

Abstract
Processing of CASP15 targets into evaluation units (EUs) and assigning them to evolutionary-based prediction classes is presented in this study. The targets were first split into structural domains based on compactness and similarity to other proteins. Models were then evaluated against these domains and their combinations. The domains were joined into larger EUs if predictors’ performance on the combined units was similar to that on individual domains. Alternatively, if most predictors performed better on the individual domains, then they were retained as EUs. As a result, 112 evaluation units were created from 77 tertiary structure prediction targets. The EUs were assigned to four prediction classes roughly corresponding to target difficulty categories in previous CASPs: TBM (template-based modeling, easy or hard), FM (free modeling), and the TBM/FM overlap category. More than a third of CASP15 EUs were attributed to the historically most challenging FM class, where homology or structural analogy to proteins of known fold cannot be detected.
Keywords: CASP15, protein structure, protein structure prediction, protein domains, evaluation units
1 |. INTRODUCTION
CASP has been monitoring progress in the protein tertiary structure prediction for over 25 years1–4. Every other year since 1994, CASP organizers contact a wide network of structural biologists in quest of targets for the upcoming protein structure modeling experiment. The latest CASP15 call yielded 93 single-sequence entries representing monomeric proteins or subunits of protein multimeric complexes (https://predictioncenter.org/casp15/targetlist.cgi?view=regular). Eighty-one entries on this list were suggested for tertiary structure prediction, while the remaining twelve were auxiliary structures for other prediction categories (see Table 1). Four targets out of the 81 could not be taken forward since an experimental structure was not available at the time, leaving 77 for the assessment. Below we discuss procedures for splitting these targets into evaluation units (EUs) and assigning them to evolutionary-based prediction classes.
Table 1.
CASP15 tertiary structure prediction targets, their split into evaluation units (EUs) and classification to homology-based prediction classes. Canceled targets are highlighted in red; targets that were released as auxiliary structures for other prediction categories (ligand, oligo, protein-RNA complex) are in yellow.
| Target | Exper. method | Number of struct. domains | Number of EUs | EU boundaries | Residues in EU | Classification |
|---|---|---|---|---|---|---|
| T1104 | X-ray | 1 | 1 | T1104-D1: 1-117 | 117 | FM |
| T1105 | X-ray | no structure | ||||
| T1105v1 | X-ray | no structure | ||||
| T1106s1 | X-ray | 1 | 1 | T1106s1-D1: 50-120 | 71 | FM/TBM |
| T1106s2 | X-ray | 1 | 1 | T1106s2-D1: 2-112 | 111 | TBM-easy |
| T1109 | X-ray | 1 | 1 | T1109-D1: 7-209, 216-226 | 214 | TBM-easy |
| T1110 | X-ray | 1 | 1 | T1110-D1: 7-227 | 221 | TBM-easy |
| T1112 | X-ray | 2 | 1 | T1112-D1: 1-460 | 460 | FM/TBM |
| T1113 | X-ray | 1 | 1 | T1113-D1: 1-192 | 192 | FM |
| T1114s1 | EM | 2 | 2 | T1114s1-D1: 20-79 | 60 | FM/TBM |
| T1114s1-D2: 80-189 | 110 | not evaluated | ||||
| T1114s2 | EM | 2 | 1 | T1114s2-D1: 48-369 | 322 | TBM-easy |
| T1114s3 | EM | 1 | 1 | T1114s3-D1: 4-516 | 513 | TBM-easy |
| T1115 | EM | 3 | 2 | T1115-D1: 25-200 | 176 | TBM-easy |
| T1115-D2: 201-272 | 72 | TBM-easy | ||||
| T1118 | X-ray | aux for ligand | ||||
| T1118v1 | X-ray | aux for ligand | ||||
| T1119 | X-ray | 1 | 1 | T1119-D1: 7-54 | 48 | TBM-hard |
| T1120 | X-ray | 2 | 2 | T1120-D1: 8-125 | 118 | FM |
| T1120-D2: 126-235 | 110 | FM | ||||
| T1121 | EM | 2 | 2 | T1121-D1: 2-204 | 203 | FM |
| T1121-D2: 205-381 | 177 | TBM-hard | ||||
| T1122 | X-ray | 1 | 1 | T1122-D1: 4-237 | 234 | FM |
| T1123 | X-ray | 1 | 1 | T1123-D1: 33-258 | 226 | FM/TBM |
| T1124 | X-ray | 2 | 1 | T1124-D1: 7-384 | 378 | TBM-easy |
| T1125 | X-ray | 5 | 6 | T1125-D1: 327-460 | 134 | FM |
| T1125-D2: 461-608 | 148 | FM | ||||
| T1125-D3: 609-797 | 189 | TBM-hard | ||||
| T1125-D4: 798-946 | 149 | FM | ||||
| T1125-D5: 947-1096 | 150 | FM | ||||
| T1125-D6: 1097-1162 | 66 | FM | ||||
| T1127 | X-ray | 1 | 1 | T1127-D1: 6-210 | 205 | TBM-hard |
| T1127v2 | X-ray | aux for ligand | ||||
| T1129s2 | EM | 1 | 1 | T1129s2-D1: 33-640 | 608 | FM |
| T1130 | X-ray | 1 | 1 | T1130-D1: 28-133, 139-195 | 163 | FM |
| T1131 | X-ray | 2 | 1 | T1131-D1: 1-161 | 161 | FM |
| T1132 | X-ray | 1 | 1 | T1132-D1: 5-102 | 98 | TBM-easy |
| T1133 | X-ray | 3 | 1 | T1133-D1: 4-427 | 424 | TBM-easy |
| T1134s1 | X-ray | 2 | 1 | T1134s1-D1: 2-230 | 229 | TBM-easy |
| T1134s2 | X-ray | 1 | 1 | T1134s2-D1: 10-313 | 304 | FM/TBM |
| T1137s1 | EM | 2 | 2 | T1137s1-D1: 20-169 | 150 | TBM-easy |
| T1137s1-D2: 170-409 | 240 | FM | ||||
| T1137s2 | EM | 1 | 2 | T1137s2-D1: 1-149 | 149 | TBM-easy |
| T1137s2-D2: 150-343 | 194 | FM | ||||
| T1137s3 | EM | 1 | 2 | T1137s3-D1: 1-149 | 149 | TBM-easy |
| T1137s3-D2: 150-313 | 164 | FM | ||||
| T1137s4 | EM | 3 | 3 | T1137s4-D1: 44-159 | 116 | TBM-easy |
| T1137s4-D2: 160-394 | 235 | FM | ||||
| T1137s4-D3: 395-468 | 74 | FM | ||||
| T1137s5 | EM | 2 | 2 | T1137s5-D1: 33-169 | 137 | TBM-easy |
| T1137s5-D2: 170-390 | 221 | FM | ||||
| T1137s6 | EM | 2 | 2 | T1137s6-D1: 1-151 | 151 | TBM-easy |
| T1137s6-D2: 152-399 | 248 | FM | ||||
| T1137s7 | EM | 1 | 1 | T1137s7-D1: 1-325 | 325 | TBM-easy |
| T1137s8 | EM | 1 | 1 | T1137s8-D1: 16-266 | 251 | TBM-easy |
| T1137s9 | EM | 1 | 1 | T1137s9-D1: 25-289 | 265 | TBM-easy |
| T1139 | X-ray | 2 | 1 | T1139-D1: 23-317 | 295 | TBM-hard |
| T1145 | X-ray | 4 | 2 | T1145-D1: 4-102 | 99 | TBM-easy |
| T1145-D2: 103-635 | 533 | FM | ||||
| T1146 | X-ray | 1 | 1 | T1146-D1: 29-307 | 279 | TBM-easy |
| T1147 | X-ray | 1 | 1 | T1147-D1: 12-103 | 92 | TBM-easy |
| T1148 | X-ray | aux for ligand | ||||
| T1150 | X-ray | 1 | 1 | T1150-D1: 3-351 | 349 | FM |
| T1151s2 | X-ray | 1 | 1 | T1151s2-D1: 28-111 | 84 | FM/TBM |
| T1152 | X-ray | 1 | 1 | T1152-D1: 1-46 | 46 | TBM-hard |
| T1153 | X-ray | 1 | 1 | T1153-D1: 3-297 | 295 | TBM-easy |
| T1154 | EM | 4 | 2 | T1154-D1: 30-234 | 205 | FM |
| T1154-D2: 235-1069 | 835 | FM | ||||
| T1155 | NMR | 1 | 1 | T1155-D1: 5-108 | 104 | FM/TBM |
| T1157s1 | EM | 3 | 3 | T1157s1-D1: 1-661 | 661 | TBM-hard |
| T1157s1-D2: 662-757, 1005-1022 | 114 | not evaluated | ||||
| T1157s1-D3: 758-1004 | 247 | not evaluated | ||||
| T1157s2 | EM | 4 | 3 | T1157s2-D1: 1-106 | 106 | TBM-easy |
| T1157s2-D2: 107-323 | 217 | TBM-easy | ||||
| T1157s2-D3: 324-464 | 141 | TBM-easy | ||||
| T1158 | EM | 5 | 2 | T1158-D1: 48-234, 347-394, 409-615, 861-974 | 556 | TBM-easy |
| T1158-D2: 235-346, 692-860, 975-1296 | 603 | TBM-easy | ||||
| T1158v1-4 | EM | aux for ligand | ||||
| T1159 | X-ray | 2 | 1 | T1159-D1: 1-160 | 160 | FM |
| T1160 | X-ray | 1 | 1 | T1160-D1: 5-33 | 29 | TBM-easy |
| T1161 | X-ray | 1 | 1 | T1161-D1: 1-48 | 48 | TBM-easy |
| T1162 | X-ray | 1 | 1 | T1162-D1: 4-28, 59-196 | 163 | TBM-easy |
| T1163 | X-ray | 1 | 1 | T1163-D1: 8-191 | 184 | TBM-easy |
| T1165 | EM | 6 | 6 | T1165-D1: 2-595 | 594 | TBM-easy |
| T1165-D2: 596-1319 | 724 | TBM-hard | ||||
| T1165-D3: 1320-2008 | 689 | TBM-hard | ||||
| T1165-D4: 2049-2130 | 82 | TBM-easy | ||||
| T1165-D5: 2621-3000 | 380 | TBM-easy | ||||
| T1165-D6: 2181-2620 | 440 | TBM-hard | ||||
| T1169 | EM | 7 | 4 | T1169-D1: 1-345 | 345 | FM |
| T1169-D2: 1302-2735 | 1434 | FM | ||||
| T1169-D3: 378-699, 1223-1301 | 401 | TBM-hard | ||||
| T1169-D4: 700-1222 | 523 | FM | ||||
| T1170 | EM | 3 | 2 | T1170-D1: 4-243 | 240 | TBM-easy |
| T1170-D2: 244-315 | 72 | TBM-easy | ||||
| T1173 | X-ray | 2 | 2 | T1173-D1: 1-62 | 62 | TBM-easy |
| T1173-D2: 63-204 | 142 | FM | ||||
| T1174 | X-ray | 2 | 2 | T1174-D1: 1-216 | 216 | FM |
| T1174-D2: 217-338 | 122 | TBM-hard | ||||
| T1175 | X-ray | 1 | 1 | T1175-D1: 1-312 | 312 | TBM-hard |
| T1176 | X-ray | 1 | 1 | T1176-D9: 1-138, 139-170 | 170 | TBM-hard |
| T1177 | X-ray | 2 | 1 | T1177-D1: 1-223 | 223 | FM |
| T1178 | X-ray | 1 | 1 | T1178-D1: 17-291 | 275 | FM |
| T1179 | X-ray | 1 | 1 | T1179-D1: 2-253 | 252 | FM |
| T1180 | X-ray | 2 | 1 | T1180-D1: 1-404 | 404 | TBM-hard |
| T1181 | X-ray | 3 | 2 | T1181-D1: 1-88 | 88 | FM/TBM |
| T1181-D2: 89-688 | 600 | FM | ||||
| T1182 | X-ray | 2 | 1 | T1182-D1: 21-544 | 524 | FM |
| T1183 | X-ray | 2 | 1 | T1183-D1: 1-195 | 195 | TBM-easy |
| T1184 | X-ray | 1 | 1 | T1184-D1: 34-63, 73-101, 106-171 | 125 | FM |
| T1185s1 | EM | 1 | 1 | T1185s1-D1: 4-71 | 68 | TBM-easy |
| T1185s2 | EM | 2 | 1 | T1185s2-D1: 11-66, 84-349 | 322 | TBM-easy |
| T1185s4 | EM | 1 | 1 | T1185s4-D1: 20-200, 222-244, 251-280 | 234 | TBM-easy |
| T1186 | X-ray | aux for ligand | ||||
| T1187 | X-ray | 1 | 1 | T1187-D1: 3-166 | 164 | FM |
| T1188 | X-ray | 2 | 1 | T1188-D1: 25-597 | 573 | TBM-easy |
| T1189 | EM | aux for RNA | ||||
| T1190 | EM | aux for RNA | ||||
| T1191 | EM | no structure | ||||
| T1192 | EM | aux for oligo | ||||
| T1193 | EM | no structure | ||||
| T1194 | X-ray | 1 | 1 | T1194-D1: 7-167 | 161 | FM |
| T1195 | NMR | 1 | 1 | T1195-D1: 3-279 | 277 | TBM-easy |
| T1196 | NMR | 1 | 1 | T1196-D1: 9-351 | 343 | TBM-easy |
| T1197 | NMR | 1 | 1 | T1197-D1: 16-277 | 262 | TBM-easy |
Defining and classifying evaluation units in CASP has been a very important and time-consuming task requiring multiple numerical tests and extensive human inspection. In five out of six recent CASPs these tasks were directed by Lisa Kinch, whose involvement with the structural classification ECOD database5 and extensive knowledge of protein structure was an invaluable asset6–10. Based on the knowledge accumulated in previous CASPs, in CASP15 we decided to develop a procedure that would mimic the procedure of previous CASPs while requiring only minimal human intervention. In the end, this allowed us to decrease the time spent on domain definition and classification by approximately fivefold.
2 |. METHODS
2.1 |. Defining evaluation units
Domains are basic folding units of proteins. They can be mobile, and their relative position can deviate depending on environmental conditions (e.g., the presence of a ligand), protein’s functional state (e.g., open or closed) or structure determination factors (e.g., crystal packing). Thus, evaluating models versus a specific domain conformation in a multi-domain structure can be too restrictive and penalizing. To address this issue, CASP adopted a domain-based approach to evaluating models, where multidomain targets are first split into smaller evaluation units. Even though the recent progress in modeling and the availability of non-rigid body structure comparison methods make splitting of targets into EUs less critical, we kept this practice to allow fair comparison of results across CASPs. A detailed procedure is described below.
Step 1: identifying varying regions.
Multi-chain and multi-model targets were checked for structural consistency by superimposing their chains /models using LGA11.
If the distance between the corresponding residues in different chains /models exceeded 3.5 Å, then the residue was marked as varying. Local regions of three or more consecutive varying residues were removed from the target. If varying regions were extensive, but superimposed well when treated separately, then they were organized into separate domains.
Step 2: parsing into domains based on structural compactness.
To define domains from structure, we consulted three automatic domain parsing programs, which identify geometrically compact substructures in a protein based on the analysis of inter-residue contacts and evolutionary preserved substructures - DomainParser12, DDomain13 and SWORD14. The programs were installed at the Prediction Center and run as:
domainparser <TARGET.pdb> ddomain <TARGET.pdb> SWORD -i <TARGET.pdb>
We also consulted a newly developed SWORD2 web server15 for analyzing two particularly large targets with elaborate domain architecture - T1165 and T1169. The SWORD /SWORD2 programs were used in CASP for the first time and proved to be especially helpful (in fact, the only option) for large structures with more than 1200 residues.
If the structure parsers agreed on domain boundaries, the consensus definition was adopted. If the programs disagreed, domain boundaries were defined upon visual inspection, evolutionary analysis (see Step 3), and /or functional information received from experimentalists.
Alternative domain definitions were considered for cases where certain regions of proteins were involved in domain swaps.
Step 3: fine-tuning domain boundaries based on similarity to other proteins.
Whole targets and suggested domains from Step 2 were run through PSI-BLAST16 and HHblits17/HHsearch18 programs to establish sequence-based similarity to proteins of known fold:
psiblast -query <TARGET.seq> -db <pdbaa_PSIBLAST> -num_iterations 3 -evalue 10.0; hhblits -i <TARGET.seq> -d <UniRef30_hhb_db> -oa3m <TARGET.a3m> -n 2 -cov 60 -id 90; hhsearch -i <TARGET.a3m> -d <PDB70_hhs_db> -o <TARGET.hhr>.
The targets were also structurally compared to proteins in the PDB with Foldseek19:
foldseek easy-search <TARGET.pdb> <./db/FSmmcif> --tmscore-threshold 0.25 --max-seqs 500 -e 0.1 -s 9.5 --alignment-type 1;
and then top 100 Foldseek hits were re-checked with LGA:
lga <hit_FS.TARGET.pdb> −4 -ie -o2 -sia -d:4 -gdc_sc -swap.
The templates discovered with PSI-BLAST and HHblits/HHsearch will almost certainly be homologous to the target. However, since Foldseek is sequence-independent and LGA was run in sequence-independent mode, the second step will potentially discover structurally analogous templates as well as homologues too remote for detection by sequence-based searches.
Template-target alignments from these searches were used to adjust domain boundaries. For example, if the domain parsing programs in Step 2 suggest termination of a domain at residue N, but templates covered the target until residue N+i, then the termination point was moved to N+i if this alteration did not contradict the alignment data for the neighboring domain.
Step 4: joining domains into larger evaluation units based on the performance.
Once domains were defined, models were trimmed accordingly and evaluated against the domains and their pair-wise combinations. GDT_TS scores from LGA’s sequence-dependent superpositions served as the numerical basis for deciding whether domains should be kept separate or combined into larger Evaluation Units (EUs) for the final evaluation.
A rationale and numeric procedure for combining domains /splitting targets into evaluation units were suggested by Nick Grishin and coworkers in CASP96. They argued that targets should be split into domains only if this can help reveal interesting predicted features in models. Rephrasing this postulate for the bottom-to-top approach (split first, then consider re-joining), domains should be merged if their separate evaluation does not provide additional benefits for the assessment. A good indicator of this scenario is the similarity of model accuracy scores on the combined and individual domains. To facilitate the decision-making, Kinch et al6 plotted GDT_TS scores for combined domains versus the weighted sum of scores for individual domains. Such a graph became later known as the Grishin plot and was adopted for defining EUs in subsequent CASPs6–10,20. If the points in such a graph line up close to the diagonal line, then joining a pair of domains into a larger evaluation unit is advised.
In CASP15, domains were joined if the slope of the zero-intercept best fit line in a Grishin plot was <1.2. Three or more domains were joined into one EU when the plots for all pairwise domain combinations supported the merger.
The process was repeated iteratively until no further combining of EUs was needed.
2.2 |. Classifying evaluation units into evolutionary prediction classes
Historically, the outcome of a protein structure modeling exercise was largely predetermined by the evolutionary relationship between the target and experimentally determined structures. Proteins with apparent homology to available structures were typically easier to model, while non-homology targets were at the harder side of the prediction difficulty spectrum. Since targets of different difficulty required different modeling approaches, yielded different degrees of model accuracy, and thus required different evaluation approaches, CASP had previously assessed modeling results separately for different target difficulties. The names of the difficulty categories changed with time, but the major factor defining the difficulty remained the same: availability of structural templates. The classical difficulty schema was shaken in CASP14, where the DeepMind group showed that highly accurate models can be built with AlphaFold 2 (AF2) for practically all targets, independently of the template availability. This breakthrough suggested that the classical division into largely homology-based difficulty categories may not be needed any more. Acting upon these developments, CASP organizers recommended assessment of tertiary structure prediction in CASP15 in one batch. This analysis is presented elsewhere in this issue21. Nevertheless, similarly to splitting targets into EUs (above), the assignment of EUs to evolutionary prediction classes is still needed for comparing CASP15 results with the earlier ones.
In previous CASPs, EUs were classified into difficulty categories based on the availability of similar structures in the PDB, as detected by sequence- and structure-based searches (reflecting estimated difficulty) and predictors’ performance (reflecting actual difficulty)9,10. Since performance has become more uniform across the whole range of targets, it is no longer useful for their discrimination. To adapt to the situation, we explored automated approaches to target classification, aiming to recapitulate the outcomes of previous CASPs as far as possible, but working solely with the results of automated PDB searches. Each EU was assigned a sequence-based and structure-based similarity score. The sequence-based score was defined as the HHscore10, which is the product of the HHsearch probability and the alignment coverage of the query for the top-ranked template identified by HHsearch. The structure-based score was the LGA_S score of the highest-ranked structural match according to the procedure described in section 2.1 , Step 3. These scores were used to automatically assign EUs to prediction classes (see Results, section 3.2).
3 |. RESULTS
3.1 |. To split or not to split
3.1.1 |. Summary
From among 77 CASP15 tertiary structure prediction targets, 43 were one-domain targets, 21 had two domains, and the rest - three domains or more (Table 1). For 52 targets no domain rearrangement was necessary, and the targets were evaluated as whole-length structures (41) or unchanged constituent domains (11). For the remaining 25 targets, in 20 cases we merged at least some domains according to Grishin plots, in two cases we merged domains according to other considerations, and in three cases we split targets in more EUs than suggested by the domain parsing programs. The domain splitting and re-joining procedure (Methods) yielded 112 evaluation units, 109 of which were included into the final tertiary structure evaluation21, while three – T1114s1-D2, T1157s1-D2 and -D3 – were cancelled due to the low resolution of the cryo-EM maps in their local areas.
Out of 34 multi-domain targets, 14 were evaluated as one EU and 20 were split into multiple EUs (Table 1). Below we discuss different scenarios of forming evaluation units and present case studies for some targets.
3.1.2 |. Multidomain-targets not requiring splitting (14)
Fourteen multi-domain targets (as defined by the automatic parsers - section 2.1, Step 2) were proposed for the evaluation without splitting into substructures.
In two cases, T1131 and T1133, we disagreed with the automatic domain parsing results and considered the targets as one-domain structures. Target T1131 is a small protein where a long central helix holds two parts of the structure together and is needed for the structural integrity of the protein; while target T1133 (PDB: 8DYS) is a nine-bladed beta-propeller that is fully and reliably covered by templates (e.g., 3WJ9_B) and well-predicted as the whole.
For eleven targets a decision to join domains into single EUs was reached based on the analysis of Grishin plots. Two examples of such targets are shown in Figure 1. Even though the targets are clearly two-domain entities, their whole structures were predicted by most groups as accurately as the constituent domains and thus did not require splitting.
Figure 1.
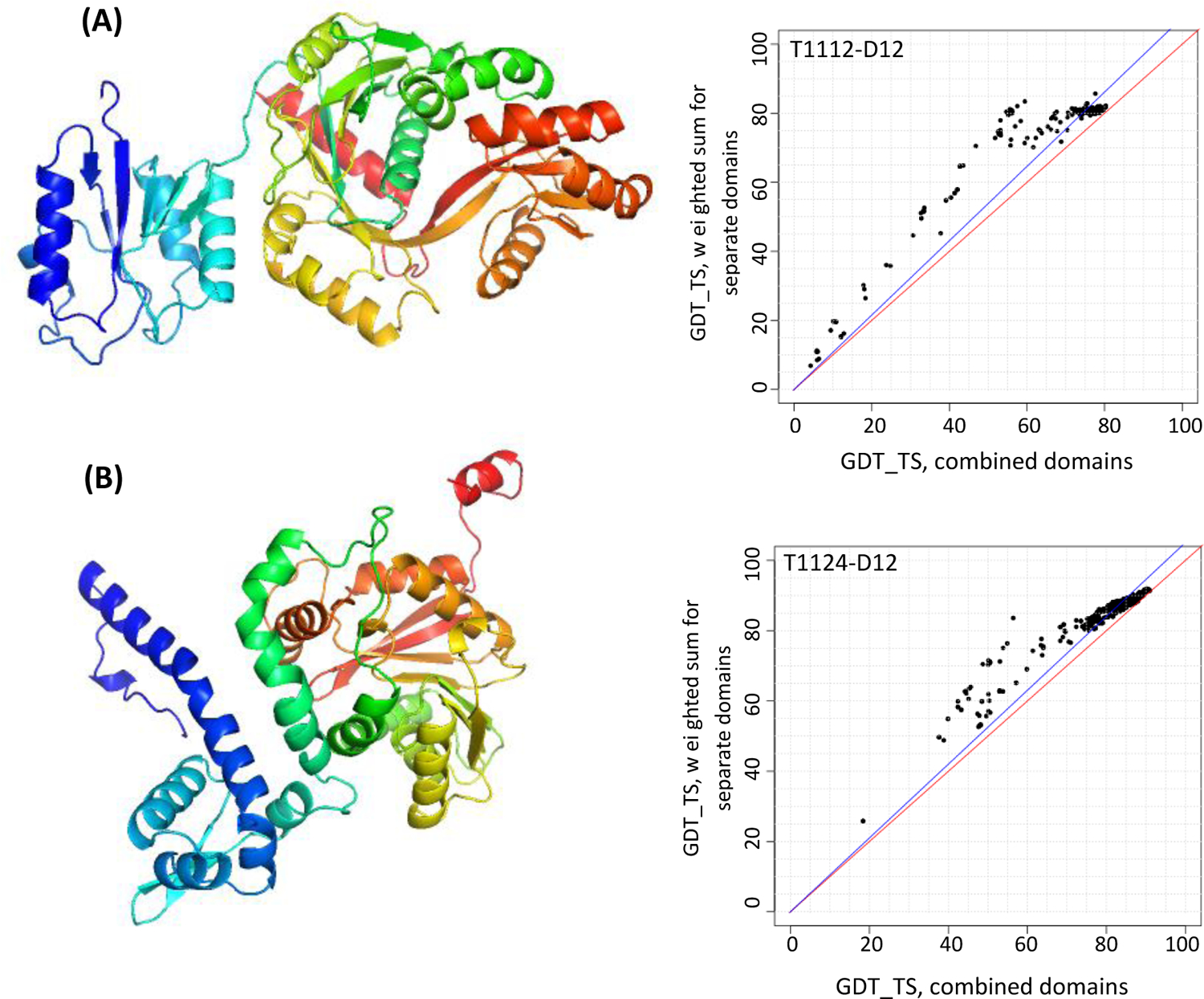
PyMOL22 target renderings (left) and Grishin plots (right) for two two-domain targets: (A) target T1112, a protein involved in the synthesis of an osmolyte involved in thermoadaptation, and (B) target T1124, a methyltransferase MfnG (PDB: 7UX8). Grishin plots are built on the GDT_TS scores for all collected models. The plots suggest evaluating domains together as the angle between the data trend line and the diagonal is small (i.e., the evaluation scores for the combined domains (X-axis) and individual domains (Y-axis) are similar for most groups).
The last target in this category, T1180, is an exception to the splitting rule (section 2.1). Even though the Grishin plot advised splitting, we did not proceed with that as the target is a fusion enzyme of two known domains, where the only prediction interest was to model inter-domain orientation.
3.1.3 |. Multidomain-targets requiring splitting (20)
For half of the 20 targets that required splitting, the number of EUs was determined by the number of structural domains (no merging was necessary), and for the other half, some domains but not all were joined.
In several cases splitting was required because different chains exhibited different folding patterns.
For example, target T1120, a DNA-binding protein DdrC, is composed of an N-terminal winged helix-turn-helix motif and a C-terminal four-helix bundle, that folds as an asymmetric domain-swapped dimer (Figure 2A)23. Superposition of the two chains revealed the distortion of the long central helix hA in chain A (cyan) into two smaller, non-colinear helices (hB1 and hB2) in chain B (green) and cause a shift in the relative position of the C-terminal domain with regards to the anchor N-terminal domain (Figure 2B). This prompted splitting of the target into two EUs at the break point (residue 125). Such a split is strongly supported by the Grishin plot (Figure 2C).
Figure 2.
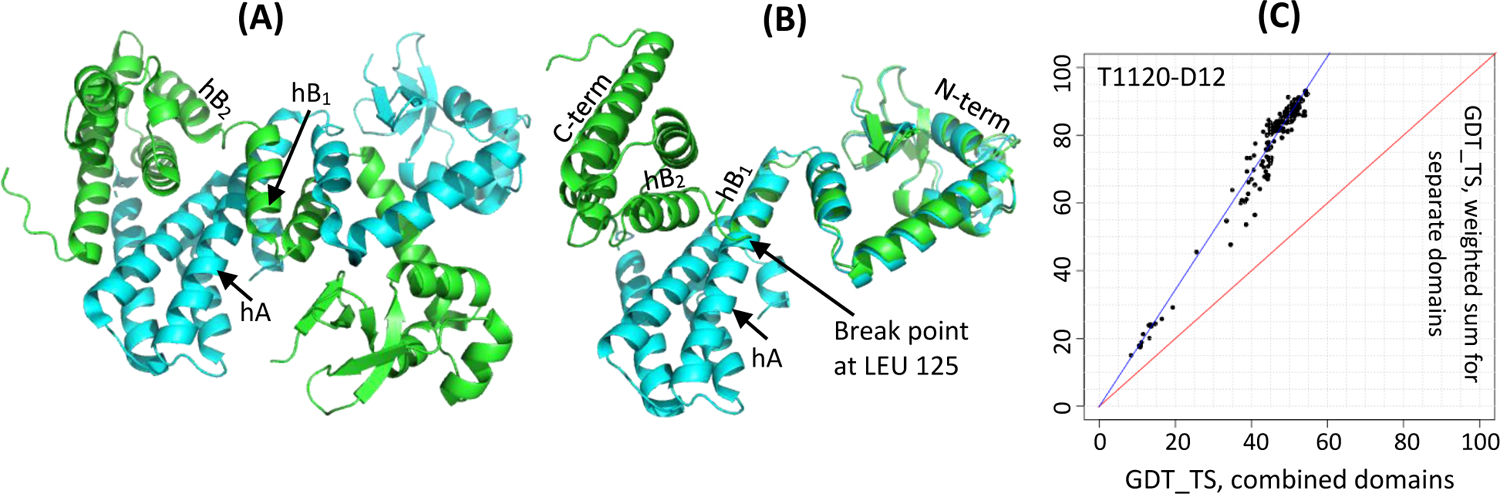
Target T1120, a DNA-binding protein DdrC (PDB: 7QVB). (A) a homodimer with two chains colored as cyan and green; (B) superposition of two chains showing the break point in the helix hA at residue LEU 125; (C) a Grishin plot showing the need for splitting (large angle between the data trend line and the diagonal). The plot was built on the GDT_TS results for all participating groups on the constituent domains D1: 8–125 and D2: 126–235 and the whole target in the chain A configuration.
Another example of such obligated splitting is target T1121, the Wadjet nuclease subunit JetD24. It is a homo-dimeric protein (Figure 3A) containing two domains that are flexibly linked and whose relative orientation differs in the two chains (Figure 3B). Because of that, the target was split into two EUs at the hinge point (residue 204). As in the previous case, such a split is strongly supported by the performance data (Figure 3C).
Figure 3.
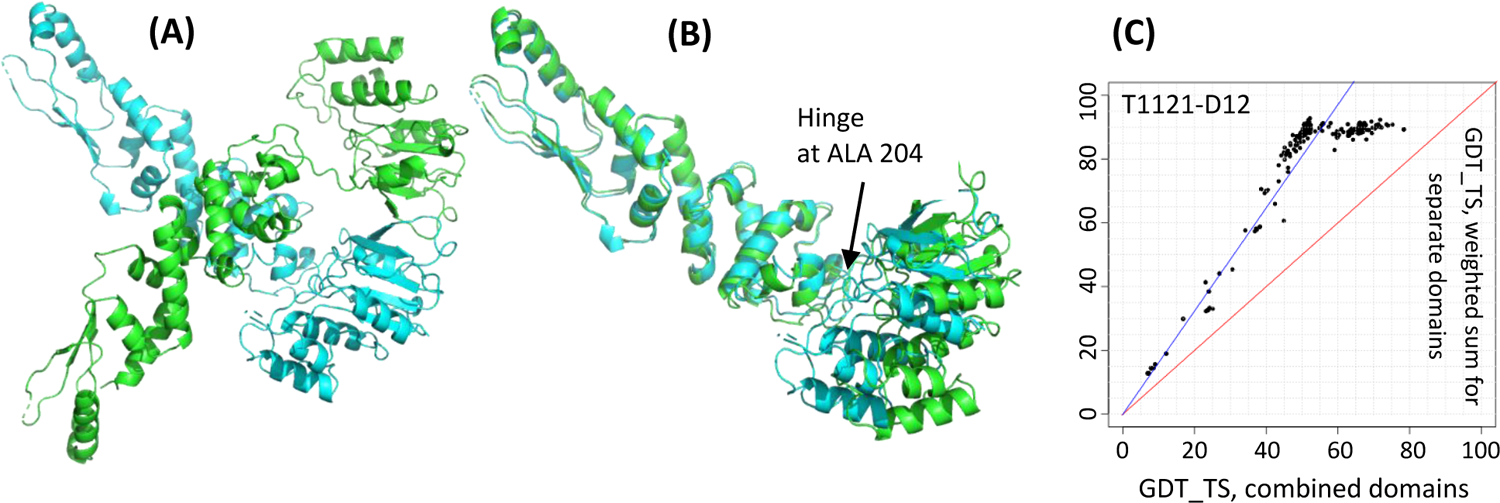
Target T1121, a DNA-cleavage protein JetD (PDB: 7TIL). (A) a homodimer with two chains colored as cyan and green; (B) superposition of its two chains showing flexibility of the C-term domain (Pfam DUF2220, right) with respect to the N-term arm-like domain (DUF3322, left); (C) a Grishin plot showing the need for splitting. The plot was built on the GDT_TS results for all groups on the constituent domains D1: 2–204 and D2: 205–381 and the whole target in the chain A configuration.
The last example in this category is target T1170, a Holliday junction hexamer where some chains deform to accommodate DNA25 in such a way that the overall structures of domains remain largely unchanged, yet their relative position varies (Figure 4AB). Non-crystallographic symmetry of the structure requires separate treatment of parts that have different relative orientation. The target was originally split into three domains (1: 4–164; 2: 165–243; 3: 244–315) and analyzed if any of those need to be merged for the final evaluation. The Grishin plots (Fig 4C) advised that domains 1 and 2 should be merged, while 3 should remain a separate evaluation entity. Thus, for the final evaluation, this target was represented by two EUs: D1: 4–243 and D2: 244–315 (encircled).
Figure 4.
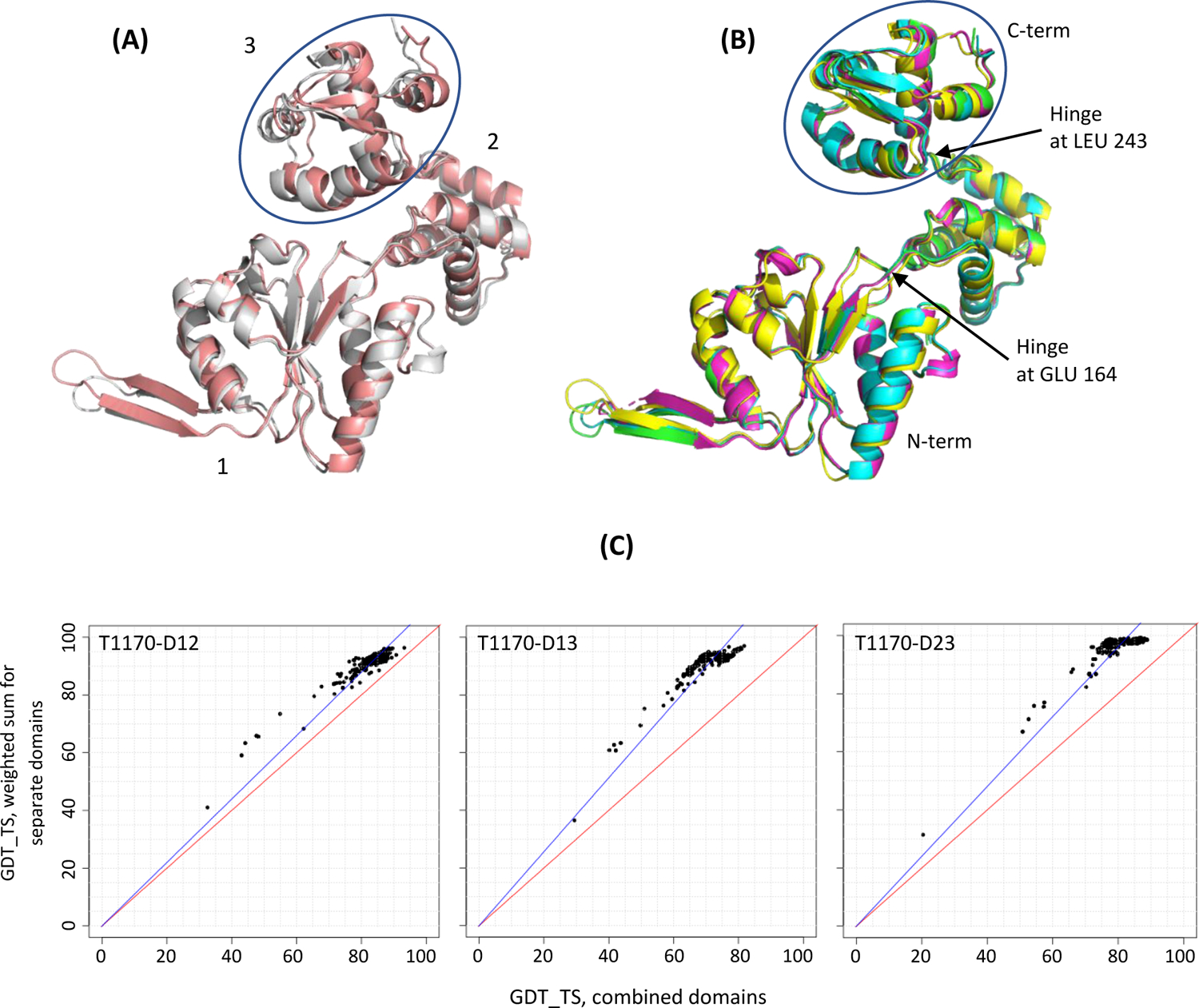
Target T1170, a Holliday junction hexamer (PDB: 7PBR). (A) superposition of two deformed chains versus (B) four undeformed chains in the same frame of reference. The domain that moves the most with respect to other two is encircled. (C) Grishin plots for the original target split into three domains show the similarity of results on domains 1, 2 and their combination 12 (left panel, points close to the diagonal), and the dissimilarity of results on the combined substructures 13 and 23 and their constituent domains (middle and right).
In all other targets, except for T1120, T1121 and T1170 discussed above, chains were largely similar, and the decision on domain splitting was dictated purely by Grishin plots. Below we discuss three cases of some of the most difficult domain rearrangements.
Target T1158 is a type IV ABC transporter, which is a common fold (see review26). In CASP15, this protein family was represented by five targets - T1158 (Figure 5A) and T1158v1-v4, which differ by rigid body movements of the two halves of the transporter with respect to one another, and no significant rearrangements within the subunits (Figure 5B). When submitted to domain parsing programs, T1158 was split in several ways, none of which made functional sense. The suggested split was either too fragmented (6 domains by DDomain, or 5/8/7 by the top three SWORD assignments) or too coarse-grained (2 domains by DomainParser: the C-terminal globular domain (red) in Figure 5A (48–1022) and the rest). We split this target into two EUs (Figure 5C) reflecting the conformational changes that the transporter undergoes performing its biological function of opening and closing gates in bound and unbound states. In other words, evaluation units for T1158 were defined not from a single structure, but from a set of structures from the same superfamily. A Grishin plot for the target (not shown) supports the suggested split.
Figure 5.
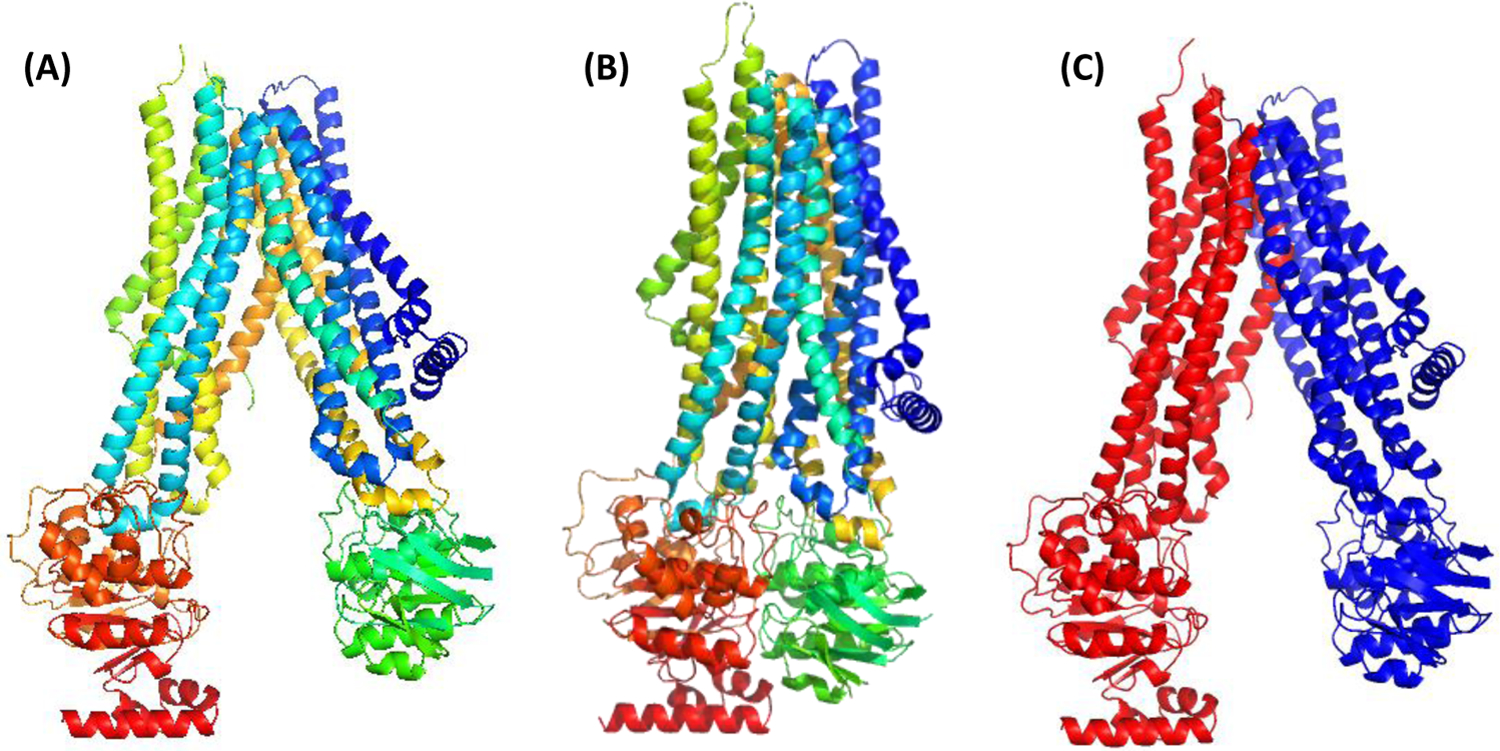
An ABC transporter (A) in apo state, T1158, colored from N-terminal (blue) to C-terminal (red); (B) in one of the bound states, T1158v4, colored from N-terminal to C-terminal; and (C) as split into two EUs: D1 (blue): 48–234,347–394,409–615,861–974 and D2 (red): 235–346,692–860,975–1296.
Target 1154 (Figure 6A) is an S-layer protein on archaeal cell surface27. DDomain classifies it into four domains (as numbered in the figure), while DomainParser and SWORD suggest a three-domain arrangement with domains 2 and 3 joined. Starting with the most disjoint 4-domain version and based on the Grishin plot analysis (B) we joined domains 2, 3 and 4 into one EU, while leaving domain 1 separate.
Figure 6.
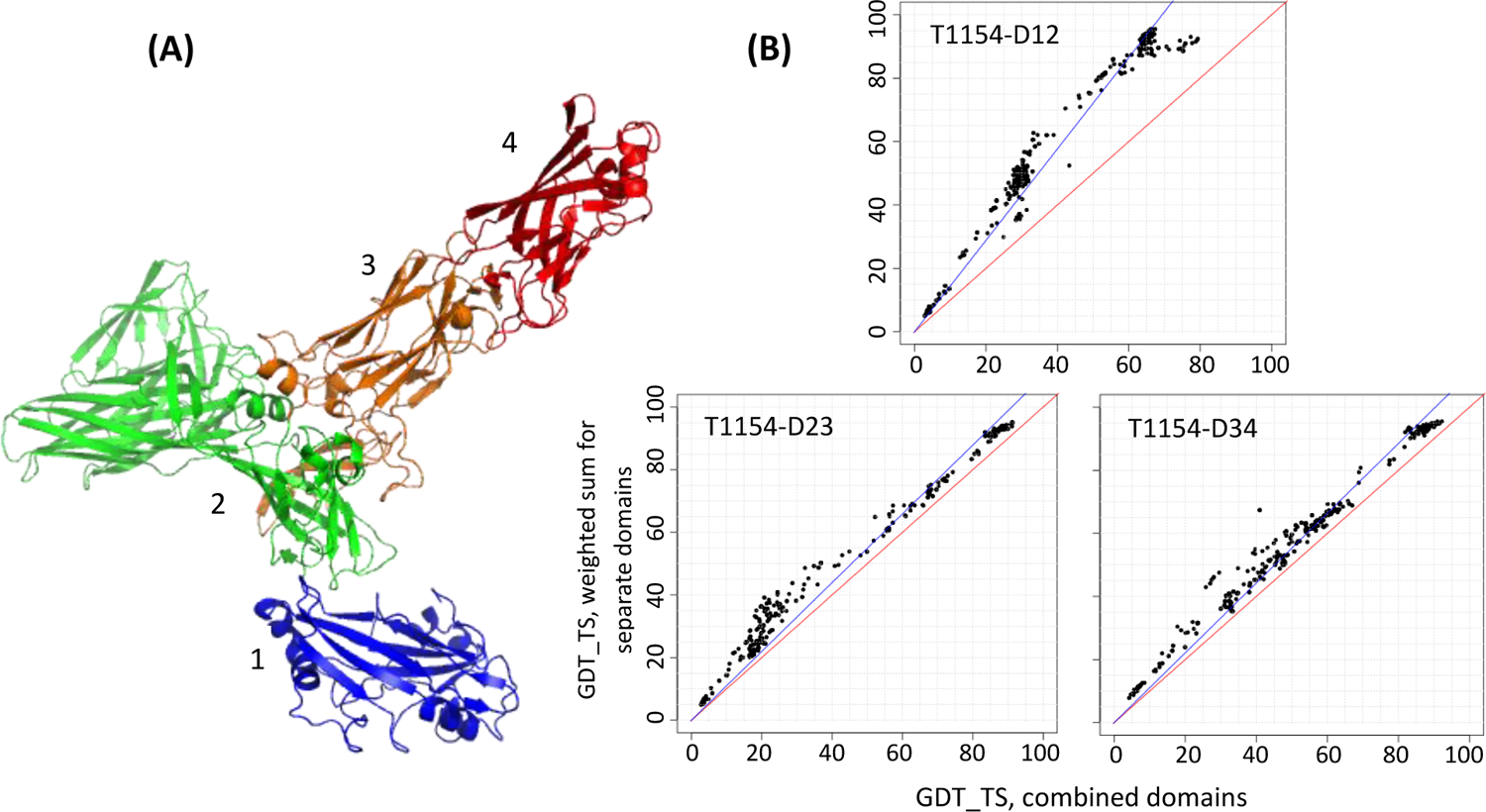
(A) A two-EU target T1154 as originally split into four domains (colored from blue to red). (B) Grishin plots for selected pairs of four original domains of T1154 as numbered in panel A. The upper panel in section (B) shows that domains 1 and 2 should remain separate for evaluation, while domains 2, 3 and 4 (bottom panels) should be joined.
The last example is target T1169, a mosquito salivary protein SGS1 involved in mosquito-borne diseases28. It is the largest monomeric target in the history of CASP (3364 residues in the sequence; 2735 residues resolved in the structure). It has a cocoon-shaped structure with multiple domains and extensive inter-domain interactions (Figure 7), thus presenting a significant challenge in defining EUs. The top-ranked SWORD/SWORD2 splitting schema suggested 7 domains; the domain definition from the authors (Figure 7B28) and the results of HHsearch homology searches (Figure 7C) offered additional help in defining domains. Domains were originally defined so that the following 7 areas were separated: the N-term β-propeller (blue in panel A, orange in panel B), region between the two β-propellers (HHsearch), β-propeller 2, region after the beta-propeller, CBM domain, lectin-CRD domain, the area containing the wedge domain up to the TM domain (HHsearch). The Grishin plot analysis suggested merging of two domains surrounding β-propeller 2, and merging of CBM, lectin-CRD and wedge-containing domains. In the end we split T1169 into four evaluation units, as colored in Figure 7A. A long linker between D1 and D4 and orphan helices in the middle of the cocoon (grey) were not assigned to any of the EUs.
Figure 7.
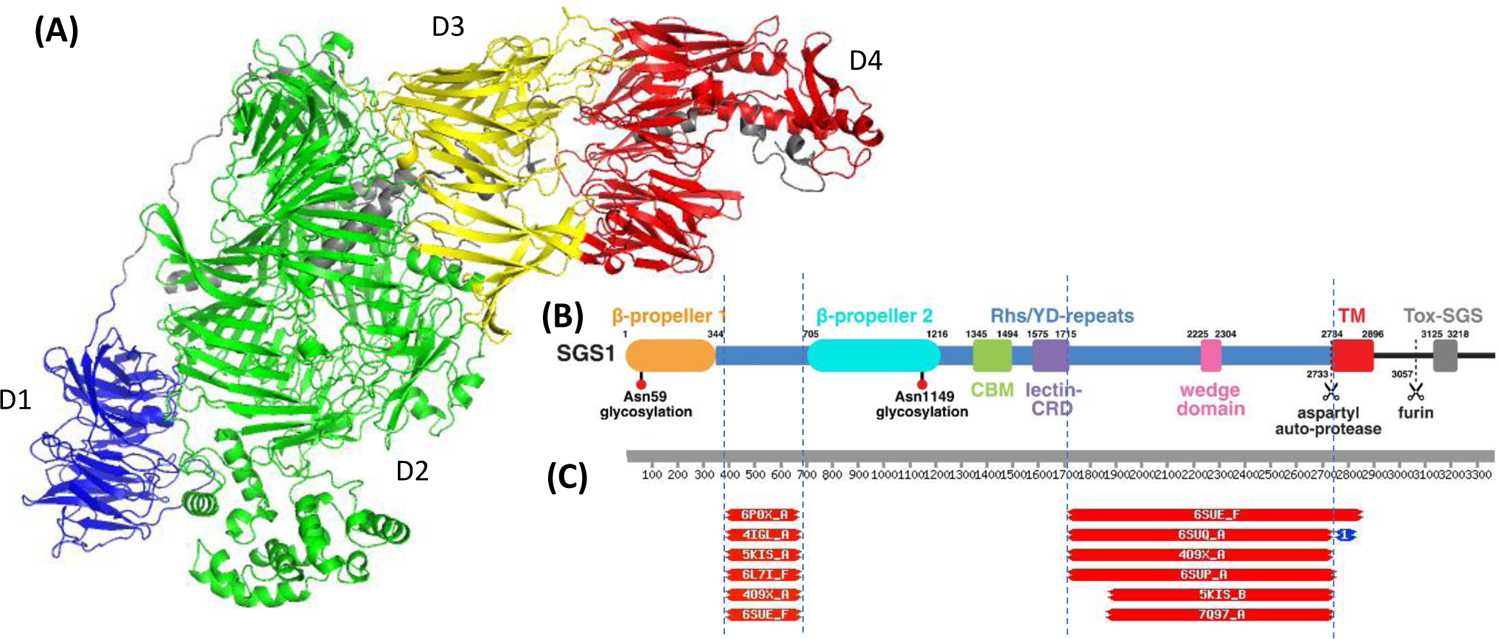
(A) Target T1169, a mosquito protein relevant to pathogen transmission (PDB:8FJP) with four evaluation units defined: D1: 1–345; D2: 1302–2735; D3: 378–699,1223–1301; D4: 700–1222. (B) Parsing of SGS1 into domains as suggested by the authors of the structure28. (C) Top HHsearch hits showing similarity of the query sequence to known folds in two areas: 395–670 (intermediate domain between the two beta-propellers - see panel B) and 1718–2735 (region after the lectin-CRD domain and up to the TM domain).
3.1.4 |. Targets that were split into more EUs than suggested by Grishin plots
Two single-domain targets as suggested by the domain parsers (T1137s2 and T1137s3) were split into two domains for consistency with the other subunits of the same heteromeric complex. Target H1137 (PDB: 8fef) is a hetero 9-mer with six subunits forming an intertwined obligatory complex. The split was made in agreement with the results of template searches and splits of other related subunits.
Another target, T1125, was split into 6 domains instead of 5 suggested by the domain parsers. In this target the C-terminal region penetrates the N-terminal part forming one structural domain, but predictors were unable to model the circular fold of the protein. Thus, for the evaluation, the N-terminal domain (#1) and C-terminal domain (#6) were considered separately.
3.1.5 |. Domain swaps
Four targets in CASP15 included domains involved in domain swaps: T1109, T1113, T1120 and T1176. Target T1120 was discussed above (3.1.3). The remaining three targets were un-swapped, and models were evaluated versus both swapped and un-swapped versions of the targets. For T1109 and T1113, models scored higher versus the original (swapped) version, and thus the original targets were used for the final evaluation; for T1176, the evaluation scores were higher for the un-swapped version, and that version was used as the target (T1176-D9: A1–138 + B139–170).
3.2 |. Prediction classes
Two scores, HHscore and LGA_S, for sequence- and structure-based relationships of the target with PDB entries, were defined in Methods. They are plotted against each other for all EUs in CASP14 and CASP15 (Figure 8). The classification of the CASP14 data resulting from the previous procedures10 – based partly on predictor performance and involving manual intervention – is indicated by symbols in panel A. This reveals that TBM-easy and FM EUs cluster in these coordinates in the upper right and lower left corners respectively, while TBM-hard and TBM/FM EUs predominantly occupy areas immediately above and below the diagonal, respectively. It also can be seen that all triangle markers but two (FM and TBM/FM targets) are below the diagonal and all squares but one (TBM-easy and TBM-hard) are above. Thus, if we consider the diagonal line (HHscore+LGA_S=100) as a boundary between the wider TBM (TBM-easy and TBM-hard together) and FM categories (FM and TBM/FM), then there are only three targets for which the prior CASP14 and current automated classifications schemes disagree.
Figure 8.
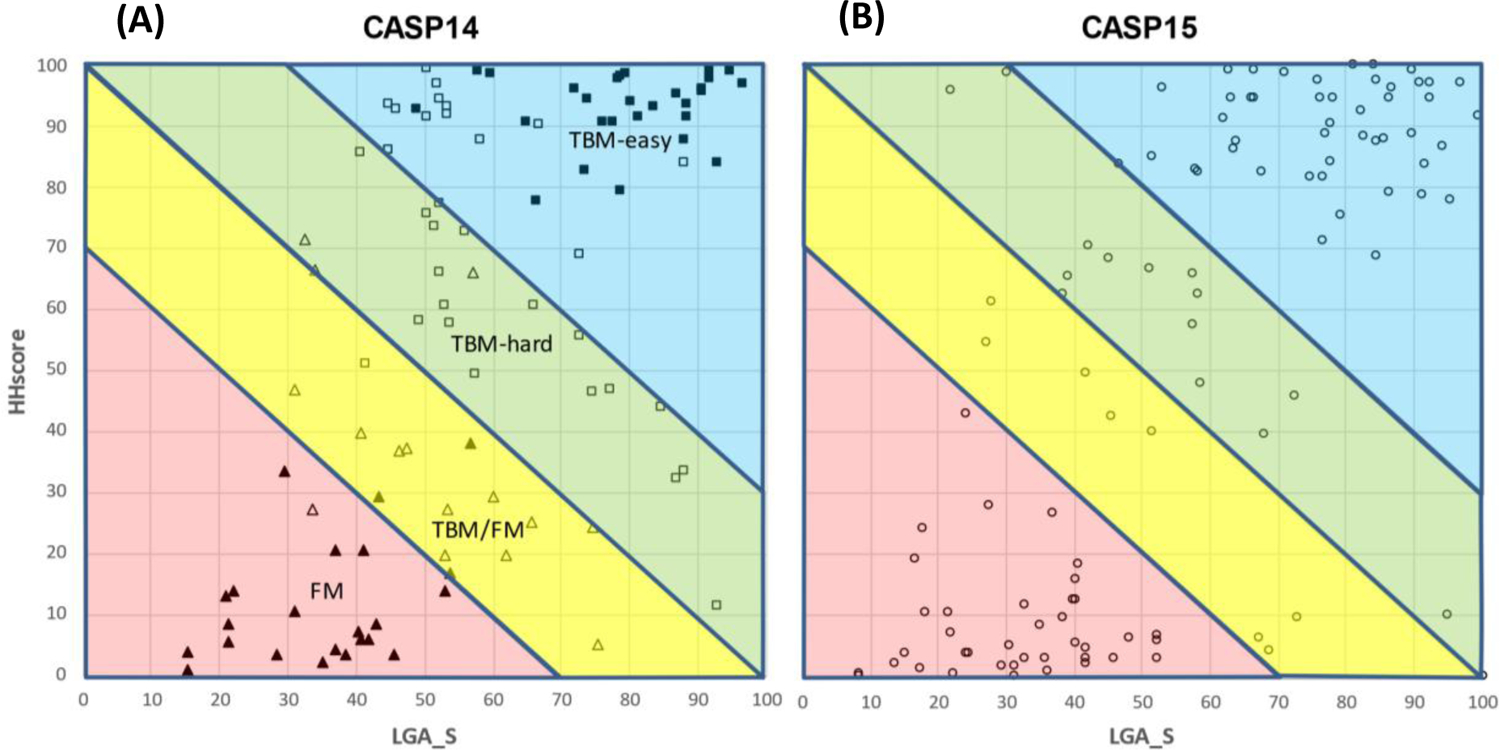
Scatter plot of evaluation units in CASP14 (A, left) and CASP15 (B, right) represented by sequence (HHscore, Y-axis) and structure (LGA_S, X-axis) scores of the top template. Evaluation units in the left panel are marked according to the difficulty categories as manually assigned in CASP14: full squares – TBM-easy; hollow squares – TBM-hard; hollow triangles –TBM/FM; full triangles – FM. Targets of the same difficulty cluster together in the suggested (X,Y) axes. An automatic delineation of EUs into four classes (X+Y<70, red; 70–100, yellow; 100–130 green; >130, blue) based on the results of sequence- and structure-based searches of the PDB is suggested to mimic the CASP14 difficulty categories. The schema is applied to define target prediction classes in CASP15 (right panel).
To further delineate TBM-easy from TBM-hard, and FM from TBM/FM we draw two lines parallel to the diagonal. These lines were drawn symmetrically so that the areas between them and the diagonal include the majority of the TBM-hard (upper) and TBM/FM (lower) EUs yet not encroaching deeply into the TBM-easy and FM territory. Based on the CASP14 data, the split lines were drawn at HHscore+LGA_S=70 and 130 levels. As a side note, we want to mention that we experimented with several other splitting schemas (like rectangular or spherical divisions) and found the linear split to be the simplest and best fitting the CASP14 and CASP13 target classifications. When the suggested schema is applied to the classification of CASP15 EUs (Figure 8B), we see that the points in the graph are nicely separated, with particularly clear clustering in the FM and TBM-easy zones.
Using this classification approach, the CASP15 EUs were automatically assigned to four largely homology-based prediction classes (see Figure 1B and Table 1). Forty-seven EUs were assigned to the TBM-easy class, 15 to TBM-hard, 8 to TBM/FM, and 39 (~35%) to FM - a class with the weakest or no evolutionary relation to available folds. These data show that the CASP15 target set was one of the most difficult (homology-wise) in the whole history of CASP. For comparison, the FM class constituted only 24% of all targets in CASP14, and 27% in CASP13. Conceivably this rise may already illustrate the impact of AF2 on target selection in structural biology: experimentalists may be switching attention to more structurally novel targets with which AF2 still struggles.
As discussed in more detail elsewhere21 29, it is clear that FM targets comprise the majority of those with which even the top predictive methods struggled, even though some FM targets were well-predicted. Thus, even though it is well known that AF2 (on which most predictive methods were based) generalizes beyond its training set, the absence of similar structural folds in the PDB still leads to a greater risk of predictive failure. Factors further predisposing a target to less accurate prediction appear to include shallow Multiple Sequence Alignment (MSA) (it is known that evolutionary covariance information extracted from MSAs is required for accurate modelling of natural proteins by AF230,31, potentially in order to obtain a sufficiently accurate initial structure estimate). Especially given the relatively small numbers of problematic targets in each CASP, however, a deeper study on this subject is needed, and deep learning methods could help with this task.
4 |. CONCLUSIONS
A key objective of CASP is to monitor progress in predictive performance on different kinds of target protein. Thus, a robust and objective classification of targets is essential. Although previous classification has benefitted from detailed consideration by experts in protein evolution, the new, purely automatic method introduced here provides a new labor-saving foundation for CASP-to-CASP comparisons. We show that it largely recapitulates previous classifications and, furthermore, may provide numerical estimates of difficulty beyond the current four classes, potentially facilitating future study of features correlating with target difficulty.
Much as a purely automatic division of targets into EUs would also be desirable, the CASP15 set illustrate why that seems not yet to be possible. For example, a satisfactory EU definition for the ABC transporter T1158 was only achieved by manual reference to a set of structures and an understating of the structure-function relationship of the target: none of the automated domain partitioning algorithms produced sensible results. Nevertheless, clear and objective guidelines were followed as far as possible relating, for example, to the gradients of the Grishin plots. Finally, it is worth noting that although consistent policy is followed for EU definition, the resulting sets may still differ from CASP to CASP as predictions improve. Thus, as more groups accurately capture domain packing there will be fewer instances of splitting and more where larger multi-domain units are retained as the EUs: this tendency towards larger EUs could tend to depress global quality metrics and should be borne in mind by future assessors. Also, as a recommendation for future assessors, we suggest taking advantage of the AlphaFold’s Predicted Aligned Error (PAE), which might naturally provide domain assignments without the need for tedious expert analysis.
Acknowledgements
This research was supported by the US National Institute of General Medical Sciences (NIGMS/NIH) grant R01GM100482 (AK) and Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/S007105/1 (DJR).
REFERENCES
- 1.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995;23(3):ii–v. [DOI] [PubMed] [Google Scholar]
- 2.Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP)-Round XII. Proteins 2018;86 Suppl 1:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019;87(12):1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins-Structure Function and Bioinformatics 2021;89(12):1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH, Grishin NV. ECOD: an evolutionary classification of protein domains. PLoS Comput Biol 2014;10(12):e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinch LN, Shi S, Cheng H, Cong Q, Pei J, Mariani V, Schwede T, Grishin NV. CASP9 target classification. Proteins 2011;79 Suppl 10:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinch LN, Li W, Schaeffer RD, Dunbrack RL, Monastyrskyy B, Kryshtafovych A, Grishin NV. CASP 11 target classification. Proteins 2016;84 Suppl 1:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abriata LA, Kinch LN, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. Definition and classification of evaluation units for tertiary structure prediction in CASP12 facilitated through semi-automated metrics. Proteins 2018;86 Suppl 1:16–26. [DOI] [PubMed] [Google Scholar]
- 9.Kinch LN, Kryshtafovych A, Monastyrskyy B, Grishin NV. CASP13 target classification into tertiary structure prediction categories. Proteins 2019;87(12):1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinch LN, Schaeffer RD, Kryshtafovych A, Grishin NV. Target classification in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins-Structure Function and Bioinformatics 2021;89(12):1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res 2003;31(13):3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Xu D, Gambow HN. Protein domain decomposition using a graph-theoretic approach. Bioinformatics 2000;16(12):1091–1104. [DOI] [PubMed] [Google Scholar]
- 13.Zhou HY, Xue B, Zhou YQ. DDOMAIN: Dividing structures into domains using a normalized domain-domain interaction profile. Protein Science 2007;16(5):947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postic G, Ghouzam Y, Chebrek R, Gelly JC. An ambiguity principle for assigning protein structural domains. Sci Adv 2017;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cretin G, Galochkina T, Vander Meersche Y, de Brevern AG, Postic G, Gelly JC. SWORD2: hierarchical analysis of protein 3D structures. Nucleic Acids Research 2022;50(W1):W732–W738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 2012;9(2):173–175. [DOI] [PubMed] [Google Scholar]
- 18.Steinegger M, Meier M, Mirdita M, Vohringer H, Haunsberger SJ, Soding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 2019;20(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Kempen M, Kim SH, Tumescheit C, Mirdita M, Gilchrist C, Söding J, Steinegger M. Foldseek: fast and accurate protein structure search. bioRxiv 2022; 10.1101/2022.02.07.479398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor TJ, Tai CH, Huang YJ, Block J, Bai H, Kryshtafovych A, Montelione GT, Lee B. Definition and classification of evaluation units for CASP10. Proteins 2014;82 Suppl 2:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigden DJ, et al. Tertiary structure assessment in CASP15. Proteins 2023(This issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrodinger L, DeLano WL. PyMOL. Available at: wwwpymolorg 2020.
- 23.Banneville AS, de la Tour CB, De Bonis S, Hognon C, Colletier JP, Teulon JM, Le Roy A, Pellequer JL, Monari A, Dehez F, Confalonieri F, Servant P, Timmins J. Structural and functional characterization of DdrC, a novel DNA damage-induced nucleoid associated protein involved in DNA compaction. Nucleic Acids Research 2022;50(13):7680–7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deep A, Gu YJ, Gao YQ, Ego KM, Herzik MA, Zhou HL, Corbett KD. The SMC-family Wadjet complex protects bacteria from plasmid transformation by recognition and cleavage of closed-circular DNA. Molecular Cell 2022;82(21):4145–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wald J, Fahrenkamp D, Goessweiner-Mohr N, Lugmayr W, Ciccarelli L, Vesper O, Marlovits TC. Mechanism of AAA plus ATPase-mediated RuvAB-Holliday junction branch migration. Nature 2022;609(7927):630–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ford RC, Beis K. Learning the ABCs one at a time: structure and mechanism of ABC transporters. Biochem Soc T 2019;47:23–36. [DOI] [PubMed] [Google Scholar]
- 27.Gambelli L, McLaren M, Conners R, Sanders K, Gaines MC, Clark L, Gold V, Kattnig D, Sikora M, Hanus C, Isupov MN, Daum B. Structure of the two-component S-layer of the archaeon Sulfolobus acidocaldarius. BioRxiv; 10.1101/2022.10.07.511299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Xia X, Calvo E, Zhou Z. Native structure of mosquito salivary protein uncovers domains relevant to pathogen transmission. Nature Communications 2023;14(899). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XV. Proteins 2023(This issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Applying and improving AlphaFold at CASP14. Proteins-Structure Function and Bioinformatics 2021;89(12):1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583. [DOI] [PMC free article] [PubMed] [Google Scholar]