

Abstract
Advanced hepatic fibrosis, driven by the activation of hepatic stellate cells (HSCs), affects millions world-wide and is the strongest predictor of mortality in non-alcoholic steatohepatitis (NASH), yet there are no approved anti-fibrotic therapies. To identify anti-fibrotic drug targets, we integrated progressive transcriptomic and morphological responses that accompany HSC activation in advanced disease using single nucleus RNA sequencing and tissue clearing in a robust murine NASH model. In advanced fibrosis, we found that an autocrine HSC signaling circuit emerged that was comprised of 68 receptor-ligand interactions conserved between murine and human NASH. These predicted interactions were supported by the parallel appearance of markedly increased direct stellate cell-cell contacts in murine NASH. As proof of principle, pharmacological inhibition of one such autocrine interaction, NTRK3-NTF3, inhibited human HSC activation in culture and reversed advanced murine NASH-fibrosis. In summary, we uncovered a repertoire of anti-fibrotic drug targets underlying advanced fibrosis in vivo. The findings suggest a therapeutic paradigm in which stage-specific therapies could yield enhanced anti-fibrotic efficacy in patients with advanced fibrosis.
One Sentence Summary:
A repertoire of autocrine signaling modalities in advanced hepatic fibrosis constitute potential therapeutic targets.
INTRODUCTION
The global prevalence of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH, a more advanced stage with inflammation and fibrosis) are rapidly rising associated with the world-wide obesity epidemic. Approximately 30 - 40% of adults in the United States have NAFLD, ~20% of whom have NASH (1). With no approved therapy, NASH is already the second most common indication for liver transplantation and the most rapidly rising etiology underlying hepatocellular carcinoma (1). Among various histologic features of NASH, fibrosis driven by the activation of hepatic stellate cells (aHSCs) into fibrogenic myofibroblasts (2) and is the strongest predictor of mortality, establishing fibrosis as a critical therapeutic target in this disease (3, 4). However, no drugs have been approved for NASH, representing an urgent unmet need (5).
Fibrosis across diverse tissue types, including liver, can persist or worsen even if the injury-driven inflammation is attenuated by treating the underlying disease. For example, fibrosis progresses in ~15-25% of patients with cirrhosis due to hepatitis C even after antiviral cure (6). Similarly, in advanced NASH, fibrosis regression has not been observed in patients with cirrhosis after bariatric surgery, even though earlier fibrosis stages may regress (7). The mechanisms underlying fibrosis persistence in advanced disease, and the specific role of activated hepatic stellate cells (aHSCs) in contributing to this response, are not known.
Using mathematical modelling we previously predicted that different cell circuits underly cellular response to acute tissue injury versus chronic injury, driving tissue repair versus fibrosis, respectively. Within the cell circuit underlying chronic injury, our model predicted that targeting the autocrine signaling loop of myofibroblasts is the most promising approach to restore the balance back to repair from fibrosis (8, 9); however, this model has not yet been validated experimentally. Based on this framework, we speculated that in liver, persistent hepatic fibrosis in advanced disease represents a state wherein myofibroblasts derived from activated hepatic stellate cells (aHSC) establish fibrosis-sustaining autocrine signaling loops mediated by a unique repertoire of receptors and ligands. If true, therapeutics targeting these receptors and ligands could represent a class of antifibrotics for patients with advanced hepatic fibrosis.
Single cell RNA-seq (scRNA-seq) technologies have transformed our understanding of human liver disease by uncovering remarkable cellular heterogeneity to clarify disease mechanisms (10). However, scRNA-seq preferentially captures immune populations at the expense of liver epithelial and fibrogenic cells (aHSCs) (11). Here, using single nucleus RNA-seq (snRNA-seq), we have profiled gene expression changes in HSCs from both human and murine NASH, the latter using a rodent model that faithfully recapitulates features of advanced disease, in particular the fibrosis and tumors (‘FAT-NASH’) model (12, 13). We describe the emergence in late-stage disease of a conserved autocrine signaling circuit in NASH-associated HSCs that comprises of ~100 predicted ligand-receptor pairs, over half of which involve short-range interactions requiring physical cell-cell proximity.
Like neurons, HSCs, which express many neuronal markers (14–17), display elaborate cellular projections in vivo that could serve important signaling functions yet their contributions have been overlooked. Using tissue clearing and 3D imaging based on iDISCO (18), we generated high-resolution 3D images of HSCs that enabled characterization of these cellular projections in situ in NASH. This approach has unveiled a marked amplification of HSC-HSC contacts linked to progressive disease severity that could support short-range HSC autocrine interactions. To establish proof of principle, we interrogated the predicted HSC-specific receptor-ligand autocrine loop mediated by Neurotrophic Receptor Tyrosine Kinase 3 (NTRK3) -Neurotrophin 3 (NTF3) in both human and murine NASH. NTRK3 protein was localized to HSC projections in NASH, but absent from normal liver. Last, we functionally validated this interaction through antagonism of NTRK3, which inhibited HSC fibrogenicity and migration in cultured human HSCs, and attenuated fibrosis in vivo in murine NASH. Our work uncovers a HSC physical interactome as a major driver of advanced hepatic fibrosis through an autocrine circuitry.
RESULTS
Single nucleus RNA-seq uncovers stellate cell autocrine signaling network in NASH patients
We performed snRNA-seq of snap-frozen liver samples from 3 control (non-tumor tissue from liver metastasis resections) and 9 patients with NASH (NAS and fibrosis score provided in Data file S1) that captured RNA expression from hepatocytes and HSCs more efficiently than those previously reported using single-cell RNA-seq (11). Representative hematoxylin and eosin and Sirius red staining of human control and NASH livers revealed steatosis and fibrosis in NASH but not in controls (Fig. 1A). Human tissues from 3 distinct sources were analyzed to ensure reproducibility, yielding snRNA-seq data from 128851 total nuclei and 3783 HSC nuclei post-QC, the largest compilation of human HSC transcriptomes to date. By informatically merging datasets from these samples and clustering using a UMAP (Uniform Manifold Approximation and Projection)-based approach, gene expression from all major cell types was tracked at the expected proportions in both control and disease samples, based on cell-specific marker genes (Fig. 1B–D, Fig. S1). Although no new cell type emerged in NASH (Fig. 1C), HSCs from patients with NASH, termed ‘NASH-associated HSCs’, expressed an altered transcriptomic profile with 169 significantly over-expressed and 291 significantly repressed genes compared to HSCs from control patients (Fig. 1E, Padj. < 0.05, gene lists provided in Data file S2). The most highly enriched pathways in NASH-associated HSCs included cell-matrix adhesion and integrin signaling as expected (Fig. 1F). Notably several genes belonging to the category “extracellular matrix organization” were downregulated, supporting HSCs not only as a secretor of ECM components but also as a key contributor to overall ECM remodeling. Using CellphoneDB software to predict cell-cell interactions in NASH livers revealed a tripartite intercellular communication network comprised of HSCs, cholangiocytes, endothelial cells, and their autocrine interactions (Fig. 1G, Data file S6, S7). These results reinforce recent observations highlighting HSCs as a hub of cell-cell communication in liver disease (19) and further establish in vivo evidence of a myofibroblast autocrine signaling circuit underlying fibrosis that was predicted using mathematical modelling (8, 9).
Figure 1. Single nucleus RNA-seq of patients with NASH uncovers an autocrine signaling loop in NASH-associated hepatic stellate cells (HSCs).
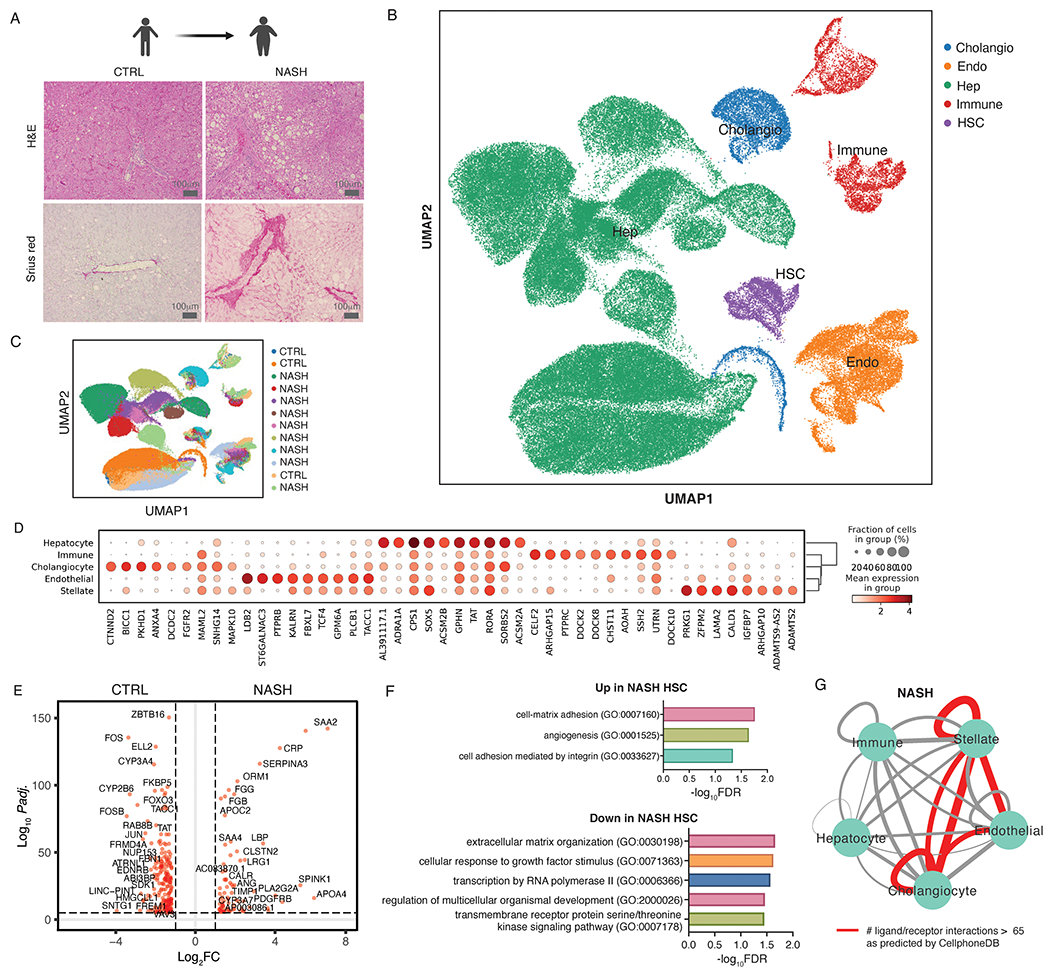
(A) Hematoxylin and Eosin and Sirius red staining of 3 control and 9 human NASH samples used for snRNA-seq. (B) UMAP visualization of all nuclei derived from control and NASH livers colored by cell type. (C) UMAP visualization of all nuclei derived from control and NASH livers colored by sample of origin. (D) Top 8 marker gene expression for each cell type. (E) Differentially expressed genes (DEGs) between HSCs from controls vs patients with NASH (red dots indicate DEG with Padj. < 0.05 and log2FC > 1). (F) All significantly enriched PANTHER pathways (FDR < 0.05) in genes up- and down-regulated in NASH-associated HSCs. (G) Total number of significant interactions between different cell types as predicted using CellphoneDB. Thickness of the connecting line is proportional to the total number of interactions, with interactions > 65 highlighted in red.
Stellate cell autocrine signaling is conserved in FAT-NASH mice
Next, we tested whether the FAT-NASH murine model of NASH faithfully recapitulated the cell-cell communication networks uncovered in human NASH (12, 13). To do so, we profiled HSC transcriptomes from control and 24-week FAT-NASH livers using snRNA-seq (Fig. 2A, Fig. S2). Three livers each from control and 24-week FAT-NASH mice were pooled into a single sample and the data were informatically merged using Seurat-LIGER algorithm to generate a single UMAP of 19249 nuclei from all samples post-QC (Fig. 2B). After annotation of different nuclei clusters as different liver cell types using canonical marker genes for mice, we recovered all known liver cell types represented at the expected proportions based on existing literature (Fig. 2C (11)). Two distinct populations of HSCs (1250 nuclei post-QC) were apparent from our data, termed ‘HSC1’ and ‘HSC2’. HSC1 was mostly comprised of nuclei from control mice and expressed classical markers of HSC quiescence including Lrat and Des, whereas HSC2 was primarily comprised of nuclei from FAT-NASH mice and expressed markers of activation such as Pdgfrb, Acta2, Col1a1, Col3a1, Mmp2, Timp1, and Timp2 (Fig. 2D,E), which represent NASH-associated HSCs. Differential gene expression analysis revealed 996 significantly up-regulated genes in HSC2 compared to HSC1, including cell adhesion and integrin signaling pathways present in patients with NASH, among other pathways that are known drivers of fibrosis including TGFβ signaling, actin cytoskeleton remodeling, and cell migration (Fig. 2F,G, Padj. < 0.05, Data file S3). As in human NASH, CellphoneDB analysis of FAT-NASH mice established a prominent autocrine signaling circuit in HSCs, with HSC2 cells harboring the greatest number of predicted ligand-receptors interactions, followed by interactions between HSC2 and cholangiocytes, and then between HSC2 with a subset of endothelial cells (Fig. 2H, Data file S4, S5). Notably, more than half of the 120 HSC2 autocrine ligand-receptor pairs involved short-range interactions (defined as either non-secreted or collagen/integrin interactions) that require cell-cell proximity between the ligand and its receptor (Fig. 2I), highlighting an autocrine circuitry that emerges in activated HSCs in advanced fibrosis.
Figure 2. Autocrine signaling in NASH-associated hepatic stellate cells is conserved in FAT-NASH mice.
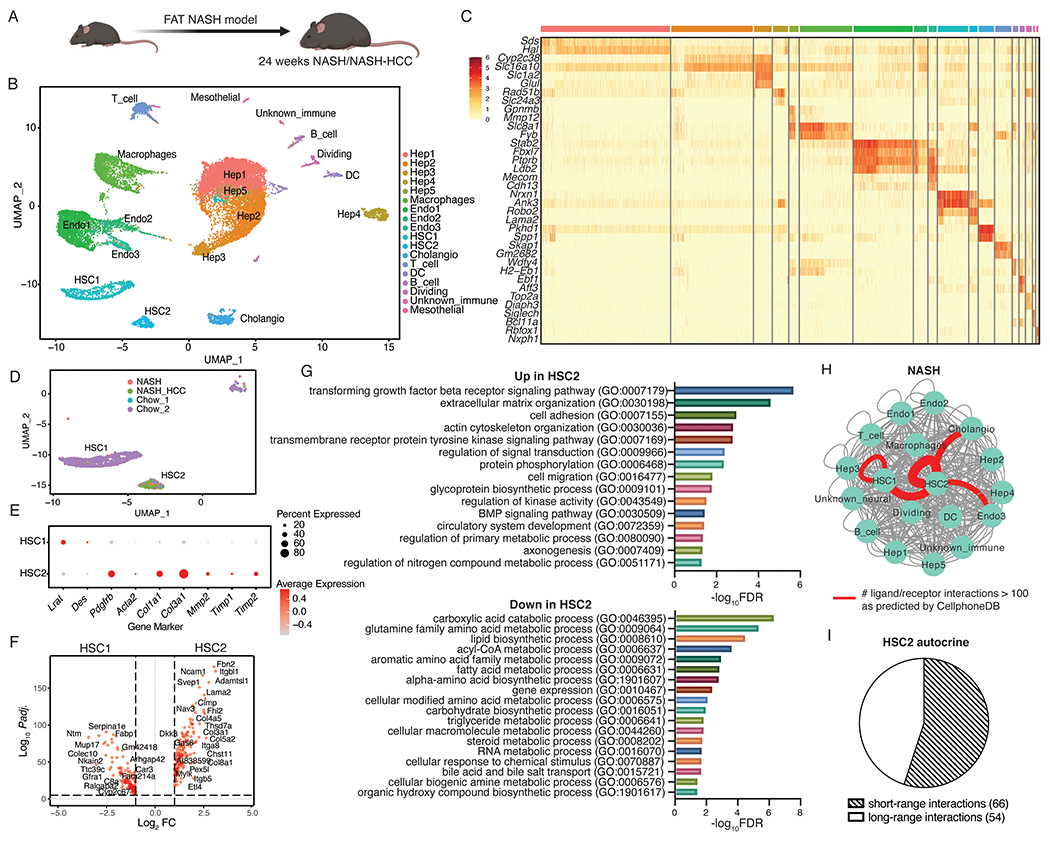
(A) Schematic of control (n=2), NASH (n=3), and NASH-HCC (n=3) mice used in this experiment. (B) UMAP visualization of nuclei combined from all liver samples colored by cell type. (C) Top 2 marker gene expression for each cell type. (D) UMAP visualization of hepatic stellate cell (HSC) clusters colored by sample origin. (E) Dot plot depicting canonical quiescence and activation marker gene expression for each HSC cluster. (F) Differentially expressed genes between HSC1 and HSC2 (red dots indicate DEG with Padj. < 0.05 and log2FC > 1). (G) All significantly enriched PANTHER pathways (FDR < 0.05) in genes up- and down-regulated in NASH-associated HSCs. (H) Total number of significant interactions between different cell types from NASH/NASH-HCC mice as predicted using CellphoneDB (P < 0.05). Thickness of the connecting line is proportional to the total number of interactions, with interactions > 70 highlighted in red. (I) HSC autocrine interactions classified as short-range or long-range based on manual curation.
Hepatic stellate cell-cell contacts increase progressively in livers of FAT-NASH mice
In the resting liver, HSCs localize to the space of Disse, interposed between hepatocytes and endothelial cells. To clarify how HSCs interact with each other within this anatomical milieu, a pipeline for iDISCO tissue clearing (18) and confocal imaging was established to generate high-resolution 2D images of HSCs using staining against DESMIN, a marker of HSCs in the murine liver (Fig. 3A,B, Fig. S3). Stacks of images were then 3D reconstructed and segmented using the IMARIS software (Figure 3F,G, Movies S1–S4). These high-resolution 3D reconstructed images enabled visualization of detailed HSC morphology including their cellular projections that closely resemble those of neurons. Individual HSCs (each represented by a different pseudocolor in Fig. 3G) were classified by IMARIS as separate DESMIN+ surface objects that overlaid a single nucleus marked by propidium iodide (modelled by pink spheres in Fig. 3G) to visualize nuclear DNA (Fig. 3G). HSCs appeared uniform and regularly spaced in the livers of healthy mice fed a chow diet.
Figure 3. Increased stellate cell-stellate cell contacts in FAT-NASH mice revealed by tissue-clearing and 3D imaging.
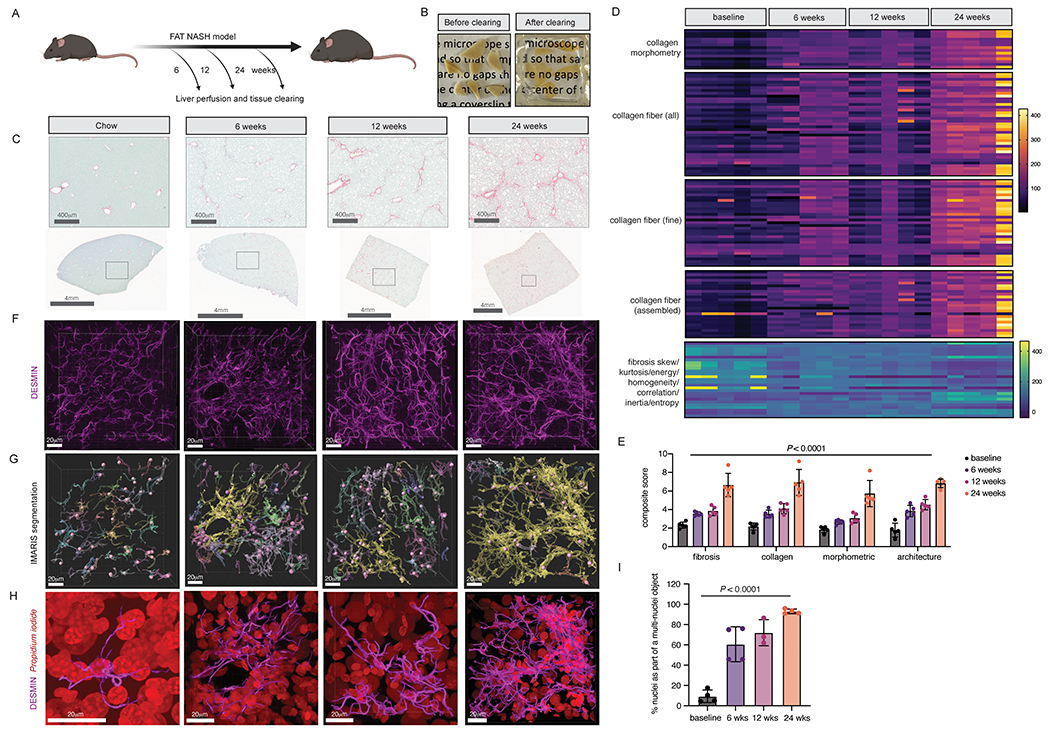
(A) Liver from FAT-NASH mice were perfused and cleared at the depicted timepoints. (B) Representative images of perfused liver pieces before and after clearing. (C) Representative images from Sirius red staining of FAT-NASH livers from different timepoints in the model. (D) AI-based quantification of Sirius red collagen staining from five mice per timepoint. (E) Composite fibrosis scores calculated based on (D). (F) Confocal imaging and 3D reconstruction of DESMIN staining in cleared livers. (G) IMARIS surface and spot segmentation of DESMIN and nuclear staining, respectively, of the same images from C. (H) Enlarged image of a single segmented DESMIN+ surface object highlighted in yellow in (G), along with nuclear staining in red. (I) Quantification of the percentage of nuclei that are found as a single nucleus per surface object or multiple nuclei per surface object (n=3-4 mice per timepoint).
To characterize potential morphological changes in HSCs in NASH, we cleared and imaged livers in mice from the FAT-NASH model at 6, 12, and 24 weeks, and correlated with amounts of fibrosis measured at the same time-points. To comprehensively quantify tissue fibrosis, we leveraged a single-fiber AI based platform, FibroNest, that extensively characterizes both absolute collagen content and important statistical features of the distributions of collagen fibers’ morphometric and architectural phenotypes. As expected, the severity of fibrosis increased progressively in our FAT-NASH model from 6 to 24 weeks as quantified using FibroNest’s severity scores coupled with a progressive increase in overall DESMIN signal (Fig. 3C–E). Correspondingly, HSCs underwent a progressive morphological transition in NASH, in which their projections appeared more elongated and initiated physical contacts with one another. We quantified these direct interactions as the number of DESMIN+ objects (as detected by IMARS) that were multi-nucleated, because HSCs are mononucleated in vivo. One such DESMIN+ object from each timepoint is highlighted yellow in Fig. 3G with original staining visualized in Fig. 3H. Using this method, we found that the number of HSC-HSC contacts (indicated by an increasing fraction of DESMIN+ objects that harbor multiple nuclei) increased as the FAT-NASH model progressed. The emergence of these interactions mirrored the increase in fibrosis severity detected using FibroNest(Fig. 3I). At 24 weeks in the FAT-NASH model, almost all HSCs made physical contacts with each other, establishing a HSC physical interactome in advanced NASH that could mediate short-range autocrine signaling (Fig. 3I). Of note, propidium iodide nuclear staining demonstrated a loss of granularity with larger nuclei during NASH progression, which most likely represents hepatocytes, suggesting a loss of heterochromatic foci in NASH-associated hepatocytes (Fig. 3H).
HSC autocrine signaling factor NTRK3 is an anti-fibrotic drug target in NASH
Our data demonstrate that HSCs create a physical interactome in NASH that parallels increased autocrine signaling predicted by snRNA-seq. To provide a proof-of-principle that HSC autocrine signaling may be mediated by HSC physical interactions in the FAT-NASH model, we sought to localize a specific autocrine receptor, NTRK3, to HSC projections in vivo. We overlapped autocrine interactions identified in human patients with NASH and FAT-NASH mice, yielding 68 autocrine receptor-ligand pairs conserved in both species (Fig. 4A). Among these candidate receptor-ligand pairs, we focused on NTRK3-NTF3 because this pathway has not been described in HSCs. Interrogation of our snRNA-seq datasets confirmed the selective expression of NTRK3 and its predicted ligand NTF3 in HSCs in both patients and mice (Fig. 4B,C). Whereas the amount of NTRK3 and NTF3 gene expression diverged between mice and patients in NASH, both genes increased in FAT-NASH mice compared to chow control. In patients with NASH NTRK3 expression was reduced compared to control patients. Using whole-liver lysates, NTRK3 protein was present in mouse livers and strongly induced in NASH (Fig. 4D), but not detected in human livers, likely because the signal was diluted by other cell types. These species differences may reflect the inherent heterogeneity of the human samples, in which patients are of diverse age, sex, and backgrounds, whereas mice are inbred and all exposed to identical conditions prior to analysis. Thus, we suggest that NTRK3 expression likely increases in NASH but further verification is necessary in a more standardized cohort of patients. More importantly, NTRK3 protein was localized by immunofluorescence to HSC projections in NASH in both species, which were visualized as an overlap with HSC-specific markers αSMA in humans (Fig. 4E–F) and DESMIN in mice (Fig. 4G–H). These key findings reinforce our proposed model that “effective signaling” between HSCs is increased through expanded physical autocrine contacts by HSCs in NASH.
Figure 4. NTRK3 is expressed on cellular projections of NASH HSCs.
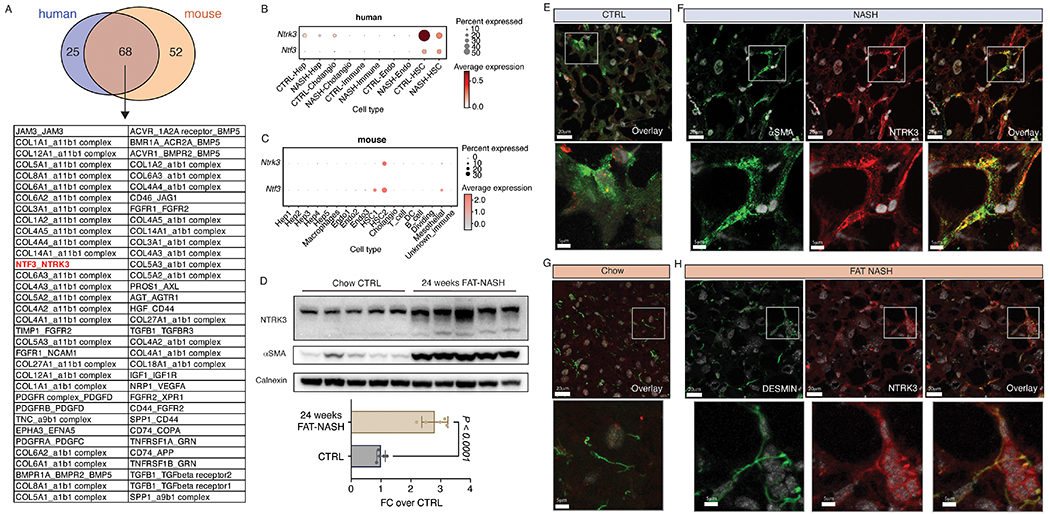
(A) Overlap of significant (P < 0.05) autocrine interactions identified in human and mouse NASH by CellphoneDB. Dot plot depicting NTRK3 gene expression across different cell types in human (B) and mouse (C) snRNA-seq data. (D) NTRK3 and αSMA protein expression in whole-liver lysates of control or 24-week FAT-NASH mice (n=5 mice per group, p-value calculated using Student’s t-test), as detected by Western blot and quantified by densitometry normalized to CALNEXIN as loading control. NTRK3 and αSMA protein expression in human control (E) and NASH (F) liver as detected by immunofluorescence. NTRK3 and DESMIN protein expression in mouse control (G) and NASH (H) liver as detected by immunofluorescence.
To establish HSC NTRK3-NTF3 autocrine signaling as a driver of NASH-fibrosis, NTRK3 was knocked down in the immortalized human stellate cell line, LX-2, using CRISPR-Cas9 and siRNA. Whereas stable NTRK3 knockdown by CRISPR reduced ERK phosphorylation and αSMA expression by Western blot, transient NTRK3 knockdown by siRNA only reduced αSMA expression (Fig. 5A). Unbiased RNAseq analyses of NTRK3 CRISPR and NTRK3 siRNA treated LX-2 documented a decrease in fibrogenic pathways including “regulation of extracellular matrix organization”, “extracellular matrix assembly”, “collagen fibril organization”, “integrin activation”, and “regulation of fibroblast migration” (Fig. 5B), with genes captured in these pathways shown in Figure 5C. We also probed a panel of established fibrogenic genes in the bulk RNA-seq datasets, which demonstrated an overall decrease in both NTRK3 CRISPR and NTRK3 siRNA treated LX-2 cells (Fig. 5D). Consistent with RNAseq analyses indicating a decrease in “regulation of fibroblast migration”, knocking out NTRK3 inhibited LX-2 cell migration based on the wound-closure assay (Fig. 5E,F). In parallel, NTRK3 activity was also blocked pharmacologically in LX-2 cells using LOXO-195, a second-generation, highly specific NTRK3 kinase domain inhibitor currently in clinical trials for the treatment of TRK fusion–positive cancers. As with CRISPR-Cas9 knockdown, LOXO-195 in LX-2 cells exhibited dose-dependent inhibition of LX-2 cell migration at nanomolar concentrations (Fig. 5G,H) without affecting cell viability or proliferation (Fig. S4). Concurrent with decreased migration, LOXO-195 treatment reduced LX-2 fibrogenicity as demonstrated by lower expression of an established panel of fibrogenic genes, compared to vehicle control (Fig. 5I).
Figure 5. NTRK3 is an HSC autocrine drug target in NASH.
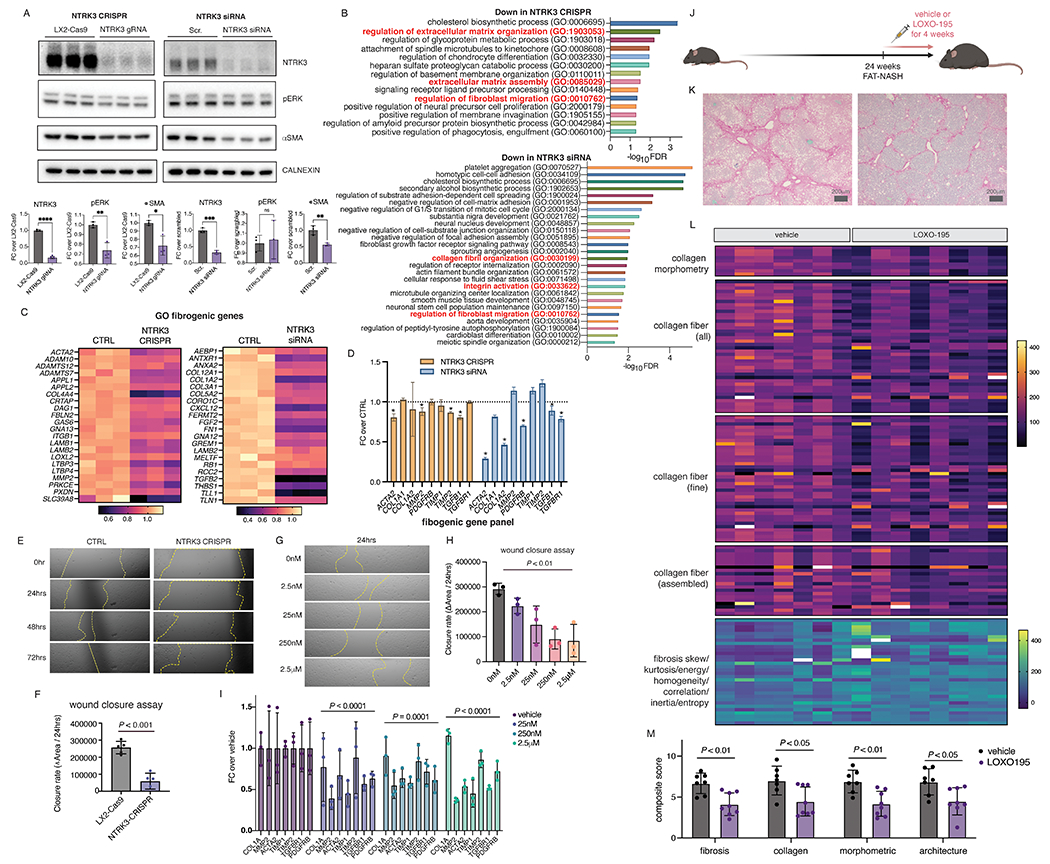
(A) ERK phosphorylation and αSMA protein expression in LX-2 cells with NTRK3 knock down by CRISPR or siRNA, quantification of Western blot by densitometry using CALNEXIN as loading control (p-value calculated using Student’s t-test). (B) Gene Ontology analysis of the top 500 most significantly down-regulated genes from RNAseq analyses of NTRK3 knockdown cells (FDR < 0.05, fold enrichment score > 5), with biological processes related to HSC fibrogenicity highlighted in red and genes from these categories shown in (C). (D) Expression of an established fibrogenic gene panel with NTRK3 knockdown from RNAseq analyses (*,Padj. < 0.05 compared to control). (E) Representative migration of NTRK3 CRISPR knockdown cells LX-2 compared to control LX-2 cells in scratch assay (F, average over 4 biological replicates, p-value calculated by Student’s t-test). (G) Pharmacological inhibition of NTRK3 using LOXO-195 dose-dependently reduced LX-2 cell migration in scratch assay (average over 3 biological replicates, representative shown in (OH, p-value represents effect of treatment calculated using one-way ANOVA). (I) Expression of an established fibrogenic gene panel in LX-2 cells treated with varying concentration of LOXO-195 for 24hrs (average over 3 replicates in a single experiment reproduced in a separate experiment, p-values represent effect of drug treatment compared to vehicle by two-way ANOVA). (J) Schematic depiction of the LOXO-195 in vivo study design. (SK) Representative Sirius red staining from FAT-NASH mice with LOXO-195 or vehicle control. (L) AI-based quantification of Sirius red collagen staining from n=7-8 mice per treatment. (M) Composite fibrosis scores calculated based on (L), p-values calculated by student’s t-test.
To provide a proof of principle that inhibition of this HSC autocrine signaling circuit could reverse fibrosis in vivo, FAT-NASH mice at 24 weeks on the model with advanced fibrosis were treated with LOXO-195 or vehicle twice daily, 5 days per week, for 4 weeks while continuing the model (Fig. 5J, Fig. S5A). There was no observed toxicity of LOXO-195 in mice compared with vehicle treated controls; the only notable difference was greater weight gain in a subset of LOXO-195 treated animals (Fig. S5B). FibroNest-based phenotypic fibrosis quantification of Sirius red stained liver sections documented significant reduction of all major features of fibrosis in mice treated with LOXO-195 compared to vehicle-treated controls (P < 0.05), which was coupled with a pruning of stellate cell processes in cleared livers (Fig. 5K–M, Fig. S6), establishing NTRK3 as a potential antifibrotic target in NASH.
DISCUSSION
In this study, we leveraged recent technological advances in snRNA-seq to capture HSC single-cell transcriptomes, coupled with tissue clearing of a mouse model to enable high resolution 3D tissue imaging. Using this strategy, we have uncovered an autocrine signaling circuitry in HSCs supported by markedly amplified cell-cell contacts underlying HSC activation in advanced NASH fibrosis.
Using snRNA-seq, we successfully captured transcripts from all liver cell types including HSCs at frequencies mirroring those found in situ (11). NASH-associated HSCs not only engaged the well-established fibrogenic pathways PDGFRB and INTEGRIN signaling, but also a previously uncharacterized CCKR signaling pathway, providing a mechanistic explanation for the observed anti-fibrotic effects of an CCKR antagonist in a murine NASH model (20). We uncovered an intercellular communication hub that emerges in NASH that consists of HSCs, endothelial cells, cholangiocytes, and their autocrine interactions catalogued in the accompanying supplemental tables that can serve as a fertile resource for future investigations into NASH therapeutics.
Mathematical modelling of growth factor exchange between different cell types during homeostasis and perturbation (for example, during pathological conditions such as tissue injury) predicted the development of a self-sustaining autocrine signaling circuit in fibroblasts that maintains fibrosis in different tissues, including liver. Using snRNA-seq to profile HSCs in human and mouse livers, we provide direct functional and morphologic evidence for the emergence of such an autocrine signaling loop in NASH fibrosis.
More than half of the HSC autocrine receptor-ligand interactions are short-range and require close physical proximity between interacting HSCs. To establish an anatomic basis for this signaling network we cleared liver tissue from mice at various stages of the FAT-NASH model using iDISCO (18), establishing that with worsening NASH-fibrosis, HSCs increased physical contact with each other through their cellular projections. In advanced disease (24 weeks on the FAT-NASH model), almost all HSCs were enmeshed within a dense interconnected network or “physical interactome” of HSCs that can support short range cell-cell signaling. Our results support a paradigm of autocrine HSC activation in disease in which physical proximity between these cells increases the effectiveness of autocrine ligand receptor interactions.
To establish HSC autocrine signaling as a driver of NASH-fibrosis, we focused on the NTRK3-NTF3 interaction, one among the 51 predicted HSC autocrine interactions that were conserved between patients with NASH and the murine FAT-NASH model. In both patients and mice, NTRK3 protein was not expressed in the healthy liver but was induced in HSCs in NASH, where its protein expression localized to HSC cellular projections. Using CRISPR and siRNA against NTRK3 in LX-2 cells, NTRK3 knockdowns were generated, which displayed markedly reduced fibrogenicity and cell migration. These effects were phenocopied by pharmacological inhibition using LOXO-195, a second generation NTRK small molecule inhibitor that is used clinically to treat tumors expressing NTRK fusion proteins (21). Migration was assessed as a key readout in culture because it most directly underlies expansion of cell-cell contacts. To validate HSC autocrine signaling as a potential therapeutic target in NASH in vivo, fibrosis was improved in FAT-NASH mice with advanced fibrosis (24 weeks) by only 4 weeks of treatment with LOXO-195. This finding not only establishes proof of principle for an autocrine pathway in advanced NASH fibrosis, but more importantly suggests that treatment of advanced fibrosis may be more effective if it leverages autocrine targets that dominate in late-stage disease.
The basis for the morphological transition of HSCs in NASH is not well understood. HSCs express a neuronal gene signature including neurotrophins and their receptors (22). In fact, the low affinity neurotrophin receptor P75NTR has been described as a marker of HSC precursors in the fetal liver and drives cytoskeletal remodeling in cultured HSCs through Rho signaling (23, 24). Future studies will investigate the potential link between increased expression of neuronal genes and morphological evolution of HSCs in vivo in NASH. In other tissues, skin fibroblasts also undergo membrane extension following injury (25), and a recent single cell transcriptomics study revealed a similar neuronal gene signature in these cells (26). These findings are in line with recent pan-tissue scRNA-seq efforts to characterize global transcriptomic signatures shared among tissue fibroblasts (27, 28) and point to conserved injury response mechanisms that incorporate both the fibroblast transcriptome and morphology. Thus, the principle of enhanced autocrine interactions during advanced fibrosis may extend to other fibrotic tissues including skin, lung, kidney and heart, but requires direct validation.
This study reveals several features of HSCs responses in vivo. We have identified genes and pathways that are selectively expressed in NASH-associated HSCs and uncover a role for autocrine ligand-receptor interactions supported by an expanded physical interactome in late-stage disease, revealing a new repertoire of antifibrotic drug targets that may be uniquely effective for treating advanced fibrosis.
There are several limitations of our study. Thus far we have optimized a tissue clearing pipeline for imaging mouse livers but not human livers. Future efforts need to optimize tissue clearing pipelines for human liver in order to verify that the HSC morphological transitions detected in murine NASH are conserved in human NASH patients. Another limitation is that we only profiled HSCs from 24 weeks FAT-NASH mouse livers, where disease is already very advanced. Future studies should profile earlier timepoints determine if different signaling circuitries dominate HSCs at earlier disease stages in order to establish stage-dependent disease vulnerabilities and therapeutics. Lastly, we validated only a single autocrine ligand-receptor pair in vivo and in culture (NTF3-NTRK3); future studies will need to explore other autocrine targets in HSCs or combinations thereof to yield a maximal antifibrotic effect.
MATERIALS AND METHODS
Study design
In this study we leveraged recent developments in single nucleus RNA-seq and tissue clearing techniques to uncover signaling circuits that emerge in NASH that can serve as therapeutic targets for advanced fibrosis. We profiled HSC transcriptomes from both human and mouse NASH livers and identified a conserved autocrine signaling circuit that consists of 68 ligand-receptor pairs. Targeting one of these HSC autocrine receptors, NTRK3, reduced HSC activation in culture and reversed advanced fibrosis in vivo in the FAT-NASH model. All results in this study have been reproduced in independent cohorts of mice and in biological replicates for cell culture. For quantifications that can be subjected to human bias, samples were randomly coded and quantification was done in a blinded fashion. Use of human tissues for single nuclei RNA-seq was IRB-exempt in accordance with guidelines of the Mount Sinai Institutional Review Board, as there were no patient identifiers and the tissue was otherwise intended to be discarded.
Patient and clinical specimens
Samples collected at Mount Sinai Hospital, New York, were freshly resected liver samples collected at time of surgery with informed consent. Samples collected at University of California, San Diego were deidentified livers declined for transplantation obtained via Lifesharing OPO. Samples collected at Gordian Biotechnology were livers not suitable for transplantation from research consented donors were perfused and transported on ice to Gordian Biotechnology from regional hospitals after transplantable organ removal. Samples sections suitably sized for snRNA-seq (~4x4x4mm) were removed from at least 5mm below the outer surface of the liver, snap frozen in liquid nitrogen, and stored at −80°C until nuclei extraction.
Mice
The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the Icahn School of Medicine at Mount Sinai, NY (IACUC-2018-0060). 6-week-old male and female C57BL/6J mice (housed separately) were purchased from Jackson Laboratories. Five mice per cage were housed in a Helicobacter-free room for 12 hours light - 12 hours dark cycle and weighed once weekly.
In this study we used the FAT-NASH rodent model of human NASH, which is a model that we have extensively used and validated (12). This model reproducibly recapitulates the stages of progression of human NASH that include advanced fibrosis and tumors (hence the term FAT – NASH for “Fibrosis And Tumors”), along with changes in the microbiome (13). Carbon tetrachloride (CCl4) was purchased from Sigma-Aldrich. CCl4 was freshly dissolved in corn oil at a final concentration of 5% before injection. The final dose of pure CCl4 was 0.2 μL/g of body weight of mice, delivered intraperitoneally once/week starting from initiation of the western diet/sugar water feeding and continued for a total period of 6, 12, or 24 weeks. Western diet containing 21.2% fat (42% Kcal), 41% sucrose and 1.25% cholesterol by weight was purchased from Envigo (Teklad Custom diet). Sugar water solution contained 18.9 g/L D-(+)-Glucose (Sigma-Aldrich) and 23.1 g/L D-(−)-Fructose (Sigma-Aldrich) dissolved in autoclaved water and filter sterilized. The diet and sugar water were replaced once weekly.
For LOXO-195 in vivo studies, LOXO-195 (TargetMol) was first dissolved in DMSO to 41.6mg/mL, before corn oil was added to a final LOXO-195 concentration of 12.5mg/mL, 100μL of this drug solution or vehicle control was given to a 25g mouse to achieve a 50mg/kg dosage (weight adjusted daily). The dosing regimen was oral gavage twice a day (separated by 8 hours), 5 days a week for a total of 4 consecutive weeks. All mice were sacrificed at 72 hours after the last dose of LOXO-195 or vehicle control.
At termination of study pieces of liver (unless used for tissue clearing), were formalin fixed for FFPE and directly frozen in Tissue-Tek OCT compound (ThermoFisher Scientific catalog no. 4583) over dry-ice then −80°C for subsequent immunohistochemistry. The remaining mouse livers were chopped into ~2 cm3 pieces, flash frozen in liquid nitrogen, and stored at −80° C until nuclei extraction.
Tissue clearing, staining, confocal imaging, and 3D image reconstruction
Mice were anesthetized and perfused through the portal vein with 20 mL of PBS followed by 20 mL of 4% PFA/PBS, liver tissues were trimmed to ~2 mm3, cleared, and stained as described in the iDISCO protocol (18). A detailed protocol that accompanies the original iDISCO publication along with an updated list of validated antibodies for this method can be found at https://idisco.info/. Antibodies used in this study were DESMIN (Abcam catalog no. ab15200, 1:400 dilution) and CD31 (R&D Systems catalog no. #AF3628, 1:400 dilution). Propidium iodide (Sigma catalog no. #P4170, 0.02 mg/mL final concentration) was used to stain DNA. Imaging was carried out on the Leica SP5 DMI confocal microscope and subsequently reconstructed using the IMARIS software at the Sinai Microscope CoRE. The IMARIS “surface” tool was used for segmenting the DESMIN staining using Surface Grain Size of 0.303 μm, diameter of largest sphere of 1.14 μm, and manual Threshold range from 13.4 to 168.4. The IMARIS “spot” tool was used to segment propidium iodide DNA staining into nuclei by setting the diameter of spots to 5μm, and these nuclei were then manually curated as belonging to HSC if surrounded by DESMIN signal in the overlay channel.
Generation of LX2-Cas9 cells and cell culture
LX-2 cells are a widely used, immortalized human HSC line previously characterized in our laboratory (29). Stable Cas9-expressing LX-2 cells were generated through lentiviral transduction of a Cas9-expressing plasmid (lentiCas9-Blast, Addgene 52962-LV). Cells were then selected based on blasticidin resistance, and homogenous Cas9-expressing LX-2 cells were isolated by single cell dilution cloning (available as LX-2 Cas9 line from Millipore-Sigima, SCC613). Scratch assay was performed in a 24-well plate by using a P200 pipet tip to scrape the LX-2 cell monolayer in a straight line. Debris were removed from the edge of the scratch by washing and replacing with fresh media. To obtain consistent field of view in imaging, markings were made on the plate using a fine tip marker. Plates were then imaged immediately after the scratch with a microscope using the phase-contrast objective at 10x magnification. Cells were returned to incubation and imaged again 24, 48, and 72 hours after initial scratch. Images acquired were analyzed quantitatively by using Photoshop. Images obtained at each time point and treatment condition were visually overlayed on top of each other and cropped to the same size. The perimeters and areas of the scratches were then delineated and quantified. For all cell culture assays, LX-2 cells were serum-starved overnight to quiesce and synchronize metabolic activity in serum-free DMEM (Thermo Fisher Scientific) supplemented with 0.1% BSA, without antibiotics at 37°C. All experiments were repeated independently 3 times, each performed in triplicates.
Immunoblotting
Flash frozen livers were homogenized using TissueLyser (Qiagen) in RIPA buffer containing protease inhibitors (Thermoscientific Pierce complete protease and phosphatase inhibitors (ThermoFisher, A32963 and 78420), 1 tablet/10 ml RIPA buffer). Bradford protein quantification was performed (Biorad, 5000006) for liver lysates. LX2 cells were directly collected in 2x Laemmli buffer (Biorad, 1610737). 50 μg liver protein or 10μL of LX2 lysate were analyzed by SDS-PAGE and immunoblotting with antibodies to hNTRK3 (Biotechne, AF373), mNTRK3 (Biotechne AF1404), αSMA (Abcam, ab5694), pERK (Cell Signaling, 4370S) and CALNEXIN (Abcam, ab75801).
Picrosirius red staining and artificial intelligence-based collagen quantification in liver sections
Liver was fixed in 10% formalin buffer and paraffin embedded liver tissues were sectioned using a 4 μm microtome. Slides containing tissue sections were baked at 60°C for 1 hour and rehydrated through xylene followed by graded ethanol (100%, 95%, 85% and 70%) into distilled water and processed for picrosirius red/Fast green staining. For collagen staining, rehydrated slides were stained for one hour in saturated picric acid with 0.1% Sirius Red (Direct Red-80; Sigma-Aldrich) followed by counterstain with 0.01% Fast Green (Sigma-Aldrich) for another hour. The slides were removed from the stain, rinsed in water, and rapidly dehydrated through graded ethanol (70%, 85%, 95%, and 100%) followed by xylene and finally placed on cover slips in Permount (ThermoFisher). Whole slides with stained sections were digitally scanned in an Aperio AT2 digital scanner (Leica Biosystems Inc.) at 40X (0.221 micron per pixel).
FibroNest was used for the quantification of Sirius red-positive collagen fibers from each image, where Sirus-red positive pixels are used to detect collagen. The whole-tissue fibrosis phenotype was described for its collagen content and structure (12 traits), the morphometric traits of the segmented collagen fibers (12), and fibrosis architecture traits (7). The collective distribution of each trait were quantified with 7 statistical parameters (quantitative Fibrosis Traits, qFTs) to account for severity, progression, distortion, and variance for both fine and assembled collagens, resulting in a total of 315 qFTs. Principal qFTs were automatically detected using the mouse FAT-NASH fibrosis progression cohort consisting of 15 liver tissues (Chow mice n=5, 6 weeks FAT-NASH n=2, 12 weeks FAT-NASH mice n=5, 24 weeks FAT-NASH mice n=5, Fig. 3D,E)). Automatically selected principal qFTs were normalized and combined to form a continuous composite score for fibrosis severity in each phenotypic layer (collagen content, fibers morphometry, fibrosis architecture) and for the whole fibrosis phenotype (Ph-CFS). In addition, Collagen fibers were classified as “fine” or “assembled” based on the complexity of their skeleton, and the morphometric phenotypes can be quantified for each subgroup. The relative changes of each individual qFTs between animals and groups were visualized in the form of a heatmap.
Single nucleus RNA-seq and data processing
For snRNA-seq of mouse samples and human samples collected at Mount Sinai (CTRL 1-3, NASH 6-9), 40-60mg of total liver tissue was chopped on ice in 1mL of 0.03% TST buffer (146mM NaCl, 10mM Tris-HCl pH 7.5, 1mM CaCl2, 21mM MgCl2 with freshly added 0.03% Tween-20, 0.01%BSA, and 0.4U/mL Ribolock RNase Inhibitor (Thermo Fisher Scientific FEREO0382) (30) with Tungsten Noyes Spring Scissors (FST 15514-12). The resulting nuclei suspension was then filtered through 40μm cell strainers (Thermo Fisher Scientific 352340) into a fresh 50mL conical tube on ice. 1 more mL of 0.03% TST was used to rinse the cell strainer and 3mL of ST buffer was added to the nuclei suspension followed by briefly mixing by gentle flicking. The nuclei suspensions were spun at 500xg for 5 minutes at 4°C in a swing-bucket centrifuge. The final nuclei pellet was suspended in 200μL of 0.4% BSA in PBS by pipetting 30 times with a regular p1000 tip. SnRNAseq datasets from 2 control mouse livers (“Chow_1” and “Chow_2) can be found at GEO accession #GSE212327. For “NASH” and “NASH_HCC” samples, 20mg of liver from 3 FAT-NASH mice were pooled for each sample and processed together in an attempt to average out heterogeneity associated with the diseased liver.
For snRNA-seq of human samples collected at Gordian (NASH 1-5), frozen human liver tissue (~60 mm3) was lysed by Dounce homogenization (Kimble 2mL: 20 times with Pestle A over ~60 seconds) in 2mL lysis buffer (10mM Tris pH 7.0, 10mM NaCl, 3mM MgCl2, 0.05% Triton X-100, 0.13% RNAse Inhibitor (Enzymatics Y9240L), 0.25% Superase RNAse Inhibitor (Thermo Fisher Scientific AM2694) with 45μM Actinomycin D, then dripped through a 40μm cell strainer (Celltreat 229481) into a protein low-binding 15mL centrifuge tube and the filter was washed into the collection tube with another 2mL of lysis buffer. After 5 minutes total exposure time to lysis buffer, samples were spun for 2 minutes at 300xg at 4°C; the supernatant was aspirated from the top first to remove fat, then the nuclei-containing pellet was resuspended in 5mL of wash buffer (DPBS + 2% BSA + 0.13% Enzymatics RNAse Inhibitor + 0.25% Superase RNAse Inhibitor + 3μM Actinomycin D) and filtered again through a 35μm strainer into a 5mL FACS tube. Nuclei were sorted into a 1.7mL a protein low-binding microcentrifuge tube pre-coated with 900μL of wash buffer on a Biorad S3e FACS using FSC/SSC > FSC-H vs FSC-W > FL4 vs. SSC gating to eliminate debris. Nuclei were pelleted at 300xg for 3 minutes at 4°C in a swing-bucket centrifuge and resuspended in wash buffer for 10x capture using an LT200 pipettor.
Nuclei preparations were processed by the Chromium 3′ Gene Expression V2 Kit (for mouse samples) or Chromium 3′ Gene Expression V3 Kit (for human samples) according to the manufacturer’s guidelines. Qubit 3 (Fisher Scientific) and 2100 Bioanalyzer (Agilent Technologies) were used for quality check of cDNA. Libraries were sequenced on the NovaSeq at Sinai or NextSeq 550 at Gordian. To generate a count matrix, the sequenced files from each independent sample were processed through 10X Genomics Cell Ranger software v6. The raw base call files were demultiplexed using Cell Ranger mkfastq pipeline to generate FASTQ files. Cellranger count pipeline was applied to the FASTQs to perform alignment against modified transcriptomes based on the mm10 and GRCh38 reference builds for mice and humans respectively that contain introns to increase mapping efficiency and the number of genes detected.
Filtered feature-barcode matrices from Cell Ranger were subsequently run through a standard Seurat pipeline for quality control. Different QC parameters were used for snRNA-seq data generated using Chromium 3′ Gene Expression V2 (mouse samples) and V3 (human samples) Kits using Seurat. For snRNA-seq generated from mouse samples, we included nuclei that fit the following criteria: 1) total number of expressed genes greater than 300 and less than 6000, 2) < 5% of which annotated as mitochondrial, and 3) total counts greater than 500 and less than 15000.
Bulk RNA-seq and data processing
Bulk RNA-seq from LX2 cells was carried out by a commercial vendor (Novogene). Briefly, RNA-seq libraries from LX-2 cells were prepared with polyA capture. RNA-seq libraries were prepared according to the Illumina NovaSeq RNA sample preparation protocol. RNA pooled from 2 wells on 24-well plates was used to generate libraries, which were analyzed on an Agilent 2100 Bioanalyzer. 150 base-pair paired-end reads that passed quality control were aligned to reference genome GRCh38. After alignment, read counts were generated and analyzed using the DESeq2 for differential gene expression. Three biological replicates were adopted for all experiments.
Dimensionality reduction, clustering, and visualization
For mouse snRNA-seq datasets we normalized and transformed the filtered count matrix as per standard Seurat pipeline. We performed dimension reduction using the top 2000 highly variable genes, performed PCA on the top 30 principle components and clustered using Louvain community detection algorithm with resolution of 0.3. Mouse samples from different conditions were merged using SeuratWrappers/LIGER into a single dataset for downstream data analysis. Processing of human snRNA-seq datasets are shown in accompanying scripts from Jupyter notebook. We identified cluster-specific gene expression by differential gene expression of nuclei in the cluster versus all other nuclei using Wilcoxon rank sum test and manually assigned cell types based on top differentially expressed genes. We visualized the reduced dimensionality data using the same principal components as previously used for clustering and UMAP. For Gene Ontology analysis, differentially expressed genes between cell clusters (Wilcoxon rank sum test, Padj. < 0.05) were input into AmiGO2 web server (http://amigo.geneontology.org/amigo) and all significantly enriched “PANTHER GO-Slim Biological Processes)” pathways (FDR < 0.05) with more than 1 gene reported.
Predicting and visualizing cell-cell interactions
Filtered and normalized feature-barcode matrices of mouse and human snRNA-seq datasets along with cell type annotation files were exported from Seurat/R and input into CellphoneDB/Python (31). Mouse gene names were converted to orthologous human gene names based on the Ensembl database, only genes with unique human orthologues were kept for downstream analysis. Default in put parameters were used for statistical analysis. For mouse datasets, significant autocrine receptor-ligand interactions (pvalue < 0.05) for all cell clusters are shown in Data file S4. For human datasets, significant autocrine ligand-receptor interactions (pvalue < 0.05) for all cell clusters from CTRL or patients with NASH are shown in Data file S5, respectively. Cytoscape was used visualize the number of ligand-receptor interactions between cell types, with the thickness of the connecting line directly proportional to the number of significant interactions between each pair of interacting cell types.
Statistical analysis
Results are shown as means ± standard deviation unless indicated otherwise. Statistical analysis was performed using Prism unless otherwise specified. Student’s t-test is used when comparing two groups with similar variance (F-test performed to compare variance, P > 0.05). Analysis involving multiple groups was performed using one-way or two-way ANOVA, followed by post hoc t test.
Supplementary Material
Table S1. NASH patient information
Table S2. Differentially expressed genes in human NASH stellate cells
Table S3. Differentially expressed genes in mouse NASH stellate cells
Table S4. CellphoneDB predicted significant receptor-ligand interactions from CTRL mouse snRNA-seq data
Table S5. CellphoneDB predicted significant receptor-ligand interactions from 24 weeks FAT-NASH mouse snRNA-seq data
Table S6. CellphoneDB predicted significant receptor-ligand interactions from CTRL patient snRNA-seq data
Table S7. CellphoneDB predicted significant receptor-ligand interactions from NASH patient snRNA-seq data
Movie S1. HSCs and endothelial cells in iDISCO cleared control livers imaged using confocal microscopy
Movie S2. HSCs and endothelial cells in iDISCO cleared control livers imaged using lightsheet microscopy
Movie S3. HSCs in iDISCO cleared chow livers imaged using confocal microscopy
Movie S4. HSCs in iDISCO cleared 24 weeks FAT-NASH livers imaged using confocal microscopy
Fig. S1. Additional analyses from CTRL and NASH patient snRNAseq data
Fig. S2. Additional analyses from CTRL and NASH mouse snRNAseq data
Fig. S3 Overview of the liver clearing and 3D imaging pipeline
Fig. S4. Effect of NTRK3 knockdown or pharmacological inhibition on LX2 cell viability or proliferation
Fig. S5. Additional data from LOXO-195 treated FAT-NASH mice
Fig. S6. Liver clearing and 3D imaging of DESMIN staining in vehicle or LOXO-195 treated FAT-NASH mice
Acknowledgments:
We thank Jill Gregory at Mount Sinai for illustrations, and Nikos Tzavaras and Deanna L. Benson at the Mount Sinai Microscope Core for assistance with Confocal and Lightsheet imaging; Kristin Beaumont, Robert Sebra, and their team at the Mount Sinai Genomics Core for assistance with single nucleus RNA sequencing; the Biorepository and Pathology CoRE for procurement of human tissue samples at Mount Sinai. We express our gratitude to all of the anonymous organ donors and their families for giving both the gift of life and the gift of knowledge by their generous donations. We thank Donor Network West for their cooperation in this project. We thank Huayi Zhao for technical support. Microscopy and image analysis was performed at the Microscopy and Advanced Bioimaging CoRE at the Icahn School of Medicine at Mount Sinai.
Funding:
Funding was provided by National Institutes of Health grants R01DK56621, R01DK128289, TR004419, and P30CA196521 (SLF); Scleroderma Research Foundation (RM); Wellcome Trust Senior Research Fellowship in Clinical Science 219542/Z/19/Z (NCH);
American Gastroenterological Association Research Scholar AGA2020-13-03 and Pilot grant AGA2021-21-02 (SW); Cancer Research UK C19767/A27145 (UA);
National Institutes of Health grants R01DK101737, R01DK099205, R01DK111866, R01AA028550, P50AA011999, P30 DK120515, U01AA029019 (TK); And a Fulbright Scholar Fellowship, the Zuckerman STEM leadership program, the Israel National Postdoctoral Award Program for Advancing Women in Science, and the European Molecular Biology Organization (EMBO) Long Term Fellowship (ALTF 304-2019) (MA).
Footnotes
Competing interests: Chris Carrico, Ian Driver, and Martin Borch Jensen are employees of Gordian Biotechnology, CA, USA. Scott Friedman is a consultant to Gordian Biotechnology, CA, USA. Scott Friedman and Shuang Wang are inventors on United States provisional patent application 63/345,236 submitted by Icahn School of Medicine at Mount Sinai claiming the use of neurotrophic tyrosine receptor kinase (Trk) inhibitors for liver disease and other diseases. Dr. Friedman is a consultant to Gordian Biotechnology. The other authors declare that they have no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials. Jupyter notebooks and scripts for processing and integrating the human patient data are available on zenodo at DOI 10.5281/zenodo.7348604.are available a All count matrices and metadata for each sample are publicly available in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under data accession no. GSE212837.
References and Notes
- 1.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwabe RF, Tabas I, Pajvani UB, Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 158, 1913–1928 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F, Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 149, 389–397 e310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, Eslam M, Gonzalez-Fabian L, Alvarez-Quiñones Sanz M, Conde-Martin AF, De Boer B, McLeod D, Hung Chan AW, Chalasani N, George J, Adams LA, Romero-Gomez M, Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 155, 443–457.e417 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Loomba R, Friedman SL, Shulman GI, Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YA, Friedman SL, Reversal, maintenance or progression: What happens to the liver after a virologic cure of hepatitis C? Antiviral Research 107, 23–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lassailly G, Caiazzo R, Ntandja-Wandji L-C, Gnemmi V, Baud G, Verkindt H, Ningarhari M, Louvet A, Leteurtre E, Raverdy V, Dharancy S, Pattou F, Mathurin P, Bariatric Surgery Provides Long-term Resolution of Nonalcoholic Steatohepatitis and Regression of Fibrosis. Gastroenterology 159, 1290–1301.e1295 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, Medzhitov R, Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757.e717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler M, Mayo A, Zhou X, Franklin RA, Meizlish ML, Medzhitov R, Kallenberger SM, Alon U, Principles of Cell Circuits for Tissue Repair and Fibrosis. iScience 23, 100841 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC, Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nature Reviews Gastroenterology & Hepatology 17, 457–472 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, Bujko A, Martens L, Thoné T, Browaeys R, De Ponti FF, Vanneste B, Zwicker C, Svedberg FR, Vanhalewyn T, Gonçalves A, Lippens S, Devriendt B, Cox E, Ferrero G, Wittamer V, Willaert A, Kaptein SJF, Neyts J, Dallmeier K, Geldhof P, Casaert S, Deplancke B, ten Dijke P, Hoorens A, Vanlander A, Berrevoet F, Van Nieuwenhove Y, Saeys Y, Saelens W, Van Vlierberghe H, Devisscher L, Scott CL, Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e338 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, Friedman SL, A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69, 385–395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carter JK, Bhattacharya D, Borgerding JN, Fiel MI, Faith JJ, Friedman SL, Modeling dysbiosis of human NASH in mice: Loss of gut microbiome diversity and overgrowth of Erysipelotrichales. PLOS ONE 16, e0244763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niki T, Pekny M, Hellemans K, De Bleser P, Van Den Berg K, Vaeyens F, Quartier E, Schuit F, Geerts A, Class VI intermediate filament protein nestin is induced during activation of rat hepatic stellate cells. Hepatology 29, 520–527 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Knittel T, Aurisch S, Neubauer K, Eichhorst S, Ramadori G, Cell-type-specific expression of neural cell adhesion molecule (N-CAM) in Ito cells of rat liver. Up-regulation during in vitro activation and in hepatic tissue repair. The American journal of pathology 149, 449–462 (1996). [PMC free article] [PubMed] [Google Scholar]
- 16.Cassiman D, van Pelt J, De Vos R, Van Lommel F, Desmet V, Yap S-H, Roskams T, Synaptophysin: A Novel Marker for Human and Rat Hepatic Stellate Cells. The American Journal of Pathology 155, 1831–1839 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neubauer K, Knittel T, Aurisch S, Fellmer P, Ramadori G, Glial fibrillary acidic protein - a cell type specific marker for Ito cells in vivo and in vitro. Journal of Hepatology 24, 719–730 (1996). [DOI] [PubMed] [Google Scholar]
- 18.Renier N, Wu Z, Simon David J., Yang J, Ariel P, Tessier-Lavigne M, iDISCO: A Simple, Rapid Method to Immunolabel Large Tissue Samples for Volume Imaging. Cell 159, 896–910 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao X-Y, Ji Y, Li C, Guo L, Zhou L, Chen Z, Leon-Mimila P, Chung MT, Kurabayashi K, Opp J, Campos-Pérez F, Villamil-Ramírez H, Canizales-Quinteros S, Lyons R, Lumeng CN, Zhou B, Qi L, Huertas-Vazquez A, Lusis AJ, Xu XZS, Li S, Yu Y, Li JZ, Lin JD, Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Molecular Cell 75, 644–660.e645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucker RD, Ciofoaia V, Nadella S, Gay MD, Cao H, Huber M, Safronenka A, Shivapurkar N, Kallakury B, Kruger AJ, Kroemer AHK, Smith JP, A Cholecystokinin Receptor Antagonist Halts Nonalcoholic Steatohepatitis and Prevents Hepatocellular Carcinoma. Digestive Diseases and Sciences 65, 189–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, Smith S, Lauriault V, Kolakowski GR, Brandhuber BJ, Larsen PD, Bouhana KS, Winski SL, Hamor R, Wu W-I, Parker A, Morales TH, Sullivan FX, DeWolf WE, Wollenberg LA, Gordon PR, Douglas-Lindsay DN, Scaltriti M, Benayed R, Raj S, Hanusch B, Schram AM, Jonsson P, Berger MF, Hechtman JF, Taylor BS, Andrews S, Rothenberg SM, Hyman DM, A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discovery 7, 963–972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cassiman D, Denef C, Desmet VJ, Roskams T, Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 33, 148–158 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Suzuki K, Tanaka M, Watanabe N, Saito S, Nonaka H, Miyajima A, p75 Neurotrophin Receptor Is a Marker for Precursors of Stellate Cells and Portal Fibroblasts in Mouse Fetal Liver. Gastroenterology 135, 270–281.e273 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Passino Melissa A, Adams Ryan A, Sikorski Shoana L, Akassoglou K, Regulation of Hepatic Stellate Cell Differentiation by the Neurotrophin Receptor p75NTR. Science 315, 1853–1856 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Marsh E, Gonzalez DG, Lathrop EA, Boucher J, Greco V, Positional Stability and Membrane Occupancy Define Skin Fibroblast Homeostasis In Vivo. Cell 175, 1620–1633.e1613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, Driskell RR, Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. eLife 9, e60066 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, Modrusan Z, Arron JR, Bourgon R, Müller S, Turley SJ, Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Eraslan G, Drokhlyansky E, Anand S, Fiskin E, Subramanian A, Slyper M, Wang J, Van Wittenberghe N, Rouhana JM, Waldman J, Ashenberg O, Lek M, Dionne D, Win TS, Cuoco MS, Kuksenko O, Tsankov AM, Branton PA, Marshall JL, Greka A, Getz G, Segrè AV, Aguet F, Rozenblatt-Rosen O, Ardlie KG, Regev A, Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ, Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54, 142 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, Vigneau S, Jané-Valbuena J, Tickle TL, Napolitano S, Su M-J, Patel AG, Karlstrom A, Gritsch S, Nomura M, Waghray A, Gohil SH, Tsankov AM, Jerby-Arnon L, Cohen O, Klughammer J, Rosen Y, Gould J, Nguyen L, Hofree M, Tramontozzi PJ, Li B, Wu CJ, Izar B, Haq R, Hodi FS, Yoon CH, Hata AN, Baker SJ, Suvà ML, Bueno R, Stover EH, Clay MR, Dyer MA, Collins NB, Matulonis UA, Wagle N, Johnson BE, Rotem A, Rozenblatt-Rosen O, Regev A, A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine 26, 792–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nature Protocols 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. NASH patient information
Table S2. Differentially expressed genes in human NASH stellate cells
Table S3. Differentially expressed genes in mouse NASH stellate cells
Table S4. CellphoneDB predicted significant receptor-ligand interactions from CTRL mouse snRNA-seq data
Table S5. CellphoneDB predicted significant receptor-ligand interactions from 24 weeks FAT-NASH mouse snRNA-seq data
Table S6. CellphoneDB predicted significant receptor-ligand interactions from CTRL patient snRNA-seq data
Table S7. CellphoneDB predicted significant receptor-ligand interactions from NASH patient snRNA-seq data
Movie S1. HSCs and endothelial cells in iDISCO cleared control livers imaged using confocal microscopy
Movie S2. HSCs and endothelial cells in iDISCO cleared control livers imaged using lightsheet microscopy
Movie S3. HSCs in iDISCO cleared chow livers imaged using confocal microscopy
Movie S4. HSCs in iDISCO cleared 24 weeks FAT-NASH livers imaged using confocal microscopy
Fig. S1. Additional analyses from CTRL and NASH patient snRNAseq data
Fig. S2. Additional analyses from CTRL and NASH mouse snRNAseq data
Fig. S3 Overview of the liver clearing and 3D imaging pipeline
Fig. S4. Effect of NTRK3 knockdown or pharmacological inhibition on LX2 cell viability or proliferation
Fig. S5. Additional data from LOXO-195 treated FAT-NASH mice
Fig. S6. Liver clearing and 3D imaging of DESMIN staining in vehicle or LOXO-195 treated FAT-NASH mice
Data Availability Statement
All data are available in the main text or the supplementary materials. Jupyter notebooks and scripts for processing and integrating the human patient data are available on zenodo at DOI 10.5281/zenodo.7348604.are available a All count matrices and metadata for each sample are publicly available in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under data accession no. GSE212837.