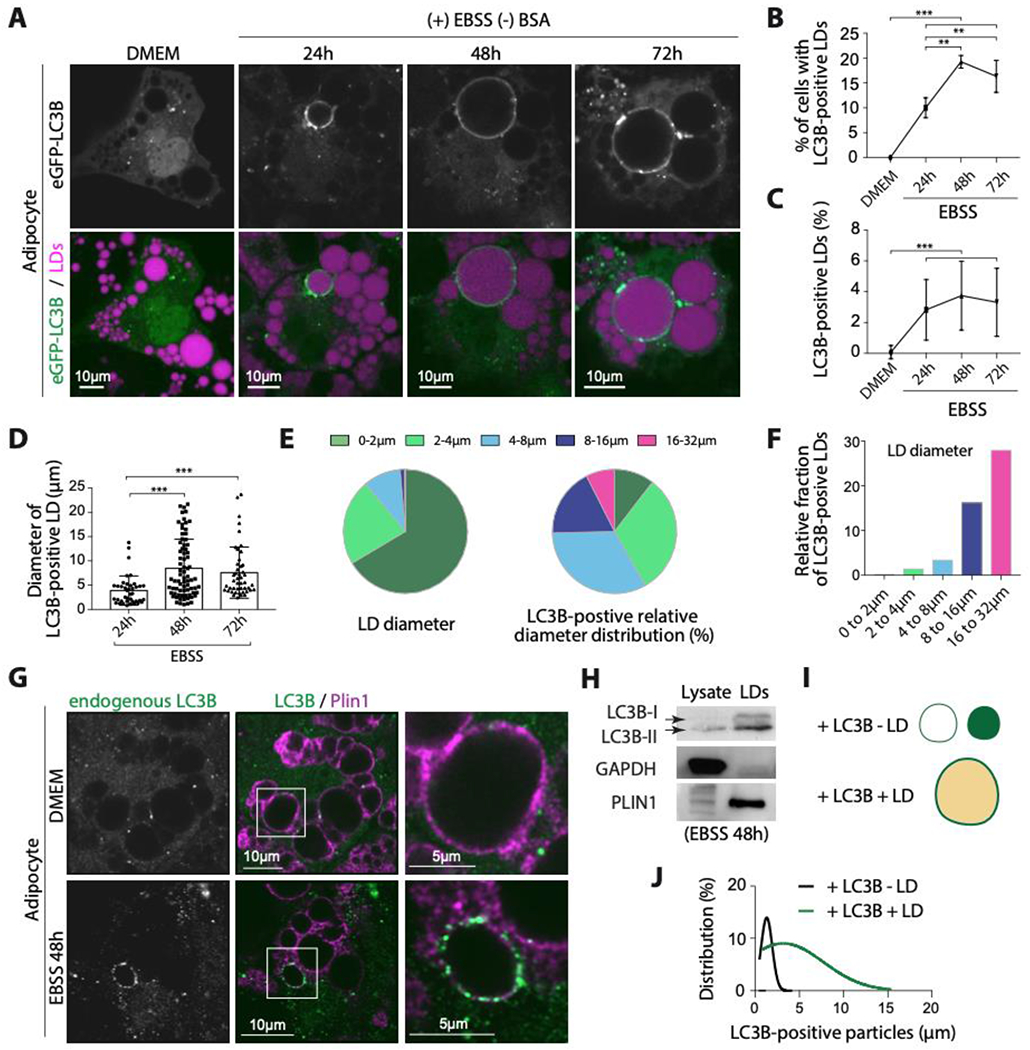
A. Confocal imaging of eGFP-LC3B and LDs in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B. Cells were incubated in EBSS for the indicated time after transfection. Scale bar, 10 μm.
B. Percentage of cells with eGFP-LC3B-positive LDs. Data were obtained from three independent experiments done as described in A. An ordinary one-way ANOVA test was used (** P < 0.001, *** P<0.0001).
C. Percentage of eGFP-LC3B-positive LDs per cell. Data were obtained from three independent experiments done as described in A. An ordinary one-way ANOVA test was used (*** P<0.0001).
D. Diameter of eGFP-LC3B-positive LDs quantified from three independent experiments done as described in A. An ordinary one-way ANOVA test was used (*** P<0.0001).
E. Left: sector graph shows the size distribution of LDs in differentiated 3T3-L1 adipocytes, transfected with eGFP-LC3B and incubated in EBSS for 48hs. Three independent experiments n=10 cells were collapsed. The right sector graph shows the size distribution of eGFP-LC3B-positive LDs. Quantifications are from 4 independent experiments.
F. The relative fraction of eGFP-LC3B-positive LDs in a size range (i.e. per size distribution, normalization of E).
G. Immunofluorescence staining of LC3B and PLIN1 in differentiated 3T3-L1 adipocytes incubated in EBSS for 48hs.
H. Western blot of lysate and LDs fractions of cells treated as described in G.
I. Schematic representation of LC3B puncta with or without LD.
J. eGFP-LC3B puncta size distribution with or without LDs. The data were obtained from 10 cells from three independent experiments.
See also Figure S1.
During nutrient starvation, the percentage of cells exhibiting the LC3B-positive LD phenotype increased, with up to 20% of cells displaying this phenotype at 48 and 72 hours (Figure 1B). Concurrently, the number of large LC3B-positive LDs also increased with starvation, reaching up to 6% at 48 and 72hs (Figures 1C and 1D). Interestingly, LC3B seemed to specifically target the subset of larger LDs, as evidenced by the relative distribution to the size of LC3-positive LDs (Figures 1E and 1F). These observations were made with overexpressed eGFP-LC3B, but endogenous LC3B was also found around larger LDs, exclusively in EBSS and not in complete media (Figure 1G). In addition, we detected the presence of lipidated LC3B-II and a smaller amount of LC3B-I on purified LDs from starved conditions (Figure 1H).
In various cell lines, the process of lipophagy involves small LDs typically of 1 μm or less in size 42–44,46. However, here, LC3B-positive LDs positive were found to be as large as ~25μm. The size distribution of LD-negative and LD-positive LC3B structures was measured and analyzed (Figures 1I and 1J). The size distribution of LD-negative LC3 puncta was found to be narrow, with a distribution centered at 1.5 μm, suggesting they were likely autophagosomes or autolysosomes. The size distribution of the LD-positive LC3B signal, on the other hand, showed a wider spread of 2-30μm (Figures 1J). Taken together, these findings suggest that the LC3B signal on the larger LDs might be distinct from LC3B on conventional autophagosomes.
To investigate whether LC3B preferentially localizes to the largest LDs in other cell lines, we also examined liver hepatoma 7 (Huh7) and HeLa cells that were treated with oleic acid (OA) for 24hs before being starved with EBSS for 48hs. While these cell lines were not capable of producing LDs as large as those found in adipocytes, the LDs that exhibited a visible LC3B ring were mostly among the largest, measuring around 2 μm and above (Figures S1D and S1E). Similar to the 3T3-L1 adipocytes, the frequency of this phenomenon increased in Huh7 cells upon starvation (Figures S1F and S1G). Also, when we transfected in Huh7 cells the eGFP-LC3B G120A construct which cannot be lipidated 47, we did not find the protein around LDs as compared to eGFP-LC3B (Figure S1H). This indicates that the LC3B signal around the LDs was the lipidated form of the protein.
Finally, to investigate whether other ATG8 proteins behave similarly to LC3B, we conducted experiments on 3T3-L1-differentiated cells and Huh7 cells. In 3T3-L1-differentiated cells, we used an antibody to detect endogenous GABARAPs, but no GABARAP ring around large LDs was observed, although the proteins were detected on smaller LDs (Figure S1I). We then delivered eGFP-plasmid constructs of GABARAP, GABARAPL1, LC3A, and LC3C to Huh7 cells, which are easier to transfect. In contrast to LC3B, none of these proteins were found in the largest LD subpopulation (Figures S1J and S1K).
Taken together, our results suggest that LC3B is recruited specifically to the surface of certain large LDs in cells that have been cultured in an EBSS starving medium for an extended period of time.
LC3B localization to LDs is not a result of nearby autophagosomes but necessitates autophagy.
We wanted to determine whether the LC3B signal on the large lipid droplets (LDs) was due to LC3B interacting with proteins on LDs or the close proximity of autophagosomes.
Proteins containing LC3B-interacting region (LIR) motifs could potentially recruit LC3B to the LD surface. ATGL, an enzyme that regulates lipophagy in mice livers, has an LIR motif 40. We tested whether ATGL could recruit LC3B by overexpressing it in Huh7 cells, but observed no change in the LC3B LD-localization phenotype under nutrient starvation conditions (Figures S2A and S2B).
Plin1 is a major adipocyte LD protein marker that has LIR sequences on its C-terminal region, specifically YVPL and YSQL, which could theoretically recruit LC3B. To test this, we expressed mCherry-Plin1 in Huh7 cells that do not express Plin148. During starvation, Plin1 was present on all LDs, but the LC3B LD localization was unchanged as compared with the control, indicating that Plin1 did not mediate LC3B recruitment to LDs (Figure S2C).
We then investigated the possibility that the LC3B ring signals around the large LDs were due to LC3B interaction with P62-bound ubiquitinated LD proteins. P62 (also known as sequestosome-1) is an adaptor protein that binds to most ubiquitinated proteins or organelles targeted for degradation. To test this model, we virally co-transfected differentiated 3T3-L1 adipocytes with eGFP-LC3 and RFP-P62 and induced 48h starvation. We found clear LC3B signals around LDs, but most were devoid of P62 (73% of cases; Figures 2A and 2B), especially larger ones. In the remaining 17%, we observed LC3 colocalizing with P62 more frequently on smaller LDs, although not completely (Figures 2A and 2B). Overexpression of mRFP-P62 did not increase the percentage of LDs or cells with LC3B-positive LDs (Figures S2D–S2F), and P62/LC3B colocalization sharply decreased on LDs compared with cytosolic puncta (Figures S2G–S2I). These observations suggest that different LC3B pools are present on LDs and autophagosome structures and that the pool on LDs is not recruited to the large LDs by the aforementioned proteins (Figure 2C).
Figure 2. LC3B is not recruited to LDs by known factors.
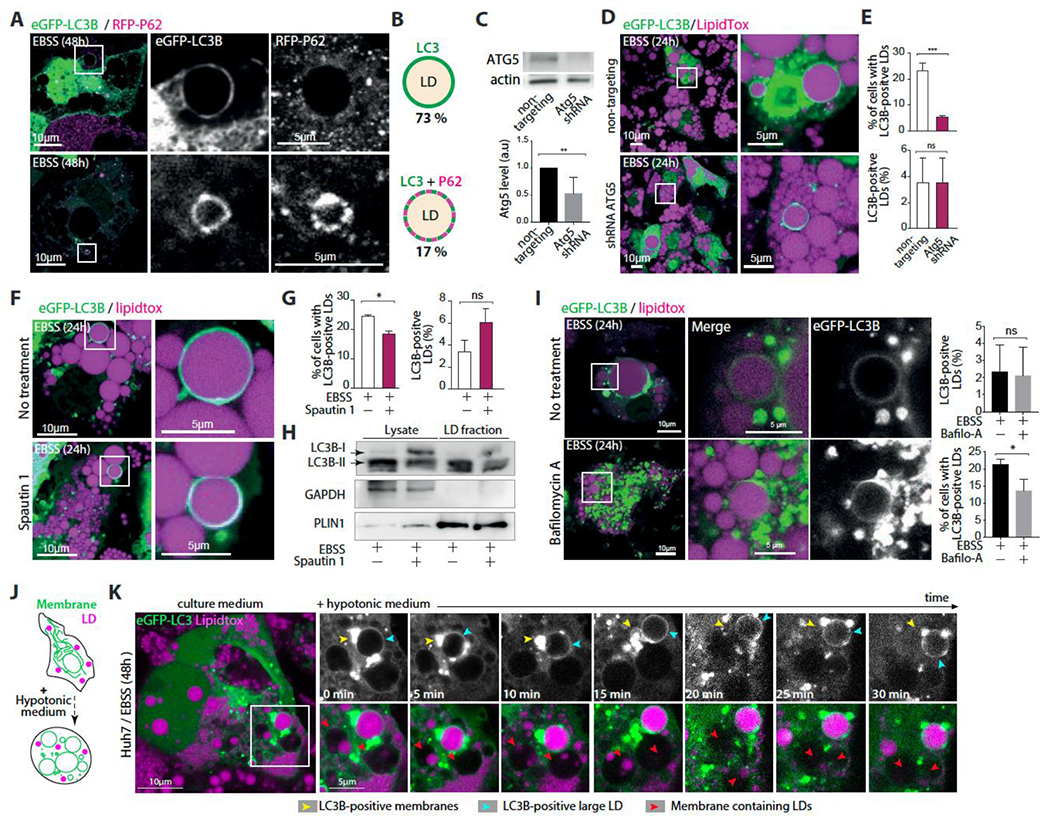
A. Confocal imaging of eGFP-LC3B, mRFP-P62 and LDs in differentiated 3T3-L1 adipocytes. Cells were virally transfected with eGFP-LC3B and mRFP-P62 and incubated in EBSS for 48hs. Scale bar,10 μm (5 μm in insets).
B. Schematic representation illustrating the recruitment of eGFP-LC3B alone or with mRFP-P62 on LDs. The percentage of LDs of each phenotype is written below the corresponding schematic representation. Quantifications are the average from three independent experiments.
C. Top: western blot of differentiated 3T3-L1 adipocytes virally co-transfected with eGFP-LC3B and with either ATG5 shRNA or non-targeting shRNA for 24h, and then incubated in EBSS for 24h. Bottom: quantification of ATG5 expression from three independent experiments Student’s unpaired t-test is used (**P<0,001).
D. Confocal imaging of LDs in differentiated 3T3-L1 adipocytes treated as described in C. Scale bar,10 μm (5 μm in insets).
E. Top: Percentage of cells with eGFP-LC3B-positive LDs. Bottom, Percentage of eGFP-LC3B-positive LDs per cell. Quantifications are from three independent experiments. Student’s unpaired t-test is used (***P<0,0001, ns P>0,05)
F. Confocal imaging of eGFP-LC3B and LDs in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B and incubated in EBSS alone or EBSS containing Spautin-1 for 48hs. Scale bar,10 μm (5 μm in insets).
G. Left, percentage of cells with eGFP-LC3B-positive LDs. Right, Percentage of eGFP-LC3B-positive LDs in the cell. Quantifications are from three independent experiments done as described in F. Student’s unpaired t-test is used (**P<0,001, ns P>0,05).
H. Western blot of lysate and LD fractions of differentiated adipocytes incubated in EBSS alone or EBSS containing Spautin-1 for 48hs.
I. Confocal imaging of eGFP-LC3B and LDs in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B and incubated in EBSS alone or EBSS containing bafilomycin A for 48hs. Scale bar,10 μm (5 μm in insets). To Right, Up: percentage of eGFP-LC3B-positive LDs in the cell. Down: Percentage of cells with eGFP-LC3B-positive LDs. Quantifications are from three independent experiments. Student’s unpaired t-test is used (* P<0,05, ns P>0,05).
J. Schematic representation of the impact of swelling of intracellular organelles by incubating the cells with a hypotonic media.
K. Time-lapse imaging experiment performed on Huh7 cells that were transfected with eGFP-LC3B and treated with oleic acid for 24hs, and then incubated in EBSS for 48 hs. At time 0, a hypotonic media was added to induce cell swelling. Imaging was done at the indicated times.
See also Figure S2.
We next conducted experiments to investigate the impact of autophagy pathways on the localization of LC3B to LDs in 3T3-L1 adipocytes. To block autophagy, we targeted ATG5 using an shRNA lentivirus and subjected the cells to starvation. The results showed a significant decrease in the percentage of cells with LC3B-positive large LDs compared to non-targeting shRNA transfected cells, indicating that ATG5 action is required for LC3B localization to large LDs (Figures 2C–2E).
To further test this hypothesis, we treated the cells with Spautin-1 to degrade the Vps34 PI3 kinase complex, which is essential for triggering autophagosome formation 45,49. The percentage of cells with large LD-localized LC3B decreased slightly compared to ATG5 knockdown, and western blot analysis confirmed a decrease in LC3B lipidation due to Spautin-1 (Figures 2F–2H). We also generated CRISPR-knockout ATG5 and Beclin1 Huh7 cells, which lack autophagy. In these cells, large LDs with an LC3B signal were almost nonexistent (Figures S2J and S2K), indicating that autophagy is required or precedes LC3B localization to large LDs during long-term starvation. Note that ATG5 activity is required for LC3 lipidation for both autophagic and non-autophagic processes, meaning that the LC3B localization to larger LDs could be still autophagosome-independent.
To enhance autophagy or autophagosome accumulation, we treated the adipocyte cells with rapamycin to inhibit mTOR or bafilomycin to block the fusion of autophagosomes with lysosomes. Rapamycin promoted LC3B localization to large LDs in the feeding state but did not significantly enhance the number of large LC3B-positive LDs or the fraction of cells displaying the phenotype (Figures S2L and S2M). In bafilomycin treatment, many autophagosomes accumulated, but the fraction of large LC3B-positive LDs per cell was also unchanged, and the percentage of cells with the phenotype even decreased (Figures 2I and S2N). These results suggest that further enhancing autophagy or autophagosome accumulation did not enhance the fraction of large LC3B-localized LDs in cells. Together, the data indicate that autophagy is required to trigger our observed phenotype and that increasing it further would not impact it. Furthermore, the LC3B signal around large LDs is distinct from autophagosomes.
Finally, to better distinguish between the LC3B signal on autophagosomes and large LDs, we used the organelle swelling approach. We exposed starved Huh7 cells containing a large LC3B-positive LD to a hypotonic medium, which causes bilayer-bounded organelles to swell and become spherical, improving spatial resolution (Figure 2J) 50–52. LDs do not swell in this process. If the hypotonicity is high enough, bilayer-bounded organelles can undergo burst and reseal cycles, or they may completely burst (Figure S2P) 53,54.
During the swelling process, the LC3B signal around the LD remained intact (Figure 2K, blue arrowhead, S2O), while the LC3B-positive membrane (autophagosome) near the LD swelled (Figure 2K, yellow arrowhead, S2O), became spherical (Figure 2K, yellow arrowhead 15min, S2O), and eventually burst (Figure 2K, yellow arrowhead, 20min). Other bilayer membrane compartments containing LDs also swelled and likely burst as well (Figure 2K, red arrowhead). These observations agree with our hypothesis that the LC3B on the surface of the large LD is distinct from that on autophagosomes, as the LC3B signal around the LD did not swell during the process.
Long-term nutrient starvation induces ATG3 recruited to the lipid droplet surface
Based on our above data, we hypothesized that LC3B could be directly ligated to the LD surface, as supported by the presence of LC3B-II in the LD fraction (Figure 1H). This hypothesis led to the prediction that ATG3, which catalyzes LC3B lipidation to PE, should also be present on LDs.
The presence of ATG3 on LDs was confirmed through Western blot analysis of LDs collected after 48 hours of starvation, which showed an enhanced ATG3 signal with LC3B-II (Figure S3A). Immunostaining of endogenous ATG3 and LC3B in differentiated 3T3-L1 adipocytes in DMEM or EBSS starvation medium further revealed ATG3 around LDs along with LC3B only during starvation (Figures 3A and S3B). The localization of ATG3 around LDs was not affected by treatment with 3-Methyladenine (3-MA) which blocks autophagosome formation via inhibiting the phosphatidylinositol 3-kinase (Figures 3B and S3C). We also detected endogenous ATG3 onto the surface of larger LDs together with eGFP-LC3B in differentiated 3T3-L1 adipocytes, or in Huh7 and HeLa cells that were initially fed with OA and then submitted to long-term starvation (Figure S3D).
Figure 3. ATG3 is recruited to lipid droplets during long-term nutrient starvation.
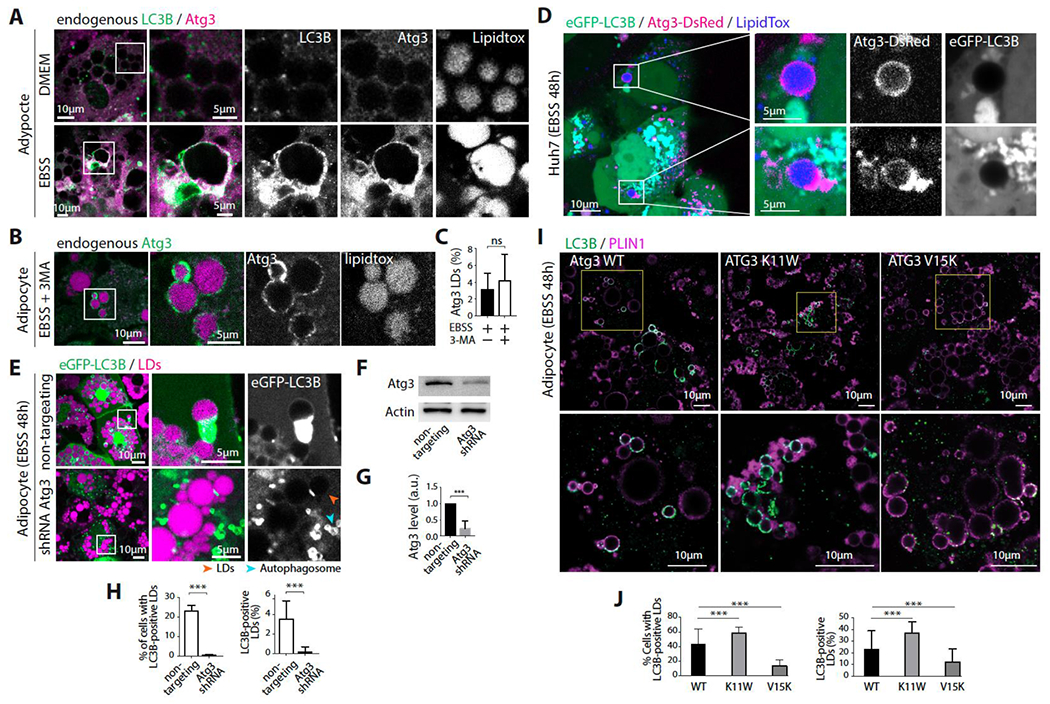
A. Immunofluorescence staining of LC3B, ATG3, and LDs in differentiated 3T3-L1 adipocytes incubated in DMEM or EBSS for 48hs.
B. Immunofluorescence staining of ATG3 and LDs in differentiated 3T3-L1 adipocytes incubated in EBSS containing 3MA for 48hs.
C. Percentage of ATG3-positive LDs per cell. Student’s unpaired t-test is used (ns P>0,05).
D. Confocal imaging of Huh7 cells co-transfected with eGFP-LC3B and ATG3-dsRED. Cells were treated with OA to induce LDs and then incubated in EBSS for 48hs. Scale bar, 10 μm (5 μm in insets).
E. Confocal imaging of LDs in differentiated 3T3-L1 adipocytes virally co-transfected with eGFP-LC3B and an ATG3 shRNA or eGFP-LC3B and the non-targeting shRNA. Cells were incubated in EBSS for 48hs after transfection. Scale bar, 10 μm (5 μm in insets).
F. Western blot of cells treated as described in E.
G. The bar graph shows the quantification of ATG3 expression from three Western blots of cells treated as described in E. Student’s unpaired t-test is used (*** P<0,0001).
H. Right, percentage of cells with eGFP-LC3B-positive LDs. Left, percentage of eGFP-LC3B-positive LDs per cell. Quantifications are from three independent experiments. Student’s unpaired t-test is used (*** P<0,0001).
I. Immunofluorescence staining of LC3B and PLIN1 in differentiated adipocytes stably transfected with mATG3 WT, mATG3 K11W, or mATG3 V15K. Cells were incubated in EBSS for 48hs then fixed and stained (LC3B in green, PLIN1 in magenta). Scale bar, 10 μm (5 μm in insets).
J. Right, percentage of cells with LC3B-positive LDs. Left, percentage of LC3B-positive LDs per cell. Quantifications are from three independent experiments. An ordinary one-way ANOVA test was used (*** P<0,0001)
See also Figure S3.
To further study ATG3 LD localization, we proceeded by overexpression. Overexpressed eGFP-ATG3 or ATG3-DsRed in Huh7 cells showed clear recruitment of ATG3 to few larger LDs only during starvation (Figure 3D), with no recruitment observed in the fed state (Figure S3E and S3F). Co-transfection of ATG3-DsRed and eGFP-LC3B showed instances where few larger LDs recruited ATG3 but not LC3B. This result is a strong piece of evidence that the ATG3 signal on LDs did not come from autophagosomes, but instead through a direct recruitment of the protein to LDs (Figure 3D). In this particular experiment, it is possible that either LC3B had not yet undergone lipidation on the LD or that the catalytic activity of ATG3-DsRed was reduced, since the lipidation process is carried out by the C-terminal domain of ATG3 17, tagged with DsRed in this case.
Inactivation of ATG3 through a specific ATG3 shRNA inhibited LC3B LD localization, as shown by the almost abolished LC3B LD localization in ATG3 shRNA-transfected 3T3-L1 and Huh7 cells maintained in EBSS for 48h (Figures 3E–3H and S3G–S3I). This data supported the requirement of ATG3 presence and activity for the localization of LC3B to LD’s surface.
ATG3 binds to membranes via its N-terminal amphipathic helix 12, a motif that is used by most cytosolic proteins to associate with LDs 55. Mutating the lysine in position 11 to tryptophan, right at the interface between the hydrophobic and hydrophilic faces of the amphipathic helix, improves the membrane association of ATG3 12. On the opposite, the valine 15-to-lysine mutation impedes binding 12. We generated 3T3-L1 adipocyte cell lines stably expressing either WT, K11W, or V15K mouse ATG3, the endogenous ATG3 remaining in the background. Compared to WT ATG3, K11W increased significantly the phenotype, percentage of cells and fraction of LC3B-positive LDs, while the V15K mutation did not impact or even decreased it (Figures 3I and 3J). These data suggest the capacity of ATG3 to bind to LDs and lipidate LC3B.
ATG3 binds to artificial LDs
To investigate the ability of ATG3 to associate with model LDs, we performed experiments using a buffer solution containing droplets made of triolein (Figure 4A) and purified ATG3-YFP. Our results indicated that ATG3 was recruited to the surface of the droplets, suggesting that it can associate itself with LDs. To further explore this interaction, we generated model LDs with different PC/PE ratios (10/0, 7/3, 5/5, and 3/7) and various phospholipid coverages ranging from 0.005% to 0.2% (w/w to triolein), mixed with Rhodamine-PE (Rh-PE) to report for the phospholipid density 56: the higher the rhodamine signal, the higher the phospholipid density (Figure 4B). We then added ATG3-YFP to the artificial LDs. Our results showed that the protein binding level decreased with the phospholipid density (Figure 4C). For each PC/PE condition, we reported the relative amount of bound ATG3 as a function of the lipid coverage and estimated the concentration of phospholipids at which half of the maximum binding was reached, i.e., concentration C½ (Figures 4D and S4A). We found that increasing PE levels led to higher ATG3 binding to the artificial LDs (Figure 4D), indicating that ATG3 associates more effectively with droplets enriched in PE, which generates more packing defects 57. Floatation assays confirmed these results, as sonicated liposomes or droplets with various PC/PE ratios showed similar ATG3 binding patterns to both the liposomal bilayers and droplet monolayers (Figure 4E).
Figure 4. ATG3 better binds to model LDs enriched in PE.
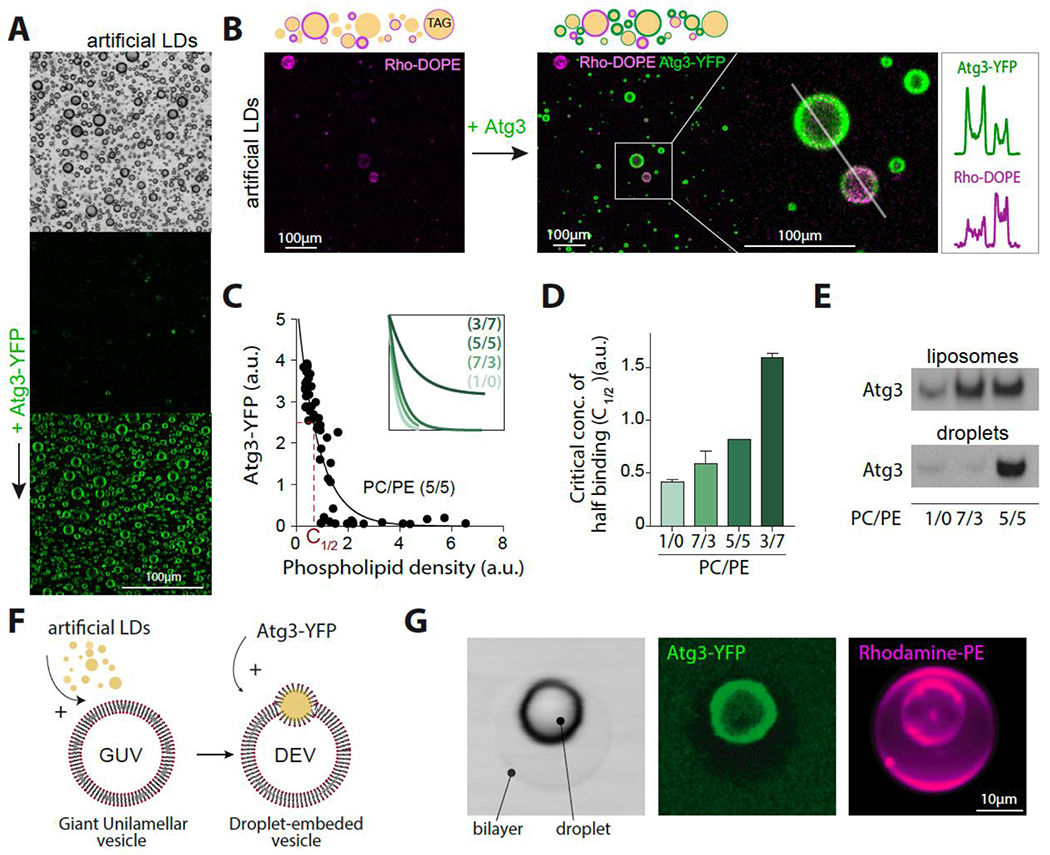
A. Confocal imaging of triolein droplets before and after ATG3-YFP addition. Scale bare (100 μm).
B. Top: Schematic illustration of triolein-in-buffer droplets decorated by different phospholipid densities ((1/1) PC/PE), reported by rhodamine-PE (Rho-PE). Bottom: confocal imaging of triolein-in-buffer droplets with different phospholipid coverage, ranging from 0.005% to 0.2% (w/w to triolein) before and after ATG3 addition. Scale bare (100 μm). Line profiles show the intensity levels of ATG3-YFP and Rho-PE on droplets depicted in the inset.
C. ATG3-YFP recruitment to triolein droplets as a function of the phospholipid density, reported by Rho-PE. The concentration at half of maximum binging is depicted in the main figure. Concentration at half of maximum binding C1/2 is shown in red. The inset figure shows the different recruitment profiles of Atg3-YPF depending on the PC/PE ratio.
D. The characteristic concentration C1/2 of ATG3-YFP binding from experiments done as described in B for the indicated PC/PE ratio.
E. Western blot of untagged ATG3 recombinant protein bound to liposomes and artificial LDs in the top fraction of flotation assays.
F. Schematic representation of the droplet-embedded vesicle (DEV) system.
G. Confocal imaging of a DEV made of 7/3 PC/PE and incubated with ATG3-YFP. Scale bare (10 μm).
See also Figure S4.
We next used a tensiometer approach to characterize the adsorption of proteins to oil/water surfaces (Figure S4B) 48,58 and studied the binding of non-tagged ATG3. To do so, we generated a triolein/buffer surface and added purified ATG3. We found that ATG3 was recruited to the triolein/buffer interface, leading to a drop in surface tension from ~32mN/m (i.e., triolein/buffer-free interface) to an equilibrium value of ~18mN/m (i.e., protein-adsorbed interface) (Figure S4C). We then compressed the protein-adsorbed interface rapidly to lateral condense the protein layer, resulting in a decrease in surface tension due to the protein being laterally compressed and better masking the interface (Figure S4D). Over time, however, surface tension re-increased to a new equilibrium as ATG3 fell off from the interface, causing the protein monolayer to relax 48,58 (Figures S4E and S4F). When we repeated this experiment with PC/PE (1/1) initially decorating the interface, we observed a less remarkable ATG3 fall-off, suggesting that PC/PE might promote ATG3 retention to the droplet surface (Figures S4G and S4H).
Finally, while the Atg16,5-12 complex is known to mediate ATG3 recruitment on bilayer membranes like the autophagosome 4, our study shows that it is not required for ATG3’s association with LDs. To further investigate the binding capacity of ATG3 on different lipid surfaces, we utilized the droplet-embedded vesicle (DEV) system, which incorporates a neutral lipid droplet into a giant bilayer vesicle (GUV) (Figure 4F) 60, made of 7/3 PC/PE. Our experiments revealed that ATG3-YFP exclusively targets the model LD and not the bilayer (Figure 4G). This observation suggests that ATG3 cannot solely target a flat bilayer with a relevant PC/PE composition. Instead, ATG3 can bind to the model LD within the same system, (Figure 4G), verily due to the lower phospholipid density on LDs than on bilayers 61.
In summary, our findings suggest that ATG3 can alone bind to the surface of lipid droplets (LDs), likely by its N-terminal amphipathic helix, independently from the ATG5/12/16 complex. The presence of PE, which is required for LC3B lipidation and promotes larger LDs 59, increases the binding of ATG3 to artificial LD surfaces.
ATG3 lipidates LC3 to LDs.
We investigated whether ATG3 recruitment could drive LC3B lipidation to LDs in vitro. We purified LDs from the differentiated 3T3-L1 adipocytes and mixed them with a buffer solution containing ATG3, Atg7, Alex488-LC3B, and ATP, or Alex488-LC3B alone as a control. We incubated the sample at 37°C and observed in real-time that Alexa488-LC3B was localized to the LDs’ surface only in the presence of the lipidation machinery (Figure 5A). We observed that after fully bleaching the LC3B signal on an LD, the signal re-increased but to a lesser extent, likely due to de novo-lipidation and the lack of a PE reservoir (Figures 5SA and S5B). We collected the samples and ran an SDS-PAGE gel, which showed an LC3B-II band corresponding to lipidation, only in the presence of the minimal ATG3 and Atg7 machinery (Figure 5B). These observations indicate that ATG3 lipidated LC3B to LDs.
Figure 5. ATG3 lipidates LC3 to purified and artificial LDs.
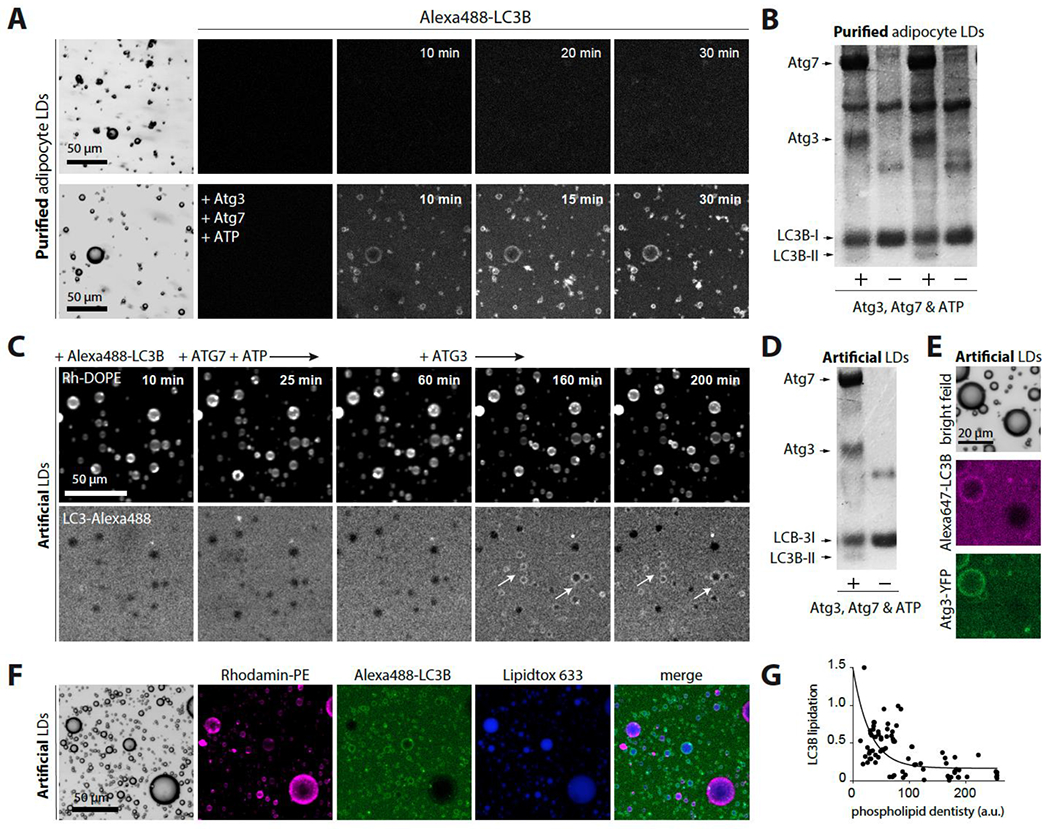
A. Confocal imaging of purified adipocyte LDs in HKM buffer containing Alexa488-LC3B, in the presence or absence of the lipidation reaction components ATG7, ATG3, ATP.
B. LDs from the previous experiment are collected and analyzed using SDS–PAGE in a stained Coomassie blue.
C. Confocal imaging of triolein-in-buffer droplets decorated by PC/PE (7/3) incubated with Alexa488-LC3B, then ATG7 and ATP. No lipidation occurred. When ATG3 was subsequently added, lipidation occurred on the artificial LDs (arrows show examples).
D. Artificial LDs from the previous experiment are collected and analyzed using SDS–PAGE in a stained Coomassie blue.
E. Triolein-in-buffer droplets decorated with PC/PE at different monolayer phospholipid densities (based on Rho-PE signal) are imaged using confocal microscopy after being incubated with Alexa647-LC3B and Atg3/Atg3-YFP (80/20), ATP, and ATG7.
F. Confocal imaging of triolein-in-buffer droplets decorated by PC/PE (7/3) at different monolayer phospholipid densities varied from 0.005% to 0.2% (w/w to triolein). They are incubated with Alexa488-LC3B and Atg3 (80/20), ATP and ATG7.
G. Quantification of F. LC3B-Alexa488 lipidation to triolein droplets as a function of the phospholipid density.
See also Figure S5.
Since the purified LDs may contain other autophagic factors recruiting LC3B-II, we switched to artificial LDs to have full control over compositions. We made triolein-in-buffer droplets decorated by PC/PE (7/3). In the reaction chamber, we first introduced Atg7 and ATP only, but after 1 hour of incubation at 37°C, no signal of LC3B was observed around the droplets (Figure 5C and S5C). We then added ATG3 to the chamber and observed that Alexa488-LC3B was around the droplets (Figure 5C and S5C). We collected the artificial LDs, analyzed them with SDS-PAGE gel, and found LC3B-II only in the presence of the lipidation machinery (Figure 5D). This result shows that ATG3 mediated LC3B lipidation to the LDs.
To study the impact of PC/PE on lipidation, we prepared artificial LDs made of PC/PE (7/3) with different monolayer phospholipid densities, as in Figure 4C, varied from 0.005% to 0.2% (w/w to triolein). We used Alexa647-LC3B and ATG3/ATG3-YFP (80/20) to correlate LC3B lipidation to ATG3 binding in the presence of Atg7 and ATP. We observed that only artificial LDs positive for ATG3 were LC3B-lipidated (Figure 5E and S5D). As shown in Figure 4B–4D, the binding of ATG3 to LDs decreased with an increase in phospholipid density. To investigate this relationship further, we used Rho-PE as a proxy for phospholipid density and analyzed the lipidated Alexa488-LC3B in the presence of ATG3, Atg7, and ATP. We found that the lipidated droplets had a lower Rh-DOPE signal (Figures 5F and 5G, and S5E), corresponding to a lower phospholipid density that facilitated ATG3 binding. These results indicate that ATG3 binding to LDs was required for LC3B lipidation and was improved by phospholipid packing defects on the LDs.
Lastly, we investigated whether membrane-bound ATG3 can lipidate GABARAPL on artificial LDs, despite the absence of GABARAPs on the large LDs in the studied cell lines (Figures S1H and S1I). Through in vitro experiments using fluorescence imaging and SDS-PAGE gel analysis, we observed that ATG3 was able to lipidate of GABARAP1L to the surface of the artificial LDs (Figure S5F–S5I). These results suggest that regulatory factors in mammalian cells differentially control the delivery of ATG8 proteins to LD-bound ATG3.
In conclusion, the findings from the reconstitution approaches suggest that ATG3 can bind to LDs with specific surface properties and subsequently lipidate Atg8 proteins to PE.
Large LC3B-positive LDs exhibit tight contact with LC3B-containing membranes.
The large LDs positive for LC3B were found to be consistently in contact with LC3B-positive membranes, in Figures 6A–B and S6A, as well as in Figures 1A and S1A–B. We surmise that LC3B on LDs enables interaction with LC3B-autophagosome-like structures.
Figure 6. LC3B-positive LDs exhibit interaction with organelles that also contain LC3B.
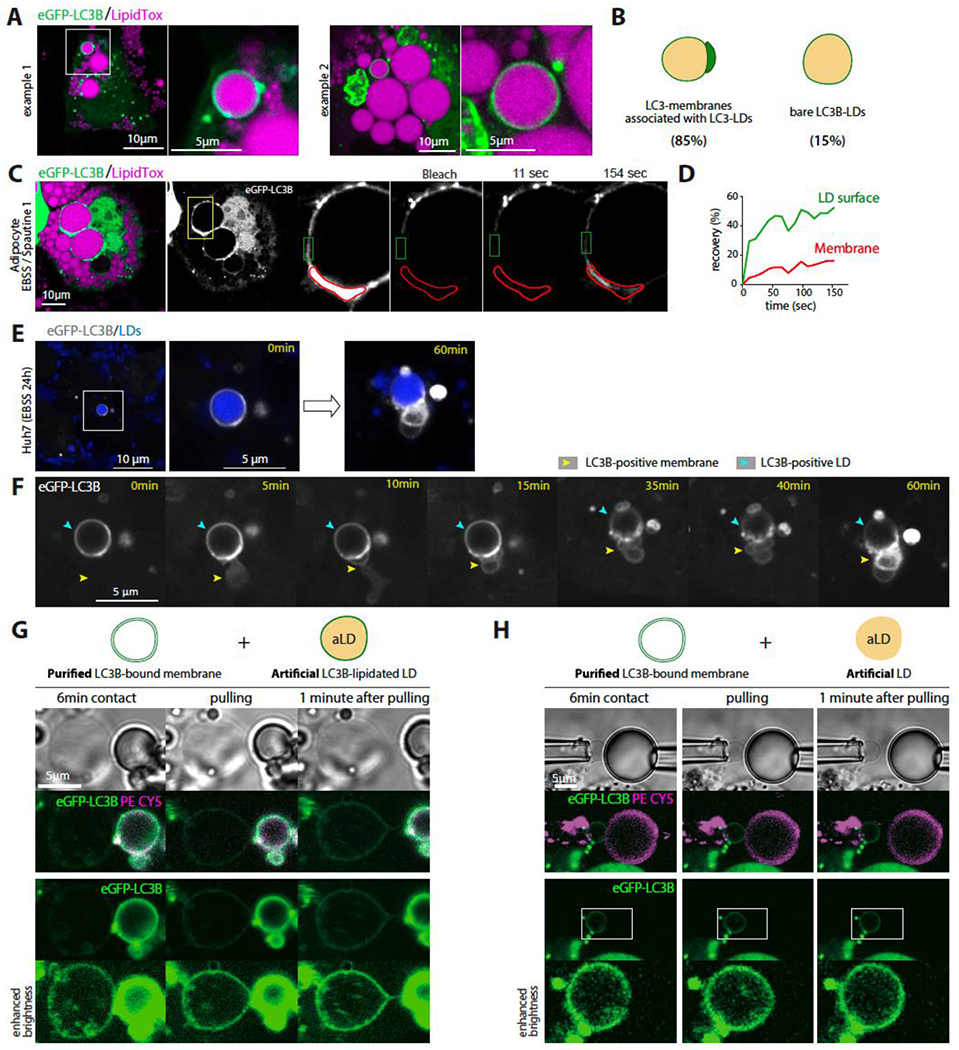
A. Confocal imaging of differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B. Cells are incubated in EBSS for 48hs after transfection. Scale bar, 10 μm.
B. Schematic illustration of eGFP-LC3B-positive LDs with or without eGFP-LC3B puncta associated. The fraction of each phenotype is indicated beneath each case. Quantifications are from three independent experiments.
C. FRAP analysis of eGFP-LC3B in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B and incubated in EBSS containing Spautin-1 for 48hs. The insets indicate the bleached region: red for the autophagosome area and green for the LD surface. Scale bar, 10 μm.
D. Recovery kinetics of eGFP-LC3B in the different regions depicted in C. The signals were corrected for the bleach.
E. Confocal imaging of Huh7 cells virally transfected with eGFP-LC3B and treated with oleic acid for 24h and then incubated in EBSS for 24h. eGFP-LC3B positive LDs are shown at 0 and 60 minutes.
F. Time-lapse from confocal live imaging of Huh7 cells presented in E at the indicated times. The cyan arrowhead indicates the eGFP-LC3B-positive LD region and the yellow one an LC3B-positive membrane being recruited to the LD.
G. Top: Schematic representation of a purified eGFP-LC3B-bound membrane and an eGFP-LC3B-lipidated artificial LD (PE-Cy5 report for phospholipids decorating the artificial LD). Bottom, confocal imaging of eGFP-LC3B bound membrane extracted from Huh7 cells and an eGFP-LC3B-lipidated artificial LD, each captured by a micropipette and put in contact for 6 minutes. Afterward, the two objects are slowly pulled away from each other.
H. Top: Schematic representation of a purified eGFP-LC3B-bound membrane and an artificial LD solely decorated by phospholipids. Bottom, confocal imaging of eGFP-LC3B-bound membrane extracted from Huh7 cells and an artificial LD with the same lipid composition as in G. Both objects are captured by a micropipette and put in contact for 6 minutes before they are slowly pulled away from each other.
See also Figure S6.
To distinguish the LC3B signal on the two compartments, FRAP experiments were conducted on LC3B on the freestanding LD surface and the membrane region in contact (Figures 6C and S6B–S6E). The LC3B displayed a much faster recovery rate on the freestanding LD region than on the contact (Figures 6C and 6D, and S6B–S6E), indicating two different states of LC3B on the membrane and the LD. This suggests that LC3Bs, both on the LD and the membrane, are involved in the contact between the organelles, leading to slower diffusion. The LC3B molecules on the LD surface that are not involved in the contact would be in a state of free diffusion, in equilibrium with the LC3B molecules involved in the contact. Over time, this LC3B pool not involved in the contact can contribute to the contact.
To test this hypothesis, we focused on an LC3B-positive LD that did not initially display a clear LC3B membrane around it, as depicted in Figure 6E–F. Over time, an LC3B-positive membrane appeared and was in contact with the LDs. The LC3B signal diminished in the freestanding LD region, while it was increasing significantly in the contact regions. This experiment provides evidence that autophagosome-like structures specifically target LC3B-positive LDs and that the LC3B-II located on the LD surface is involved in these interactions. These membrane structures may also recruit the LC3B-II pool from the LD surface to their own membrane.
An in vitro approach was taken to further test our hypothesis. LC3B-positive bilayer vesicles, likely autophagosomes or nascent autophagosomes, were collected with micropipettes after the plasma membrane of starved and swollen Huh7 cells was aspirated and broken, as shown in Figure 2K. On the other hand, artificial LDs were prepared, either lipidated with LC3B, as in Figure 5C, or not, as a control (Figures 6G and 6H). The artificial LDs were brought into contact with the cell-derived LC3B vesicles for several minutes. The two compartments were then pulled apart to examine the interactions. An interaction was only observed in the case of the LC3B-lipidated LD (Figures 6G and 6H). Pulling the two objects apart led to the deformation of the vesicle into a tubule connecting it to the LC3B-lipidated droplet (Figure 6G), indicating the tethering of the two interfaces, as previously observed 62, and, eventually, their subsequent merge and physical contiguity 65.
Collectively, the data obtained from in vitro and cellular studies suggest that LC3B located on the surface of LDs acts as a co-factor, attracting LC3B-positive intracellular membranes towards the LDs, and potentially being transferred to them.
Lysosomes are recruited to the membranes associated with LC3B-positive LDs.
We investigated whether the interaction between lipid droplets (LDs) and autophagosome-like structures leads to degradation. To do this, we subjected 3T3-L1 adipocytes to a 48-hour incubation in EBSS and observed a faint surface signal of LC3B on large LDs (Figure 7A, S7A). This signal was likely the freely diffusive form of LC3B-II (Figure 6C). However, we did not observe lysotracker activity in the area of the LD where the LC3B-positive signal was detected, indicating that degradation was not occurring there. On the other hand, the LD region that was in contact with the LC3B-positive autophagosome-like membrane was lysotracker-positive, suggesting that degradation was occurring in this region of the LD. This mechanism is distinct from degradation processes involving LDs such as lipophagy 43 or lysosome-mediated LD degradation 63.
Figure 7. LC3B-positive LDs interact with acidified autophagosome-like membranes.
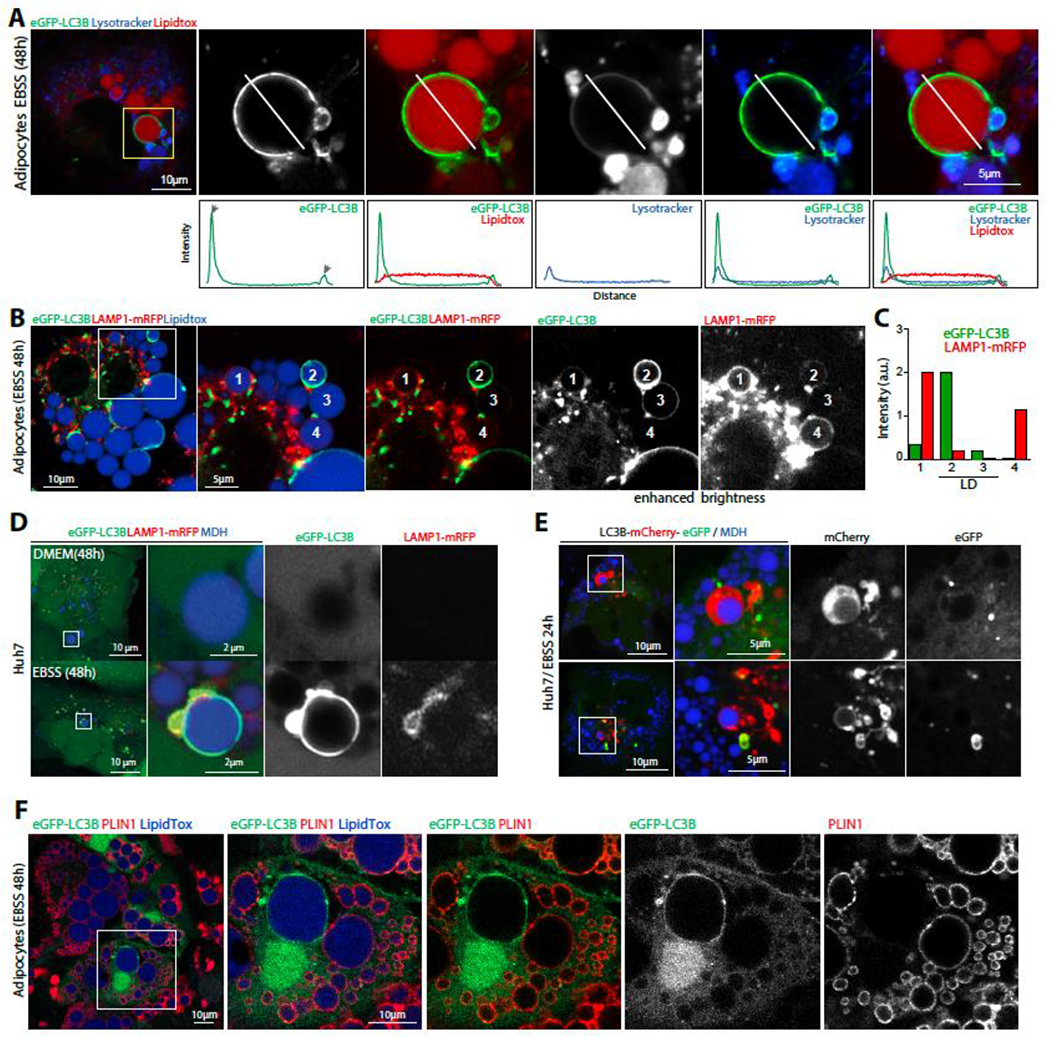
A. Confocal imaging of eGFP-LC3B, lysotracker (blue) and LDs (LipidTox) in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B. Cells are incubated in EBSS for 48hs after transfection. Scale bar,10 μm (5 μm in insets). Bottom panels are intensity profiles of the line drawn in each image.
B. Confocal imaging of eGFP-LC3B (green), LAMP1-mRFP (red) and LDs (LipidTox) in differentiated 3T3-L1 adipocytes virally transfected with eGFP-LC3B and LAMP1-mRFP, incubated in EBSS for 48hs. Example LDs at different stages of eGFP-LC3B and LAMP1-mRFP recruitment are numbered.
C. Relative intensity of eGFP-LC3B and LAMP1-mRFP on the different LDs.
D. Confocal imaging of eGFP-LC3B, LAMP1-mRFP and LDs in Huh7 virally transfected with eGFP-LC3B and LAMP1-mRFP, loaded with oleic acid for 24hs, and starved or not. Scale bar,10 μm (2 μm in insets)
E. Confocal imaging of LC3B-mCherry-eGFP and LDs in Huh7 loaded with oleic acid for 24h and then placed in EBSS for 24h. Scale bar,10 μm (5 μm in insets).
F. Immunofluorescence staining of PLIN1 in differentiated 3T3-L1 adipocytes transfected with eGFP-LC3B and incubated in EBSS for 48hs.
See also Figure S7.
To better visualize lysosomes, we transfected cells with LAMP1-mRFP and induced long-term starvation. We observed the lysosomal signal on LC3-positive LDs, some of which were at different stages of contact with autophagosome/lysosome (Figures 7B and 7C). One large LC3B-positive LD was locally interacting with an LC3B-membrane that was colocalizing with LAMP1, indicating that it was at a later stage of autophagosome/lysosome recruitment (Figures 7B and 7C, LD1). In contrast, another LC3B-positive LD at an earlier stage did not show the lysosomal signal (Figures 7B and 7C, LD3).
We observed similar findings in Huh7 cells, where some large LC3B-positive LDs interacted partially with LC3-positive membranes that were LAMP1-positive (Figure 7D) or lysotracker-positive (Figures S7B and S7C). We found that the area of contact with the acidified membrane only modestly increased within a time course of 40 minutes (Figure S7C), suggesting that the full contact of the membrane with large LDs would take a much longer time if it would ever happen. Furthermore, we found that the LAMP1 signal only colocalized on the large LDs positive for LC3B and not LC3A or LC3C (Figure S7D). When we transfected Huh7 cells with LC3B-mCherry-eGFP and incubated them in EBSS for 24hs, we found that the larger LDs were specifically displaying the mCherry signal alone around them, indicating lysosomal-mediated turnover around this LD (Figure 7E).
To determine whether lipids were being degraded from the few large LC3B-positive LDs, Bodipy-labeled fatty acids were fed to Huh7 cells or 3T3-L1 cells differentiated into adipocytes. Long-term nutrient starvation was induced to determine whether fatty acids were liberated by the LC3B-positive LDs. However, we failed to observe the fluorescent fatty acids transfer to the autolysosome-like membranes close to the LC3B-positive LDs (Figure S7E). Then, we examined Plin1, which is the major marker of LDs in 3T3-L1 adipocytes. We found that large LC3-positive LDs frequently lacked PLIN1 partially or totally (Figure 7F and S7F). This suggests that PLIN1 was degraded for or by LC3B lipidation to the large LDs and the subsequent autolysosome-like recruitment. This degradation could occur prior, during, or after LC3B lipidation to the LD. Yet, we observed instances where Atg3/LC3B and PLIN1 were present on the same large LD, but the proteins were laterally excluding each other (Figure S7F and S7G). The finding suggests that the lipidation of LC3B triggers the removal of Plin1 from the LD (Figure 7E). This removal could have facilitated the LD’s interaction with the autophagosome-like structures and their acidification, or the contact with the autophagosome-like structures could have excluded Plin1 from the contact, leading to its degradation.
Taken together, our data support the existence of a pathway in which LC3B-positive LDs become in contact with LC3B-positive autophagosome-like structures that are then acidified to mediate local degradations.
Discussion.
ATG3 bound more strongly to model LDs that exhibit larger phospholipid packing defects than bilayers 61, as depicted in Figure 4. This probably explains that the recruitment of ATG3 to bilayers in cells typically requires additional machinery, such as the ATG5-Atg12-Atg16 complex. In vitro, the recruitment of ATG3 to LDs was enhanced by PE, which suggests that larger LDs may contain more PE 59. During long-term nutrient starvation, the monolayer of large LDs may become enriched in specific lipids, such as PE, or remodeled, such as by the removal of proteins and phospholipids, to enable ATG3 binding and subsequent LC3B lipidation.
Our findings suggest that there are at least two possible mechanisms underlying our observed phenotype. The first model suggests that prolonged starvation leads to the remodeling of the LD surface, allowing for LC3B lipidation to support the biogenesis of autophagosomes from LDs. Indeed, we consistently observed increasing amounts of LC3-membranes near LC3-lipidated LDs. Also, the LC3B-positive LDs represented a small fraction of LDs, despite they belonged to the larger LDs population, containing a significant amount of lipids. Finally, we did not observe clear evidence of lipid transfer from the LDs to the membranes. These observations raise questions about the occurrence of this mechanism for degrading only a few large LDs. The second model suggests that LC3-bound LDs recruit autophagosomes that ultimately lead to the degradation of the LDs. We observed that the LC3B-membranes surrounding LDs were acidified, which supports the idea of LC3B-positive LDs being degraded. However, because this mechanism is only triggered during prolonged starvation, it was difficult to timely capture the occurrence of the events to discriminate between these two models. Yet, It is possible that both models are correct and that both mechanisms occur simultaneously.
It may not be surprising that both ATG3 and LC3B localize to LDs, as several autophagic proteins, including Atg2, Atg14, and DFCP1, have been found on LDs 22–27. This suggests that autophagic processes can take place directly at the surface of LDs. Our data suggest that the LD surface may serve as a lipidation platform to support certain autophagic processes, such as autophagosome biogenesis and the local degradation of cellular components near the LD surface. It is tempting to speculate that during prolonged nutrient deprivation, canonical autophagic pathways may be overwhelmed, and autophagic processes may become more easily organized at the surface of some LDs to alleviate the system. Indeed, the process of LC3B lipidation and autophagosome biogenesis typically involves multiple steps and autophagic proteins before Atg3 can bind to nascent phagophores and lipidate LC3B. However, our findings suggest that prolonged starvation allows for direct binding of Atg3 to LDs and subsequent LC3B lipidation on the LD surface. This LD-lipidated LC3B may then be transferred to nearby autophagosomes, e.g. via monolayer-bilayer bridges formed between these two organelles. In this scenario, PE lipids could be supplied to the LDs from the ER, via lipid transfer proteins or ER-LD bridges, to support continuous LC3B lipidation on LDs in prolonged starvation. While our data show agreements with this model, further investigation is needed to confirm it fully.
The catabolic pathways of LDs are interdependent, and several interactions have been shown between these pathways, as previously reported. For instance, lipolysis requires chaperone-mediated autophagy for the degradation of Plin2,3 to facilitate the access of ATGL to LDs 65. ATGL interacts with LC3B and regulates Sirtuin 1 activity, which modulates autophagy. Such an interaction couples lipolysis and lipophagy 40,41. Ongoing lipolysis reduces the size of LDs until they can fit into autophagosomes and be degraded by lipophagy 43. Finally, ATGL lipolytic activity is required for the delivery of tiny LDs directly from donor LDs to lysosomes 63. Based on these findings, the localization of LCB3 to large LDs in our case might cross-talk with other autophagic pathways, especially with autophagy (Figure 2). For example, classical autophagy may primarily act during starvation for a certain time, after which it triggers cues inducing the remodeling of large LDs’ surface, allowing ATG3 binding and LC3B lipidation. In this model, blocking autophagy would prevent the release of the cues remodeling LDs, which could explain the reduction of LC3B localization to large LDs in ATG5 or Beclin1 KO.
Autophagosome-like structures fused with lysosomes while the large LDs were partially in contact with the LC3-positive membranes (Figure 7). This indicates that the mechanism mediating the degradation of the large LC3B-positive LDs is unique and is not lipophagy-related 42,64. In murine 3T3-L1 adipocyte and human Huh7 cells, LC3B-positive membranes adhered to LC3B-positive LDs. Although Atg8 proteins may trans-dimerize 62,66, it cannot be concluded yet from our study that LC3B trans-homodimerization was responsible for the docking of the autophagosome-like membranes to the LDs, and their possible hemifusion. At least, in vitro, our data showed that LC3B is an adaptor on LDs mediating the interaction with LC3B cell membranes (Figure 6). In the LC3B-associated phagocytosis pathway, LC3B lipidated to endosomes mediates interaction and fusion with lysosomes. Hence, it is highly plausible that LC3B lipidated to the large LDs mediates interaction and hemifusion of the LDs with the autolysosome-like membranes. In the case of lipophagy, which targets smaller LDs, other protein adaptors, such as Rab10 46 or spartin 44, might govern the LD interaction with autophagosomes.
The mechanism by which LDs are delivered and degraded in the lumen of phospholipid bilayer vesicles is currently unknown. For example, a recent study proposes that lysosomes can pinch off small LDs from larger ones in hepatocytes 63 but how such a process happens is unknown. In the eventuality that the large LDs targeted by LC3B are destined for degradation, the acidified autophagosomes in contact with large LDs would likely mediate such degradation locally. This raises questions about how the monolayer of LDs interacts and delivers content to the lumen of a bilayer vesicle, as well as how LC3B could mediate such processes in our case. At this stage, we can only speculate that LC3B on LDs mediates the tethering and possibly the hemifusion of the LD with the autolysosome-like structures. This hemifusion would expose neutral lipids to the lumen of the autolysosomes. Alternatively, the autolysosome-like structures could locally deform the donor LD and pinch off small LDs 63. Protein degradation may also occur in this process, with proteins like PLIN1 potentially being delivered to the autolysosomes via mechanisms similar to chaperone-mediated degradation, at the autolysosome-like membrane and large LD contact. Further research is needed to fully understand the mechanisms involved in LD delivery and degradation within bilayer-bounded vesicles.
Limitations of the study
Despite multiple attempts using correlative electron microscopy, it was not possible to obtain a clear visualization of the membranes adjacent to the large LC3B-decorated LDs. A structural understanding of the interaction between LC3B-membranes and LC3-LDs would have been gained from such visualization.
The mechanism of ATG3 binding to LDs and its specificity towards larger LDs is currently unknown, and the physiological significance of this localization is not yet fully clear.
The relationship between our identified pathway and autophagy is not well defined, as both pathways depend on autophagic proteins, which makes it challenging to distinguish them from each other.
The precise function of LC3B on LDs is still unknown, and its role in tethering and possibly facilitating fusion is only speculated based on the previously identified capacity of Atg8 proteins in trans-dimerization.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to all the group members for their valuable comments and critical read of the manuscript. This work was supported by the Fondation pour la Recherche Médicale (FRM : EQU202103012564), the Programme Emergence de la Ville de Paris (DDEEES 165), the ANR-16-TERC-0002-01 (LDEN) to ART, and the National Institutes of Health (GM100930) to TJM. MO was partly financed by the Fondation de l’Ecole Normale Supperieure with QBio program and AS was funded by Q-Life.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity
We support the inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- 2.Feng Y, He D, Yao Z, and Klionsky DJ (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Ohsumi Y, and Yoshimori T (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct 27, 421–429. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N (2020). The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol 63, 1–10. [DOI] [PubMed] [Google Scholar]
- 5.Sawa-Makarska J, Baumann V, Coudevylle N, von Bülow S, Nogellova V, Abert C, Schuschnig M, Graef M, Hummer G, and Martens S (2020). Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamb CA, Dooley HC, and Tooze SA (2013). Endocytosis and autophagy: shared machinery for degradation. Bioessays 35, 34–45. [DOI] [PubMed] [Google Scholar]
- 7.Klionsky DJ, and Eskelinen E-L (2014). The vacuole vs. the lysosome: When size matters. Autophagy 10, 185–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yim WW-Y, and Mizushima N (2020). Lysosome biology in autophagy. Cell Discov. 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melia TJ, Lystad AH, and Simonsen A (2020). Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakatogawa H (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol 21, 439–458. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132. [DOI] [PubMed] [Google Scholar]
- 12.Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, and Melia TJ (2014). Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat. Cell Biol 16, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shpilka T, Weidberg H, Pietrokovski S, and Elazar Z (2011). Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, and Tooze SA (2014). WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5–16L1. Mol. Cell 55, 238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gammoh N, Florey O, Overholtzer M, and Jiang X (2013). Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex–dependent and–independent autophagy. Nat. Struct. Mol. Biol 20, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, and Mizushima N (2013). FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 14, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 18.Martens S, and Fracchiolla D (2020). Activation and targeting of ATG8 protein lipidation. Cell Discov. 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heckmann BL, and Green DR (2019). LC3-associated phagocytosis at a glance. J. Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herb M, Gluschko A, and Schramm M (2020). LC3-associated phagocytosis-the highway to hell for phagocytosed microbes. In Seminars in cell & developmental biology (Elsevier; ), pp. 68–76. [DOI] [PubMed] [Google Scholar]
- 21.Galluzzi L, and Green DR (2019). Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao G, Sheng Y, Yang H, Chua BT, and Xu L (2019). DFCP1 associates with lipid droplets. Cell Biol. Int 43, 1492–1504. [DOI] [PubMed] [Google Scholar]
- 23.Li D, Zhao YG, Li D, Zhao H, Huang J, Miao G, Feng D, Liu P, Li D, and Zhang H (2019). The ER-localized protein DFCP1 modulates ER-lipid droplet contact formation. Cell Rep. 27, 343–358. [DOI] [PubMed] [Google Scholar]
- 24.Pfisterer SG, Bakula D, Frickey T, Cezanne A, Brigger D, Tschan MP, Robenek H, and Proikas-Cezanne T (2014). Lipid droplet and early autophagosomal membrane targeting of Atg2A and Atg14L in human tumor cells. J. Lipid Res 55, 1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, and Uchiyama Y (2009). The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun 382, 419–423. [DOI] [PubMed] [Google Scholar]
- 26.Shibata M, Yoshimura K, Tamura H, Ueno T, Nishimura T, Inoue T, Sasaki M, Koike M, Arai H, and Kominami E (2010). LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem. Biophys. Res. Commun 393, 274–279. [DOI] [PubMed] [Google Scholar]
- 27.Velikkakath AKG, Nishimura T, Oita E, Ishihara N, and Mizushima N (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capitanio C, Bieber A, and Wilfling F (2023). How Membrane Contact Sites Shape the Phagophore. Contact 6, 25152564231162496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiam AR, Farese RV Jr, and Walther TC (2013). The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol 14, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welte MA, and Gould AP (2017). Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1260–1272. 10.1016/j.bbalip.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhiman R, Caesar S, Thiam AR, and Schrul B (2020). Mechanisms of protein targeting to lipid droplets: A unified cell biological and biophysical perspective. In Seminars in Cell & Developmental Biology (Elsevier; ). [DOI] [PubMed] [Google Scholar]
- 32.Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng J-X, Graham M, Christiano R, and Fröhlich F (2013). Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh R, and Cuervo AM (2012). Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, and Deretic V (2014). Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol 24, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D, Song J-Z, Li H, Shan M-H, Liang Y, Zhu J, and Xie Z (2015). Storage lipid synthesis is necessary for autophagy induced by nitrogen starvation. FEBS Lett. 589, 269–276. [DOI] [PubMed] [Google Scholar]
- 36.Wilfling F, Thiam AR, Olarte M-J, Wang J, Beck R, Gould TJ, Allgeyer ES, Pincet F, Bewersdorf J, and Farese RV Jr (2014). Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 3, e01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schütter M, Giavalisco P, Brodesser S, and Graef M (2020). Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell 180, 135–149. [DOI] [PubMed] [Google Scholar]
- 38.Shpilka T, Welter E, Borovsky N, Amar N, Mari M, Reggiori F, and Elazar Z (2015). Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 34, 2117–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velázquez AP, Tatsuta T, Ghillebert R, Drescher I, and Graef M (2016). Lipid droplet–mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol 212, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ, and Singh R (2016). Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 23, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sathyanarayan A, Mashek MT, and Mashek DG (2017). ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 19, 1–9. 10.1016/j.celrep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, and McNiven MA (2019). Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol 218, 3320–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung J, Park J, Lai ZW, Lambert TJ, Richards RC, Farese RV, and Walther TC (2021). The Troyer syndrome protein spartin mediates selective autophagy of lipid droplets. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, and Acevedo-Arozena A (2021). Guidelines for the use and interpretation of assays for monitoring autophagy. autophagy, 1–382. [Google Scholar]
- 46.Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, Casey CA, Liu J, Stöckli J, and James DE (2016). A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci. Adv 2, e1601470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, and Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ajjaji D, Ben M’barek K, Mimmack ML, England C, Herscovitz H, Dong L, Kay RG, Patel S, Saudek V, and Small DM (2019). Dual binding motifs underpin the hierarchical association of perilipins1–3 with lipid droplets. Mol. Biol. Cell 30, 703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, and Furuya T (2011). Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaiswal A, Hoerth CH, Pereira AMZ, and Lorenz H (2019). Improved spatial resolution by induced live cell and organelle swelling in hypotonic solutions. Sci. Rep 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King C, Sengupta P, Seo AY, and Lippincott-Schwartz J (2020). ER membranes exhibit phase behavior at sites of organelle contact. Proc. Natl. Acad. Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santinho A, Salo VT, Chorlay A, Li S, Zhou X, Omrane M, Ikonen E, and Thiam AR (2020). Membrane Curvature Catalyzes Lipid Droplet Assembly. Curr. Biol 30, 2481–2494.e6. 10.1016/j.cub.2020.04.066. [DOI] [PubMed] [Google Scholar]
- 53.Santinho A, Chorlay A, Foret L, and Thiam AR (2021). Fat Inclusions Strongly Alter Membrane Mechanics. Biophys. J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chabanon M, Ho JC, Liedberg B, Parikh AN, and Rangamani P (2017). Pulsatile lipid vesicles under osmotic stress. Biophys. J 112, 1682–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thiam AR, and Dugail I (2019). Lipid droplet–membrane contact sites–from protein binding to function. J. Cell Sci 132, jcs230169. [DOI] [PubMed] [Google Scholar]
- 56.Chorlay A, and Thiam AR (2020). Neutral lipids regulate amphipathic helix affinity for model lipid droplets. J. Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caillon L, Nieto V, Gehan P, Omrane M, Rodriguez N, Monticelli L, and Thiam AR (2020). Triacylglycerols sequester monotopic membrane proteins to lipid droplets. Nat. Commun 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Small DM, Wang L, and Mitsche MA (2009). The adsorption of biological peptides and proteins at the oil/water interface. A potentially important but largely unexplored field. J. Lipid Res 50, S329–S334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ben M’barek K, Ajjaji D, Chorlay A, Vanni S, Forêt L, and Thiam AR (2017). ER Membrane Phospholipids and Surface Tension Control Cellular Lipid Droplet Formation. Dev. Cell 41, 591–604.e7. 10.1016/j.devcel.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 60.Chorlay A, Santinho A, and Thiam AR (2020). Making Droplet-Embedded Vesicles to Model Cellular Lipid Droplets. STAR Protoc, 100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chorlay A, Forêt L, and Thiam AR (2021). Origin of gradients in lipid density and surface tension between connected lipid droplet and bilayer. Biophys. J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Motta I, Nguyen N, Gardavot H, Richerson D, Pincet F, and Melia TJ (2018). GABARAP Like-1 enrichment on membranes: Direct observation of trans-homo-oligomerization between membranes and curvature-dependent partitioning into membrane tubules. bioRxiv, 348730. [Google Scholar]
- 63.Schulze RJ, Krueger EW, Weller SG, Johnson KM, Casey CA, Schott MB, and McNiven MA (2020). Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu K, and Czaja MJ (2013). Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaushik S, and Cuervo AM (2015). Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol 17, 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakatogawa H, Ichimura Y, and Ohsumi Y (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178. [DOI] [PubMed] [Google Scholar]
- 67.Imam S, Talley S, Nelson RS, Dharan A, O’Connor C, Hope TJ, and Campbell EM (2016). TRIM5α Degradation via Autophagy Is Not Required for Retroviral Restriction. J. Virol 90, 3400–3410. 10.1128/JVI.03033-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Engelenburg SB, and Palmer AE (2010). Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat. Methods 7, 325–330. 10.1038/nmeth.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agrotis A, Pengo N, Burden JJ, and Ketteler R (2019). Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 15, 976–997. 10.1080/15548627.2019.1569925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.N’Diaye E-N, Kajihara KK, Hsieh I, Morisaki H, Debnath J, and Brown EJ (2009). PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 10, 173–179. 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.