

Abstract
Background
Multiple sclerosis (MS) is a chronic disease of the central nervous system that affects mainly young adults (two to three times more frequently in women than in men) and causes significant disability after onset. Although it is accepted that immunotherapies for people with MS decrease disease activity, uncertainty regarding their relative safety remains.
Objectives
To compare adverse effects of immunotherapies for people with MS or clinically isolated syndrome (CIS), and to rank these treatments according to their relative risks of adverse effects through network meta‐analyses (NMAs).
Search methods
We searched CENTRAL, PubMed, Embase, two other databases and trials registers up to March 2022, together with reference checking and citation searching to identify additional studies.
Selection criteria
We included participants 18 years of age or older with a diagnosis of MS or CIS, according to any accepted diagnostic criteria, who were included in randomized controlled trials (RCTs) that examined one or more of the agents used in MS or CIS, and compared them versus placebo or another active agent. We excluded RCTs in which a drug regimen was compared with a different regimen of the same drug without another active agent or placebo as a control arm.
Data collection and analysis
We used standard Cochrane methods for data extraction and pairwise meta‐analyses. For NMAs, we used the netmeta suite of commands in R to fit random‐effects NMAs assuming a common between‐study variance. We used the CINeMA platform to GRADE the certainty of the body of evidence in NMAs. We considered a relative risk (RR) of 1.5 as a non‐inferiority safety threshold compared to placebo. We assessed the certainty of evidence for primary outcomes within the NMA according to GRADE, as very low, low, moderate or high.
Main results
This NMA included 123 trials with 57,682 participants.
Serious adverse events (SAEs)
Reporting of SAEs was available from 84 studies including 5696 (11%) events in 51,833 (89.9%) participants out of 57,682 participants in all studies. Based on the absolute frequency of SAEs, our non‐inferiority threshold (up to a 50% increased risk) meant that no more than 1 in 18 additional people would have a SAE compared to placebo.
Low‐certainty evidence suggested that three drugs may decrease SAEs compared to placebo (relative risk [RR], 95% confidence interval [CI]): interferon beta‐1a (Avonex) (0.78, 0.66 to 0.94); dimethyl fumarate (0.79, 0.67 to 0.93), and glatiramer acetate (0.84, 0.72 to 0.98).
Several drugs met our non‐inferiority criterion versus placebo: moderate‐certainty evidence for teriflunomide (1.08, 0.88 to 1.31); low‐certainty evidence for ocrelizumab (0.85, 0.67 to 1.07), ozanimod (0.88, 0.59 to 1.33), interferon beta‐1b (0.94, 0.78 to 1.12), interferon beta‐1a (Rebif) (0.96, 0.80 to 1.15), natalizumab (0.97, 0.79 to 1.19), fingolimod (1.05, 0.92 to 1.20) and laquinimod (1.06, 0.83 to 1.34); very low‐certainty evidence for daclizumab (0.83, 0.68 to 1.02).
Non‐inferiority with placebo was not met due to imprecision for the other drugs: low‐certainty evidence for cladribine (1.10, 0.79 to 1.52), siponimod (1.20, 0.95 to 1.51), ofatumumab (1.26, 0.88 to 1.79) and rituximab (1.01, 0.67 to 1.52); very low‐certainty evidence for immunoglobulins (1.05, 0.33 to 3.32), diroximel fumarate (1.05, 0.23 to 4.69), peg‐interferon beta‐1a (1.07, 0.66 to 1.74), alemtuzumab (1.16, 0.85 to 1.60), interferons (1.62, 0.21 to 12.72) and azathioprine (3.62, 0.76 to 17.19).
Withdrawals due to adverse events
Reporting of withdrawals due to AEs was available from 105 studies (85.4%) including 3537 (6.39%) events in 55,320 (95.9%) patients out of 57,682 patients in all studies. Based on the absolute frequency of withdrawals, our non‐inferiority threshold (up to a 50% increased risk) meant that no more than 1 in 31 additional people would withdraw compared to placebo.
No drug reduced withdrawals due to adverse events when compared with placebo.
There was very low‐certainty evidence (meaning that estimates are not reliable) that two drugs met our non‐inferiority criterion versus placebo, assuming an upper 95% CI RR limit of 1.5: diroximel fumarate (0.38, 0.11 to 1.27) and alemtuzumab (0.63, 0.33 to 1.19).
Non‐inferiority with placebo was not met due to imprecision for the following drugs: low‐certainty evidence for ofatumumab (1.50, 0.87 to 2.59); very low‐certainty evidence for methotrexate (0.94, 0.02 to 46.70), corticosteroids (1.05, 0.16 to 7.14), ozanimod (1.06, 0.58 to 1.93), natalizumab (1.20, 0.77 to 1.85), ocrelizumab (1.32, 0.81 to 2.14), dimethyl fumarate (1.34, 0.96 to 1.86), siponimod (1.63, 0.96 to 2.79), rituximab (1.63, 0.53 to 5.00), cladribine (1.80, 0.89 to 3.62), mitoxantrone (2.11, 0.50 to 8.87), interferons (3.47, 0.95 to 12.72), and cyclophosphamide (3.86, 0.45 to 33.50).
Eleven drugs may have increased withdrawals due to adverse events compared with placebo: low‐certainty evidence for teriflunomide (1.37, 1.01 to 1.85), glatiramer acetate (1.76, 1.36 to 2.26), fingolimod (1.79, 1.40 to 2.28), interferon beta‐1a (Rebif) (2.15, 1.58 to 2.93), daclizumab (2.19, 1.31 to 3.65) and interferon beta‐1b (2.59, 1.87 to 3.77); very low‐certainty evidence for laquinimod (1.42, 1.01 to 2.00), interferon beta‐1a (Avonex) (1.54, 1.13 to 2.10), immunoglobulins (1.87, 1.01 to 3.45), peg‐interferon beta‐1a (3.46, 1.44 to 8.33) and azathioprine (6.95, 2.57 to 18.78); however, very low‐certainty evidence is unreliable.
Sensitivity analyses including only studies with low attrition bias, drug dose above the group median, or only patients with relapsing remitting MS or CIS, and subgroup analyses by prior disease‐modifying treatments did not change these figures.
Rankings
No drug yielded consistent P scores in the upper quartile of the probability of being better than others for primary and secondary outcomes.
Authors' conclusions
We found mostly low and very low‐certainty evidence that drugs used to treat MS may not increase SAEs, but may increase withdrawals compared with placebo. The results suggest that there is no important difference in the occurrence of SAEs between first‐ and second‐line drugs and between oral, injectable, or infused drugs, compared with placebo.
Our review, along with other work in the literature, confirms poor‐quality reporting of adverse events from RCTs of interventions. At the least, future studies should follow the CONSORT recommendations about reporting harm‐related issues. To address adverse effects, future systematic reviews should also include non‐randomized studies.
Keywords: Adolescent, Adult, Female, Humans, Male, Young Adult, Alemtuzumab, Azathioprine, Cladribine, Daclizumab, Dimethyl Fumarate, Fingolimod Hydrochloride, Glatiramer Acetate, Immunosuppressive Agents, Immunosuppressive Agents/adverse effects, Immunotherapy, Interferon beta-1a, Interferon beta-1a/adverse effects, Interferon beta-1b, Multiple Sclerosis, Multiple Sclerosis/drug therapy, Natalizumab, Network Meta-Analysis, Rituximab
Plain language summary
What are the risks of therapies for treating multiple sclerosis?
Key messages
‐ Immunotherapies used to treat multiple sclerosis appear not to increase serious health events, compared to sham drugs (placebo).
‐ Many of these drugs have unwanted effects and, for some of them, more people included in studies dropped out because of side effects compared to sham drugs.
‐ These results are only partly, or are not, reliable since serious health events are relatively rare in people with multiple sclerosis, meaning that the issue is difficult to study, and serious health events were also not well reported in the studies.
What is the condition?
Multiple sclerosis (MS) affects the brain and the spinal cord. MS affects more women than men. In MS, the immune system attacks the sheath that covers our body's nerves and weakens their function. Some people with severe MS may even not be able to use their arms or legs well for some time, but they usually recover. Disability, for example in walking, can arise in some people who have many attacks over the years.
How is the condition treated?
Several treatments that modulate the immune system are available that can help speed recovery from attacks and improve the course of the disease.
What did we want to find out?
We aimed to investigate the risks of the drugs used to treat MS. We wanted to assess all types of health events that are serious, for example, admissions to hospital, or events that made people stop taking the medication. We also wanted to investigate health events in specific body organs.
What did we do?
We searched for studies that investigated drugs aiming to improve the course of MS, compared with other drugs or sham drugs, in people with recurrent episodes of the disease.
What did we find?
Serious health events were found in about one in nine people receiving a sham drug during one or two years. The following drugs were found not to increase these events: interferon beta‐1a (Avonex), dimethyl fumarate, glatiramer acetate, teriflunomide, ocrelizumab, ozanimod, interferon beta‐1b, interferon beta‐1a (Rebif), natalizumab, fingolimod, and laquinimod. We cannot tell whether the following drugs cause more serious health events than sham because the studies were small or there were few events (for cladribine, siponimod, ofatumumab, and rituximab). We were very unsure about daclizumab, immunoglobulins, diroximel fumarate, peg‐interferon beta‐1a, alemtuzumab, interferons and azathioprine because the evidence regarding serious health events was of very poor quality.
Unwanted effects causing people to stop taking the medication were found in one in 16 people receiving a sham drug for one or two years. The following drugs may have increased these dropouts: teriflunomide, glatiramer acetate, fingolimod, interferon beta‐1a (Rebif), daclizumab and interferon beta‐1b. We cannot tell whether ofatumumab causes more dropouts than sham because the studies were small or there were few events. We are very unsure about diroximel fumarate, alemtuzumab, methotrexate, corticosteroids, ozanimod, natalizumab, ocrelizumab, dimethyl fumarate, siponimod, rituximab, cladribine, mitoxantrone, interferons, cyclophosphamide, laquinimod, interferon beta‐1a (Avonex), immunoglobulins, peg‐interferon beta‐1a and azathioprine because the evidence regarding dropouts was of very poor quality.
What are the limitations of the evidence?
Most of the evidence came from studies conducted in ways that may have introduced errors into their results, including the fact that harms were not well reported. Moreover, serious health events and unwanted effects are rare in people with MS and, thus, difficult to study.
How up‐to‐date is the evidence?
This review is up‐to‐date until March 2022.
Summary of findings
Summary of findings 1. Summary of findings 1. Immunotherapies compared to placebo for adults with multiple sclerosis.
| Immunotherapies compared to placebo for adults with multiple sclerosis | |||||||
|
Population: adults with multiple sclerosis Interventions: immunosuppressive and immunomodulatory drugs Comparator: placebo Outcome: serious adverse events (SAEs), mostly at 1 or 2 years Setting: specialist setting Equivalence criterion: RR between 0.67 and 1.50, also meaning that non‐inferiority with placebo was achieved if RR ≤ 1.50, with no more than 1 in 18 additional people having a SAE compared to placebo, at a baseline SAE occurrence of 1 in 9 patients (11.3%) | |||||||
| Anticipated absolute effects (95% CI) | |||||||
| Drug (vs. placebo) | No. of studies for network meta‐analysis (no. of participants in the drug‐specific arm) | No. of studies with direct comparison to placebo (total no. of participants in the drug‐specific arm and in the placebo arm) | Assumed placebo risk (per 1000) | Corresponding intervention risk (95% CI) | Mixed RR (95% CI) | Certainty of evidence | P score |
| Interferon beta‐1a (Avonex) | 11 (3776) | 5 (1885) | 113 | 88 (73, 105) | 0.78 (0.66, 0.94) |
Low Due to risk of bias1 |
0.87 |
| Dimethyl fumarate | 6 (2109) | 5 (2834) | 113 | 89 (77, 105) | 0.79 (0.67, 0.93) |
Low Due to risk of bias1 |
0.86 |
| Daclizumab | 2 (1336) | 1 (621) | 113 | 93 (76, 114) | 0.83 (0.68, 1.02) |
Very low Due to risk of bias1 and incoherence2 |
0.79 |
| Glatiramer acetate | 13 (4688) | 8 (4984) | 113 | 95 (81, 111) | 0.84 (0.72, 0.98) |
Low Due to risk of bias1 |
0.78 |
| Ocrelizumab | 4 (1421) | 2 (889) | 113 | 96 (76, 121) | 0.85 (0.67, 1.07) |
Low Due to risk of bias1 |
0.76 |
| Ozanimod | 2 (1774) | 0 (0) | 113 | 99 (66, 149) | 0.88 (0.59, 1.33) |
Low Due to risk of bias3 and imprecision4 |
0.67 |
| Interferon beta‐Ib | 6 (2674) | 1 (939) | 113 | 106 (88, 127) | 0.94 (0.78, 1.12) |
Low Due to risk of bias3 |
0.62 |
| Interferon beta‐1a (Rebif) | 17 (3692) | 7 (2384) | 113 | 110 (91, 131) | 0.96 (0.80, 1.15) |
Low Due to risk of bias1 |
0.57 |
| Natalizumab | 5 (1309) | 4 (2134) | 113 | 111 (90, 136) | 0.97 (0.79, 1.19) |
Low Due to risk of bias1 |
0.55 |
| Diroximel fumarate | 1 (253) | 0 (0) | 113 | 119 (2 6, 5 27 ) | 1.05 (0.23, 4.6 6 ) |
Very low Due to risk of bias1 and imprecision5 |
0.50 |
| Immunoglobulins | 3 (234) | 3 (407) | 113 | 119 (37, 375) | 1.05 (0.33, 3.32) |
Very low Due to risk of bias1 and imprecision5 |
0.49 |
| Peg‐interferon beta‐1a | 1 (1012) | 1 (1512) | 113 | 121 (75, 200) | 1.07 (0.66, 1.74) |
Very low Due to risk of bias1 and imprecision5 |
0.43 |
| Fingolimod | 10 (4088) | 5 (3774) | 113 | 119 (104, 136) | 1.05 (0.92, 1.20) |
Low Due to risk of bias1 |
0.41 |
| Teriflunomide | 7 (3207) | 4 (3044) | 113 | 122 (99, 148) | 1.08 (0.88, 1.31) |
Moderate Due to risk of bias3 |
0.39 |
| Cladribine | 2 (1294) | 2 (1935) | 113 | 123 (89, 171) | 1.10 (0.79, 1.52) |
Low Due to risk of bias3 and imprecision4 |
0.39 |
| Rituximab | 3 (404) | 2 (543) | 113 | 127 (82,195) | 1.12 (0.73, 1.73) |
Low Due to risk of bias3 and imprecision4 |
0.38 |
| Interferons | 1 (77) | 0 (0) | 113 | 183 (24, 1000) | 1.62 (0.21, 12.72) |
Very low Due to risk of bias1 and imprecision5 |
0.35 |
| Laquinimod | 7 (2278) | 7 (4360) | 113 | 12 7 (104, 155) | 1.12 (0. 92, 1.37) |
Low Due to risk of bias1 |
0.33 |
| Alemtuzumab | 3 (1188) | 0 (0) | 113 | 132 (96, 182) | 1.16 (0.85, 1.60) |
Very low Due to risk of bias1 and imprecision4 |
0.31 |
| Siponimod | 2 (1334) | 2 (1941) | 113 | 133 (105, 168) | 1.20 (0.95, 1.51) |
Low Due to risk of bias3 and imprecision4 |
0.26 |
| Ofatumumab | 4 (1153) | 2 (295) | 113 | 142 (99, 202) | 1.26 (0.88, 1.79) |
Low Due to risk of bias3 and imprecision4 |
0.24 |
| Azathioprine | 2 (243) | 1 (354) | 113 | 409 (86, 1000) | 3.62 (0.76, 17.19) |
Very low Due to risk of bias1 and imprecision5 |
0.07 |
Mixed RR: risk ratio obtained from network meta‐analysis
P score: the mean extent to which a treatment is likely to be better than an alternative intervention averaged over all interventions
Explanations for certainty of evidence: averaged over all interventions
Explanations for certainty of evidence:
- Major concerns regarding risk of bias in most studies (downgrade ‐2)
- Some concerns regarding incoherence (downgrade ‐1)
- Some concerns regarding risk of bias in most studies (downgrade ‐1)
- Some concerns regarding imprecision (downgrade ‐1)
- Major concerns regarding imprecision (downgrade ‐2)
- Some concerns regarding heterogeneity (downgrade ‐1)
GRADE Working Group grades of evidence
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate quality: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect.
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of the effect.
Summary of findings 2. Summary of findings 2: Immunotherapies compared to placebo for adults with multiple sclerosis.
| Immunotherapies compared to placebo for adults with multiple sclerosis | ||||||||
|
Population: adults with multiple sclerosis Interventions: immunosuppressive and immunomodulatory drugs Comparator: placebo Outcome: withdrawals due to adverse effects, mostly at 1 or 2 years Setting: specialist setting Equivalence criterion: RR between 0.67 and 1.50, also meaning that non‐inferiority with placebo was achieved if RR ≤ 1.50, with no more than 1 in 31 additional people withdrawing compared to placebo, at a baseline withdrawal occurrence of 1 in 16 patients (6.5%) | ||||||||
| Anticipated absolute effects (95% CI) | ||||||||
| Drug (vs. placebo) | No. of studies for network meta‐analysis (no. of participants in the drug‐specific arm) | No. of studies with direct comparison to placebo (total no. of participants in the drug‐specific arm and in the placebo arm) | Assumed placebo risk (per 1000) | Corresponding intervention risk (95% CI) |
Mixed RR (95% CI) |
Certainty of evidence | P score | |
| Diroximel fumarate | 1 (253) | 0 (0) | 65 | 25 (7, 84) | 0.38 (0.11, 1.27) |
Very low Due to risk of bias1, imprecision2 and incoherence6 |
0.95 | |
| Alemtuzumab | 3 (1188) | 0 (0) | 65 | 40 (21, 75) | 0.63 (0.33, 1.19) |
Very low Due to risk of bias1, imprecision2 and incoherence6 |
0.92 | |
| Ozanimod | 2 (1774) | 0 (0) | 65 | 67 (37, 123) | 1.06 (0.58, 1.93) |
Very low Due to risk of bias4, imprecision5 and incoherence6 |
0.76 | |
| Natalizumab | 5 (1309) | 4 (2134) | 65 | 80 (51, 124) | 1.20 (0.77, 1.85) |
Very Low Due to risk of bias4, imprecision2 and incoherence3 |
0.71 | |
| Corticosteroids | 2 (87) | 0 (0) | 65 | 68 (10, 464) | 1.05 (0.16, 7.14) |
Very low Due to risk of bias1, imprecision2 and incoherence6 |
0.66 | |
| Ocrelizumab | 4 (1421) | 2 (889) | 65 | 84 (51, 136) | 1.32 (0.81, 2.14) |
Very low Due to risk of bias1 and imprecision2 |
0.64 | |
| Dimethyl fumarate | 6 (1948) | 4 (2578) | 65 | 88 (63, 123) | 1.34 (0.96, 1.86) |
Very low Due to risk of bias1, imprecision2 and incoherence6 |
0.64 | |
| Teriflunomide | 7 (3207) | 4 (3044) | 65 | 89 (66, 120) | 1.37 (1.01, 1.85) |
Low Due to risk of bias4 and heterogeneity7 |
0.63 | |
| Methotrexate | 1 (31) | 1 (60) | 65 | 61 (1, 1000) | 0.94 (0.02, 46.7) |
Very low Due to risk of bias4, imprecision5 and incoherence6 |
0.61 | |
| Laquinimod | 7 (2278) | 7 (4360) | 65 | 83 (56, 124) | 1.42 (1.01, 2.00) |
Very low Due to risk of bias1 and imprecision7 |
0.60 | |
| Ofatumumab | 4 (1153) | 2 (295) | 65 | 99 (57, 171) | 1.50 (0.87, 2.59) |
Low Due to risk of bias4, imprecision2 and incoherence5 |
0.55 | |
| Interferon beta‐1a (Avonex) | 13 (4007) | 6 (2169) | 65 | 98 (72, 134) | 1.54 (1.13, 2.10) |
Very low Due to risk of bias1 and heterogeneity7 |
0.54 | |
| Rituximab | 3 (404) | 2 (543) | 65 | 106 (37, 303) | 1.63 (0.53, 5.00) |
Very low Due to risk of bias1 and imprecision5 |
0.50 | |
| Siponimod | 2 (1334) | 2 (1941) | 65 | 97 (55, 171) | 1.63 (0.96, 2.79) |
Very low Due to risk of bias4, incoherence5 |
0.49 | |
| Cladribine | 2 (1294) | 2 (1935) | 65 | 111 (59, 213) | 1.80 (0.89, 3.62) |
Very low Due to risk of bias4, imprecision2 and incoherence3 |
0.43 | |
| Glatiramer acetate | 15 (4752) | 9 (5032) | 65 | 114 (88, 147) | 1.76 (1.36, 2.26) |
Low Due to risk of bias1 |
0.43 | |
| Fingolimod | 11 (4118) | 5 (3774) | 65 | 115 (90, 147) | 1.79 (1.40, 2.28) |
Low Due to risk of bias1 |
0.41 | |
| Immunoglobulins | 7 (533) | 7 (1003) | 65 | 122 (66, 224) | 1.87 (1.01, 3.45) |
Very low Due to risk of bias1, heterogeneity7, incoherence3 |
0.40 | |
| Mitoxantrone | 3 (182) | 2 (242) | 65 | 137 (33, 575) | 2.11 (0.50, 8.87) |
Very low Due to risk of bias1, imprecision5 and inconsistency3 |
0.39 | |
| Interferon beta‐1a (Rebif) | 16 (3886) | 7 (2693) | 65 | 135 (99, 185) | 2.15 (1.58, 2.93) |
Low Due to risk of bias1 |
0.29 | |
| Daclizumab | 2 (1336) | 1 (621) | 65 | 139 (84, 232) | 2.19 (1.31, 3.65) |
Low Due to risk of bias1 |
0.29 | |
| Cyclophosphamide | 1 (72) | 0 (0) | 65 | 251 (29, 1000) | 3.86 (0.45, 33.50) |
Very low Due to risk of bias1 and imprecision5 |
0.24 | |
| Interferons | 2 (124) | 0 (0) | 65 | 226 (62, 826) | 3.47 (0.95, 12.72) |
Very low Due to risk of bias1 and incoherence3 |
0.21 | |
| Interferon beta‐1b | 12 (3615) | 6 (2601) | 65 | 177 (122, 258) | 2.59 (1.87, 3.77) |
Low Due to risk of bias1 |
0.20 | |
| Peg‐interferon beta‐1a | 1 (1012) | 1 (1512) | 65 | 225 (94, 540) | 3.46 (1.44, 8.33) |
Very low Due to risk of bias1 and incoherence3 |
0.16 | |
| Azathioprine | 6 (369) | 4 (513) | 65 | 452 (167, 1000) | 6.95 (2.57, 18.78) |
Very low Due to risk of bias1 and incoherence3 |
0.04 | |
Mixed RR: risk ratio obtained from network meta‐analysis
P score: the mean extent to which a treatment is likely to be better than an alternative intervention averaged over all interventions
Explanations for certainty of evidence:
- Major concerns regarding risk of bias in most studies (downgrade ‐2)
- Some concerns regarding imprecision (downgrade ‐1)
- Major concerns regarding incoherence (downgrade ‐2)
- Some concerns regarding risk of bias in most studies (downgrade ‐1)
- Major concerns regarding imprecision (downgrade ‐2)
- Some concerns regarding incoherence (downgrade ‐1)
- Some concerns regarding heterogeneity (downgrade ‐1)
GRADE Working Group grades of evidence
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate quality: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect.
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of the effect.
Background
Description of the condition
Multiple sclerosis (MS) is a chronic immune‐mediated disease of the central nervous system. A total of 2.8 million people are estimated to live with MS worldwide (35.9 per 100,000 population). MS prevalence has increased in every world region in the last decade but gaps in prevalence estimates persist. The pooled incidence rate across 75 reporting countries is 2.1 per 100,000 persons/year, and the mean age of diagnosis is 32 years. Females are twice as likely to live with MS as males (Walton 2020).
MS is pathologically characterized by inflammation, demyelination, axonal and neuronal loss. Clinically, it is characterized by recurrent relapses or progression, or both, typically striking young adults and ultimately leading to severe disability. In 1996, the clinical course of MS was classified as relapsing‐remitting MS (RRMS), secondary progressive MS (SPMS), primary progressive MS (PPMS), and progressive relapsing MS (PRMS) (Lublin 1996). These forms of MS were used to design trials of interventions over two decades and to approve disease‐modifying treatments (DMTs) for relapsing MS. In 2013, an updated classification of MS forms was produced (Lublin 2014). The concept of disease activity was added based on the presence of clinical relapse or new magnetic resonance imaging (MRI) lesions. The new classification included: (i) active or inactive relapsing MS, with or without worsening; and (ii) active or inactive primary or secondary progressive disease, with or without progression. Two new forms were also added, clinically isolated syndrome (CIS) and radiologically isolated syndrome (RIS), and PRMS was eliminated.
Twenty‐two DMTs have been approved over the past 20 years for treatment of RRMS. The Food and Drug Administration (FDA) approved intravenous infusion of ocrelizumab also for PPMS, oral siponimod and oral ozanimod for CIS, RRMS and active SPMS (aSPMS), oral cladribine for RRMS and aSPMS. For the first time, by the beginning of 2019, these FDA's approvals allowed people with SPMS to be treated with DMTs.
Several national and international guidelines on the use of DMTs for MS have been produced after the 2013 classification (Lublin 2014). Recommendations vary amongst guidelines concerning specific drugs, reflecting — amongst other things — the differences in the regulatory agency's recommendations and different regional or local health policies (Montalban 2018; Rae‐Grant 2018).
Description of the intervention
We considered all immunotherapies that are used, whether approved or off‐label, for people with MS or CIS up to September 30, 2020.
Approved
Injectable medications
Beta interferons (Betaferon®; Extavia®; Rebif®; Avonex®) and glatiramer acetate (Copaxone®, Brabio® or generic) were the first medicines approved for RRMS by the European Medicines Agency (EMA) and the US FDA in the years 1993 to 2002. Betaferon® and Extavia® are injected subcutaneously every other day. Rebif® is injected subcutaneously three times a week. Avonex® is injected into a muscle once a week. Copaxone® or Brabio® are injected subcutaneously daily, or three times a week at a higher dose. Glatiramer acetate generic (Glatopa®) is injected subcutaneously daily.
Peginterferon beta‐1a (Plegridy®) was approved for RRMS in 2014 by EMA and FDA. It is injected subcutaneously at a dose of 125 μg every two weeks.
Daclizumab (Zenapax® or Zinbryta®) was approved by the FDA and EMA in 2016 for treatment of RRMS. It is injected subcutaneously once monthly. The medicine was withdrawn in the European Union in 2018 due to the risk of serious and potentially fatal immune reactions affecting the brain, liver and other organs.
Ofatumumab (Kesimpta®) was approved by the FDA in 2020 for CIS, RRMS and active SPMS. It is injected subcutaneously at an initial dose of 20 mg at weeks 0, 1, and 2, followed by a dose of 20 mg, once monthly.
Oral medications
Fingolimod (Gilenya®) was approved for RRMS by the FDA in 2010 and EMA in 2011. It is taken as a capsule of 0.5 mg, once daily. The first dose is taken under medical supervision to monitor heart rate and blood pressure.
Teriflunomide (Aubagio®) was approved for RRMS by the FDA in 2012 and EMA in 2013. It is taken as a tablet at a dose of 7 or 14 mg, once daily.
Dimethyl fumarate (Tecfidera®) was approved for RRMS by the FDA in 2013 and EMA in 2014. It is taken as a capsule of 120 mg, twice daily.
Laquinimod (Nerventra®) was approved for RRMS by the Russian Ministry of Health in 2013. EMA refused marketing authorisation in 2014 because the benefits of the medicine at the dose studied were not sufficient to outweigh the potential risks in people with MS. The FDA also refused approval.
Cladribine (Mavenclad® orMovectro®) was approved by EMA in 2017 and the FDA in 2019 for the treatment of highly‐active RRMS and active SPMS. It is taken as a pill at a dose of 1.75 mg/kg for up to five consecutive days in the first month and for up to five consecutive days in the second month, with the same course repeated a year later. This may need to be repeated at some point in the future.
Siponimod (Mayzent®) was approved by the FDA in 2019 and EMA in 2020 for treatment of CIS, RRMS and active SPMS. It is taken as a tablet and the maintenance dose is 1 mg or 2 mg daily.
Diroximel fumarate (Vumerity®) was approved by the FDA in 2019 for CIS, RRMS and active SPMS. It is administered as two 231 mg capsules a day.
Ozanimod (Zeposia®) was approved by the FDA and EMA in 2020 for CIS, RRMS and active SPMS. It is taken as a capsule at a maintenance dose of 0.92 mg, once daily.
Monomethyl fumarate (Bafiertam®) was approved by the FDA in 2020 for CIS, RRMS and active SPMS. It is taken as a capsule at a maintenance dose of 190 mg (administered as two 95 mg capsules), twice a day orally.
Infused medications
Mitoxantrone (Novantrone®) was approved in 2000 by the FDA and EMA for the treatment of people with active RRMS and progressive MS. It is taken as a short intravenous infusion (approximately 5 to 15 minutes) of 12 mg/m2 every 3 months.
Natalizumab (Tysabri®) was approved in 2006 for people with highly active RRMS by EMA and the FDA. It is taken as an intravenous infusion via a drip at a dose of 300 mg, once every four weeks.
Alemtuzumab (Lemtrada®) was approved for RRMS by EMA in 2013 and the FDA in 2014. It is taken as two treatment courses. The first course consists of intravenous infusions at a dose of 12 mg on five consecutive days (60 mg total dose). The second course is taken 12 months later and consists of intravenous infusions on three consecutive days (36 mg total dose). Some people may need a third or further infusion.
Ocrelizumab (Ocrevus®) was approved for RRMS and PPMS by the FDA in 2017 and by EMA in 2018. It is taken as an intravenous infusion at a dose of 300 mg/10 mL (30 mg/mL) in a single‐dose vial and then further infusions every six months.
Used off‐label
Azathioprine is used for the treatment of MS in many countries. The American guidelines (Rae‐Grant 2018) recommend the use of azathioprine for people with MS who, for financial or geographical reasons, do not have access to approved DMTs. The German guidelines recommend that, for people with MS who have a stable course under existing therapy with azathioprine, the therapy can be continued as long as the duration of the therapy is not exceeded by ten years. Azathioprine is taken as a tablet at a maintenance dose of 2 mg/kg per day.
Rituximab is not officially approved for treatment of MS, but its off‐label use for active RRMS and active SPMS is widely used in high‐, medium‐ and low‐income countries (Bourdette 2016). It is taken as an intravenous infusion in single doses of 500‐1000 mg two weeks apart, then, every 6 months, 500 to 1000 mg or 375 mg/m2 every week for four weeks. However, a treatment protocol has not been established.
Methotrexate is used in progressive forms of MS. It is taken as a tablet at a dose of 7.5 mg weekly (with 1 mg daily folic acid supplementation).
Cyclophosphamide has been administered to people with MS since 1991 on various schedules as an intravenous infusion at a dose of 1 g over three days, or 400 to 500 mg once daily over five days. The medicine has also been given orally at 2 mg/kg, once daily.
Intravenous immunoglobulins have been used for people with severe and frequent relapses, for whom other treatments have been contraindicated.
Long‐term corticosteroids have been proposed for the treatment of patients with MS since 1961, with controversial results. They have been administered by different schedules as pulsed periodic high‐dose methylprednisolone or oral continuous low‐dose prednisolone.
How the intervention might work
The harm profile of an intervention is strictly related to its mechanism of action, its modality of administration and pharmacokinetic, pharmacodynamic and possibly pharmacogenetic aspects of drug response (Goodman 2006).
According to the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH 2015), adverse events (AEs) are classified in terms of system organ class (SOC), that is, by identifying the anatomical or physiological system affected by the AE itself.
Immunotherapies for MS belong to different pharmacological categories, have different modalities of administration (by intramuscular or subcutaneous injection, by infusion or by mouth) and have different metabolism; although all target the immune system, they are characterized by different effects, as follows: (1) immunomodulation (interferons, glatiramer acetate, pegylated interferon beta‐1a, dimethyl fumarate, monomethyl fumarate, diroximel fumarate, laquinimod, siponimod, ozanimod, immunoglobulins); (2) systemic immunosuppression, inducing a reduction in activation or efficacy of the immune system through cytostatic or cytotoxic effects (mitoxantrone, methotrexate, cyclophosphamide, long‐term corticosteroids, cladribine, azathioprine, teriflunomide); and (3) selective immunosuppression, as with monoclonal antibodies or biological agents directed towards exactly defined antigens (natalizumab, fingolimod, alemtuzumab, daclizumab, rituximab, ocrelizumab, ofatumumab).
These aspects must be considered when the safety profile of a drug is determined, because safety is usually a consequence of the drug’s primary pharmacological effect.
We might classify the main types and the etiopathogenesis of AEs of MS immunotherapies according to the Medical Dictionary for Regulatory Activities System Organ Classes (MedDRA SOC), as follows.
Immune system disorders. All immunotherapies may cause acute or delayed systemic reactions due to allergic response, anaphylaxis, autoimmune disorder, cytokine release syndrome and serum sickness. Such reactions occur in particular during monoclonal antibody treatment (Lycke 2015) but also with immunomodulating agents, such as interferons. The exact process of flu‐like interferon syndrome is poorly understood but probably is related to increased endogenous pyrogens such as interleukin‐6 (IL‐6) and tumor necrosis factor‐alpha (TNF‐α) (Martìnez‐Càceres 1998). Autoimmune diseases such as thyroiditis, psoriasis and rheumatoid arthritis are more frequent in people treated with immunomodulatory or immunosuppressive drugs than in naive patients (Chouhfeh 2015).
Blood and lymphatic system disorders. Cytostatic effects or selective antagonism versus critical cell antigens might cause complete or partial myelosuppression, or lymphopenia. This latter AE occurs, for example, in fingolimod‐treated people, as the result of prevention of egress from secondary lymphoid tissues or following use of alemtuzumab, which selectively causes depletion of T and B lymphocytes. The mechanisms of these AEs during immunomodulating therapies (interferons, dimethyl fumarate) remain uncertain.
Infections and infestations. These might occur during immunosuppressive therapies that impair the immune system and induce immunosurveillance depression. Opportunistic infection such as progressive multi‐focal leukoencephalopathy (PML) in people treated with natalizumab seems to be due to inhibition of effector T‐cell trafficking from blood to CNS, which might favour local John Cunningham virus (JCV) replication (Van Assche 2005). PML has also been reported in people treated with fingolimod or dimethyl fumarate, probably resulting from similar causes. Other opportunistic infections such as herpes virus reactivation and tuberculosis are associated with immunosuppressive or immunomodulatory therapies (Williamson 2015).
Pregnancy, puerperium and perinatal conditions. Pregnancy and fetal damage have been reported with all therapies, although with different severity of harm or risk for reproductive potential and pregnancy category rating (Federal Register 2015). They are probably related to pharmacological effects on DNA and RNA replication (Amato 2015).
Neoplasms benign, malignant and unspecified. The association between MS and cancer has long been investigated but has led to conflicting results. No studies have reported an increased risk of cancer after long‐term exposure to injectable immunomodulatory drugs (interferons and glatiramer acetate). Several reports suggest an increase in cancer risk amongst MS patients treated with immunosuppressant drugs such as mitoxantrone, azathioprine and cyclophosphamide. Because of their action on the immune system, and due to a lack of available long‐term data, a special warning of the potential risk of cancer accompanies the use of cladribine, fingolimod, natalizumab or alemtuzumab. Regulatory agencies recommend using risk management plans for fingolimod, natalizumab, alemtuzumab, dimethyl fumarate, teriflunomide, daclizumab and ocrelizumab (Lebrun 2018).
AEs such as hepatic disorders are common to all types of drugs; others seem to be strictly related to a specific compound. Fingolimod causes transient activation of sphingosine‐1‐phosphate receptor 1 (S1P1) in atrial myocytes, which is associated with a transient reduction in heart rate, while lung hyperreactivity leading to bronchospasms and airway constriction is mediated by S1P1 and sphingosine‐1‐phosphate receptor 3 (S1P3) activation. Alemtuzumab treatment is associated with risk of secondary autoimmunity due to reconstitution of the lymphocyte repertoire. Dimethyl fumarate‐treated people have experienced flushing and gastrointestinal problems, although the causes of these events remain uncertain (Bomprezzi 2015).
Many of these AEs are known and expected on the basis of a drug’s mechanism of action and pharmacodynamic aspects; other reactions remain of uncertain origin or appear during long‐term monitoring of people. Familiarity with the safety profile of each drug is critical for identification of potential mitigation strategies (Farber 2015).
Why it is important to do this review
Although it is accepted that immunotherapies for people with MS may decrease disease activity, uncertainty regarding their relative safety remains. This uncertainty is due to the limited number of direct comparison trials, which provide the most rigorous and valid research evidence on the relative safety of different, competing treatments. A summary of the results, including both direct and indirect comparisons, may help to clarify the stated uncertainty.
There is uncertainty about what early treatment approach is best in MS, particularly in relapsing MS. Recently, there is a tendency to advocate the use of an early intensive approach starting high‐efficacy treatments earlier in relapsing MS (Hartung 2021; Prosperini 2020; Simpson 2021). However, this approach is limited by safety concerns and the preferred approach in clinical practice is the use of moderately effective drugs initially and switching to more efficacious and potentially higher risk agents if MS activity is insufficiently controlled. Consequently, there is an urgent need to evaluate if there are significant differences in the occurrence of serious adverse effects between first‐line (e.g. interferons beta or glatiramer acetate) and second‐line disease treatments (e.g. natalizumab, rituximab, or ocrelizumab).
Network meta‐analysis (NMA) is the most recent and best method that summarizes the evidence of multiple interventions within a single analysis and allows researchers to estimate the relative treatment effect between each two treatments, also those that have never been compared in a trial, by using direct and indirect evidence (Nikolakopoulou 2018). NMA also allows ranking interventions by benefits and harms (Salanti 2011), and thus is used in clinical guidelines to support recommendations (Kanters 2016).
Objectives
To compare the adverse effects of immunotherapies for people with MS or CIS, and to provide a ranking of these treatments according to their relative risks of adverse effects through NMA.
Methods
Criteria for considering studies for this review
Types of studies
We included all RCTs that examined one or more of the agents used in MS or CIS and compared them versus placebo or another active agent. We excluded RCTs in which a drug regimen was compared with a different regimen of the same drug without another active agent or placebo as a control arm. We excluded RCTs that compared treatment‐switch strategy versus continuing treatment.
Types of participants
We included participants 18 years of age or older with a diagnosis of MS or CIS according to any accepted diagnostic criteria (Lublin 1996; McDonald 2001; Polman 2005; Polman 2011; Poser 1983). We included all participants regardless of sex, degree of disability or disease duration.
We considered MS type (relapsing‐remitting MS or clinically isolated syndrome, versus primary or secondary progressive MS) to be the main participant characteristics that could potentially threaten the transitivity assumption in NMAs.
Types of interventions
We included the following immunotherapies (even if they were not licenced in any country) used as monotherapies (i.e. we excluded combination treatments). We excluded interventions administered by a non‐approved route and not used in clinical practice. For example, cladribine is approved and used in clinical practice as an oral medication for the treatment of highly‐active relapsing or active progressive MS; we excluded studies in which cladribine was given by intravenous infusions.
Interferon beta‐1b;
Interferon beta‐1a (Avonex, Rebif);
Glatiramer acetate;
Pegylated interferon beta‐1a;
Ofatumumab;
Fingolimod;
Teriflunomide;
Dimethyl fumarate;
Cladribine;
Siponimod;
Diroximel fumarate;
Ozanimod;
Monomethyl fumarate;
Mitoxantrone;
Natalizumab;
Alemtuzumab;
Ocrelizumab;
Azathioprine;
Rituximab;
Methotrexate;
Cyclophosphamide;
Immunoglobulins;
Long‐term corticosteroids;
Daclizumab;
Laquinimod.
We included regimens as defined in the primary studies, irrespective of their dose and treatment duration. We considered that drug doses could be a source of heterogeneity and lead to violation of the transitivity assumption. We took a pragmatic approach and pooled all dosages in primary analyses, and conducted a sensitivity analysis restricted to dosages higher than the median of the study arms for each drug. We did not expect variation due to route of administration and treatment duration, since these are specific to each drug.
Types of outcome measures
Primary outcomes
We estimated the relative risks of adverse effects at longest follow‐up of competing interventions according to the following primary outcomes:
Number of participants with any (one or more) serious adverse events (SAEs)
Number of withdrawals due to adverse events (AEs)
In this chapter, we use the term adverse event for an unfavourable or harmful outcome that occurs during, or after, the use of a drug, but is not necessarily caused by it (Peryer 2020), and a serious adverse event as any event or reaction, occurring at any dose, that results in death, a life‐threatening adverse event, inpatient hospitalisation or prolongation of existing hospitalisation, a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions, a congenital anomaly or birth defect, or important medical events based on appropriate medical judgment (ICH 2015; FDA 2020).
We considered that study duration post hoc would not be a source of heterogeneity since two‐thirds of the studies lasted between one and two years and about one fifth between two years and three years, which would allow the detection of most short‐ or medium‐term harms. Thus, we did not adopt a specific time frame for outcome collection.
Secondary outcomes
We estimated the relative risks of adverse effects at longest follow‐up of competing interventions according to the following secondary outcomes, as classified by the Medical Dictionary for Regulatory Activities System Organ Classes (MedDRA SOC) (version 18.0) (ICH 2015).
Cardiac disorders (SAEs and AEs, separately);
Infections and infestations (SAEs and AEs, separately);
Infusion and injection site reactions (SAEs and AEs, separately) ; for intravenous medications, the number of infusion reactions were extracted and for subcutaneous or intramuscular medications, injection site reactions were extracted;
Nervous system disorders (SAEs and AEs, separately);
Psychiatric disorders (SAEs and AEs, separately);
Gastrointestinal disorders (SAEs and AEs, separately);
Blood and lymphatic system disorders (SAEs and AEs, separately);
Hepatobiliary disorders (SAEs and AEs, separately);
Immune system disorders (SAEs and AEs, separately);
Pregnancy, puerperium and perinatal conditions;
Deaths;
Neoplasms.
We expressed all outcomes for each SAE category as percentages of participants with any (one or more) SAEs.
Search methods for identification of studies
This review fully incorporates the results of searches conducted until March 2022.
Electronic searches
We conducted systematic searches in the following databases for RCTs and controlled clinical trials without language, publication year or publication status restrictions up to 04 March 2022:
PubMed (1946 to 04 March 2022);
Embase.com (Elsevier) (1974 to 04 March 2022);
Cochrane Central Register of Controlled Trials (CENTRAL; 2022, Issue 2) in the Cochrane Library;
CINAHL Complete EBSCOhost (Cumulative Index to Nursing and Allied Health Literature; 1981 to 04 March 2022);
LILACS Bireme (Latin American and Caribbean Health Science Information Database; 1982 to 04 March 2022).
To identify RCTs and controlled clinical trials in the databases, we used the Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision); PubMed format (Lefebvre 2022) with a modification to truncate the search line for trial[ti] (line 70, PubMed strategy, Appendix 1). The modification to trial*[ti] increased the sensitivity of the filter slightly and enabled the search to capture a known study reference (Miller 1961) and post hoc or pooled analyses with eligible studies. We also used the Cochrane Embase RCT filter for Embase.com (Glanville 2019), the Cochrane CINAHL Plus RCT filter (Glanville 2019a), and the highly sensitive search strategy for clinical trials in LILACS (Manríquez 2008).
We searched the following trial registers on March 04, 2022:
World Health Organization International Clinical Trials Registry Platform (trialsearch.who.int);
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov).
Search strategies for databases and trial registers are provided in Appendix 1.
Searching other resources
We also searched the following agency websites for pre‐ and post‐ marketing reports up to 04 March 2022:
United States Food and Drug Administration (fda.gov);
European Medicines Agency (ema.eurpoa.eu);
Australian Medicines Regulatory Authority ‐ Therapeutic Goods Administration (tga.gov.au).
Finally, we reviewed the references from relevant systematic reviews and included studies.
Data collection and analysis
Selection of studies
We used Cochrane’s Screen4Me workflow to help assess the search results. Screen4Me comprises three components: known assessments – a service that matches records in the search results to records that have already been screened in Cochrane Crowd and been labeled as an RCT or as Not an RCT; the RCT classifier – a machine learning model that distinguishes RCTs from non‐RCTs; and, if appropriate, Cochrane Crowd – Cochrane’s citizen science platform where the Crowd help to identify and describe health evidence.
For more information about Screen4Me and the evaluations that have been done, please go to the Screen4Me webpage on the Cochrane Information Specialist’s portal (community.cochrane.org/organizational‐info/resources/resources‐ groups/information‐specialists‐portal). In addition, more detailed information regarding evaluations of the Screen4Me components is available (Marshall 2018; McDonald 2017; Noel‐Storr 2018; Thomas 2017).
After using the search strategy described above and the Screen4Me workflow to obtain titles and abstracts of studies that may be relevant to the review, two teams of two review authors each (GC and SF; MGL and MC) independently screened titles and abstracts and discarded studies that were not applicable; however, we retained studies and reviews that might include relevant data or information on trials. Two teams of two review authors each (GC and SF; MGL and MC) independently assessed the retrieved abstracts and, when necessary, the full text of these studies to determine which studies satisfied the inclusion criteria. We compared multiple reports of the same study and used the most comprehensive report. We resolved discrepancies in judgment by discussion with a third review author (IT).
Data extraction and management
Two teams of two review authors each (GC and SF; MGL and MC) independently extracted data using a predefined data extraction form within an Excel spreadsheet. Disagreements were solved by discussion with a third review author (IT).
Outcome data
We extracted from each included study the number of participants who:
had any SAE;
withdrew because of any AE;
experienced any specific AE or SAE according to the MedDRA SOC (ICH 2015), as defined in the Types of outcome measures section;
were randomized; and
took one or more doses of the interventions included in the review.
We extracted arm‐level data. When data were not reported or were unclear in the primary studies, we consulted reports from FDA, EMA and TGA.
Data on potential effect modifiers
We extracted from each included study data on the following potential effect modifiers:
Population: age (range), forms of MS (CIS, RRMS, SPMS, PPMS and PRMS), disease duration (mean if provided or median), days since symptom onset and randomisation for CIS, baseline Expanded Disability Status Scale (EDSS) score (mean), previous treatment with immunotherapies (no or yes/possible);
Duration of follow‐up;
Intervention: dose, frequency or duration of treatment;
Risk of bias: blinding of participants, blinding of outcome assessors, incomplete outcome data;
Funding source.
Other data
We extracted data from each included study on the following additional information.
Study: first author or acronym, year of publication, recruitment period, publication type (full‐text publication, abstract publication, unpublished data);
Study design: inclusion criteria, sequence generation, allocation concealment, selective outcome reporting, early termination of trial.
Assessment of risk of bias in included studies
We assessed the risk of bias of each included study by using the Cochrane criteria (Higgins 2011). These include random sequence generation, allocation concealment, blinding of participants, blinding of outcome assessors, incomplete outcome data, selective outcome reporting, and other potential sources of bias. We judged the risk of bias in each study on the basis of each criterion and classified the study as having ’low’, ’high’, or ’unclear’ risk of bias. We judged incomplete outcome data as showing a low risk of bias when numbers and causes of dropouts were balanced (i.e. in the absence of a significant difference) between arms and appeared to be unrelated to studied outcomes. We judged selective outcome reporting as showing a low risk of bias when study results included the three outcome categories relevant to the review, i.e. SAEs, AEs and withdrawals due to AEs.
To summarize the quality of studies across the two primary outcomes, we considered blinding of participants, blinding of outcome assessors and incomplete outcome data to classify each study as having low risk of bias when we judged all the selected criteria as having low risk of bias; high risk of bias when we judged at least one criterion amongst those selected as having high risk of bias; and moderate risk of bias in the remaining cases.
We assessed characteristics associated with monitoring and reporting AEs by considering two qualitative components that may have a large influence on the completeness of AE data: (1) whether authors defined SAEs according to an accepted international classification and reported the number of each specific type of SAE per arm; and (2) whether authors actively monitored for AEs asking participants about the occurrence of specific AEs in structured questionnaires or interviews or predefined laboratory tests at prespecified time intervals, or simply provided AEs that the study participants spontaneously reported on their own initiative (Ioannidis 2004; Peryer 2020). Passive surveillance of AEs leads to fewer recorded adverse events than active surveillance (Ioannidis 2004).
Two teams of two review authors each (GC and SF; MGL and MC) assessed the risk of bias of each study independently and resolved disagreements by discussion to reach consensus.
Measures of treatment effect
Relative treatment effects
We estimated, through pairwise meta‐analysis, the safety of competing interventions by using the risk or rate ratio (RR) with a 95% confidence interval (95% CI) for each outcome. We presented results from the NMA as summary relative effect sizes (RR) with 95% CIs for each possible pair of treatments.
Relative treatment ranking
We estimated ranking probabilities for all treatments at each possible rank for each intervention for each outcome. In the protocol, we had planned to determine a treatment hierarchy by using the surface under the cumulative ranking curve (SUCRA) and mean ranks (Salanti 2011). Since in the review phase we used the R package netmeta for analyses (see below for further details), we estimated ranking by means of P scores, a frequentist version of SUCRA (Rucker 2015). By definition, the P score of a treatment is the mean extent to which a treatment is likely to be better than an alternative intervention averaged over all interventions. More specifically, such an extent of certainty is calculated, under a normality assumption, as one minus the P value of the one‐sided test rejecting the null that the treatment is not better than the alternative intervention. As such, a P score gives the rank of a treatment within the range of all interventions, with 0 corresponding to the worst treatment and 1 to the best.
Unit of analysis issues
Cluster and cross‐over trials have not been carried out to evaluate immunotherapies for the treatment of people with MS or CIS.
Studies with multiple treatment groups
For multi‐arm trials, the intervention groups of relevance are those that could be included in a pairwise comparison of intervention groups, which, if investigated alone, would have met the criteria for inclusion of studies in the review. For example, if we identify a study comparing 'interferon beta versus natalizumab versus interferon beta plus natalizumab', only one comparison (interferon beta vs natalizumab) addresses the review objective, and no comparison involving combination therapy does. Thus, the 'interferon beta plus natalizumab' therapy group is not relevant to the review. However, if the study compared 'interferon beta‐1b versus interferon beta‐1a (Rebif) versus interferon beta‐1a (Avonex)', all three pairwise comparisons of interventions are relevant to the review. In this case, we treated multi‐arm studies as multiple independent two‐arm studies in pairwise meta‐analysis and accounted for the correlation between effect sizes in multi‐arm studies through NMA. Due to inclusion of multi‐arm studies, for treatment comparisons where direct evidence is available, the results (estimates) derived from pairwise meta‐analyses and NMA may differ.
Dealing with missing data
A likely scenario for assessment of effects of missing data on AE outcomes (i.e. rates of AEs) is not feasible, and on SAE outcomes is nonsense (i.e. assuming that participants who contributed to missing outcome data had a SAE); therefore, we performed a sensitivity analysis including only trials with low risk of attrition bias and discussed the extent to which missing data could have altered results or conclusions of the review.
Assessment of heterogeneity
Assessment of clinical heterogeneity within treatment comparisons
To evaluate the presence of heterogeneity derived from different characteristics of study participants, we had planned to assess differences in age, gender, MS type, and disease duration across trials using information reported in the Characteristics of included studies table. Since age, gender and disease duration were similar within MS type subgroups (relapsing‐remitting MS vs. progressive MS), we considered MS type (relapsing or progressive MS) only in a subgroup analysis.
Assessment of transitivity across treatment comparisons
We considered the following participants’ characteristics as a source of heterogeneity potentially threatening the transitivity assumption in the NMAs: MS type (relapsing‐remitting MS or clinically isolated syndrome, versus primary or secondary progressive MS), and prior use of disease‐modifying drugs (naive versus non‐naive).
Assessment of reporting biases
Given that it is not mandatory for investigators to publish results of clinical trials, it is difficult for review authors to obtain an estimate of the number of unpublished trials on MS. We presented the proportion of participants for whom each primary and secondary outcome was reported.
In the protocol, we had planned to evaluate the possibility of reporting bias by creating contour‐enhanced funnel plots (Peters 2008), which show areas of statistical significance and can help to distinguish reporting bias from other possible reasons for asymmetry. In this review, since each study estimated the relative effects of different interventions, we used the comparison‐adjusted funnel plot (Chaimani 2012; Chaimani 2013).
Data synthesis
Methods for direct treatment comparisons
We performed conventional pairwise meta‐analyses for each primary outcome using a random‐effects model for each treatment comparison with at least two studies (DerSimonian 1986). We have used the Mantel‐Haenszel method for pooling, adding an increment of 0.5 to each cell counts for studies with a zero cell count. Because of the large number of drugs included in the review, we presented pairwise meta‐analyses in the upper part of league tables to allow comparisons of direct and mixed estimates.
Methods for indirect and mixed comparisons
We performed NMAs using random‐effects models within a frequentist setting, assuming common heterogeneity across all comparisons, and we accounted for correlations induced by multi‐arm studies (Miladinovic 2014; Salanti 2012). These models enabled us to estimate the probability that each intervention is at each possible rank for each outcome, given the relative effect sizes as estimated in the NMA.
We had planned to perform NMA in Stata using the 'mvmeta' command (Chaimani 2013; Multiple‐Treatments Meta‐analysis (MTM); White 2011; White 2012). In the review phase, we performed NMAs using random‐effects models within a frequentist setting using the R package netmeta (Rucker 2015; Schwrtzer 2015). netmeta is based on a graph‐theoretical approach for NMA that was found to be equivalent to methods based on weighted least squares regression (Rucker 2012). We used forest plots to visualize mixed estimates of pairwise comparisons with placebo as a reference, and network graphs to represent the evidence network with edge widths proportional to the number of studies comparing two treatments and node sizes proportional to the number of studies assessing a treatment. A common between‐study variance was assumed in NMA models.
Subgroup analysis and investigation of heterogeneity
Assessment of statistical heterogeneity
Assumptions when heterogeneity is estimated
In NMA, we assumed a common estimate for the heterogeneity variance across different comparisons.
Measures and tests for heterogeneity
Assessment of statistical heterogeneity in the entire network was based on the magnitude of the heterogeneity variance parameter (τ2), estimated by using NMA models (Jackson 2014).
Assessment of statistical inconsistency
We assumed that any patient who met the inclusion criteria was, in principle, equally likely to have been randomized to any of the eligible interventions.
Local approaches for evaluating inconsistency
To evaluate the presence of inconsistency locally, we used the method proposed by Dias (Dias 2010) and implemented in the netmeta package. This method is based on back‐calculation and infers the contribution of indirect evidence from the direct evidence and the output of a NMA.
Global approaches for evaluating inconsistency
To test global heterogeneity and inconsistency, we used the method proposed by Rucker (Rucker 2012) and implemented in the netmeta package. This method calculates the Q statistic measuring the deviation from consistency. The global Q statistic can be decomposed into the sum of within‐design Q statistics (corresponding to individual pairwise meta‐analyses) and the between‐designs Q‐statistic corresponding to the remaining design inconsistency between comparisons.
Subgroup analyses
We conducted subgroup analysis by prior disease‐modifying treatments to assess whether SAEs or withdrawals due to AEs varied between naive and non‐naive participants.
Other sources of heterogeneity
In the protocol, we had planned to take into account the predefined effect modifiers by performing meta‐regression or, if any, by discussing the extent to which they could have altered the results or conclusions of the review (age, gender, disease duration). Since age, gender and disease duration were similar within MS type subgroups (relapsing‐remitting MS versus progressive MS), we considered MS type (relapsing or progressive MS) only in subgroup analyses.
Sensitivity analysis
For each primary outcome, we performed planned sensitivity analyses with the inclusion of only trials with low risk of attrition bias. We also conducted two additional sensitivity analyses, one including only studies with doses that were higher than the median dose of each treatment across all studies and one including only studies on relapsing‐remitting MS or clinically isolated syndrome.
We had planned a sensitivity analysis on the exclusion of trials with a total sample size of fewer than 50 randomized participants, to detect potential small study effects. In the review, we explored the possibility of small‐study effects using the comparison‐adjusted funnel plot.
Summary of findings and assessment of the certainty of the evidence
In the protocol, we had planned to present seven outcomes in the SoF. In the review phase, due to the large number of outcomes and treatments, we decided to present two SoFs, one for each primary outcome (Table 1; Table 2). Comparisons of all drugs versus placebo were the focus of these SoFs.
In the two SoFs, we presented the main results of this review, according to recommendations provided in Chapter 11 of the Cochrane Handbook for Systematic Reviews of Interventions (version 5.1.0) (Schünemann 2011) and according to Yepes‐Nuñez 2019. We provided estimates derived from the NMA in accordance with the methods of the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) Working Group (GRADE Working Group 2004). We included in the SoF tables the outcomes at longest available follow‐up for each drug.
We had planned to grade the certainty of evidence for each outcome by considering study limitations, indirectness, inconsistency, imprecision of effect estimates and risk of reporting bias. In the review phase, we used the Confidence in Network Meta‐Analysis (CINeMA) as a methodological framework to evaluate the confidence in the results from NMAs (Nikolakopoulou 2020). This approach required further steps with respect to assess the certainty of evidence, and it covers six domains: (i) within‐study bias (referring to the impact of risk of bias in the included studies), (ii) reporting bias (referring to publication and other reporting bias), (iii) indirectness, (iv) imprecision, (v) heterogeneity, and (vi) incoherence. Heterogeneity and incoherence are two dimensions of inconsistency which refer, respectively, to the extent to which the prediction interval overlapped with the confidence interval, and the significance testing of the difference between direct and indirect evidence when both were available for comparison.
Decisions regarding the imprecision, heterogeneity, and incoherence require the specification of a range of equivalence for relative effects (RR) based on absolute effects. We selected a range of equivalence between RR = 0.67 and RR = 1.50. This choice was made post hoc by the authors after discussion of its implications on relative and absolute effects of the primary outcomes, since the CINeMA platform had been made available only after our protocol was published. The use of thresholds for clinically important effects of different sizes has recently been recommended also by the GRADE Working Group to rate imprecision in NMAs (Brignardello‐Petersen 2021).
Summary of Findings tables were not constructed for secondary outcomes, but we used the same threshold to contextualize the impact of our RR estimates on secondary outcomes. For some events that were very rare, we also presented the impact of doubling the risk of harms (RR = 2), also in terms of absolute estimates of effect.
Reporting
We reported the results of the review by completing the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) harms checklist (Zorzela 2016, available on the Open Science Framework at https://osf.io/vujxa/?view_only=d90fac4ebe994de9985a2fb3acc21d84).
Results
Description of studies
For a full description of studies, see the Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
The search identified a total of 16,643 records. After removing duplicates in EndNote and the Cochrane Register of Studies, Cochrane's Screen4Me workflow helped to identify potential reports of randomized trials for the remaining 11,825 records. The results of the Screen4Me assessment process can be seen in Figure 1. 6934 records were rejected as describing studies with ineligible designs. The remaining 4891 records were assessed for eligibility by Cochrane Crowd. The Crowd rejected an additional 675 records, and we evaluated the remaining 4216 records for reported data on adverse effects.
1.
Screen4Me assessment process for eligible study designs.
From 4891 possible RCT records, Cochrane Crowd rejected an additional 675 records, and we evaluated the remaining 4216 records for reported data on adverse effects.
We provisionally selected a total of 191 studies as potentially fulfilling the inclusion criteria. After full‐text assessment, we included 123 studies and excluded 59 studies, together with nine ongoing studies. For a further description of our screening process, see the PRISMA study flow diagram (Figure 2).
2.
Prisma flow diagram
Included studies
One hundred and twenty‐three trials (57,682 participants; median sample size: 278; range: 13 to 2220) were included in the review. Included studies were published between 1961 and 2022 (median 2009). Ten (8.1%) trials included CIS only, seven (6.0%) trials PP only, 71 (57.7%) trials RR only, 11 (8.9%) trials SP only and, in 24 (19.5%) trials, different forms of MS. Forty (32.5%) trials included only naive patients. One hundred (81.3%) trials were funded by pharmaceutical companies. Forty‐six (37.4%) trials included three or more study arms, which were generally different doses or regimens, since only five studies included three interventions. Eighty‐four (68.3%) trials used a placebo comparator, 36 (29.3%) trials used an active comparator, and the remaining three (2.4%), both a placebo and an active comparator. Median follow‐up was 24 months (< 12‐month follow‐up from 22 studies, 12‐ < 24‐month follow‐up from 29 studies, 24‐month follow‐up from 51 studies, and > 24‐36‐month follow‐up from 21 studies).
Nine studies were excluded from the statistical analysis since they did not report any data on the predefined selected outcomes (Ashtari 2011; BPSM 1995; Calabrese 2012; Etemadifar 2006; Koch‐Henriksen 2006; Miller 1961; Mokhber 2014; Motamed 2007; Tubridy 1999).
We identified nine ongoing trials (Characteristics of ongoing studies). We will include these studies in a future update of this review.
Excluded studies
After full‐text review, we excluded 59 studies: six studies on interventions administered by route not approved/not used in clinical practice; 44 studies which did not meet the PICO of the review (38 wrong intervention, 3 wrong participants and 3 wrong study design), eight articles reporting secondary or pooled analyses of included studies, and one withdrawn study. See Characteristics of excluded studies.
Risk of bias in included studies
The risks of bias in the included studies are summarized in Figure 3 and Figure 4. Considering our predefined criteria (blinding of participants, blinding of outcome assessors and incomplete outcome data) to assess the overall risk of bias of a study, we judged 8 (7%) out of 123 trials as having low risk of bias (ASCLEPIOS II 2020; CLARITY 2010; Comi 2001; Comi 2008; Fazekas 2008; GATE 2015; MIRROR 2018; Ziemssen 2017); we judged 42 (34%) as having moderate risk of bias (Achiron 1998; Achiron 2004; AFFIRM 2006; APEX 2019; APOLITOS 2021; BOLD 2013; Bornstein 1991; Boyko 2016; CHAMPS 2000; ETOMS 2001; EXPAND 2018; FUMAPMS 2021; GALA 2013; Goodkin 1995; IFNB MS Group 1993; IMPROVE 2010; Johnson 1995; Kappos 2006; Knobler 1993; Leary 2003; Lewanska 2002; Miller 1961; Miller 2003; Montalban 2009; Noseworthy 2000; O'Connor 2006; OLYMPUS 2009; Pakdaman 2007; Polman 2005; PRISMS 1998; RADIANCE 2019; REFLEX 2012; Saida 2012; Saida 2017; SELECT 2013; SPECTRIMS 2001; SUNBEAM 2019; TEMSO 2011; TOPIC 2014; TRANSFORMS 2010; Tubridy 1999; Wolinsky 2007); and we judged 73 (59%) as having high risk of bias (ADVANCE 2014; ALLEGRO 2012; Andersen 2004; ARPEGGIO 2020; ASCEND 2018; ASCLEPIOS I 2020; Ashtari 2011; ASSESS 2020; AVANTAGE 2013; BECOME 2009; BENEFIT 2006; BEYOND 2009; Bornstein 1987; Boyko 2017; BPSM 1995; BRAVO 2014; British and Dutch 1988; Calabrese 2012; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; CCMSSG 1991; Cheshmavar 2021; CombiRx 2013; CONCERTO 2022; CONFIRM 2012; DECIDE 2015; DEFINE 2012; Ellison 1989; Etemadifar 2006; Etemadifar 2007; European Study Group 1998; EVIDENCE 2002; EVOLVE‐MS‐2 2020; Fazekas 1997; FREEDOMS 2010; FREEDOMS II 2014; Ghezzi 1989; GOLDEN 2017; Goodkin 1991; Hartung 2002; Hauser 2008; Hommes 2004; IMPACT 2002; INCOMIN 2002; INFORMS 2016; Kappos 2008; Kappos 2011; Koch‐Henriksen 2006; Likosky 1991; MAIN TRIAL 2014; Masjedi 2021; Milanese 1993; Millefiorini 1997; Mokhber 2014; Motamed 2007; MOVING 2020; MSCRG 1996; NASP 2004; OPERA I 2017; OPERA II 2017; ORACLE 2014; ORATORIO 2017; OWIMS 1999; Pohlau 2007; PreCISe 2009; PROMESS 2017; REFORMS 2012; REGARD 2008; REVEAL 2020; TENERE 2014; TOWER 2014; Van de Wyngaert 2001).
3.

Risk of bias graph: review authors' judgments about each risk of bias item presented as percentages across all included studies. Serious AE definitions were not applicable when the study did not report serious AE (empty row).
4.

Risk of bias summary: review authors' judgments about each risk of bias item for each included study. Serious AE definitions were not applicable when the study did not report serious AE (empty cells).
Allocation
No study was considered at high risk of bias regarding sequence generation, 29 (24%) were unclear, and 94 (76%) at low risk. Three (2%) studies were considered at high risk of bias (mainly for open allocation), 59 (48%) were unclear, and 61 (50%) at low risk for allocation concealment.
Blinding
Thirty‐three studies (27%) were considered at high risk of performance bias (mainly for single‐blinding), 51 (41%) were unclear, and 39 others (32%) trials were at low risk. Twenty‐seven (23%) studies were considered at high risk for detection bias (mainly for open‐label design), 79 (64 %) were unclear, and 14 (12%) trials were at low risk. Overall, we judged seven studies (6%) as having low risk in both domains.
Incomplete outcome data
Forty‐seven trials (38%) were considered at high risk of attrition bias (because of unbalanced numbers, reasons for dropouts, or both between the comparison groups); 15 (12%) were at unclear risk, and 61 trials (50%) at low risk.
Selective reporting
Reporting of our primary outcomes, SAEs and withdrawals due to AEs, was explicit in most studies, totalling respectively 51,833 (89.9%) and 55,320 (95.9%) participants out of 57,682 patients in all studies. On the other hand, many studies did not report explicitly our secondary outcomes, types of SAEs or AEs. In Figure 5, we report the total number of participants in studies reporting each type of AEs. We used available data in analyses and did not attempt missing imputation techniques.
5.
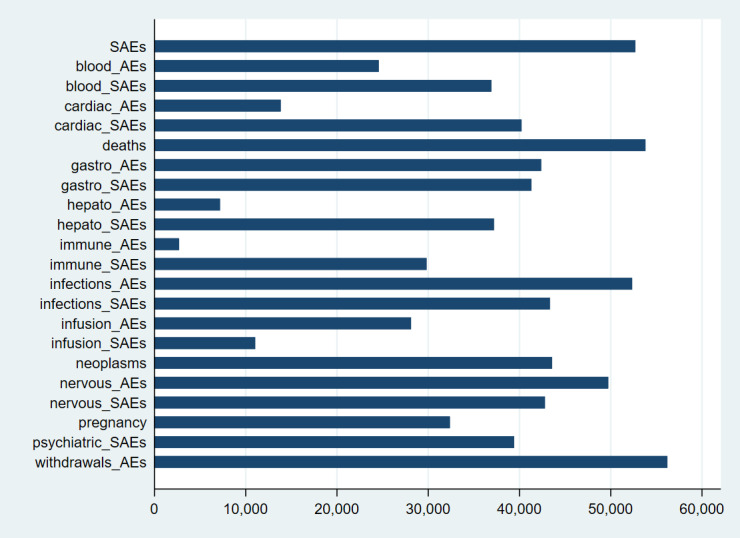
Reporting of adverse events in studies: bar length corresponds to the number of participants for which an adverse event was reported.
Definition of serious adverse events
Only 37% of trials had adequate definition and reporting of SAEs according to the guidelines of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). In ClinicalTrials.gov (Study Results), these trials reported the total number of serious clinical or laboratory‐determined adverse effects and gave numbers of specific types of serious adverse events per arm. There was an improvement in reporting after the release of the CONSORT checklist, with new recommendations about reporting harms‐related issues in randomized trials (Ioannidis 2004).
Forty‐eight trials (39%) did not report any definition of SAEs and key information is missing on criteria used to assess and select SAEs per arm. The majority of these trials reported the total number of SAEs but did not specify their types. Two trials (2%) reported only generic statements without specific numbers.
Twenty‐seven trials (22%) did not provide data on SAEs.
Method of AE monitoring
We assessed whether authors actively monitored for AEs, or simply provided AEs that the study participants spontaneously reported on their own initiative. We judged that the majority (58%) of trials specified the time frame of surveillance and did active monitoring for AEs because participants were asked about the occurrence of specific adverse events in questionnaires or interviews, or predefined laboratory tests were performed at prespecified time intervals. Different methods were adopted for monitoring the adverse effects of each drug with variable reliability of the different approaches. The median duration of the surveillance period was 24 months. We found data to assess our judgment in the published article, in the study protocol, or in ClinicalTrials.gov. Forty‐four (36%) of included studies did not report the method used to monitor adverse events, and so we classified them as having 'unclear risk'. Seven studies were classified as having high risk because the recorded adverse events were those that the study participants spontaneously reported on their own initiative.
The majority of studies reported only the adverse events observed at a certain frequency or rate threshold (for example, > 3%, > 5%, or > 10% of participants).
Other potential sources of bias
We judged 117 (95%) trials as having low risk of other sources of bias.
Effects of interventions
The dataset used in the analyses is available on the Open Science Framework at https://osf.io/vujxa/?view_only=d90fac4ebe994de9985a2fb3acc21d84.
Primary outcome: SAEs
Eighty‐four (68%) studies, including 5696 (11%) events in 51,833 (89.9%) of 57,682 participants, provided data for this analysis (Figure 6). The raw overall SAE frequency was 10.3% and was used as an assumed risk in Table 1. Adopting a 1.5 RR threshold for clinical importance, the corresponding increase in absolute risk would be 5.5% (1 more SAE in 18 participants). Table 1 shows the number of studies (participants or events) for each drug in the NMA on SAEs, together with relative and absolute effects (95% CIs) with an assumed risk estimate of 110 SAEs per 1000 people. This table also presents the certainty of evidence for each drug versus placebo and reasons for downgrading it.
6.
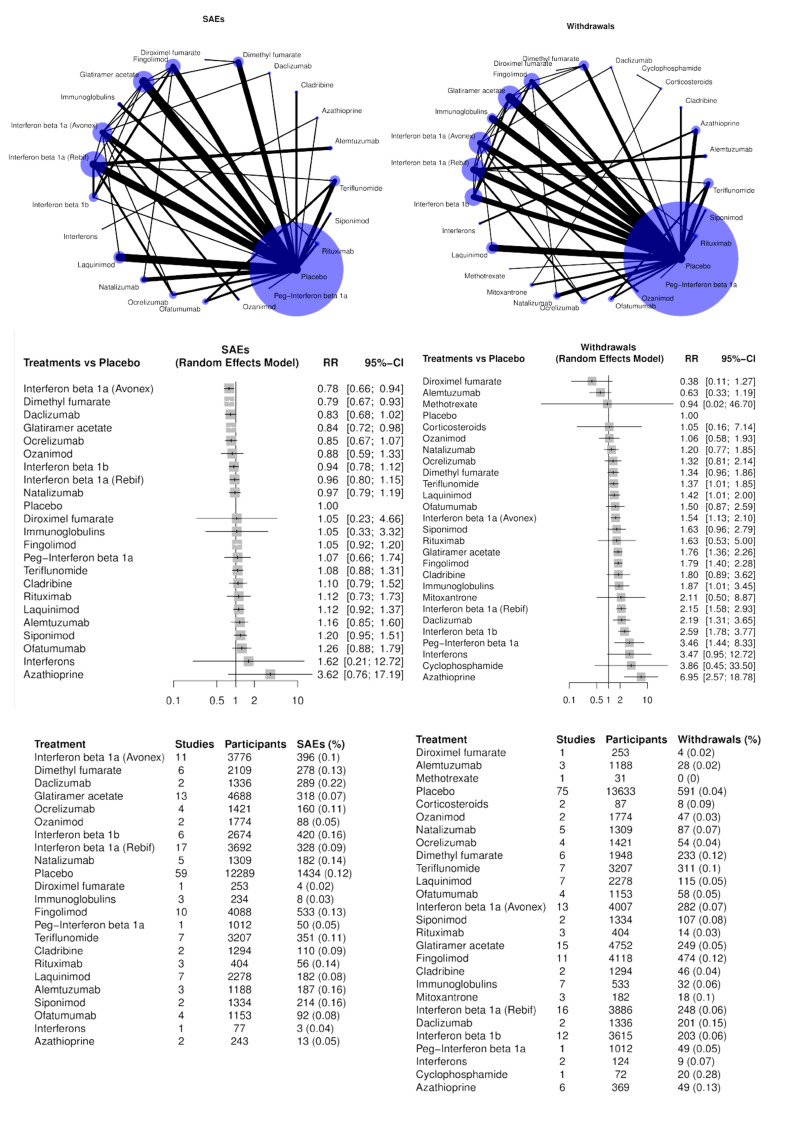
NMA estimates for primary outcomes serious adverse events (SAEs, left) and withdrawals (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Three drugs may decrease SAEs compared to placebo (RR, 95% CI):
low‐certainty evidence for interferon beta‐1a (Avonex) (0.78, 0.66 to 0.94), dimethyl fumarate (0.79, 0.67 to 0.93) and glatiramer acetate (0.84, 0.72 to 0.98).
Several drugs met our non‐inferiority criterion versus placebo (an upper 95% CI RR limit of 1.5 or lower):
moderate‐certainty evidence for teriflunomide (1.08, 0.88 to 1.31);
low‐certainty evidence for ocrelizumab (0.85, 0.67 to 1.07), ozanimod 0.88 (0.88, 0.59 to 1.33), interferon beta‐1b (0.94, 0.78 to 1.12), interferon beta‐1a (Rebif) (0.96, 0.80 to 1.15), natalizumab (0.97, 0.79 to 1.19), fingolimod (1.05, 0.92 to 1.20) and laquinimod (1.06, 0.83 to 1.34);
very low‐certainty evidence for daclizumab (0.83, 0.68 to 1.02).
Non‐inferiority with placebo was not met due to imprecision for the following drugs, although none of the drugs with effects in the direction of more SAEs increased harm to a statistically significant extent:
low‐certainty evidence for cladribine (1.10, 0.79 to 1.52), siponimod (1.20, 0.95 to 1.51), ofatumumab (1.26, 0.88 to 1.79) and rituximab (1.01, 0.67 to 1.52);
very low‐certainty evidence for immunoglobulins (1.05, 0.33 to 3.32), diroximel fumarate (1.05, 0.23 to 4.69), peg‐interferon beta‐1a (1.07, 0.66 to 1.74), alemtuzumab (1.16, 0.85 to 1.60), interferons (1.62, 0.21 to 12.72) and azathioprine (3.62, 0.76 to 17.19).
Table 1 demonstrates the probability that a drug had fewer SAEs than other drugs. interferon beta‐1a (Avonex), dimethyl fumarate, daclizumab, glatiramer acetate, and ocrelizumab were in the upper quartile with P scores between 87% and 76%, in decreasing order.
Table 2 illustrates the RR point estimates of mixed and pairwise comparisons between all drugs, sorted according to RR point estimates from the most to the least effective drug. In this league table, cells below the diagonal contain RR mixed estimates comparing the treatment in the column versus the treatment in the row, whereas cells above the diagonal contain RR direct estimates comparing the treatment in the row versus the treatment in the column. If the (mixed or direct) RR A versus B is available, the B versus A comparison can be easily calculated as 1/RR of A versus B. There was no overall inconsistency between direct and indirect evidence (tau2: 0.0; test of global inconsistency: P = 0.665). Loop‐specific inconsistency was only detected regarding the effect of daclizumab versus placebo (direct RR 0.84; 0.63 to 1.11; 1 study; indirect: RR 1.00; 0.76 to 1.30; P = 0.026) and the effect of daclizumab versus interferon beta‐1a (Avonex) (direct: RR 0.87; 0.74 to 1.04; 1 study; indirect: RR 0.72; 0.49 to 1.05; P = 0.026).
Finally, we explored the possibility of small‐study effects using the comparison‐adjusted funnel plot, which did not suggest important concerns (Figure 7).
7.
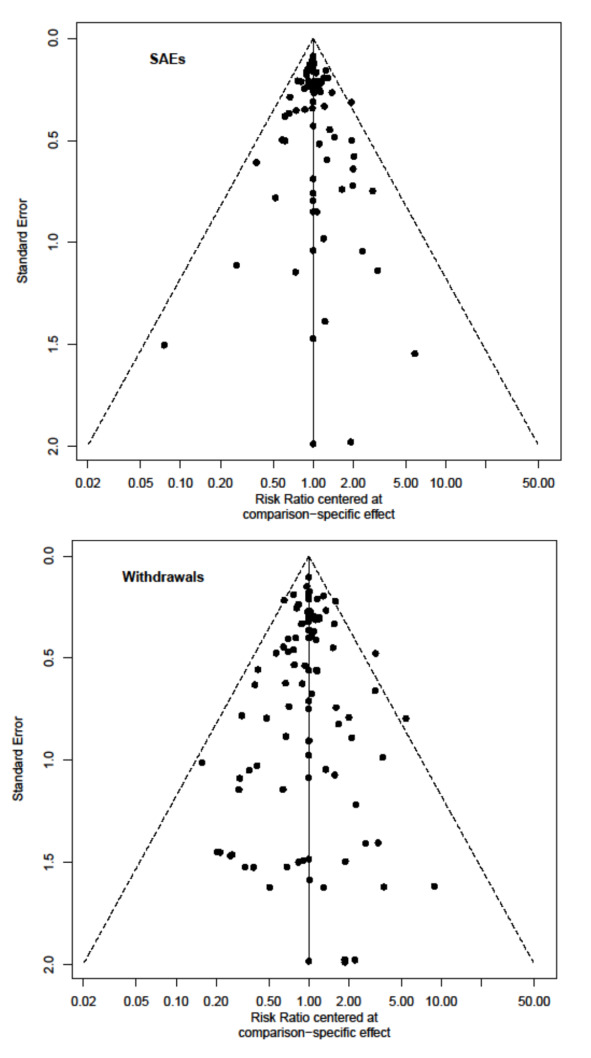
Comparison‐adjusted funnel‐plot for SAEs (top) and withdrawals (bottom)
Sensitivity analyses
The sensitivity analyses including only doses that were higher than the median of each drug group (68 studies, 4653 events (12%) in 38,743 participants), as well as those excluding studies at high risk of attrition bias (35 studies, 1621 events (8.18%) in 19,819 participants), are shown in Figure 8. These sensitivity analyses had no substantial impact on the interpretation of our results, and estimates were much less precise for all drugs in the sensitivity analysis excluding trials with attrition bias concerns. This was also found in the sensitivity analysis including only studies on relapsing‐remitting MS or clinically isolated syndrome (61 studies, 4033 events (9.93%) in 40,613 participants) (Figure 8).
8.

Sensitivity analysis for SAEs including only studies at low risk of attrition bias (top row), doses above the median of each drug group (middle row) or studies including relapsing‐remitting MS or clinically isolated syndrome (bottom row). In the summary tables on the right, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Subgroup analyses
We conducted subgroup analyses by prior disease‐modifying treatments, comparing 19 trials (9893 participants) including only naive participants with 61 studies (40,613 participants) including previously treated participants. Figure 9 shows the results of these subgroup analyses for SAEs and withdrawals. The naive subgroup included few drugs and 95% CIs were very large and overlapped with those of the non‐naive subgroup.
9.

Subgroup analyses by prior use of disease‐modifying drugs: non‐naive patients (top) and naive patients (bottom)
Primary outcome: withdrawals due to adverse events
One‐hundred and five (85.4%) studies with 3537 (6.39%) events in 55,320 (95.9%) of 57,682 participants provided data for this analysis (Figure 6). Withdrawals due to AEs imply a direct decision made by study investigators on causality. We used a 1.5 RR threshold for clinical importance, which translated into an overall increase of 3.21% in withdrawals (about 1 more withdrawal in 31 participants). The Table 2 shows the number of studies (participants or events) for each drug in the NMA, together with relative and absolute effects (95% CIs) compared to an overall assumed risk estimate of 65 withdrawals due to AEs per 1000 participants. This table also presents the certainty of evidence for each drug versus placebo and reasons for downgrading it.
No drug was found to reduce withdrawals due to adverse events when compared with placebo.
There was very low‐certainty evidence (meaning that estimates are not reliable) that two drugs met our non‐inferiority criterion versus placebo (an upper 95% CI RR limit of 1.5 or lower):
diroximel fumarate (0.38, 0.11 to 1.27) and alemtuzumab (0.63, 0.33 to 1.19).
Non‐inferiority with placebo was not met due to imprecision for the following drugs:
low‐certainty evidence for ofatumumab (1.50, 0.87 to 2.59);
very low‐certainty evidence for methotrexate (0.94, 0.02 to 46.70), corticosteroids (1.05, 0.16 to 7.14), ozanimod (1.06, 0.58 to 1.93), natalizumab (1.20, 0.77 to 1.85), ocrelizumab (1.32, 0.81 to 2.14), dimethyl fumarate (1.34, 0.96 to 1.86), siponimod (1.63, 0.96 to 2.79), rituximab (1.63, 0.53 to 5.00), cladribine (1.80, 0.89 to 3.62), mitoxantrone (2.11, 0.50 to 8.87), interferons (3.47, 0.95 to 12.72), and cyclophosphamide (3.86, 0.45 to 33.50).
Eleven drugs may have increased withdrawals due to adverse events when compared with placebo:
low‐certainty evidence for teriflunomide (1.37, 1.01 to 1.85), glatiramer acetate (1.76, 1.36 to 2.26), fingolimod (1.79, 1.40 to 2.28), interferon beta‐1a (Rebif) (2.15, 1.58 to 2.93), daclizumab (2.19, 1.31 to 3.65) and interferon beta‐1b (2.59, 1.87 to 3.77);
very low‐certainty evidence for laquinimod (1.42, 1.01 to 2.00), interferon beta‐1a (Avonex) (1.54, 1.13 to 2.10), immunoglobulins (1.87, 1.01 to 3.45), peg‐interferon beta‐1a (3.46, 1.44 to 8.33) and azathioprine (6.95, 2.57 to 18.78); the precision of this estimate confirmed a clinically important increase in withdrawals (lower 95% CI limit above RR = 1.5) for interferon beta‐1a (Rebif), interferon beta‐1b and azathioprine; however, estimates with low or very low‐certainty are not reliable.
Table 1 shows the probability that a drug caused fewer withdrawals than other drugs (P score). Diroximel fumarate, alemtuzumab, placebo, ozanimod and ozanimod were in the upper P score quartile between 95% and 76% in descending order.
Table 3 displays the RR point estimates of mixed comparisons between drugs that are sorted from the least to the most harmful in the rows, together with the effects in direct meta‐analyses. In this league table, cells below the diagonal contain RR mixed estimates comparing the treatment in the column versus the treatment in the row, whereas cells above the diagonal contain RR direct estimates comparing the treatment in the row versus the treatment in the column. If the (mixed or direct) RR A versus B is available, the B versus A comparison can be easily calculated as 1/RR of A versus B. Direct meta‐analyses were consistent, but less precise, than mixed estimates of effects. There was borderline overall incoherence between direct and indirect evidence (tau2: 0.040; global test of inconsistency: P = 0.084). Loop‐specific inconsistency was detected regarding the effects of dimethyl fumarate versus placebo (direct: RR 0.88, 0.62 to 1.25; 4 studies; indirect: RR 0.24, 0.09 to 0.61, P = 0.011), dimethyl fumarate versus glatiramer acetate (direct: RR 1.21, 0.71 to 2.08; 1 study; indirect: RR 0.49; 0.29 to 0.83; P = 0.019), fingolimod versus glatiramer acetate (direct: RR 0.60; 0.35 to 1.02; 1 study; indirect: RR 1.32; 0.91 to 1.91; P = 0.018) and fingolimod versus interferon beta‐1a (Avonex) (direct: RR 2.09; 1.08 to 4.06; 1 study; indirect: RR 0.92; 0.60 to 1.40; P = 0.039).
Finally, we explored the possibility of small‐study effects using the comparison‐adjusted funnel plot, which did not suggest important concerns (Figure 7).
Sensitivity analyses
The sensitivity analyses, including only doses that were higher than the median of each drug group (82 studies, 2803 (6.8%) withdrawals in 41,462 participants), as well as that excluding studies at high risk of attrition bias (46 studies, 874 (4.25%) withdrawals in 20,588 participants), are shown in Figure 10. These sensitivity analyses did not change our findings and were generally less precise. This was also found in the sensitivity analysis including only studies on relapsing‐remitting MS or clinically isolated syndrome (67 studies, 2659 (6.36%) events in 41,803 participants).
10.
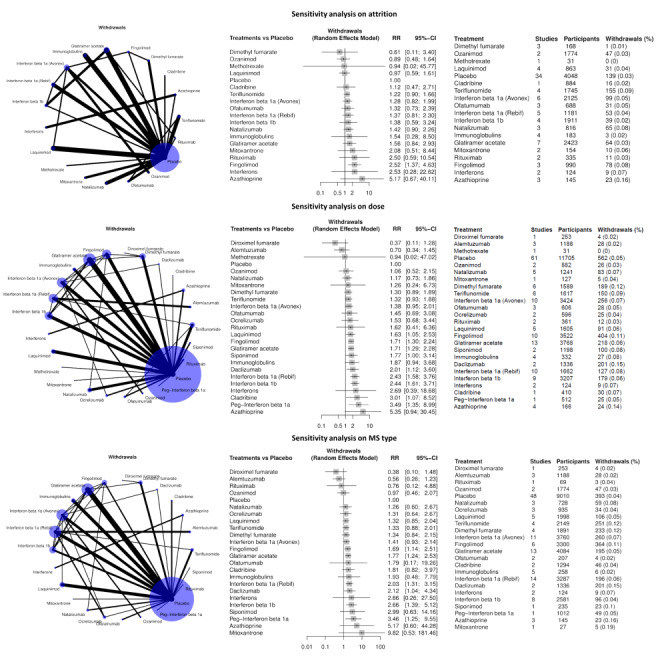
Sensitivity analysis for withdrawals including only studies at low risk of attrition bias (top row), doses above the median of each drug group (middle row), or studies including relapsing‐remitting MS or clinically isolated syndrome (bottom row). In the summary tables on the right, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Subgroup analyses
The subgroup analyses by prior disease‐modifying treatments were not different in the naive versus previously treated subgroups (Figure 9), except for alemtuzumab; however, the corresponding findings came from indirect comparisons and very low‐certainty evidence.
Secondary outcomes
Cardiac adverse events
Figure 11 shows NMA results for cardiac SAEs and AEs.
11.
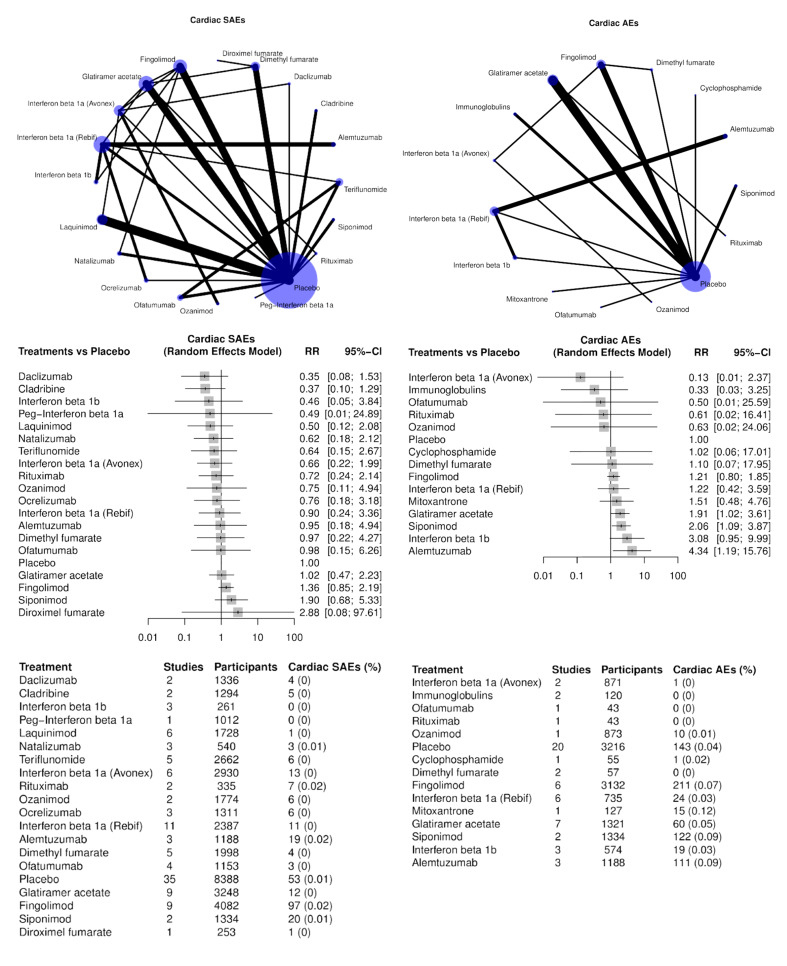
Cardiac SAEs and AEs. In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Cardiac SAEs were analyzed in 56 studies (271 events in 39,214 participants, 0.69%). All confidence intervals from the NMA were wide, and no conclusion could be drawn regarding SAEs compared to placebo, with only cladribine meeting the criteria for equivalence.
Cardiac AEs were analyzed in 29 studies (717 events in 13,689 participants, 5.24%). Glatiramer acetate, siponimod and alemtuzumab may have increased cardiac AEs compared with placebo, but 95% CIs were large and approached equivalence, especially for glatiramer acetate.
Infections
Figure 12 shows NMA results for infection SAEs and AEs.
12.
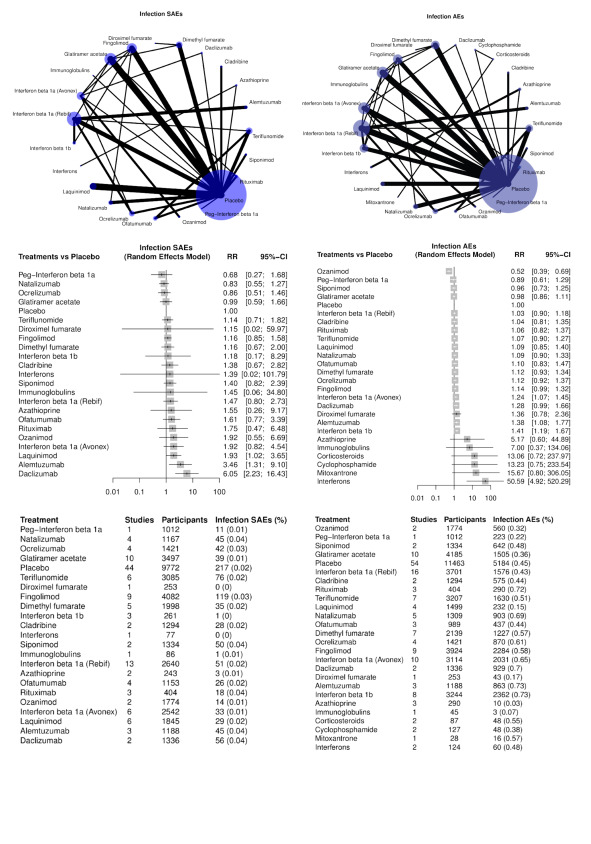
Infection SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Infection SAEs were analyzed in 66 studies (939 events in 42,464 participants, 2.21%). Laquinimod (RR 1.93, 95% CI 1.02 to 3.65), alemtuzumab (RR 3.46, 95% CI 1.31 to 9.10) and daclizumab (RR 6.05, 95% CI 2.23 to 16.43) significantly increased infection SAEs compared to placebo. The upper 95% CI limit was below RR = 1.5 for natalizumab and ocrelizumab, which can be considered non‐inferior to placebo. The upper 95% CI limit for glatiramer acetate, fingolimod, and teriflunomide passed this threshold but was still below RR = 2, which is consistent with a potential increase in infection SAES by only about 1%.
Infection AEs were reported in 81 trials and were very common (24,251 events in 49,491 participants, 49.6%). Compared with placebo, ozanimod may have decreased infection AEs (RR 0.52, 95% CI 0.39 to 0.69); interferon beta‐1a (Avonex) (RR 1.24, 95% CI 1.07 to 1.45), alemtuzumab (RR 1.38, 95% CI 1.08 to 1.77), and interferon beta‐1b (RR 1.41, 95% CI 1.19 to 1.67) may have increased infection AEs. Interferons significantly increased infection AEs (RR 50.59, 95% CI 4.92 to 520.29), but this estimate was very imprecise and was based on only two studies with 124 participants. For most of the other drugs, the RR estimate crossed equivalence but, given the high frequency of infection AEs, non‐inferiority was not considered. A limitation of the analyses of infection AEs was that several studies used events, not participants, as the unit of analysis, and the number of AEs exceeded that of participants in some studies, which could not be included in the NMA. This may have led to underestimated RRs of infection AEs for some drugs.
Infusion and injection site reactions
Figure 13 shows NMA results for infusion and injection site reactions (SAEs and AEs).
13.
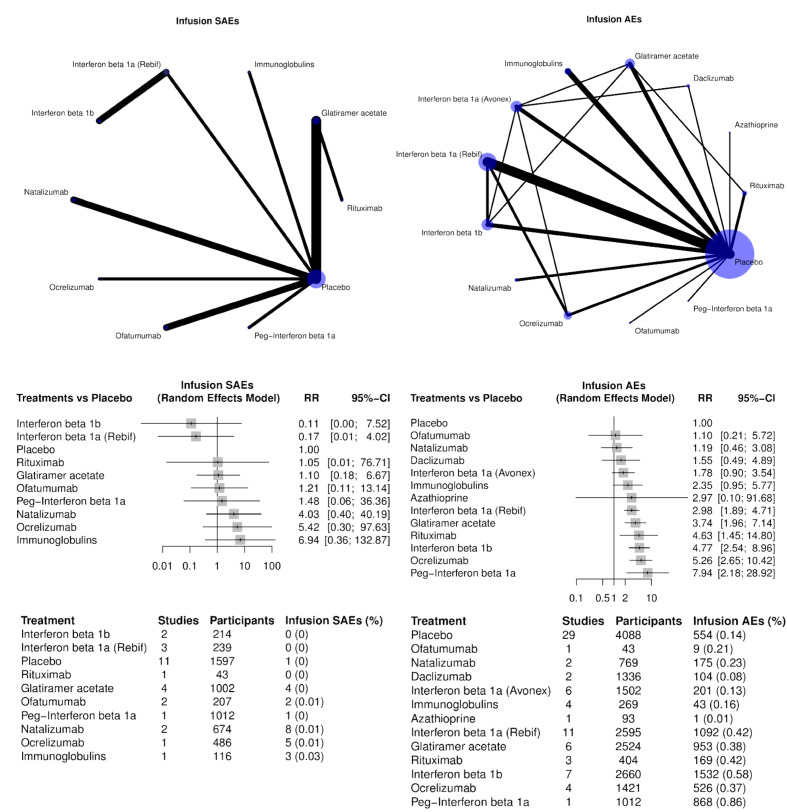
Infusion SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
SAEs were recorded in 14 studies with 24 events (0.4%) in 5590 participants. There was no evidence that any drug increased SAEs when compared to placebo due to large uncertainty of the estimates.
AEs were recorded in 38 studies with 6627 events (33.3%) among 18,716 participants. Interferon beta‐1a (Rebif), glatiramer acetate, rituximab, interferon beta‐1b, ocrelizumab, and peg‐interferon beta‐1a increased AEs compared to placebo. A limitation of the analyses was that several studies used events, not participants, as the unit of analysis, and the number of AEs exceeded that of participants. Thus, they could not be included in the NMA.
Nervous system adverse events
Figure 14 shows NMA results for nervous system SAEs and AEs.
14.
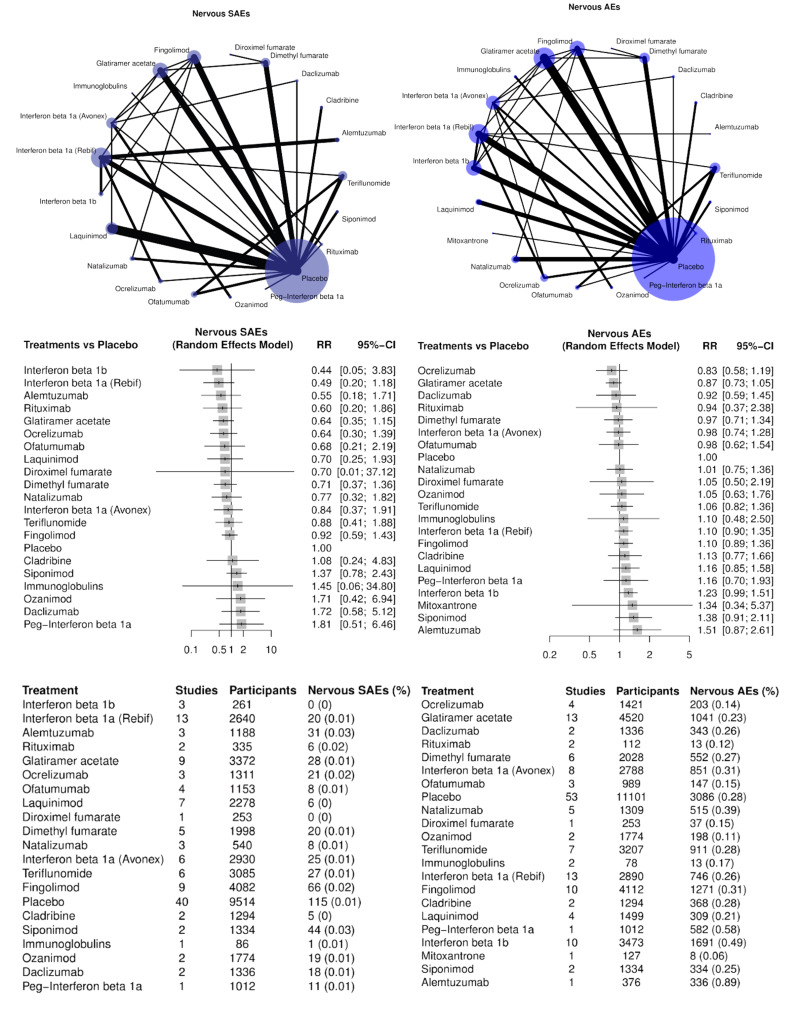
Nervous SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Nervous system SAEs were recorded in 61 studies (479 events (1.15%) in 41,776 participants). There was uncertainty about these events since 95% CIs included zero for all drugs. The upper 95% CI limit was below RR = 2, with point estimates in the direction of fewer SAEs, for the following drugs: interferon beta‐1a (Rebif), alemtuzumab, rituximab, glatiramer acetate, laquinimod, ocrelizumab, dimethyl fumarate, natalizumab, interferon beta‐1a (Avonex), teriflunomide, and fingolimod.
Nervous system AEs were recorded in 75 studies (13,555 events (28.8%) in 47,033 participants). No analyses found that any drug may have decreased or increased nervous system AEs. We avoided descriptions based on upper 95% CI limits given the high frequency and potential overlapping with MS‐related symptoms in this category.
Psychiatric adverse events
Figure 15 shows NMA results for psychiatric SAEs and AEs.
15.
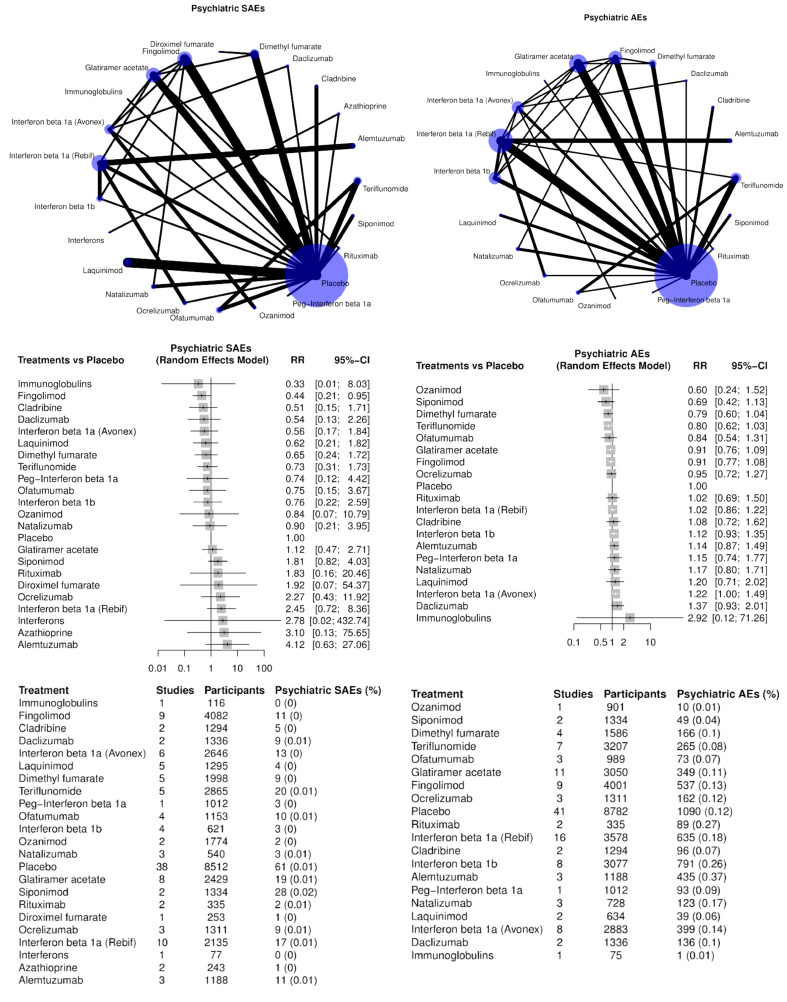
Psychiatric SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Psychiatric SAEs were recorded in 59 studies (241 events (0.63%) in 38,549 participants). Fingolimod may have decreased psychiatric SAEs (RR 0.44, 95% CI 0.21 to 0.95). No conclusion could be drawn on whether other drugs were different from placebo and 95% CI upper limits were below RR = 2 for cladribine, interferon beta‐1a (Avonex), laquinimod, dimethyl fumarate, and teriflunomide.
Psychiatric AEs were recorded in 64 studies (5,538 events (13.4%) in 41,301 participants). Interferon beta‐1a (Avonex) seemed to slightly increase these events (RR 1.22, 95%CI 1.00 to 1.49). No analyses found that any other RRs were not different from placebo, with various degrees of imprecision, which was very large for immunoglobulins.
Gastrointestinal adverse events
Figure 16 shows NMA results for gastrointestinal SAEs and AEs.
16.
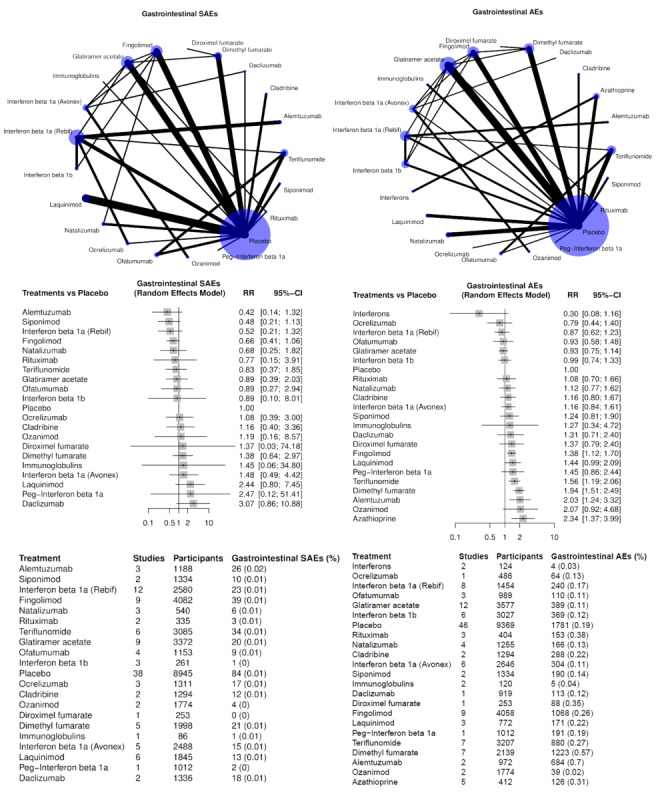
Gastrointestinal SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Gastrointestinal SAEs were recorded in 59 studies (358 events (0.89%) in 40,272 participants). No analyses found that any drug could increase or decrease gastrointestinal SAEs compared to placebo. Estimates were reasonably precise (RR 95% CI upper limit < 2) for alemtuzumab, siponimod, interferon beta‐1a (Rebif), fingolimod, natalizumab, and teriflunomide.
Gastrointestinal AEs were recorded in 67 studies (8,646 events (20.8%) in 41,597 participants). No analyses found that any drug decreased AEs. Fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, and azathioprine may have increased the risk of gastrointestinal AEs.
Blood adverse events
Figure 17 shows NMA results for blood SAEs and AEs.
17.
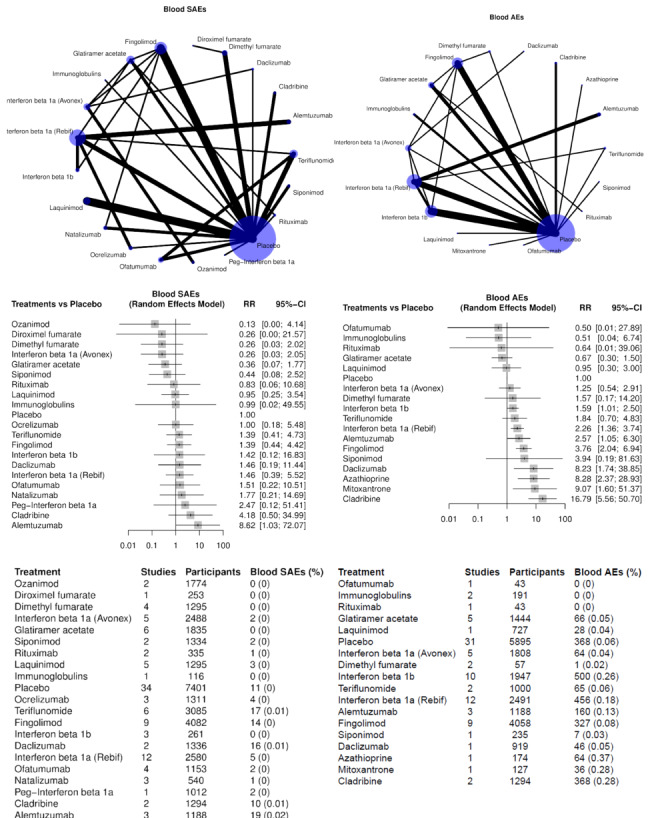
Blood SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Blood SAEs were recorded in 55 studies (109 events (0.30%) in 35,968 participants). There was large uncertainty in all RR estimates, with an acceptable safety precision (upper 95% CI RR < 2) achieved only for glatiramer acetate.
Blood AEs were recorded in 45 studies (2556 events (10.8%) in 23,641 participants). A statistically significant increase in blood AEs was recorded for interferon beta‐1b, interferon beta‐1a (Rebif), alemtuzumab, fingolimod, daclizumab, azathioprine, mitoxantrone, and cladribine.
Hepatic adverse events
Figure 18 shows NMA results for hepatic SAEs and AEs.
18.
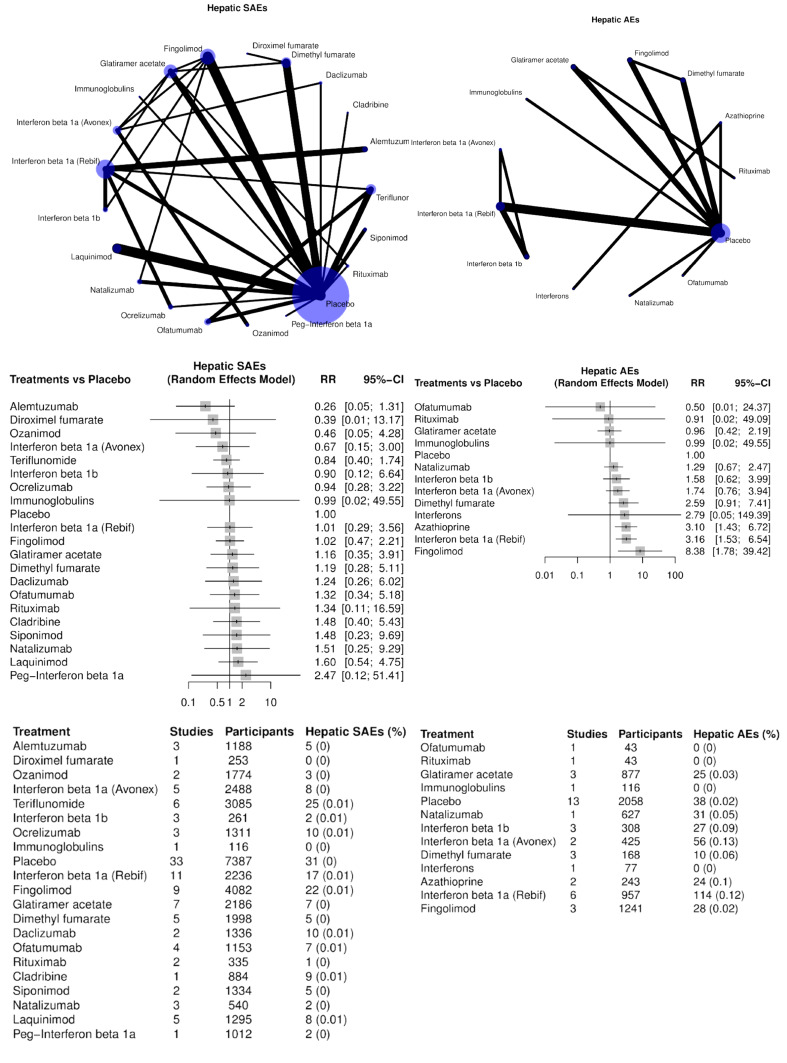
Hepatic SAEs (left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Hepatic SAEs were recorded in 54 studies (179 events (0.49%) in 36,254 participants). There was large uncertainty for all comparisons, except for alemtuzumab and teriflunomide (upper 95% CI limit of RR < 2).
Hepatic AEs were recorded in 20 studies (353 events (4.91%) in 7183 participants). Hepatic AEs may have been increased by azathioprine, interferon beta‐1a (Rebif), and fingolimod. There was a lot of uncertainty about all other comparisons.
Immune system adverse events
Figure 19 shows NMA results for immune system SAEs and AEs.
19.
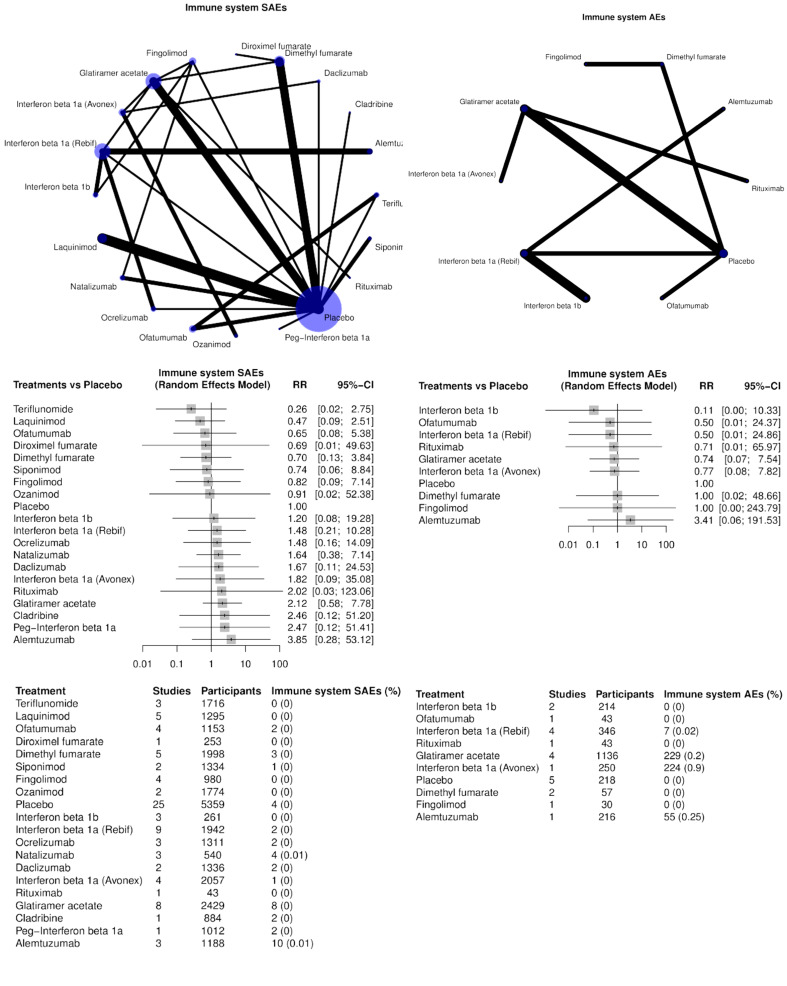
Immune system SAEs( left) and AEs (right). In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Immune system SAEs were recorded in 44 studies (43 events (0.15%) in 28,865 participants). There was a lot of uncertainty about all drugs.
Immune system AEs were recorded in 11 studies (515 events (20.2%) in 2553 participants), but no events were reported for placebo, interferon beta‐1b, ofatumumab, rituximab, dimethyl fumarate, and fingolimod. On the contrary, these events were very common in trials using interferon beta‐1a (Avonex) (89.6%), glatiramer acetate (20.2%) and alemtuzumab (25.0 %), which raises the issue of comparability of trials with such different AE definitions or risks.
Pregnancy, puerperal and perinatal adverse events
Figure 20 shows NMA results for pregnancy, puerperal and perinatal adverse events.
20.
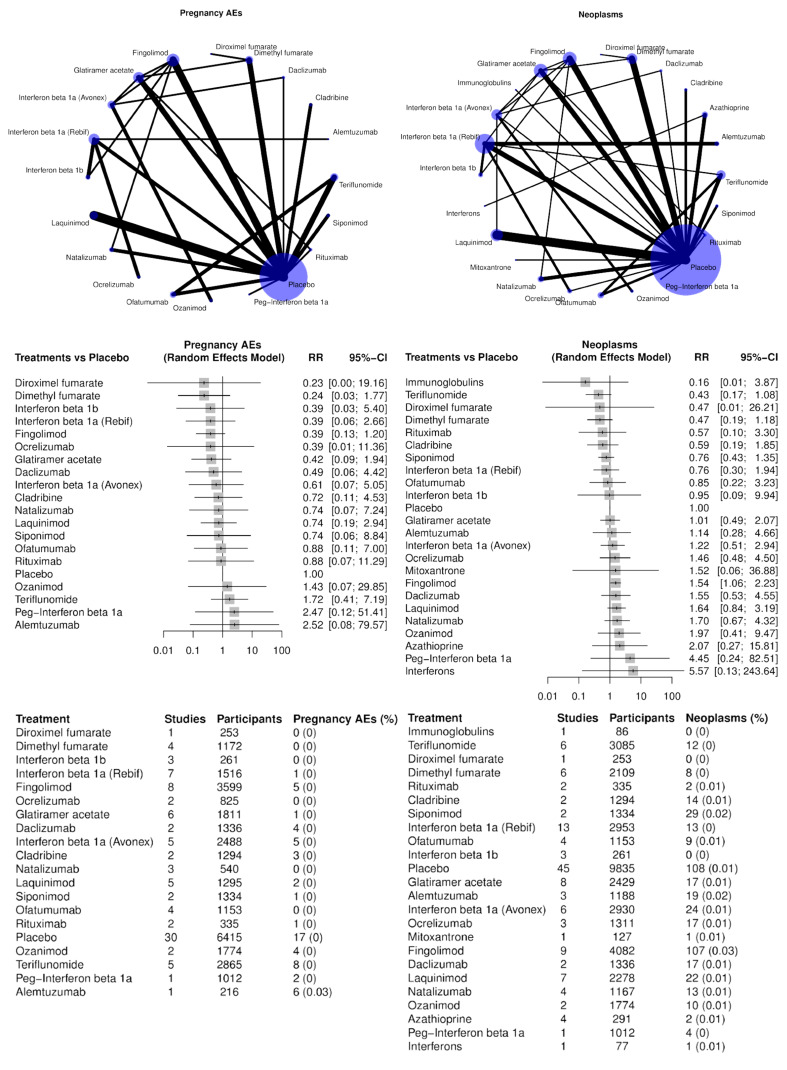
Pregnancy, puerperal, perinatal (left) and neoplasm (right) AEs. In the summary tables at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Pregnancy, puerperal and perinatal adverse events were recorded in 47 studies and were very rare (60 events (0.19%) in 31,494 participants). There was much uncertainty for all drugs, and an upper 95% CI limit of RR < 2 was recorded only for dimethyl fumarate, fingolimod, and glatiramer acetate. It is important to note that pregnancy is usually amongst the exclusion criteria in trials on DMTs.
Neoplasms
Figure 20 shows NMA results for neoplasms.
Neoplasms were recorded in 67 studies (449 events (1.05%) in 42,700 participants). There was uncertainty for all drugs, with estimates including no difference. An upper 95% CI limit of RR < 2 versus placebo was recorded for teriflunomide, dimethyl fumarate, cladribine, siponimod, interferon beta‐1a (Rebif), with glatiramer acetate approaching this threshold.
Death
Figure 21 shows NMA results for death.
21.
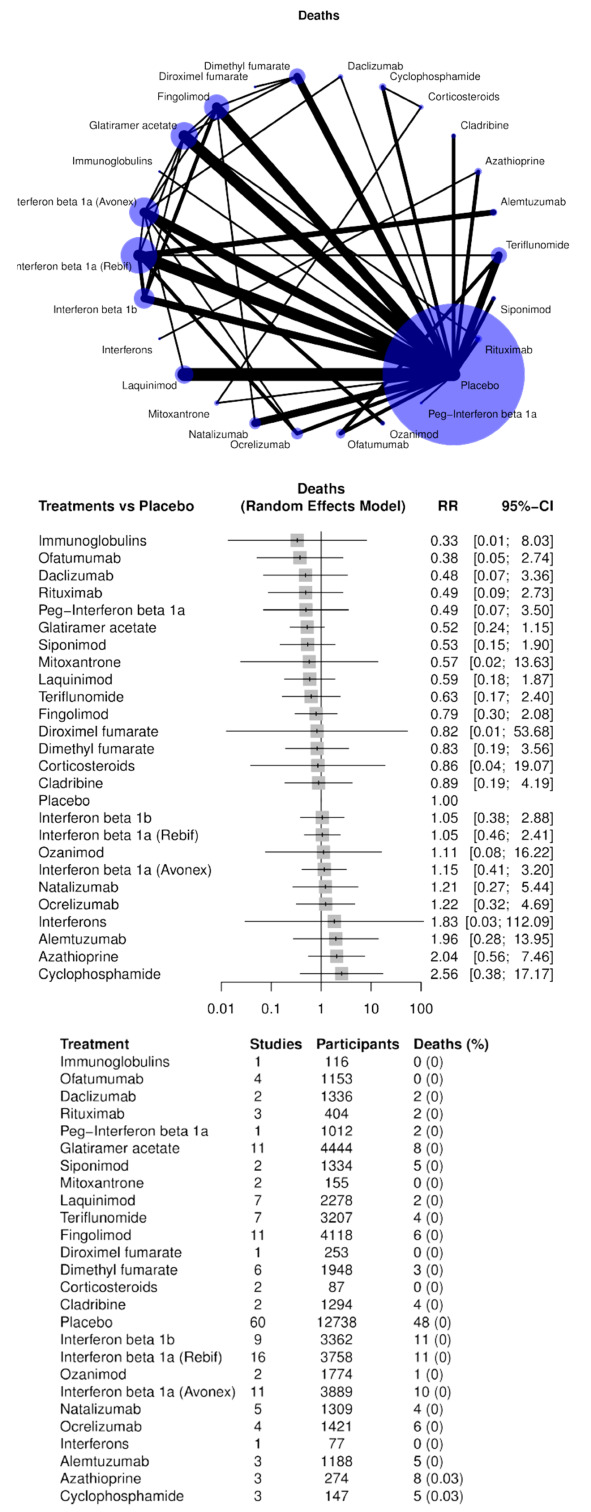
Death. In the summary table at the bottom, % stands for the ratio between the number of events and the number of participants comprised between 0 and 1.
Death was recorded in 88 studies (147 events (0.28%) in 53,077 participants). There was a lot of uncertainty about all drugs, with all estimates including no difference. An upper 95% CI limit of RR < 2 was recorded only for glatiramer acetate, siponimod and laquinimod.
Rankings for all SAEs
Table 1 shows the P scores for all drugs and all SAE outcomes, sorted in descending order according to the score for SAE. Data are sorted from the best drug regarding SAEs. Cells in bold character highlight treatments with a P score ≥ 0.75 (upper probability quartile). Empty cells mean that a given treatment was not included in the NMA.
Interferon beta‐1a (Avonex), dimethyl fumarate, daclizumab, glatiramer acetate, and ocrelizumab were in the upper probability quartile of being the best drugs for all SAEs. Dimethyl fumarate was in the upper quartile also regarding blood SAEs, pregnancy‐puerperal‐perinatal and neoplasm AEs.
Diroximel fumarate, alemtuzumab, placebo and ozanimod were in the upper quartile regarding withdrawals due to AEs. However, this is no surprise for alemtuzumab which is infused yearly; this may improve compliance.
Discussion
Summary of main results
This review has summarized the evidence on the relative safety of immunotherapies for treating MS. Most of the studies were short‐term trials, with the median duration being 24 months; therefore, any harmful effects of these treatments beyond two years remain uncertain.
Serious adverse events are a heterogeneous category that mainly includes all‐cause hospitalization, which may not necessarily be treatment‐related. As such, SAEs may trade both harms and benefits, and the fact that we found some drugs to actually reduce SAEs (interferon beta‐1a (Avonex), dimethyl fumarate, glatiramer acetate, and possibly daclizumab) may be due to a real beneficial effect or to chance, given that this evidence was low or very low‐certainty. We found low‐certainty evidence that several drugs are non‐inferior to placebo in terms of safety concerning SAEs.
Withdrawals due to adverse events may be a better generic indicator of safety and have direct implications on treatment compliance. We found low‐certainty evidence that several drugs increased withdrawals compared to placebo.
Using rankings, interferon beta‐1a (Avonex), dimethyl fumarate, daclizumab, glatiramer acetate, and ocrelizumab were in the upper quartile of a probability of having fewer SAEs. Diroximel fumarate, alemtuzumab, and ozanimod were in the upper quartile of having fewer withdrawals.
We found that there was no evidence of a dose‐effect for any of the included treatments in a sensitivity analysis restricted to drug doses above the median. Sensitivity analyses including only studies with low attrition bias and including only patients with relapsing‐remitting MS or CIS showed consistent results with the overall analyses. The subgroup analysis by prior disease‐modifying treatments did not show differences between naive and non‐naive participants both for SAEs and withdrawals.
Overall completeness and applicability of evidence
A total of 123 eligible RCTs were included. We analyzed two primary outcomes and twelve secondary outcomes, as prespecified in our published protocol (Tramacere 2016). Sixty‐eight per cent and 85.4% of the included trials reported the number of participants who experienced SAEs or withdrew due to AEs over 12 or 24 months’ follow up. However, few studies with low numbers of events reported data on the secondary outcomes, leading to uncertainty about the risk profile of the treatments included in the review. Our literature search identified a number of ongoing trials which could provide valuable data in addition to that presented in this review; we will include these in future updates.
SAEs and withdrawals due to AEs did not differ between participants who had been previously treated with disease‐modifying treatments and naive participants. SAEs and withdrawals due to AEs also did not differ between participants with relapsing or progressive forms of MS.
Several factors limit the applicability of the evidence in our review. First, the evidence for SAEs and withdrawals due to AEs was derived from RCTs on DMTs mostly compared with placebo. Few data are available on DMT prescribing patterns in the real‐world MS population. However, the North American Research Committee on Multiple Sclerosis (NARCOMS) registry and the US Department of Veterans Affairs Multiple Sclerosis Surveillance Registry (MSSR) reported that about 60 to 70% of young adults with MS are treated with at least one of these medicines (Zhang 2021). Therefore, there is uncertainty about whether the results of adverse events in the review could be applied to current practice. Secondly, this review included data from RCTs, in which selected populations of MS participants (with the exclusion of several comorbidities, concomitant treatments, and young age range) that were managed in highly‐controlled settings were followed up for a relatively short period (average of two years). Moreover, the pressures on participants and investigators under trial conditions to reduce the number of withdrawals and dropouts can result in rates that do not reflect the experience of adverse events within the wider population (Peryer 2020). The extent to which RCT safety can be extrapolated to real‐life MS populations and routine care settings is limited. Third, the administration of co‐therapies during follow‐up was poorly reported amongst the included studies, and this is another limitation of the evidence. Fourth, the short duration of the studies did not enable us to determine the long‐term harms and rare adverse events of DMTs for people with MS.
Quality of the evidence
We frequently downgraded the certainty of the evidence for SAEs and withdrawals due to AEs from the initial level of high certainty due to study limitations and then either due to inconsistency, imprecision or incoherence, resulting in low or very low‐certainty evidence for most of the comparisons. We judged 59% of included trials to be at high risk of bias, when criteria for blinding of participants and personnel, blinding of outcome assessors, or complete outcome data were unmet. Blinding was described in the majority of included studies; however, 23% of included RCTs were at a high risk of detection bias as they were described as ‘open‐label’. A 'treating' physician assessed SAEs or made a clinical decision to withdraw treatment due to AEs in most of the included studies. Thus, both outcomes were potentially influenced by knowledge of the intervention received, leading to a judgment of "unclear" risk of detection bias for 64% of included trials. Half of the trials were at low risk for incomplete outcome data, and one‐third were at high risk due to unbalanced numbers or reasons for dropouts, or both, between the comparison groups. The frequency of downgrading the certainty of the evidence regarding SAEs and withdrawals due to AEs was respectively 32% and 21% of treatment estimates for imprecision; 0% and 42% for inconsistency or incoherence.
One‐third of trials did not report any definition of SAEs and key information was missing on criteria used to assess and select SAEs per arm, and one‐third of studies did not report whether results were based on active monitoring or spontaneously reported AEs. This is another limitation of the quality of the evidence, because results based on spontaneously reported adverse outcomes may lead to concerns that these were selected post hoc based on the finding being noteworthy (Peryer 2020). Moreover, passive surveillance of harms leads to fewer recorded SAEs than active surveillance (Ioannidis 2004).
Data collection and reporting of SAEs and their attribution to study treatment were assigned by the 'treating' physician or the nurse (investigator) in all the included trials, but methods and factors used to assign attribution were not reported in the majority of articles or study protocols, and the process was likely highly subjective. Another reason for concern that makes the reliability of attribution questionable is that several trials reported only a composite measure of SAEs, e.g. "Any serious adverse event" or "Number of patients with serious adverse events", which does not give information on what exactly the events were in the comparison groups. The subjective decision of potentially unblinded investigators to include and count SAEs not related to study interventions may have led to bias in selection of the reported result or differential reporting of SAEs between the comparison groups.
About 40% of included trials reported SAEs and AEs according to the terminology of the Medical Dictionary for Regulatory Activities (MedDRA); however, they did not categorize each adverse outcome as to its potential attribution to study interventions or, instead, possibly related to MS, other medical conditions, or to concomitant treatments. For example, severe relapse of MS, i.e. one of the beneficial outcomes in included trials, was also counted as an SAE in about one‐third of them and, not surprisingly, relapse was more frequent in the placebo arm. The same is true for another potential event overlapping with MS‐related symptoms, e.g. disability worsening, severe fatigue, depression considered as SAEs.
Potential biases in the review process
1. Transitivity assumption
We assumed that any participant who met the inclusion criteria was, in principle, equally likely to have been randomized to any of the eligible interventions. However, several participant characteristics have changed in newer trials, and thus a transitivity hypothesis may not have been a reasonable assumption to make, due to differences in participant or trial characteristics. Characteristics of MS participants in trials have changed over time, as a result of changes in inclusion criteria. Since 2010, the successive revisions of the McDonald criteria have broadened the diagnostic criteria. Thus, many patients were diagnosed early and participants in new trials presented lower disease activity and slower clinical progression compared with participants in older trials (Zhang 2019). However, we simply don't know whether these changes acted as effect modifiers on adverse events.
2. Heterogeneity, inconsistency, incoherence
We did not find any strong evidence of the presence of heterogeneity either in direct pairwise comparisons or in the entire network. Similarly, the loop‐specific approach and the ’design‐by‐treatment’ model did not provide any clear indication of the presence of inconsistency either locally or in the entire network. Thus, we believe that the consistency assumption is reasonable for this type of data. However, the power of these tests and approaches to detect inconsistency is low, particularly for networks with few included studies per comparison. Because only direct or only indirect evidence was available for many treatments regarding withdrawals, we could not verify incoherence for some indirect comparisons and this was an additional source of concern.
3. Subgroup and sensitivity analyses
None of the analyses performed on any of the hypothesized effect modifiers provided any significantly different results compared to the overall analyses.
4. Multiple testing and multiple interval estimation
Adjustments for multiple tests are not routinely used in systematic reviews, nevertheless, issues of multiplicity apply just as much to systematic reviews as to other types of research (Bender 2008; Chen 2005). There is no simple or completely satisfactory solution to the problem of multiple testing in systematic reviews; however, outcome classification in advance as primary and secondary outcomes, keeping subgroup analyses to a minimum and selecting results for emphasis on estimating intervention effects rather than testing for them, should have reduced the effect of this potential bias.
5. Selective non‐reporting
SAEs and withdrawals due to AEs were reported by 90% and 97% of participants; therefore, we judged that non‐reporting did not cause bias in the results for the two primary outcomes. However, selective non‐reporting may have caused bias in the results of many secondary outcomes which were reported only in subsets of studies (Figure 5).
Agreements and disagreements with other studies or reviews
In our review, we found mostly low or very‐low certainty evidence that DMTs used to treat MS may not increase SAEs but may increase withdrawals due to AEs compared with placebo during a median two years' follow‐up period. All the agents were associated with a higher rate of total withdrawals due to AEs compared with placebo. Using rankings, diroximel fumarate, alemtuzumab, ozanimod were in the upper quartile of having fewer withdrawals. These findings did not change in a sensitivity analysis including only studies on relapsing MS or clinically isolated syndrome.
Another systematic review with NMA (Śladowska 2022) focused on the safety profile of DMTs in relapsing MS. This study found no differences between drugs in terms of SAEs except for cladribine (3.5 mg, 17.3%) versus ocrelizumab (10.3%) and ofatumumab (16.6%) versus ocrelizumab. These are higher rates than we demonstrated (9% with cladribine and 8% with ofatumumab). They did not find significant differences in AEs leading to the discontinuation of study drugs, except for ponesimod (10.1%) versus alemtuzumab (12 mg, 3.0%) and placebo (4.2%); the numbers of discontinuations for alemtuzumab and placebo were consistent with ours (2% with alemtuzumab and 4% with placebo). Ponesimod was not included in our review. Śladowska 2022 also showed a significant increase in serious infections and urinary tract infections with alemtuzumab.
Other reviews have reported on the benefits and safety profiles of DMTs. One Cochrane review with NMA (Filippini 2013) estimated the relative efficacy and acceptability of interferon ß‐1b, interferon ß‐1a (Rebif and Avonex), glatiramer acetate, natalizumab, mitoxantrone, methotrexate, cyclophosphamide, azathioprine, intravenous immunoglobulins, and long‐term corticosteroids versus placebo or another active agent in participants with all phenotypes of MS. All the agents included in the review were associated with a significantly higher rate of withdrawals due to AEs compared with placebo, without differences from each other. All of them, except interferons, were associated with a non‐significantly higher rate of total SAEs compared with placebo during a median two years' follow‐up period. Authors commented in their discussion that there was overall poor reporting of AEs and SAEs, and short follow‐up in the included studies. A Cochrane review with NMA (Tramacere 2015) compared the benefit and acceptability of interferon beta‐1b, interferon beta‐1a (Avonex, Rebif), glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, pegylated interferon beta‐1a, daclizumab, laquinimod, azathioprine and immunoglobulins for the treatment of relapsing MS and provided a ranking of these treatments according to the proportion of participants who withdrew due to any AE. Almost all the included agents were associated with a higher proportion of withdrawals compared with placebo at 12 and 24 months. All the treatments were associated with a non‐significantly higher proportion of people with at least one SAE compared with placebo during a median two‐years' follow‐up period. The authors commented in the discussion that information on SAEs was scanty, poorly reported and characterized by heterogeneous results. Pooled data from 28 RCTs in a NMA included interferon beta, peginterferon beta, glatiramer acetate, mitoxantrone, natalizumab, fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, cyclophosphamide, laquinimod, ocrelizumab, cladribine, azathioprine, rituximab, ozanimod and ofatumumab for relapsing MS. Despite that discontinuation due to AEs of most DMTs was higher than placebo, nearly all of them did not meet statistical significance. In Xu 2018 (a NMA of 14 RCTs), natalizumab, natalizumab plus INFβ‐1a, alemtuzumab, daclizumab, ocrelizumab, and placebo were compared with INFβ‐1a. All biological treatments had a similar incidence rate of SAEs, except that placebo had a tendency of more serious adverse events, which could be explained by more hospitalizations for treatment of relapses of MS.
Most of our findings cannot be compared to most previous reviews, other than those described above, because they focused primarily on efficacy outcomes (Hamidi 2018; Li 2020; Lucchetta 2019; McCool 2019; Samjoo 2020; Siddiqui 2018; Silva 2022).
As reported in one recent systematic review (Lopez‐Leon 2020), in terms of pregnancy and perinatal adverse events, we did not find informative data because pregnancy is usually amongst the exclusion criteria in trials on DMTs. Lopez‐Leon 2020 is a systematic review aiming to measure the effects of DMTs on pregnancy and fetal outcomes. They searched for relevant publications from the period January 2000 to August 2019 and identified only six small observational studies on interferon, glatiramer acetate, and natalizumab. These drugs did not appear to increase the risk of spontaneous abortions, pre‐term birth or major congenital malformations. There was inconclusive information found on the AEs of all other DMTs used in clinical practice.
Authors' conclusions
Implications for practice.
We found mostly low‐certainty evidence that drugs used to treat MS may not increase SAEs, but may increase withdrawals compared with placebo, which may affect treatment compliance.
The results of our review suggest that there is no important difference in the occurrence of serious adverse effects between injectable first‐line (interferons beta and glatiramer acetate) and second‐line disease‐modifying treatments (natalizumab, rituximab, or ocrelizumab) compared with placebo; and no important difference between oral drugs (fingolimod, teriflunomide, dimethyl fumarate, laquinimod, ozanimod), injectable drugs (interferons beta, glatiramer acetate, daclizumab), or infused drugs (natalizumab, rituximab, or ocrelizumab), compared with placebo.
Regarding withdrawals due to adverse events, the very‐poor quality of the evidence collected for several drugs prevents us from making positive statements on which drugs may have better compliance. No drug was found to reduce withdrawals due to adverse events when compared with placebo.
Implications for research.
Our review, along with other work in the literature, confirms that RCTs of relevant interventions have poor‐quality reporting of adverse events (serious and not serious). In order to draw robust conclusions about the harmful effects and the risk/benefit profile of different disease‐modifying treatments for MS, studies should follow the CONSORT recommendations about reporting harm‐related issues (Ioannidis 2004).
Medium and long‐term adverse effects, particularly of early intensive treatment with drugs, which have different mechanisms of action but the same therapeutic indication (natalizumab, alemtuzumab, mitoxantrone, fingolimod, cladribine, rituximab, or ocrelizumab), should be comprehensively evaluated considering their risk/benefit profile in clinical care.
Importantly, there is a need for head‐to‐head comparison(s) trials in order to draw solid conclusions on the comparative safety of disease‐modifying treatments. In the present review, two‐thirds of included trials adopted placebo as comparator, rather than an active comparator, which we and others find an unwelcome development (Garattini 2013).
To address adverse effects, it may be necessary to also seek non‐randomized studies, because the effects are unlikely to be seen in randomized trials due to their small size, short duration and selected eligibility of participants (Peryer 2020). Future systematic reviews should therefore include national and international registries and other types of large non‐randomized studies which are relevant sources for providing complementary data on the long‐term safety of disease‐modifying treatments for MS.
History
Protocol first published: Issue 5, 2016
Acknowledgements
We would like to acknowledge and thank the following people for their help in assessing the search results for this review via Cochrane’s Screen4Me workflow: Nikolaos Sideris, Eleanor McKean, Didier Ruedin, Devon Hone, Mohith Kumar Abaka, Catherine Batias, Yuna Shun‐hin Wong, Susanna Wisniewski, Kaveh Haji‐Allahverdipoor, Mohammad Aloulou, Luis Coloma, Hadi Keshavarz, Shivangi Srivastava, Yuan Chi, Linda Hart, Cheryl Chan, Niwanda Yogiswara, Basavaraj Poojar, Lucas Henrique Caetano Carmona dos Santos, Adrià Bermudo‐Gallaguet, Jens Thorsen, Akhilanand Chaurasia, Jehath Syed, Shammas Mohammed, Avishek Mukherjee, Mary MacCara, Shirley Hall, Lai Ogunsola, Hebatullah Abdulazeem, Oscar Ngongo, Luma Haj Kassem, Michele Tognetti, Amin Sharifan, Anna Noel‐Storr, Ilkin Mengusoglu, and Andrea Paola Ramos Mora.
Cochrane Multiple Sclerosis and Rare Diseases of the CNS supported the authors in the development of this review. We thank Andrea Fittipaldo, Maria Domenica Camerlingo, and Maria Chiara Bassi for contributing to the search strategies and methods used to identify studies. Graziella Filippini is a member of Cochrane Multiple Sclerosis and Rare Diseases of the CNS but was not involved in the editorial process or decision‐making for this review.
The following people conducted the editorial process for this review:
Sign‐off Editor (final editorial decision): Robert Boyle, Imperial College London
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial comments/guidance to authors, edited the article): Joey Kwong, Cochrane Central Editorial Service
Editorial Assistant (conducted editorial policy checks, collated peer‐reviewer comments, supported editorial team): Leticia Rodrigues, Cochrane Central Editorial Service
Copy Editor (copy‐editing and production): Anne Lethaby, c/o Cochrane Central Production Service
Peer‐reviewers (provided comments and recommended an editorial decision): Georgios Tsivgoulis, Second Department of Neurology, School of Medicine, National & Kapodistrian University of Athens, Athens, Greece (clinical/content review); Emilio Portaccio, University of Florence, Florence, Italy (clinical/content review); Kristin Osika (consumer review); Guy Peryer, University of East Anglia (methods review); Nuala Livingstone, Kerry Dwan and Sofia Tsokani, Cochrane Evidence Production & Methods Directorate (methods reviews); Farhad Shokraneh, Cochrane Neuromuscular Group, University College London Hospitals and Yuan Chi, Cochrane Campbell Global Ageing Partnership (search reviews)*. *Robin Featherstone, formerly of Cochrane Evidence Production & Methods Directorate, provided search review prior to her involvement as a co‐author.
Appendices
Appendix 1. Appendix: Search strategies for databases and trial registers
Database: PubMed
Date search conducted: March 4, 2022
| # | Query | Results |
| 1 | "adverse effects"[MeSH Subheading] AND "multiple sclerosis/drug therapy"[MeSH Major Topic] | 2843 |
| 2 | "demyelinating autoimmune diseases, cns"[MeSH Terms:noexp] | 525 |
| 3 | "Demyelinating Diseases"[MeSH Terms:noexp] | 12,549 |
| 4 | "Multiple Sclerosis"[MeSH Terms] | 65,411 |
| 5 | "myelitis, transverse"[MeSH Terms] | 5116 |
| 6 | "Optic Neuritis"[MeSH Terms] | 9678 |
| 7 | "clinically isolated syndrome*"[Title/Abstract] | 1767 |
| 8 | "devic"[Title/Abstract] OR "devic s"[Title/Abstract] OR "devics"[Title/Abstract] | 560 |
| 9 | "disseminated sclerosis*"[Title/Abstract] | 638 |
| 10 | "demyelinating disease*"[Title/Abstract] OR "demyelinating disorder*"[Title/Abstract] | 8871 |
| 11 | "demyelinating myelitis*"[Title/Abstract] OR "necrotising myelitis*"[Title/Abstract] OR "necrotizing myelitis*"[Title/Abstract] OR "transverse myel*"[Title/Abstract] | 2845 |
| 12 | "multiple sclerosis*"[Title/Abstract] OR "MS"[Title] | 122,360 |
| 13 | "neuropapilliti*"[Title/Abstract] OR "optic neuriti*"[Title/Abstract] OR "retrobulbar neuriti*"[Title/Abstract] | 7007 |
| 14 | "neuromyelitis optica*"[Title/Abstract] OR "nmo spectrum disorder*"[Title/Abstract] | 4997 |
| 15 | #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 | 150,219 |
| 16 | "adrenal cortex hormones/adverse effects"[MeSH Terms:noexp] OR "adrenal cortex hormones/drug effects"[MeSH Terms:noexp] OR "adrenal cortex hormones/drug therapy"[MeSH Terms:noexp] OR "adrenal cortex hormones/therapeutic use"[MeSH Terms:noexp] | 39,585 |
| 17 | "Alemtuzumab"[MeSH Terms] | 2182 |
| 18 | "Azathioprine"[MeSH Terms] | 15,061 |
| 19 | "Cladribine"[MeSH Terms] | 1634 |
| 20 | "Cyclophosphamide"[MeSH Terms:noexp] | 51,980 |
| 21 | "Daclizumab"[MeSH Terms] | 714 |
| 22 | "Dimethyl Fumarate"[MeSH Terms] | 894 |
| 23 | "Fingolimod Hydrochloride"[MeSH Terms] | 2513 |
| 24 | "Glatiramer Acetate"[MeSH Terms] | 1449 |
| 25 | "immunoglobulins/adverse effects"[MeSH Terms:noexp] OR "immunoglobulins/drug effects"[MeSH Terms:noexp] OR "immunoglobulins/therapeutic use"[MeSH Terms:noexp] OR "immunoglobulins, intravenous"[MeSH Terms] | 17,042 |
| 26 | "Interferon‐beta"[MeSH Terms] | 10,127 |
| 27 | "Interferon Type I"[MeSH Terms:noexp] | 15,451 |
| 28 | "Methotrexate"[MeSH Terms] | 39,919 |
| 29 | "Methylprednisolone"[MeSH Terms:noexp] | 19,778 |
| 30 | "Mitoxantrone"[MeSH Terms] | 4356 |
| 31 | "Natalizumab"[MeSH Terms] | 1808 |
| 32 | "Prednisolone"[MeSH Terms:noexp] | 33,796 |
| 33 | "Rituximab"[MeSH Terms] | 17,048 |
| 34 | "adrenal cortex hormone*"[Title/Abstract] OR "corticosteroid*"[Title] OR "cortico steroid*"[Title] OR "corticoid*"[Title/Abstract] | 42,663 |
| 35 | "alemtuzumab*"[Title/Abstract] OR "campath*"[Title/Abstract] OR "lemtrada*"[Title/Abstract] | 3435 |
| 36 | "avonex*"[Title/Abstract] OR "rebif*"[Title/Abstract] | 477 |
| 37 | "aubagio*"[Title/Abstract] OR "teriflunomide*"[Title/Abstract] | 662 |
| 38 | "azathioprine*"[Title/Abstract] OR "azothioprine*"[Title/Abstract] OR "imurel*"[Title/Abstract] OR "imuran*"[Title/Abstract] OR "immuran*"[Title/Abstract] | 16,599 |
| 39 | "bafiertam*"[Title/Abstract] OR "monomethyl fumarate*"[Title/Abstract] OR "methyl hydrogen fumarate*"[Title/Abstract] OR "methylhydrogenfumarate*"[Title/Abstract] | 114 |
| 40 | "beta interferon*"[Title/Abstract] OR "beta 1 interferon*"[Title/Abstract] OR "interferon beta*"[Title/Abstract] OR "fiblaferon*"[Title/Abstract] OR "fibroblast interferon*"[Title/Abstract] OR "ifnbeta*"[Title/Abstract] OR "ifn beta*"[Title/Abstract] OR "interferon*"[Title] | 69,947 |
| 41 | "betaferon*"[Title/Abstract] OR "betaseron*"[Title/Abstract] OR "beta seron*"[Title/Abstract] OR "extavia*"[Title/Abstract] | 315 |
| 42 | "copaxone*"[Title/Abstract] OR "Cop 1"[Title/Abstract] OR "copolymer 1"[Title/Abstract] OR "glatiramer*"[Title/Abstract] OR "glatopa*"[Title/Abstract] OR "TV 5010"[Title/Abstract] OR "TV5010"[Title/Abstract] | 2169 |
| 43 | "cladribine*"[Title/Abstract] OR "leustatin*"[Title/Abstract] OR "mavenclad*"[Title/Abstract] OR "movectro*"[Title/Abstract] | 1567 |
| 44 | "cyclophosphamide*"[Title/Abstract] OR "cyclophosphane*"[Title/Abstract] OR "cytophosphan*"[Title/Abstract] OR "cytoxan*"[Title/Abstract] OR "endoxan*"[Title/Abstract] OR "neosar*"[Title/Abstract] OR "procytox*"[Title/Abstract] OR "sendoxan*"[Title/Abstract] | 53,432 |
| 45 | "daclizumab*"[Title/Abstract] OR "zinbryta*"[Title/Abstract] OR "zenapax*"[Title/Abstract] | 1016 |
| 46 | "dimethylfumarate"[Title/Abstract] OR "dimethyl fumarate*"[Title/Abstract] OR "BG 00012"[Title/Abstract] OR "BG00012"[Title/Abstract] OR "BG 12"[Title/Abstract] OR "diroximel fumarate*"[Title/Abstract] OR "tecfidera*"[Title/Abstract] OR "vumerity*"[Title/Abstract] | 1522 |
| 47 | "fingolimod*"[Title/Abstract] OR "gilenya*"[Title/Abstract] OR "gilenia*"[Title/Abstract] OR "FTY 720"[Title/Abstract] OR "FTY720"[Title/Abstract] | 3643 |
| 48 | "immunoglobulin*"[Title] OR "intravenous immunoglobulin*"[Title/Abstract] OR "iv immunoglobulin*"[Title/Abstract] OR "IVIG"[Title/Abstract] | 60,363 |
| 49 | "kesimpta*"[Title/Abstract] OR "ofatumumab*"[Title/Abstract] OR "HUMAX CD20 2F2"[Title/Abstract] OR "GSK 1841157"[Title/Abstract] OR "GSK1841157"[Title/Abstract] | 614 |
| 50 | "laquinimod*"[Title/Abstract] OR "ABR 215062"[Title/Abstract] OR "ABR215062"[Title/Abstract] | 196 |
| 51 | "mayzent*"[Title/Abstract] OR "siponimod*"[Title/Abstract] OR "BAF 312"[Title/Abstract] OR "BAF312"[Title/Abstract] | 174 |
| 52 | "methotrexate*"[Title/Abstract] OR "amethopterin*"[Title/Abstract] OR "mexate*"[Title/Abstract] | 45,329 |
| 53 | "methylprednisolone*"[Title/Abstract] OR "metipred*"[Title/Abstract] | 18,358 |
| 54 | "mitoxantrone*"[Title/Abstract] OR "mitozantrone*"[Title/Abstract] OR "ralenova*"[Title/Abstract] OR "novantron*"[Title/Abstract] OR "onkotrone*"[Title/Abstract] | 5522 |
| 55 | "natalizumab*"[Title/Abstract] OR "tysabri*"[Title/Abstract] OR "antegren*"[Title/Abstract] | 2692 |
| 56 | "ocrelizumab*"[Title/Abstract] OR "ocrevus*"[Title/Abstract] OR "R 1594"[Title/Abstract] OR "PR070769"[Title/Abstract] | 585 |
| 57 | "ozanimod*"[Title/Abstract] OR "zeposia*"[Title/Abstract] OR "RPC1063"[Title/Abstract] | 134 |
| 58 | "peginterferon*"[Title/Abstract] OR "pegylated interferon*"[Title/Abstract] OR "plegridy*"[Title/Abstract] OR "peg ifn beta*"[Title/Abstract] | 9070 |
| 59 | "prednisolone*"[Title/Abstract] OR "predonine*"[Title/Abstract] | 29,863 |
| 60 | "rituximab*"[Title/Abstract] OR "rituxan*"[Title/Abstract] OR "mabthera*"[Title/Abstract] OR "IDEC C2B8"[Title/Abstract] | 24,433 |
| 61 | #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #44 OR #45 OR #46 OR #47 OR #48 OR #49 OR #50 OR #51 OR #52 OR #53 OR #54 OR #55 OR #56 OR #57 OR #58 OR #59 OR #60 | 441,628 |
| 62 | #15 AND #61 | 17,698 |
| 63 | #1 OR #62 | 18,232 |
| 64 | "randomized controlled trial"[Publication Type] | 561,490 |
| 65 | "controlled clinical trial"[Publication Type] | 651,322 |
| 66 | "randomized"[Title/Abstract] | 601,526 |
| 67 | "placebo"[Title/Abstract] | 232,512 |
| 68 | "Clinical Trials as Topic"[MeSH Terms:noexp] | 199,385 |
| 69 | "randomly"[Title/Abstract] | 378,097 |
| 70 | "trial*"[Title] | 342,500 |
| 71 | #64 OR #65 OR #66 OR #67 OR #68 OR #69 OR #70 | 1,471,816 |
| 72 | "animals"[MeSH Terms] NOT "humans"[MeSH Terms] | 4,966,901 |
| 73 | #71 NOT #72 | 1,355,187 |
| 74 | #63 AND #73 | 3044 |
Database: Embase.com (Elsevier)
Date search conducted: March 4, 2022
| # | Query | Results |
| 1 | 'demyelinating disease'/de | 17,188 |
| 2 | 'multiple sclerosis'/de | 147,781 |
| 3 | 'optic neuritis'/de | 12,139 |
| 4 | 'transverse myelitis'/exp | 671 |
| 5 | 'clinically isolated syndrome*':ab,ti | 3860 |
| 6 | devic:ab,ti OR 'devic s':ab.ti OR devics:ab,it | 350 |
| 7 | 'disseminated sclerosis*':ab,ti | 620 |
| 8 | (demyelinating NEAR/1 (disease* OR disorder*)):ab,ti | 13,394 |
| 9 | ((demyelinating OR necrotising OR necrotizing OR transverse) NEAR/1 myelitis*):ab,ti | 4453 |
| 10 | 'multiple sclerosis*':ab,ti OR 'MS':ti | 180,386 |
| 11 | neuropapilliti*:ab,ti OR ((optic OR retrobulbar) NEAR/1 neuriti*"):ab,ti | 11,150 |
| 12 | 'neuromyelitis optica*':ab,ti OR 'nmo spectrum disorder*':ab,ti | 9298 |
| 13 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 | 231,754 |
| 14 | 'alemtuzumab'/de | 18,001 |
| 15 | 'azathioprine'/de | 102,692 |
| 16 | 'beta interferon'/exp | 324,261 |
| 17 | 'cladribine'/de | 7994 |
| 18 | 'corticosteroid'/dd_ae OR 'corticosteroid'/dd_dt | 154,759 |
| 19 | 'cyclophosphamide'/dd_ae OR 'cyclophosphamide'/dd_dt | 144,522 |
| 20 | 'daclizumab'/de | 6275 |
| 21 | 'dimethyl fumarate'/de | 5371 |
| 22 | 'fingolimod'/de | 11,470 |
| 23 | 'glatiramer'/de | 9874 |
| 24 | 'immunoglobulin'/dd_ae OR 'immunoglobulin'/dd_dt OR 'immunoglobulin'/dd_iv | 54,102 |
| 25 | 'methotrexate'/dd_ae OR 'methotrexate'/dd_dt | 117,944 |
| 26 | 'methylprednisolone'/de | 109,285 |
| 27 | 'mitoxantrone'/de | 24,591 |
| 28 | 'natalizumab'/de | 12,030 |
| 29 | 'prednisolone'/de | 142,902 |
| 30 | 'rituximab'/de | 93,776 |
| 31 | 'adrenal cortex hormone*':ab,ti OR 'corticosteroid*':ti OR 'cortico steroid*':ti OR 'corticoid*':ab,ti | 47,123 |
| 32 | 'alemtuzumab*':ab,ti OR 'campath*':ab,ti OR 'lemtrada*':ab,ti | 8121 |
| 33 | avonex*:ab,ti OR rebif*:ab,ti | 862 |
| 34 | 'aubagio*':ab,ti OR 'teriflunomide*':ab,ti | 1783 |
| 35 | 'azathioprine*':ab,ti OR 'azothioprine*':ab,ti OR 'imurel*':ab,ti OR 'imuran*':ab,ti OR 'immuran*':ab,ti | 29,441 |
| 36 | 'bafiertam*':ab,ti OR 'monomethyl fumarate*':ab,ti OR 'methyl hydrogen fumarate*':ab,ti OR 'methylhydrogenfumarate*':ab,ti | 233 |
| 37 | 'beta interferon*':ab,ti OR 'beta 1 interferon*':ab,ti OR 'interferon beta*':ab,ti OR 'fiblaferon*':ab,ti OR 'fibroblast interferon*':ab,ti OR 'IFNbeta*':ab,ti OR 'IFN beta*':ab,ti OR 'interferon':ti | 77,366 |
| 38 | 'betaferon*':ab,ti OR 'betaseron*':ab,ti OR 'beta seron*':ab,ti OR 'extavia*':ab,ti | 555 |
| 39 | 'copaxone*':ab,ti OR 'Cop 1':ab,ti OR 'copolymer 1':ab,ti OR 'glatiramer*':ab,ti OR 'glatopa*':ab,ti OR 'TV 5010':ab,ti OR 'TV5010':ab,ti | 4722 |
| 40 | 'cladribine*':ab,ti OR 'leustatin*':ab,ti OR 'mavenclad*':ab,ti OR 'movectro*':ab,ti | 2969 |
| 41 | 'cyclophosphamide*':ab,ti OR 'cyclophosphane*':ab,ti OR 'cytophosphan*':ab,ti OR 'cytoxan*':ab,ti OR 'endoxan*':ab,ti OR 'neosar*':ab,ti OR 'procytox*':ab,ti OR 'sendoxan*':ab,ti | 85,190 |
| 42 | 'daclizumab*':ab,ti OR 'zinbryta*':ab,ti OR 'zenapax*':ab,ti | 1682 |
| 43 | 'dimethylfumarate*':ab,ti OR 'dimethyl fumarate*':ab,ti OR 'BG 00012':ab,ti OR 'BG00012':ab,ti OR 'BG 12':ab,ti OR 'diroximel fumarate*':ab,ti OR 'tecfidera*':ab,ti OR 'vumerity*':ab,ti | 3482 |
| 44 | 'fingolimod*':ab,ti OR 'gilenya*':ab,ti OR 'gilenia*':ab,ti OR 'FTY 720':ab,ti OR 'FTY720':ab,ti | 7329 |
| 45 | 'immunoglobulin*':ti OR 'intravenous immunoglobulin*':ab,ti OR "IV immunoglobulin*":ab,ti OR "IVIG":ab,ti | 83,207 |
| 46 | 'kesimpta*':ab,ti OR 'ofatumumab*':ab,ti OR 'HUMAX CD20 2F2':ab,ti OR 'GSK 1841157':ab,ti OR 'GSK1841157':ab,ti | 1409 |
| 47 | 'laquinimod*':ab,ti OR 'ABR 215062':ab,ti OR 'ABR215062':ab,ti | 456 |
| 48 | 'mayzent*':ab,ti OR 'siponimod*':ab,ti OR 'BAF 312':ab,ti OR 'BAF312':ab,ti | 395 |
| 49 | 'methotrexate*':ab,ti OR 'amethopterin*':ab,ti OR 'mexate*':ab,ti | 75,153 |
| 50 | 'methylprednisolone*':ab,ti OR 'metipred*':ab,ti | 30,108 |
| 51 | 'mitoxantrone*':ab,ti OR 'mitozantrone*':ab,ti OR 'ralenova*':ab,ti OR 'novantron*':ab,ti OR 'onkotrone*':ab,ti | 7718 |
| 52 | 'natalizumab*':ab,ti OR 'tysabri*':ab,ti OR 'antegren*':ab,ti | 6321 |
| 53 | 'ocrelizumab*':ab,ti OR 'ocrevus*':ab,ti OR 'R 1594':ab,ti OR 'PR070769':ab,ti | 1481 |
| 54 | 'ozanimod*':ab,ti OR 'zeposia*':ab,ti OR 'RPC1063':ab,ti | 335 |
| 55 | 'peginterferon*':ab,ti OR 'pegylated interferon*':ab,ti OR 'plegridy*':ab,ti OR 'peg ifn beta*':ab,ti | 16,796 |
| 56 | 'prednisolone*':ab,ti OR 'predonine*':ab,ti | 44,076 |
| 57 | 'rituximab*':ab,ti OR 'rituxan*':ab,ti OR 'mabthera*':ab,ti OR 'IDEC C2B8':ab,ti | 54,117 |
| 58 | #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #44 OR #45 OR #46 OR #47 OR #48 OR #49 OR #50 OR #51 OR #52 OR #53 OR #54 OR #55 OR #56 OR #57 | 1,237,635 |
| 59 | #13 AND #58 | 50,011 |
| 60 | 'randomized controlled trial'/de | 700,747 |
| 61 | 'controlled clinical trial'/de | 436,388 |
| 62 | random*:ti,ab,tt | 1,761,577 |
| 63 | 'randomization'/de | 93,310 |
| 64 | 'intermethod comparison'/de | 282,564 |
| 65 | placebo:ti,ab,tt | 338,208 |
| 66 | (compare:ti,tt OR compared:ti,tt OR comparison:ti,tt) | 581,965 |
| 67 | ((evaluated:ab OR evaluate:ab OR evaluating:ab OR assessed:ab OR assess:ab) AND (compare:ab OR compared:ab OR comparing:ab OR comparison:ab)) | 2,455,296 |
| 68 | (open NEXT/1 label):ti,ab,tt | 94,850 |
| 69 | ((double OR single OR doubly OR singly) NEXT/1 (blind OR blinded OR blindly)):ti,ab,tt | 255,920 |
| 70 | 'double blind procedure'/de | 193,441 |
| 71 | (parallel NEXT/1 group*):ti,ab,tt | 29,030 |
| 72 | (crossover:ti,ab,tt OR 'cross over':ti,ab,tt) | 115,242 |
| 73 | ((assign* OR match OR matched OR allocation) NEAR/6 (alternate OR group OR groups OR intervention OR interventions OR patient OR patients OR subject OR subjects OR participant OR participants)):ti,ab,tt | 410,683 |
| 74 | (assigned:ti,ab,tt OR allocated:ti,ab,tt) | 440,657 |
| 75 | (controlled NEAR/8 (study OR design OR trial)):ti,ab,tt | 409,692 |
| 76 | (volunteer:ti,ab,tt OR volunteers:ti,ab,tt) | 266,467 |
| 77 | 'human experiment'/de | 570,108 |
| 78 | trial:ti,tt | 358,650 |
| 79 | #60 OR #61 OR #62 OR #63 OR #64 OR #65 OR #66 OR #67 OR #68 OR #69 OR #70 OR #71 OR #72 OR #73 OR #74 OR #75 OR #76 OR #77 OR #78 | 5,730,637 |
| 80 | (((random* NEXT/1 sampl* NEAR/8 ('cross section*' OR questionnaire* OR survey OR surveys OR database or databases)):ti,ab,tt) NOT ('comparative study'/de OR 'controlled study'/de OR 'randomised controlled':ti,ab,tt OR 'randomized controlled':ti,ab,tt OR 'randomly assigned':ti,ab,tt)) | 2807 |
| 81 | ('cross‐sectional study'/de NOT ('randomized controlled trial'/de OR 'controlled clinical study'/de OR 'controlled study'/de OR 'randomised controlled':ti,ab,tt OR 'randomized controlled':ti,ab,tt OR 'control group':ti,ab,tt OR 'control groups':ti,ab,tt)) | 322,217 |
| 82 | ('case control*':ti,ab,tt AND random*:ti,ab,tt NOT ('randomised controlled':ti,ab,tt OR 'randomized controlled':ti,ab,tt)) | 19,453 |
| 83 | ('systematic review':ti,tt NOT (trial:ti,tt OR study:ti,tt)) | 201,814 |
| 84 | (nonrandom*:ti,ab,tt NOT random*:ti,ab,tt) | 17,624 |
| 85 | 'random field*':ti,ab,tt | 2622 |
| 86 | ('random cluster' NEAR/4 sampl*):ti,ab,tt | 1530 |
| 87 | (review:ab AND review:it) NOT trial:ti,tt | 957,720 |
| 88 | ('we searched':ab AND (review:ti,tt OR review:it)) | 40,569 |
| 89 | 'update review':ab | 120 |
| 90 | (databases NEAR/5 searched):ab | 52,543 |
| 91 | ((rat:ti,tt OR rats:ti,tt OR mouse:ti,tt OR mice:ti,tt OR swine:ti,tt OR porcine:ti,tt OR murine:ti,tt OR sheep:ti,tt OR lambs:ti,tt OR pigs:ti,tt OR piglets:ti,tt OR rabbit:ti,tt OR rabbits:ti,tt OR cat:ti,tt OR cats:ti,tt OR dog:ti,tt OR dogs:ti,tt OR cattle:ti,tt OR bovine:ti,tt OR monkey:ti,tt OR monkeys:ti,tt OR trout:ti,tt OR marmoset*:ti,tt) AND 'animal experiment'/de) | 1,143,365 |
| 92 | ('animal experiment'/de NOT ('human experiment'/de OR 'human'/de)) | 2,404,608 |
| 93 | #80 OR #81 OR #82 OR #83 OR #84 OR #85 OR #86 OR #87 OR #88 OR #89 OR #90 OR #91 OR #92 | 3,923,340 |
| 94 | #77 NOT #93 | 5,081,724 |
| 95 | #59 AND #94 | 11,002 |
| 96 | ([medline]/lim OR [pubmed‐not‐medline]/lim) | 29,998,057 |
| 97 | #95 NOT #96 | 6515 |
Database: Cochrane Central Register of Controlled Trials (CENTRAL; 2022, Issue 2) in the Cochrane Library
Date search conducted: March 4, 2022
Note: strategy conducted in Advanced search, Search manager
| # | Query | Results |
| #1 | MeSH descriptor: [Demyelinating Autoimmune Diseases, CNS] this term only | 5 |
| #2 | MeSH descriptor: [Demyelinating Diseases] this term only | 84 |
| #3 | MeSH descriptor: [Multiple Sclerosis] explode all trees | 3863 |
| #4 | MeSH descriptor: [Myelitis, Transverse] explode all trees | 55 |
| #5 | MeSH descriptor: [Optic Neuritis] explode all trees | 187 |
| #6 | ("clinically isolated" NEXT syndrome*):ti,ab | 213 |
| #7 | (devic OR "devic s" OR devics):ti,ab | 16 |
| #8 | (disseminated NEXT sclerosis*):ti,ab | 2 |
| #9 | (demyelinating NEXT (disease* OR disorder*)):ti,ab | 256 |
| #10 | ((demyelinating OR necrotising OR necrotizing OR transverse) NEXT myelitis*):ti,ab | 51 |
| #11 | multiple sclerosis:ti,ab OR MS:ti | 11,495 |
| #12 | (neuropapilliti* OR ((optic OR retrobulbar) NEXT neuriti*)):ti,ab | 417 |
| #13 | ((neuromyelitis NEXT optica*) OR ("nmo spectrum" NEXT disorder*)):ti,ab | 257 |
| #14 | {OR #1‐#13} | 12,233 |
| #15 | MeSH descriptor: [Adrenal Cortex Hormones] this term only and with qualifier(s): [therapeutic use ‐ TU, adverse effects ‐ AE] | 1626 |
| #16 | MeSH descriptor: [Alemtuzumab] explode all trees | 154 |
| #17 | MeSH descriptor: [Azathioprine] explode all trees | 1248 |
| #18 | MeSH descriptor: [Cladribine] explode all trees | 103 |
| #19 | MeSH descriptor: [Cyclophosphamide] explode all trees | 5648 |
| #20 | MeSH descriptor: [Daclizumab] explode all trees | 203 |
| #21 | MeSH descriptor: [Dimethyl Fumarate] explode all trees | 104 |
| #22 | MeSH descriptor: [Fingolimod Hydrochloride] explode all trees | 167 |
| #23 | MeSH descriptor: [Glatiramer Acetate] explode all trees | 184 |
| #24 | MeSH descriptor: [Immunoglobulins] this term only and with qualifier(s): [therapeutic use ‐ TU, adverse effects ‐ AE, drug effects ‐ DE] | 125 |
| #25 | MeSH descriptor: [Immunoglobulins, Intravenous] explode all trees | 896 |
| #26 | MeSH descriptor: [Interferon‐beta] explode all trees | 783 |
| #27 | MeSH descriptor: [Interferon Type I] this term only | 507 |
| #28 | MeSH descriptor: [Methotrexate] explode all trees | 4318 |
| #29 | MeSH descriptor: [Methylprednisolone] this term only | 2797 |
| #30 | MeSH descriptor: [Mitoxantrone] explode all trees | 524 |
| #31 | MeSH descriptor: [Natalizumab] explode all trees | 93 |
| #32 | MeSH descriptor: [Prednisolone] this term only | 3157 |
| #33 | MeSH descriptor: [Rituximab] explode all trees | 1419 |
| #34 | (("adrenal cortex" NEXT hormone*) OR corticoid*):ti,ab | 747 |
| #35 | (corticosteroid* OR (cortico NEXT steroid*)):ti | 6855 |
| #36 | (alemtuzumab* OR campath* OR lemtrada*):ti,ab | 668 |
| #37 | (avonex* OR rebif*):ti,ab | 400 |
| #38 | (aubagio* OR teriflunomide*):ti,ab | 341 |
| #39 | (azathioprine* OR azothioprine* OR imurel* OR imuran* OR immuran*):ti,ab | 2683 |
| #40 | (bafiertam* OR (monomethyl NEXT fumarate*) OR ("methyl hydrogen" NEXT fumarate*) OR methylhydrogenfumarate*):ti,ab | 35 |
| #41 | ((beta* NEAR/2 interferon*) OR fiblaferon* OR (fibroblast NEXT interferon*) OR IFNbeta* OR (IFN NEXT beta*)):ti,ab OR interferon*:ti | 9262 |
| #42 | (betaferon* OR betaseron* OR (beta NEXT seron*) OR extavia*):ti,ab | 145 |
| #43 | (copaxone* OR "Cop 1" OR "copolymer 1" OR glatiramer* OR glatopa* OR "TV 5010" OR "TV5010"):ti,ab | 692 |
| #44 | (cladribine* OR leustatin* OR mavenclad* OR movectro*):ti,ab | 395 |
| #45 | (cyclophosphamide* OR cyclophosphane* OR cytophosphan* OR cytoxan* OR endoxan* OR neosar* OR procytox* OR sendoxan*):ti,ab | 10,718 |
| #46 | (daclizumab* OR zinbryta* OR zenapax*):ti,ab | 468 |
| #47 | (dimethylfumarate* OR (dimethyl NEXT fumarate*) OR "BG 00012" OR "BG00012" OR "BG12" OR (diroximel NEXT fumarate*) OR tecfidera* OR vumerity*):ti,ab | 465 |
| #48 | (fingolimod* OR gilenya* OR gilenia* OR "FTY 720" OR "FTY720"):ti,ab | 614 |
| #49 | (kesimpta* OR ofatumumab* OR "HUMAX CD20 2F2" OR "GSK 1841157" OR "GSK1841157"):ti,ab | 284 |
| #50 | (immunoglobulin*):ti OR ((intravenous NEXT immunoglobulin*) OR (IV NEXT immunoglobulin*) OR IVIG):ti,ab | 3196 |
| #51 | (laquinimod* OR "ABR 215062" OR "ABR215062"):ti,ab | 90 |
| #52 | (mayzent* OR siponimod* OR "BAF 312" OR "BAF312"):ti,ab | 130 |
| #53 | (methotrexate* OR amethopterin* OR mexate*):ti,ab | 10,672 |
| #54 | (methylprednisolone* OR metipred*):ti,ab | 3923 |
| #55 | (mitoxantrone* OR mitozantrone* OR ralenova* OR novantron* OR onkotrone*):ti,ab | 1300 |
| #56 | (natalizumab* OR tysabri* OR antegren*):ti,ab | 417 |
| #57 | (ocrelizumab* OR ocrevus* OR "R 1594" OR "PR070769"):ti,ab | 269 |
| #58 | (ozanimod* OR zeposia* OR "RPC1063"):ti,ab | 150 |
| #59 | (peginterferon* OR (pegylated NEXT interferon*) OR plegridy* OR ("peg ifn" NEXT beta*)):ti,ab | 3592 |
| #60 | (prednisolone* OR predonine*):ti,ab | 5738 |
| #61 | (rituximab* OR rituxan* OR mabthera* OR "IDEC C2B8"):ti,ab | 5209 |
| #62 | {OR #15‐#61} | 61,706 |
| #63 | #14 AND #62 | 4633 |
| #64 | #14 AND #62 in Trials | 4581 |
Database: CINAHL Complete via EBSCOhost
Date search conducted: March 4, 2022
Notes: Boolean/Phrase search mode selected; search options disabled for "Apply related words" and "Apply equivalent subjects"
| # | Query | Results |
| S1 | (MH "Demyelinating Diseases") | 2213 |
| S2 | (MH "Multiple Sclerosis+") | 21,813 |
| S3 | (MH "Myelitis, Transverse+") | 814 |
| S4 | (MH "Optic Neuritis+") | 877 |
| S5 | TI "clinically isolated syndrome*" OR AB "clinically isolated syndrome*" | 435 |
| S6 | TI (devic OR "devic s" OR devics) OR AB (devic OR "devic s" OR devics) | 76 |
| S7 | TI "disseminated sclerosis*" OR AB "disseminated sclerosis*" | 20 |
| S8 | TI (demyelinating N1 (disease* OR disorder*)) OR AB (demyelinating N1 (disease* OR disorder*)) | 1120 |
| S9 | TI ((demyelinating OR necroti#ing OR transverse) N1 myelitis*) OR AB ((demyelinating OR necroti#ing OR transverse) N1 myelitis*) | 606 |
| S10 | TI ("multiple sclerosis*" OR MS) OR AB "multiple sclerosis*" | 23,842 |
| S11 | TI neuropapilliti* OR ((optic OR retrobulbar) N1 neuriti*) OR AB neuropapilliti* OR ((optic OR retrobulbar) N1 neuriti*) | 1108 |
| S12 | TI "neuromyelitis optica*" OR "nmo spectrum disorder*" | 832 |
| S13 | S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 | 31,485 |
| S14 | (MH "Adrenal Cortex Hormones/AE/DE/TU") | 10,396 |
| S15 | (MH "Azathioprine") | 1,589 |
| S16 | (MH "Cyclophosphamide/AE/DE/TU") | 3051 |
| S17 | (MH "Immunoglobulins/AE/DE/TU") OR (MH "Immunoglobulins, Intravenous") | 5213 |
| S18 | (MH "Interferons") | 9635 |
| S19 | (MH "Methotrexate") | 6571 |
| S20 | (MH "Mitoxantrone") | 457 |
| S21 | (MH "Prednisolone+") | 6886 |
| S22 | (MH "Natalizumab") | 303 |
| S23 | (MH "Rituximab") | 2210 |
| S24 | TI ("adrenal cortex hormone*" OR corticosteroid* OR "cortico steroid*" OR corticoid*) OR AB ("adrenal cortex hormone*" OR corticoid*) | 6597 |
| S25 | TI (alemtuzumab* OR campath* OR lemtrada*) OR AB (alemtuzumab* OR campath* OR lemtrada*) | 568 |
| S26 | TI (avonex* OR rebif*) OR AB (avonex* OR rebif*) | 148 |
| S27 | TI (aubagio* OR teriflunomide*) OR AB (aubagio* OR teriflunomide*) | 156 |
| S28 | TI (azathioprine* OR azothioprine* OR imurel* OR imuran* OR immuran*) OR AB (azathioprine* OR azothioprine* OR imurel* OR imuran* OR immuran*) | 1776 |
| S29 | TI (bafiertam* OR "monomethyl fumarate*" OR "methyl hydrogen fumarate*" OR methylhydrogenfumarate*) OR AB (bafiertam* OR "monomethyl fumarate*" OR "methyl hydrogen fumarate*" OR methylhydrogenfumarate*) | 11 |
| S30 | TI (betaferon* OR betaseron* OR "beta seron*" OR extavia*) OR AB (betaferon* OR betaseron* OR "beta seron*" OR extavia*) | 111 |
| S31 | TI (copaxone* OR "Cop 1" OR "copolymer 1" OR glatiramer* OR glatopa* OR "TV 5010" OR ""TV5010") OR AB (copaxone* OR glatiramer* OR glatopa* OR "TV 5010" OR ""TV5010") | 193 |
| S32 | TI (cladribine* OR leustatin* OR mavenclad* OR movectro*) OR AB (cladribine* OR leustatin* OR mavenclad* OR movectro*) | 297 |
| S33 | TI (cyclophosphamide* OR cyclophosphane* OR cytophosphan* OR cytoxan* OR endoxan* OR neosar* OR procytox* OR sendoxan*) OR AB (cyclophosphamide* OR cyclophosphane* OR cytophosphan* OR cytoxan* OR endoxan* OR neosar* OR procytox* OR sendoxan*) | 6472 |
| S34 | TI (daclizumab* OR zinbryta* OR zenapax*) OR AB (daclizumab* OR zinbryta* OR zenapax*) | 131 |
| S35 | TI (dimethylfumarate* OR "dimethyl fumarate*" OR "BG 00012" OR "BG00012" OR "BG 12" OR "diroximel fumarate*" OR tecfidera* OR vumerity*) OR AB (dimethylfumarate* OR "dimethyl fumarate*" OR "BG 00012" OR "BG00012" OR "BG 12" OR "diroximel fumarate*" OR tecfidera* OR vumerity*) | 271 |
| S36 | TI (fiblaferon* OR "fibroblast interferon*" OR IFNbeta* OR "IFN beta*" OR interferon*) OR AB ("beta interferon*" OR "beta 1 interferon*" OR "interferon beta*" OR fiblaferon* OR "fibroblast interferon*" OR IFNbeta* OR "IFN beta*") | 5093 |
| S37 | TI (fingolimod* OR gilenya* OR gilenia* OR "FTY 720" OR "FTY720") OR AB (fingolimod* OR gilenya* OR gilenia* OR "FTY 720" OR "FTY720") | 670 |
| S38 | TI (immunoglobulin* OR IVIG) OR AB ("intravenous immunoglobulin*" OR "IV immunoglobulin* OR IVIG) | 6059 |
| S39 | TI (kesimpta* OR ofatumumab* OR "HUMAX CD20 2F2" OR "GSK 1841157" OR "GSK1841157") OR AB (kesimpta* OR ofatumumab* OR "HUMAX CD20 2F2" OR "GSK 1841157" OR "GSK1841157") | 195 |
| S40 | TI (laquinimod* OR "ABR 215062" OR "ABR215062") OR AB (laquinimod* OR "ABR 215062" OR "ABR215062") | 42 |
| S41 | TI (mayzent* OR siponimod* OR "BAF 312" OR "BAF312") OR AB (mayzent* OR siponimod* OR "BAF 312" OR "BAF312") | 69 |
| S42 | TI (methotrexate* OR amethopterin* OR mexate*) OR AB (methotrexate* OR amethopterin* OR mexate*) | 7292 |
| S43 | TI (methylprednisolone* OR metipred*) OR AB (methylprednisolone* OR metipred*) | 2822 |
| S44 | TI (mitoxantrone* OR mitozantrone* OR ralenova* OR novantron* OR onkotrone*) OR AB (mitoxantrone* OR mitozantrone* OR ralenova* OR novantron* OR onkotrone*) | 493 |
| S45 | TI (natalizumab* OR tysabri* OR antegren*) OR AB (natalizumab* OR tysabri* OR antegren*) | 806 |
| S46 | TI (ocrelizumab* OR ocrevus* OR "R 1594" OR "PR070769") OR AB (ocrelizumab* OR ocrevus* OR "R 1594" OR "PR070769") | 165 |
| S47 | TI (ozanimod* OR zeposia* OR "RPC1063") OR AB (ozanimod* OR zeposia* OR "RPC1063") | 51 |
| S48 | TI (peginterferon* OR "pegylated interferon*" OR plegridy* OR "peg ifn beta*") OR AB (peginterferon* OR "pegylated interferon*" OR plegridy* OR "peg ifn beta*") | 1500 |
| S49 | TI (prednisolone* OR predonine*) OR AB (prednisolone* OR predonine*) | 3257 |
| S50 | TI (rituximab* OR rituxan* OR mabthera* OR "IDEC C2B8") OR AB (rituximab* OR rituxan* OR mabthera* OR "IDEC C2B8") | 6387 |
| S51 | S14 OR S15 OR S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S32 OR S33 OR S34 OR S35 OR S36 OR S37 OR S38 OR S39 OR S40 OR S41 OR S42 OR S43 OR S44 OR S45 OR S46 OR S47 OR S48 OR S49 OR S50 | 66,690 |
| S52 | S13 AND S51 | 4249 |
| S53 | MH randomized controlled trials | 127,041 |
| S54 | MH double‐blind studies | 52,694 |
| S55 | MH single‐blind studies | 15,474 |
| S56 | MH random assignment | 73,072 |
| S57 | MH pretest‐posttest design | 48,775 |
| S58 | MH cluster sample | 4924 |
| S59 | TI (randomised OR randomized) | 125,116 |
| S60 | AB (random*) | 367,935 |
| S61 | TI (trial) | 161,758 |
| S62 | MH (sample size) AND AB (assigned OR allocated OR control) | 4311 |
| S63 | MH (placebos) | 13,636 |
| S64 | PT (randomized controlled trial) | 140,462 |
| S65 | AB (control W5 group) | 131,489 |
| S66 | MH (crossover design) OR MH (comparative studies) | 449,242 |
| S67 | AB (cluster W3 RCT) | 444 |
| S68 | MH animals+ | 100,916 |
| S69 | MH (animal studies) | 145,244 |
| S70 | TI (animal model*) | 3344 |
| S71 | S68 OR S69 OR S70 | 237,342 |
| S72 | MH (human) | 2,524,082 |
| S73 | S71 NOT S72 | 204,471 |
| S74 | S53 OR S54 OR S55 OR S56 OR S57 OR S58 OR S59 OR S60 OR S61 OR S62 OR S63 OR S64 OR S65 OR S66 OR S67 | 945,899 |
| S75 | S74 NOT S73 | 900,730 |
| S76 | S52 AND S75 | 943 |
Database: LILACS (Latin American and Caribbean Health Sciences Literature)
Date search conducted: March 4, 2022
Note: strategy conducted in Advanced search (iAH)
1. Ti MS OR Tw "multiple sclerosis" OR Tw "optic neuritis" OR Tw "demyelinating disease" OR Tw "demyelinating diseases" OR Tw "clinically isolated syndrome" OR Tw "clinically isolated syndromes"
AND
2. Tw interferon$ OR Tw corticosteroid$ OR Tw immunoglobulin$ OR Tw avonex$ OR Tw rebif$ OR Tw betaferon$ OR IFNbeta$ OR Tw alemtuzumab$ OR Tw cladribine$ OR Tw teriflunomide$ OR Tw azathioprine$ OR Tw "monomethyl fumarate" OR Tw copaxone$ OR Tw "Cop 1" OR Tw "Copolymer 1" OR Tw glatiramer$ OR Tw cladribine$ OR Tw cyclophosphamide$ OR Tw daclizumab$ OR Tw "dimethyl fumarate" OR Tw "BG 12" OR Tw tecfidera$ OR Tw fingolimod$ OR Tw IVIG OR Tw ofatumumab$ OR Tw laquinimod$ OR Tw siponimod$ OR Tw methotrexate$ OR Tw methylprednisolone$ OR Tw mitoxantrone$ OR Tw natalizumab$ OR Tw ocrelizumab$ OR Tw ozanimod$ OR Tw peginterferon$ OR Tw "pegylated interferon" OR Tw prednisolone$ OR Tw rituximab$
AND
3. Tw estud$ OR Tw clin$ OR Ab grupo$ OR Pt "comparative study" OR Tw placebo$ OR Tw random$ OR Ti compara$ OR Ti tratamiento OR Tw control$ OR MH /dt (134)
Trial register: World Health Organization International Clinical Trials Registry Platform (trialsearch.who.int)
Date search conducted: March 4, 2022
Note: strategy conducted in Advanced search
Condition: "demyelinating disease" OR "multiple sclerosis" OR "optic neuritis" OR "transverse myelitis"
AND
Intervention: alemtuzumab OR avonex OR azathioprine OR cladribine OR copaxone OR corticosteroid OR cyclophosphamide OR daclizumab OR "dimethyl fumarate" OR fingolimod OR glatiramer OR "interferon beta" OR "intravenous immunoglobulins" OR laquinimod OR methotrexate OR methylprednisolone OR mitoxantrone OR natalizumab OR ocrelizumab OR ofatumumab OR ozanimod OR peginterferon OR rebif OR rituximab OR siponimod OR teriflunomide
AND
Recruitment status: ALL (822)
Trial register: US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov)
Date search conducted: March 4, 2022
Note: strategy conducted in Expert search
AREA[StudyType] EXPAND[Term] COVER[FullMatch] "Interventional" AND AREA[ConditionSearch] EXPAND[Concept] ( "Demyelinating Diseases" OR "Multiple Sclerosis" OR "Myelitis, Transverse" OR "Neuromyelitis Optica" OR "Optic Neuritis" ) AND AREA[InterventionSearch] EXPAND[Concept] ( Alemtuzumab OR Azathioprine OR Cladribine OR Cyclophosphamide OR Daclizumab OR "Dimethyl Fumarate" OR "Fingolimod Hydrochloride" OR "Glatiramer Acetate" OR Interferons OR "Immunoglobulins, Intravenous" OR laquinimod OR Methotrexate OR Methylprednisolone OR Mitoxantrone OR Natalizumab OR Ocrelizumab OR Ofatumumab OR Ozanimod OR "Peginterferon alfa‐2a" OR "Peginterferon alfa‐2b" OR Prednisolone OR Rituximab OR Siponimod OR Teriflunomide ) (604)
Data and analyses
Comparison 1. Treatment safety: pairwise comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 SAEs | 85 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 Interferon beta‐1b versus placebo | 1 | 939 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.79, 1.23] |
| 1.1.2 Interferon beta‐1a (Avonex) versus placebo | 5 | 1885 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.58, 1.40] |
| 1.1.3 Interferon beta‐1a (Rebif) versus placebo | 7 | 2384 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.68, 1.33] |
| 1.1.4 Glatiramer acetate versus placebo | 8 | 4984 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.69, 1.08] |
| 1.1.5 Natalizumab versus placebo | 4 | 2134 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.78, 1.25] |
| 1.1.6 Azathioprine versus placebo | 1 | 354 | Risk Ratio (M‐H, Random, 95% CI) | 3.62 [0.76, 17.19] |
| 1.1.7 Immunoglobulins versus placebo | 3 | 407 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.22, 7.90] |
| 1.1.8 Fingolimod versus placebo | 5 | 3774 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.86, 1.22] |
| 1.1.9 Teriflunomide versus placebo | 4 | 3044 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.87, 1.31] |
| 1.1.10 Dimethyl fumarate versus placebo | 5 | 2834 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.66, 0.93] |
| 1.1.11 Daclizumab versus placebo | 1 | 621 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.46, 0.86] |
| 1.1.12 Laquinimod versus placebo | 7 | 4360 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.91, 1.37] |
| 1.1.13 Pegylated interferon beta‐1a versus placebo | 1 | 1512 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.66, 1.74] |
| 1.1.14 Cladribine versus placebo | 2 | 1935 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.62, 1.79] |
| 1.1.15 Rituximab versus placebo | 2 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.73, 1.75] |
| 1.1.16 Ocrelizumab versus placebo | 2 | 889 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.69, 1.23] |
| 1.1.17 Siponimod versus placebo | 2 | 1941 | Risk Ratio (M‐H, Random, 95% CI) | 2.05 [0.33, 12.59] |
| 1.1.18 Ofatumumab versus placebo | 2 | 295 | Risk Ratio (M‐H, Random, 95% CI) | 2.74 [0.33, 23.06] |
| 1.1.19 Glatiramer acetate versus interferon beta‐1b | 1 | 2220 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.73, 1.25] |
| 1.1.20 Interferon beta‐1a(Rebif) versus interferon beta‐1a (Avonex) | 1 | 676 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.63, 2.14] |
| 1.1.21 Glatiramer acetate versus interferon beta‐1a (Avonex) | 1 | 509 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.19] |
| 1.1.22 Fingolimod versus interferon beta‐1a (Avonex) | 1 | 1280 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.98, 2.36] |
| 1.1.23 Daclizumab versus interferon beta‐1a (Avonex) | 1 | 1841 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.96, 1.35] |
| 1.1.24 Laquinimod versus interferon beta‐1a (Avonex) | 1 | 875 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.82, 1.94] |
| 1.1.25 Ocrelizumab versus interferon beta‐1a (Avonex) | 1 | 164 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.19, 5.19] |
| 1.1.26 Glatiramer acetate versus interferon beta‐1a (Rebif) | 1 | 756 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.57, 1.57] |
| 1.1.27 Teriflunomide versus interferon beta‐1a (Rebif) | 1 | 321 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.51, 2.74] |
| 1.1.28 Alemtuzumab versus interferon beta‐1a (Rebif) | 3 | 1684 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.90, 1.65] |
| 1.1.29 Azathioprine versus interferons | 1 | 146 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [0.58, 8.59] |
| 1.1.30 Dimethyl fumarate versus glatiramer acetate | 1 | 1054 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.72, 1.27] |
| 1.1.31 Cyclophosphamide versus corticosteroids | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.59, 2.16] |
| 1.1.32 Natalizumab versus fingolimod | 1 | 108 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.07] |
| 1.1.33 Fingolimod versus interferon beta‐1b | 2 | 164 | Risk Ratio (M‐H, Random, 95% CI) | 3.86 [0.71, 20.94] |
| 1.1.34 Interferon beta 1a (Rebif) versus interferon beta‐1b | 2 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.28, 2.63] |
| 1.1.35 Ocrelizumab versus interferon beta 1a (Rebif) | 2 | 1651 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.57, 1.11] |
| 1.1.36 Rituximab versus glatiramer acetate | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.1.37 Fingolimod versus glatiramer acetate | 1 | 1035 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.79, 2.12] |
| 1.1.38 Ozanimod versus interferon beta‐1a (Avonex) | 2 | 2659 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.78, 1.63] |
| 1.1.39 Ofatumumab versus teriflunomide | 2 | 1882 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.85, 1.55] |
| 1.1.40 Diroximel fumarate versus dimethyl fumarate | 1 | 504 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.30, 5.85] |
| 1.2 Withdrawals due to AEs | 105 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.2.1 Interferon beta‐1b versus placebo | 6 | 2601 | Risk Ratio (M‐H, Random, 95% CI) | 3.13 [1.77, 5.53] |
| 1.2.2 Interferon beta‐1a (Avonex) versus placebo | 6 | 2169 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.84, 2.80] |
| 1.2.3 Interferon beta‐1a (Rebif) versus placebo | 7 | 2693 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [0.93, 3.56] |
| 1.2.4 Glatiramer acetate versus placebo | 9 | 5032 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [1.07, 2.26] |
| 1.2.5 Natalizumab versus placebo | 4 | 2134 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.87, 1.81] |
| 1.2.6 Mitoxantrone versus placebo | 2 | 242 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.35, 18.66] |
| 1.2.7 Azathioprine versus placebo | 4 | 513 | Risk Ratio (M‐H, Random, 95% CI) | 6.98 [2.65, 18.42] |
| 1.2.8 Immunoglobulins versus placebo | 7 | 1003 | Risk Ratio (M‐H, Random, 95% CI) | 1.91 [1.07, 3.41] |
| 1.2.9 Methotrexate versus placebo | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.2.10 Fingolimod versus placebo | 5 | 3774 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [1.50, 2.25] |
| 1.2.11 Teriflunomide versus placebo | 4 | 3044 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [1.02, 2.15] |
| 1.2.12 Dimethyl fumarate versus placebo | 4 | 2578 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.93, 1.46] |
| 1.2.13 Daclizumab versus placebo | 1 | 621 | Risk Ratio (M‐H, Random, 95% CI) | 3.67 [0.85, 15.89] |
| 1.2.14 Laquinimod versus placebo | 7 | 4360 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [0.97, 2.06] |
| 1.2.15 Pegylated interferon beta‐1a versus placebo | 1 | 1512 | Risk Ratio (M‐H, Random, 95% CI) | 3.46 [1.58, 7.58] |
| 1.2.16 Cladribine versus placebo | 2 | 1935 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [0.69, 4.82] |
| 1.2.17 Rituximab versus placebo | 2 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.28, 10.35] |
| 1.2.18 Ocrelizumab versus placebo | 2 | 889 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.63, 2.97] |
| 1.2.19 Ofatumumab versus placebo | 2 | 295 | Risk Ratio (M‐H, Random, 95% CI) | 3.71 [0.20, 67.95] |
| 1.2.20 Siponimod versus placebo | 2 | 1941 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.06, 2.35] |
| 1.2.21 Interferon beta‐1a (Avonex) versus interferon beta‐1b | 1 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.03, 1.79] |
| 1.2.22 Glatiramer acetate versus interferon beta‐1b | 2 | 2295 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.48, 1.81] |
| 1.2.23 Interferon beta‐1a (Rebif) versus interferon beta‐1a (Avonex) | 1 | 676 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.56, 1.96] |
| 1.2.24 Glatiramer acetate versus interferon beta‐1a (Avonex) | 1 | 509 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.32, 1.31] |
| 1.2.25 Fingolimod versus interferon beta‐1a (Avonex) | 1 | 1280 | Risk Ratio (M‐H, Random, 95% CI) | 2.09 [1.23, 3.57] |
| 1.2.26 Daclizumab versus interferon beta‐1a (Avonex) | 1 | 1841 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.67] |
| 1.2.27 Laquinimod versus interferon beta‐1a (Avonex) | 1 | 875 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.47, 1.44] |
| 1.2.28 Ocrelizumab versus interferon beta‐1a (Avonex) | 1 | 164 | Risk Ratio (M‐H, Random, 95% CI) | 2.45 [0.29, 20.49] |
| 1.2.29 Glatiramer acetate versus interferon beta‐1a (Rebif) | 1 | 756 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.46, 1.52] |
| 1.2.30 Teriflunomide versus interferon beta‐1a (Rebif) | 1 | 321 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.25, 0.76] |
| 1.2.31 Alemtuzumab versus interferon beta‐1a (Rebif) | 3 | 1684 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.12, 0.58] |
| 1.2.32 Azathioprine versus interferons | 2 | 240 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.87, 4.62] |
| 1.2.33 Dimethyl fumarate versus glatiramer acetate | 1 | 1054 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.84, 1.76] |
| 1.2.34 Fingolimod versus dimethyl fumarate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.2.35 Natalizumab versus fingolimod | 1 | 108 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.10] |
| 1.2.36 Fingolimod versus interferon beta‐1b | 2 | 164 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.19, 3.02] |
| 1.2.37 Interferon beta 1a (Rebif) versus interferon beta‐1b | 1 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 12.80 [0.74, 222.66] |
| 1.2.38 Mitoxantrone versus corticosteroids | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.60, 6.64] |
| 1.2.39 Cyclophosphamide versus corticosteroids | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 3.67 [1.46, 9.21] |
| 1.2.40 Ocrelizumab versus interferon beta‐1a (Rebif) | 2 | 1651 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.37, 0.89] |
| 1.2.41 Rituximab versus glatiramer acetate | 1 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.14, 6.46] |
| 1.2.42 Fingolimod versus glatiramer acetate | 1 | 1035 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.41, 0.86] |
| 1.2.43 Ozanimod versus interferon beta‐1a (Avonex) | 1 | 1313 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.42, 1.36] |
| 1.2.44 Ozanimod versus interferon beta‐1a (Avonex) | 1 | 1346 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.32, 1.18] |
| 1.2.45 Ofatumumab versus teriflunomide | 2 | 1882 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.75, 1.59] |
| 1.2.46 Diroximel fumarate versus dimethyl fumarate | 1 | 504 | Risk Ratio (M‐H, Random, 95% CI) | 0.28 [0.09, 0.85] |
1.1. Analysis.

Comparison 1: Treatment safety: pairwise comparisons, Outcome 1: SAEs
1.2. Analysis.

Comparison 1: Treatment safety: pairwise comparisons, Outcome 2: Withdrawals due to AEs
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Achiron 1998.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 19‐60 years; clinically definite RRMS; mean disease duration 4 years; mean EDSS 3.0; prior use of DMT not reported | |
| Interventions | Loading dose of immunoglobulins 0.4 g/kg body weight intravenously daily for 5 consecutive days followed by additional booster doses of immunoglobulins 0.4 g/kg body weight intravenously daily every 2 months for 24 months (n = 20) Placebo consisting of 0.9% saline administered with the same schedule as the active treatment (n = 20) |
|
| Outcomes | Withdrawals due to AEs over 24 months | |
| Notes | Funding: Miles Inc. Cutter Biological, Bayer and Promedico | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Patients were assigned to receive immunoglobulin or placebo by a block‐stratified randomisation procedure, designed to ensure groups balanced for YER, age, and disease duration" (page 399). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Randomization was performed at the pharmacy, and the bottles of immunoglobulin or placebo were wrapped in sealed opaque bags and brought to the patients' rooms. The entire IV set was covered by an opaque plastic bag to ensure that any possible fluid turbidity or frothing would not be evident to the investigators or patients" (page 399). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo consisting of 0.9% saline...Taking into account the different physical properties of the two solutions and the theoretic possibility of identifying the solutions by physical means such as heating, electrophoresis, or whipping, considerable precautions were taken to ensure blindability.All patients ... were blinded to treatment" (page 399). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Evaluators were blinded to treatment" (page 399). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 5.0% was lost‐to follow‐up (5.0% in immunoglobulins, and 5.0% in placebo), without indications of different reasons between the comparison groups. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Not reported |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Achiron 2004.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 15‐50 years; CIS; mean time since neurological event 35 days; mean EDSS 2.2; prior use of DMT not reported. | |
| Interventions | Loading dose of immunoglobulins 0.4 g/kg body weight intravenously daily for 5 consecutive days followed by additional booster doses of immunoglobulins 0.4 g/kg body weight intravenously daily every 6 weeks for 12 months (n = 45) Placebo in the form of intravenously administered 0.9% sodium chloride for 12 months (n = 46) |
|
| Outcomes | Withdrawals due to AEs; AEs over 12 months | |
| Notes | Funding: Omrix Biopharmaceuticals | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Patients’ allocation was based on a block‐stratified randomization procedure, and accordingly, they were randomly assigned to each of the 2 treatment groups" (page 1516). |
| Allocation concealment (selection bias) | Low risk | Quoted: "At the pharmacy, containers and tubing of IVIg or saline were wrapped in sealed opaque bags" (page 1516). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo was in the form of intravenously administered 0.9% sodium chloride in identical settings and regime. At the pharmacy, containers and tubing of IVIg or saline were wrapped in sealed opaque bags" (page 1516). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Each patient was evaluated by an examining neurologist who was unaware of the patient’s treatment assignment" (page 1516). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 1517): Immunoglobulins: 41 (91.1%) of 45 participants (1 lost to follow‐up, 3 discontinued because of consent withdrawal) Placebo: 43 (95.5%) of 45 participants (2 discontinued because of protocol violation) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Not reported |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ADVANCE 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.5; prior use of any MS medication at any time prior to the start of study: 17% | |
| Interventions | PegIFNß‐1a 125 μg subcutaneously once every 2 weeks for 12 months (n = 512) PegIFNß‐1a 125 μg subcutaneously once every 4 weeks for 12 months (n = 500) Placebo subcutaneously once every 2 weeks for 12 months (n = 500) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Patients were randomly assigned (1:1:1) to receive subcutaneous injections with pre‐filled syringes of placebo, peginterferon beta‐1a at a dose of 125 μg once every 2 weeks, or peginterferon beta‐1a 125 μg once every 4 weeks, stratified by site" (page 658). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Randomisation was done by a centralised interactive voice response and web system" (page 658). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo was a matched diluent, given with a matched pre‐filled syringe. Patients received either study drug or placebo every 2 weeks to maintain masking; those assigned to receive study drug every 4 weeks received alternate injections of placebo and peginterferon beta‐1a every 2 weeks. All study management and site personnel, and patients were masked to treatment assignment" (page 658). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Each site had separate examining and treating neurologists, thereby maintaining rater masking for all treatment groups." (page 658). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment (Figure 1, page 660): Peginterferon beta‐1a 125 μg every 2 weeks: 438 (85.5%) of 512 participants (4.7% adverse events) Peginterferon beta‐1a 125 μg every 4 weeks: 438 (87.6%) of 500 participants (4.8% adverse events) Placebo: 456 (91.2%) of 500 participants: (1.0% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00906399). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Method of AE monitoring | Unclear risk | "Time frame: Screening through week 96 (treatment period), plus 4 weeks (±5 days) follow‐up.... Adverse events were collected by systematic assessment" (NCT00906399). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
AFFIRM 2006.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS; median disease duration 5 years (range, 0‐34 years); mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | Natalizumab 300 mg intravenously once every 4 weeks for up to 116 weeks (n = 627) Placebo (unspecified) (n = 315) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Biogen Idec, Inc. and Elan Pharmaceutica | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Patients were randomly assigned to treatment that was stratified according to study site in blocks of three (two active, one placebo) with the use of a computer‐generated block randomization schedule" (page 900). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Amultidigit identification number, implemented by an interactive voice‐response system was used" (page 900). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Treating neurologists were responsible for all aspects of patient care, including the management of adverse events and the treatment of relapsing disease. Examining neurologists performed objective evaluation with use of the EDSS and neurologic examination during all study visits; they were not in contact with patients in any other capacity, so as to reduce the possibility of being unblinded by side effects or laboratory assessments." (page 901) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 906): Natalizumab: 551 (87.9%) of 627 participants (2.4% adverse events, 3.8% discontinued treatment, 1.9% requested withdrawal) Placebo: 269 (85.4%) of 315 participants (1.9% adverse events, 4.8% discontinued treatment, 4.1% requested withdrawal) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00027300). |
| Serious AE definitions | Unclear risk | Insufficient information on the definition of SAEs |
| Method of AE monitoring | Low risk | Quoted: "Treating neurologists were responsible for all aspects of patient care, including the management of adverse events". "Patients visited the clinic every 12 weeks for ... blood chemical and hematologic analyses, evaluation of adverse events..." (page 901). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ALLEGRO 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 9 years; mean EDSS 2.6; prior use of DMT at any time prior to the start of study: 39.0% (38.2% in laquinimod, and 39.7% in placebo) | |
| Interventions | Laquinimod 0.6 mg oral capsule once daily for 24 months (n = 550) Placebo oral capsule once daily for 24 months (n = 556) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "The randomization list, stratified according to study center, was computer‐generated" (page 1002). |
| Allocation concealment (selection bias) | Low risk | Quoted: "The subject was allocated a screening number by the investigator using an Interactive Voice Response System (IVRS)" (page 44 of the trial protocol). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: "Subjects will be instructed to contact the study site in the event of any change in their medical condition, or the appearance of any AEs.A mandatory phone call will be performed by the treating physician or the site’s nurse/study coordinator during predefined times. A list of predefined questions relating to signs or symptoms suggestive of vascular thrombosis will be presented to the subject. In case of positive response to one of the presented questions, the subject should be immediately invited to the site for examination and further evaluation of his/her condition in accordance to the guidance of safety monitoring" (Protocol page 46). Quoted: "The treating physician was unaware of the study‐group assignment" (page 1002). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Supplemental Figure 1, page 10): Laquinimod: 437 (79.4%) of 550 participants (7.6% adverse events, 8.0% consent withdrawn) Placebo 427 (76.8%) of 556 participants (5.0% adverse events, 10.8% consent withdrawn) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified safety and tolerability outcome measures (Trial protocol). |
| Serious AE definitions | Low risk | Quoted: "A Serious Adverse Event (SAE) is defined as an AE that results in any of the following: death; life‐threatening; requires hospitalization or prolongs existing inpatients’ hospitalization; results in persistent or significant disability or incapacity; results in a congenital abnormality or birth defect; an important medical event which requires medical intervention to prevent any of the above outcomes". (Trial protocol; page 73). |
| Method of AE monitoring | Low risk | Quoted: "Safety assessments were performed at screening, at baseline, and at months 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24" (page 1002). "Safety assessments included measurement of vital signs and weight, physical examination, electrocardiography, clinical laboratory tests, and recording of adverse events" (page 1003). "SAEs were collected by systematic assessment" (NCT00509145). "A mandatory phone call will be performed by the treating physician or the site’s nurse/study coordinator fourteen (± 2) days after month 1, and month 2 visits. A list of predefined questions relating to signs or symptoms suggestive of vascular thrombosis will be presented to the subject" (Protocol; page 47). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Andersen 2004.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite SPMS; mean disease duration 14 years; mean EDSS 4.8; prior use of DMT not reported | |
| Interventions | IFNß‐1a (Rebif) 22 μg subcutaneously weekly for 36 months (n = 188) Placebo (unspecified) for 36 months (n = 183) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Serono International, Geneva, Switzerland The study had ended prematurely due to negative results from SPECTRIMS study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomised in equal allocation to receive IFN beta‐1a (Rebif; Serono), 22 mg SC once weekly, or matching placebo, for 3 years" (page 707). |
| Allocation concealment (selection bias) | Unclear risk | "equal allocation" (page 707) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Lack of clarity in the method used to assess adverse effects data. Quoted: "Adverse events and concomitant medications were recorded throughout the study, and clinical laboratory evaluation was performed at months 1, 3, and 6, and then at 6 monthly evaluation visits or as needed." (page 707) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 17.3% was lost‐to follow‐up (20.4% in IFNß‐1a, and 14.0% in placebo), with some indications of differences in reasons: adverse events of 9.8% in IFNß‐1a and 3.8% in placebo. |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. Types and measures of adverse effects were not prespecified in the "Methods" section of the article. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Quoted: "Adverse events and concomitant medications were recorded throughout the study, and clinical laboratory evaluation was performed at months 1, 3, and 6, and then at 6 monthly evaluation visits or as needed" (page 707). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
APEX 2019.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration not reported; mean EDSS 2.0; prior use of DMT at any time prior to the start of study: 56.7% (56.8% in dimethyl fumarate, and 56.6% in placebo) | |
| Interventions | Dimethyl fumarate 240 mg orally two times daily for 6 months (n = 111) Placebo orally two times daily for 6 months (n = 113) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 6 months | |
| Notes | Funding: Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A stratified block randomization procedure was used for Part I, with 5 strata, 1 for each country (block size: 4; 50 blocks per country)" (page 1, Additional file 1). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Randomization was performed using a centralized interactive voice/web response system" (page 2). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo capsules matched DMF capsules in size, shape, color, and taste. All patients (including those receiving placebo) were dosed with the same number of capsules twice daily (Additional file 1). Quoted: "Patients, their families, and all study staff were blinded to patient treatment assignments. In addition, separate study personnel were designated to treat patients and to conduct efficacy and relapse assessments" (page 2). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1, page 3): Dimethyl fumarate: 105 (94.6%) of 111 treated participants (0.9% adverse events, 0.9% consent withdrawn, 3.6% other reasons) Placebo: 107 (94.7%) of 113 treated participants (2.6% adverse events, 2.6% consent withdrawn) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes reported in the Additional file 1 (pages 2‐4). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 9). |
| Method of AE monitoring | Low risk | Quoted: "...laboratory/safety evaluations were conducted every 4 weeks (±5 days)" (page 2). "AEs of special interest were defined based on Standardized Medical Dictionary for Regulatory Activities (MedDRA) Queries (SMQs), Custom MedDRA Queries (CMQs), System Organ Classes (SOCs), High Level Group Terms (HLGTs), High Level Terms, and/or Preferred Terms (PTs), as appropriate" (Appendix pages 2‐3). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
APOLITOS 2021.
| Study characteristics | ||
| Methods | RCT. Parallel assignment. 12 centers in Japan and 2 in Russia. The study was conducted from March 15, 2018 to July 29, 2020. | |
| Participants | Patients aged 18–55 years, diagnosed with relapsing MS, according to the 2010 revised McDonald criteria, with an EDSS score of 0 to 5.5, >= 1 appearance of a new neurological abnormality or worsening of a pre‐existing neurological abnormality during the 2 years before screening and an MRI activity in the brain during the previous year, and neurologically stable within 1 month before randomization | |
| Interventions | Ofatumumab 20 mg (0.4 mL) subcutaneous injections on days 1, 7, and 14 (initial dosing), and subsequent dosing every 4 weeks starting at week 4 for 6 months (n = 43) Placebo (unspecified) subcutaneous injection matching ofatumumab every 4 weeks for 6 months (n = 21) |
|
| Outcomes | Adverse events including death and non‐fatal serious adverse events (SAEs) measured at 6 months | |
| Notes | Funding: This study was funded by Novartis Pharma AG, Basel, Switzerland. Ten co‐authors were employees of Novartis Pharma. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: "Randomization was stratified by geographical region (Japan or Russia) and the baseline number of gadolinium‐enhancing (Gd+) T1 lesions (0 or >= 1)" (page 2). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure e‐1): Ofatumumab: 40 (93.0%) of 43 participants (2.3% lack of efficacy, 2.3% patient/guardian decision, 2.3% lost to follow‐up) Placebo: 19 (90.5%) of 21 participants (4.8% lack of efficacy, 4.8% patient/guardian decision) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse events were not prespecified in the protocol (NCT03249714). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | Quoted: "The safety assessments were primarily based on the frequency of adverse events, including death and non‐fatal serious adverse events" (Page 3). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ARPEGGIO 2020.
| Study characteristics | ||
| Methods | RCT. Patients were randomized from January 2015 to April 2016 at 85 sites in 10 countries. | |
| Participants | Age: 25‐55 years; clinically definite PPMS; mean disease duration 8 years; mean EDSS 4.5; prior use of DMT at any time prior to the start of study: 22.5% | |
| Interventions | Laquinimod 1.5 mg oral capsule once daily for 12 months (n = 95) [This arm was discontinued as of 01 January 2016 and no participants reached the 12 months time frame] Laquinimod 0.6 mg oral capsule once daily for 12 months (n = 139) Placebo (unspecified) oral capsule once daily for 12 months (n = 140) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A capped randomization procedure ensured that ≤ 20% of all enrolled patients had a baseline EDSS score of 6.0 and 6.5" (pages 1028‐9). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Randomization was performed centrally using an independent interactive Web‐based or voice response system" (page 1029). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Efficacy assessments were performed by an examining neurologist who remained unaware of the patient’s safety status and was strictly instructed not to discuss safety issues with the treating physician, to assure an accurate and objective evaluation" (page 1029). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment (figure e‐2): Laquinimod 0.6 mg: 93 (66.9%) of 139 participants (6.5% adverse events) Placebo: 109 (77.9%) of 140 participants (1.4% adverse events) Laquinimod 1.5 mg arm was discontinued early by the sponsor for safety reasons. |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (NCT02284568). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in full accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice Consolidated Guideline (E6)" (page e1028). |
| Method of AE monitoring | Low risk | Quoted: "Safety endpoints included assessment of AEs throughout the study and vital signs, ECGs, and clinical laboratory parameters and concomitant medication usage" (page e1030). "Time frame: Day 1 up to week 130 (longest duration of treatment)... Term from vocabulary, MedDRA (19.0). AEs collected by systematic assessment" (NCT02284568). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ASCEND 2018.
| Study characteristics | ||
| Methods | RCT. Study conducted between Sept 13, 2011, and July 16, 2015. Participants from 163 sites in 17 countries | |
| Participants | Age: 18‐58 years; clinically definite SPMS; mean disease duration 16 years; median EDSS 6; prior use of DMT not reported | |
| Interventions | Natalizumab 300 mg intravenously once every 4 weeks for up to 24 months (n = 440) Placebo (unspecified) for 24 months (n = 449) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "In part 1 of ASCEND, eligible patients were randomly assigned (1:1) to receive natalizumab or placebo of identical appearance. Patients were stratified by site and by EDSS score (3·0–5·5 vs 6·0–6·5)" (page 407). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Patients were randomly assigned by an interactive voice/web response system (IXRS, Bracket Global LLC, San Francisco, CA, USA)" (page 407). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "The treating neurologist completed the equivalent of a regular clinic visit" (page 408). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 408): Natalizumab: 326 (74.3%) of 439 participants (1.8% unsatisfactory therapeutic effect, 4.3% adverse events) Placebo: 312 (69.5%) of 449 participants (3.6% unsatisfactory therapeutic effect, 3.3% adverse events) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified primary safety outcomes (NCT01416181). |
| Serious AE definitions | Low risk | Quoted: "The study was done according to the International Conference on Harmonisation, and good clinical practice guidelines" (page 407). |
| Method of AE monitoring | Low risk | Quoted: "Treatment‐emergent adverse events and serious adverse events were recorded in the safety population" (page 407). "Treatment‐emergent adverse events only are presented. Time frame: Adverse events are captured through the last study visit; participants were followed through week 228, or 24 weeks following last dose of study treatment, or premature withdrawal" (NCT01416181). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ASCLEPIOS I 2020.
| Study characteristics | ||
| Methods | RCT, from October 2016 through March 2018, participants were enrolled at 385 sites in 37 countries | |
| Participants | Age: 18‐55 years; clinically definite RRMS or SPMS; mean disease duration 8 years; mean EDSS 2.9; prior use of DMT: 59.8% (58.9% in ofatumumab, and 60.6% in teriflunomide) | |
| Interventions | Ofatumumab 20 mg subcutaneously every 4 weeks for up to 30 months (n = 465) Teriflunomide 14 mg orally once daily for up to 30 months (n = 462) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 30 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Randomization was stratified according to geographic region and subtype of multiple sclerosis" (page 548). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Eligible patients were randomly assigned in a 1:1 ratio through interactive response technology" (page 548). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients in the ofatumumab group also received matching placebo capsule orally once daily, and patients in the teriflunomide group also received matching placebo subcutaneous injections corresponding to the active drug in the other group" (page 548) and "The identity of the treatments will be concealed by the use of investigational treatment that are all identical in packaging, labeling, schedule of administration, appearance, taste and odor" (Protocol, page 34). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: "Investigator staff, persons performing the assessments, will remain blinded to the identity of the treatment from the time of randomization until database lock" (Protocol, page 33) and "The Investigator will be responsible for...management of adverse events ... ensuring access to appropriate expertise for consultation (e.g. infectious disease, ECG interpretation, mental health care) during the study as needed)" (Protocol, page 43). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 30 months on study treatment (Figure S4): Ofatumumab: 400 (86.0%) of 465 participants (0.2% lack of efficacy, 3.0% adverse events, 2.1% lost to follow‐up) Teriflunomide 359 (77.7%) of 462 participants (2.6% lack of efficacy, 3.0% adverse events, 1.1% lost to follow‐up) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (NCT02792218). |
| Serious AE definitions | Low risk | Quoted: "The trial was conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice" (page 547). "Adverse events were recorded at all visits and graded according to the Common Terminology Criteria for Adverse Events (CTCAE)" (page 548). Definition of SAEs reported in the study protocol |
| Method of AE monitoring | Low risk | Quoted: "AE were recorded at all visits" (page 548). "The occurrence of AE must be sought by non‐directive questioning of the patient at each visit during the study. AE also may be detected when they are volunteered by the patient during or between visits or through physical examination findings, laboratory test findings, or other assessments...Clinically significant abnormal laboratory values or test results must be identified through a review of values outside of normal ranges or clinically notable ranges, significant changes from baseline or the previous visit, or values which are considered to be non‐typical in patient with underlying disease. Clinically notable laboratory findings are defined according to the Common Terminology Criteria for Adverse Events (CTCAE, version 4.03). (Protocol, page 63‐5). "Time frame: Adverse events were reported from first dose of study treatment until end of study treatment plus 100 days post treatment, up to a maximum duration of 2.7 years" (NCT02792218). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ASCLEPIOS II 2020.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS or SPMS; mean disease duration 8 years; mean EDSS 2.9; prior use of DMT: 60.6% (59.5% in ofatumumab, and 61.8% in teriflunomide) | |
| Interventions | Ofatumumab 20 mg subcutaneously every 4 weeks for up to 30 months (n = 481) Teriflunomide 14 mg orally once daily for up to 30 months (n = 474) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 30 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Randomization was stratified according to geographic region and subtype of multiple sclerosis" (page 548). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Eligible patients were randomly assigned in a 1:1 ratio through interactive response technology" (page 548). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients in the ofatumumab group also received matching placebo capsule orally once daily, and patients in the teriflunomide group also received matching placebo subcutaneous injections corresponding to the active drug in the other group" (page 548) and "The identity of the treatments will be concealed by the use of investigational treatment that are all identical in packaging, labeling, schedule of administration, appearance, taste and odor" (Protocol, page 34). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: "Investigator staff, persons performing the assessments, will remain blinded to the identity of the treatment from the time of randomization until database lock" (Protocol, page 33) and "The Investigator will be responsible for...management of adverse events ... ensuring access to appropriate expertise for consultation (e.g. infectious disease, ECG interpretation, mental health care) during the study as needed" (Protocol, page 43). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 30 months on study treatment (Figure S4): Ofatumumab: 383 (79.6%) of 481 participants (1.5% lack of efficacy, 3.3% adverse events, 1.9% lost to follow‐up) Teriflunomide 370 (78.1%) of 474 participants (1.9% lack of efficacy, 2.7% adverse events, 1.1% lost to follow‐up) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (NCT02792231). |
| Serious AE definitions | Low risk | Quoted: "The trial was conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice" (page 547). "Adverse events were recorded at all visits and graded according to the Common Terminology Criteria for Adverse Events (CTCAE)" (page 548). Definition of SAEs reported in the study protocol |
| Method of AE monitoring | Low risk | Quoted: "AEs were recorded at all visits" (page 548). "The occurrence of AEs must be sought by non‐directive questioning of the patient at each visit during the study. AEs also may be detected when they are volunteered by the patient during or between visits or through physical examination findings, laboratory test findings, or other assessments...Clinically significant abnormal laboratory values or test results must be identified through a review of values outside of normal ranges/clinically notable ranges, significant changes from baseline or the previous visit, or values which are considered to be non‐typical in patient with underlying disease. Clinically notable laboratory findings are defined according to the Common Terminology Criteria for Adverse Events (CTCAE, version 4.03). (Protocol, page 63‐5). "Time frame: Adverse events were reported from first dose of study treatment until end of study treatment plus 100 days post treatment, up to a maximum duration of 2.7 years" (NCT02792218). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ashtari 2011.
| Study characteristics | ||
| Methods | RCT. Participants were recruited from neurology outpatient clinics of Isfahan University of Medical Sciences in 2007. | |
| Participants | Age: 15‐55 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 2.2; prior use of DMT not reported | |
| Interventions | Methotrexate 7.5 mg orally weekly for 12 months (n = 40) IFNß‐1a (Avonex) 30 µg intramuscularly once a week for 12 months (n = 40) |
|
| Outcomes | AEs or SAEs were not reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Patients were randomized according to a preexisting list produced by a computer program that differed from a random number generator only in that it assigned equal numbers of patients into each treatment group" (page 458). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "The trial was single‐blinded" (page 459). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "Patients were evaluated at baseline and 12 months after the start of the therapy by a neurologist to evaluate the development of side effects of the medications, compliance of the patients, and efficacy parameters" (page 459). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, there was no lost to follow‐up. Quoted: "There was not any discontinuation of drugs among patients of the two groups" (page 460). |
| Selective reporting (reporting bias) | High risk | The published report did not report withdrawals due to AEs, AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: "Patients were evaluated at baseline and 12 months after the start of the therapy by a neurologist to evaluate the development of side effects of the medications, compliance of the patients, and efficacy parameters" (page 459). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ASSESS 2020.
| Study characteristics | ||
| Methods | RCT; the study was conducted between August 9, 2012, and April 30, 2018 (including the time required to recruit participants) in 127 sites. | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 4.5 years; mean EDSS 2.7; prior use of DMT at any time prior to the start of study: 53.1% (51.7% in fingolimod 0.5 mg, 52.7% in fingolimod 0.25 mg, and 55.0% in glatiramer acetate) | |
| Interventions | Fingolimod 0.5 mg orally once daily for 12 months (n = 352) Fingolimod 0.25 mg orally once daily for 12 months (n = 370) Glatiramer acetate 20 mg subcutaneously daily for 12 months (n = 342) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A patient randomization list will be produced by the provider using a validated system that automates the random assignment of patient numbers to randomization numbers. These randomization numbers are linked to the different treatment arms, which in turn are linked to medication numbers. A separate medication list will be produced by or under the responsibility of the contract research organization (CRO) using a validated system that automates the random assignment of medication numbers to study drug packs containing each of the study drugs" (Protocol, page 41). |
| Allocation concealment (selection bias) | Low risk | Quoted:"By an interactive voice response system" (page 8) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "The study design was deemed unsuitable to blinding of participants because administering daily placebo injections for participants in the 2 fingolimod groups was not considered ethical practice for a postmarketing study" (page 15). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment(Figure 1, page 20): Fingolimod 0.5 mg: 283 (80.4%)of 352 participants (5.7% consent withdrawn, 0.3% lack of efficacy, 4.5% adverse events) Fingolimod 0.25 mg: 296 (80.0%) of 370 participants (3.2% consent withdrawn, 1.6% lack of efficacy, 4.9% adverse events) Glatiramer acetate: 223 (65.2%) of 342 participants (12.0% consent withdrawn, 3.8% lack of efficacy, 5.9% adverse events) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (Protocol). |
| Serious AE definitions | Low risk | Quoted: "SAEs defined according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use)"(page 9 and Protocol) |
| Method of AE monitoring | Low risk | Quoted: "The occurrence of AEs should be sought by nondirective questioning of the patient at each visit during the study. Adverse events also may be detected when they are volunteered by the patient during or between visits or through physical examination, laboratory test, or other assessments...Unlike routine safety assessments, SAEs are monitored continuously and have special reporting requirements" (Protocol). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
AVANTAGE 2013.
| Study characteristics | ||
| Methods | Parallel RCT conducted at 61 centres in France from March 2006 to April 2008 | |
| Participants | Relapsing MS or a first demyelinating event suggestive of MS (CIS). Diagnostic criteria unclear. Age ≥ 18 years. Mean (SD) EDSS score: 1.8 (1.3). Mean (SD) time from diagnosis of MS: 3.3 (6.4) years | |
| Interventions |
|
|
| Outcomes | Adverse events, serious adverse events, mortality, measured at 3 months’ follow‐up | |
| Notes | Funding: Bayer HealthCare AG, Germany. There is an agreement between Principal Investigators and the Sponsor (or its agents) that restricts the PI's rights to discuss or publish trial results after the trial is completed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blind (open‐label). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Assessors of adverse events were not blind (open‐label). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 3‐month follow‐up study (NCT00317941). IFNβ‐1b group 66 (90.4%) of 73 participants (unknown reasons for 7 not completed) IFNβ‐1b light group 74 (93.7%) of 79 participants (unknown reasons for 5 not completed) IFN‐β‐1a group 60 (88.2%) of 68 (unknown reasons for 8 not completed) |
| Selective reporting (reporting bias) | Low risk | Adverse events were reported as selected in the protocol (NCT00317941). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | Quoted. " Some adverse events reported by participants and some reported by physicians" (NCT00317941) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BECOME 2009.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55; clinically definite RRMS or CIS; median time since MS onset 1 year; mean EDSS 2.0; all participants (except one) were previously untreated patients | |
| Interventions | IFNß‐1b (Betaseron) 250 μg subcutaneously every other day for 24 months (n = 36) Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 39) |
|
| Outcomes | Withdrawals due to AEs over 24 months | |
| Notes | Funding: Bayer Schering Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Randomization was stratified by clinical site (Newark or Teaneck) and the presence of enhancement on screening MRI" (page 1977). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients could not be blinded because of the characteristic injection reactions to IFN‐1b or GA" (page 1977). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Characteristic injection reactions to IFN‐1b or GA did not allow blinding of adverse effects assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 1977): IFNß‐1b: 29 (80.5%) of 36 participants (19.4 lost to follow‐up, 11.1% discontinued treatment) Glatiramer acetate: 35 (89.7%) of 39 participants (10.3 lost to follow‐up, 10.3% discontinued treatment) |
| Selective reporting (reporting bias) | High risk | The published report did not report either AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Not reported |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BENEFIT 2006.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐45 years; CIS; time since neurological event within 60 days; mean EDSS 1.5; all participants were previously untreated patients | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 305) Placebo for 24 months (n = 182) |
|
| Outcomes | Withdrawals due to AEs; AEs over 24 months | |
| Notes | Funding: Schering AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A minimization procedure with an element of chance was applied to minimize imbalance of treatment groups" (page 1243). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Patients were centrally randomized" (page 1243). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "To ensure blinding, the study medications were identical in appearance, packaging, and labeling" (page 1243). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "A treating physician was responsible for the overall medical care of the patient" (page 1243). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 1244): IFNß‐1b: 227 (77.7%) of 292 treated participants (11.0% adverse events) Placebo: 148 (84.1%) of 176 treated participants (0.6% adverse events) |
| Selective reporting (reporting bias) | High risk | SAEs were not clearly reported. |
| Serious AE definitions | High risk | Quoted: "The study was conducted in agreement with Good Clinical Practice (GCP) principles according to the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use E6.11" (page 1243). However, results are reported as follows: "Serious AEs were reported in equal proportions of patients in the two treatment groups (6.8%)" (page 1246). |
| Method of AE monitoring | Unclear risk | Quoted: "Regular visits were scheduled for safety assessments at months 3, 6, 9, 12, 18, and 24" (page 1243). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BEYOND 2009.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 5 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 897) IFNß‐1b (Betaseron) 500 µg subcutaneously every other day for 24 months (n = 899) Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 448) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Bayer HealthCare Pharmaceuticals | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Use of SAS‐based block randomisation with regional stratification" (page 890) |
| Allocation concealment (selection bias) | Low risk | Quoted: "Patients were randomly assigned in a 2:2:1 ratio... by the central randomisation group..." (page 890). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Physicians and patients were double‐blind to comparisons between the two doses of IFNß‐1b... Ibuprofen or acetaminophen were given at the same time as random assignment to IFNß‐1b, at least during the first 3 months, to reduce flu‐like symptoms. The treating physicians and the patients were therefore aware of treatment assignments...The occurrence of adverse events was assessed by telephone, 6 weeks after each visit" (page 891). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "The treating physicians and the patients were aware of treatment assignments...The unmasked, treating physicians were responsible for overall medical care" (page 891). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 890): IFNß‐1b 500 µg: 726 (81.8%) of 887 treated participants (6.0% withdrew, 2.2% adverse events, 5.4% other reasons) IFNß‐1b 250 µg: 784 (88.3%) of 888 treated participants (4.3% withdrew, 1.5% adverse events, 3.0% other reasons) Glatiramer acetate: 374 (84.0%) of 445 treated participants (4.0% withdrew, 1.8% adverse events, 5.4% other reasons) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00099502). |
| Serious AE definitions | High risk | Quoted: "BEYOND was done according to good clinical practice and the International Conference on Harmonisation (ICH) guidelines" (page 890). "Categorisation of serious adverse events conformed to ICH guidelines" (page 892). However, the results are reported as follows: "The incidence of serious adverse events was similar among the groups" (page 894). |
| Method of AE monitoring | High risk | Quoted: "Clinic visits were scheduled every 3 months to assess ... safety, and tolerability. The occurrence of new neurological symptoms and adverse events was assessed by telephone, 6 weeks after each visit" (page 891). "Treating physicians recorded the intensity and frequency of adverse events and assessed whether they were related to treatment" (page 892) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BOLD 2013.
| Study characteristics | ||
| Methods | Adaptive dose‐ranging parallel RCT, conducted at 73 medical centers in Canada, USA, Russia, and nine European countries (Finland, Germany, Hungary, Italy, Norway, Poland, Spain, Switzerland, and Turkey) between 30 March 2009 and 22 October 2010 | |
| Participants | Relapsing MS according to Polman 2005 diagnostic criteria, aged 18–55 years, who had at least one documented relapse during the previous year, at least two documented relapses during the previous 2 years, or one or more gadolinium‐enhancing lesions on MRI at screening, and an EDSS score of 0–5·0 | |
| Interventions | Participants were treated in cohorts sequentially, separated by an interim analysis. Cohort 1: Siponimod 10 mg (n = 50), 2 mg (n = 49), 0.5 mg (n = 43), placebo (unspecified) (n = 45), oral once‐daily for 6 months Cohort 2: Siponimod 1.25 mg (n = 42), 0.25 mg (n = 51), placebo (unspecified) (n = 16), oral once‐daily for 3 months |
|
| Outcomes | Adverse events including cardiac events measured by electrocardiography and Holter monitoring for the first 24 h after receipt of the first dose. Clinical laboratory parameters (assessed at a central laboratory [CoreLab Partners, Princeton, NJ, USA]) | |
| Notes | Funding: Novartis Pharma AG was involved in the study design, and some authors of the article are employed by Novartis and contributed to its preparation. Novartis Pharma AG provided funding for editorial assistance by Oxford PharmaGenesis (Oxford, UK), handling of data by Quintiles, and central laboratory monitoring by CoreLab Partners. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A central, automated system" |
| Allocation concealment (selection bias) | Low risk | Quoted: "A central interactive voice‐response system automated the random assignment of patient numbers to randomisation numbers". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients, investigator staff, the independent assessing physician, the independent first dose administrator, and sponsor staff involved in the conduct of the study remained masked to treatment allocation from the time of randomisation until database lock". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Cohort 1. Completed 6 months on study treatment (Figure 2, page 759):
Cohort 2. Completed 3‐month follow‐up study:
|
| Selective reporting (reporting bias) | Unclear risk | Adverse events were reported by the MedDRA dictionary version 13.1 (ClinicalTrials.gov: NCT00879658); however, no information on selection criteria was available. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | AEs were not prespecified in the protocol (NCT00879658). Quoted: "AEs are collected from first patient first visit until last patient last visit. All AEs reported are from date of first patient first treatment until last patient last visit up to approximately 2.5 years (NCT00879658). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bornstein 1987.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 20‐35 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 3.1; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 25) Placebo bacteriostatic saline subcutaneously daily for 24 months (n = 25) |
|
| Outcomes | Withdrawals due to AEs; AEs over 24 months | |
| Notes | Funding: Grants from the NINCDS and the NIH, Bethesda, Md | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The random assignment of the first patient of a pair determined the assignment of both" (page 409). |
| Allocation concealment (selection bias) | High risk | An open allocation schedule was used: "Treatment assignments were made known to the clinical assistant responsible for the production, labelling and distribution of medication" (page 409). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "The patient's self evaluation of... side effects were reported to the clinical assistant, who was not blinded to the treatment" (page 409). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Patients visited the clinic every three months for two years. At each visit, a neurologist unaware of the patient's treatment group completed a neurologic examination and status evaluation" (page 409). "Patients reported such effects to the unblinded clinical coordinator" (page. 412). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment Glatiramer acetate: 25 (100%) of 25 participants Placebo: 23 (92%) of 25 participants |
| Selective reporting (reporting bias) | High risk | The published report did not report SAEs. |
| Method of AE monitoring | High risk | "Self‐evaluation reported to a clinical assistant" (page 409) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bornstein 1991.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 20‐60 years; clinically definite progressive MS; disease duration not reported; mean EDSS 5.6; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 30 mg subcutaneously (15 mg twice a day) for 24 months (n = 51) Placebo, saline alone, subcutaneously twice a day for 24 months (n = 55) | |
| Outcomes | AEs over 24 months | |
| Notes | Funding: Grants from the NINCDS and the NIH, Bethesda, Md | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted:"Randomisation within centres was accomplished by randomised block design" (page 534). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: "The investigator notified the statistical center, which assigned a randomization code number. Shipment of glatiramer acetate to the patients at their individual centers was totally at random and was dictated by the patients' date of entry into the trial. Only the statistician and the clinical assistant at Albert Einstein College of Medicine, who distributed medication, were aware of patients' assignments" (page 534). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The placebo was described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: "Sterile single‐dose vials containing 0.75 mL of bacteriostatic saline alone or the Cop 1 solution" (page 534) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "Side effects and problems with injections or compliance were not discussed with the study neurologist, but were reported to a clinical assistant. Another blinded neurologist was available to examine patients with severe or unusual side effects (page 534). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 24 months on study treatment (page 535): Glatiramer acetate: 41 (80.4%) of 51 treated participants Placebo: 45 (81.8%) of 55 treated participants Reasons for early withdrawal were pooled for the two arms. |
| Selective reporting (reporting bias) | High risk | The published report did not report either withdrawals due to AEs or SAEs. |
| Method of AE monitoring | Low risk | Quoted: "At each routine visit, patients were asked to complete a questionnaire that reported any local or systemic symptoms or side effects they might have experienced during the previous 3 months" (page 538). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Boyko 2016.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 6 years; median EDSS 2.0; all participants were previously untreated patients | |
| Interventions | Generic glatiramer acetate 20 mg subcutaneously daily for 12 months (n = 62) Brand glatiramer acetate 20 mg subcutaneously daily for 12 months (n = 64) Placebo subcutaneously daily for 12 months (n = 32) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: CJSC BIOCAD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Randomization in the study was central. Enrolled and stratified subjects were randomized within each stratum (block randomization). Each block comprised five symbols: two “1”, two “2", and one ”3" (2:2:1 ratio). A random number generator software created random sequences comprising numbers from 1 to 30. Each of 30 numbers corresponded to one of 30 possible blocks. As the study design did not define in advance the exact number of patients in each stratum, the patients were randomized within each stratum to assure the equal distribution between the arms; therefore, each stratum contained its own block sequence" (information provided by the authors on request). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Central allocation" and "Each syringe was labeled as required by the double‐blind design. For the purpose of blinding, pre‐filled syringes with BCD‐063, Copaxone®‐Teva and placebo had equivalent labels. In addition to the general information, this label contained the code of the drug lot to be used. Each lot ID corresponded to certain batches of the test drug, reference drug and placebo. Drug lots were numbered successively starting from 01001 (test drug arm), 02001 (comparator arm), and 03001 (placebo arm) for the first patient randomized to each arm, and up to the number corresponding to the last patient randomized to each arm (according to his/her order number in the study)" (information provided by the authors on request). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: "After signing an informed consent form, each patient received a 5‐digit screening number consisting of a 2‐digit center number and a 3‐digit patient’s number (continuing numbering in sequence). This was recorded in source documents and a patients’ screening log. Only a CJSC BIOCAD employee (Clinical Study Manager or an authorized Medical Expert) who performed randomization and entered the data to the Randomization Table knew what patient was assigned to which study arm. Information from the Randomization Table could be disclosed if a lot had to be unblinded" (information provided by the authors on request). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "After signing an informed consent form, each patient received a 5‐digit screening number consisting of a 2‐digit center number and a 3‐digit patient’s number (continuing numbering in sequence). This was recorded in source documents and a patients’ screening log. Only a CJSC BIOCAD employee (Clinical Study Manager or an authorized Medical Expert) who performed randomization and entered the data to the Randomization Table knew what patient was assigned to which study arm. Information from the Randomization Table could be disclosed if a lot had to be unblinded" (information provided by the authors on request). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 13.9% was lost‐to follow‐up (13.5% in glatiramer, and 15.6% in in placebo), without indications of differences in reasons. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT02753088). |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Active monitoring for AEs (information provided by the authors on request) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Boyko 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; median disease duration 2 years; median EDSS 2.2; all participants were previously untreated patients | |
| Interventions | Generic IFNß‐1a 44 µg subcutaneously once a week for 12 months (n = 53) Brand IFNß‐1a (Rebif) 44 µg subcutaneously once a week for 12 months (n = 56) Placebo subcutaneously once a week for 12 months (n = 54) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: CJSC BIOCAD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization in the study was central. Enrolled and stratified subjects were randomized within each stratum (block randomization). Patients enrolled in the study were adaptively allocated to treatment groups at a ratio of 1:1:1 within each block. A random number generator software created random sequences of 6 numbers. During the randomization, the clinical research manager of BIOCAD CJSC distributed the patient to the appropriate stratum, assigned him the first free group number in the block and encoded this three‐digit random number. After randomization, the clinical research manager assigned the numbers of the series of drugs/placebo corresponding to the group to which the patient was exposed. The researcher was only informed of the randomization number and numbers of the series of drugs that the patient was supposed to receive" (information provided by the authors on request). |
| Allocation concealment (selection bias) | Low risk | "Central allocation" and "To keep the treatment blinded, syringes with BCD‐033, Rebif and placebo had similar labels. Besides general information, the label contained the lot number. Each lot number corresponded to definite batches of BCD‐033, Rebif or placebo. Each patient was assigned an individual lot number of the drug, which was a random number in the range of 0 to 999. The patient was then given an individual kit containing 12 syringes (the amount needed for therapy for a month) of the study drug, reference drug or placebo. The number of the individual kit consisted of the lot number, as well as the serial number" (information provided by the authors on request). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "After signing an informed consent form, each patient received a 5‐digit screening number consisting of a 2‐digit center number and a 3‐digit patient’s number (continuing numbering in sequence). This was recorded in source documents and a patients’ screening log. Only a CJSC BIOCAD employee (Clinical Study Manager or an authorized Medical Expert) who performed randomization and entered the data to the Randomization Table knew what patient was assigned to which study arm. Information from the Randomization Table could be disclosed if a lot had to be unblinded" (information provided by the authors on request). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "After signing an informed consent form, each patient received a 5‐digit screening number consisting of a 2‐digit center number and a 3‐digit patient’s number (continuing numbering in sequence). This was recorded in source documents and a patients’ screening log. Only a CJSC BIOCAD employee (Clinical Study Manager or an authorized Medical Expert) who performed randomization and entered the data to the Randomization Table knew what patient was assigned to which study arm. Information from the Randomization Table could be disclosed if a lot had to be unblinded" (information provided by the authors on request). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 15.3% was lost‐to follow‐up (13.8% in IFNß‐1a, and 18.5% in in placebo), with some indications of differences in reasons: unsatisfactory therapeutic effect of 0.9% in IFNß‐1a, and 13.0% in placebo. |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Method of AE monitoring | Low risk | Active monitoring for AEs (information provided by the authors on request) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BPSM 1995.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Mean age: 35 years; clinically definite RRMS; disease duration: not reported; EDSS < 5.5; prior use of DMT not reported | |
| Interventions | Methylprednisolone 2 g in saline solution intravenously for 12 hours, every 45‐60 days for 24 months or until relapse (n = 17) Placebo saline solution intravenously at the same schedule (n = 19) |
|
| Outcomes | AEs, SAEs and withdrawals due to AEs not reported | |
| Notes | Funding: None The study had ended prematurely due to organisational reasons and lack of funding. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Randomisation and allocation were generated by computer" (page 6). |
| Allocation concealment (selection bias) | Low risk | Quoted: "The method of randomisation was centralised, providing adequate concealment of allocation" (page 6). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "There were 10 patients (7 treated) lost to follow‐up (28% overall). Moreover, patients experiencing an exacerbation were not followed up although a two year follow‐up was planned" (page 6). |
| Selective reporting (reporting bias) | High risk | The published report did not report SAEs or AEs. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Not reported |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
BRAVO 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; median disease duration 5 years; median EDSS 2.5; prior use of DMT at any time prior to the start of study: 7.4% (6.9% in laquinimod, 9.4% in interferon Beta‐1a, and 6.0% in placebo) | |
| Interventions | Laquinimod 0.6 mg oral capsule once daily for 24 months (n = 434) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 24 months (n = 447) Placebo oral capsule once daily for 24 months (n = 450) |
|
| Outcomes | Withdrawals due to AEs and SAEs over 24 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "The computer‐generated randomization scheme prepared by the Teva Global Biostatistics Unit" (page 775) |
| Allocation concealment (selection bias) | Unclear risk | Quoted: "1:1:1 treatment assignment ratio stratified by study center, to laquinimod 0.6 mg capsule once‐daily, matching oral placebo, or IFNß‐1a IM 30 µg once‐weekly injection" (page 775) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients and treating neurologists were blinded to oral treatment assignment (laquinimod or placebo), but not to IFNb‐1a IM assignment", and "All patients, including those receiving oral treatment, wore clothing and/or a robe that ensured coverage of all potential IM injection sites during examination and were instructed not to discuss adverse events (AEs), routes of administration, or treatment assignments with the examining neurologist" (page 775). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Patients and treating neurologists were blinded to oral treatment assignment (laquinimod or placebo), but not to IFNb‐1a IM assignment" (page 775). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 776): Laquinimod: 353 (81.3%) of 434 treated participants (4.8% adverse events, 8.5% consent withdrawn) IFNβ‐1a: 378 (84.6%) of 447 treated participants (5.8% adverse events, 6.0% consent withdrawn) Placebo: 359 (79.8%) of 450 treated participants (4.2% adverse events, 8.7% consent withdrawn) |
| Selective reporting (reporting bias) | High risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00605215) and AEs were not reported in the article. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Low risk | Quoted: "Patients were evaluated at 12 scheduled visits: months ‐1 (screening), 0 (baseline), 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24. Safety assessments (laboratory measures, vital signs) were performed at all visits, and electrocardiograms (ECGs) were performed at months ‐1, 0, 1, 2, 3, 6, 12, 18, and 24/early termination" (page 775). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
British and Dutch 1988.
| Study characteristics | ||
| Methods | RCT. Study conducted from April, 1983, until October, 1987; 20 hospitals in the United Kingdom and Holland | |
| Participants | Age: 15‐50 years; clinically definite RRMS, SPMS or PPMS; mean disease duration 9 years; mean EDSS 3,7; prior use of DMT not reported | |
| Interventions | Azathioprine 2.5 mg/kg body weight oral daily for 36 months (n = 174) Placebo for 36 months (n = 180) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Wellcome Company. Wellcome Research Laboratories provided the medications. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Quoted: "...assigned patients with MS to receive azathiopirine or placebo by the trial randomisation code" (page 180) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "The patients were seen by a non‐masked doctor who had access to laboratory results and prescribed the trial medication. The occurrence of side‐effects, mean red cell volume (MCV), and dose changes were notified to the trial centre every 3 months. The dose of azathiopirine was reduced in the presence of intolerable side‐effects, leucopenia, anemia, or abnormal liver function tests" (page 180). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Table II and Table III, pages 180‐181): Azathioprine: 128 (73.6%) of 174 treated participants (10.9% adverse events, 8.6% patient preference) Placebo: 151 (83.9%) of 180 treated participants (1.1% adverse events, 9.4% patient preference) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Quoted: "The occurrence of side‐effects, mean red cell volume (MCV), and dose changes were notified to the trial centre every 3 months" (page 180). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Calabrese 2012.
| Study characteristics | ||
| Methods | RCT. A single‐centre study conducted from 1 January 2007 to 30 June 2008 | |
| Participants | Relapsing MS according to the McDonald‐Polman diagnostic criteria for MS, age range 18–55 years and an EDSS score of ≤ 5.0 | |
| Interventions | Subcutaneous IFN beta‐1a, 44 mcg three times weekly (n = 55) Intramuscular IFN beta‐1a, 30 mcg once weekly (n = 55) Subcutaneous glatirame acetate, 20 mcg once daily (n = 55) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs not reported | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "The random allocation sequence was computer generated" (page 419). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It was likely that study participants and personnel were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quoted: "Completed the 2‐year follow‐up study: 46 (83.6%)of 55 participants in the subcutaneous IFN beta‐1a group, 47 (85.4%) of 55 participants in the intramuscular IFN beta‐1a group, and 48 (87.3%) of 55 participants in the Glatiramer acetate group" (page 420). Reasons for study discontinuation not reported |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either AEs or SAEs. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CAMMS223 2008.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50; clinically definite RRMS; median time since first relapse 1 year; mean EDSS 1.9; all participants were previously untreated patients | |
| Interventions | Alemtuzumab 24 mg intravenously daily on 5 consecutive days during the first month and on 3 consecutive days in months 12 and 24 (n = 110) Alemtuzumab 12 mg intravenously daily on 5 consecutive days during the first month and on 3 consecutive days in months 12 and 24 (n = 113) IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 36 months (n = 111) All participants received 1 g of methylprednisolone intravenously for 3 days at baseline and at months 12 and 24. |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Genzyme (a Sanofi company) and Bayer Schering Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Eligible patients were randomly assigned in a 1:1:1 ratio to receive alemtuzumab (at a dose of either 12 mg per day or 24 mg per day) or interferon beta‐1a with the use of the Pocock and Simon minimization algorithm to balance the study groups with regard to age (< 30 years or ≥ 30 years), sex, and baseline EDSS score (< 2.0 or ≥ 2.0)" (page 1787). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Patients were allocated via an interactive voice response system (IVRS)" (Information provided on request by Genzyme). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "The infusion‐related syndrome associated with alemtuzumab precluded double‐blinding" (page 1799). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "Safety was assessed quarterly by the treating neurologist, who was aware of study‐group assignment" (page 1787). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 25.1% was lost‐to follow‐up (16.4% in alemtuzumab 24 mg, 18.6% in alemtuzumab 12 mg, and 40.5% in IFNβ‐1a), with some indications of differences in reasons: adverse event of 0.01% in alemtuzumab 24 mg, 1.8% in alemtuzumab 12 mg, and 11.7% in IFNβ‐1a; and lack of efficacy of 1.8% in alemtuzumab 24 mg, 1.8% in alemtuzumab 12 mg, and 14.4% in IFNβ‐1a. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00050778). |
| Serious AE definitions | Low risk | SAEs defined in the notes of Table 3 (page 1798) |
| Method of AE monitoring | Low risk | Quoted: "Safety was assessed quarterly by the treating neurologist, who was aware of study‐group assignment" (page 1787); "Thyroid function and levels of antithyrotropin receptor antibodies and lymphocyte subpopulations were measured quarterly at a central laboratory"; and "All adverse events with an onset up to 36 months are reported. In addition, all serious adverse events and autoimmune‐associated disorders occurring before March 1, 2008, are listed" (page 1788). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CARE‐MS I 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration 2 years; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | Alemtuzumab 12 mg intravenously daily on 5 consecutive days in month 0 and on 3 consecutive days in month 12 (n = 386) IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 195) Participants in both groups received 1 g per day of methylprednisolone intravenously on 3 consecutive days at baseline and at month 12. After a protocol amendment in January, 2009, alemtuzumab patients received oral aciclovir 200 mg twice daily during alemtuzumab infusion and for 28 days thereafter as prophylaxis against herpes infection. |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "We randomly allocated patients in a 2:1 ratio" and "Randomisation was stratified by site" (page 1820). |
| Allocation concealment (selection bias) | Low risk | Quoted: "We randomly allocated patients using an interactive voice response system" (page 1820). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Because both study drugs have adverse effects that precluded masking of patients and treating clinicians to treatment assignment, and because subcutaneous interferon beta 1a was available only in proprietary prefilled syringes that could not effectively be duplicated for placebo..." (page 1820), and "Because of the different schedules and routes of administration, and side‐effect profiles of the study drugs, as in the phase 2 study, masking of patients and treating clinicians to treatment assignment was not feasible" (page 1826). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "Because both study drugs have adverse effects that precluded masking of patients and treating clinicians to treatment assignment, and because subcutaneous interferon beta 1a was available only in proprietary prefilled syringes that could not effectively be duplicated for placebo..." (page 1820), and "Because of the different schedules and routes of administration, and side‐effect profiles of the study drugs, as in the phase 2 study, masking of patients and treating clinicians to treatment assignment was not feasible" (page 1826). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 7.1% was lost‐to follow‐up (4.9% in alemtuzumab 12 mg, and 11.3% in IFNβ‐1a), with some indications of differences in reasons: adverse events of 2.6% in alemtuzumab, and 0% in IFNβ‐1a. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00530348). |
| Serious AE definitions | Low risk | Quoted: "The study was done in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice 11" (page 1820). |
| Method of AE monitoring | Low risk | Quoted: "To assess safety, we undertook monthly questionnaire follow‐up of patients, and did complete blood counts, serum creatinine, urinalysis, and microscopy monthly (every three months in patients in the interferon beta 1a group), and thyroid function tests every 3 months”; “Circulating lymphocyte subsets were assessed every 3 months in all patients and 1 month after alemtuzumab administration. We screened for anti‐alemtuzumab antibodies with a bridging ELISA before and at 1 month, 3 months, and 12 months after each dosing”; and “We measured interferon beta 1a‐neutralising antibodies at baseline and at 24 months with a cytopathic effect inhibition assay” (page 1821). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CARE‐MS II 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 5 years; mean EDSS 2.7; all patients were previously treated: "at least one relapse while on interferon beta or glatiramer after at least 6 months of treatment" | |
| Interventions | Alemtuzumab 24 mg intravenously daily on 5 consecutive days in month 0 and on 3 consecutive days in month 12 (n = 173; data presented for safety assessment, only) Alemtuzumab 12 mg intravenously daily on 5 consecutive days in month 0 and on 3 consecutive days in month 12 (n = 436) IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 231) Participants in both groups received 1 g per day of methylprednisolone intravenously on 3 consecutive days at baseline and at month 12. After a protocol amendment in December, 2008, alemtuzumab patients received oral aciclovir 200 mg twice daily during alemtuzumab infusion and for 28 days thereafter as prophylaxis against herpes infection. |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "2:1 randomisation allocation stratified by site" (pages 1830‐1831). |
| Allocation concealment (selection bias) | Low risk | Quoted: "We randomly allocated patients with an interactive voice response system" (page 1830). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Because both study drugs had adverse effects that precluded double‐blinding, and interferon beta 1a proprietary syringes could not effectively be duplicated for placebo..." (page 1831) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "Because both study drugs had adverse effects that precluded double‐blinding, and interferon beta 1a proprietary syringes could not effectively be duplicated for placebo..." (page 1831) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 11.4% was lost‐to follow‐up (4.6% in alemtuzumab 12 mg, and 24.2% in IFNβ‐1a), with some indications of differences in reasons: lack of efficacy of 0% in alemtuzumab 12 mg, and 2.6% in IFNβ‐1a. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00548405). |
| Serious AE definitions | Low risk | Quoted: "The study was done in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 1830). |
| Method of AE monitoring | Low risk | Quoted: “To assess safety, we undertook monthly questionnaire follow‐up of patients, and did complete blood counts, serum creatinine, and urinalysis with microscopy monthly (every 3 months in patients in the interferon beta 1a group), and thyroid function tests every 3 months”, “We assessed circulating lymphocyte subsets every 3 months in all patients and 1 month after every course of alemtuzumab. We screened for anti‐ alemtuzumab antibodies with ELISA before and at 1 month, 3 months and 12 months after each dosing”, and “We measured interferon beta 1a‐neutralising antibodies at baseline and at 24 months with a cytopathic effect inhibition assay” (page 1832). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CCMSSG 1991.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: at least 15 years; clinically or laboratory‐supported SPMS, PPMS or PRMS; mean disease duration 10 years; mean EDSS 5.8; prior use of DMT not reported | |
| Interventions | Cyclophosphamide 1 g intravenously three times weekly for 36 months + 40 mg prednisone tapered for 16 days (total dose ≤ 9 g) (n = 55) Placebo for 36 months (n = 56) | |
| Outcomes | AEs over 36 months | |
| Notes | Funding: BRISTOL Myers; Upjohn | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “A randomisation sequence was generated separately for each centre. Patients were stratified by centre and EDSS score” (page 442). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Single‐masked” |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “Each patient was followed by both a monitoring neurologist who was aware of treatment allocation and an evaluating neurologist who was not. The monitoring neurologist supervised the experimental treatments” (page 443). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quoted: “All patient were followed until death or until the end of the study period” (page 443). |
| Selective reporting (reporting bias) | High risk | The published report did not report either withdrawals due to AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: “The external safety monitoring committee monitored the progress of the trial every 6 months (severe adverse experiences, deaths, clinical status)” (page 443). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CHAMPS 2000.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; CIS; median time since neurological event 19 days; mean EDSS not reported; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Avonex) 30 µg intramuscularly weekly for 36 months (n = 193) Placebo intramuscularly weekly for 36 months (n = 190) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “We used a minimisation procedure to assign patients randomly in approximately equal numbers to the two treatment groups” (page 899). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: “Patients and site personnel were unaware of the treatment assignments” (page 899). "The occurrence of the influenza‐like syndrome related to interferon beta‐1a therapy could have provided some patients with a clue to the treatment assignment" (page 902). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “The treating neurologist was responsible for asking the patient about adverse events” (page 899). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 14.9% was lost‐to follow‐up (15.5% in IFNß‐1a, 14.2% in placebo). No information on the reasons for the study discontinuation |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | High risk | Quoted: “Each centre was instructed to report all adverse events during the first six months of treatment, but thereafter to report only serious adverse events, as well as depression, seizures, cardiac events, and injection‐site reactions, whether they were serious” (page 899). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Cheshmavar 2021.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite progressive MS; mean disease duration 14 years; mean EDSS 3.2; prior use of DMT not reported | |
| Interventions | Rituximab 1000 mg intravenously every 6 months for 12 months (n = 43) Glatiramer acetate 40 mg subcutaneously three times a week for 12 months (n = 41) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Isfahan University of Medical Sciences | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “We assigned a random number to each participant using Microsoft Excel function to generate random numbers and randomized patients based on their numbers (we allocated even and odd numbers to RTX and GA group, respectively)” (page 1, Supporting Information). |
| Allocation concealment (selection bias) | High risk | Quoted: “We assigned a random number to each participant using Microsoft Excel function to generate random numbers and randomized patients based on their numbers (we allocated even and odd numbers to RTX and GA group, respectively)” (page 1, Supporting Information). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 13.1% was lost‐to follow‐up (14.0% in rituximab, and 12.2% in glatiramer), without indications of differences in reasons. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT03315923). |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Quoted: “We interviewed patients every other month through phone‐calls to ask for adverse events” (page 180). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CLARITY 2010.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 9 years; mean EDSS 2.9; prior use of DMT: 30.3% (26.1% in cladribine 3.5 mg, 32.2% in cladribine 5.25 mg and 32.5% in placebo) | |
| Interventions | Cladribine 3.5 mg/kg of body weight orally in two short courses for the first 12 months and two short courses for the second 12 months (for a total of 8 to 20 days per year) (n = 433) Cladribine 5.25 mg/kg of body weight orally in four short courses for the first 12 months and two short courses for the second 12 months (for a total of 8 to 20 days per year) (n = 456) Placebo for 24 months (n = 437) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: EMD Serono | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “A computer‐generated treatment randomization code” (page 417) |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed with the use of a central system” (page 417). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “To maintain the double‐blind nature of the study, all patients within a weight range received the same number of tablets (cladribine or matched placebo)” (page 418). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "At each study site, a treating physician reviewed clinical laboratory results and assessed treatment‐emergent adverse events and safety information, and an independent evaluating physician who was unaware of study‐group assignments performed neurologic examinations” (page 418). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Supplemental Figure 2, page 3): Cladribine 5.25 mg: 406 (89.0%) of 456 participants (2.4% lost to follow‐up, 2.0% adverse events, 0.9% protocol violation, 0.9% unsatisfactory efficacy, 0.2% died, 4.6% consent withdrawn for administrative, convenience and personal reasons) Cladribine 3.5 mg: 398 (91.9%) of 433 participants (1.8% lost to follow‐up, 1.1% adverse events, 0.9% protocol violation, 1.1% unsatisfactory efficacy, 0.2% died, 2.8% consent withdrawn for administrative, convenience and personal reasons) Placebo: 380 (87.0%) of 437 participants (0.9% lost to follow‐up, 1.1% adverse events, 2.3% protocol violation, 4.8% unsatisfactory efficacy, 0.5% died, 3.4% consent withdrawn for administrative, convenience and personal reasons) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (Supplementary Appendix). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with International Conference on Harmonization Tripartite Guidelines for Good Clinical Practice" (Supplementary Appendix, page 1). |
| Method of AE monitoring | Low risk | Quoted: “A treating physician reviewed clinical laboratory results and assessed treatment‐emergent adverse events and safety information...The safety assessment included a review of the incidence of treatment‐emergent adverse events in each study group, physical examination, and laboratory measurements” (page 418). "Time frame: Baseline up to week 96. AEs collected by non‐systematic assessment, Term from vocabulary, MedDRA (11.0)" (NCT00213135). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CombiRx 2013.
| Study characteristics | ||
| Methods | RCT. The trial enrolled participants from January 2005 through April 2009, with study follow‐up closing in April 2012 when the final participant enrolled completed 36 months on study. The trial was conducted at 68 sites, both private practice and academic, in the US and Canada. | |
| Participants | Age: 18‐60 years; clinically definite RRMS; mean disease duration 1 year; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | IFNβ‐1a (Avonex) 30 µg intramuscular once a week with matched placebo preparation for 36 months (n = 250) Glatiramer acetate 20 mg subcutaneously daily with matched placebo preparation for 36 months (n = 259) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: National Institutes of Health (NIH) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Participants were randomized via a computerised data entry system using a permuted block design within sites with permuted block sizes of 6 and 12” (page 328). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Participants were randomized via a computerised data entry system that masked treatment arm allocation” (page 328). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Participants were randomized via a computerised data entry system that masked drug dispensing to participants and all site personnel for the entire duration of the trial period. All participants administered the same number of injections” (page 328). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Treating clinician and an examining clinician were both blinded to treatment assignment” (page 328). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Figure 3, page 14): IFNβ‐1a: 194 (77.6%) of 250 treated participants (7.2% adverse events) Glatiramer acetate: 223 (86.1%) of 259 treated participants (4.6% adverse events) Quoted: "A higher proportion of participants in the glatiramer acetate treatment completed month 36 (P = 0.029, Figure 3). |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (Lindsey 2012; NCT00211887). |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition. "Adverse events were only collected with regard to the affected organ system" (NCT00211887). |
| Method of AE monitoring | Unclear risk | Quoted: “Safety was assessed by recording all adverse events, serious and non‐serious” (page 329) and "Adverse events were only collected with regard to the affected organ system. AEs collected by systematic assessment (NCT00211887) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Comi 2001.
| Study characteristics | ||
| Methods | RCT. Twenty‐nine centers in six European countries and Canada participated in the trial. Enrollment started in February 1997 and concluded in November 1997. | |
| Participants | Age 18‐50 years; clinically definite RRMS; mean disease duration 8 years; mean EDSS 2.4; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneously daily for 9 months (n = 119) Placebo (not described) (n = 120) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 9 months | |
| Notes | Funding: Teva Pharmaceutical | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The randomization list, stratified by centres, was computer‐generated by the TEVA Statistical Data Management Department. Equal allocation of the two treatment groups was used” (page 291). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “All personnel involved in the study were unaware of the treatment allocation” (page 291). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “A treating neurologist was responsible for the overall medical management of the patient including safety monitoring...An examining neurologist was responsible for all scheduled neurological examinations and exacerbation follow‐up...The treating neurologist and the patient were informed of the importance of not discussing safety issues with the examining neurologist” (page 291). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 9 months on study treatment (page 292): Glatiramer acetate: 110 (92.4%) of 119 participants Placebo: 113 (94.2%) of 120 participants Quoted: "7 patients dropped out in each arm. 7 patients dropped out in the first trimester, 5 in the second trimester, and 2 in the third trimester. 2 subjects in the placebo group and 3 in the gòatirame group discontinued treatment because of adverse experiences. One patient in the placebo arm discontinued treatment that he considered ineffective, another left because of poor compliance, one was lost to follow‐up, and 2 refused to continue MRI monitoring. One subject discontinued glatiramer treatment when he moved away from the center, and another after a severe exacerbation. 4 glatiramer subjects withdrew their consent without providing a reason" (page 292). |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Quoted:“The treating physician monitored safety...Vital signs and adverse effects were assessed monthly...Safety evaluations that included vital signs, hematology, and biochemical tests were performed every 3 months at all regularly scheduled clinical visits.” (page 291). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Comi 2008.
| Study characteristics | ||
| Methods | RCT. The study was undertaken in 51 centers in nine countries. Enrollment started in March 2005, and was completed in October 2005. | |
| Participants | Age 18‐50 years; clinically definite RRMS; disease duration not reported; mean EDSS 2.4; prior use of DMT not reported | |
| Interventions | Laquinimod 0.6 mg oral capsule once daily for 9 months (n = 106) Laquinimod 0.3 mg oral capsule once daily for 9 months (n = 98) Placebo oral capsule once daily for 9 months (n = 102) |
|
| Outcomes | Withdrawals due to AEs and SAEs over 9 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The randomisation list, stratified by study centre, was computer‐generated” (page 2086). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “The drug preparations were identical except for laquinimod concentration. Patients and all personnel were blinded to treatment assignment” (page 2086). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “At each study site, a treating neurologist was responsible for the overall medical management of patients, including safety monitoring...An independent external data safety monitoring board met six times via teleconference and three times in face‐to‐face meetings during the trial period, to review the study conduct and the unblinded safety results” (page 2086). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 36 weeks on study treatment (Figure 1, page 2088): Laquinimod 0.6 mg 100 (94.3%) of 106 participants (1.9% adverse events, 1.9% request of primary care) Laquinimod 0.3 mg 92 (93.9%) of 98 participants (3.0% withdrew consent, 2.0% adverse events) Placebo 91 (89.2%) of 102 participants (2.0% lost to follow‐up, 2.0% withdrew consent, 4.9% adverse events, 1.0% request of primary care) |
| Selective reporting (reporting bias) | High risk | The published report did not clearly report AEs. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Low risk | Quoted: “Vital signs, adverse events, and concomitant medications were assessed at baseline, then at months 1, 3, 6, 7, 8, and 9 or early termination. Blood samples for laboratory safety assessments (haematology, serum chemistry, and urinalysis) were obtained at baseline, then every 3 months or at early termination. ECG was done at baseline then at months 1, 3, and 9 or early termination” (page 2086). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
CONCERTO 2022.
| Study characteristics | ||
| Methods | RCT. The study was conducted at 215 sites in 29 countries. The study was conducted from February 20, 2013 to July 4, 2017. | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 5.8 years; mean EDSS 2.7; prior use of DMT at any time prior to the start of study: 29.2% (27.0% in laquinimod 0.6 mg, and 31.5% in placebo). The majority (89%) of the participants were from eastern European countries; Russia (20.3%), Ukraine (18.7%), Bulgaria (9.3%), and Poland (7.5%). | |
| Interventions | Laquinimod 1.2 mg oral capsule once daily for 24 months (n = 732) [This arm was discontinued 8 months after the study start due to findings of cardiovascular events and patients exposed to laquinimod 1.2 mg were encouraged to continue follow‐up of the drug for 24 months]. Laquinimod 0.6 mg oral capsule once daily for 24 months (n = 727) Placebo oral capsule once daily for 24 months (n = 740) |
|
| Outcomes | AEs, SAEs, withdrawals due to AEs, vital signs, electrocardiograms, and clinical laboratory parameters at specific scheduled site visits over 24 months | |
| Notes | Funding: This study was sponsored by Teva Pharmaceutical Industries, Petach Tikva, Israel. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Moreover, authors reported "double blind"; however, many, if not most, participants treated with 1.2 mg of laquinimod had become probably aware of the treatment they were receiving during the course of the trial. Quoted: "The laquinimod 1.2‐mg dose arm was discontinued as of 1 January 2016 due to findings of cardiovascular events at laquinimod doses above 0.6 mg daily. All patients re‐consented with disclosure of the risk observed with higher doses" (page 609). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Investigators were blinded to treatment assignments" (page 609). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information for laquinimod 1.2 mg. Completed 24 months on study treatment (Figure 1, page 610): Laquinimod 0.6 mg group 587 (80.7%) of 727 participants (8.8% consent withdrawn, 4.4% adverse events, 2.6% other reasons, 1.8% unsatisfactory efficacy) Placebo group 556 (75.1%) of 740 participants (11.9% consent withdrawn, 2.6% adverse events, 3.8% other reasons, 3.0% unsatisfactory efficacy) |
| Selective reporting (reporting bias) | Low risk | Outcomes were those reported in the protocol (ClinicalTrials.gov: NCT01707992). |
| Serious AE definitions | Low risk | Quoted: "Serious adverse event was an AE resulting in any of the following outcomes or deemed significant for any other reason: death; initial or prolonged inpatient hospitalization; life‐threatening experience (immediate risk of dying); persistent or significant disability/incapacity; congenital anomaly" (ClinicalTrials.gov: NCT01707992). |
| Method of AE monitoring | Low risk | Quoted:" An AE was any untoward medical occurrence in a participant who received study drug without regard to possibility of causal relationship"(ClinicalTrials.gov: NCT01707992). Clinically significant vital signs abnormalities, abnormal serum chemistry and hematology values, and ECG abnormalities, were all pre‐defined. (ClinicalTrials.gov: NCT01707992). AEs were collected for the procedure and at each follow‐up visit at 2 weeks, 6 weeks, 12 weeks, 24 weeks. Quoted: "ECG findings assessed as “abnormal, clinically significant” were evaluated by the data monitoring committee cardiologist". |
| Other bias | Low risk | Study appeared free of other sources of bias. |
CONFIRM 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration (time since diagnosis) 5 years; mean EDSS 2.6; prior use of any MS medication at any time prior to the start of study: 40% to 41% across study groups | |
| Interventions | Dimethyl fumarate 240 mg orally three times daily for 24 months (n = 345) Dimethyl fumarate 240 mg orally two times daily for 24 months (n = 362) Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 350) Placebo orally three times daily for 24 months (n = 363) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned in a 1:1:1:1 ratio to receive oral placebo, BG‐12 at a dose of 240 mg two times daily, BG‐12 at a dose of 240 mg three times daily, or subcutaneous daily injections of 20 mg of glatiramer acetate for 96 weeks” (page 1088); and “The randomization was stratified by site” (page 33 of the Protocol). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization took place across all study sites using a centralized Interactive Voice Response System (IVRS)” (page 33 of Protocol). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Patients receiving glatiramer acetate were aware of their treatment assignment. All study management and site personnel, investigators, and patients were unaware of assignment to the BG‐12 and placebo groups”, and “To ensure that the assignments to the BG‐12 and placebo groups would not be revealed, patients in those groups were instructed not to take the study medication within 4 hours before each study visit, since a flushing reaction is known to be more common with BG‐12” (page 1088). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Each site used separate examining and treating neurologists, thereby maintaining rater blinding for all study groups, including the group that received glatiramer acetate" (page 1088). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Supplementary appendix; Figure S2, page 10): Dimethyl fumarate 240 mg three times daily: 248 (71.9%) of 345 treated participants (7.5% adverse events, 7.2% discontinued study drug) Dimethyl fumarate 240 mg two times daily: 253 (70.5%) of 359 treated participants (5.8% adverse events, 8.6% discontinued study drug) Glatiramer acetate: 263 (75.1%) of 350 treated participants (2.9% adverse events, 8.3% discontinued study drug) Placebo: 234 (64.5%) of 363 treated participants (3.0% adverse events, 12.1% discontinued study drug) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (Appendix, Protocol). |
| Serious AE definitions | Low risk | Definition of SAEs reported in the Protocol (page 64) |
| Method of AE monitoring | Low risk | Quoted: “Throughout the course of the study, every effort was made to remain alert to possible adverse events (AEs)”; and “Any AE or SAE experienced by the subject was recorded on the CRF, regardless of the severity of the event or its relationship to study treatment” (pages 66‐67, Protocol). Relationship of events to study treatment reported (pages 68 Protocol). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
DECIDE 2015.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.5; prior use of DMT at any time prior to the start of study: 41.1% (41.3% in daclizumab 150 mg and 40.8% in IFNβ‐1a 30 µg) | |
| Interventions | Daclizumab 150 mg subcutaneously every 4 weeks for 36 months (n = 919) IFNβ‐1a (Avonex) 30 µg intramuscular once weekly for 36 months (n = 922) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization was...stratified according to study site and prior use of interferon beta with the use of permuted‐block randomization” (page 1420). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was conducted with the use of a centralized interactive voice response system” (page 1420). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “All the patients and study personnel, including the treating neurologists, were unaware of the treatment assignments” (page 1420). "To prevent unblinding based on influenza‐like symptoms following interferon beta‐1a injection, patients were instructed to take nonsteroidal anti‐inflammatory drugs at the dose and frequency according to local labels before and for 24 hours after each injection of interferon beta‐1a or matching placebo" (pages 1419‐20). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “All the patients and study personnel, including the treating neurologists, were unaware of the treatment assignments” (page 1420); and “The treating neurologist was responsible for routine neurological care of the patient, assessment and treatment of adverse events and multiple sclerosis relapse, review of hematology and laboratory assessments, and monitoring and follow‐up of abnormal hepatic tests.” (Page 6 Supplementary file) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Supplementary appendix; Figure S2, page 17): Daclizumab: 653 (71.1%) of 919 participants (14.1% adverse events, 3.4% lack of efficacy, 7.4% consent withdrawn) IFNβ‐1a: 644 (69.8%) of 922 participants (9.0% adverse events, 7.4% lack of efficacy, 9.9% consent withdrawn) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (Supplementary Appendix; NCT01064401). |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Low risk | Quoted: “Study visits occurred every 4 weeks and included ... clinical and safety assessments” (page 1420). "A protocol amendment, dated May 27, 2011, increased monitoring for liver‐function tests to monthly...provided a treatment algorithm for the evaluation and management of cutaneous events" (Appendix page 5). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
DEFINE 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration (time since diagnosis) 6 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 40.7% (40.4% in dimethyl fumarate 240 mg three times daily, 39.5% in dimethyl fumarate 240 mg two times daily, and 42.2% in placebo). | |
| Interventions | Dimethyl fumarate 240 mg orally three times daily for 24 months (n = 416) Dimethyl fumarate 240 mg orally two times daily for 24 months (n = 411) Placebo orally three times daily for 24 months (n = 410) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned, in a 1:1:1 ratio, to receive BG‐12 at a dose of 240 mg twice daily, BG‐12 at a dose of 240 mg three times daily, or placebo. Randomization was performed centrally and was stratified according to site” (page 1100). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally” (page 1100), and “Randomization took place across all study sites using a centralized Interactive Voice Response System (IVRS)” (page 33 of Protocol). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “To maintain concealment of the study‐group assignments, each study centre used separate examining and treating neurologists“ (page 1100). "The treating neurologist will be responsible for...assessment (including assignment of causality) and treatment of adverse events and MS relapses... and review of selected hematology and blood chemistry results from the central laboratory to assess whether the subject’s study treatment should be temporarily withheld or permanently discontinued" (Appendix Protocol; page 44). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 23 months on study treatment (Supplementary appendix; Figure S2, page 6): Dimethyl fumarate 240 mg three times daily: 288 (69.2%) of 416 treated participants (8.9% adverse events, 7.7% discontinued study treatment, 4.6% consent withdrawn) Dimethyl fumarate 240 mg two times daily: 281 (68.5%) of 410 treated participants (9.8% adverse events, 8.3% discontinued study treatment, 5.4% consent withdrawn) Placebo: 264 (64.7%) of 408 treated participants (5.4% adverse events, 13.0% discontinued study treatment, 7.6% consent withdrawn) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (Appendix Protocol; pages 42‐3). |
| Serious AE definitions | Low risk | SAEs defined in the Protocol (page 64) |
| Method of AE monitoring | Low risk | Quoted: “The primary treating neurologist was responsible for assessment (including assignment of causality) and treatment of adverse events... Study visits were scheduled every 4 weeks for safety assessments, including the monitoring of laboratory values” (page 1100). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ellison 1989.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18 years or older; clinically definite SPMS or PPMS; mean disease duration 15 years; mean DSS 5.5; prior use of DMT not reported | |
| Interventions | Azathioprine 3 mg/kg body weight orally daily for 36 months (n = 33) Placebo for 36 months (n = 34) |
|
| Outcomes | Withdrawals due to AEs over 36 months | |
| Notes | Funding: Wellcome Company and Upjohn Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Treatment would be allocated by a randomization process to block of 4 successive patients. A master list was computed in which treatments were assigned according to patient sequence number. Patient sequence was the order of presenting the initial prescription to the pharmacy". (page 1019) |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “The statistician told the examining neurologists that the treatments would be allocated by a randomisation process to blocks of 4 successive patients, but the assignment rules were not revealed” (page 1019). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “The monitor assessed patients for non‐MS abnormalities (which included potential adverse effects)” (page 1019). "He also monitored the laboratory results for adverse effects, adjusted the azathioprine dosage, and prescribed symptomatic treatment" (page 1020). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Table 1, page 1021): Azathioprine: 18 (58.1%) of 31 treated participants (16.1% adverse events) Placebo: 22 (64.7%) of 34 treated participants (5.9% adverse events) |
| Selective reporting (reporting bias) | High risk | Protocol was not available. The published report did not report SAEs. AEs were not clearly reported. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Quoted: “The patients were instructed to call the clinic anytime they suspected an adverse event and then actively monitored by neurologist. At each clinic visit non‐MS abnormalities were sought by open‐ended and focused questions by the study nurse and the monitoring neurologist...once a month for the first three months and every three months thereafter. The monitoring neurologist reviewed the effects discovered by the nurse, interviewed and examined the patient, confirmed the non‐MS abnormality, assigned the date of onset, severity, presumed cause, action taken (e.g. adjust dose of medication, order a test, prescribe a treatment), and duration. He also monitored the laboratory results for adverse effects...” (page 1020). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Etemadifar 2006.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 15‐50 years; clinically definite RRMS; mean disease duration 3 years; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | IFNβ‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 30) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 24 months (n = 30) IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 30) | |
| Outcomes | AEs, SAEs and withdrawals due to AEs were not reported. | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: "Patients were assigned randomly and equally to one of the three treatment groups” (page 284). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “The trial was single‐blinded in that patients were aware” (page 284) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Physicians who assessed the outcome were unaware of the treatment type that the patient had received. Patients were evaluated at 6, 12, and 24 months after the start of the therapy by a qualified neurologist to evaluate the development of side effects of the medications and compliance of the patients.” (page 284). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information |
| Selective reporting (reporting bias) | High risk | The published report did not report either withdrawals due to AEs, AEs or SAEs. |
| Method of AE monitoring | High risk | Quoted: “Given the lack of safety assessment of this trial, it is important to recall that the safety of IFN‐b products in the treatment of relapsing MS had already been established for the three drugs in previous studies” (page 286). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Etemadifar 2007.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 13‐50 years; clinically definite RRMS; mean disease duration not reported ("short duration"); mean EDSS 1.5; all participants were previously untreated patients | |
| Interventions | Azathioprine 3 mg/kg body weight orally daily for 12 months (n = 47) IFNß (Betaseron, Avonex, or Rebif) for 12 months (n = 47: 15 Betaseron 250 μg subcutaneously every other day, 19 Avonex 30 µg intramuscularly once a week, 13 Rebif 44 µg subcutaneously three times a week) |
|
| Outcomes | Withdrawals due to AEs and AEs over 12 months | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomized according to a pre‐existing list produced by a computer program that differed from a random number generator only in that it assigned equal numbers of patients into each treatment group” (page 1724). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “The first treatment group received IFNβ products regimen. The second group received AZA” (page 1724). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “The trial was single blinded in that patients were aware..." (page 1724) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “...physicians who assessed the outcome were unaware of treatment type that the patient was receiving”, and “Two neurologists who do not know which patients had received which treatment clinically evaluated all patients” (pages 1724‐1725). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 1724): Azathioprine: 44 (93.6%) of 47 treated participants (6.4% adverse events) Interferon beta: 44 (93.6%) of 47 treated participants (6.4% adverse events) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: “Adverse events, vital signs and blood tests were monitored monthly...The patients were available for follow‐up at 3, 6, and 12 months” (pages 1724‐1725). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ETOMS 2001.
| Study characteristics | ||
| Methods | RCT. 57 centers in 14 European countries took part in this study. Patients were enrolled between August 1995, and July 1997. | |
| Participants | Age: 18‐40 years; CIS; time since neurological event within 3 months; mean EDSS 1.2; all participants were previously untreated patients | |
| Interventions | IFNß‐1a (Rebif) 22 µg by subcutaneously once weekly for 24 months (n = 154) Placebo for 24 months (n = 155) |
|
| Outcomes | AEs and SAEs over 24 months | |
| Notes | Funding: Serono | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The treatment was assigned according to a computer‐generated randomisation list stratified by centre” (page 1577). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “At each study site, a treating physician was responsible for the overall management of the patient, including safety monitoring ... An evaluating physician was responsible for all scheduled neurological examinations and exacerbation follow‐up visits ... Masking of the study preparation given during the double‐blind phase was maintained for both patients and physicians” (page 1577). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 24 months on study treatment. Quoted: "241 (78%) patients received study treatment until the end of the study" (page 1578). Reasons for study discontinuation in each arm were not described. |
| Selective reporting (reporting bias) | High risk | Protocol was not available. The published report did not report withdrawals due to AEs. |
| Serious AE definitions | Unclear risk | Quoted: "Serious adverse events (defined according to the guidelines of the International Conference on Harmonisation"(page 1580) but SAEs are reported as an aggregate outcome, as follows: "SAEs were reported in five patients in the placebo group and six in the interferon beta‐1a group" (page 1580). |
| Method of AE monitoring | Low risk | Quoted: “Neurological and safety assessments, including vital signs, haematology, and biochemical tests, were done at the end of months 1, 6, 12, 18, and 24” (page 1577). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
European Study Group 1998.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age 18‐55 years; clinically definite SPMS; mean disease duration 13 years; mean EDSS 5.1; prior use of DMT not reported | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 36 months (n = 360) Placebo (unspecified) for 36 months (n = 358) | |
| Outcomes | Withdrawals due to AEs and AEs over 36 months | |
| Notes | Funding: Schering AG, Berlin | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “A central randomisation schedule assigned placebo or interferon ‐1b to blocks of six patients in a 1/1 ratio” (page 1492). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “Access to the code was strictly limited according to study protocol” (page 1492). Study protocol was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Interferon ‐1b was indistinguishable from placebo” (page 1492). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “Designated treating physicians were responsible only for general medical care, safety assessments and treatment of relapses" (page 1492). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Figure 1, page 1493 and Table 3, page 1494): IFNß‐1b: 270 (75.0%) of 360 treated participants (1.4% adverse events, 6.4% inefficacy of trial medication) Placebo: 261 (72.9%) of 358 treated participants (1.1% adverse events, 12.3% inefficacy of trial medication) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Low risk | Quoted: "Regular visits were scheduled for days 1, 3, 5, and 15, months 1–3, and thereafter every 3 months until month 36 (end of treatment) and month 39 (end of drug‐free follow‐up)" (page 1491‐2). “Safety assessments included adverse events, vital signs, physical examinations, and concomitant medication. Standard laboratory tests were done at all regular visits by a central laboratory. An electrocardiogram was done at the beginning and end of the study. The Montgomery Alsberg Depression Rating Scale (MADRS), an observer rating scale, was used to assess mood changes and suicidal risk at all regular quarterly visits...” (pages 1492‐1493). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
EVIDENCE 2002.
| Study characteristics | ||
| Methods | RCT. Participants were enrolled at 56 centers (15 in Europe, 5 in Canada, and 36 in the United States). | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 12 months (n = 339) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 12 months (n = 338) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Serono Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Treatment assignments were determined using a computer‐generated randomization list...Randomization was stratified by center, with an initial block size of six followed by block sizes of four in order to reduce the ability of sites to determine subsequent treatment allocation based on prior allocation” (page 1498). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Treatment assignments were allocated through a centralized telephone randomization system to unblinded site personnel. Patients were allocated equally to the two treatment groups" (page 1498). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “It would have been very difficult to keep the patients blinded to their treatment allocation, due to the markedly different injection frequencies and adverse event (AE) profiles of the 2 regimens”(page 2033). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "It would also have been difficult to keep treating physicians blinded while dealing with AEs. Even when attempts are made to have full double‐blinding in IFN studies, AEs such as skin reactions, flu‐like symptoms, and laboratory abnormalities may lead to unintentional corruption of blinding.” (Schwid 2007, page 2033) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 1499): Subcutaneous IFNβ‐1a: 314 (92.6%) of 339 treated participants (4.7% adverse events, 0.9% lack of efficacy) Intramuscular IFNβ‐1a: 317 (93.8%) of 338 treated participants (4.1% adverse events, 0.3% lack of efficacy) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00292266). |
| Serious AE definitions | Low risk | Quoted: "Guidelines were provided for the treating physician based on the World Health Organization (WHO) side‐effect severity scale" (page 1497). |
| Method of AE monitoring | High risk | “Adverse events were determined by spontaneous reporting and monthly laboratory testing during the comparative phase” (page 2031). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
EVOLVE‐MS‐2 2020.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 10 years; mean EDSS 2.7; prior use of DMT at any time prior to the start of study: 66.5% (66.8% in diroximel fumarate, and 66.1% in dimethyl fumarate) | |
| Interventions | Diroximel fumarate 462 mg orally two times daily for 1 month (n = 254) Dimethyl fumarate 240 mg orally two times daily for 1 month (n = 252) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 1 month. | |
| Notes | Funding: Alkermes Inc. and Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Block randomization was performed using a block size of 4" (page 186). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “Patients were randomized 1:1 into one of the two treatment groups, and all patients received two capsules twice daily for all doses to maintain blinding” (page 186). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “The treatment period was double‐blind; DMF capsules were over‐encapsulated to create the blinded stud drug” (page 186). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “Patients were instructed to assess gastrointestinal tolerability with two self‐administered questionnaires (IGISIS and GGISIS) twice per day within 9 h of taking the study drug, using an eDiary", "Adverse events were assessed by the investigator at weekly visits and recorded by severity and relatedness" and "...investigators, and sites remained blinded" (page 188). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 5.2% was lost‐to follow‐up (3.2% in diroximel fumarate, and 7.2% in dimethyl fumarate), with some indications of differences in reasons: adverse events 1.6% in diroximel fumarate, and 6.0% in dimethyl fumarate (Table 2, page 190). |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (NCT03093324). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Council on Harmonisation Guidelines for Good Clinical Practice" (page 187), and SAEs term from vocabulary, MedDRA 20.1 (NCT03093324). |
| Method of AE monitoring | Low risk | Quoted: “Patients utilized two eDiary symptom scales to evaluate the duration and severity of GI symptoms on a daily basis: IGISIS and GGISIS... Adverse events were assessed by the investigator at weekly visits and recorded by severity and relatedness.” (page 188). "Safety assessments included AEs (including GI AEs), vital signs, clinical laboratory tests (chemistry, hematology, and urinalysis), and electrocardiogram findings" (page 188). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
EXPAND 2018.
| Study characteristics | ||
| Methods | RCT. The study was undertaken at 292 hospital clinics and specialised multiple sclerosis centers in 31 countries. | |
| Participants | Age: 18‐60 years; clinically definite progressive MS; mean disease duration 17 years; mean EDSS 5.4; prior use of DMT: 21.7% (22.2% in siponimod, and 20.9% in placebo) | |
| Interventions | Siponimod 2 mg orally once daily for at least 12 months (n = 1105) Placebo orally once daily for at least 12 months (n = 546) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Novartis Pharma AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Eligible patients were randomly assigned (2:1) to receive once daily oral siponimod 2 mg or matching placebo by blocked randomisation with a block size of 6. Randomisation was stratified for each of the 31 countries” (page 1265). |
| Allocation concealment (selection bias) | Low risk | Quoted: “...the randomisation list was produced by an interactive response technology provider (Parexel, Billerica, MA, USA) using a validated system automating the random assignment of patient numbers to randomisation numbers. Randomisation numbers were linked to the different treatment groups, which in turn were linked to medication numbers. A separate medication list was produced by Novartis drug supply management using a validated system that automated the random assignment of medication numbers to packs containing the study drugs” (page 1265). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Patients and study staff remained masked to treatment assignment for the duration of the core part of the study” and “Study drug and placebo were identical in packaging, labelling, schedule of administration, appearance, taste, and odour” (page 1265). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “To maintain masking, an independent doctor monitored patients during dose titration, and the counts for the total number of leucocytes, neutrophils, and lymphocytes were normally withheld by the central laboratory and only reported to the investigator in case of notable abnormalities” (page 1265). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 12 months on study treatment (Figure 1, page 1266): Siponimod 839 (76.3%) of 1100 participants (17.9% discontinued study, 8% adverse events) Placebo 399 (73.1%) of 546 participants (22.3% discontinued study, 5% adverse events) Reasons for study treatment discontinuation were not clearly reported. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT01665144). |
| Serious AE definitions | Low risk | Quoted: "The trial adhered to the International Conference on Harmonization Guidelines for Good Clinical Practice" (page 1265). |
| Method of AE monitoring | Low risk | Quoted: "Adverse events were coded according to the Medical Dictionary for Regulatory Activities, version 19.0. To characterise cardiac safety during dose titration, patients underwent continuous mobile cardiac telemetry. For patients from countries where mobile cardiac telemetry technology was not approved as a medical device, holter electrocardiograms were recorded on 3 days (appendix p 8)." (page 1265) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Fazekas 1997.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 15‐64 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 3.3; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.15‐0.20 g/kg body weight intravenously monthly for 24 months (n = 75) Placebo intravenously monthly for 24 months (n = 75) |
|
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: Sero‐Merieux (Vienna, Austria) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Centralised computer‐generated randomisation schedule with stratification by centre, age, sex, and deterioration rate” (page 590) |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomly and centrally allocated” (page 590) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Infusions of IVIg and saline placebo were identical in appearance and were stored in plastic bags for concealment during administration" (page 590). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “At each monthly visit a neurologist who was aware of treatment allocation (treating physician) administered the study medication and asked the patient about any side‐effects" (page 590). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 24 months on study treatment (Figure 1, page 590): Immunoglobulins 64 (85.3%) of 75 participants Placebo 56 (76.7%) of 73 participants Reasons for study treatment discontinuation were not reported. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | High risk | Quoted: "The treating physician asked the patient about any side‐effects" (page 590). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Fazekas 2008.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 3 years; mean EDSS 2.0; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.2 g/kg body weight intravenously monthly for 12 months (n = 45) Immunoglobulins 0.4 g/kg body weight intravenously monthly for 12 months (n = 42) Placebo intravenously monthly for 12 months (n = 41) | |
| Outcomes | Withdrawals due to AEs and SAEs over 12 months | |
| Notes | Funding: Bayer HealthCare AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The random code number was computer generated by the Statistics and Data System Department of Bayer” (page 266). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomisation performed by an unblinded pharmacist who assigned code numbers from sealed envelopes sequentially” (page 266) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: “Considerable effort was made to achieve optimal blinding, including the provision that all patients received a total volume of 4 mL/kg body weight per infusion, which was adjusted by the addition of dextrose 5%” (page 266). No information was reported on personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “Considerable effort was made to achieve optimal blinding" (page 266). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 267): Immunoglobulins 0.2: 38 (84.4%) of 45 participants (6.7% unsatisfactory efficacy, 2.2% adverse event, 2.2% consent withdrawn, 2.2% non‐compliance) Immunoglobulins 0.4: 38 (90.5%) of 42 participants (9.5% consent withdrawn) Placebo 37 (90.2%) of 41 participants (4.9% pregnancy, 2.4% unsatisfactory efficacy, 2.4% consent withdrawn) |
| Selective reporting (reporting bias) | High risk | The protocol was not available. The published report did not report AEs. |
| Serious AE definitions | Unclear risk | Definition of SAEs was not reported. Quoted: "Both the design and the execution of the trial, ... followed the International Conference on Harmonisation–Good Clinical Practices requirements" (page 269). |
| Method of AE monitoring | Unclear risk | No information |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
FREEDOMS 2010.
| Study characteristics | ||
| Methods | RCT. Participants were assigned to study treatment from January 2006 through August 2007, at 138 centers in 22 countries. | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 8 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 40.9% (39.6% in fingolimod 1.25 mg, 42.6% in fingolimod 0.5 mg, and 40.4% in placebo). | |
| Interventions | Fingolimod 1.25 mg orally once daily for 24 months (n = 429) Fingolimod 0.5 mg orally once daily for 24 months (n = 425) Placebo orally once daily for 24 months (n = 418) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned, in a 1:1:1 ratio, to receive oral fingolimod capsules in a dose of 0.5 mg or 1.25 mg or matching placebo ... Randomization was performed ... with the use of stratification according to site, with a block size of six within each site” (page 388). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally, with the use of a validated system” (page 388). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Participants assigned to fingolimod 1.25 mg/day became aware. Quoted: "Following a recommendation from the independent data and safety monitoring board, participants randomized to the fingolimod 1.25 mg dose were converted to the fingolimod 0.5 mg dose. Randomization numbers for the fingolimod 1.25 mg dose group were provided by Novartis to the study sites then individual participants were identified and invited to the study site for an unscheduled visit in order to be converted to fingolimod 0.5 mg/day" (Supplementary data, page 1). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 18.8% was lost‐to follow‐up (22.6% in fingolimod 1.25 mg, 13.2% in fingolimod 0.5 mg, and 20.6% in placebo), with some indications of differences in reasons: unsatisfactory therapeutic effect of 3.0% in fingolimod 1.25 mg, 1.4% in fingolimod 0.5 mg, and 6.0% in placebo; and abnormal laboratory values(s) of 4.7% in fingolimod 1.25 mg, 2.1% in fingolimod 0.5 mg, and 0.2% in placebo (Figure 1, page 391). |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (Supplementary Appendix). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 388). |
| Method of AE monitoring | Low risk | Quoted: “Study visits, including safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization” (page 389).“The examination included an assessment of skin, head and neck, lymph nodes, breast, heart, lungs, abdomen, and back, and comments on general appearance. Participants were recommended to continue to perform skin self‐examination on a monthly basis” (Appendix). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
FREEDOMS II 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 11 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 74.8% (77.6% in fingolimod 1.25 mg, 73.7% in fingolimod 0.5 mg, and 73.0% in placebo). | |
| Interventions | Fingolimod 1.25 mg orally once daily for 24 months (n = 370) Fingolimod 0.5 mg orally once daily for 24 months (n = 358) Placebo orally once daily for 24 months (n = 355) "After review of data from the FREEDOMS and TRANSFORMS phase 3 studies, completed on Nov 12, 2009, after consultation with and at the recommendation of the data and safety monitoring board, we decided to stop the 1.25 mg dose. Patients on the high dose were subsequently switched to the 0.5 mg dose in a blinded manner" (page 546). |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “We randomly allocated patients (1:1:1; stratified by study centre) to receive oral fingolimod capsules in a dose of 0.5 mg or 1.25 mg or matching placebo, once daily for 24 months. The randomisation sequence was generated with an automated system under the supervision of the Novartis Drug Supply Management team” (page 546). |
| Allocation concealment (selection bias) | Low risk | Quoted: “To mask treatment allocation, both fingolimod and placebo were dispensed in hard gelatin capsules of identical colour and size and packed in identical bottles” (page 546). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "To mask treatment allocation, both fingolimod and placebo were dispensed in hard gelatin capsules of identical colour and size and packed in identical bottles"(page 546). "Patients, site personnel, and other personnel were blinded to the study medication assignments" (Supplementary web appendix). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 28.2% was lost‐to follow‐up (32.2% in fingolimod 1.25 mg, 24.0% in fingolimod 0.5 mg, and 28.2% in placebo), with some indications of differences in reasons: unsatisfactory therapeutic effect of 2.7% in fingolimod 1.25 mg, 1.7% in fingolimod 0.5 mg, and 4.8% in placebo; and adverse events or abnormal laboratory values(s) of 12.7% in fingolimod 1.25 mg, 10.1% in fingolimod 0.5 mg, and 5.1% in placebo (Figure 1, page 547). |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (Supplementary web appendix). |
| Serious AE definitions | Low risk | Quoted: "The study was done in accordance with International Conference on Harmonisation Good Clinical Practice guidelines" (page 546). |
| Method of AE monitoring | Low risk | Quoted: “...safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization” (Appendix, page 3). "We recorded adverse events, serious adverse events, serious adverse events of special interest, 24‐h Holter electrocardiography (ECG) post first‐dose and at 3 months, first‐dose bradycardia events, infections, laboratory tests, vital signs, ECG, echocardiography, pulmonary function tests, chest high‐resolution CT, chest radiographs, ophthalmic examinations, including serial optical coherence tomography, and dermatological assessments" (pages 548‐9). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
FUMAPMS 2021.
| Study characteristics | ||
| Methods | RCT. A single‐center study executed at the Danish Multiple Sclerosis Center, Copenhagen University Hospital—Rigshospitalet in Denmark from December 6, 2016, to January 16, 2019 | |
| Participants | Primary progressive MS. Eligible participants aged 18–65 years, had an EDSS score between 0 and 6.5, and a CSF concentration of NFL above 380 ng/L. | |
| Interventions | Oral dimethyl fumarate 240 mg twice daily. From days 1–21, the study drug dose was titrated from 120 to 480 mg as a daily maintenance dose for 12 months (n = 27) Placebo for 12 months (n = 27) |
|
| Outcomes | Primary. Change in CSF concentration of NFL from screening to week 48 Adverse events were reported using Common Terminology Criteria for Adverse Events (CTCAE). Safety assessments included physical examination by the trial investigators every 24 weeks, along with vital parameters assessment and safety blood tests every 12 weeks. |
|
| Notes | Funding: the study was funded by a grant from Biogen (Cambridge, MA) and grants from the Danish Multiple Sclerosis Society. The funders did not contribute to study design or implementation, data analyses, data interpretation, or writing. Biogen provided trial medicine at no cost. The corresponding author had full access to the data and had final responsibility for the decision to submit for publication. ClinicalTrials.gov Identifier: NCT02959658 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Low risk | Quoted: "The randomization and masking were performed by the Regional Capital Pharmacy in Copenhagen". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: "Study drug and placebo capsules were identical and with identical packaging, labeling, expiration date, taste, and odor... There was a high frequency of flushing and gastrointestinal pain in the treatment arm, which may have compromised blinding of investigators and patients". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "Common side effects of the study drug were evaluated by the investigators". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 4): Dimethyl fumarate: 26 (96.3%) of 27 participants (1 discontinued study) Placebo: 24 (88.9%) of 27 participants (3 discontinued study) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events were reported using Common Terminology Criteria for Adverse Events (CTCAE); however, no information on pre‐selected adverse events was available (NCT02959658). |
| Serious AE definitions | Low risk | SAEs were defined according to Common Terminology Criteria for Adverse Events (CTCAE). |
| Method of AE monitoring | Low risk | Quoted: "Safety assessments included physical examination by the trial investigators every 24 weeks, along with vital parameters assessment and safety blood tests every 12 weeks...The frequent occurrence of common AEs in the dimethyl fumarate group, resulting in more frequent contact with trial personnel, may also have led to increased reporting of other adverse events, for example, infections". |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
GALA 2013.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 8 years; mean EDSS 2.8; prior use of DMT at any time prior to the start of study: 13.6% (13.6% in glatiramer acetate, and 13.7% in placebo) | |
| Interventions | Glatiramer acetate 40 mg subcutaneously three times a week for 12 months (n = 943) Placebo subcutaneously three times a week for 12 months (n = 461) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Eligible patients were assigned to treatment groups in a 2:1 ratio (GA 40 mg tiw or placebo) according to the randomization scheme produced. The randomization scheme used constrained blocks stratified by centre” (page 706). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "...matching placebo (40 mg of mannitol dissolved in water). Study drugs were packaged and labeled in a way that maintained the masked nature of the study; the appearance, shape, color, and smell were identical" (page 706). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Patients’ general medical assessments were performed separately from the neurological assessments by 2 neurologists or physicians. The examining neurologist or physician was responsible for all neurological assessments” (page 706). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months' follow‐up study (Fig. 1, page 708): Glatiramer acetate: 859 (91.1%) of 943 participants (3.1% adverse events, 3.6% consent withdrawn, 0.5% lost to follow‐up, 0.4% no re‐consent after relapse, 0.5% other reasons) Placebo: 430 (93.3%) of 461 participants (1.3% adverse events, 3.7% consent withdrawn, 0.2% lost to follow‐up, 0.2% no re‐consent after relapse, 0.4% other reasons) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | Quoted: “Safety assessments included adverse events, standard clinical laboratory tests, vital signs, and electrocardiographic measurements” (page 707). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
GATE 2015.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 2.7; prior use of DMT not reported | |
| Interventions | Generic glatiramer acetate 20 mg subcutaneously daily for 9 months (n = 355) Brand glatiramer acetate 20 mg subcutaneously daily for 9 months (n = 357) Placebo subcutaneously daily for 9 months (n = 84) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 9 months | |
| Notes | Funding: Synthon BV | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted:“Randomization was performed centrally and stratified according to geographical region (European Union, North America, or the rest of the world) and the number of gadolinium‐enhancing lesions at screening” (page 1434). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Study group assignments were performed using an interactive web and voice response system" (page 1434). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Copaxone® and placebo will be identical in appearance and will be packaged identically and will only be identified by means of a medication number. The randomization list that relates medication number to type of treatment is located at a central location and not available to trial personnel (sponsor, investigator, evaluators)" (Protocol; page 25). Quoted: "During the trial, participants, study personnel, MRI evaluators, steering committee members, and the study statistician (R.M.) were unaware of study group assignments” (page 1434). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: "During the trial, participants, study personnel, steering committee members, and the study statistician were unaware of study group assignments" (page 1434) and “Investigators are responsible for monitoring the safety and for providing appropriate medical care in subjects who have entered this trial. In addition, the investigator remains responsible for following AEs that are serious or that caused the subject to discontinue before completing the trial until resolution or stabilization" (Protocol; page 41). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 9 months on study treatment (Figure 1, page 1435): Generic glatiramer acetate: 324 (91.8%) of 353 treated participants (3.1% adverse events, 3.1% consent withdrawn) Brand glatiramer acetate: 324 (90.8%) of 357 treated participants (1.1% adverse events, 5.3% consent withdrawn) Placebo: 81 (96.4%)of 84 treated participants (2.4% adverse events, 1.2% consent withdrawn) |
| Selective reporting (reporting bias) | Unclear risk | Quoted: "There are no pre‐specified safety endpoints in this trial" (Protocol; page 41). |
| Serious AE definitions | Low risk | "The study was conducted in accord with International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines for good clinical practice" (page 1434). |
| Method of AE monitoring | Low risk | Quoted: “Safety assessments were performed at screening, baseline, and months 1, 3, 6, and 9” (page 1434). "Safety assessments included monitoring of adverse events, local injection site reactions, vital signs, and laboratory test results. Neurological symptoms related to confirmed relapses and local injection site reactions recorded in the tolerability diaries were not additionally reported as adverse events" (page 1435). Quoted: "Each subject will be carefully questioned and/or examined by the investigator or a medically qualified delegate (i.e. authorized by the investigator, in a separate form, to record adverse events) to obtain information regarding adverse events (AEs, including serious AEs) at each visit until the last protocol specified visit or contact. All adverse events will be reported and documented" (Protocol; page 41). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ghezzi 1989.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS or SPMS; mean disease duration 5 years (RRMS) and 7 years (SPMS); mean EDSS 2.1 (RRMS) 3.7 (SPMS); prior use of DMT not reported | |
| Interventions | Azathioprine 2.5 mg/kg body weight orally daily for 18 months (n = 93) Placebo for 18 months (n = 92) |
|
| Outcomes | AEs over 18 months | |
| Notes | Sponsor: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 27.0% was lost‐to follow‐up (26.0% in azathioprine, and 28.0% in placebo). Nothing was said about reasons for the treatment discontinuation. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either withdrawals due to AEs or SAEs. |
| Method of AE monitoring | Unclear risk | No information |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
GOLDEN 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS; mean disease duration 5 years; mean EDSS 2.6; prior use of DMT at any time prior to the start of study: 50.9% (52.5% in fingolimod, and 46.4% in IFNß‐1b) | |
| Interventions | Fingolimod 0.5 mg orally once daily for 18 months (n = 106) IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 18 months (n = 51) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 18 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “At baseline, eligible patients were randomised (2:1) to receive oral fingolimod (0.5 mg/day) or subcutaneous IFN β‐1b (250 μg every other day; Fig. 1)” (page 2438). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding of participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information on blinding of adverse events assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 19.1% was lost‐to follow‐up (8.5% in fingolimod, and 23.4% in IFNβ‐1b), with some indications of differences in reasons: unsatisfactory therapeutic effect of 0.9% in fingolimod, and 13.7% in IFNβ‐1b. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT01333501). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 2438). |
| Method of AE monitoring | Low risk | Quoted: “Safety assessments included reporting of adverse events (AEs), serious AEs (SAEs), vital signs, physical/neurological examinations, skin examination, laboratory examinations, electrocardiogram (ECG) monitoring (as required) and ophthalmologic examinations. AEs, SAEs and vital signs were assessed at each study visit. Physical examinations were performed at screening and months 6, 12 and 18; ophthalmologic examinations were performed at screening and months 3, 6 and 18; and skin examinations were performed at screening and month 18.” (page 2439). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Goodkin 1991.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 3.5; prior use of DMT not reported | |
| Interventions | Azathioprine 3.0 mg/kg body weight orally daily for 24 months (n = 30) Placebo orally daily for 24 months (n = 29) | |
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: Wellcome Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomised by the statistician using random number tables” (page 21) |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unmasked treating neurologist. It was unclear whether participants were blinded. Quoted: “Group PLC received indistinguishable placebo”; and “whenever the treating physician made a dose change for an AZA patient, a similar dose change was simultaneously made for a matched placebo patient to preserve the blind” (pages 20‐21). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “Each patient had the same masked examining neurologist and unmasked treating neurologist for the duration of the study” (page 21). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 11.9% was lost‐to follow‐up (10.0% in azathioprine, and 13.8% in placebo), without indications of differences in reasons. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | High risk | Quoted: “Side effect were reported to the treating neurologist every 6 months” (page 21). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Goodkin 1995.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 21‐60 years; clinically definite PPMS or SPMS; median disease duration 10 years; mean EDSS 5.4; prior use of DMT not reported | |
| Interventions | Methotrexate 7.5 mg orally weekly for 24 months (n = 31) Placebo for 24 months (n = 29) | |
| Outcomes | Withdrawals due to AEs over 24 months | |
| Notes | Funding: none | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “The randomisation scheme was developed for each strata prior the initiation of the study and was blocked in groups of 10” (page 32). |
| Allocation concealment (selection bias) | High risk | “Treatment assignments were made by the unblinded study coordinator once the eligibility of the patients was confirmed” (page 32). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on blinding of participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “The treating neurologist was permitted access to the treatment code if clinical status suggested toxicity that would potentially require cessation of treatment" (page 32). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 3.3% was lost‐to follow‐up (6.5% in methotrexate, and 0% in placebo). This is likely a chance result due to the small sample size. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. AEs were not clearly reported. |
| Method of AE monitoring | Unclear risk | “All participants maintained a daily diary of undesirable events… The adverse event diary was checked every 3 months by the study nurse during a clinical visit” (page 32). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hartung 2002.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Progressive relapsing MS, also termed worsening relapsing MS, or gradual progression of disability with or without superimposed clinical relapses (secondary progressive MS); age: 18‐55 years; mean disease duration 10 years; mean EDSS 4.6; all participants were previously untreated patients | |
| Interventions | Mitoxantrone 5 mg/m² body surface area intravenously every 3 months for 24 months (n = 66) Mitoxantrone 12 mg/m² body surface area intravenously every 3 months for 24 months (n = 63) Placebo for 24 months (n = 65) | |
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: Wyeth‐Lederle Benelux and Germany | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation was done by means of a computer generated schedule prepared for each site with a block size of three, without stratification” (page 2019). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: “Placebo solution (15 mL) was mixed with 3 mg methylene blue to match the colour of active‐drug infusions” (page 2019). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | “A treating physician was aware of treatment assignment. This physician carried out all medical assessments, reviewed laboratory data, adjusted the dose of study drug according to protocol, provided treatments for symptoms” (page 2019). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 2020): Mitoxantrone 5 mg: 54 (83.1%) of 65 participants (6.2% lost to follow‐up, 4.6% lack of efficacy, 4.6% refused) Mitoxantrone 12 mg:48 (77.4%) of 62 participants (8.1% adverse events, 6.5% lack of efficacy, 3.2% lost to follow‐up, 3.2% refused) Placebo: 47 (73.4%) of 64 participants (12.5% lack of efficacy, 9.4% refused, 3.1% adverse events, 1.6% lost to follow‐up) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. SAEs were not clearly reported. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Quoted: "Cardiac monitoring was done before treatment and then once a year. The monitoring included electrocardiography with rhythm‐control printout and measurement of left‐ventricular ejection fraction by echocardiography or radionuclide scan" (page 2020). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hauser 2008.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 10 years; median EDSS 2.5; prior use of DMT at any time prior to the start of study: 77.9% (78.3% in rituximab and 77.1% in placebo) | |
| Interventions | Rituximab 1000 mg intravenously on days 1 and 15 followed up to 12 months (n = 69) Placebo on days 1 and 15 followed up to 12 months (n = 35) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Biogen Idec and Genentech, Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned in a 2:1 ratio to receive rituximab or placebo (Fig. 1), and they were hierarchically stratified according to study site, status with respect to previous treatment with interferon beta or glatiramer acetate (either no treatment or discontinuation of medication ≥ 6 months previously vs. treatment within the previous 6 months), and baseline disease severity according to the Expanded Disability Status Scale (EDSS) score (≤ 2.5 vs. > 2.5)” (page 679). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on whether placebo infusion was indistinguishable from rituximab infusion in terms of taste, appearance, and duration of infusion |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "The treating investigator was the safety assessor and made all treatment decisions based on the patient's clinical response and laboratory findings" (page 680). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment (Table 2, page 681): Rituximab: 58 (84.1%) of 69 treated participants (0 patient’s decision, 4.3% physician’s decision, 2.9% lost to follow‐up, 2.9% relapse, 2.9% initiation of excluded therapy) Placebo: 21 (60.0%) of 35 treated participants (11.4% patient’s decision, 8.6% physician’s decision, 5.7% lost to follow‐up, 5.7% relapse, 5.7% initiation of excluded therapy) |
| Selective reporting (reporting bias) | Low risk | Outcomes were those reported in the protocol (ClinicalTrials.gov: NCT00097188). |
| Serious AE definitions | Low risk | Quoted: "The Common Toxicity Criteria, version 3.0, were used to grade adverse events (page 680)". "Serious adverse events were defined as life‐threatening, resulting in death, requiring prolonged inpatient hospitalization, disabling, resulting in a congenital anomaly or malignant condition, or requiring surgical intervention to prevent one of these outcomes" (page 686) |
| Method of AE monitoring | Low risk | Quoted: “At regularly scheduled visits over a period of 48 weeks, neurologic and physical examinations, MRI, and routine laboratory tests were performed and adverse events were recorded” (page 680). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hommes 2004.
| Study characteristics | ||
| Methods | RCT. A European‐Canadian multicenter study | |
| Participants | Clinically and laboratory supported definite diagnosis of MS and a secondary progressive form of the disease for at least 1 year with a disease duration of at least 3 years; age: 18‐55 years; mean time since first symptoms 14 years; EDSS score from 3.0 to 6.5, prior use of DMT not reported | |
| Interventions | Immunoglobulins 1 g/kg body weight intravenously monthly for 30 months (n = 159) Placebo for 30 months (n = 159) |
|
| Outcomes | Withdrawals due to AEs over 27 months | |
| Notes | Funding: Bayer Corporation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients are assigned to immunoglobulins or placebo according to the randomization list generated by the biometrical department of Bayer, Germany in a 1/1 ratio. The randomisation was done centrally as block randomisation with stratification by centre” (Hommes 2000; Protocol, page S28). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: "Access to the code is strictly limited according to the sponsors Standard Operation Procedures" (Hommes 2000; Protocol, page S28) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Concealment of treatment was guaranteed by use of an albumin solution identical in appearance to the study medication, with identical labelling and opaque plastic wrapping. In addition vials have identical labeling and are covered by a white opaque plastic wrap.” (Hommes 2000; Protocol, page S28). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Blinding is guaranteed by operating with a treating and evaluating neurologist” (Hommes 2000; Protocol, page S28). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 27 months on study treatment (Figure 1, page 1152): Immunoglobulins: 120 (75.5%) of 159 treated participants (6.3% adverse events, 6.3% consent withdrawn, 5.0% insufficient therapeutic effect, 3.1% non‐compliance) Placebo: 140 (88.0%) of 159 treated participants (3.1% adverse events, 1.9% consent withdrawn, 3.1% insufficient therapeutic effect, 0.6% non‐compliance) |
| Selective reporting (reporting bias) | High risk | Quoted: "Safety assessments include vital signs, adverse events, physical examination, concomitant medication, ECG, standard laboratory tests and serology" (Hommes 2000; Protocol, page S31). SAEs and AEs were not clearly reported in the article. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: "Regular visits for neurological evaluation are scheduled every 3 months even after a premature termination of the treatment unless the patient withdraws consent or is lost to follow‐up" (Hommes 2000; Protocol, page S27). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
IFNB MS Group 1993.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration (time since diagnosis) 4 years; mean EDSS 2.9; prior use of DMT not reported | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 124) IFNß‐1b (Betaseron) 50 µg subcutaneously every other day for 24 months (n = 125) Placebo subcutaneously every other day for 24 months (n = 123) |
|
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: Triton Biosciences, Inc., Alameda, CA and Berlex Laboratories Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Each placebo vial contained only similar quantity of albumin and dextrose”; “All personnel were blinded to treatment categories” (page 656). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “One treating neurologist who knew about side effects, reviewed laboratory findings for toxicity, and was responsible for overall care" (page 656) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 9.1% was lost‐to follow‐up (7.3% in IFNß‐1b 250 µg, 11.2% in IFNß‐1b 50 µg, and 8.9% in placebo). Nothing was said about the reasons for the study discontinuation. Withdrawals and losses to follow‐up were difficult to find, and different data were given in different articles about the same trial. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: “Treating neurologist reviewed side effects, laboratory findings for toxicity ...” (page 656). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
IMPACT 2002.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite SPMS; mean disease duration 16 years; mean EDSS 5.2; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Avonex) 60 µg intramuscularly weekly for 24 months (n = 217) Placebo for 24 months (n = 219) | |
| Outcomes | Withdrawals due to AEs over 24 months | |
| Notes | Funding: BIOGEN INC. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The contract research organisation computer generated two minimisation schemes, one for North America and one for Europe and Israel” (page 680). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "The treating nurse and neurologist were responsible for clinical management of the subjects." (page 680). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 681): IFNβ‐1a: 156 (71.9%) of 217 treated participants (10.1% adverse events, 2.8% perceived worsening, 9.7% subject request, 3.7% treatment failure EDSS) Placebo: 165 (75.3%) of 219 treated participants (3.6% adverse events, 10.5% perceived worsening, 4.1% subject request, 1.4% treatment failure EDSS) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs and AEs were not clearly reported. |
| Method of AE monitoring | Low risk | Quoted: “Routine safety blood samples were obtained every 3 months", and "An independent external Data and Safety Monitoring Committee reviewed safety data at three time points during the trial and performed a preplanned interim analysis after all subjects had been followed for 15 months" (page 681). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
IMPROVE 2010.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS; disease duration at least 12 months; EDSS not reported; prior use of DMT not reported | |
| Interventions | A new formulation of IFNβ‐1a (Rebif) 44 µg subcutaneously three times weekly for 4 months (n = 120) Placebo subcutaneously three times weekly for 4 months (n = 60) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 4 months | |
| Notes | Funding: Merck Serono S.A. – Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Quoted: “Patients were randomised centrally” (page 888). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 4 months' follow‐up: IFN‐b1a 112 (93.3%) of 120 participants; placebo 57 (95.0%) of 60 participants. Reasons for withdrawals not reported |
| Selective reporting (reporting bias) | Low risk | SAEs and AEs were reported in NCT00441103. |
| Serious AE definitions | Low risk | Definition of SAEs reported: Quoted: "A serious adverse event (SAE) was an AE that resulted in any of the following outcomes: death; life‐threatening; persistent/significant disability/incapacity; initial or prolonged inpatient hospitalization; congenital anomaly/birth defect"(NCT00441103). |
| Method of AE monitoring | Unclear risk | Quoted: “Following treatment completion or early termination, patients underwent a 4‐week safety observation period” (page 888). "Time frame: Baseline up to week 40. Term from vocabulary, MedDRA (9.1)" (NCT00441103). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
INCOMIN 2002.
| Study characteristics | ||
| Methods | RCT. Study conducted at 15 centers throughout Italy. Participants were recruited between October 1997, and June 1999. | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration (time since diagnosis) 6 years; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 96) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 24 months (n = 92) |
|
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: The Italian Ministry of Health and the Italian MS Society | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation followed computer‐generated random sequences of digits that were different for each centre and for each sex, to achieve centre and sex stratification” (page 1454). |
| Allocation concealment (selection bias) | Low risk | Quoted: “The codes were randomly assigned to treatments by an independent team of statisticians unaware of the patient’s clinical characteristics” (page 1454). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded to treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “All clinical outcomes were assessed in an open‐label manner” (page 1454). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 1455): IFNß‐1b: 85 (88.5%) of 96 treated participants (5.2% adverse events, 3.1% lack of efficacy, 2.1% lost to follow‐up) IFNß‐1a: 73 (79.3%) of 92 treated participants (1.1% adverse events, 10.9% lack of efficacy, 4.3% lost to follow‐up) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Low risk | Quoted: “Safety assessments included adverse events, vital signs, physical examination, and concomitant medications. Patients underwent haematology and biochemical tests, including liver‐function tests, every 2 weeks for the first 8 weeks, and then every 3 months” (page 1455). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
INFORMS 2016.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Clinically definite PPMS according to the 2005 revised McDonald criteria; age: 25‐65 years; mean disease duration 6 years; mean EDSS 4.7; prior use of DMT at any time prior to the start of study: 21.2% (18.4% in fingolimod 1.25/0.5 mg, 19.0% in fingolimod 0.5 mg, and 23.6% in placebo) | |
| Interventions | Fingolimod 1.25/0.5 mg orally once daily for at least 36 months and a maximum of 60 months (n = 147) Fingolimod 0.5 mg orally once daily for at least 36 months and a maximum of 60 months (n = 336) Placebo orally once daily for at least 36 months and a maximum of 60 months (n = 487) "Patients were initially assigned to fingolimod 1.25 mg per day or placebo (cohort 1); however, after a protocol amendment on Nov 19, 2009, patients were switched in a masked manner to fingolimod 0.5 mg, whereas those on placebo continued on matching placebo. From then onwards, patients were assigned to receive fingolimod 0.5 mg/day or placebo (cohort 2)" (page 1077). |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned (1:1) with computer generated blocks to receive either fingolimod or placebo. The randomisation sequence was automatically generated by a validated system under the responsibility of Novartis Drug Supply Management. The randomisation occurred in blocks of four within centre in a 1:1 ratio to fingolimod or placebo” (page 1077). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Allocation was concealed through the use of blinded code‐break cards with removable, scratch‐off cover for the whole double‐blind treatment period” (page 1077). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “All randomised drug assignments remained masked to patients, investigator staff, people performing the assessments, and data analysts for the whole double‐blind treatment period (at least 36 months and up to 5 years)” and "We achieved masking by use of identical packaging and identical capsule colour and size for treatment and placebo” (page 1077). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Heart rate reduction is a known pharmacological effect of fingolimod that can potentially unmask study participants. To maintain masking, an independent first dose administrator monitored pulse rate after the first dose of study drug. Employees of the funder who were independent of the study team monitored first dose safety and were masked to study allocation" (page 1077). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Figure 1, page 1078): Fingolimod 1.25/0.5 mg: 78 (53.1%) of 147 treated participants (22.4% adverse events or abnormal laboratory values, 7.5% unsatisfactory therapeutic effect) Fingolimod 0.5 mg: 211 (62.8%) of 336 treated participants (14.3% adverse events or abnormal laboratory values, 6.8% unsatisfactory therapeutic effect) Placebo: 315 (64.7%) of 487 treated participants (7.4% adverse events or abnormal laboratory values, 13.1% unsatisfactory therapeutic effect) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00731692). |
| Serious AE definitions | Low risk | Quoted: "The study was done in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 1077). Definition of SAEs reported (Appendix page 11) |
| Method of AE monitoring | Low risk | Quoted: “...safety assessments at 2 weeks and 1 month, 2 months, 3 months, 6 months, 9 months, and 12 months during the first year after randomisation and then every 3 months until month 36” (page 1077). "The occurrence of adverse events should be sought by non‐directive questioning of the patient at each visit during the study. Adverse events also may be detected when they are volunteered by the patient during or between visits or through physical examination, laboratory test, or other assessments" (Appendix page 11). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Johnson 1995.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐45 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.6; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 125) Placebo (unspecified) for 24 months (n = 126) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Teva Pharmaceutical | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “A centralized randomization scheme was used” (page 1270). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “At each monthly visit, patients received medication and reported adverse events and use of concomitant medications. A treating neurologist evaluated symptoms and adverse events.A nurse coordinator at each center distributed medication, noted concomitant treatments, and obtained blood and urine specimens for laboratory analysis. The nurse coordinator and neurologist were blinded to study medication assignment throughout the trial" (page 1270). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 24 months on study treatment (Table 3, page 1271): Glatiramer acetate: 106 (84.8%) of 125 participants Placebo: 109 (86.5%) of 126 participants Reasons for treatment discontinuation were partly reported. |
| Selective reporting (reporting bias) | Unclear risk | Protocol was not available. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: “At each monthly visit, patients received medication and reported adverse events and use of concomitant medications...A second treating neurologist evaluated symptoms and adverse events...Urinalysis, hematologic studies, a serum chemistry panel, and anti‐copolymer 1 antibodies were evaluated at 3‐month intervals” (page 1270). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kappos 2006.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS or SPMS; mean disease duration 9 years; mean EDSS 2.6; prior use of DMT not reported | |
| Interventions | Fingolimod 5.0 mg orally once daily for 6 months (n = 94) Fingolimod 1.25 mg orally once daily for 6 months (n = 94) Placebo orally once daily for 6 months (n = 93) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 6 months | |
| Notes | Funding: Novartis Pharma, Basel, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization was stratified according to disease course (relapsing–remitting or secondary progressive) with the use of a centralized automated system. The medication was prepackaged on the basis of a block size of 3 (1.25 mg, 5.0 mg, and placebo)” (page 1125). |
| Allocation concealment (selection bias) | Low risk | Quoted: “...use of a centralized automated system that provided randomized packages of the study drug to each centre” (page 1125) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Adverse events were assessed and reported at each visit (scheduled and unscheduled) by the treating physicians. Laboratory evaluations were undertaken at a central laboratory. Laboratory values that might have revealed the treatment assignment (e.g., lymphocyte counts) were not disclosed to treating physicians unless they exceeded prespecified safety limits. In cases of clinical adverse events or notable laboratory abnormalities, the dose of study medication was reduced or withheld at the discretion of the treating neurologist" (page 1126). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1, page 1128): Fingolimod 5 mg: 81 (88.0%) of 92 participants (8.7% adverse events) Fingolimod 1.25 mg: 88 (94.6%) of 93 participants (5.4% adverse events) Placebo: 86 (93.5%) of 92 participants (4.3% adverse events) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (NCT00333138). |
| Serious AE definitions | Low risk | Quoted: "The study adhered to the International Conference on Harmonization Guidelines for Good Clinical Practice" (page 1125). |
| Method of AE monitoring | Low risk | Quoted: “Study visits took place at screening, at baseline, on days 1 and 7, and then monthly for 6 months. Adverse events were assessed and reported at each visit (scheduled and unscheduled) by the treating physicians” "Vital signs were obtained at each visit, and laboratory and hematologic measures were obtained at baseline, day 1, and months 1, 3, 6, 9, and 12. Electrocardiograms were obtained at baseline, on days 1 and 7, and at months 1, 3, 6, and 12, and 24 hours. Holter electrocardiographic monitoring was performed at selected sites at baseline, day 1, and month 3. Pulmonary‐function tests were performed at screening and months 6 and 12. These tests were introduced by means of a protocol amendment and thus were performed in a subgroup of patients" (page 1126). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kappos 2008.
| Study characteristics | ||
| Methods | Parallel RCT conducted at 43 centres in the Czech Republic, Germany, Hungary, Netherlands, Poland, Russia, Sweden, Switzerland, Turkey, and UK, between 24 Nov 2004 and 22 May 2006. | |
| Participants | Relapsing MS by McDonald criteria 2001, age 18–55 years, an EDSS score between 0 and 5, and either at least one relapse within 12 months of randomization and a previous brain MRI scan showing lesions consistent with MS, or gadolinium‐enhancing lesions on MRI scans done within 6 weeks of randomization | |
| Interventions | Dimetyl fumarate 240 mg orally three times daily for 5.5 months (n = 64) Dimetyl fumarate 120 mg orally three times daily for 5.5 months (n = 64) Dimetyl fumarate 120 mg orally once daily for 5.5 months (n = 64) Placebo for 5.5 months (n = 65) |
|
| Outcomes | Adverse events at 24 weeks. Hematology, blood chemistry, and urinalysis were done every 4 weeks. Electrocardiographs were done at screening and at weeks 12 and 24. | |
| Notes | Funding: Biogen Idec, Inc. Some authors of the included article were employed by Biogen IDec Inc and contributed to the study design and statistical analysis of the data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "BG00012 and placebo were administered as enteric coated microtablets in gelatin capsules, which had identical appearance and taste. Daily medication was given in blister packs of six tablets, with different numbers of tablets containing placebo or the active drug to preserve the blinding" (page 1464). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "To prevent unblinding of treatment assignment, separate study personnel were assigned to treat patients and to assess drug efficacy. A treating neurologist was responsible for routine neurological care, assessing and treating adverse events, and analysing laboratory test results" (page 1464). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 5.5 months on study treatment (Figure 2, page 1465): Dimetyl fumarate 240 mg three times daily: 47 (74.6%) of 63 treated participants (15.9% discontinued treatment, 6.3% adverse events) Dimetyl fumarate 120 mg three times daily: 51 (79.7%) of 64 participants (12.5% discontinued treatment, 3.1% adverse events) Dimetyl fumarate 120 mg orally once daily: 54 (84.4%) of 64 participants (9.4% discontinued treatment, 1.6% adverse events) Placebo: 53 (81.5%) of 65 participants (9.2% discontinued treatment, 0 adverse events) |
| Selective reporting (reporting bias) | Unclear risk | No information on selection criteria was available (NCT00168701). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | Quoted: "Participants attended clinics every 4 weeks during 24‐weeks follow‐up...All adverse events were documented throughout the study, regardless of severity or relation to study drug" (page 1465). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kappos 2011.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 3.3; prior use of DMT at any time prior to the start of study: 41.3% (52.7% in ocrelizumab 600 mg, 50.9% in ocrelizumab 2000 mg, 31.5% interferon beta‐1a 30 µg and 29.6% placebo) | |
| Interventions | Ocrelizumab 600 mg intravenously with a dual infusion of 300 mg on days 1 and 15 (n = 56) Ocrelizumab 2000 mg intravenously with a dual infusion of 1000 mg on days 1 and 15 (n = 55) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for the first 24 weeks (n = 55) Placebo intravenously on days 1 and 15 (n = 54) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs at 6 months | |
| Notes | Funding: F Hoffmann‐La Roche Ltd, Biogen Idec Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “A randomisation list was generated by an independent group within Roche” and “Randomised patients (1:1:1:1) to one of the four treatment groups stratified by geographical region” (page 1782). |
| Allocation concealment (selection bias) | Low risk | Quoted: “This list was provided to an interactive voice response system” (page 1782). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “All individuals directly involved in this study remain blinded to the dose of ocrelizumab. We masked treatment assignment for patients in both the placebo and ocrelizumab groups throughout the study. In the interferon beta‐1a group, only the raters were masked to allocation" (page 1782). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “The treating investigator had access to safety and efficacy data, and made all treatment decisions on the basis of patients’ clinical responses and laboratory findings" (page 1780). "In the interferon beta‐1a group, only the efficacy raters were masked to allocation” (page 1782). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 5.5 months on study treatment (Figure 2, page 1782): Ocrelizumab 2000 mg: 48 (87.3%) of 55 participants (3.6% adverse events, 3.6% consent withdrawn, 1.8% died, 1.8% violated selection criteria, 1.8% failure to return) Ocrelizumab 600 mg: 51 (92.7%) of 55 participants (3.6% adverse events, 3.6% consent withdrawn) Interferon beta‐1a: 51 (94.4%) of 54 participants (1.8% adverse events, 3.7% consent withdrawn) Placebo: 54 (100.0%) of 54 participants |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00676715). |
| Serious AE definitions | Low risk | Quoted: "We did the study in accordance with International Conference on Harmonization Good Clinical Practice guidelines" (page 1780). |
| Method of AE monitoring | Low risk | Quoted: “Safety was assessed at weeks 2, 4, 8, 12, 16, 20, 24, and 48 with regular neurological and physical examinations, vital signs, electrocardiograph, and the occurrence of adverse events” (page 1781). "The treating investigator made all treatment decisions on the basis of patients’ clinical responses and laboratory findings" (page 1780). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Knobler 1993.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 3.1; prior use of DMT not reported | |
| Interventions | IFNβ‐1b (Betaseron) 25 µg subcutaneously three times weekly for 36 months (n = 6) IFNβ‐1b (Betaseron) 125 µg subcutaneously three times weekly for 36 months (n = 6) IFNβ‐1b (Betaseron) 250 µg subcutaneously three times weekly for 36 months (n = 6) IFNβ‐1b (Betaseron) 500 µg subcutaneously three times weekly for 36 months (n = 6) Placebo for 36 months (n = 7) |
|
| Outcomes | Withdrawals due to AEs and AEs over 36 months | |
| Notes | Funding: Triton Biosciences, Inc., Alameda, CA and Berlex Laboratories Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Were randomized into five equal groups of 6 patients each, after signing an informed consent” (page 335). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Patients and investigators had no prior knowledge of the relationship between the injection volume delivered and the dosage group to which patients were assigned...The supplies of Betaseron and placebo were identical in appearance.” (page 334). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “To secure double‐blinding, one neurologist at each centre performed the neurological examination for each patient and verified clinical exacerbations. A second neurologist independently evaluated the battery of clinical laboratory tests of hematological, renal, and hepatic functions performed at regular 3‐month intervals to identify adverse reactions” (page 335). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 36 months on study treatment (Table 6; page 338): Betaseron 25 µg: 4 (66.7%) of 6 participants Betaseron 125 µg: 3 (50.0%) of 6 participants Betaseron 250 µg: 4 (66.7%) of 6 participants Betaseron 500 µg: 4 (66.7%) of 6 participants Placebo: 4 (57.1%) of 7 participants |
| Selective reporting (reporting bias) | High risk | Protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Low risk | Quoted: “A second neurologist independently evaluated the battery of clinical laboratory tests of hematological, renal, and hepatic functions performed at regular 3‐month intervals to identify adverse reactions... At each patient visit, a nurse coordinator collected patient diaries of daily events and documented adverse events noted in these records” (page 335). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Koch‐Henriksen 2006.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 8 years; mean EDSS 2.9; prior use of DMT not reported | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 158) IFNβ‐1a (Rebif) 22 µg subcutaneously once a week for 24 months (n = 143) |
|
| Outcomes | The published report did not report either AEs or SAEs. | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The randomization algorithm was adjusted to reduce deviations from a 50/50 result in each centre” (page 1057). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “A central computerised randomization schedule assigned patients to treatment” (page 1057). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Blinding was abandoned because it could not be maintained owing to the different administration schemes of the two study drugs” (page 1057). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “Open‐label trial” (page 1057). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 25.6% was lost‐to follow‐up (27.8% in IFNß‐1b, and 23.1% in IFNβ‐1a), with some indications of differences in reasons: Quoted: “The main cause of withdrawal in the IFN‐1b 250 g arm was side effects (24/158, 15.2%), and treatment failure was the most frequent cause in the IFN‐1a arm (15/143, 10.5%)” (page 1057). |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either AEs or SAEs. |
| Method of AE monitoring | Low risk | Quoted: “Patients were interviewed about side effects and had routine blood tests including hematology and liver function tests every 3 months and thyroid tests and neutralizing antibodies every 6 months” (page 1057). |
| Other bias | Unclear risk | Rebif administered at a very low dose |
Leary 2003.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite PPMS; mean disease duration 8 years; median EDSS 5.3; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Avonex) 30 µg intramuscularly weekly for 24 months (n = 15) IFNβ‐1a (Avonex) 60 µg intramuscularly weekly for 24 months (n = 15) Placebo for 24 months (n = 20) | |
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: BIOGEN | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The randomization was carried out off‐site by Biogen using a randomization block method” (page 44). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “The study drug was blinded off‐site by Biogen and delivered to the study centre with the study numbers already allocated. Subjects were allocated by study number consecutively as they were entered into the study. A copy of the randomization codes was kept in pharmacy and by Biogen, but no codes were broken until the study and analysis was completed”. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Subjects and study personnel were blinded to treatment status” (page 44). "Nonsteroidal anti‐inflammatory drugs or paracetamol were recommended for prophylaxis of interferon‐associated flulike reactions" (page 45). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Adverse events, physical examination findings, and hematologic and biochemical parameters were monitored throughout the study” (page 45). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Table 1, page 45): IFNβ‐1a 30 µg: 14 (93.3%) of 15 participants (6.7% adverse event) IFNβ‐1a 60 µg: 11 (73.3%) of 15 participants (26.7% adverse event) Placebo: 18 (90%) of 20 participants (10% lack of benefit) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Serious AE definitions | Unclear risk | Quoted: "There were no significant differences in serious adverse events requiring hospital admission between the treatment groups (data not shown)" (page 49). |
| Method of AE monitoring | Low risk | Quoted: “Adverse events, physical examination findings, and hematologic and biochemical parameters were monitored throughout the study. An interim safety review by an independent assessor was performed at the midpoint of the trial” (page 45). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Lewanska 2002.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 9 years; mean EDSS 3.0; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.2 g/kg body weight intravenously monthly for 12 months (n = 17) Immunoglobulins 0.4 g/kg body weight intravenously monthly for 12 months (n = 16) Placebo intravenously monthly for 12 months (n = 18) | |
| Outcomes | Withdrawals due to AEs over 12 months | |
| Notes | Funding: Supported by the KBN (State Research Committee) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The generation of allocation sequence was based on random‐number table” (page 566). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo consisted of saline to avoid nonspecific protein effect. Infusions of IVIG and placebo were stored in identical opaque plastic bags for concealment during administration" (page 566). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Monitoring and recording of relapses, concomitant treatment, side effects or other medical events were documented throughout the study” (page 566). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 3.9% was lost‐to follow‐up (6.3% in immunoglobulins 0.4 g/kg, 0% in immunoglobulins 0.2 g/kg, and 5.6% in placebo), without indications of differences in reasons. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either AEs or SAEs. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: “Laboratory safety examinations were made at the beginning and at the end of the study period and included hemoglobin, complete blood cell count, hepatitis B and C serologies, serum creatinine, blood urea, nitrogen, electrolytes, blood glucose, liver enzymes, and urine analysis” (page 566). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Likosky 1991.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite SPMS, PPMS or PRMS; mean disease duration 10 years; mean EDSS 5.8; prior use of DMT not reported | |
| Interventions | Cyclophosphamide 500 mg intravenously five days per week until the leucocyte count fell below 4000/mm3 (n = 22) Folic acid (1 mg) was administered intravenously five times weekly for two weeks (n = 21). |
|
| Outcomes | Adverse events and SAEs were not reported. | |
| Notes | Funding: The Community Service Program of Kaiser Foundation Hospitals. Bristol Myers Company provided the medications. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Single‐blinded” (page 1055) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “Evaluating physicians were unaware of the treatment status of the patients they evaluated” (page 1056). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 14.0% was lost‐to follow‐up (13.6% in cyclophosphamide and 14.3% in placebo). |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either withdrawals due to AEs, AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: "Follow‐up examinations were conducted 12, 18, and 24 months.Complete blood cell count, urine analysis, and serum sodium results were monitored throughout the treatment period" (page 1056). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
MAIN TRIAL 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 1.9; prior use of DMT at any time prior to the start of study: 6.0% (6.5% in azathioprine, and 5.5% in interferon beta) | |
| Interventions | Azathioprine 3 mg/kg body weight orally daily for 24 months (n = 77) IFNß (Betaseron, Avonex, or Rebif) for 24 months (n = 73: 5 Betaseron 250 μg subcutaneously every other day, 26 Avonex 30 µg intramuscularly once a week, 35 Rebif 22 µg subcutaneously three times a week, 7 Rebif 44 µg subcutaneously three times a week) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: AIFA (Italian Medicines Agency) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were selected for AZA or IFNs using a randomization list (1:1 ratio), in blocks of four and stratified by disability score (EDSS ≤ 3.5 or > 3.5)” (page 2). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Patients were selected for AZA or IFNs using a computer generated central randomization list” (page 2). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Single‐blinded” (page 2) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “Patients were assessed by an unblinded treating and a blinded examining neurologist at their centres” (page 2). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 15.3% was lost‐to follow‐up (19.5% in azathioprine, and 11.0% in IFNß), without indications of differences in reasons. |
| Selective reporting (reporting bias) | Low risk | Quoted: "The published report included all prespecified primary safety outcomes" (Protocol). |
| Serious AE definitions | Low risk | Quoted: "Definition of SAEs according to the National Cancer Institute Common Terminology Criteria for AE" (Protocol) |
| Method of AE monitoring | Low risk | Quoted: “At scheduled (quarterly) and unscheduled (i.e., at the onset of new symptoms or complications) follow‐up visits the treating neurologist recorded symptoms, blood test results, clinical AEs and their management... Data was collected on: 1) AEs and serious AEs (SAEs); 2) patients with any AE; 3) patient withdrawal after any AE; 4) severity of any AE and their correlation with treatments as judged by the treating neurologist. Frequency and severity of AEs were actively assessed every three months or upon patient request" (page e113371). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Masjedi 2021.
| Study characteristics | ||
| Methods | RCT. The study was conducted at the clinics affiliated with the Isfahan University of Medical Sciences since March 2016. | |
| Participants | Newly diagnosed patients with MS using McDonald 2010 criteria. 18‐55‐years‐old who had not received any immunomodulatory therapy except for corticosteroids | |
| Interventions | Dimethyl fumarate oral 240 mg twice daily (n = 33) Fingolimod oral 0.5 mg daily (n = 34) |
|
| Outcomes | Adverse events at 24 months | |
| Notes | Funding; not reported. The study protocol was approved by the Ethics Committee of the Isfahan University of Medical Sciences (IR.MUI.REC.1396.3.786) and by Vice Chancellor for Research of the Isfahan University of Medical Sciences (code number: 396786). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Random allocation software" (page 87) |
| Allocation concealment (selection bias) | Unclear risk | Quoted: "Study population was selected through convenience sampling method until achieving the required number of study population. Then, they were randomly divided into groups of treatment" (page 87). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Assessment of adverse events was unblinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (page 88): Dimethyl fumarate: 30 (90.9%) of 33 participants (9.1% unwillingness for continuing the follow‐up visits) Fingolimod: 30 (88.2%) of 34 participants (5.9% non‐adherence, 5.9% severe relapses) |
| Selective reporting (reporting bias) | High risk | Protocol was not available. SAEs were not reported. |
| Method of AE monitoring | Unclear risk | AEs were not prespecified. Quoted:"The patients were visited every 3 months and were evaluated for drug related adverse effects" (page 88). |
| Other bias | Unclear risk | Quoted: "The two comparison groups were statistically different in terms of educational level, habitat, and smoking". |
Milanese 1993.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: mean 30 years; clinically definite RRMS, SPMS or PPMS, mean disease duration 7.5 years, mean EDSS 3.3; prior use of DMT not reported | |
| Interventions | Azathioprine 2.5 mg/kg body weight orally daily for 36 months (n = 19) Placebo (lactose) in identical form (50 mg tablets) for 36 months (n = 21) |
|
| Outcomes | Withdrawals due to AEs over 36 months | |
| Notes | Funding: Wellcome Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Patients were allocated to the azathioprine or placebo groups according to a list of random code numbers” (page 295). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “Placebo (lactose) was supplied in identical form (50 mg tablets)” (page 295). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "Adverse events were checked by a non‐blinded physician, who was also responsible for any change in treatment” (page 295). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (Table 1, page 296): Azathioprine: 7 (36.8%) of 19 treated participants (36.8% required the double‐blind regimen to be interrupted, mostly within a year of starting treatment, 21.1% adverse events) Placebo: 12 (57.1%) of 21 treated participants (28.6% required the double‐blind regimen to be interrupted, mostly within a year of starting treatment, 0 adverse events, 9.5% other reasons) |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. In the published report, SAEs and AEs were not clearly reported. |
| Method of AE monitoring | Unclear risk | Quoted: “Laboratory investigations (complete and differential blood count, erythrocyte sedimentation rate, creatinine, transaminases, γ glutamyl transferase, electrophoresis and urinalysis) were performed weekly during the first 2 months and every 3 months thereafter” (page 295). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Millefiorini 1997.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐45 years; clinically definite RRMS; mean disease duration 5 years; mean EDSS 3.6; prior use of DMT not reported | |
| Interventions | Mitoxantrone 8 mg/m² of body surface intravenously monthly for 12 months (total dose of 96 mg/m² of body surface over 12 months) (n = 27) Placebo intravenously monthly for 12 months (n = 24) | |
| Outcomes | Withdrawals due to AEs over 12 months | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomized to MTX or placebo using a scheme stratified on age, sex and EDSS which resulted in eight different age/sex/EDSS strata. According to the study protocol, within each stratum the allocation of patients to treatment or placebo was balanced by using a block design of size eight” (page 154). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Central allocation" (page 154) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “The intravenous bag and tubing were black to ensure no differences between the treatment groups. Placebo group patients received a solution containing the vehicle alone” (page 154). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "The treating physician, not blinded to study treatment, was responsible for the subject’s overall medical care, including physical examinations, evaluation of the patient’s subjective findings, prescribing and monitoring the study medication, and evaluating and managing adverse events" (page 154). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. Incomplete reporting of AEs or SAEs |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: “The safety of the treatment was assessed on the basis of adverse events volunteered by the patient either spontaneously or on questioning and monitoring of the main laboratory parameters. Blood and urine samples were taken and ECG carried out upon entry to the trial and at each monthly visit. To assess the potential cardiac toxicity of MTX, each patient had an echocardiographic study performed at enrolment and 6 and 12 months later.” (page 155) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Miller 1961.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: mean 33 years; clinically definite SPMS or PRMS; mean disease duration 12 years; EDSS not reported; all participants were previously untreated patients. | |
| Interventions | Prednisolone tablets 15 mg orally daily for 8 months then 10 mg daily for 18 months (n = 29) Calcium aspirin 9 tablets (54 g) orally daily for 18 months (n = 27) Placebo: corresponding number of “dummy” tablets for 18 months (n = 30) |
|
| Outcomes | ‐‐ | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “On admission to the trial, each patient was randomly allocated to one of the three treatment groups, and the initial routine was continued unchanged throughout” (page 128). |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not clearly described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 8.1% was lost‐to follow‐up (10.3% in prednisolone, 11.1% in aspirin, and 3.3% in placebo). |
| Selective reporting (reporting bias) | High risk | The published report did not report either withdrawals due to AEs, AEs or SAEs. |
| Method of AE monitoring | Unclear risk | Not reported |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Miller 2003.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS or SPMS; mean disease duration 12 years; mean EDSS 4.3; prior use of DMT not reported | |
| Interventions | Natalizumab 3 mg/kg of body weight intravenously every 28 days for 6 months (n = 68) Natalizumab 6 mg/kg of body weight intravenously every 28 days for 6 months (n = 74) Placebo (unspecified) every 28 days for 6 months (n = 71) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Elan Pharmaceuticals and Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Patients were randomly assigned to one of three treatments with use of a computer‐generated block randomization schedule” (page 16). |
| Allocation concealment (selection bias) | Low risk | “Randomization was performed centrally by an independent organization (PPD) Development” (page 16). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Neither the study personnel nor the patients were aware of the blinded treatment assignments” (page 16). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “The treating neurologist, unaware of the patients’ treatment assignments, obtained a medical history and, at each monthly visit, conducted physical and neurologic examinations and recorded adverse events” (page 17). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (page 18): Natalizumab 6 mg/kg: 66 (89.2%) of 74 treated participants (4.0% adverse events) Natalizumab 3 mg/kg: 63 (92.6%) of 68 treated participants (5.9% adverse events) Placebo: 66 (93.0%) of 71 treated participants (4.2% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: “All adverse events were recorded. Patients were examined at scheduled intervals... The treating neurologist obtained a medical history and, at each monthly visit, conducted physical and neurologic examinations and recorded adverse events” (page 17). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
MIRROR 2018.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 4 years; EDSS ≤ 5.5; prior use of DMT not reported | |
| Interventions | Ofatumumab 3 mg subcutaneously every 12 weeks for 3 months (n = 34) Ofatumumab 30 mg subcutaneously every 12 weeks for 3 months (n = 32) Ofatumumab 60 mg subcutaneously every 12 weeks for 3 months (n = 34) Ofatumumab 60 mg subcutaneously every 4 weeks for 3 months (n = 64) Placebo every 4 weeks for 3 months (n = 67) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 3 months | |
| Notes | Funding: GlaxoSmithKline | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization was computer‐generated by the project statistician” (e‐supplement, page 2). |
| Allocation concealment (selection bias) | Low risk | Quoted: “A registration and medication ordering system interactive voice response system (provided by the sponsor) served as a central system to sequentially allocate and maintain randomization numbers for each subject as they were randomized by Investigators” (e‐supplement, page 2). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: “An unblinded pharmacist at the investigational site prepared the ofatumumab injections and made the placebo to match the ofatumumab doses using normal saline" (e‐supplement, page 2). "Given higher rates of injection‐related symptoms in the ofatumumab treatment arms than in the placebo arm, there was potential that blinding could have been compromised" (e‐supplement, page 7). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: "Given higher rates of injection‐related symptoms in the ofatumumab treatment arms than in the placebo arm, there was potential that blinding could have been compromised" (e‐supplement, page 7). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 5.2% was lost‐to follow‐up (8.8% in ofatumumab 3 mg every 12 weeks, 6.3% in ofatumumab 30 mg every 12 weeks, 2.9% in ofatumumab 60 mg every 12 weeks, 6.3% in ofatumumab 60 mg every 4 weeks, and 3.0% in placebo), without indications of differences in reasons. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT01457924). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: “Safety was assessed on the basis of adverse event (AE) reporting, the Columbia Suicidality Severity Rating Scale, vital signs, physical and neurologic examinations, laboratory analyses, and immunogenicity (development of human anti‐human antibody [HAHA]) with the Meso Scale electro chemiluminescence” (page 1807). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Mokhber 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Mean age 29 years; clinically definite RRMS; disease duration not reported; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | IFNß‐1a (Avonex) 30 µg intramuscularly once per week for 12 months (n = 23) IFNß‐1a (Rebif) 44 µg subcutaneously three times per week for 12 months (n = 23) IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 12 months (n = 23) | |
| Outcomes | AEs, SAEs and withdrawals due to AEs not reported | |
| Notes | Funding: none | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “The study neurologist (MRA) enrolled the participants and allocated the subjects using a computer‐generated list of random numbers to the 3 treatment groups of three distinct commercially available forms of interferon beta” (page 17). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and neurologists were not blinded. Quoted: "Avonex was administered 30 mcg once per week via intramuscular injection; Rebif was administered 44 mcg three times per week via subcutaneous injection; and Betaferon was administered 0.25 mg every other day via subcutaneous injection. Injections were performed by either the patients themselves or their caregivers after being trained by the study neurologists" (pages 17‐18). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 17): Avonex 20 (87.0%) of 23 participants Rebif 23 (100%) of 23 participants Betaferon 22 (95.6%) of 23 participants Reasons for withdrawals not reported |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either AEs or SAEs. |
| Method of AE monitoring | Unclear risk | No information |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Montalban 2009.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite primary progressive MS or "transitional" MS (defined as those patients with a progressive course and a single relapse before or during progression); mean disease duration 11 years; mean EDSS 5.3; all participants were previously untreated patients | |
| Interventions | IFNß‐1b (Betaseron) 250 µg subcutaneously every other day for 24 months (n = 36) Placebo (unspecified) for 24 months (n = 37) |
|
| Outcomes | Withdrawals due to AEs and AEs over 24 months | |
| Notes | Funding: SCHERING ESPANA S.A. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “... Using a randomisation list. This randomization was performed in blocks of 6 and for each treatment was assigned in a 1:1 ratio” (page 1196). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “A dual physician scheme (treating/examining neurologist/neuropsychologist) was utilized" (page 1197). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 1200): Betaferon: 34 (94.4%) of 36 participants Placebo: 33 (89.2%) of 37 participants |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Serious AE definitions | Low risk | Quoted: "The following frequency distributions were investigated in both study arms: the presence of at least one adverse event, the presence of serious adverse events, the presence of adverse events causing death, the presence of adverse events requiring hospitalisation, the presence of adverse events causing discontinuation, adverse events by body system and preferred term, and adverse events’ relationship with the study drug" (page 1199). |
| Method of AE monitoring | Low risk | Quoted: "Safety issues during the study were monitored by an independent Safety Committee. Participants were asked to report any adverse event... Safety variables (Ashworth Scale and Beck Depression Inventory were recorded on screening, baseline (visits 1 and 2), during treatment initiation (only Ashworth scale), on visit 5 and every 3 months; the presence of adverse events and intercurrent illnesses was assessed at all visits... Laboratory tests (including liver and renal function and hematological parameters) were performed on visits 1, 2 and 5 to 14; thyroid function (TSH and free T4) was also tested every 6 months" (page 1197). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Motamed 2007.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: range 17–39 years; clinically isolated syndromes (CIS); time since neurological event not reported; mean EDSS 1.7; all participants were previously untreated patients | |
| Interventions | IFNß‐1a (Rebif) 22 µg subcutaneously three times a week for 21 months (n = 11) Control: participants did not receive disease‐modifying treatment for 21 months (n = 14). |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs not reported | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Patients were randomly assigned in approximately equal numbers to the two treatment groups” (page 345). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information |
| Selective reporting (reporting bias) | High risk | Study protcol was not available. The published report did not report withdrawals due to AEs, AEs and SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: “Neurological and safety assessments were performed at the end of months 1, 2, 3, 9, 15, and 21 by a neurologist” (page 345). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
MOVING 2020.
| Study characteristics | ||
| Methods | Parallel RCT. Study conducted between June 2013 and April 2015 at two German MS university hospitals | |
| Participants | A diagnosis of acute unilateral optic neuritis, with clinical onset within 30 days before screening; participants who had to fulfil diagnostic criteria of RRMS according to the 2010 McDonald criteria or of clinically isolated syndrome (CIS) with at least two typical lesions on brain or spinal MRI. Participants were 18 to 55 years of age, with an EDSS score <= 6.0, and had received either no disease‐modifying treatment in the previous 3 months or to have been on stable immunomodulation using interferon beta or glatiramer acetate for at least 6 months. | |
| Interventions | Fingolimod 0.5 mg once‐daily orally for 6 months (n = 6) Interferon beta 1b 250 μg subcutaneous injections every other day for 6 months (n = 7) |
|
| Outcomes | Adverse events (AE) and serious adverse events (SAE), vital signs including heart rate, systolic, diastolic blood pressure, body weight and laboratory tests were recorded and a complete physical examination was performed at 3 and 6 months. | |
| Notes | Following initiation of the study, additional treatment options for relapsing MS were licenced, including oral medications. In consequence, use of an injectable comparator made the study unattractive to many patients. Recruitment was significantly slowed and stopped prematurely at the request of the funding source, Novartis Pharma. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Pre‐specified randomization lists were generated for each study site by the central study pharmacy, stratified by residual vision (≤ 0.5 vs. > 0.5). Each allocation sequence used block permutation with a block size of 4" (page 3). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Participants and treating physicians were not blinded regarding the treatment arm" (page 3). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "A treating neurologist at each site was responsible for assessing eligibility, obtaining informed consent and supervising study procedures in an unblinded manner, including drug treatment, safety assessments, validation of co‐medications, and handling of adverse events" (page 3). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1, page 6): Fingolimod: 5 (83.3%) of 6 participants (16.7% recurrent optic neuritis) IFNβ‐1b: 4 (57.1%) of 7 participants (42.9% ipsilateral recurrent optic neuritis) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events were not reported in the study protocol and no information on selection criteria was available (NCT01647880). |
| Serious AE definitions | Unclear risk | No information. Quoted: "Hospitalization for the sole purpose of treating an MS relapse was not defined as a SAE" (Additional file 1 Supplementary methods and results; page 5). |
| Method of AE monitoring | Low risk | Quoted: "Vital signs including heart rate, systolic, diastolic blood pressure and body weight were recorded and a complete physical examination was performed at each visit. At baseline, a standard electrocardiogram was obtained and evaluated by a qualified physician. In the fingolimod arm, a 6 h first dose cardiac monitoring was performed at the baseline visit. Adverse events and serious adverse events (SAE) were recorded at each visit. Laboratory studies including blood cell count, sodium, potassium, calcium, phosphate, creatinine, urea, aminotransferases and urine analysis were performed at each visit. In addition, leukocyte differential counts, c‐reactive protein, thyroid stimulating hormone and immune fixation were included at baseline, and a hCG urine dipstick test was performed for women of childbearing age at all planned visits. For patients in the fingolimod arm, follow‐up laboratory studies including blood count, leukocyte differentiation, c‐reactive protein, creatinine and transaminases were scheduled two months after the last dose" (Additional file 1 Supplementary methods and results; page 5). |
| Other bias | High risk | Study prematurely stopped. High possibility of selection bias |
MSCRG 1996.
| Study characteristics | ||
| Methods | RCT. Study conducted at 4 centers in the United States. Recruitment period from November 1990 to early 1993 | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.4; all participants were previously untreated patients | |
| Interventions | IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 24 months (n = 158) Placebo intramuscularly once a week for 24 months (n = 143) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Quoted: "Supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke (NINDS) and Biogen, Inc, Cambridge, MA. Personnel of the study sponsor (Biogen) were involved in the conduct and data analysis" (page 293). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Randomisation performed at statistical centre of Buffalo General Hospital, one of the participating centres (biased coin assignment used for sequence generation)” (page 286) |
| Allocation concealment (selection bias) | Low risk | Quoted: “Schedule sent to each clinical centre, ... included patients were sequentially assigned the next ID number from the schedule“ (page 286). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo was human albumin. Quoted: “Personnel and participants were blinded to treatment status” (page 286). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Treating physicians reviewed toxicity test results, examined patients, and made all medical decisions” (page 286). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months' follow‐up (Table 2; page 288): Interferon beta 1a: 85 (53.8%) of 158 participants (8.9% treatment discontinuation) Placebo: 87 (60.8%) of 143 participants (6.3% treatment discontinuation) Withdrawals and losses to follow‐up: poorly described The study stopped early for benefit without a formal‐stopping rule. 73 (46.2%) of 158 patients in the treatment group and 56 (39.3%) of 143 controls had not completed the scheduled 2 years of follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Adverse events: criteria for monitoring and recording clearly described (according to Food and Drug Administration, HHS 21 CFR, Chapters 1,312.32, part c, 4/1/90) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
NASP 2004.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite SPMS; mean disease duration 15 years; mean EDSS 5.1; prior use of DMT not reported | |
| Interventions | IFNβ‐1b (Betaseron) 250 µg subcutaneously every other day for 36 months (n = 317) INFβ‐1b (Betaseron) 160 µg/m2 body surface area (mean administered dose 220 µg) every other day for 36 months (n = 314) Placebo for 36 months (n = 308) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Berlex Laboratories (Richmond, CA) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization allocation was by blocks of six... The randomization schedule was generated by the Biostatistics and Data Management Group of Berlex Laboratories (Richmond, CA) using an SAS program” (page 1789). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization allocation was by blocks of six, such that subjects received IFN beta 1b 250 μg, IFN beta 1b 160 μg/m2 body surface area, or placebo in a ratio of 1:1:1. At the start of the study, each site received an adequate number of blocks, based on assumed patient recruitment, to ensure sequential patient numbering within the site. The biostatistician and supporting programmers were the only individuals with access to the randomisation codes” (page 1789). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quoted: “Placebo lacked active drug but were otherwise identical in composition, appearance, and volume to the corresponding IFN beta 1b dosing arm” (page 1789). No other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “Treating physicians were responsible for the general medical care of each subject, safety assessments, and treatment of relapses” (page 1789). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 11.1% was lost‐to follow‐up (13.9% in INFβ‐1b 250 μg, 8.9% in INFβ‐1b 160 μg, and 10.4% in placebo), with some indications of differences in reasons. Quoted: “The study had ended prematurely based on the results of a planned interim analysis indicating that continuing the trial was unlikely to change the results. The study initiated on August 2, 1995, interrupted November 22, 1999. The last patient enrolled on April 1, 1997. The final patient visit occurred on November 15, 1999”. (FDA page 21). “Only 72% of randomised patients completed 33 months or more on study and could be included in analysis” (FDA page 26). |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Quoted: “At each scheduled visit, patients underwent physical and neurologic examinations, assessment for adverse events, concomitant medications, and basic laboratory testing for safety assessment... An Independent Data and Safety Monitoring Board (IDSMB) reviewed interim safety and efficacy data every 6 months” (page 1789). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Noseworthy 2000.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS and SPMS; mean disease duration 11 years; mean EDSS 5.3; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.4 g/kg body weight intravenously daily for 5 consecutive days and single infusion every 2 weeks for 3 months (total 11 infusions) (n = 33) Placebo for 5 days and single infusion every 2 weeks for 3 months (total 11 infusions) (n = 34) | |
| Outcomes | AEs over 6 months | |
| Notes | Funding: grant from the NIH, the National Multiple Sclerosis Society and the Italian Committee for the Myelin Project | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Patients were randomized in blocks of four” (page 1136). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “All personnel and patients were blinded to treatment status” (page 1136). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “The Internal Safety Monitoring Committee reviewed all clinical and laboratory events that suggested possible drug‐related toxicities and made immediate recommendations about management” (page 1137). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure, page 1138): Immunoglobulins: 30 (90.1%) of 33 treated participants Placebo: 29 (85.3%) of 34 treated participants |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report either withdrawals due to AEs or SAEs. |
| Method of AE monitoring | Low risk | “The trial was monitored for performance and safety by National Institutes of Health (NIH)–approved Internal and External Safety Monitoring Committees. The Internal Safety Monitoring Committee reviewed all clinical and laboratory events that suggested possible drug‐related toxicities and made immediate recommendations about management. All significant IVIg toxicities were referred immediately to the External Safety Monitoring Committee” (page 1137). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
O'Connor 2006.
| Study characteristics | ||
| Methods | RCT. The study was conducted between April 2001 and March 2003 at 10 MS clinics in Canada and 6 in France. | |
| Participants | Age: 18‐65 years; clinically definite RRMS or SPMS; mean disease duration 9 years; median EDSS 2.3; prior use of DMT not reported | |
| Interventions | Teriflunomide 14 mg orally once daily for 9 months (n = 57) Teriflunomide 7 mg orally once daily for 9 months (n = 61) Placebo (unspecified) orally once daily for 9 months (n = 61) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 9 months | |
| Notes | Funding: Sanofi‐Aventis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “A 1:1:1 randomization to placebo, teriflunomide 7 mg, and teriflunomide 14 mg was stratified by baseline EDSS score” (page 895). |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Safety was assessed by physical and neurologic examination, clinical laboratory analysis, and vital signs assessment. Spontaneously reported adverse events were recorded at clinic visits" (page 895). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 9 months on study treatment (Figure 1; page 895): Teriflunomide 14 mg: 45 (78.9%) of 57 participants (14.0% adverse events, 3.5% lack of efficacy, 3.5% did not wish to continue) Teriflunomide 7 mg: 58 (95.1%) of 61 participants (4.9% adverse events) Placebo: 57 (93.4%) of 61 participants (6.6% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: "Safety was assessed by physical and neurologic examination, clinical laboratory analysis, and vital signs assessment. Spontaneously reported adverse events were recorded at clinic visits" (page 895). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
OLYMPUS 2009.
| Study characteristics | ||
| Methods | RCT. Study conducted at 60 centers in the US and Canada. Recruitment period: start not reported, end October 2007 | |
| Participants | Age: 18‐65 years; clinically definite PPMS; mean disease duration 9 years; mean EDSS 4.8; prior use of DMT at any time prior to the start of study: 35.1% (35.3% in rituximab and 34.7% in placebo) | |
| Interventions | Rituximab 1000 mg intravenously every 6 months for 22 months (n = 292) Placebo every 6 months for 22 months (n = 147) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Genentech, Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomly assigned 2:1 to receive rituximab or placebo, and hierarchically stratified according to study site, previous MS therapies with interferon‐beta or glatiramer acetate, and baseline disease severity according to the Expanded Disability Status Scale (EDSS) score (≤ 4.0 vs > 4.0)" (page 461). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | There was no information to determine if placebo infusion was indistinguishable from rituximab infusion in terms of taste, appearance, and duration of infusion. Quoted: "Participants received 4 courses of rituximab or placebo infusions". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted “An independent Data Monitoring Committee met formally every 3 months to review unblinded safety data and adverse events” (page 462). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 2, page 463): Rituximab: 241 (82.5%) of 292 treated participants (2.7% adverse events, 9.2% patient's decision, 1.4% disease progression) Placebo: 124 (84.3%) of 147 treated participants (0% adverse events, 7.5% patient's decision, 3.4% disease progression) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00087529). |
| Serious AE definitions | Low risk | Quoted: "Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0" (page 462). SAEs were collected by non‐systematic assessment (NCT00087529). |
| Method of AE monitoring | Unclear risk | Quoted: “Data Monitoring Committee met formally every 3 months to review unblinded safety data, including MRI outcomes, clinical outcomes, and adverse events” (page 462). "Time frame: Up to 122 weeks (from start of first infusion until study completion or early termination). AEs collected by non‐systematic assessment" (NCT00087529) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
OPERA I 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 4 years; mean EDSS 2.8; prior use of DMT in the 2 years prior to the start of study: 27.4% (26.2% in ocrelizumab, and 28.6% in IFNβ‐1a) | |
| Interventions | Ocrelizumab 600 mg intravenously every 6 months for 24 months, with a dual infusion of 300 mg on days 1 and 15 for the first dose and as a single 600 mg infusion thereafter (n = 410) IFNβ‐1a (Rebif) 44 µg subcutaneously three times weekly for 24 months (n = 411) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Hoffmann‐La Roche | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization of patients was stratified by region (US/rest of the world) and baseline EDSS score (less than 4/greater than or equal to 4)” (Appendix, page 5). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally with the use of an independent interactive web‐response system” (page 223). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients in each group received a matching subcutaneous or intravenous placebo, as appropriate" (page 223). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “Each trial centre had separate treating and examining investigators, all of whom were unaware of the treatment assignments throughout the trial” (page 223). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Appendix; Figure S2a, page 13): Ocrelizumab: 366 (89.7%) of 408 treated participants (3.2% adverse events, 2.0% lack of efficacy) IFNβ‐1a: 340 (83.1%) of 409 treated participants (6.1% adverse events, 2.9% lack of efficacy) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (NCT01247324). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Quoted: "Adverse events will be graded according to Common Terminology Criteria for Adverse Events (CTCAE), version 4 and is provided to the investigator in a separate handout entitled “Common Terminology Criteria for Adverse Events v4.0". |
| Method of AE monitoring | Low risk | Quoted: "During the study, investigators are requested to promptly investigate patients reporting signs or symptoms of infection, to take appropriate specimens for identification of the pathogen and to treat infections aggressively" (Supplementary Appendix). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
OPERA II 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 4 years; mean EDSS 2.8; prior use of DMT in the 2 years prior to the start of study: 25.9% (27.1% in ocrelizumab, and 24.7% in IFNβ‐1a) | |
| Interventions | Ocrelizumab 600 mg intravenously every 6 months for 24 months, with a dual infusion of 300 mg on days 1 and 15 for the first dose and as a single 600 mg infusion thereafter (n = 417) IFNβ‐1a (Rebif) 44 µg subcutaneously three times weekly for 24 months (n = 418) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Hoffmann‐La Roche | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization of patients was stratified by region (US/rest of the world) and baseline EDSS score (less than 4/greater than or equal to 4)” (Appendix, page 5). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally with the use of an independent interactive Web‐response system” (page 223). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients in each group received a matching subcutaneous or intravenous placebo, as appropriate" (page 223). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “Each trial centre had separate treating and examining investigators, all of whom were unaware of the treatment assignments throughout the trial” (page 223). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Appendix; Figure S2b, page 13): Ocrelizumab: 360 (86.3%) of 417 treated participants (3.8% adverse events, 1.4% lack of efficacy, 2.9% consent withdrawn) IFNβ‐1a: 320 (76.7%) of 417 treated participants (6.0% adverse events, 3.6% lack of efficacy, 6.0% consent withdrawn) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (NCT01412333). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Quoted: "Adverse events will be graded according to Common Terminology Criteria for Adverse Events (CTCAE), version 4 and is provided to the investigator in a separate handout entitled “Common Terminology Criteria for Adverse Events v4.0". |
| Method of AE monitoring | Low risk | Quoted: "During the study, investigators are requested to promptly investigate patients reporting signs or symptoms of infection, to take appropriate specimens for identification of the pathogen and to treat infections aggressively" (Supplementary Appendix). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ORACLE 2014.
| Study characteristics | ||
| Methods | RCT. Study conducted at 160 hospitals and private clinics or treatment centers in 34 countries between October 21 2008, and October 11 2010, with the last patient visit on April 19 2012 | |
| Participants | Age: 18‐55 years; CIS; mean time since neurological event 79 days; median EDSS 1.5; prior use of DMT not reported | |
| Interventions | Cladribine orally at cumulative doses of 3.5 mg/kg for 24 months (n = 206) Cladribine orally at cumulative doses of 5.25 mg/kg for 24 months (n = 205) Placebo (unspecified) for 24 months (n = 206) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Serono SA Geneva | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation was done using a central web‐based randomisation system and was stratified by geographic region (Americas, Western Europe, Eastern Europe, Russia, Asia, and rest of world)” (page 258). |
| Allocation concealment (selection bias) | Low risk | Quoted: "A central web‐based randomisation system" (page 258) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Patients were unaware of their treatment assignment; tablet numbers were standardised across groups” (page 258). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Masking was maintained using a two‐physician model (both doctors were masked). The treating physician supervised study medication administration, and recorded and treated adverse events” (page 258) and "Dosing with study drug at each scheduled visit was initiated only after evaluation of corresponding safety laboratory assessments; the treating physician decided whether it was safe to proceed with dosing" (page 259). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Truncated. The study period was planned to be 22 months. It was stopped early following the sponsor’s decision to stop the cladribine programme (Supplementary web‐appendix). Patients who converted to clinically definite MS during the double‐blind period were excluded because they left the double‐blind period to enter the open‐label maintenance period (Table 2, page 261). Completed 24 months on study treatment (Figure 2, page 259): Cladribine 5.25 mg/kg: 134 (65.7%) of 204 participants (9.8% adverse events, 15.7% patients’ decision or trial or programme termination, 8.8% other) Cladribine 3.5 mg/kg: 158 (76.7%) of 206 participants (4.8% adverse events, 14.6% patients’ decision or trial or programme termination, 3.9% other) Placebo: 175 (84.9%) of 206 participants (2.4% adverse events, 8.7% patients’ decision or trial or programme termination, 2.9% other) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified safety outcomes (NCT00725985). |
| Serious AE definitions | Low risk | Quoted: "Any AE that resulted in death; was life threatening; resulted in persistent/significant disability/incapacity; resulted in/prolonged an existing in‐patient hospitalization; was a congenital anomaly/birth defect; or was a medically important condition..." (NCT00725985) |
| Method of AE monitoring | Low risk | Quoted: “Neurological and MRI assessments, adverse events, and laboratory findings were recorded at study visits and at regularly scheduled interim visits” (page 259). "Time frame: Baseline up to 96 weeks. Term from vocabulary, MedDRA (11.0). AEs were collected by non‐systematic assessment"(NCT00725985). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
ORATORIO 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite PPMS; mean disease duration 7 years; mean EDSS 4.7; prior use of DMT in the 2 years prior to the start of study: 11.6% (11.3% in ocrelizumab, and 12.3% in placebo) | |
| Interventions | Ocrelizumab 600 mg intravenously with a dual infusion of 300 mg every 24 weeks for 30 months (n = 488) Placebo (unspecified) intravenously with a dual infusion of 300 mg every 24 weeks for 30 months (n = 244) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 30 months | |
| Notes | Funding: Hoffmann‐La Roche | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization that was stratified according to geographic region and age was performed centrally” (page 211). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally by an independent interactive web‐response system” (page 211). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Each trial centre had separate treating and examining investigators” (page 211). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 30 months on study treatment (Supplementary Appendix; Figure S2, page 10): Ocrelizumab: 387 (80.3%) of 482 treated participants (4.4% lack of efficacy, 3.7% adverse events, 4.6% consent withdrawn) Placebo: 162 (66.7%) of 243 treated participants (11.1% lack of efficacy, 4.9% adverse events, 8.6% consent withdrawn) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified safety outcomes (Supplementary Appendix; pages 7‐8). |
| Serious AE definitions | Low risk | Quoted: "Definition of SAEs according to ICH Guideline for Clinical Safety Data Management, Definitions and Standards for Expedited Reporting, Topic E2"... "Adverse events will be graded according to Common Terminology Criteria for Adverse Events (CTCAE), version 4”..."SAE fulfils at least one of the following criteria: is fatal; is life‐threatening; required in‐patient hospitalization or prolongation of existing hospitalization; results in persistent or significant disability/incapacity; is a congenital anomaly/birth defect; is medically significant"..."Clinical relapses resulting in hospitalization will be reported as adverse events... "Particular attention should be directed toward early identification and treatment of infections" (clinicaltrials.gov, NCT01194570) (Supplementary Appendix; page 7). |
| Method of AE monitoring | Low risk | Quoted: "Adverse events, vital signs, weight, physical and neurological examination, clinical laboratory tests, 12 lead ECG, locally reviewed MRI for safety (non MS CNS pathology), concomitant medications". (Protocol. Eudract N. 2010‐020338‐25). “Time frame: From the first infusion up to the study clinical cut‐off date 24 July 2015 (up to 229 weeks)” (NCT01194570). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
OWIMS 1999.
| Study characteristics | ||
| Methods | RCT. Study conducted at 11 centers in Canada, Netherlands, Italy, Israel and France. Enrollment began in March 1995, was completed in November 1995, and the last study visit for the 1‐year time point occurred in November 1996. | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.6; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 5.5 or 12 months (n = 98) IFNβ‐1a (Rebif) 22 µg subcutaneously three times a week for 5.5 or 12 months (n = 95) Placebo subcutaneously three times a week for 5.5 or 12 months (n = 100) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Ares‐Serono International SA, Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation performed at Corporate Biometrics Department of Ares‐Serono (computer‐generated list)” (page 680) |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “The randomization code for each patient was delivered to the investigator in sealed envelopes” (page 680), however it was unclear whether envelopes were sequentially numbered and opaque. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo was human serum albumin and mannitol. Participants were blinded to treatment" (page 681). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Two physicians at each center assessed all patients. The treating physician supervised drug administration, recorded and treated adverse events, and monitored safety assessments" (page 681). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment (Figure 1, page 684): IFNβ‐1a 44 µg: 85 (86.7%) of 98 treated participants (5.1% adverse events, 5.1% patient's decision) IFNβ‐1a 22 µg: 87 (91.6%) of 95 treated participants (1.0% adverse events, 5.3% patient's decision) Placebo: 97 (97.0%) of 100 treated participants (0% adverse events, 3.0% patient's decision) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: “The treating physician recorded and treated AEs...” (page 680). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Pakdaman 2007.
| Study characteristics | ||
| Methods | RCT. Study conducted in four centers in Iran from February 2002 to August 2005 | |
| Participants | Age: 19‐50 years; CIS; time since neurological event within 3 months; EDSS not reported; prior use of DMT not reported | |
| Interventions | IFNß‐1a (Avonex) 30 µg intramuscular once a week for 36 months (n = 104) Placebo (unspecified) once a week for 36 months (n = 98) |
|
| Outcomes | SAEs and AEs over 36 months | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Each patient was examined by a treating and an evaluating neurologist" (page 430). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quoted: "Of the 217 participants randomised, 202 (93%) completed the study; 104 received interferon beta 1a and 98 received placebo" (page 430). |
| Selective reporting (reporting bias) | High risk | Protocol was not available. The published report did not report withdrawals due to AEs and AEs apart from influenza‐like syndrome. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: “Neurological and safety assessments, including vital signs, hematologic, and serum biochemical tests, were performed at the end of months 1, 6, 12, 18, 24, 30, and 36” (page 430). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Pohlau 2007.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite active SPMS or PPMS; mean disease duration 15 years; mean EDSS 5.6; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.4 g/kg body weight intravenously monthly for 24 months (n = 116) Placebo for 24 months (n = 115) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Novartis Pharma GmbH and ZLB Behring | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation was performed by the Biometric Department Novartis Germany using a scheme, which provided balanced blocks of patient numbers for both treatment groups and the two diagnostic layers” (page 1109). |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: "Placebo was albumin, 0.1% solution in saline.IVIG and placebo could not visually be distinguished” (page 1109). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1; page 1110): Immunoglobulins: 56 (48.3%) of 116 participants Placebo: 57 (49.6%) of 115 participants Quoted: "Patients who withdrew for lack of efficacy were more frequent in the placebo group and patients discontinuing for adverse events were more numerous in the immunoglobulin group. An analysis of the EDSS score at baseline showed that the decision to withdraw was likely to be associated with the severity of the disease, i.e. the number of dropout patients with an initial EDSS score of ≤ 5.0 was 16%, compared with 35% of patients with an EDSS score of > 5.0) (page 1110). |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: “Safety and tolerability of the treatment were assessed by recording adverse events, vital signs and by laboratory findings. All adverse events and clinical symptoms related to the disease or the study medication were recorded every 4 weeks” (page 1109). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Polman 2005.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS or SPMS; mean disease duration 6 years; mean EDSS 3.1; prior use of DMT not reported | |
| Interventions | Laquinimod 0.3 mg oral capsule once daily for 6 months (n = 74) Laquinimod 0.1 mg oral capsule once daily for 6 months (n = 68) Placebo oral capsule once daily for 6 months (n = 67) |
|
| Outcomes | Withdrawals due to AEs and SAEs over 6 months | |
| Notes | Funding: Active Biotech | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Individual centres were issued with blocks of randomization numbers” (page 988). |
| Allocation concealment (selection bias) | Unclear risk | “Individual centres were issued with corresponding tablet blisters with randomization numbers to balance the treatment allocation within each centre” (page 988). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: "Investigators and sponsor personnel remained blinded throughout the study". (page 988) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1; page 988): Laquinimod: 0.3 mg 69 (93.2%) of 74 participants (2.7% adverse events, 2.7% voluntary withdrew) Laquinimod: 0.1 mg 65 (95.6%) of 68 participants (0 adverse events, 2.9% voluntary withdrew) Placebo: 64 (95.5%) of 67 participants (1.5% adverse event, 1.5% voluntary withdrew) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. The published report did not clearly report AEs. |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Unclear risk | Quoted: “Safety evaluations consisted of vital signs, physical examinations, and a variety of laboratory measures...safety evaluations were performed every eighth week. Safety evaluations were also scheduled 2 and 4 weeks after treatment initiation” (page 988). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
PreCISe 2009.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐45 years; CIS, mean time since neurological event 74 days; mean EDSS 1.0; all participants were previously untreated patients | |
| Interventions | Glatiramer acetate 20 mg subcutaneously daily for 36 months (n = 243) Placebo for 36 months (n = 238) | |
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Teva Pharmaceuticals | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “The randomisation scheme used SAS‐based blocks with block size of 4, stratified by centre” (page 1504). |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Study drugs were packaged and labelled in a way that maintained the masked nature of the study; the appearance, shape, colour, and smell were identical. Patients and all personnel were masked to the treatment assignment” (page 1504). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “The treating neurologist assessed patients within 7 days of the patient notification to the site.” (page 1504) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Truncated. The study period was planned to be 3 years. Based on the results of a planned interim analysis of efficacy and on the recommendations of the data monitoring committee (unmasked), the trial was stopped early and all participants were switched to glatiramer acetate. At the time of the interim analysis, 230 (47.8%) of 481 randomised participants completed the study. 98 (40.3%) of 243 treated participants and 132 (55.5%) of 238 placebo participants completed the study. Proportion and reasons of incomplete data differed between the groups. 39 (16.0%) of 243 participants in the glatiramer group and 23 (8.8%) of 238 in the placebo group discontinued treatment early (table 1, page 1505) and the proportion of termination because of adverse events differed between the two treatment groups (5.8% in the glatiramer group and 1.7% in the placebo group). |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00666224). |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | "Tolerability and safety assessments included adverse events, standard clinical laboratory tests, vital signs, weight, physical examinations, and electrocardiograph measurements" (page 1506). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
PRISMS 1998.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.5; prior use of DMT: "Only 3% of patients had received previous immunosuppressive therapy" | |
| Interventions | IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 184) IFNβ‐1a (Rebif) 22 µg subcutaneously three times a week for 24 months (n = 189) Placebo subcutaneously three times a week for 24 months (n = 187) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Ares‐Serono International SA, Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation at Corporate Biometrics Department of Ares‐Serono (computer‐generated list, stratified by centre, equal allocation of the treatment groups by a block size of 6)” (page 1499). |
| Allocation concealment (selection bias) | Unclear risk | Quoted: “The study drug was packed accordingly to the randomisation list and delivered to the centres so that treatment allocation remained concealed” (page 1499). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “All personnel involved in the study were unaware of treatment allocation” (page 1499). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “Patients were assessed by two physicians. A “treating” neurologist was responsible for overall medical management of the patient, including treatment of any side‐effects” (page 1499). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 1499): IFNβ‐1a 44 µg: 165 (89.7%) of 184 treated participants (4.9% adverse events, 3.3% patient's decision) IFNβ‐1a 22 µg: 167 (88.4%) of 189 treated participants (3.2% adverse events, 5.3% patient's decision) Placebo: 170 (90.9%) of 187 treated participants (1.1% adverse events, 5.3% patient's decision) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Low risk | Quoted: " If WHO grade II or III toxic effects occurred, study medication was decreased to half dosage or temporarily discontinued. For WHO grade IV toxic effects and for protocol violations including non‐compliance and unacceptable adverse events, patients were withdrawn from treatment" (page 1499). |
| Method of AE monitoring | Low risk | Quoted: " Patients had haematology and biochemical tests, including liver‐function tests, every 2 weeks for the first 8 weeks, and then every 3 months. Thyroid‐function tests were done every 6 months. Serum samples were tested for antibodies to interferon beta every 6 months" (page 1499). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
PROMESS 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite SPMS; mean disease duration 13 years; median EDSS; prior use of DMT not reported | |
| Interventions | Cyclophosphamide 750 mg/m² body surface area intravenously every 4 weeks during the first 12 months and every 8 weeks during the second 12 months (n = 72) Methyprednisolone 1 g intravenously every 4 weeks during the first 12 months and every 8 weeks during the second 12 months (n = 66) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: French Ministry of Health (Programme Hospitalier de Recherche Clinique 2004) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were allocated to receive either CPM or MP using a web‐based secured system according to a randomization list generated and kept confidential by the statistician of the Clinical Trials Unit (CTU, CHU Bordeaux) using SAS 9.1” (page 3). |
| Allocation concealment (selection bias) | Low risk | Quoted: “A web‐based secured system” (page 3) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “All study personnel were blinded to group allocation, including the neurologists and nurses administrating the treatments... Study drugs were prepared in hospital pharmacies in similar infusion vials that precluded the identification of the group assignment by patients and study personnel... The procedures for the administration of the study drugs were similar in both groups.” (page 3) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “The treating neurologist evaluated the clinical state, safety and tolerability" (page 3). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 24 months on study treatment (Figure 1, page 6): Cyclophosphamide: 39 (54.2%) of 72 treated participants (27.8% adverse events) Methyprednisolone: 44 (66.7%) of 66 treated participants (7.6% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | The published report included prespecified primary safety outcomes (NCT00241254). |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: "Adverse events were collected according to a standardized method using the EudraVigilance database" (page 4). |
| Other bias | Low risk | Quoted: "One hundred and forty‐eight patients were included between 11/16/2005 and 07/16/2009, when recruitment closed. Due to a low recruitment rate, it was decided to stop the study at this stage" (page 5). |
RADIANCE 2019.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; relapsing–remitting, progressive–relapsing, or secondary progressive; mean disease duration 6 years; mean EDSS 2.5; prior use of DMT: 28.9% (28.4% in ozanimod 1 mg, 29.8% in ozanimod 0.5 mg, and 28.6% in IFNβ‐1a) | |
| Interventions | Ozanimod 1 mg orally daily for 24 months (n = 434) Ozanimod 0.5 mg orally daily for 24 months (n = 443) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 24 months (n = 443) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Celgene International II | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “The randomisation sequence was generated by the contract research organisation and based on a blocked algorithm stratified by baseline EDSS score (≤ 3·5 vs > 3·5) and country” (page 1023). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Participants were randomised (1:1:1) via an interactive voice response system” (page 1023). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Placebos consisting of daily oral capsules identical in appearance to ozanimod were given to participants in the interferon beta‐1a group and weekly intramuscular injections identical to interferon beta‐1a were given to participants in the ozanimod group”; and “Participants, investigators, study personnel, and the funder were masked to treatment and total and differential white blood cell counts” (page 1023). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “An assessor masked to treatment investigated participants using the EDSS at all visits. The assessor was not involved in treatment and participants were instructed to not discuss clinical symptoms or adverse effects with them. A treating neurologist, the investigator, handled all other assessments and supervised medical management" (page 1023). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1; page 1024): Ozanimod 1.0 mg: 388 (89.6%) of 433 participants (4.4% voluntarily withdrew, 3.0% adverse event, 1.1% physician decision) Ozanimod 0.5 mg: 374 (85.2%) of 439 participants (7.1% voluntarily withdrew, 3.0% adverse event, 1.4% physician decision) Interferon beta‐1a: 376 (85.3%) of 441 participants (6.8% voluntarily withdrew, 4.1% adverse event, 1.6% physician decision) |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified primary safety outcomes (NCT02047734). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Quoted: "Treatment‐emergent adverse events (TEAEs) were assessed by system organ class (MedDRA, version 18.1), severity, and causality"(page 1025) (NCT02047734). |
| Method of AE monitoring | Low risk | Quoted: “Safety analyses included the incidence and type of treatment‐emergent adverse events (TEAEs), serious TEAEs, and TEAEs leading to discontinuation of study treatment. TEAEs were assessed by system organ class (using MedDRA, version 18.1), severity, and causality. Participants were monitored for the following AEs of special interest: cardiac abnormalities (bradycardia, conduction abnormalities, and new ischaemic changes on ECG); serious or opportunistic infections; hepatotoxicity (confirmed alanine aminotransferase or aspartate aminotransferase at least three times the ULN, with or without raised bilirubin); ophthalmic abnormalities; pulmonary function test abnormalities; and cutaneous and other malignancies. Suicidality, which is common among those with multiple sclerosis, was assessed at each visit.” (page 1023) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
REFLEX 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐50 years; CIS; time since neurological event 58 days; mean EDSS 1.5; all participants were previously untreated patients | |
| Interventions | Serum‐free IFNß‐1a (Rebif) 44 µg subcutaneously once a week (plus placebo twice a week for masking) for 24 months (n = 175) Serum‐free IFNß‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 171) Placebo three times a week for 24 months (n = 171) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Merck Serono SA | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Dynamic treatment allocation, by use of a minimisation algorithm with an element of chance, was applied to minimise imbalance between the treatment groups” (page 34). |
| Allocation concealment (selection bias) | Low risk | Quoted: “The study centre dialled a centralised interactive voice response system (provided by S‐Clinica, Brussels, Belgium) to randomly assign patients in a 1:1:1 ratio” (page 34). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Patients assigned to the once a week regimen were given two additional subcutaneous injections of placebo per week for masking purposes, and patients assigned to placebo received three subcutaneous injections per week. Placebo was supplied as a transparent sterile solution for injection in prefilled syringes matching the interferon beta‐1a prefilled syringes, each containing 0.5 mL" (page 34). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quoted: “A two‐physician (treating and assessing) model was used to assist with study masking. The treating physician was responsible for supervision of study drug administration and for recording adverse events and safety assessments” (page 34). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 35): IFNß‐1a 44 µg three times a week: 146 (85.9%) of 170 treated participants (2.9% adverse events) IFNß‐1a 44 µg once a week: 156 (89.6%) of 174 treated participants (2.3% adverse events) Placebo: 146 (85.9%) of 170 treated participants (3.5% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00404352). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: “Visits were scheduled every 3 months from baseline to month 24 to ... report safety. An additional safety visit was made at month 1 for all patients” (page 34). "The safety and tolerability of subcutaneous interferon beta‐1a were assessed by documentation of adverse events, laboratory tests, vital signs, electrocardiograms, physical examination, and neutralising antibodies to interferon beta. The preferred terms pertaining to prespecified adverse events that were known to be related to interferon beta—flu‐like syndrome (influenza‐like illness), cytopenia, hepatic disorders, thyroid disorders, hypersensitivity reactions, skin rashes, depression and suicidal ideation, and injection‐site reactions—were grouped and analysed together" (pages 35‐6). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
REFORMS 2012.
| Study characteristics | ||
| Methods | Parallel RCT conducted at 27 clinical sites in the US between May 2006 and July 2009 | |
| Participants | Diagnosis of relapsing MS as defined by the Poser or 2005 revised McDonald criteria. Participants were 18–60 years of age, and had not previously received IFNβ treatment. | |
| Interventions | Serum‐free IFNβ‐1a 44 μg subcutaneously three times weekly for 3 months (n = 65) IFNβ‐1b 250 μg subcutaneously every other day for 3 months (n = 64) |
|
| Outcomes | Serious adverse events (SAEs); adverse events (AEs); withdrawn due to AEs at 3 months | |
| Notes | The primary objective was to compare subject‐reported injection‐site pain between the serum‐free IFNβ‐1a and IFNβ‐1b, during a 12‐week period. Funding: the study was sponsored by EMD Serono, Inc., Rockland MA, USA, an affiliate of Merck KGaA, Darmstadt, Germany, and Pfizer Inc, New York, NY, USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "Treatments were allocated using a computer generated randomization code" (page 2). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Participants and personnel were not blinded (open‐label)" (page 2). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted. "The study was open‐label, except for blinded assessments of injection‐site reactions"(page 2). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 3 months on study treatment (Figure 2, page 5): IFNβ‐1a: 56 (86.1%) of 65 treated participants (10.7% adverse events, 3.1% lost to follow‐up) IFNβ‐1b: 63 (98.4%) of 64 treated participants (0 adverse events, 1.6% lost to follow‐up) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00428584). |
| Serious AE definitions | Unclear risk | Quoted: "AEs were coded to system organ class and preferred term using the MedDRA dictionary version 9.1 and summarized by severity and relationship, vital signs, hematology, and serum chemistry" (page 3). |
| Method of AE monitoring | Unclear risk | Quoted: "Safety endpoints included adverse events, laboratory tests, physical examinations, vital signs, and concomitant medications". Injection‐site pain was spontaneously reported by participants" (NCT00428584). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
REGARD 2008.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS; mean disease duration 6 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Rebif) 44 µg subcutaneously three times a week for 24 months (n = 386) Glatiramer acetate 20 mg subcutaneously daily for 24 months (n = 378) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: EMD Serono and Pfizer | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Computer‐generated randomisation list stratified by centre” (page 904) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Neither the patients nor the treating physicians were blinded to treatment” (page 904). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “Neither the patients nor the treating physicians were blinded to treatment” (page 904). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 904): IFNβ‐1a: 301 (78.0%) of 386 treated participants (6.0% adverse events, 3.9% discontinued, 1.0% disease progression, 7.2% other reasons) Glatiramer acetate: 324 (85.7%) of 378 treated participants (5.0% adverse events, 0.5% discontinued, 1.8% disease progression, 4.2% other reasons) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00078338). |
| Serious AE definitions | Unclear risk | Insufficient information on SAEs definition |
| Method of AE monitoring | Low risk | Quoted: “Adverse events (including pregnancy), withdrawals owing to adverse events, serious adverse events, and laboratory results were obtained for safety comparisons...Follow‐up clinical assessments were done at 4, 12, and 24 weeks, and every 24 weeks thereafter up to 96 weeks” (page 905). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
REVEAL 2020.
| Study characteristics | ||
| Methods | Parallel RCT. The study was conducted at 43 sites in nine countries between October 2014 and May 2016. | |
| Participants | Clinically definite active RRMS; age: 18‐60 years; mean disease duration 7.5 years; mean EDSS 2.6; prior use of DMT (most commonly glatiramer acetate or interferons beta) at any time prior to the start of study: 50.0% (48.1% in natalizumab, and 51.9% in fingolimod) | |
| Interventions | Natalizumab 300 mg intravenously once every 4 weeks for up to 12 months (n = 56) Fingolimod 0.5 mg orally once daily for 12 months (n = 55) |
|
| Outcomes | Adverse events, laboratory measurements, vital signs and physical examinations over 12 months | |
| Notes | Funding: Biogen. Quoted: "The study was designed to include approximately 540 patients. However, after 1 year of enrolling patients, only 111 patients had been enrolled. The decision to terminate the study due to slow enrolment was made by the sponsor (Biogen) in November 2015". | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: "Neither the patients nor the treating physicians were blinded to treatment" (page 904). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: "Neither the patients nor the treating physicians were blinded to treatment" (page 904). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Supplementary Figure 1): Natalizumab: 1 (1.8%) of 56 participants (90.7% sponsor study termination, 3.7% lost to follow‐up, 1.9% adverse events, 1.9% consent withdrawn) Fingolimod: 3 (5.4%) of 55 participants (79.6% sponsor study termination, 1.9% lost to follow‐up, 5.6% adverse events, 5.6% physician decision, 1.9% other) Quoted: "After 1 year of enrolling patients, only 111 patients had been enrolled. The decision to terminate the study due to slow enrolment was made by the sponsor (Biogen) in November 2015" (page 2). |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT02342704). The protocol was first posted on 21 January 2015, after the study start day (30 November 2014). |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | Quoted: "Safety was assessed based on AEs, laboratory measurements, vital signs and physical examinations" (page 2). |
| Other bias | Unclear risk | Study prematurely stopped. High possibility of selection bias |
Saida 2012.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐60 years; clinically definite RRMS or SPMS; mean disease duration 8 years; mean EDSS 2.1; prior use of DMT not reported | |
| Interventions | Fingolimod 1.25 mg orally once daily for 6 months (n = 57) Fingolimod 0.5 mg orally once daily for 6 months (n = 57) Placebo orally once daily for 6 months (n = 57) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 6 months | |
| Notes | Funding: Novartis Pharma KK and Mitsubishi Tanabe Pharma Corp | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization was performed by a central centre (Bell system 24 Inc., Tokyo), with the use of a validated system that assigned randomization numbers to patients and automated the dynamic allocation of treatment arms to randomization numbers” (Appendix, page 5). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Central allocation system" (Appendix, page 5) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Patients, investigators, site personnel, first‐dose administrators, MRI evaluators and data analysts (i.e. all study personnel) remained blinded during the six‐month core study” (page 1270). "The identity of treatments was concealed by the use of study drugs that were identical in appearance, packaging, labelling and schedule of administration” (Appendix, page 5). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1, page 1271): Fingolimod 1.25 mg: 48 (88.9%) of 54 treated participants (11.1% adverse events) Fingolimod 0.5 mg: 48 (84.2%) of 57 treated participants (10.5% adverse events) Placebo: 51 (89.5%) of 57 treated participants (5.3% adverse events) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00537082) |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice" (page 1270). |
| Method of AE monitoring | Low risk | Quoted: “Adverse events, serious adverse events and MS relapse assessments were conducted at screening, baseline, days 1 and 15, and months 1, 2, 3, 4, 5 and 6” (page 1270), and “Safety assessments included recording of adverse events, serious adverse events, haematology values, vital signs, results of dermatological and ophthalmological examinations, and results of pulmonary and liver function tests” (Appendix, page 5). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Saida 2017.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐65 years; clinically definite RRMS; mean disease duration 8 years; median EDSS 2.3; prior use of DMT at any time prior to the start of study: 88.3% (91.5% in natalizumab and 85.1% in placebo) | |
| Interventions | Natalizumab 300 mg intravenously once every 4 weeks for 6 months (n = 47) Placebo for 6 months (n = 47) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 6 months | |
| Notes | Funding: Biogen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization of patients to treatment was stratified using a computer‐generated randomization schedule and a multi‐digit identification number” (page 26). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Allocation was implemented by an interactive voice and web response system” (page 26). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment (Figure 1, page 27): Natalizumab: 46 (97.9%) of 47 treated participants Placebo: 43 (91.5%) of 47 treated participants |
| Selective reporting (reporting bias) | Low risk | The published report included all prespecified safety outcomes (NCT01440101). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice" (page 26). |
| Method of AE monitoring | Low risk | Quoted: “AEs were monitored throughout the study, and assessment of vital signs occurred every 4 weeks. Physical and neurological exams, laboratory testing, patients’ self‐rating of global well‐being on a VAS were collected every 12 weeks. Serum samples were evaluated for antibodies to natalizumab and for anti–JC virus (JCV) antibody testing for progressive multifocal leukoencephalopathy risk assessment" (page 26). "Time frame: AEs were collected from baseline (week 0) through week 24 (treatment period) + 20 weeks (or 24 weeks after last infusion)" (NCT01440101). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
SELECT 2013.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; median disease duration (since diagnosis) 3 years; mean EDSS 2.7; prior use of DMT at any time prior to the start of study: 23.7% (22.5% in daclizumab 300 mg, 25.5% in daclizumab 150 mg, and 24.0% in placebo) | |
| Interventions | Daclizumab 300 mg subcutaneously once every 4 weeks for 12 months (n = 209) Daclizumab 150 mg subcutaneously once every 4 weeks for 12 months (n = 208) Placebo subcutaneously once every 4 weeks for 12 months (n = 204) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Biogen Idec and AbbVie Biotherapeutics Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quoted: “Patients were randomly assigned in a 1:1:1 ratio” (page 2168). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Patients were randomly assigned via a centralised interactive voice response system” (page 2168). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “All personnel and patients were masked to treatment assignment” (page 2168). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 2; page 2169): Daclizumab 300 mg: 197 (97.0%) of 203 participants (4.4% adverse events, 2.5% consent withdrawn) Daclizumab 150 mg: 192 (95.5%) of 201 participants (3.0% adverse events, 4.5% consent withdrawn) Placebo: 150 mg: 188 (95.9%) of 196 participants (1.0% adverse events, 5.6% consent withdrawn) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00390221). |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Unclear risk | Quoted: “Safety parameters were assessed at all visits” (page 2168). "Time frame: AEs and SAEs were collected from the screening visit (≤ 21 days prior to baseline) through the follow‐up visit (week 72 ± 5 days) or early discontinuation" (NCT00390221). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
SPECTRIMS 2001.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite SPMS; mean disease duration 13 years; mean EDSS 5.4; prior use of DMT not reported | |
| Interventions | IFNβ‐1a (Rebif) 22 µg subcutaneously three times/week for 36 months (n = 209) IFNβ‐1a (Rebif) 44 µg subcutaneously three times/week for 36 months (n = 204) Placebo (unspecified) for 36 months (n = 205) |
|
| Outcomes | Withdrawals due to AEs and AEs over 36 months | |
| Notes | Funding: Serono International, Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “"Computer generated randomisation list provided by Serono, stratified by center; treatments were equally allocated with a block size of six. The block size was not revealed to the investigators" (page 1497). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Solutions of IFNb‐1a and placebo were physically indistinguishable, and packaging and labeling were prepared to preserve blinding. Treatment assignments were provided to investigators in sealed envelopes for emergency use" (page 1497). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "Because IFN side effects are well recognized, a treating physician supervised drug administration, monitored safety, and managed adverse events, and a separate evaluating physician conducted neurologic assessments and followed‐up exacerbations" (page 1497). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completed 36 months on study treatment (Figure 1; page 1499): IFNβ‐1a 44μg: 161 (78.9%) of 204 treated participants IFNβ‐1a 22 μg: 172 (82.3%) of 209 treated participants Placebo: 173 (84.4%) of 205 treated participants Reasons for treatment discontinuation were not clearly reported. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. The published report did not report SAEs. |
| Method of AE monitoring | Unclear risk | Quoted: “Two MS specialists who were otherwise uninvolved in the study reviewed safety data and supervised analyses... Adverse events and changes in concomitant medications were followed throughout the study, and clinical laboratory evaluation was performed at the 3‐month evaluation visits or as needed” (page 1497). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
SUNBEAM 2019.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; relapsing–remitting, secondary progressive, or progressive–relapsing; mean disease duration 7 years; mean EDSS 2.6; prior use of DMT: 30.5% (28.6% in ozanimod 1 mg, 29.3% in ozanimod 0.5 mg, and 33.7% in IFNβ‐1a) | |
| Interventions | Ozanimod 1 mg orally daily for 12 months (n = 447) Ozanimod 0.5 mg orally daily for 12 months (n = 451) IFNβ‐1a (avonex) 30 µg intramuscularly once a week for 12 months (n = 448) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Celgene International II | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation was based on a blocked algorithm stratified by country and baseline EDSS score (≤ 3·5 vs > 3·5)” (page 1011). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomisation was...done through interactive voice and web‐based response technology” (page 1011). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “To maintain masking, participants assigned to interferon beta‐1a received daily oral placebo capsules identical in appearance to ozanimod; those assigned to ozanimod received weekly intramuscular placebo injections”; and “Treating investigators, study personnel, participants, and the sponsor were masked to treatment and total and differential white blood cell counts” (pages 1011‐1012). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “To prevent potential unmasking due to observed efficacy, adverse events, or laboratory changes, an independent assessor masked to treatment assessed participants using the EDSS at all visits. Participants were advised not to discuss clinical symptoms or adverse events with the EDSS assessor. The treating investigator did all other study assessments.” (pages 1012) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study treatment (Figure 1, page 1011): Ozanimod 1 mg: 418 (93.5%) of 447 treated participants (2.9% adverse events, 2.9% voluntarily withdrew) Ozanimod 0.5 mg: 425 (94.2%) of 451 treated participants (1.5% adverse events, 3.1% voluntarily withdrew) IFNβ‐1a: 412 (92.0%) of 448 treated participants (3.6% adverse events, 2.2% voluntarily withdrew) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified primary safety outcomes (NCT02294058). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Method of AE monitoring | Low risk | Quoted: “On day 1, vital signs were measured before administration of the drug and hourly for the first 6 h after, and electrocardiograms were done before and at 6 h after. At the end of the 6‐h period, if heart rate was less than 45 bpm or at its lowest value since administration of the drug, or if ECG showed a prolonged QTcF interval or second‐degree or third‐degree atrioventricular block, then monitoring was continued until resolution. Monitoring was repeated on days 5 and 8 at the investigators’ discretion for participants with a cardiac safety issue on the previous day of dose escalation. Study visits occurred at screening, baseline, month 1, and every 3 months between month 3 and the end of treatment. Adverse events and liver function tests were assessed at each visit. General retinal exams, including eye history, visual acuity, and dilated ophthalmoscopy, were obtained if macular oedema was suspected. Pulmonary function tests were done at screening and months 3, 6, and 12. Skin examinations were done at screening and month 12. (page 1012) |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
TEMSO 2011.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; relapsing–remitting, secondary progressive, or progressive–relapsing; mean disease duration 9 years; mean EDSS 2.7; prior use of DMT in the previous 2 years: 27.0% (28.4% in teriflunomide 14 mg, 27.9% in teriflunomide 7 mg, and 24.8% in placebo) | |
| Interventions | Teriflunomide 14 mg orally once daily for 24 months (n = 359) Teriflunomide 7 mg orally once daily for 24 months (n = 366) Placebo orally once daily for 24 months (n = 363) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 24 months | |
| Notes | Funding: Sanofi‐Aventis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Eligible patients were randomly assigned (in a 1:1:1 ratio) to receive a once‐daily oral dose of placebo, 7 mg of teriflunomide, or 14 mg of teriflunomide for 108 weeks. Randomization was stratified according to the baseline EDSS score (≤ 3.5 or > 3.5) and according to trial site, with a block size of 6.” (page 1294). |
| Allocation concealment (selection bias) | Low risk | Quoted: “The treatment allocation was determined according to the randomization code provided by an interactive voice response system (IVRS). Treatment codes were maintained by the IVRS and no code‐breaking material was provided on site” (page 45 of Medical Review of FDA). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. Quoted: “Each medication kit was labelled with a two‐part tear‐off label...” (Protocol, page 39). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “A treating neurologist at each site was responsible for evaluating patient eligibility, supervising the administration of study medication, recording and managing adverse events, assessing relapses, and monitoring safety assessments. The treating neurologist was aware of any side effects that could potentially be related to active therapy” (pages 1294‐1295). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 24 months on study treatment (Figure 1, page 1296): Teriflunomide 14 mg: 263 (73.5%) of 358 participants (10.6% adverse events, 7.2% patient request, 4.7% lack of efficacy, 7.2% progressive disease) Teriflunomide 7 mg: 274 (75.1%) of 365 participants (10.1% adverse events, 8.7% patient request, 3.8% lack of efficacy, 8.7% progressive disease) Placebo: 259 (71.3%) of 363 participants (8.0% adverse events, 9.1% patient request, 6.6% lack of efficacy, 3.0% progressive disease) |
| Selective reporting (reporting bias) | Low risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00134563). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Method of AE monitoring | Low risk | Quoted: “A treating neurologist at each site was responsible for recording and managing adverse events and monitoring safety assessments” and “Safety was evaluated on the basis of adverse events reported by study participants or investigators. Laboratory tests were performed at the time of screening, at baseline, every 2 weeks for the first 24 weeks, and then every 6 weeks until study completion. Physical and neurologic examinations were performed at week 12 and then every 24 weeks. An abdominal ultrasonographic examination to assess for pancreatic abnormalities was performed before the study and then every 24 weeks, because of previous infrequent reports of pancreatitis associated with teriflunomide use” (pages 1294‐1295). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
TENERE 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18 years and older; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.1; prior use of DMT in the previous 2 years: 18.8% (11.7% in teriflunomide 14 mg, 21.1% in teriflunomide 7 mg, and 24.0% in interferon Beta‐1a) | |
| Interventions | Teriflunomide 14 mg orally once daily for at least 12 months (n = 111) Teriflunomide 7 mg orally once daily for at least 12 months (n = 109) IFNβ‐1a (Rebif) 44 µg ("when the 44 μg dose was not tolerated, the dose was reduced to 22 μg") subcutaneously three times a week for at least 12 months (n = 104) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Patients were randomised 1:1:1 to teriflunomide 7 mg or 14 mg or IFNβ‐1a, and stratified by country (Americas, Eastern Europe, Western Europe and Africa) and baseline EDSS score (≤ 3.5 or > 3.5)” (page 706). |
| Allocation concealment (selection bias) | Low risk | Quoted: “A phone interactive voice response system was used to randomize patients”. Information provided on request by Genzyme |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quoted: “Patients were randomised 1:1:1 to teriflunomide 7 mg or 14 mg (double‐blind) or IFNβ‐1a (open‐label)” (page 706). “It may also be noted that while the examining neurologist was blinded to treatment, patients were unblinded, which could have introduced a potential bias.” (page 715) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quoted: “The treating neurologist was responsible for patient selection, medication administration, managing AEs, and relapse and safety assessments, while an examining neurologist scored the Functional Systems (FS) and EDSS. The examining neurologist remained blinded to treatment and associated AEs” (page 706). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, at 1 year, 17.9% was lost‐to follow‐up (17.1% in teriflunomide 14 mg, 10.1% in teriflunomide 7 mg, and 26.9% in IFNβ‐1a) (data provided on request by Genzyme), with some indications of differences in reasons. |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00883337). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Information provided on request by Genzyme |
| Method of AE monitoring | Low risk | Quoted: “Safety and tolerability were assessed using AE reporting, vital signs and laboratory assessments. Adverse event reports were collected at randomisation, weeks 2, 6, 12, 18, 24, 36 and every 12 weeks thereafter. Vital signs were documented at screening, randomisation and every 12 weeks thereafter; clinical laboratory results were assessed throughout the study. Adverse events and vital signs were also recorded during unscheduled relapse visits” (page 707). |
| Other bias | Unclear risk | The study was completed 48 weeks after the last patient was randomized, resulting in a variable duration of follow‐up. |
TOPIC 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; CIS; time since neurological event 2 months; mean EDSS: 1.7; all participants were previously untreated patients | |
| Interventions | Teriflunomide 14 mg orally once daily for 25 months (n = 216) Teriflunomide 7 mg orally once daily for 25 months (n = 205) Placebo orally once daily for 25 months (n = 197) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 27 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “...using a permuted‐block randomisation schedule (block size of six) with stratification by baseline mono focal or multifocal status” (page 978) |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomisation was done centrally, by an interactive voice recognition system that generated an allocation sequence”; “...investigators called the interactive voice recognition system to receive a random, masked treatment assignment for each patient” (page 978). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Placebo identical in taste and appearance...Patients, staff administering the interventions, and outcome assessors were masked to treatment assignment” (page 978). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: “A treating neurologist at each site assessed participant eligibility, supervised study drug administration, and did the safety assessments” (page 979). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 27 months on study treatment or entered extension study (Figure 1, page 979): Teriflunomide 14 mg: 163 (76.2%) of 214 participants (8.4% adverse events; 5.6% lack of efficacy; 0.5% lost to follow‐up; 9.3% other reason) Teriflunomide 7 mg: 150 (73.9%) of 203 participants (12.3% adverse events; 2.9% lack of efficacy; 0.5% lost to follow‐up; 0.5% progressive disease; 9.8% other reason) Placebo: 141 (71.6%) of 197 participants (9.1% adverse events; 9.6% lack of efficacy; 0.5% lost to follow‐up; 1.5% progressive disease; 0.5% death; 7.1% other reason) Quoted: "Following an amendment to the protocol on May 24, 2011, patients who had a relapse which defined clinically definite multiple sclerosis, and had been treated for at least 6 months, could also enter the extension study, which is still ongoing". |
| Selective reporting (reporting bias) | Low risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00622700). |
| Serious AE definitions | Low risk | Definition of SAEs according to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). CTCAE, common terminology criteria for adverse events |
| Method of AE monitoring | Low risk | “Adverse events were reported by study participants or investigators throughout the study; investigators recorded all such events on case report forms” (page 979). "Time frame: All AEs were collected from signature of the informed consent form up to the final visit (up to 390 weeks [maximum exposure in core treatment period: 120 weeks and maximum exposure in extension treatment period: 283 weeks]) regardless of seriousness or relationship to investigational product. Reported adverse events are treatment‐emergent adverse events, that is, AEs that developed or worsened during the 'on treatment period'. AEs collected by systematic assessment. Term from vocabulary, MedDRA‐18.1" (NCT00622700). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
TOWER 2014.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 8 years; mean EDSS 2.7; prior use of DMT in the previous 2 years: 32.8% (33.9% in teriflunomide 14 mg, 30.1% in teriflunomide 7 mg, and 34.7% in placebo) | |
| Interventions | Teriflunomide 14 mg orally once daily for at least 12 months (n = 372) Teriflunomide 7 mg orally once daily for at least 12 months (n = 408) Placebo orally once daily for at least 12 months (n = 389) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomisation was done using a permuted‐block randomisation schedule with stratification according to study site and baseline EDSS score (≤ 3.5 or > 3.5)” (page 248). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomisation was done centrally, via an interactive voice recognition system that generated an allocation sequence”; and “investigators used the allocation sequence to randomly assign eligible patients in a 1:1:1 ratio to receive once‐daily oral placebo, teriflunomide 7 mg, or teriflunomide 14 mg” (page 248). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: “Patients and individuals administering the interventions were masked to treatment assignment. Oral placebo, teriflunomide 7 mg, or teriflunomide 14 mg were identical in taste and appearance" (page 248). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether outcome assessors were blinded to adverse events' assessment. Quoted: “A treating neurologist was responsible for assessment of patient eligibility, supervision of administration of study drug or placebo, recording of adverse events, and assessment of relapses” (page 248). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 12 months on study treatment (Figure 1, page 249): Teriflunomide 14 mg: 244 (65.9%) of 370 participants (15.6% adverse events; 5.4% lack of efficacy) Teriflunomide 7 mg: 273 (67.1%) of 407 participants (13.2% adverse events; 7.4% lack of efficacy) Placebo: 263 (67.8%) of 388 participants (6.7% adverse events; 9.5% lack of efficacy) |
| Selective reporting (reporting bias) | Low risk | The published report included prespecified primary safety outcomes (NCT00751881). |
| Serious AE definitions | Low risk | Quoted: "The study was done in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 248). "A serious adverse event was defined as an event that resulted in death, was life‐threatening, needed inpatient hospital admission or prolonged an existing hospital stay, resulted in persistent or significant disability or incapacity, was a congenital anomaly or birth defect, or was a medically important event" (pages 248‐9). |
| Method of AE monitoring | Low risk | Quoted: “Safety was assessed through adverse event reporting (upon occurrence), clinical laboratory tests (every 2 weeks until week 24, then every 6 weeks while still on treatment), vital signs (at weeks 2 and 6, then every 6 weeks until week 24, then every 12 weeks while still on treatment), abdominal ultrasonography (at week 24, then every 24 weeks), and electrocardiogram (at baseline and end of treatment)” (page 248). |
| Other bias | Unclear risk | The study was completed 48 weeks after the last patient was randomised, resulting in a variable duration of follow‐up. |
TRANSFORMS 2010.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 18‐55 years; clinically definite RRMS; mean disease duration 7 years; mean EDSS 2.2; prior use of DMT at any time prior to the start of study: 56.7% (58.5% in fingolimod 1.25 mg, 55.2% in fingolimod 0.5 mg, and 56.3% in interferon beta‐1a) | |
| Interventions | Fingolimod 1.25 mg orally once daily for 12 months (n = 426) Fingolimod 0.5 mg orally once daily for 12 months (n = 431) IFNβ‐1a (Avonex) 30 µg intramuscularly once a week for 12 months (n = 435) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 12 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: “Randomization was performed in blocks of six within each site and was stratified according to site” (page 403). |
| Allocation concealment (selection bias) | Low risk | Quoted: “Randomization was performed centrally”; and “Study‐group assignments were performed with the use of an interactive voice‐response system” (page 403). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants were assigned to a daily dose of oral fingolimod or to intramuscular interferon beta‐1a at a weekly dose. Masking was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "At each site, a treating neurologist supervised medical management" (page 403) and: "Ophthalmic examinations and pulmonary function tests were performed at screening and months 1, 3, 6, and 12...Skin examination was included in the general physical examination performed by the treating neurologist at screening, and months 6 and 12...Monthly self‐examination by participants and examination by a dermatologist at screening and month 12" (Appendix page 7). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 12 months on study drug (Figure 1, page 405): Fingolimod 1.25 mg: 358 (85.2%) of 420 treated participants (7.6% adverse events; 2.4% consent withdrawn; 1.2% unsatisfactory therapeutic effect; 1.9% abnormal laboratory value) Fingolimod 0.5 mg: 385 (89.7%) of 429 treated participants (3.7% adverse events; 2.1% consent withdrawn; 1.2% unsatisfactory therapeutic effect; 1.6% abnormal laboratory value) Interferon beta‐1a: 380 (88.2%) of 431 treated participants (2.8% adverse events; 3.7% consent withdrawn; 1.6% unsatisfactory therapeutic effect; 0.7% abnormal laboratory value) |
| Selective reporting (reporting bias) | Unclear risk | Types and measures of adverse effects were not prespecified in the protocol (NCT00340834). |
| Serious AE definitions | Low risk | Quoted: "The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice" (page 403). |
| Method of AE monitoring | Low risk | Quoted: “Safety assessments were conducted during screening, at baseline, and at months 1, 2, 3, 6, 9, and 12... An independent data and safety monitoring board evaluated overall safety in the fingolimod phase 3 program” (page 404). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Tubridy 1999.
| Study characteristics | ||
| Methods | Parallel RCT conducted at eight UK centers. Study period not reported | |
| Participants | Active relapsing‐remitting and secondary progressive MS according to the criteria of Poser 1983, age 18‐55 years, EDSS score 2.0‐7.0, two or more clinical relapses in the previous 18 months, > 4 weeks since the onset of the last relapse | |
| Interventions | Natalizumab (Antegren): two endovenous infusions at weeks 0 and 4; 3 mg/kg of study drug diluted to a 100‐mL solution with normal saline (n = 37) Placebo: two endovenous infusions of a 100‐mL normal saline solution at weeks 0 and 4 (n = 35) |
|
| Outcomes | Adverse events measured at 6 months. SAEs not reported | |
| Notes | Funding: the study was sponsored by Elan Pharmaceuticals Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | It was unclear if study participants, clinicians, and other personnel were blinded. Quoted: "The patients received 3 mg/kg of study drug or placebo diluted to a 100‐mL solution with normal saline" (page 4). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was unclear whether assessors were blinded to adverse events' assessment. Quoted: "At each visit, any change in the patients' well‐being was recorded...There was also a brief clinical examination, and any adverse events, new medication, or MS exacerbations the patients may have had since the previous visit were recorded" (page 4). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed 6 months on study treatment: Natalizumab: 37 (100%) of 37 participants Placebo: 33 (94.3%) of 35 participants |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Definitions of SAEs were not reported. |
| Method of AE monitoring | Low risk | Quoted: "Adverse events were registered at follow‐up visits (weeks 1, 2, 4, 6, 8, 12, 16, 20, and 24). On each occasion, blood was taken for hematology, biochemistry, immune function profile and Antegren and anti‐Antegren antibody levels. In addition, a full clinical examination was performed at weeks 0, 12, and 24. An electrocardiogram was performed at weeks ‐4, 4, 12, and 24" (Page 4) and "Independent safety committee was appointed to review adverse and other safety events throughout the trial" (page 5). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Van de Wyngaert 2001.
| Study characteristics | ||
| Methods | RCT. The study was conducted in two centers between June 1992 and December 1994. | |
| Participants | A definite clinical diagnosis of MS according to the criteria of Poser 1983 and a relapsing, active secondary progressive form of MS. Participants were 18‐50 years old, with an EDSS score of 3 to 6, must have recovered, at least partially, from their last disease relapse at least one month before study entry, and displayed worsening of their EDSS of 1 point during the last 12 months. | |
| Interventions | Induction treatment:
Maintenance treatment: Both treatments (MP and methylene blue versus alizapride and MTX) were given once every three months, ten times until month 32. The complete treatment thus consisted of 13 infusions. |
|
| Outcomes | Adverse events assessed every three month up to 36 months. Quoted: " hematological parameters, blood was tested for immunoglobulin (I g) electrophoresis and routine clinical chemistry before each injection. Hematology was also checked ten days after each infusion". | |
| Notes | Funding: The study was supported by the “Fondation Charcot Belgique”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "A table of random numberswas used to generate the random sequence" (page 212). |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Information about concealment of allocation from participants and personnel at the point of assignment was missing, however. they were likely unblinded due to different adverse events between the comparison drugs. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completed 36 months on study treatment (page 214): MTX: 10 (35.7%) of 28 participants (4 gastro‐intestinal effects, 4 lack of efficacy, 4 personal reasons, 2 lost to follow‐up, 1 breast cancer, 2 fall of myocardial ejection fraction below 50%, 1 brachial phlebitis with pulmonary embolism) MP: 14 (66.7%) of 21 participants (2 depression, 1 anaphylactoid reaction, 2 lack of efficacy, 1 participated in another study, 1 other). The reasons for discontinuation were different between the comparison groups. |
| Selective reporting (reporting bias) | High risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Unclear risk | No information on pre‐specified AEs. Protocol was not available. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Wolinsky 2007.
| Study characteristics | ||
| Methods | RCT | |
| Participants | Age: 30‐65 years; clinically definite PPMS; mean disease duration 11 years; mean EDSS 4.9; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneously every day for 36 months (n = 627) Placebo (unspecified) for 36 months (n = 316) |
|
| Outcomes | Withdrawals due to AEs; AEs and SAEs over 36 months | |
| Notes | Funding: Teva Pharmaceutical | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The placebo was not described in sufficient detail to judge whether blinding of participants and personnel was sufficient. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: “All patients were attended by a treating neurologist and an examining neurologist who were blinded to treatment. The treating neurologist supervised drug administration, recorded and treated adverse events, and coordinated MRI testing” (page 16). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study stopped early for futility. 376/627 (60%) of participants in the glatiramer acetate group and 186/315 (59%) in the placebo group had received study drugs for 24 months. Insufficient information to judge withdrawals and lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | Not reported |
| Method of AE monitoring | Low risk | Quoted: “The treating neurologist supervised drug administration, recorded and treated adverse events” and “Safety was assessed by adverse event reporting, vital signs, electrocardiograms, and laboratory tests...Neurological, laboratory, and vital sign evaluations were conducted during on‐site visits at months 1 and 3 and every 3 months thereafter until month 36, and continued every 3 months for patients in the double‐blind extension trial.” (page 16). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ziemssen 2017.
| Study characteristics | ||
| Methods | RCT. A dose‐escalation study performed at 7 centers in Germany from August 2009 to March 2012 | |
| Participants | Relapsing MS (revised McDonald criteria 2005), at least one documented relapse in the 3 years prior to screening. Mean (SD) age 37.8 (9.0); baseline EDSS score 0‐5.5 | |
| Interventions | Oral laquinimod administered daily for one month (n = 84)
Placebo for 4 weeks (n = 28) |
|
| Outcomes | Adverse events, clinical laboratory (biochemistry, hematology, and urinalysis) assessments, vital signs, and electrocardiograms, measured at one month | |
| Notes | In January 2016, after the completion of the study, the Data Monitoring Committee recommended discontinuation of laquinimod doses greater than 1.0 mg per day due to an imbalance in cardio‐ and cerebrovascular adverse events in emerging safety data in the MS clinical studies (Teva Pharmaceutical Industries Ltd). Funding: The study was funded by Teva Pharmaceutical Industries Ltd. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quoted: "The Teva Global Biostatistics unit prepared a computer‐generated randomization scheme for each cohort using a SAS® PLAN procedure. Each scheme used a block design; however, due to the small number of patients recruited at each center for each cohort, there was no stratification by center" (page 2). |
| Allocation concealment (selection bias) | Low risk | Quoted: "Patients were randomized to comparison groups by an Interactive Web Response System according to the randomization algorithm" (page 2‐3). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quoted: "Laquinimod capsules and their matching placebo capsules were of identical appearance and packaged in aluminum‐silver/aluminum‐soft blister cards to maintain study blinding. All patients were administered laquinimod or matching placebo capsules, taken at the same hour every day, with water...The investigators, the sponsor, and any personnel involved in patient assessment, monitoring, analysis, and data management were blinded to patient assignment" (page 3). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quoted: "The investigators remained blinded to the patients’ treatment assignment" (page 3). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Completed one month on study treatment (Figure 1, page 4): Laquinimod group:
Placebo group: 27 (96.4% ) of 28 participants (1 MS relapse) |
| Selective reporting (reporting bias) | Unclear risk | Study protocol was not available. |
| Serious AE definitions | Unclear risk | No information |
| Method of AE monitoring | Low risk | Quoted: "Scheduled in‐clinic visits, during which safety evaluations were performed, occurred at screening (day 7), baseline (day 0), and on days 7, 14, 21, and 28...Safety assessments included evaluation of adverse events (AEs), clinical laboratory (biochemistry, hematology, and urinalysis) assessments, vital signs, and electrocardiograms" (page 4). |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
AE: adverse event; CIS: clinically isolated syndrome; CNS: central nervous system; CPM: Cyclophosphamide; CRF: case report form; CRO: contract research organization; CSF: cerebrospinal fluid; CT: clinical trial; CTCAE: Common Terminology Criteria for Adverse Events; DMF: dimethyl fumarate; DMT: disease‐modifying treatment; ECG: electrocardiogram; EDSS: Expanded Disability Status Scale; FS: functional system; GA: glatiramer acetate; GGISIS: Global Gastrointestinal Symptom and Impact Scale; GI: gastrointestinal; HLGT: high level group term; ICH: intracerebral hemorrhage; IFN: interferon; IGISIS: Individual Gastrointestinal Symptom and Impact ScaleI; IM: intramuscular; IV: intravenous; IVIG: IV immunoglobulin; IVRS: interactive voice response system; JCV: John Cunningham virus; MADRS: Montgomery‐Asberg depression rating scale; MCV: mean corpuscular volume; MedDRA: medical dictionary for regulatory activities; MP: methylprednisolone; MRI: magnetic resonance imaging; MS: multiple sclerosis; MTX: mitoxantrone; NFL: neurofilament light chain; PPMS: primary progressive MS; PRMS: progressive relapsing MS; PT: physical therapy; QTcF: Fridericia‐corrected QT; RCT: randomised controlled trial; RRMS: relapsing remitting MS; RTX: rituximab; SAE: serious adverse event; SC: subcutaneous; SD: standard deviation; SMQ: standardised MedDRA queries; SOC: standard of care; SPMS: secondary progressive MS; TEAE: treatment‐emergent AE; tiw: three times a week; ULN: the upper limit of the normal range; VAS: visual analogue scale; WHO: World Health Organization; YER: yearly exacerbation rate.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Barkhof 2018 | A pooled analysis of studies included in the review OPERA I and OPERA II |
| Beutler 1996 | Cladribine is approved and used in clinical practice as an oral medication for the treatment of highly‐active relapsing or active progressive MS. In this study, cladribine was given by intravenous infusions. |
| Boiko 2018 | Wrong intervention: comparison of Teberif (a bioanalog of Rebif) and Rebif (interferon β‐1a) |
| Boyko 2019 | Wrong intervention: Study evaluating a treatment‐switch strategy from interferonβ 1a (Rebif) to biosimilar Teberif |
| Boyko 2022 | Type of intervention outside inclusion criteria (Sampeginterferon β‐1a) |
| Cohen 2009 | Wrong intervention: interferon beta‐1a combined with methotrexate, IV methylprednisolone |
| Cohen 2019 | Wrong intervention: comparison of natalizumab administered at standard interval dosing (every 4 weeks) with natalizumab administered at extended interval dosing (every 6 weeks) |
| Comi 2011 | Wrong intervention: comparison of glatiramer 20 mg with glatiramer 40 mg |
| Comi 2016 | The study reported analyses of safety outcomes from studies included in the review (TEMSO; TOWER; and TOPIC). |
| CORAL 2006 | Wrong intervention: oral glatiramer acetate is not used in clinical practice. |
| DELIVER 2016 | Wrong treatment: a study evaluating intravenous, subcutaneous, intramuscular Natalizumab or a non‐natalizumab reference treatment (or no treatment) |
| Edan 1997 | Wrong intervention: comparison of mitoxantrone and methylprednisolone with methylprednisolone alone over six months |
| EPOC 2014 | Wrong intervention: Study evaluating a treatment‐switch strategy from an injectable DMT (IFNβ‐1b, IFNβ‐1a, glatiramer acetate) to Fingolimod |
| EVOLVE‐MS‐1 2022 | Wrong intervention: Study evaluating a treatment‐switch strategy from dimethyl fumarate (in EVOLVE‐MS‐2) or from interferons and glatiramer acetate, to diroximel fumarate |
| Freedman 2012 | Wrong intervention: Teriflunomide as add‐on therapy to interferons beta compared with interferons beta |
| Freedman 2015 | Wrong intervention: Teriflunomide as add‐on therapy to glatiramer acetate compared with glatiramer acetate |
| Gobbi 2013 | Wrong intervention: Study evaluating a treatment‐switch strategy from natalizumab to subcutaneous interferonβ 1b |
| Goodman 2009 | Wrong intervention: comparison of IV natalizumab or placebo in addition to glatiramer acetate for up to 24 weeks (“combination therapy” and “GA alone” groups) |
| Hartung 2020 | Wrong intervention: single‐arm study aiming to evaluate effectiveness and safety of ocrelizumab. No control group |
| Hauser 2018 | The study reported safety evaluations from ocrelizumab clinical trials included in the review (OPERA I, OPERA II, ORATORIO) and associated open‐label extension periods up to September 2017. |
| Havrdova 2009 | Wrong intervention: comparison between intramuscular IFNβ‐1a, intramuscular IFNβ‐1a plus azathioprine, intramuscular IFNβ‐1a plus azathioprine and prednisone |
| Hu 2016 | Wrong participants: healthy subjects |
| Hughes 2018 | Wrong participants: chronic inflammatory demyelinating polyradiculoneuropathy |
| Kappos 2014 | Wrong intervention: Atacicept is not included in the review. |
| Kappos 2016 | Wrong intervention: Extension study. Participants taking siponimod in the BOLD trial continued at the originally assigned dose and participants taking placebo were re‐randomized to the 5 siponimod doses. |
| Kastrukoff 1990 | Wrong intervention: Lymphoblastoid interferon is not used in clinical practice. |
| Khoury 2010 | Wrong intervention: Albuterol treatment as an add‐on therapy to glatiramer acetate treatment |
| Komori 2016 | Wrong intervention: intrathecal rituximab |
| Le Page 2015 | Wrong intervention: oral versus intravenous high‐dose methylprednisolone |
| Mancardi 2015 | Wrong intervention: Autologous hematopoietic stem cell transplantation |
| Mayer 2019 | Analyses of studies included in the review (OPERA I, OPERA II, and ORATORIO) |
| Montalban 2019 | Wrong intervention: Extension study. Participants were randomised to evobrutinib, placebo, or open‐label dimethyl fumarate. The comparison of interest was dimethyl fumarate vs placebo. However, after 24 weeks of the placebo‐controlled phase of the trial, participants in the placebo group were switched to receive evobrutinib for a further 24‐week blinded extension phase and adverse events were reported only at 52 weeks. |
| NCT00206648 | Wrong intervention: Study evaluating a treatment‐switch strategy from avonex to betaferon |
| NCT01058005 | Wrong intervention: Study evaluating a treatment‐switch strategy from glatiramer acetate or interferon beta‐1a to natalizumab. Terminated due to significantly slower than expected enrolment; the Sponsor decided to terminate the study. |
| NCT01065727 | The study did non measure adverse events or serious adverse events. Wrong intervention: mitoxantrone followed by immunomodulator |
| NCT01337427 | Withdrawn (The study was not feasible to conduct in the US and abroad). |
| Okai 2019 | Analyses of studies included in the review (CARE MS I and II) |
| Perumal 2019 | A single‐arm study of natalizumab in anti–JC virus (JCV) seronegative patients with early relapsing MS |
| PREFERMS 2018 | Wrong interventions: comparison of fingolimod with interferon β‐1a, interferon β‐1b or glatiramer acetate. For patients previously treated with glatiramer, an interferon was preselected during consultation with their physician before randomization. Similarly, glatiramer was preselected for patients previously treated with an interferon. Interferons or glatiramer were preselected for treatment‐naïve patients. Aggregate data were reported for adverse events in the comparison group. |
| Rahimdel 2015 | Wrong intervention: comparison of mitoxantrone plus methylprednisolone monthly with mitoxantrone for six months |
| Ramo‐Tello 2014 | Wrong intervention: comparison of oral with intravenous methylprednisolone |
| RESTORE 2014 | Wrong intervention: Study evaluating a treatment‐switch strategy from natalizumab to natalizumab, intramuscular interferon b‐1a (IFN‐b‐1a), glatiramer acetate (GA), methylprednisolone (MP), or placebo. In the other‐therapies group, participants and their neurologist selected the immunomodulatory therapy on an individual basis; as such, the distribution of participants receiving IFN‐b‐1a, GA, and MP was not randomized. |
| Rice 2000 | Cladribine is approved and used in clinical practice as an oral medication for the treatment of highly‐active relapsing or active progressive MS. In this study, cladribine was used subcutaneously. |
| Rieckmann 2012 | Wrong intervention: Study evaluating a treatment‐switch strategy from mitoxantrone to subcuteneous IFN beta‐1a |
| RIVITALISE 2016 | Rituximab is used off‐label in clinical practice for treatment of active relapsing or progressive MS. It is administered as an intravenous infusion. In this study, participants received intrathecal injection of rituximab. |
| Romine 1999 | Cladribine is approved and used in clinical practice as an oral medication for the treatment of highly‐active relapsing or active progressive MS. In this study, cladribine was used subcutaneously. |
| Saida 2016 | Wrong intervention: comparison of intramuscular IFN beta‐1a at dosages of either 30 mcg once weekly (full‐dose) or 15 mcg once weekly for 2 weeks then 30 mcg once weekly thereafter (titration group) |
| SELECTION 2014 | Wrong design: Extension study. Patients who received placebo in SELECT were randomly assigned (1:1) to receive 150 mg or 300 mg subcutaneous daclizumab every 4 weeks for 52 weeks (treatment initiation group); those who had received daclizumab were randomly assigned (1:1) to continue their present dose with (washout and re‐initiation group) or without (continuous treatment group) a washout period of 20 weeks. |
| SENTINEL 2006 | Wrong intervention: interferon beta‐1a in combination with natalizumab compared with interferon beta‐1a and placebo |
| Sipe 1994 | Cladribine is approved and used in clinical practice as an oral medication for the treatment of highly‐active relapsing or active progressive MS. In this study, cladribine was used intravenously via a surgically implanted central line. |
| Sorensen 2014 | Ofatumumab is approved and used in clinical practice as a once‐monthly subcutaneous injection for the treatment of relapsing forms of MS in adults. In this study, participants received two intravenous infusions of ofatumumab two weeks apart. |
| Sorensen 2017 | Analyses of studies included in the review (ALLEGRO and BRAVO trials) |
| Stelmasiak 2000 | Wrong study design: case series |
| Tahara 2020 | Wrong participants: neuromyelitis optica spectrum disorders |
| Trojano 2015 | Wrong Intervention: participants were randomised to 1 of 6 natalizumab regimens. No comparison group |
| Turner 2019 | Subgroup analyses of efficacy endpoints from studies included in the review (OPERA I and OPERA II) |
| Wolinsky 2015 | Wrong intervention: comparison of two doses of glatiramer acetate |
| Wray 2019 | Pooled analyses of studies included in the review (CAMMS223, CARE‐MS I, and CARE‐MS II) |
| Wynn 2010 | Wrong intervention: add‐on of high‐ or low‐dose daclizumab to interferon beta |
DMT: disease‐modifying treatment; GA: glatiramer acetate; IV: intravenous; JCV: John Cunningham virus; MP: methylprednisolone; MS: multiple sclerosis.
Characteristics of ongoing studies [ordered by study ID]
EudraCT 2018‐000284‐93.
| Study name | Mapi_GADepotPhaseIII‐001 |
| Methods | RCT |
| Participants | Relapsing MS. Age: 18‐64 years; subjects should be ambulatory with an EDSS score of 0‐5.5 at screening and baseline visits |
| Interventions | Glatiramer acetate 40 mg intramuscularly once every four weeks for 12 months Placebo for 12 months Planned number of subjects to be included in the whole clinical trial: 960 |
| Outcomes | Safety and tolerability during 12 months of treatment |
| Starting date | (first received 2 October 2019) |
| Contact information | Uri Danon at Mapi Pharma Ltd., Israel. Email: Uri@mapi‐pharma.com. |
| Notes |
NCT04035005.
| Study name | A study to evaluate the efficacy and safety of ocrelizumab in adults with primary progressive multiple sclerosis (O'HAND) |
| Methods | Parallel RCT |
| Participants | Primary progressive MS. Age: 18‐65 years; EDSS score at screening and baseline ≥ 3.0 to 8.0 |
| Interventions | Ocrelizumab 600 mg intravenously every 6 months for 30 months Placebo intravenously every 6 months for 30 months Estimated enrollment: 1000 participants |
| Outcomes | Safety during 30 months of treatment |
| Starting date | 12 August 2019 |
| Contact information | Reference Study ID Number: WA40404 www.roche.com/about_roche/roche_worldwide.htm; 888‐662‐6728 (U.S. and Canada); global‐roche‐genentech‐trials@gene.com. |
| Notes |
NCT04121403.
| Study name | Norwegian study of oral cladribine and rituximab in multiple sclerosis (NOR‐MS) |
| Methods | Parallel RCT |
| Participants | Active relapsing MS according to the 2017 McDonald criteria. Age: 18‐65 years; EDSS between 0 and 5.5 |
| Interventions | Oral cladribine (Mavenclad tabler 10 mg) Rituximab biosimilar concentrate as solution for infusion Estimated enrollment: 264 participants. Randomization is 1:1. |
| Outcomes |
|
| Starting date | 16 October 2019 |
| Contact information | Gro Owren Nygaard, MD, PhD; 91757192 ext +47; uxgryg@ous‐hf.no; Helle Stangeland, MSc; 90029660 ext +47; stahel@ous‐hf.no. |
| Notes |
NCT04578639.
| Study name | Ocrelizumab versus rituximab off‐label at the onset of relapsing MS disease (OVERLORD‐MS) |
| Methods | Parallel RCT |
| Participants | Relapsing MS according to the 2017 revised diagnostic criteria of McDonald within the last 12 months. Age: 18‐60 years; clinically definite RRMS; EDSS score ≤ 4.0 |
| Interventions | Rituximab 1000 mg intravenously every 6 months for 24 months Ocrelizumab 600 mg intravenously every 6 months for 24 months Estimated enrollment: 211 participants. Randomization rituximab: ocrelizumab is 2:1. |
| Outcomes | Safety during 24 months of treatment. |
| Starting date | 2 November 2020 |
| Contact information | Øivind Torkildsen, MD; +47 5597 5045; oivind.fredvik.grytten.torkildsen@helse‐bergen.no; Kjell‐Morten Myhr, MD; +47 55975045; kjell‐morten.myhr@helse‐bergen.no. |
| Notes |
NCT04688788.
| Study name | Non‐inferiority study of ocrelizumab and rituximab in active multiple sclerosis (DanNORMS) |
| Methods | Parallel RCT |
| Participants | Active relapsing or progressive MS, according to the 2017 McDonald criteria, never treated, or no DMT in the previous 2 years. Age: 18‐65 years; EDSS score ≤ 6.5 |
| Interventions | Rituximab biosimilar (Ruxience®) 1000 mg intravenously given every 6th month (first 2 infusions 1000 mg/1000 mg given 2 weeks apart) for 24 months Ocrelizumab 600 mg intravenously every 6 months (first 2 infusions 300 mg/300 mg given 2 weeks apart) for 24 months Estimated enrollment: 594 participants. Randomization rituximab: ocrelizumab 2:1 |
| Outcomes | Safety during 24 months of treatment. |
| Starting date | 28 April 2021 |
| Contact information | Jeppe Romme Christensen, MD, PhD; 0045 38633379; jeppe.romme.christensen@regionh.dk; Finn Sellebjerg, Prof., MD, PhD; 0045 38633236; finn.thorup.sellebjerg@regionh.dk. |
| Notes |
NCT04695080.
| Study name | ChariotMS ‐ Cladribine to halt deterioration in people with advanced multiple sclerosis (ChariotMS) |
| Methods | Parallel RCT |
| Participants | Advanced relapsing or progressive MS according to the McDonald Criteria (2017) Thompson (2018). Age: 18 years and older; EDSS score of 6.5‐8.5 (inclusive) |
| Interventions | Cladribine (MAVENCLAD®) 3.5 mg/kg, administered as weight‐adjusted 10 mg tablets in two treatment courses (12 months apart) lasting 8‐10 days each, for 24 months Placebo administered as weight‐adjusted tablets in two treatment courses (12 months apart) lasting 8‐10 days each, for 24 months Estimated enrollment: 200 participants. Randomization is 1:1. |
| Outcomes |
Time frame: Through study completion, an average of 24 months |
| Starting date | 25 June 2021 |
| Contact information | Klaus Schmierer, PhD, FRCP; +44 (0)20 7882 6246; k.schmierer@qmul.ac.uk; Harpreet Mangat; +44 (0)75 9750 0255; chariot@qmul.ac.uk. |
| Notes |
NCT04788615.
| Study name | NCT04788615 |
| Methods | RCT open‐label, rater‐blind, multi‐center, prospective, parallel‐arm, active comparator |
| Participants | Relapsing MS. Participants are newly diagnosed or have never been on active treatment at the time of study entry with ≤ 3 years from first MS symptoms. Estimated enrollment: 236 participants |
| Interventions |
|
| Outcomes | Adverse events reports until safety follow‐up [Time frame: baseline to 15 months and 6 months safety follow‐up]. Number of SAEs, and SAEs with hospitalizations [Time frame: baseline to 15 months] |
| Starting date | 23 July 2021 |
| Contact information | Novartis Pharmaceuticals +41613241111 novartis.email@novartis.com |
| Notes |
NCT05090371.
| Study name | NCT05090371 |
| Methods | Parallel RCT |
| Participants | Relapsing MS according to McDonald diagnostic criteria (2017). Age 18‐45 years; EDSS score 0‐5.5 |
| Interventions | Ofatumumab 20 mg, 3 loading doses followed by administration every 4 weeks as per label Other disease‐modifying treatment with approved label use for treatment which participants were on at least 6 months prior to screening |
| Outcomes | Treatment‐emergent adverse events and serious adverse events (Time frame: baseline up to month 15) |
| Starting date | 2 March 2022 |
| Contact information | Novartis Pharmaceuticals 1‐888‐669‐6682; novartis.email@novartis.com |
| Notes |
RAMBLE 2021.
| Study name | RAMBLE |
| Methods | RCT |
| Participants | Diagnosis of relapsing MS meeting 2017 McDonald criteria. Diagnosed with MS within the previous 10 years. Expanded disability status scale (EDSS) score < 5.0 |
| Interventions | Rituimab 100 mg/m2 of estimated body surface area, administered intravenously via an infusion over 30 minutes Placebo |
| Outcomes | Primary: frequency of autoimmune adverse events (time to event) assessed via monitoring visits, MRI scans, blood tests, liver function tests and urine tests. Time point: at 3 years post‐trial commencement Monitoring visits (6 months): intercurrent illness, relapse history and concomitant medications |
| Starting date | 31/12/2024 |
| Contact information | Not available |
| Notes |
AE: adverse event; DMT: disease‐modifying treatment; EDSS: Expanded Disability Status Scale; MRI: magnetic resonance imaging; MS: multiple sclerosis; RCT: randomised controlled trial; RRMS: relapsing remitting multiple sclerosis; SAE: serious adverse event.
Differences between protocol and review
1) Relative treatment ranking. In the protocol, we had planned to determine a treatment hierarchy by using the surface under the cumulative ranking curve (SUCRA) and mean ranks (Salanti 2011). In the review phase we used the R package netmeta for analyses and estimated ranking by means of P‐scores, a frequentist version of SUCRA (Rucker 2015).
2) Studies with multiple treatment groups. In the review, we decided to merge agents administered at different doses by summing numbers of events and sample sizes for each agent in each study, and then we performed a sensitivity analysis including only studies with doses that were higher than the median dose of each treatment across all studies.
3) Methods for indirect and mixed comparisons. We had planned to perform NMA in Stata using the 'mvmeta' command. In the review phase, we performed NMAs using random‐effects models within a frequentist setting using the R package netmeta (Rucker 2015; Schwrtzer 2015).
4) Local approaches for evaluating inconsistency. In the protocol, we had planned to use the loop‐specific approach (Veroniki 2013). In the review phase we used the method proposed by Dias (Dias 2010) which is implemented in the netmeta package.
5) Global approaches for evaluating inconsistency. In the protocol, we had planned to use the 'design‐by‐treatment' model to evaluate the assumption of consistency across the entire network (Higgins 2012). In the review phase, we used the method proposed by Rucker (Rucker 2012) which is implemented in the netmeta package.
6) Other sources of heterogeneity. In the protocol, we had planned to assess differences in age, gender, and disease duration of participants across trials. Since age, gender and disease duration were similar within MS type subgroups (relapsing‐remitting MS vs. progressive MS), we considered only MS type (relapsing or progressive MS) in a subgroup analysis.
7) Sensitivity analysis. We had planned a sensitivity analysis on the exclusion of trials with a total sample size of fewer than 50 randomized participants, to detect potential small‐study effects. In the review we explored the possibility of small‐study effects using the comparison‐adjusted funnel plot.
8) 'Summary of findings' table. In the protocol, we had planned to present seven outcomes in the SoF. In the review phase, due to the large number of outcomes and treatments, we decided to present two SoFs, one for each primary outcome (SAEs and withdrawals due to AEs).
For each SoF, we had planned to choose two values for the assumed risk with placebo (i.e. second highest and second lowest placebo group risks) in the included studies; in the review, we used only the overall raw frequency of AEs.
We had planned to grade the certainty of evidence for each outcome by considering study limitations, indirectness, inconsistency, imprecision of effect estimates and risk of reporting bias. In the review phase, we used the Confidence in Network Meta‐Analysis (CINeMA) as a methodological framework to evaluate the confidence in the results from NMAs, a tool which was not available when the protocol was prepared. CINEMA requires the specification of an equivalence range for primary outcomes. The Methods section has been re‐written and an explanation has been given, highlighting that this was a post hoc decision marked as a change to the protocol.
9) Electronic searches. In the protocol, we had planned to search the specialized register of the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group. As this source was no longer being maintained when we conducted the review, we executed bespoke strategies of PubMed, Embase.com, CENTRAL, CINAHL, LILACS and trial registers instead.
10) Types of studies. In the review, we specified that we excluded RCTs that compared treatment‐switch strategy vs continuing treatment.
11) Types of interventions. In the review, we specified that we excluded interventions administered by a route not approved and not used in clinical practice.
Contributions of authors
Concept development ‐ GF, IT, MDB.
Title registration ‐ GF, IT, MDB, GC, SF, MGL, MC, GV, VP, EL.
Drafting of protocol ‐ IT, MDB.
Editing of protocol ‐ GF, IT, MDB, GC, SF, MGL, MC, GV, VP, EL.
Title and abstract review: GC, SF, MGL, MC.
Search methods and record screening: RF.
Data abstraction: GC, SF, MGL, MC, IT.
Data entry: GC, SF, MGL, MC, IT.
Data analysis: GV, VP, EL.
Data interpretation: GF, IT, MDB, GC, SF, MGL, MC, GV, VP, EL.
Drafting the review: GV.
Editing and revising the review drafts: GF, IT, VP, RF (search‐related content).
Commenting on the review drafts: MDB, GC, SF, MGL, MC, GV, VP, EL.
All authors approved the final version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Sources of support
Internal sources
-
Fondazione I.R.C.C.S. Istituto Neurologico Carlo Besta, Milan, Italy
The Neurological Institute Carlo Besta hosted and supported the Editorial Base of the Group up to June 2020
External sources
-
No sources of support supplied, Italy
No sources of support supplied
Declarations of interest
IT ‐ none known.
GV ‐ no relevant interests; published research on this topic; works as a health professional at University of Florence and AOU Careggi, Italy; since 1 Jan 2021, Queen's University Belfast; local PI of the RHINE trial on faricimab for DMO on behalf of the University of Florence and Florence Careggi Hospital (did not sign the contract and funding was not under direct control; Roche funded the multicentre trial and Florence was a trial site).
VP ‐ none known.
EL ‐ none known.
MDB ‐ none known.
MC ‐ none known.
GC ‐ none known.
SF ‐ none known.
MGL ‐ none known.
RF ‐ financial assistance with personal prescription costs from Biogen Inc. (2018 to 2022); former member of Cochrane Central Editorial Service.
GF ‐ no relevant interests; Co‐ordinating Editor of Cochrane Multiple Sclerosis and Rare Diseases of the CNS but was excluded from the editorial and decision‐making process of this review.
These authors should be considered joint first author
New
References
References to studies included in this review
Achiron 1998 {published data only}
- Achiron A, Gabbay U, Gilad R, Hassin B, Barak Y, Gornish M, et al. Intravenous immunoglobulin treatment in multiple sclerosis. Effect on relapses. Neurology 1998;50(2):398-402. [Google Scholar]
Achiron 2004 {published data only}
- Achiron A, Kishner I, Sarova-Pinhas I, Raz H, Faibel M, Stern Y, et al. Intravenous immunoglobulin treatment following the first demyelinating event suggestive of multiplesclerosis: a randomized, double-blind, placebo-controlled trial. Archives of Neurology 2004;61(10):1515-20. [Google Scholar]
ADVANCE 2014 {published data only}
- Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J, et al. Pegylated interferon beta-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. The Lancet Neurology 2014;13:657-65. Supplementary webappendix; p. 1-16. [Google Scholar]
- NCT00906399. Efficacy and safety study of Peginterferon beta-1a in participants with relapsing multiple sclerosis (ADVANCE). clinicaltrials.gov/show/NCT00906399 (first posted May 21, 2009).
- Newsome SD, Guo S, Altincatal A, Proskorovsky I, Kinter E, Phillips G, et al. Impact of peginterferon beta-1a and disease factors on quality of life in multiple sclerosis. Multiple Sclerosis and Related Disorders 2015;4(4):350-7. [Google Scholar]
AFFIRM 2006 {published data only}
- Lublin FD, Cutter G, Giovannoni G, Pace A, Campbell NR, Belachew S. Natalizumab reduces relapse clinical severity and improves relapse recovery in MS. Multiple Sclerosis and Related Disorders 2014;3(6):705-11. [Google Scholar]
- NCT00027300. Safety and efficacy of natalizumab in the treatment of multiple sclerosis [A randomized, double-blind, placebo-controlled, parallel-group, multicenter study to determine the safety and efficacy of natalizumab in subjects with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00027300 (first received 3 December 2001).
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. New England Journal of Medicine 2006;354(9):899-910. [Google Scholar]
ALLEGRO 2012 {published data only}
- Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. New England Journal of Medicine 2012;366(11):1000-9. Supplementary Appendix; Trial protocol; p. 1-352. [Google Scholar]
- NCT00509145. Safety and efficacy of orally administered laquinimod versus placebo for treatment of relapsing remitting multiple sclerosis (RRMS) (ALLEGRO) [A multinational, multicenter, randomized, double-blind, parallel-group, placebo-controlled study, to evaluate the safety, tolerability and efficacy of daily oral administration of Laquinimod 0.6 mg in subjects with RRMS]. clinicaltrials.gov/show/NCT00509145 (first received 31 July 2007).
Andersen 2004 {published data only}
- Andersen O, Elovaara I, Farkkila M, Hansen HJ, Mellgren SI, Myhr KM, et al. Multicentre, randomised, double blind, placebo controlled, phase III study of weekly, low dose, subcutaneous interferon beta-1a in secondary progressive multiple sclerosis. Journal of Neurology, Neurosurgery and Psychiatry 2004;75(5):706-10. [Google Scholar]
APEX 2019 {published data only}
- Kondo T, Kawachi I, Onizuka Y, Hiramatsu K, Hase M, Yun J, et al. Efficacy of dimethyl fumarate in Japanese multiple sclerosis patients: interim analysis of randomized, double-blind APEX study and its open-label extension. Multiple Sclerosis Journal – Experimental, Translational and Clinical 2019;5(3):2055217319864974. [Google Scholar]
- Mori M, Ohashi T, Onizuka Y, Hiramatsu K, Hase M, Yun J, et al. Efficacy and safety of delayed-release dimethyl fumarate in treatment-naïve Japanese patients with relapsing-remitting multiple sclerosis: A post-hoc subgroup analysis of the APEX study. Journal of the Neurological Sciences 2017;381:795-6. [Google Scholar]
- Mori M, Ohashi T, Onizuka Y, Hiramatsu K, Hase M, Yun J, et al. Efficacy and safety of dimethyl fumarate in Japanese MS patients who had the history of treatment with fingolimod: APEX part 1+2 interim analysis. Multiple Sclerosis 2019;25(3):457-8. [Google Scholar]
- Mori M, Ohashi T, Onizuka Y, Hiramatsu K, Hase M, Yun J, et al. Efficacy and safety of dimethyl fumarate in treatment-naïve Japanese patients with multiple sclerosis: Interim analysis of the randomized placebo-controlled study. Multiple Sclerosis Journal – Experimental, Translational and Clinical 2019;5(2):2055217319852727. [Google Scholar]
- NCT01838668. An efficacy and safety study of BG00012 (dimethyl fumarate) in Asian subjects with relapsing remitting multiple sclerosis (RRMS) [A multicenter, randomized, double-blind, placebo-controlled, efficacy and safety study of BG00012 in subjects from the Asia-Pacific region and other countries with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT01838668 (first received 24 April 2013).
- Ochi H, Niino M, Onizuka Y, Hiramatsu K, Hase M, Yun J, et al. 72-week safety and tolerability of dimethyl fumarate in Japanese patients with relapsing-remitting multiple sclerosis: analysis of the randomised, double blind, placebo-controlled, phase III APEX study and its open-label extension. Advances in Therapy 2018;35(10):1598-611. [Google Scholar]
- Saida T, Yamamura T, Kondo T, Yun J, Yang M, Li J, et al. A randomized placebo-controlled trial of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis from East Asia and other countries. BMC Neurology 2019;19(1):5. Appendix 1; p. 1-4. [Google Scholar]
APOLITOS 2021 {published data only}
- Kira JI, Nakahara J, Sazonov DV, Kurosawa T, Tsumiyama I, Willi R, et al. Effect of ofatumumab versus placebo in relapsing multiple sclerosis patients from Japan and Russia: Phase 2 APOLITOS study. Multiple sclerosis (Houndmills, Basingstoke, England) 2022;28(8):1229-38. Supplemental material [Figure e-1; Table e-1].
- NCT03249714. Efficacy and safety of ofatumumab compared to placebo in patients with relapsing multiple sclerosis followed by extended treatment with open-label ofatumumab [A 24-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter study to evaluate the efficacy, safety and pharmacokinetics of Ofatumumab in patients with relapsing multiple sclerosis followed by an extended treatment of at least 24 weeks with open-label Ofatumumab i]. clinicaltrials.gov/show/NCT03249714 (first received 15 August 2017).
ARPEGGIO 2020 {published data only}
- Giovannoni G, Barkhof F, Hartung HP, Cree B, Krieger S, Montalban X, et al. Arpeggio: a placebo-controlled trial of oral laquinimod in primary progressive multiple sclerosis. Neurology 2018;90(15 Supplement):S8.003. [Google Scholar]
- Giovannoni G, Knappertz V, Steinerman JR, Tansy AP, Li T, Krieger S, et al. A randomized, placebo-controlled, phase 2 trial of laquinimod in primary progressive multiple sclerosis. Neurology 2020;95(8):e1027-40. Figure e-2. [Google Scholar]
- NCT02284568. A phase 2 clinical study in subjects with primary progressive multiple sclerosis to assess the efficacy, safety and tolerability of two oral doses of laquinimod either of 0.6 mg/day or 1.5mg/day (experimental drug) as compared to placebo (ARPEGGIO) [A 24-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter study to evaluate the efficacy, safety and pharmacokinetics of Ofatumumab in patients with relapsing multiple sclerosis followed by an extended treatment of at least 24 weeks with open-label Ofatumumab]. clinicaltrials.gov/show/NCT02284568 (first received 6 November 2014).
ASCEND 2018 {published data only}
- Cano S, Cleanthous S, Marquis P, Petrillo J, Steiner D, Watson C, et al. Measuring the impact of secondary progressive multiple sclerosis (Spms) in the Ascend trial: equating the Msis-29, Msws-12, Abilhand-56 and Sf-36. Value Health 2015;18(7):A713. [Google Scholar]
- Kapoor R, Ho PR, Campbell N, Chang I, Deykin A, Forrestal F, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurology 2018;17(5):405-15. Appendix, p. 1-14. [Google Scholar]
- NCT01416181. A clinical study of the efficacy of natalizumab on reducing disability progression in participants with secondary progressive multiple sclerosis (ASCEND in SPMS) [A multicenter, randomized, double-blind, placebo-controlled study of the efficacy of natalizumab on reducing disability progression in subjects with secondary progressive multiple sclerosis, with optional open-label extension]. clinicaltrials.gov/show/NCT01416181 (first received 12 August 2011).
ASCLEPIOS I 2020 {published data only}
- Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. New England Journal of Medicine 2020;383(6):546-57. Appendix; Figure S4. Protocol; p. 1-61. [Google Scholar]
- NCT02792218. Efficacy and safety of Ofatumumab compared to Teriflunomide in patients with relapsing multiple sclerosis (ASCLEPIOS I) [A randomized, double-blind, double-dummy, parallel-group study comparing the efficacy and safety of Ofatumumab versus Teriflunomide in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT02792218 (first received 7 June 2016).
ASCLEPIOS II 2020 {published data only}
- Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. New England Journal of Medicine 2020;383(6):546-57. Figure S4. [Google Scholar]
- NCT02792231. Efficacy and safety of ofatumumab compared to teriflunomide in patients with relapsing multiple sclerosis. (ASCLEPIOS II) [A randomized, double-blind, double-dummy, parallel-group study comparing the efficacy and safety of Ofatumumab versus Teriflunomide in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT02792231 (first received 7 June 2016).
Ashtari 2011 {published data only}
- Ashtari F, Savoj MR. Effects of low dose methotrexate on relapsing-remitting multiple sclerosis in comparison to Interferon β-1α: A randomized controlled trial. Journal of Research in Medical Sciences 2011;16(4):457-62. [Google Scholar]
ASSESS 2020 {published data only}
- Cree BAC, Goldman MD, Corboy JR, Singer BA, Fox EJ, Arnold DL, et al. Efficacy and safety of 2 fingolimod doses vs glatiramer acetate for the treatment of patients with relapsing-remitting multiple sclerosis: a randomized clinical trial. JAMA Neurology 2020;78(1):1-13. Appendix, [Protocol]; p.1-96. [Google Scholar]
- NCT01633112. MS study evaluating safety and efficacy of two doses of fingolimod versus copaxone (ASSESS) [A 12-month, randomized, rater- and dose-blinded study to compare the efficacy and safety of Fingolimod 0.25 mg and 0.5 mg administered orally once daily with Glatiramer acetate 20 mg administered subcutaneously once daily in patients with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT01633112 (first received 4 July 2012).
AVANTAGE 2013 {published data only}
- International Conference on Harmonisation (ICH GCP). The AVANTAGE study - A randomized, multicenter, phase iv, open-label prospective study comparing injection site reaction and injection site pain in patients with relapsing remitting multiple sclerosis (RRMS) or after a first demyelinating event suggestive of MS newly started on interferon beta-1b (Betaferon®) or interferon beta-1a (Rebif®). https://ichgcp.net/clinical-trials-registry/NCT00317941 (accessed 6 June 2022) 2013.
- NCT00317941. Safety study in relapsing-remitting multiple sclerosis (RRMS) patients receiving Betaferon or Rebif [The AVANTAGE study - A randomized, multicenter, phase iv, open-label prospective study comparing injection site reaction and injection site pain in patients with relapsing remitting multiple sclerosis (RRMS) or after a first demyelinating event suggestive of MS newly started on interferon beta-1b (Betaferon®) or interferon beta-1a (Rebif®)]. ClinicalTrials.gov: NCT00317941 (first received: 25 April 2006).
BECOME 2009 {published data only}
- Cadavid D, Wolansky LJ, Skurnick J, Lincoln J, Cheriyan J, Szczepanowski K, et al. Efficacy of treatment of MS with IFNbeta-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology 2009;72(23):1976-83. [Google Scholar]
- NCT00176592. Phase IV study, betaseron versus copaxone for relapsing remitting or CIS forms of MS using triple dose Gad 3 T MRI (BECOME) [Phase IV, rater-blinded, randomized study, comparing 250 mg of Betaseron with 20 mg of Copaxone in patients with the relapsing-remitting(RR) or CIS forms of MS using 3 tesla (3t) magnetic resonance imaging (MRI) with triple-dose gadolinium]. clinicaltrials.gov/show/NCT00176592 (first received 15 September 2005).
BENEFIT 2006 {published data only}
- Kappos L, Polman CH, Freedman MS, Edan G, Hartung HP, Miller DH, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006;67(7):1242-9. [Google Scholar]
BEYOND 2009 {published data only}
- NCT00099502. BEYOND: Betaferon/Betaseron efficacy yielding outcomes of a new dose in multiple sclerosis (MS) patients [International, randomized, multicenter, phase IIIB study in patients with relapsing-remitting multiple sclerosis comparing over a treatment period of at least 104 weeks: 1. double-blinded safety, tolerability, and efficacy of Betaseron/Betaferon 250 µg (8 miu) and Betaseron/Betaferon 500 µg (16 MIU), both given subcutaneously every other day, and 2. rater-blinded safety, tolerability, and efficacy of Betaseron/Betaferon s.c. every other day with Copaxone 20 mg s.c. once daily]. clinicaltrials.gov/show/NCT00099502 (first received 16 December 2004).
- O'Connor P, Filippi M, Arnason B, Comi G, Cook S, Goodin D, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurology 2009;8(10):889-97. [Google Scholar]
- Schippling S, O'Connor P, Knappertz V, Pohl C, Bogumil T, Suarez G, et al. Incidence and course of depression in multiple sclerosis in the multinational BEYOND trial. Journal of Neurology 2016;263(7):1418-26. [Google Scholar]
BOLD 2013 {published data only}
- NCT00879658. Safety, tolerability, efficacy and optimal dose finding study of BAF312 in patients with relapsing-remitting multiple sclerosis [A phase II, double-blind, randomized, multi-center, adaptive dose-ranging, placebo-controlled, parallel-group study evaluating safety, tolerability and efficacy on MRI lesion parameters and determining the dose response curve of BAF312 given orally once daily in patients with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov: NCT00879658 (first received 10 April 2009).
- Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. The Lancet. Neurology 2013;12(8):756-67. [Google Scholar]
Bornstein 1987 {published data only}
- Bornstein MB, Miller A, Slagle S, Weitzman M, Crystal, Drexler E, et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. New England Journal of Medicine 1987;317(7):408-14.
Bornstein 1991 {published data only}
- Bornstein MB, Miller A, Slagle S, Weitzman M, Drexler E, Keilson M. A placebo-controlled, double-blind, randomised, two-center, pilot trial of Cop 1 in chronic progressive multiple sclerosis. Neurology 1991;41:533-9. [Google Scholar]
Boyko 2016 {published and unpublished data}
- Boyko AN, Lashch NY, Sharanova SN, Zakharova MN, Trifonova OV, Simaniv TO, et al. Comparative, placebo-controlled clinical study of efficacy and safety of glatiramer acetate 20 mg in patients with relapsing-remitting multiple sclerosis: results of the first year of the study [Sravnitel'noe platsebo-kontroliruemoe klinicheskoe issledovanie effektivnosti i bezopasnosti preparatov glatiramera atsetata 20 mg u patsientov s remittiruyushchim rasseyannym sklerozom: rezul'taty pervogo goda nablyudeniya]. Zhurnal Nevrologii I Psikhiatrii Imeni S.S. Korsakova 2016;116(10):61-7. [Google Scholar]
- NCT02753088. Efficacy and safety of BCD-063 and Copaxone-Teva in patients with relapsing-remitting multiple sclerosis [International, multicentre, double-blind, placebo-controlled, comparative, randomized study to compare efficacy and safety of the generic drug BCD-063 (CJSC "BIOCAD", Russia) and Copaxone®-Teva ("Teva Pharmaceutical Industries Limited", Israel) in patients with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT02753088 (first received 27 April 2016).
Boyko 2017 {published data only}
- Boyko AN, Bosenko LP, Vasilovskiy VV, Volkova LI, Zakharova MN, Kotov SV, et al. A comparative placebo-controlled clinical study on the efficacy and safety of interferon beta-1a for subcutaneous injections in patients with remitting multiple sclerosis: results of the first year of observations [Sravnitel'noe platsebo-kontroliruemoe klinicheskoe issledovanie éffektivnosti i bezopasnosti preparatov interferona beta-1a dlia podkozhnogo vvedeniia u patsientov s remittiruiushchim rasseiannym sklerozom: rezul'taty pervogo goda nabliudeniia]. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova 2017;117(2):107-13. [Google Scholar]
BPSM 1995 {published data only}
- Ciccone A, Beretta S, Brusaferri F, Galea I, Protti A, Spreafico C. Corticosteroids for the long-term treatment in multiple sclerosis. Cochrane Database of Systematic Reviews 2008, Issue 1. Art. No: CD006264. [DOI: 10.1002/14651858.CD006264.pub2] [DOI] [Google Scholar]
BRAVO 2014 {published data only}
- NCT00605215. BRAVO study: Laquinimod double blind placebo controlled study in RRMS patients with a rater blinded reference arm of Interferon β-1a (Avonex®) (BRAVO) [A multinational, multicenter, randomized, parallel-group study performed in subjects with relapsing-remitting multiple sclerosis (RRMS) to assess the efficacy, safety and tolerability of Laquinimod over placebo in a double-blind design and of a reference arm of Interferon β-1a (Avonex®) in a rater-blinded design]. clinicaltrials.gov/show/NCT00605215 (first received 30 January 2008).
- Nakamura K, Vollmer TL, Gorfine T, Knappertz V, Arnold DL. Effect of laquinimod on gray matter and white matter atrophy in relapsing-remitting multiple sclerosis: Analysis of the BRAVO phase III trial. Multiple Sclerosis 2014;20(1):84-5. [Google Scholar]
- Vollmer TL, Sorensen PS, Selmaj K, Zipp F, Havrdova E, Cohen JA, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. Journal of Neurology 2014;261(4):773-83. Appendix, p. 1-10. [Google Scholar]
British and Dutch 1988 {published data only}
- The British and Dutch MSATG. Double-masked trial of azathioprine in multiple sclerosis. Lancet 1988;2(8604):179-83. [Google Scholar]
Calabrese 2012 {published data only}
- Calabrese M, Bernardi V, Atzori M, Mattisi I, Favaretto A, Rinaldi F, et al. Effect of disease-modifying drugs on cortical lesions and atrophy in relapsing-remitting multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2012;18(4):418-24. [Google Scholar]
CAMMS223 2008 {published data only}
- CAMMS223 TI, Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. New England Journal of Medicine 2008;359(17):1786-801. [Google Scholar]
- Fox EJ, Wynn D, Coles AJ, Palmer J, Margolin DH. ALEMTUZUMAB improves neurological functional systems in treatment-naive relapsing-remitting multiple sclerosis patients. Journal of the Neurological Sciences 2016;363:188-94. [Google Scholar]
- NCT00050778. A phase II study comparing low- and high-dose alemtuzumab and high-dose Rebif® in patients with early, active relapsing-remitting multiple sclerosis [A phase II, randomized, open-label, three-arm study comparing low- and high-dose Alemtuzumab and high-dose subcutaneous Interferon beta-1a (Rebif®) in patients with early, active relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00050778 (first received 23 December 2002).
CARE‐MS I 2012 {published data only}
- Arnold DL, Fisher E, Brinar VV, Cohen JA, Coles AJ, Giovannoni G, et al. Superior MRI outcomes with ALEMTUZUMAB compared with subcutaneous INTERFERON beta-1a in MS. Neurology 2016;87(14):1464-72. [Google Scholar]
- Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012;380(9856):1819-28. [Google Scholar]
- Krieger S, Lubetzki C, Palmer J, Margolin DH. Alemtuzumab reduces disease activity in treatment naive patients with highly active relapsing-remitting multiple sclerosis. Multiple Sclerosis 2014;20(1):106-7. [Google Scholar]
- Montalban X, Arnold DL, Fisher E, Margolin DH, Palmer J. Improvement in MRI outcomes across subgroups with alemtuzumab versus interferon beta-1a in treatment naive relapsing-remitting multiple sclerosis. Multiple Sclerosis 2014;20(1):83-4. [Google Scholar]
- NCT00530348. Comparison of Alemtuzumab and Rebif® efficacy in multiple sclerosis, study one (CARE-MS I) [A phase 3 randomized, rater-blinded study comparing two annual cycles of intravenous Alemtuzumab to three-times weekly subcutaneous Interferon beta-1a (Rebif®) in treatment-naïve patients with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00530348 (first received 17 September 2007).
CARE‐MS II 2012 {published data only}
- Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012;380(9856):1829-39. [Google Scholar]
- NCT00548405. Comparison of alemtuzumab and Rebif® efficacy in multiple sclerosis, study two (CARE-MS II) [A phase 3, randomized, rater- and dose-blinded study comparing two annual cycles of intravenous low- and high-dose Alemtuzumab to three-times weekly subcutaneous Interferon beta 1a (Rebif®) in patients with relapsing remitting multiple sclerosis who have relapsed on therapy]. clinicaltrials.gov/show/NCT00548405 (firstreceived 24 October 2007).
- Steinman L, Wang H, Liu Y, Palmer J, Zhang Q. Defining clinical meaning of patient-reported outcomes with disability assessment in multiple sclerosis: An analysis of the CARE-MS II study. Multiple Sclerosis 2014;20(1):419. [Google Scholar]
CCMSSG 1991 {published data only}
- CCMSSG. The Canadian cooperative trial of cyclophosphamide and plasma exchange in progressive multiple sclerosis. Lancet 1991;337(8739):441-6. [Google Scholar]
CHAMPS 2000 {published data only}
- Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. New England Journal of Medicine 2000;343(13):898-904. [Google Scholar]
Cheshmavar 2021 {published data only}
- Cheshmavar M, Mirmosayyeb O, Badihian N, Badihian S, Shaygannejad V. Rituximab and glatiramer acetate in secondary progressive multiple sclerosis: A randomized clinical trial. Acta Neurologica Scandinavica 2021;143(2):178-87. [Google Scholar]
- NCT03315923. Comparison of clinical effects of rituximab and glatiramer acetate in secondary progressive multiple sclerosis patients [Comparison of expanded disability status scale, GAD-enhanced brain lesions, annualized relapse rate, and side effects between active secondary progressive multiple sclerosis patients on Rituximab and Glatiramer acetate]. clinicaltrials.gov/show/NCT03315923 (first received 20 October 2017).
CLARITY 2010 {published data only}
- Cook S, Leist T, Comi G, Montalban X, Giovannoni G, Nolting A, et al. Safety of cladribine tablets in the treatment of patients with multiple sclerosis: An integrated analysis. Multiple Sclerosis and Related Disorders 2019;29:157-67. [Google Scholar]
- Cook S, Leist T, Comi G, Montalban X, Sylvester E, Hicking C, et al. Infections during periods of grade 3 or 4 lymphopenia in patients taking CLADRIBINE tablets 3.5 mg/kg: Data from an integrated safety analysis. Multiple Sclerosis 2017;23(3):599. [Google Scholar]
- Cook S, Vermersch P, Comi G, Giovannoni G, Rammohan K, Rieckmann P, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Multiple Sclerosis (Houndmills, Basingstoke, England) 2011;17(5):578-93. [Google Scholar]
- Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sørensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):416-26. Appendix, p. 1-17. [Google Scholar]
- NCT00213135. A safety and efficacy study of oral cladribine in subjects with relapsing-remitting multiple sclerosis (RRMS) (CLARITY) [A phase III, randomized, double-blind, three-arm, placebo-controlled, multi-center study to evaluate the safety and efficacy of oral Cladribine in subjects with relapsing-remitting multiple sclerosis (RRMS)]. clinicaltrials.gov/show/NCT00213135 (first received 21 September 2005).
- Rammohan K, Comi G, Cook S, Giovannoni G, Rieckmann P, Soelberg S, et al. Consistent efficacy of short-course CLAdRIbine tablets therapy across differing prognostic indicators for relapsing-remitting multiple sclerosis: Results from the phase III, double-blind, placebo-controlled, 96-week CLARITY study. Multiple Sclerosis 2010;16(10):S146. [Google Scholar]
- Rammohan K, Comi G, Cook S, Giovannoni G, Rieckmann P, Soelberg S, et al. Efficacy of cladribine tablets for relapsing-remitting multiple sclerosis in patients with high disease activity: Results from the phase III, 96-week CLARITY study. Journal of Neurology 2011;258:S21. [Google Scholar]
- Rammohan K, Giovannoni G, Comi G, Cook S, Rieckmann P, Soelberg S, et al. Cladribine tablets for relapsing-remitting multiple sclerosis: Efficacy across patient subgroups from the phase III CLARITY study. Multiple Sclerosis and Related Disorders 2012;1(1):49-54. [Google Scholar]
- Rieckmann P, Comi G, Cook S, Giovannoni G, Rammohan K, Soelberg S, et al. Consistent efficacy of cladribine tablets across the spectrum of patients with relapsing-remitting multiple sclerosis of differing severity: Data from the 96-week, double-blind CLARITY study. European Journal of Neurology 2010;17:56. [Google Scholar]
- Rieckmann P, Giovannoni G, Comi G, Cook S, Rammohan K, Sorensen P, et al. Efficacy of short-course oral therapy with cladribine tablets for relapsing-remitting multiple sclerosis in the 96-week, Phase III, double-blind, placebo-controlled CLARITY study. Multiple Sclerosis 2010;16(10):1283. [Google Scholar]
- Rieckmann P, Soelberg S, Sorensen P, Comi G, Cook S, Giovannoni G, et al. Exploring correlations between changes in lymphocyte counts and clinical/magnetic resonance imaging outcomes in cladribine-treated patients with relapsing-remitting multiple sclerosis: Analyses from the double blind, 96-week CLARITY study. Journal of Neurology 2011;258:S48. [Google Scholar]
- Vermersch P, Comi G, Cook S, Giovannoni G, Rammohan K, Rieckmann P, et al. Tolerability and retention on treatment with cladribine tablets for relapsing-remitting multiple sclerosis over 96 weeks in the phase III, double-blind, placebo-controlled CLARITY study. Multiple Sclerosis 2010;16(10):S298-9. [Google Scholar]
- Vermersch P, Comi G, Cook S, Giovannoni G, Rammohan K, Rieckmann P, et al. Tolerability profile of cladribine tablets therapy for patients with relapsing-remitting multiple sclerosis: Factors contributing to treatment completion overall and in patients with high disease activity in the 96-week CLARITY study. Journal of Neurology 2011;258:S259. [Google Scholar]
CombiRx 2013 {published data only}
- Lindsey J, Scott T, Lynch S, Cofield S, Nelson F, Conwit R, et al. The CombiRx trial of combined therapy with interferon and glatiramer cetate in relapsing remitting MS: Design and baseline characteristics. Multiple Sclerosis and Related Disorders 2012;1(2):81-6. [Google Scholar]
- Lublin FD, Cofield SS, Cutter GR, Conwit R, Narayana PA, Nelson F, et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Annals of Neurology 2013;73(3):327-40. Table, [Serious Adverse Events (Safety Population)]; p. 24-25. [Google Scholar]
- NCT00211887. Combination therapy in patients with relapsing-remitting multiple sclerosis (MS) CombiRx [A multi-center, double-blind, randomized study comparing the combined use of Interferon beta-1a and Glatiramer acetate to either agent alone in patients with relapsing-remitting multiple sclerosis (CombiRx)]. clinicaltrials.gov/show/NCT00211887 (first received 21 September 2005).
Comi 2001 {published data only}
- Comi G, Filippi M, Wolinsky JS. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging--measured disease activity and burden in patients with relapsing multiple sclerosis. European/Canadian Glatiramer Acetate Study Group. Annals of Neurology 2001;49(3):290-7. [Google Scholar]
Comi 2008 {published data only}
- Comi G, Pulizzi A, Rovaris M, Abramsky O, Arbizu T, Boiko A, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008;371(9630):2085-92. [Google Scholar]
- NCT00349193. A study to evaluate the effectiveness, tolerability and safety of laquinimod [A multinational, multicenter randomized double-blind, parallel-group, placebo-controlled study, to evaluate the efficacy, tolerability and safety of two doses of, oral Laquinimod in relapsing remitting (R-R) multiple sclerosis (MS) subjects]. clinicaltrials.gov/show/NCT00349193 (first received 6 July 2006).
CONCERTO 2022 {published data only}
- Comi G, Dadon Y, Sasson N, Steinerman JR, Knappertz V, Vollmer TL, et al. CONCERTO: a randomized, placebo-controlled trial of oral laquinimod in relapsing-remitting multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2022;28(4):608-19. [Google Scholar]
- NCT01707992. The efficacy, safety, and tolerability of Laquinimod in participants with relapsing remitting multiple sclerosis (RRMS) (CONCERTO) [A multinational, multicenter, randomized, double-blind, parallel-group, placebo-controlled study followed by an active treatment period, to evaluate the efficacy, safety and tolerability of two doses of oral administration of Laquinimod (0.6 mg/day or 1.2 mg/day) in patients with relapsing remitting multiple sclerosis (RRMS)]. clinicaltrials.gov/show/NCT01707992 (first received 16 October 2012).
CONFIRM 2012 {published data only}
- Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. New England Journal of Medicine 2012;367(12):1087-97. Appendix, Protocol; p. 1-34. [Google Scholar]
- Miller DH, Fox RJ, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Effects of delayed-release dimethyl fumarate on MRI measures in the phase 3 CONFIRM study. Neurology 2015;84(11):1145-52. [Google Scholar]
- NCT00451451. Efficacy and safety study of oral BG00012 with active reference in relapsing-remitting multiple sclerosis (CONFIRM) [A randomized, multicenter, placebo-controlled and active reference (Glatiramer acetate) comparison study to evaluate the efficacy and safety of BG00012 in subjects with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00451451 (first received 23 March 2007).
DECIDE 2015 {published data only}
- Benedict RH, Cohan S, Lynch SG, Riester K, Wang P, Castro-Borrero W, et al. Improved cognitive outcomes in patients with relapsing-remitting multiple sclerosis treated with Daclizumab beta: Results from the DECIDE study. Multiple Sclerosis 2018;24(6):795-804. [Google Scholar]
- Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. New England Journal of Medicine 2015;373(15):1418-28. Supplementary Appendix; p. 1-36. [Google Scholar]
- Krueger JG, Kircik L, Hougeir F, Friedman A, You X, Lucas N, et al. Cutaneous adverse events in the randomized, double-blind, active-comparator DECIDE study of daclizumab high-yield process versus intramuscular interferon beta-1a in relapsing-remitting multiple sclerosis. Advances in Therapy 2016;33(7):1231-45. [Google Scholar]
- NCT01064401. Efficacy and safety of BIIB019 (daclizumab high yield process) versus interferon β 1a in participants with relapsing-remitting multiple sclerosis (DECIDE) [Multicenter, double-blind, randomized, parallel-group, monotherapy, active-control study to determine the efficacy and safety of Daclizumab high yield process (DAC HYP) versus Avonex® (interferon β 1a) in patients with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT01064401 (first received 8 February 2010).
DEFINE 2012 {published data only}
- Gold R, Arnold DL, Bar-Or A, Fox RJ, Kappos L, Chen C, et al. Safety and efficacy of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: 9 years' follow-up of DEFINE, CONFIRM, and ENDORSE. Therapeutic Advances in Neurological Disorders 2020;13:1756286420915005. [Google Scholar]
- Gold R, Giovannoni G, Phillips JT, Fox RJ, Yang L, Taylor C. Efficacy of delayed-release dimethyl FUMARATE in newly diagnosed patients with relapsing-remitting multiple sclerosis: Eight-year follow-up of an integrated analysis of DEFINE, CONFIRM, and ENDORSE. Multiple Sclerosis 2017;23(3):313-4. [Google Scholar]
- Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. New England Journal of Medicine 2012;367(12):1098-107. Appendix Protocol; p. 1-88. [Google Scholar]
- NCT00420212. Efficacy and safety of oral BG00012 in relapsing-remitting multiple sclerosis (DEFINE) [A randomized, multicenter, double-blind, placebo-controlled, dose-comparison study to determine the efficacy and safety of BG00012 in subjects with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00420212 (first received 11 January 2007).
Ellison 1989 {published data only}
- Ellison G, Myers L, Mickey M, Graves M, Tourtellotte W, Syndulko K, et al. A placebo-controlled, randomized, double-masked, variable dosage, clinical trial of azathioprine with and without methylprednisolone in multiple sclerosis. Neurology 1989;39(8):1018-26. [Google Scholar]
Etemadifar 2006 {published data only}
- Etemadifar M, Janghorbani M, Shaygannejad V. Comparison of Betaferon, Avonex, and Rebif in treatment of relapsing-remitting multiple sclerosis. Acta Neurologica Scandinavica 2006;113(5):283-7. [Google Scholar]
Etemadifar 2007 {published data only}
- Etemadifar M, Janghorbani M, Shaygannejad V. Comparison of interferon beta products and azathioprine in the treatment of relapsing-remitting multiple sclerosis. Journal of Neurology 2007;254(12):1723-8. [Google Scholar]
ETOMS 2001 {published data only}
- Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Early Treatmentof Multiple Sclerosis Study Group. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet 2001;357(9268):1576-82. [Google Scholar]
European Study Group 1998 {published data only}
- European SG. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet 1998;352(9139):1491-7. [Google Scholar]
- Kappos L, Polman C, Pozzilli C, Thompson A, Beckmann K, Dahlke F, European Study Group in Interferon beta-1b in Secondary-Progressive MS. Final analysis of the European multicenter trial on IFNbeta-1b in secondary-progressive MS. Neurology 2001;57(11):1969-75. [Google Scholar]
EVIDENCE 2002 {published data only}
- Coyle PK, Reder AT, Freedman MS, Fang J, Dangond F. Early MRI results and odds of attaining 'no evidence of disease activity' status in MS patients treated with interferon β-1a in the EVIDENCE study. Journal of the Neurological Sciences 2017;379:151-6. [Google Scholar]
- NCT00292266. A study of Rebif® compared with Avonex® in the treatment of relapsing-remitting multiple sclerosis (MS) [An open label, randomized, multicenter, comparative, parallel group study of Rebif® 44 mcg administered three times per week by subcutaneous injection, compared with Avonex® 30 mcg administered once per week by intramuscular injection in the treatment of relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT00292266 (first received 15 February 2006).
- Panitch H, Goodin DS, Francis G, Chang P, Coyle PK, O'Connor P, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: The EVIDENCE Trial. Neurology 2002;59(10):1496-506. [Google Scholar]
- Schwid S, Panitch H. Full results of the Evidence of Interferon Dose-Response-European North American Comparative Efficacy (EVIDENCE) study: a multicenter, randomized, assessor-blinded comparison of low-dose weekly versus high-dose, high-frequency interferon beta-1a for relapsing multiple sclerosis. Clinical Therapeutics 2007;29(9):2031-48. [Google Scholar]
EVOLVE‐MS‐2 2020 {published data only}
- NCT03093324. A tolerability study of ALKS 8700 in subjects with relapsing remitting multiple sclerosis (RRMS) EVOLVE-MS-2 [A phase 3 study in subjects with relapsing remitting multiple sclerosis to evaluate the tolerability of ALKS 8700 and Dimethyl fumarate]. clinicaltrials.gov/show/NCT03093324 (first received 28 March 2017).
- Naismith RT, Wundes A, Ziemssen T, Jasinska E, Freedman MS, Lembo AJ, et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs 2020;34(2):185-96. [Google Scholar]
EXPAND 2018 {published data only}
- Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 2018;391(10127):1263-73. Supplementary appendix; p. 1-20. [Google Scholar]
- NCT01665144. Exploring the efficacy and safety of siponimod in patients with secondary progressive multiple sclerosis (EXPAND) [A multicenter, randomized, double-blind, parallel-group, placebo-controlled variable treatment duration study evaluating the efficacy and safety of Siponimod (BAF312) in patients with secondary progressive multiple sclerosis followed by extended treatment with open-label BAF312]. clinicaltrials.gov/show/NCT01665144 (first received 15 August 2012).
Fazekas 1997 {published data only}
- Fazekas F, Deisenhammer F, Strasser-Fuchs S, Nahler G, Mamoli B. Randomised placebo-controlled trial of monthly intravenous immunoglobulin therapy in relapsing-remitting multiple sclerosis. Lancet 1997;349(9052):589-93. [Google Scholar]
Fazekas 2008 {published data only}
- Fazekas F, Lublin F, Li D, Freedman M, Hartung H, Rieckmann P, et al. Intravenous immunoglobulin in relapsing-remitting multiple sclerosis: a dose-finding trial. Neurology 2008;71(4):265-71. [Google Scholar]
FREEDOMS 2010 {published data only}
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):387-401. Supplementary Appendix; p. 1-9. [Google Scholar]
- NCT00289978. Efficacy and safety of Fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS) [A 24-month, double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing the efficacy and safety of Fingolimod 1.25 mg and 0.5 mg administered orally once daily versus placebo in patients with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00289978 (first received 10 February 2006).
FREEDOMS II 2014 {published data only}
- Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurology 2014;13(6):545-56. Supplementary webappendix; p. 1-12. [Google Scholar]
- NCT00355134. Efficacy and safety of fingolimod (FTY720) in patients with relapsing-remitting multiple sclerosis (FREEDOMS II) [24-month double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing the efficacy and safety of 0.5 mg and 1.25 mg Fingolimod (FTY720) administered orally once daily versus placebo in patients with relapsing-remitting multiple sclerosis with optional extension phase]. clinicaltrials.gov/show/NCT00355134 (first received 21 July 2006).
- Radue EW, Sprenger T, Vera A, Francis G, Rochotte E, Tomic D, et al. Effect of fingolimod on evolution of baseline enhancing MRI lesions into persistent T1 hypointense lesions: Post hoc analysis of the FREEDOMS study. Multiple Sclerosis 2014;20(1):112-3. [Google Scholar]
FUMAPMS 2021 {published data only}
- Højsgaard Chow H, Talbot J, Lundell H, Gøbel Madsen C, Marstrand L, Lange T, et al. Dimethyl fumarate treatment in patients with primary progressive multiple sclerosis: a randomized, controlled trial. Neurology (R) Neuroimmunology & Neuroinflammation 2021;8(5):e1037. [Google Scholar]
- NCT02959658. Dimethyl fumarate treatment of primary progressive multiple sclerosis (FUMAPMS) [Dimethyl Fumarate treatment of primary progressive multiple sclerosis]. clinicaltrials.gov/show/NCT02959658 (first received 9 November 2016).
GALA 2013 {published data only}
- Davis MD, Ashtamker N, Steinerman JR, Knappertz V. Time course of glatiramer acetate efficacy in patients with RRMS in the GALA study. Neurology Neuroimmunology & Neuroinflammation 2017;4(2):e327. [Google Scholar]
- Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R, GALA Study Group. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Annals of Neurology 2013;73(6):705-13. Supplementary Table 3; Serious Adverse Events by System Organ Class, p.3. [Google Scholar]
GATE 2015 {published data only}
- Cohen J, Belova A, Selmaj K, Wolf C, Sormani MP, Oberyé J, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurology 2015;72(12):1433-41. Appendix 1, Protocol; p. 1-64. Appendix 2, [eMethods; eTables 1-5; eFigure]. [Google Scholar]
- Cohen J, Belova A, Selmaj K, Wolf C, Sormani MP, Oberyé J, et al. Multi-centre, randomized, double-blind, placebo-controlled, parallel-group, 9 month, equivalence trial comparing the efficacy and safety and tolerability of GTR (Synthon BV) to Copaxone® (Teva) in subjects with relapsing remitting multiple sclerosis followed by an open-label 15 month GTR treatment part evaluating the long-term GTR treatment effects. JAMA Neurology 2015;72(12):1433-41. Clinical trial protocol (version 4.0); p. 1-64.
- NCT01489254. Efficacy and safety of GTR in comparison to Copaxone® (GATE) [Multi-centre, randomized, double-blind, placebo-controlled, parallel-group, 9 month, equivalence trial comparing the efficacy and safety and tolerability of GTR (Synthon BV) to Copaxone® (Teva) in subjects with relapsing remitting multiple sclerosis followed by an open-label 15 month GTR treatment part evaluating the long-term GTR treatment]. clinicaltrials.gov/show/NCT01489254 (first received 9 December 2011).
Ghezzi 1989 {published data only}
- Ghezzi A, Di Falco M, Locatelli C. Clinical controlled randomized trial of azathioprine in multiple sclerosis. Elsevier, 1989. [Google Scholar]
GOLDEN 2017 {published data only}
- Comi G, Patti F, Rocca MA, Mattioli FC, Amato MP, Gallo P, et al. Efficacy of fingolimod and interferon beta-1b on cognitive, MRI, and clinical outcomes in relapsing-remitting multiple sclerosis: an 18-month,open-label, rater-blinded, randomised, multicentre study (the GOLDEN study). Journal of Neurology 2017;264(12):2436-49. [Google Scholar]
- NCT01333501. Fingolimod versus interferon beta 1b in cognitive symptoms (cognition) [A 18-month, open-label, rater-blinded, randomized, multi-center, active-controlled, parallel-group pilot study to assess efficacy and safety of Fingolimod in comparison to Interferon beta 1b in treating the cognitive symptoms associated to relapsing-remitting multiple sclerosis and to assess possible relationship of these effects to regional brain atrophy]. clinicaltrials.gov/show/NCT01333501 (first received 12 April 2011).
Goodkin 1991 {published data only}
- Goodkin D, Bailly R, Teetzen M, Hertsgaard D, Beatty W. The efficacy of azathioprine in relapsing-remitting multiple sclerosis. Neurology 1991;41:20-5. [Google Scholar]
Goodkin 1995 {published data only}
- Goodkin D, Rudick R, VanderBrug Medendorp S, Daughtry M, Schwetz K, Fischer J, et al. Low-dose (7.5 mg) oral methotrexate reduces the rate of progression in chronic progressive multiple sclerosis. Annals of Neurology 1995;37(1):30-40. [Google Scholar]
Hartung 2002 {published data only}
- Hartung H, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey S, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002;360(9350):2018-25. [Google Scholar]
Hauser 2008 {published data only}
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New England Journal of Medicine 2008;358(7):676-88. [Google Scholar]
- NCT00097188. A study to evaluate rituximab in adults with relapsing remitting multiple sclerosis [A phase II, randomized, double-blind, parallel-group, placebo-controlled, multicenter study to evaluate the safety and efficacy of Rituximab (Mabthera/Rituxan) in adults with relapsing remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00097188 (first received 19 November 2004).
Hommes 2004 {published data only}
- Hommes O, Sorensen P, Fazekas F, Enriquez M, Koelmel H, Fernandez O, et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo-controlled trial. Lancet 2004;364(9440):1149-56. [Google Scholar]
- Hommes OR, Maas-Enriquez M. ESIMS--an ongoing clinical trial in secondary progressive multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2000;6(Suppl 2):S27-32. [Google Scholar]
IFNB MS Group 1993 {published data only}
- IFNB MSG. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology 1993;43(4):655-61. [Google Scholar]
IMPACT 2002 {published data only}
- Cohen J, Cutter G, Fischer J, Goodman A, Heidenreich F, Kooijmans M, et al. Benefit of interferon beta-1a on MSFC progression in secondary progressive MS. Neurology 2002;59(5):679-87. [Google Scholar]
IMPROVE 2010 {published data only}
- De Stefano N, Curtin F, Stubinski B, Blevins G, Drulovic J, Issard D, et al. Rapid benefits of a new formulation of subcutaneous interferon beta-1a in relapsing-remitting multiple sclerosis. Multiple Sclerosis 2010;16(7):888-92. [Google Scholar]
- NCT00441103. A study to evaluate Rebif® new formulation (interferon-beta-1a) in relapsing remitting multiple sclerosis (IMPROVE) [A two-arm, randomized, double-blind, control group-compared, multicenter, phase IIIB study with monthly MRI and biomarker assessments to evaluate the efficacy, safety, and tolerability of Rebif® new formulation (IFN beta-1a) in subjects with relapsing remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00441103 (first received 28 February 2007).
INCOMIN 2002 {published data only}
- Durelli L, Verdun E, Barbero P, Bergui M, Versino E, Ghezzi A, et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet 2002;359:1453-60. [Google Scholar]
INFORMS 2016 {published data only}
- Fox E, Lublin F, Wolinsky J, Cohen J, Meng X, Ziehn M, et al. Analysis of lymphocyte counts and infection rates with fingolimod in patients with primary progressive multiple sclerosis over the INFORMS trial. Neurology 2018;90(15 Supplement):P1.387. [Google Scholar]
- Lublin F, Miller DH, Freedman MS, Cree BA, Wolinsky JS, Weiner H, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016;387(10023):1075-84. Supplementary appendix; p. 1-17. [Google Scholar]
- NCT00731692. This was an open-label, single-arm extension study (CFTY720D2306E1) to a double-blind, randomized multicenter, placebo-controlled, parallel-group core study (CFTY720D2306) in PPMS. (INFORMS) [A double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing the efficacy and safety of 0.5mg Fingolimod administered orally once daily versus placebo in patients with primary progressive multiple sclerosis and an open-label, single-arm extension study to the double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing the efficacy and safety of 0.5 mg FTY720 administered orally once daily versus placebo in patients with primary progressive multiple sclerosis]. clinicaltrials.gov/show/NCT00731692 (first received 11 August 2008).
Johnson 1995 {published data only}
- Johnson K, Brooks B, Cohen J, Ford C, Goldstein J, Lisak R, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995;45(7):1268-76. [Google Scholar]
- Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. 1998 [classical article]. Neurology 2001;57(12 Suppl 5):S46-53. [Google Scholar]
- Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology 1998;50(3):701-8. [Google Scholar]
Kappos 2006 {published data only}
- Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. New England Journal of Medicine 2006;355(11):1124-40. Supplementary Appendix; p. 1. [Google Scholar]
- NCT00333138. Efficacy and safety of FTY720 in patients with relapsing multiple sclerosis [Double-blind, randomized, placebo-controlled, parallel-group, multicenter study evaluating the safety, tolerability and effect on MRI lesion parameters of FTY720 vs placebo in patients with relapsing multiple sclerosis including 18-month extension phase]. clinicaltrials.gov/show/NCT00333138 (first received 2 June 2006).
Kappos 2008 {published data only}
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008;372(9648):1463-72. [Google Scholar]
- NCT00168701. Efficacy and safety of BG00012 in MS [Double-blind, placebo-controlled, dose-ranging study to determine the efficacy and safety of BG00012 in subjects with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/ NCT00168701 (first received 15 September 2005).
Kappos 2011 {published data only}
- Kappos L, Li D, Calabresi PA, O'Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis:a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011;378(9805):1779-87. [Google Scholar]
- NCT00676715. A study of the efficacy and safety of ocrelizumab in patients with relapsing-remitting multiple sclerosis [Phase II, multicenter, randomized, parallel-group, partially blinded, placebo and Avonex controlled dose finding study to evaluate the efficacy as measured by brain MRI lesions, and safety of 2 dose regimens of Ocrelizumab in patients with RRMS]. clinicaltrials.gov/show/NCT00676715 (first received 13 May 2008).
Knobler 1993 {published data only}
- Knobler R, Greenstein J, Johnson K, Lublin F, Panitch H, Conway K, et al. Systemic recombinant human interferon-beta treatment of relapsing-remitting multiple sclerosis: pilot study analysis and six-year follow-up. Journal of Interferon & Cytokine Research 1993;13:333-40. [Google Scholar]
Koch‐Henriksen 2006 {published data only}
- Koch-Henriksen N, Sørensen P, Christensen T, Frederiksen J, Ravnborg M, Jensen K, et al. A randomised study of two interferon-beta treatments in relapsing-remitting multiple sclerosis. Neurology 2006;66(7):1056-60. [Google Scholar]
Leary 2003 {published data only}
- Leary S, Miller D, Stevenson V, Brex P, Chard D, Thompson A. Interferon beta-1a in primary progressive MS: an exploratory, randomized, controlled trial. Neurology 2003;60(1):44-51. [Google Scholar]
Lewanska 2002 {published data only}
- Lewanska M, Siger-Zajdel M, Selmaj K. No difference in efficacy of two different doses of intravenous immunoglobulins in MS: clinical and MRI assessment. European Journal of Neurology 2002;9(6):565-72. [Google Scholar]
Likosky 1991 {published data only}
- Likosky W, Fireman B, Elmore R, Eno G, Gale K, Goode G, et al. Intense immunosuppression in chronic progressive multiple sclerosis: the Kaiser study. Journal of Neurology, Neurosurgery and Psychiatry 1991;54(12):1055-60. [Google Scholar]
MAIN TRIAL 2014 {published data only}
- EudraCT 2006-004937-13. Multicentee randomized controlled study of azathioprine versus iterferon beta in relapsing remitting multiple sclerosis. www.clinicaltrialsregister.eu/ctr-search/search?query=2006-004937-13 (first received 17 August 2006).
- Massacesi L, Tramacere I, Amoroso S, Battaglia MA, Benedetti MD, Filippini G, et al. Azathioprine versus beta interferons for relapsing-remitting multiple sclerosis: a multicentre randomized non-inferiority trial. PLoS One 2014;9(11):e113371. Tables S1 and S2; p. 1-3.
- Massacesi L, Tramacere I, Amoroso S, Battaglia MA, Benedetti MD, Filippini G, et al. Azathioprine versus beta interferons for relapsing-remitting multiple sclerosis: a multicentre randomized non-inferiority trial. PLoS One 2014;9(11):e113371. [Google Scholar]
- Massacesi L, Tramacere I, Amoroso S, Battaglia MA, Benedetti MD, Filippini G, et al. Multicenter randomized controlled study of Azathioprine versus Interferon beta in relapsing-remitting multiple sclerosis. PLoS One 2014;9(11):e113371. Study protocol; p. 1-32.
Masjedi 2021 {published data only}
- Masjedi SS, Etemadifar M, Zadeh NM, Afzali M. Assessment of fingolimod versus dimethyl fumarate for the treatment of multiple sclerosis; a 24-month follow-up study. American Journal of Clinical and Experimental Immunology 2021;10(3):86-92. [Google Scholar]
Milanese 1993 {published data only}
- Milanese C, La Mantia L, Salmaggi A, Eoli M. A double blind study on azathioprine efficacy in multiple sclerosis: final report. Journal of Neurology 1993;240(5):295-8. [Google Scholar]
Millefiorini 1997 {published data only}
- Millefiorini E, Gasperini C, Pozzilli C, D'Andrea F, Bastianello S, Trojano M, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. Journal of Neurology 1997;244(3):153-9. [Google Scholar]
Miller 1961 {published data only}
- Miller H, Newell D, Ridley A. Multiple sclerosis. Trials of maintenance treatment with prednisolone and soluble aspirin. Lancet 1961;1(7169):127-9. [Google Scholar]
Miller 2003 {published data only}
- Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. New England Journal of Medicine 2003;348(1):15-23. [Google Scholar]
MIRROR 2018 {published data only}
- Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology 2018;90(20):e1805-14. e-supplement; links.lww.com/WNL/A437. [Google Scholar]
- NCT01457924. Ofatumumab subcutaneous administration in subjects with relapsing-remitting multiple sclerosis (MIRROR) [A randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study to investigate the MRI efficacy and safety of six months' administration of Ofatumumab in subjects with relapsing-remitting multiple sclerosis (RRMS)]. clinicaltrials.gov/show/NCT01457924 (first received 24 October 2011).
- Sorenson PS, Kavanagh ST, Austin DJ, Grove RA, Lopez MC, Tolson JM, et al. Follow-up data from the Mirror study: A dose-ranging study of subcutaneous ofatumumab in subjects with relapsing-remitting multiple sclerosis. Multiple Sclerosis 2014;20(1):P048. [Google Scholar]
Mokhber 2014 {published data only}
- Mokhber N, Azarpazhooh A, Orouji E, Rao SM, Khorram B, Sahraian MA, et al. Cognitive dysfunction in patients with multiple sclerosis treated with different types of interferon beta: A randomized clinical trial. Journal of the Neurological Sciences 2014;342(1-2):16-20. [Google Scholar]
Montalban 2009 {published data only}
- Montalban X, Sastre-Garriga J, Tintore M, Brieva L, Aymerich F, Rio J, et al. A single-center, randomized, double-blind, placebo-controlled study of interferon beta-1b on primary progressive and transitional multiple sclerosis. Multiple Sclerosis 2009;15(10):1195-205. [Google Scholar]
Motamed 2007 {published data only}
- Motamed MR, Najimi N, Fereshtehnejad SM. The effect of interferon-beta1a on relapses and progression of disability in patients with clinically isolate syndromes (CIS) suggestive of multiple sclerosis. Clinical Neurology and Neurosurgery 2007;109(4):344-9. [Google Scholar]
MOVING 2020 {published data only}
- Albert C, Mikolajczak J, Liekfeld A, Piper SK, Scheel M, Zimmermann HG, et al. Fingolimod after a first unilateral episode of acute optic neuritis (MOVING) - preliminary results from a randomized, rater-blind, active-controlled, phase 2 trial. BMC Neurology 2020;20(1):75. [Google Scholar]
- Albert C, Mikolajczak J, Liekfeld A, Piper SK, Scheel M, Zimmermann HG, et al. Fingolimod after a first unilateral episode of acute optic neuritis (MOVING) - preliminary results from a randomized, rater-blind, active-controlled, phase 2 trial. BMC Neurology 2020;20(1):75. 2020;20(1):75. Additional file 1 Supplementary methods and results; p. 1-14.
- NCT01647880. MOdification of VIsual Outcomes after Optic Neuritis in CIS or MS by Gilenya (MOVING Study) [Phase II/III study to investigate the effects of Fingolimod versus Interferon beta-1b on visual recovery after optic neuritis]. clinicaltrials.gov/show/NCT01647880 (first received 24 July 2012).
MSCRG 1996 {published data only}
- Jacobs L, Cookfair D, Rudick R, Herndon R, Richert J, Salazar A, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Annals of Neurology 1996;39:285-94. [Google Scholar]
NASP 2004 {published data only}
- Panitch H, Miller A, Paty D, Weinshenker B. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology 2004;63(10):1788-95. [Google Scholar]
Noseworthy 2000 {published data only}
- Noseworthy JH, O'Brien PC, Weinshenker BG, Weis JA, Petterson TM, Erickson BJ, et al. IV immunoglobulin does not reverse established weakness in MS. Neurology 2000;55(8):1135-43. [Google Scholar]
O'Connor 2006 {published data only}
- O'Connor PW, Li D, Freedman MS, Bar-Or A, Rice GP, Confavreux C, et al. A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology 2006;66(6):894-900. [Google Scholar]
OLYMPUS 2009 {published data only}
- Hawker K, O'Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Annals of Neurology 2009;66(4):460-71. [Google Scholar]
- NCT00087529. A study to evaluate the safety and efficacy of rituximab in adults with primary progressive multiple sclerosis (OLYMPUS) [A phase II/III, randomized, double-blind, parallel-group, placebo-controlled, multicenter study to evaluate the safety and efficacy of Rituximab in adults with primary progressive multiple sclerosis]. clinicaltrials.gov/show/NCT00087529 (first received 13 July 2004).
- Zhang J, Waubant E, Cutter G, Wolinsky J, Glanzman R. EDSS variability before randomization may limit treatment discovery in primary progressive MS. Multiple Sclerosis 2013;19(6):775-81. [Google Scholar]
OPERA I 2017 {published data only}
- De Seze J, Hauser SL, Kappos L, Montalban X, Pozzilli C, Chognot C, et al. Infusion-related reactions with OCRELIZUMAB in phase III studies. Multiple Sclerosis 2017;23(3):878-9. [Google Scholar]
- Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus Interferon beta-1a in relapsing multiple sclerosis. New England Journal of Medicine 2017;376(3):221-34. Supplementary appendix; p. 1-36. [Google Scholar]
- NCT01247324. A study of ocrelizumab in comparison with interferon beta-1a (Rebif) in participants with relapsing multiple sclerosis [A randomized, double-blind, double-dummy, parallel-group study to evaluate the efficacy and safety of Ocrelizumab in comparison to Interferon beta-1a (Rebif®) in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT01247324 (first received 24 November 2010).
OPERA II 2017 {published data only}
- Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. New England Journal of Medicine 2017;376(3):221-34. [Google Scholar]
- NCT01412333. A study of ocrelizumab in comparison with interferon beta-1a (Rebif) in participants with relapsing multiple sclerosis [A randomized, double-blind, double-dummy, parallel-group study to evaluate the efficacy and safety of Ocrelizumab in comparison to Interferon beta-1a (Rebif) in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT01412333 (first received 9 August 2011).
ORACLE 2014 {published data only}
- Leist T, Comi G, Cree B, Coyle P, Freedman M, Hartung H, et al. Oral cladribine delays time to conversion to clinically definite MS in patients with a first demyelinating event: top line results from the phase III ORACLE MS Study. Neurology 2013;80(7 Supplement):P07.114. [Google Scholar]
- Leist TP, Comi G, Cree BA, Coyle PK, Freedman MS, Hartung HP, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurology 2014;13(3):257-67. [Google Scholar]
- NCT00725985. Oral cladribine in early multiple sclerosis (MS) (ORACLE MS) [A phase III, randomized, double-blind, placebo-controlled, multi-center clinical trial of oral Cladribine in subjects with a first clinical event at high risk of converting to MS]. clinicaltrials.gov/show/NCT00725985 (first received 31 July 2008).
ORATORIO 2017 {published data only}
- Fox EJ, Markowitz C, Applebee A, Montalban X, Wolinsky JS, Belachew S, et al. Effect of OCRELIZUMAB on upper limb function in patients with primary progressive multiple sclerosis in the ORATORIO study. Multiple Sclerosis 2017;23(3):658-9. [Google Scholar]
- Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. New England Journal of Medicine 2017;376(3):209-20. Supplementary appendix; p. 1-27. [Google Scholar]
- NCT01194570. A study of Ocrelizumab in participants with primary progressive multiple sclerosis [A phase III, multicentre, randomized, parallel-group, double-blind, placebo controlled study to evaluate the efficacy and safety of Ocrelizumab in adults with primary progressive multiple sclerosis]. clinicaltrials.gov/show/NCT01194570 (first received 3 September 2010).
OWIMS 1999 {published data only}
- OWIMS. Evidence of interferon beta-1a dose response in relapsing-remitting MS: the OWIMS Study. The Once Weekly Interferon for MS Study Group. Neurology 1999;53(4):679-86. [Google Scholar]
Pakdaman 2007 {published data only}
- Pakdaman H, Sahraian MA, Fallah A, Pakdaman R, Ghareghozli K, Ghafarpour M, et al. Effect of early interferon beta-1a therapy on conversion to multiple sclerosis in Iranian patients with a first demyelinating event. Acta Neurologica Scandinavica 2007;115(6):429-31. [Google Scholar]
Pohlau 2007 {published data only}
- Pohlau D, Przuntek H, Sailer M, Bethke F, Koehler J, Konig N, et al. Intravenous immunoglobulin in primary and secondary chronic progressive multiple sclerosis: a randomized placebo controlled multicentre study. Multiple Sclerosis 2007;13(9):1107-17. [Google Scholar]
Polman 2005 {published data only}
- Polman C, Barkhof F, Sandberg-Wollheim M, Linde A, Nordle O, Nederman T, et al. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology 2005;64(6):987-91. [Google Scholar]
PreCISe 2009 {published data only}
- Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet 2009;374(9700):1503-11. [Google Scholar]
- NCT00666224. Evaluate early glatiramer acetate treatment in delaying conversion to clinically definite multiple sclerosis of subjects presenting with clinically isolated syndrome (PreCISe) [A multinational, multicenter, randomized, double-blind, placebo controlled, parallel group study to evaluate the effect of early Glatiramer acetate treatment in delaying the conversion to clinically definite multiple sclerosis (CDMS) of subjects presenting with clinically isolated syndrome (CIS)]. clinicaltrials.gov/show/NCT00666224 (first received 24 April 2008).
PRISMS 1998 {published data only}
- PRISMS. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet 1998;352(9139):1498-504. [Google Scholar]
PROMESS 2017 {published data only}
- Brochet B, Deloire MS, Perez P, Loock T, Baschet L, Debouverie M, et al. Double-blind controlled randomized trial of cyclophosphamide versus methylprednisolone in secondary progressive multiple sclerosis. PLoS One 2017;12(1):e0168834. Protocol synopsys; S2 File. CONSORT 2010 checklist promess; S4 File. [Google Scholar]
- NCT00241254. Efficacy of cyclophosphamide versus methylprednisolone in patients with secondary progressive multiple sclerosis (PROMESS) [A double-blind, two-arm, multicenter, randomized trial to evaluate efficacy of Cyclophosphamide versus Methylprednisolone in patients with recent secondary progressive multiple sclerosis: P.R.OM.E.S.S study]. clinicaltrials.gov/show/NCT00241254 (first received 18 October 2005).
RADIANCE 2019 {published data only}
- Cohen J, Comi G, Selmaj K, Bar-Or A, Arnold D, Steinman L, et al. Clinical and magnetic resonance imaging results from Radiance Part B, a multicenter, randomized, double-blind, phase 3 trial of Ozanimod versus intramuscular Interferon β-1a in relapsing multiple sclerosis (RMS). Neurology 2018;90(15 Supplement):P3.410. [Google Scholar]
- Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurology 2019;18(11):1021-33. Supplementary appendix; p. 1-32. [Google Scholar]
- NCT02047734. Efficacy and safety study of ozanimod in relapsing multiple sclerosis (RADIANCE) [A phase 2/3, multi-center, randomized, double-blind, placebo-controlled (part a) and double-blind, double-dummy, active-controlled (part b), parallel group study to evaluate the efficacy and safety of RPC1063 administered orally to relapsing multiple sclerosis patients]. clinicaltrials.gov/show/NCT02047734 (first received 28 January 2014).
REFLEX 2012 {published data only}
- Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BM, et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurology 2012;11(1):33-41. [Google Scholar]
- NCT00404352. REbif FLEXible dosing in early multiple sclerosis (MS) (REFLEX) [A phase III, randomized, double-blind, placebo-controlled, multicenter clinical trial of Rebif new formulation (44 microgram [mcg] three times weekly [tiw] and 44 mcg once weekly [ow]) in subjects at high risk of converting to multiple sclerosis (REFLEX)]. clinicaltrials.gov/show/NCT00404352 (first received 28 November 2006).
REFORMS 2012 {published data only}
- NCT00428584. RNF and Betaseron® Tolerability Study (REFORMS) [A randomized, multicenter, two arm, open label, twelve week phase IIIB study to evaluate the tolerability of Rebif (new formulation) (IFN beta-1a) and Betaseron (IFN beta-1b) in IFN-naive subjects with relapsing remitting multiple sclerosis (RRMS) followed by a single arm, eighty-two week minimum, Rebif (new formulation) only safety extension]. clinicaltrials.gov/show/NCT00428584 (first received 30 January 2007).
- Singer B, Bandari D, Cascione M, LaGanke C, Huddlestone J, Bennett R, et al. Comparative injection-site pain and tolerability of subcutaneous serum-free formulation of interferonβ-1a versus subcutaneous interferonβ-1b: results of the randomized, multicenter, Phase IIIb REFORMS study. BMC Neurology 2012;12:154. [Google Scholar]
REGARD 2008 {published data only}
- Mikol D, Barkhof F, Chang P, Coyle P, Jeffery D, Schwid S, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurology 2008;7(10):903-14. [Google Scholar]
- NCT00078338. Rebif® versus Copaxone® in the treatment of relapsing remitting multiple sclerosis [Phase IV, multicenter, open label, randomized study of Rebif® 44 mcg administered three times per week by subcutaneous injection compared with Copaxone® 20 mg administered daily by subcutaneous injection in the treatment of relapsing remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00078338 (first received 26 February 2004).
REVEAL 2020 {published data only}
- Butzkueven H, Licata S, Jeffery D, Arnold DL, Filippi M, Geurts JJ, et al. Natalizumab versus fingolimod for patients with active relapsing-remitting multiple sclerosis: results from REVEAL, a prospective, randomised head-to-head study. British Medical Journal Open 2020;10(10):e038861. Online supplementary figure 1; Patient flow. [Google Scholar]
- NCT02342704. Impact of Natalizumab versus Fingolimod in relapsing-remitting multiple sclerosis (RRMS) participants (REVEAL) [A multicenter, randomized, open-label study to assess the impact of Natalizumab versus Fingolimod on central nervous system tissue damage and recovery in active relapsing-remitting multiple sclerosis subjects]. clinicaltrials.gov/show/NCT02342704 (first received 21 January 2015).
Saida 2012 {published data only}
- NCT00537082. Efficacy and safety of FTY720 in patients with relapsing multiple sclerosis (MS) [A 6-month, double-blind, randomized, placebo-controlled, parallel-group, multicenter study comparing efficacy and safety of FTY720 0.5 mg and 1.25 mg administered orally once daily in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT00537082 (first received 28 September 2007).
- Saida T, Kikuchi S, Itoyama Y, Hao Q, Kurosawa T, Nagato K, et al. A randomized, controlled trial of fingolimod (FTY720) in Japanese patients with multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2012;18(9):1269-77. Online appendix; p. 1-14. [Google Scholar]
Saida 2017 {published data only}
- NCT01440101. Natalizumab (BG00002, Tysabri) study in Japanese participants with relapsing-remitting multiple sclerosis (RRMS) (Tysabri Japan) [Multicenter study of BG00002 in Japanese subjects with RRMS, consisting of a multiple-dose, open-label evaluation of its safety, tolerability, pharmacokinetics and pharmacodynamics (part a) and a randomized, double-blind, placebo-controlled, multiple-dose evaluation of safety and efficacy (part b)]. clinicaltrials.gov/show/NCT01440101 (first received 26 September 2011).
- Saida T, Kira JI, Kishida S, Yamamura T, Sudo Y, Ogiwara K, et al. Efficacy, safety, and pharmacokinetics of natalizumab in Japanese multiple sclerosis patients: A double-blind, randomized controlled trial and open-label pharmacokinetic study. Multiple Sclerosis and Related Disorders 2017;11:25-31. [Google Scholar]
SELECT 2013 {published data only}
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet 2013;381(9884):2167-75. Supplementary appendix; p. 1-6. [Google Scholar]
- NCT00390221. Safety and efficacy study of daclizumab high yield process (DAC HYP) to treat relapsing-remitting multiple sclerosis (SELECT) [Multicenter, double-blind, placebo-controlled, dose-ranging study to determine the safety and efficacy of Daclizumab HYP (DAC HYP) as a monotherapy treatment in subjects with relapsing-remitting multiple sclerosis]. clinicaltrials.gov/show/NCT00390221 (first received 19 October 2006).
- Phillips G, Guo S, Bender R, Havrdova E, Proskorovsky I, Vollmer T. Assessing the impact of multiple sclerosis disease activity and DACLIZUMAB HYP treatment on patient-reported outcomes: Results from the SELECT trial. Multiple Sclerosis and Related Disorders 2016;6:66-72. [Google Scholar]
SPECTRIMS 2001 {published data only}
- SPECTRIMS. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology 2001;56(11):1496-504. [Google Scholar]
SUNBEAM 2019 {published data only}
- Comi G, Arnold D, Cree B, Kappos L, Selmaj K, Bar-Or A, et al. Ozanimod demonstrates efficacy and safety in a multicenter, randomized, double-blind, double-dummy, active-controlled phase 3 trial of relapsing multiple sclerosis (SUNBEAM). Neurology 2018;90(15 Supplement):P3.396. [Google Scholar]
- Comi G, Kappos L, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurology 2019;18(11):1009-20. Supplementary appendix; p. 1-26. [Google Scholar]
- Cree B, Bar-Or A, Comi G, Selmaj K, Arnold D, Steinman L, et al. Safety of Ozanimod versus Interferon β-1a in two multicenter, randomized, double-blind, parallel-group, active-controlled, double-dummy phase 3 studies in relapsing multiple sclerosis (SUNBEAM and RADIANCE Part B). Neurology 2018;90(15 Supplement):S36.006. [Google Scholar]
- NCT02294058. Study of ozanimod (RPC1063) in relapsing multiple sclerosis (MS) (SUNBEAM) [A phase 3, multi-center, randomized, double-blind, double-dummy, active controlled, parallel group study to evaluate the efficacy and safety of RPC1063 administered orally to relapsing multiple sclerosis patients]. clinicaltrials.gov/show/NCT02294058 (first received 19 November 2014).
TEMSO 2011 {published data only}
- NCT00134563. Study of teriflunomide in reducing the frequency of relapses and accumulation of disability in patients with multiple sclerosis (TEMSO) [A randomized, double-blind, placebo-controlled, parallel group design study to evaluate the efficacy and safety of teriflunomide in reducing the frequency of relapses and delaying the accumulation of physical disability in subjects with multiple sclerosis with relapses]. ClinicalTrials.gov/show/NCT00134563 (first received 25 August 2005).
- O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. New England Journal of Medicine 2011;365(14):1293-303. [Google Scholar]
- Sprenger T, Kappos L, Radue EW, Gaetano L, Mueller-Lenke N, Wuerfel J, et al. Teriflunomide significantly slows brain volume loss in MS patients irrespective of disability progression. Neurology 2016;86(Suppl 1):16. [Google Scholar]
TENERE 2014 {published data only}
- NCT00883337. A study comparing the effectiveness and safety of teriflunomide and interferon beta-1a in patients with relapsing multiple sclerosis (TENERE) [A multi-center, randomized, parallel-group, rater-blinded study comparing the effectiveness and safety of Teriflunomide and Interferon beta-1a in patients with relapsing multiple sclerosis plus a long term extension period]. clinicaltrials.gov/show/NCT00883337 (first received17 April 2009).
- Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Multiple Sclerosis 2014;20(6):705-16. [Google Scholar]
TOPIC 2014 {published data only}
- Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurology 2014;13(10):977-86. Supplementary webappendix; p. 1-14. [Google Scholar]
- NCT00622700. Phase III study with Teriflunomide versus placebo in patients with first clinical symptom of multiple sclerosis (TOPIC) [An international, multi-center, randomized, double-blind, placebo-controlled, parallel group study to evaluate the efficacy and safety of two year treatment with Teriflunomide 7 mg once daily and 14 mg once daily versus placebo in patients with a first clinical episode suggestive of multiple sclerosis plus a long term extension period]. clinicaltrials.gov/show/NCT00622700 (first received 25 February 2008).
TOWER 2014 {published data only}
- Confavreux C, O'Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurology 2014;13(3):247-56. [Google Scholar]
- Freedman MS, Morawski J, Thangavelu K. Clinical efficacy of Teriflunomide over a fixed 2-year duration in the TOWER study [A multi-center double-blind parallel-group placebo-controlled study of the efficacy and safety of Teriflunomide in patients with relapsing multiple sclerosis]. Multiple Sclerosis Journal – Experimental, Translational and Clinical 2018;4(2):2055217318775236. [Google Scholar]
- Miller AE, Macdonell R, Comi G, Freedman MS, Kappos L, Maurer M, et al. Teriflunomide reduces relapses with sequelae and relapses leading to hospitalizations: results from the TOWER study. Journal of Neurology 2014;261(9):1781-8. [Google Scholar]
- Miller AE, Xu X, Macdonell R, Vucic S, Truffinet P, Benamor M, et al. Efficacy and safety of teriflunomide in Asian patients with relapsing forms of multiple sclerosis: a subgroup analysis of the phase 3 TOWER study. Journal of Clinical Neuroscience 2019;59:229-31. [Google Scholar]
- NCT00751881. An efficacy study of Teriflunomide in participants with relapsing multiple sclerosis (TOWER) [A multi-center double-blind parallel-group placebo-controlled study of the efficacy and safety of Teriflunomide in patients with relapsing multiple sclerosis]. clinicaltrials.gov/show/NCT00751881 (first received 12 September 2008).
- Qiu W, Huang DH, Hou SF, Zhang MN, Jin T, Dong HQ, et al. Efficacy and safety of teriflunomide in Chinese patients with relapsing forms of multiple sclerosis: a subgroup analysis of the phase 3 TOWER study. Chinese Medical Journal 2018;131(23):2776-84. [Google Scholar]
TRANSFORMS 2010 {published data only}
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):402-15. Appendix, p. 1-11. [Google Scholar]
- NCT00340834. Efficacy and safety of fingolimod in patients with relapsing-remitting multiple sclerosis with optional extension phase (TRANSFORMS) [A 12-month double-blind, randomized, multicenter, active-controlled, parallel-group study comparing the efficacy and safety of 0.5 mg and 1.25 mg Fingolimod (FTY720) administered orally once daily versus Interferon ß-1a (Avonex) administered im once weekly in patients with relapsing-remitting multiple sclerosis with optional extension]. clinicaltrials.gov/show/NCT00340834 (first received 21 June 2006).
Tubridy 1999 {published data only}
- Tubridy N, Behan PO, Capildeo R, Chaudhuri A, Forbes R, Hawkins CP, et al. The effect of anti-alpha4 integrin antibody on brain lesion activity in MS. The UK Antegren Study Group. Neurology 1999;53(3):466-72. [Google Scholar]
Van de Wyngaert 2001 {published data only}
- de Wyngaert FA, Beguin C, D'Hooghe MB, Dooms G, Lissoir F, Carton H, et al. A double-blind clinical trial of mitoxantrone versus methylprednisolone in relapsing, secondary progressive multiple sclerosis. Acta Neurologica Belgica 2001;101(4):210-6. [Google Scholar]
Wolinsky 2007 {published data only}
- Steinerman JR, Davis MD, Knappertz V, Giovannoni G, Wolinsky JS. Disability progression and cerebrospinal fluid status in PPMS: Re-analysis of the ProMiSe clinical trial data set. Neurology 2017;88:S16. [Google Scholar]
- Wolinsky J, Narayana P, O'Connor P, Coyle P, Ford C, Johnson K, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-controlled trial. Annals of Neurology 2007;61(1):14-24. [Google Scholar]
Ziemssen 2017 {published data only}
- EudraCT: 2009-011234-99. A Phase I, sequential group, randomized, double-blind, placebo-controlled study to assess the tolerability and safety of escalating doses of oral laquinimod administereddaily in subjects with relapsing remitting multiple sclerosis (RRMS). www.clinicaltrialsregister.eu/ctr-search/trial/2009-011234-99/DE Registered 23 June 2009.
- Ziemssen T, Tumani H, Sehr T, Thomas K, Paul F, Richter N, et al. Safety and in vivo immune assessment of escalating doses of oral laquinimod in patients with RRMS. Journal of Neuroinflammation 2017;14(1):172. Additional file 1 [Figure S1, Figure S2, Figure S3, Table S1 Distribution of study drug termination reasons, Table S2, Table S3). [Google Scholar]
References to studies excluded from this review
Barkhof 2018 {published data only}
- Barkhof F, Kappos L, Bar-Or A, Li D, Belachew S, Julian L, et al. Rapid onset of ocrelizumab suppression of brain MRI activity in relapsing-remitting multiple sclerosis. Multiple Sclerosis 2018;24:NP14-5. [Google Scholar]
Beutler 1996 {published data only}
- Beutler E, Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J. The treatment of chronic progressive multiple sclerosis with cladribine. Proceedings of the National Academy of Sciences 1996;93(4):1716-20. [Google Scholar]
Boiko 2018 {published data only}
- Boiko AN, Bosenko LP, Vasilovskii VV, Volkova LI, Zakharova MN, Kotov SV, et al. Comparative placebo-controlled clinical trial of the efficacy and safety of Interferon β-1a formulations for S.C. administration in patients with remitting multiple sclerosis: first-year results. Neuroscience and Behavioral Physiology 2018;48(7):883-9. [Google Scholar]
- Boyko AN, Bosenko LP, Vasilovskiy VV, Volkova LI, Zakharova MN, Kotov SV, et al. Efficacy, tolerability and safety of the treatment with teberif: the results of a 2-year randomized clinical trial of treatment naïve patients with remitting multiple sclerosis, who have not received DMT, after switching from other Interferon β-1a. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova 2019;119(2):73-85. [Google Scholar]
Boyko 2019 {published data only}
- Boyko AN, Bosenko LP, Vasilovskiy VV, Volkova LI, Zakharova MN, Kotov SV, et al. Efficacy, tolerability and safety of the treatment with teberif: the results of a 2-year randomized clinical trial of treatment naïve patients with remitting multiple sclerosis, who have not received DMT, after switching from other interferon β-1a [Éffektivnost', perenosimost' i bezopasnost' terapii preparatom teberif: rezul'taty dvukhletnego klinicheskogo issledovaniia u patsientov s remittiruiushchim rasseiannym sklerozom, ranee ne poluchavshikh PITRS, i pri perekliuchenii s drugogo interferona β-1a]. Zhurnal Nevrologii I Psikhiatrii Imeni S.S. Korsakova 2019;119(2. Vyp. 2):73-85. [Google Scholar]
Boyko 2022 {published data only}
- Boyko AN, Bakhtiyarova KZ, Dudin VA, Zaslavsky LG, Malkova NA, Parshina YV, et al. The new pegylated Interferon beta-1a (sampegInterferon beta-1a, BCD-054) in the treatment of remitting multiple sclerosis. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova 2019;119(10):100-9. [Google Scholar]
- Boyko AN, Boyko OV, Bakhtiyarova KZ, Gusev EI, Dudin VA, Zaslavsky LG, et al. Efficacy and safety of sampeginterferon β-1a in the treatment of relapsing remitting multiple sclerosis: results of 52 weeks of therapy in a randomized, double-blind clinical trial [Effektivnost' i bezopasnost' sampeginterferona β-1a dlya lecheniya remittiruyushchego rasseyannogo skleroza: rezul'taty 52-nedel'nogo randomizirovannogo dvoinogo slepogo klinicheskogo issledovaniya]. Zhurnal Nevrologii i Psikhiatrii Imeni S.S. Korsakova 2022;122(1):62-71. [Google Scholar]
Cohen 2009 {published data only}
- Cohen JA, Imrey PB, Calabresi PA, Edwards KR, Eickenhorst T, Felton WL 3rd, et al. Results of the Avonex Combination Trial (ACT) in relapsing-remitting MS. Neurology 2009;72(6):535-41. [Google Scholar]
Cohen 2019 {published data only}
- Cohen JA, Campbell N, Wiendl H, Foley J, Butzkueven H, Ryerson LZ, et al. Evaluating the efficacy and safety of 6-week extended interval dosing of Natalizumab via a prospective, controlled, randomized phase 3B study. Multiple Sclerosis 2019;25:48-9. [Google Scholar]
- Foley J, Defer G, Zhovtis Ryerson L, Cohen JA, Arnold DL, Butzkueven H, et al. Baseline characteristics of multiple sclerosis patients enrolled in NOVA, a multicentre, randomised trial to assess the efficacy of Natalizumab every-6-weeks dosing. European Journal of Neurology 2020;27:212. [Google Scholar]
- NCT03689972. A study to evaluate efficacy, safety, and tolerability of EID of Natalizumab (BG00002) in participants with RRMS switching from treatment with Natalizumab SID in relation to continued SID treatment- followed by extension study comprising sc and iv Natalizumab administration [A randomized, controlled, open-label, rater-blinded, phase 3B study of the efficacy, safety, and tolerability of 6-week extended interval dosing (EID) of Natalizumab (BG00002) in subjects with relapsing-remitting multiple sclerosis switching from treatment with 4-week Natalizumab standard interval dosing (SID) in relation to continued SID treatment - followed by an open-label crossover extension study comprising subcutaneous and intravenous Natalizumab administration]. ClinicalTrials.gov/show/NCT03689972 (first received 1 October 2018).
Comi 2011 {published data only}
- Comi G, Cohen JA, Arnold DL, Wynn D, Filippi M, FORTE Study Group. Phase III dose-comparison study of glatiramer acetate for multiple sclerosis. Annals of Neurology 2011;69(1):75-82. [Google Scholar]
Comi 2016 {published data only}
- Comi G, Freedman MS, Kappos L, Olsson TP, Miller AE, Wolinsky JS, et al. Pooled safety and tolerability data from four placebo-controlled teriflunomide studies and extensions. Multiple Sclerosis and Related Disorders 2016;5:97-104. [Google Scholar]
CORAL 2006 {published data only}
- Filippi M, Wolinsky JS, Comi G, CORAL Study Group. Effects of oral glatiramer acetate on clinical and MRI-monitored disease activity in patients with relapsing multiple sclerosis: a multicentre, double-blind, randomised, placebo-controlled study. The Lancet Neurology 2006;5(3):213-20. [Google Scholar]
DELIVER 2016 {published data only}
- NCT00559702. A randomized, open-label, dose-ranging study to evaluate the pharmacokinetics and initial safety of subcutaneous and intramuscular Natalizumab in subjects with multiple sclerosis. ClinicalTrials.gov/show/NCT00559702 (first received 16 November 2007).
- Plavina T, Fox EJ, Lucas N, Muralidharan KK, Mikol D. A randomized trial evaluating various administration routes of natalizumab in multiple sclerosis. Journal Of Clinical Pharmacology 2016;56(10):1254-62. [Google Scholar]
Edan 1997 {published data only}
- Edan G, Miller D, Clanet M, Confavreux C, Lyon-Caen O, Lubetzki C, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. Journal of Neurology, Neurosurgery and Psychiatry 1997;62(2):112-8. [Google Scholar]
EPOC 2014 {published data only}
- Fox E, Edwards K, Burch G, Wynn DR, LaGanke C, Crayton H, et al. Outcomes of switching directly to oral fingolimod from injectable therapies: Results of the randomized, open-label, multicenter, Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Multiple Sclerosis And Related Disorders 2014;3(5):607-19. [Google Scholar]
- NCT01216072. A 6-month, randomized, open-label, patient outcomes, safety and tolerability study of fingolimod (FTY720) 0.5 mg/day vs. comparator in patients with relapsing forms of multiple sclerosis (EPOC) [A 6-month, randomized, active comparator, open-label, multi-center study to evaluate patient outcomes, safety and tolerability of Fingolimod (FTY720) 0.5 mg/day in patients with relapsing forms of multiple sclerosis who are candidates for MS therapy change from previous disease modifying therapy (EPOC)]. ClinicalTrials.gov/show/NCT01216072 (first received 7 October 2010).
EVOLVE‐MS‐1 2022 {published data only}
- NCT026 34307. A study of ALKS 8700 in adults with relapsing remitting multiple sclerosis (MS) EVOLVE-MS-1. ClinicalTrials.gov: NCT02634307 (first received 18 December 2015).
- Wray S, Then Bergh F, Wundes A, Arnold DL, Drulovic J, Jasinska E, et al. Efficacy and safety outcomes with diroximel fumarate after switching from prior therapies or continuing on DRF: results from the phase 3 EVOLVE-MS-1 study. Advances In Therapy 2022;39(4):1810-31. [Google Scholar]
Freedman 2012 {published data only}
- Freedman MS, Wolinsky JS, Wamil B, Confavreux C, Comi G, Kappos L, et al. Teriflunomide added to interferon- in relapsing multiple sclerosis: a randomized phase II trial. Neurology 2012;78(23):1877-85. [Google Scholar]
Freedman 2015 {published data only}
- Freedman MS, Wolinsky JS, Truffinet P, Comi G, Kappos L, Miller AE, et al. A randomized trial of teriflunomide added to glatiramer acetate in relapsing multiple sclerosis. Multiple Sclerosis Journal – Experimental, Translational and Clinical 2015;1:1-10. [Google Scholar]
Gobbi 2013 {published data only}
- Gobbi C, Meier DS, Cotton F, Sintzel M, Leppert D, Guttmann CR, et al. Interferon beta 1b following natalizumab discontinuation: one year, randomized, prospective, pilot trial. BMC Neurology 2013;13:10. [Google Scholar]
- NCT01144052. Natalizumab de-escalation with Interferon beta-1b [De-escalation after Natalizumab treatment with Interferon-beta-1b in patients with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT01144052 (first received 15 June 2010).
- Zecca C, Riccitelli GC, Calabrese P, Pravatà E, Candrian U, Guttmann CR, et al. Treatment satisfaction, adherence and behavioral assessment in patients de-escalating from natalizumab to interferon β. BMC Neurology 2014;14:38. [Google Scholar]
Goodman 2009 {published data only}
- Goodman AD, Rossman H, Bar-Or A, Miller A, Miller DH, Schmierer K, et al. Glance: Results of a phase 2, randomized, double-blind, placebo-controlled study. Neurology 2009;72(9):808-12. [Google Scholar]
Hartung 2020 {published data only}
- Hartung HP. Ocrelizumab shorter infusion: Primary results from the ENSEMBLE PLUS substudy in patients with MS. Neurology Neuroimmunology & Neuroinflammation 2020;7:5. [Google Scholar]
Hauser 2018 {published data only}
- Hauser SL, Kappos L, Montalban X, Koendgen H, Li C, Marcillat C, et al. Safety of Ocrelizumab in Multiple sclerosis: Updated Analysis in Patients With Relapsing and Primary Progressive Multiple sclerosis. European Journal of Neurology 2018;25:334. [Google Scholar]
Havrdova 2009 {published data only}
- Havrdova E, Zivadinov R, Krasensky J, Dwyer MG, Novakova I, Dolezal O, et al. Randomized study of interferon beta-1a, low-dose azathioprine, and low-dose corticosteroids in multiple sclerosis. Multiple Sclerosis 2009;15(8):965-76. [Google Scholar]
Hu 2016 {published data only}
- Hu X, Shang S, Nestorov I, Hasan J, Seddighzadeh A, Dawson K, et al. COMPARE: Pharmacokinetic profiles of subcutaneous pegINTERFERON beta-1a and subcutaneous INTERFERON beta-1a over 2 weeks in healthy subjects. British Journal of Clinical Pharmacology 2016;82(2):380-8. [Google Scholar]
Hughes 2018 {published data only}
- Hughes R, Dalakas MC, Merkies I, Latov N, Léger JM, Nobile-Orazio E, et al. Oral fingolimod for chronic inflammatory demyelinating polyradiculoneuropathy (FORCIDP Trial): a double-blind, multicentre, randomised controlled trial. Lancet Neurology 2018;17(8):689-98. [Google Scholar]
Kappos 2014 {published data only}
- Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurology 2014;13(4):353-63. [Google Scholar]
- NCT00642902. A phase 2 study of Atacicept in subjects with relapsing multiple sclerosis (ATAMS) [A four-arm randomized, double-blind, placebo-controlled, multicenter phase ii study to evaluate the safety, tolerability and efficacy as assessed by frequent MRI measures of 3 doses of Atacicept monotherapy in subjects with relapsing multiple sclerosis (RMS) over a 36 week treatment course]. ClinicalTrials.gov/show/NCT00642902 (first received 25 March 2008).
Kappos 2016 {published data only}
- Kappos L, Li DK, Stüve O, Hartung HP, Freedman MS, Hemmer B, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD study. JAMA Neurology 2016;73(9):1089-98. [Google Scholar]
- NCT01185821. Long-term safety, tolerability and efficacy of BAF312 given orally in patients with relapsing-remitting multiple sclerosis [A dose blinded extension study to the CBAF312A2201 study to evaluate long-term safety, tolerability and efficacy of BAF312 given orally once daily in patients with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT01185821 (first received 20 August 2010).
Kastrukoff 1990 {published data only}
- Kastrukoff LF, Oger JJ, Hashimoto SA, Sacks SL, Li DK, Palmer MR, et al. Systemic lymphoblastoid interferon therapy in chronic progressive multiple sclerosis. I. Clinical and MRI evaluation. Neurology 1990;40(3 Pt 1):479-86. [Google Scholar]
Khoury 2010 {published data only}
- Khoury SJ, Healy BC, Kivisäkk P, Viglietta V, Egorova S, Guttmann CR, et al. A randomized controlled double masked trial of albuterol add-on therapy in patients with multiple sclerosis. Archives of Neurology 2010;67(9):1055-61. [Google Scholar]
Komori 2016 {published data only}
- Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, et al. Insufficient disease inhibition by intrathecal RITUXIMAB in progressive multiple sclerosis. Annals of Clinical and Translational Neurology 2016;3(3):166-79. [Google Scholar]
- NCT01212094. Double blind combination of rituximab by intravenous and intrathecal injection versus placebo in patients with low-inflammatory secondary progressive multiple sclerosis (RIVITaLISe). ClinicalTrials.gov/show/NCT01212094 (first received 30 September 2010).
Le Page 2015 {published data only}
- Le Page E, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): A randomised, controlled, double-blind, non-inferiority trial. Lancet 2015;386:974-81. [Google Scholar]
- NCT00984984. Efficacy and safety of Methylprednisolone per os versus iv for the treatment of multiple sclerosis (MS) relapses (COPOUSEP) [Randomised double-blinded trial comparing efficacy and safety of Methylprednisolone per os versus iv for the treatment of multiple sclerosis relapses]. ClinicalTrials.gov/show/NCT00984984 (first received 25 September 2009).
Mancardi 2015 {published data only}
- Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology 2015;84(10):981-8. [Google Scholar]
Mayer 2019 {published data only}
- Mayer L, Kappos L, Racke MK, Rammohan K, Traboulsee A, Hauser SL, et al. Ocrelizumab infusion experience in patients with relapsing and primary progressive multiple sclerosis: Results from the phase 3 randomized OPERA I, OPERA II, and ORATORIO studies. Multiple Sclerosis and Related Disorders 2019;30:236-43. [Google Scholar]
Montalban 2019 {published data only}
- Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Evobrutinib phase 2 study group. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. The New England Journal of Medicine 2019;380(25):2406-17. [Google Scholar]
- NCT02975349. A study of efficacy and safety of M2951 in participants with relapsing multiple sclerosis [A randomized, double-blind, placebo-controlled phase ii study of M2951 with a parallel, open-label, active control group (Tecfidera), in patients with relapsing multiple sclerosis to evaluate efficacy, safety, tolerability, pharmacokinetics, and biological activity]. ClinicalTrials.gov/show/NCT02975349 (first received 29 November 2016).
NCT00206648 {published data only}
- NCT00206648. An efficacy and safety comparison study of two marketed drugs in patients with relapsing-remitting MS (ABOVE) [A randomized, rater-blinded, multicenter, parallel-group study comparing the efficacy and safety of Betaseron 250 µg subcutaneously every other day with Avonex 30 µg intramuscularly once per week in relapsing-remitting multiple sclerosis patients previously treated with Avonex]. ClinicalTrials.gov/show/NCT00206648 (first received 21 September 2005).
NCT01058005 {published data only}
- NCT01058005. Study evaluating Rebif, Copaxone, and Tysabri for active multiple sclerosis (SURPASS) [A multicenter, randomized, open-label, parallel-group, active-controlled study to evaluate the benefits of switching therapy (glatiramer acetate or interferon beta-1a) to natalizumab in subjects with relapsing remitting multiple sclerosis (SURPASS)]. ClinicalTrials.gov/show/NCT01058005 (first received 28 January 2010).
NCT01065727 {published data only}
- NCT01065727. Impact study of 2 therapeutic strategy for aggressive remitting multiple sclerosis (IQUALYSEP) [Study impact, on clinical outcomes, quality of life and costs of 2 therapeutic strategy (monthly Natalizumab versus Mitoxantrone then immunomodulator) at 3 years of follow-up for aggressive remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT01065727 (first received 9 February 2010).
NCT01337427 {published data only}
- NCT01337427. Using optical coherence tomography (OCT) to evaluate the efficacy and safety of Pegylated Interferon beta-1a (BIIB017) in patients with relapsing multiple sclerosis [Optical Coherence Tomography (OCT) in a multicenter, randomized,double-blind, parallel-group, placebo-controlled study to evaluate the efficacy and safety of Pegylated interferon beta-1a (BIIB017) in subjects with relapsing multiple sclerosis]. ClinicalTrials.gov/show/NCT01337427 (first received 18 April 2011).
Okai 2019 {published data only}
- Okai AF, Amezcua L, Berkovich RR, Chinea AR, Edwards KR, Steingo B, et al. Efficacy and safety of alemtuzumab in patients of African descent with relapsing-remitting multiple sclerosis: 8-year follow-up of CARE MS I and II (TOPAZ study). Neurology and Therapy 2019;8(2):376-81. [Google Scholar]
Perumal 2019 {published data only}
- Perumal J, Fox RJ, Balabanov R, Balcer LJ, Galetta S, Makh S, et al. Outcomes of Natalizumab treatment within 3 years of relapsing-remitting multiple sclerosis diagnosis: A prespecified 2-year interim analysis of STRIVE. BMC Neurology 2019;19(1):116. [Google Scholar]
PREFERMS 2018 {published data only}
- Cascione M, Tenenbaum N, Wendt J, Meng X, Schofield L, Cree BACT. Treatment retention on Fingolimod compared with injectable multiple sclerosis therapies in African-American patients: a subgroup analysis of a randomized phase 4 study. Multiple Sclerosis and Related Disorders 2018;25:50-6. [Google Scholar]
- Cree B, Cohen J, Silva D, Ritter S, Piani MD, Tomic D, et al. Confirmed disability improvement in patients treated with FINGOLIMOD in phase 3 and extension trial programmes for up to 96 months. Multiple Sclerosis 2017;23(3):322. [Google Scholar]
- Cree BAC, Arnold DL, Cascione M, Fox EJ, Williams IM, Meng X, et al. Phase IV study of retention on fingolimod versus injectable multiple sclerosis therapies: a randomized clinical trial.. Therapeutic Advances In Neurological Disorders 2018;11:1756286418774338. [Google Scholar]
- NCT01623596. Evaluation of patient retention of Fingolimod vs. currently approved disease modifying therapy in patients with relapsing remitting multiple sclerosis. (PREFERMS) [A 12-month, prospective, randomized, active-controlled, open-label study to evaluate the patient retention of Fingolimod vs. approved first-line disease modifying therapies in adults with relapsing remitting multiple sclerosis (PREFERMS)]. ClinicalTrials.gov/show/NCT01623596 (first arrived 20 June 2012).
Rahimdel 2015 {published data only}
- Rahimdel A, Zeinali A, Mellat A. Evaluating the role of corticosteroid pulse therapy in patients with secondary progressive multiple sclerosis receiving mitoxantrone: a double blind randomized controlled clinical trial. Iranian Red Crescent Medical Journal 2015;17:e30618. [Google Scholar]
Ramo‐Tello 2014 {published data only}
- NCT00753792. Oral corticotherapy in megadoses to treat multiple sclerosis during relapse [Multicenter, randomized, double blind, clinical trial to compare the clinical and radiological efficacy of equivalent doses of Methylprednisolone administered orally or intravenously in patients with multiple sclerosis during relapse]. ClinicalTrials.gov/show/NCT00753792 (first received 17 September 2008).
- Ramo-Tello C, Grau-López L, Tintoré M, Rovira A, Ramió i Torrenta L, Brieva L, et al. A randomized clinical trial of oral versus intravenous methylprednisolone for relapse of MS. Multiple Sclerosis (Houndmills, Basingstoke, England) 2014;20(6):717-25. [Google Scholar]
- Ramo-Tello C, Tintoré M, Rovira A, Ramió-Torrenta L, Brieva L, Saiz A, et al. Baseline clinical status as a predictor of methylprednisolone response in multiple sclerosis relapses. Multiple Sclerosis 2016;22:117-21. [Google Scholar]
RESTORE 2014 {published data only}
- Fox RJ, Cree BAC, De Sèze J, Gold R, Hartung HP, Jeffery D, et al. MS disease activity in RESTORE. A randomized 24-week natalizumab treatment interruption study. Neurology 2014;82: 2014;82:1491–8. [Google Scholar]
- NCT01071083. Treatment interruption of Natalizumab (RESTORE) [Randomized treatment interruption of Natalizumab]. ClinicalTrials.gov/show/NCT01071083 (first received 19 February 2010).
Rice 2000 {published data only}
- Rice GP, Filippi M, Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Cladribine MRI Study Group. Neurology 2000;54(5):1145-55. [Google Scholar]
Rieckmann 2012 {published data only}
- NCT01142466. A phase IV study of Rebif ® 44mcg administered three times per week by subcutaneous injection compared with no treatment in the therapy of relapsing multiple sclerosis after mitoxantrone (REMAIN) [Phase IV, multicenter, open label, randomized study of Rebif® 44mcg administered three times per week by subcutaneous injection compared with no treatment in the therapy of relapsing multiple sclerosis after Mitoxantrone]. ClinicalTrials.gov/show/NCT01142466 (first received 11 June 2010).
- Rieckmann P, Heidenreich F, Sailer M, Zettl UK, Zessack N, Hartung HP, et al. Treatment de-escalation after mitoxantrone therapy: results of a phase IV, multicentre, open-label, randomized study of subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis. Therapeutic Advances In Neurological Disorders 2012;5(1):3-12. [Google Scholar]
RIVITALISE 2016 {published data only}
- Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Annals of Clinical and Translational Neurology 2016;3(3):166-79. [Google Scholar]
- NCT01212094. Double blind combination of rituximab by intravenous and intrathecal injection versus placebo in patients with low-inflammatory secondary progressive multiple sclerosis (RIVITaLISe). clinicaltrials.gov/show/NCT01212094 (firstreceived 30 September 2010).
Romine 1999 {published data only}
- Romine JS, Sipe JC, Koziol JA, Zyroff J, Beutler E. A double-blind, placebo-controlled, randomized trial of cladribine in relapsing-remitting multiple sclerosis. Proceedings of the Association of American Physicians 1999;111(1):35-44. [Google Scholar]
Saida 2016 {published data only}
- Saida T, Kira JI, Ueno Y, Harada T. Long-term efficacy and safety of intramuscular interferon beta-1a: Randomized postmarketing trial of two dosing regimens in Japanese patients with relapsing-remitting multiple sclerosis. Multiple Sclerosis and Related Disorders 2016;7:102-8. [Google Scholar]
SELECTION 2014 {published data only}
- Giovannoni G, Gold R, Selmaj K, Havrdova E, Montalban X, Radue EW, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): a multicentre, randomised, double-blind extension trial. The Lancet. Neurology 2014;13(5):472-81. [Google Scholar]
- Gold R, Radue EW, Giovannoni G, Selmaj K, Havrdova E, Stefoski D, et al. Safety and efficacy of daclizumab in relapsing-remitting multiple sclerosis: 3-year results from the SELECTED open-label extension study. BMC Neurology 2016;16:117. [Google Scholar]
- Gold R, Radue EW, Giovannoni G, Selmaj K, Havrdova EK, Montalban X, et al. Long-term safety and efficacy of daclizumab beta in relapsing-remitting multiple sclerosis: 6-year results from the SELECTED open-label extension study. Journal of Neurology 2020;267(10):2851-64.. [Google Scholar]
- Gold R, Stefoski D, Selmaj K, Havrdova E, Hurst C, Holman J, et al. Pregnancy experience: nonclinical studies and pregnancy outcomes in the daclizumab clinical study program. Neurology and Therapy 2016;5(2):169-82. [Google Scholar]
- NCT00870740. Safety and efficacy extension study of daclizumab high yield process (DAC HYP) in participants with multiple sclerosis who have completed study 205MS201 (NCT00390221) to treat relapsing-remitting multiple sclerosis (SELECTION) [A double-blind, multicenter, extension study to evaluate the safety and efficacy of DAC HYP in subjects with multiple sclerosis who have completed treatment in study 205MS201 (SELECT)]. ClinicalTrials.gov/show/NCT00870740 (first received 27 March 2009).
- NCT01051349. Safety and efficacy extension study of daclizumab high yield process (DAC HYP) (BIIB019) in participants who have completed study 205MS202 (NCT00870740) to treat relapsing remitting multiple sclerosis (SELECTED) [A multicenter, open-label, extension study to evaluate the long term safety and efficacy of Daclizumab high yield process (DAC HYP) monotherapy in subjects with multiple sclerosis who have completed treatment in study 205MS202 (SELECTION)]. ClinicalTrials.gov/show/NCT01051349 (first received 18 January 18).
SENTINEL 2006 {published data only}
- NCT00030966. Safety and efficacy of Natalizumab in combination with Avonex in the treatment of multiple sclerosis [A randomized, double-blind, placebo-controlled, parallel-group, multicenter study to determine the safety and efficacy of Natalizumab, when added to Avonex® (Interferon beta-1a), in subjects with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT00030966 (first received 18 February 2002).
- Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. New England Journal of Medicine 2006;354(9):911-23. [Google Scholar]
Sipe 1994 {published data only}
- Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, Beutler E. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet 1994;344(8914):9-13. [Google Scholar]
Sorensen 2014 {published data only}
- NCT00640328. Ofatumumab dose-finding in relapsing remitting multiple sclerosis (RRMS) patients (OMS115102) [A double-blind, randomized, placebo controlled, multicenter, dose-finding trial of Ofatumumab in relapsing remitting multiple sclerosis (RRMS) patients]. clinicaltrials.gov/show/NCT00640328 (first received 21 March 2008).
- Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology 2014;82(7):573-81. [Google Scholar]
Sorensen 2017 {published data only}
- Sorensen PS, Comi G, Vollmer TL, Montalban X, Kappos L, Dadon Y, et al. Laquinimod safety profile: pooled analyses from the ALLEGRO and BRAVO trials. International Journal of MS Care 2017;19(1):16-24. [Google Scholar]
Stelmasiak 2000 {published data only}
- Stelmasiak Z, Bartosik-Psujek H, Belniak-Legiec E, Mitosek-Szewczyk K. The effect of cladribine on some parameters of blood and cerebrospinal fluid in patients with relapsing-remitting multiple sclerosis (RR-MS). Annales Universitatis Mariae Curie-Sklodowska 2000;55:221-5. [Google Scholar]
Tahara 2020 {published data only}
- Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, et al. Safety and efficacy of Rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurology 2020;19(4):298-306. [Google Scholar]
Trojano 2015 {published data only}
- Trojano M, Ramio-Torrenta L, Grimaldi L, Lubetzki C, Schippling S, Evans K, et al. A randomized, blinded, parallel-group phase-2 study exploring the efficacy, safety, and tolerability of multiple natalizumab dosing regimens in patients with relapsing multiple sclerosis (REFINE). European Journal of Neurology 2015;22:49. [Google Scholar]
Turner 2019 {published data only}
- Turner B, Cree BAC, Kappos L, Montalban X, Papeix C, Wolinsky JS, et al. Ocrelizumab efficacy in subgroups of patients with relapsing multiple sclerosis. Journal of Neurology 2019;266(5):1182-93. [Google Scholar]
Wolinsky 2015 {published data only}
- NCT01874145. Safety and tolerability of Glatiramer acetate (GLACIER) [An open-label, randomized, multi-center, parallel-arm study to assess the safety and tolerability of Glatiramer acetate 40 mg/ml three times a week compared to 20 mg/ml daily subcutaneous injections in subjects with relapsing-remitting multiple sclerosis]. ClinicalTrials.gov/show/NCT01874145 (first received 10 June 2013).
- Wolinsky JS, Borresen TE, Dietrich DW, Wynn D, Sidi Y, Steinerman JR, et al. GLACIER: An open-label, randomized, multicenter study to assess the safety and tolerability of glatiramer acetate 40 mg three-times weekly versus 20 mg daily in patients with relapsing-remitting multiple sclerosis. Multiple Sclerosis and Related Disorders 2015;4:370-6. [Google Scholar]
- Wynn D, Kolodny S, Rubinchick S, Steinerman J, Knappertz V, Wolinsky J. Patient experience with glatiramer acetate 40 mg/1 ml three-times weekly treatment for relapsing-remitting multiple sclerosis: Results from the GLACIER extension study. Neurology 2015;84:S14. [Google Scholar]
Wray 2019 {published data only}
- Wray S, Havrdova E, Snydman DR, Arnold DL, Cohen JA, Coles AJ, et al. Infection risk with alemtuzumab decreases over time: pooled analysis of 6-year data from the CAMMS223, CARE-MS I, and CARE-MS II studies and the CAMMS03409 extension study. Multiple Sclerosis 2019;25(12):1605-17. [Google Scholar]
Wynn 2010 {published data only}
- NCT00109161. Study of subcutaneous Daclizumab in patients with active, relapsing forms of multiple sclerosis [A phase II randomized, double-blinded, placebo-controlled, multi-center study of subcutaneous Daclizumab in patients with active, relapsing forms of multiple sclerosis]. ClinicalTrials.gov/show/NCT00109161 (first received 25 April 2005).
- Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurology 2010;9(4):381-90. [Google Scholar]
References to ongoing studies
EudraCT 2018‐000284‐93 {published data only}
- A multinational, multicenter, randomized, Phase III, double blind, parallel group, placebo controlled study in subjects with relapsing forms of MS to assess the efficacy, safety and tolerability of GA Depot, a long acting IM injection of glatiramer acetate, administered once every four weeks. https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-000284-93/EE/.
NCT04035005 {published data only}
- A study to evaluate the efficacy and safety of Ocrelizumab in adults with primary progressive multiple sclerosis (O'HAND) [A phase IIIB multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of Ocrelizumab in adults with primary progressive multiple sclerosis]. ClinicalTrials.gov/show/NCT04035005 (first received 29July 2019).
NCT04121403 {published data only}
- Norwegian study of oral Cladribine and Rituximab in Multiple Sclerosis (NOR-MS) [Norwegian study of oral Cladribine and Rituximab in multiple sclerosis (NOR-MS) a prospective randomized open-label blinded endpoint (PROBE) multicenter non-inferiority study]. ClinicalTrials.gov/show/NCT04121403 (first received 9 October 2019).
NCT04578639 {published data only}
- Ocrelizumab VErsus Rituximab Off-Label at the Onset of Relapsing MS Disease (OVERLORD-MS) [Ocrelizumab versus Rituximab off-label at the onset of relapsing MS]. ClinicalTrials.gov/show/NCT04578639 (first received 8 October 2020).
NCT04688788 {published data only}
- Non-inferiority study of Ocrelizumab and Rituximab in active Multiple Sclerosis (DanNORMS) [Danish non-inferiority study of Ocrelizumab and Rituximab in MS (DanNORMS): a randomized study comparing the efficacy of Ocrelizumab and Rituximab in active multiple sclerosis]. ClinicalTrials.gov/show/NCT04688788 (first received 30 December 2020).
NCT04695080 {published data only}
- ChariotMS - Cladribine to halt deterioration in people with advanced multiple sclerosis (ChariotMS) [ChariotMS - A national (UK) phase IIB, multi-centre, randomised, double-blind, placebo controlled (1:1) efficacy trial with cost-utility analysis of Cladribine tablets (3.5mg/kg over two years) in people with advanced multiple sclerosis. Is Cladribine superior to placebo in protecting upper limb function?]. ClinicalTrials.gov/show/NCT04695080 (first received 5 January 2021).
NCT04788615 {published data only}
- NCT04788615. Open label randomized multicenter to assess efficacy & tolerability of Ofatumumab 20mg vs. first line DMT in RMS (STHENOS) [Open-label rater-blind randomized multi-center parallel-arm active- comparator study to assess the efficacy and tolerability of Ofatumumab 20mg sc monthly vs. first line DMT - physician's choice in the treatment of newly diagnosed RMS]. ClinicalTrials.gov/show/NCT04788615 (first received 9 March 2021).
NCT05090371 {published data only}
- NCT05090371. A multicenter study of continued current therapy vs transition to Ofatumumab after neurofilament (NFL) elevation (SOSTOS) [A randomized, open label, multi-center, active-comparator study to assess efficacy, safety & tolerability of Ofatumumab 20mg sc monthly versus continued current therapy in relapsing-remitting multiple sclerosis after elevation of serum neurofilament light levels (SOSTOS)]. ClinicalTrials.gov/show/NCT05090371 (first received 22 October 2021).
RAMBLE 2021 {published data only}
- ACTRN12621001502820. Reducing the frequency of autoimmune adverse events in the treatment of multiple sclerosis with alemtuzumab using B-cell depletion (RAMBLE): a phase II, randomised, placebo-controlled clinical trial. https://trialsearch.who.int/Trial2.aspx?TrialID=ACTRN12621001502820 2021.
Additional references
Amato 2015
- Amato MP, Portaccio E. Fertility, pregnancy and childbirth in patients with multiple sclerosis: impact of disease-modifying drugs. CNS Drugs 2015;29(3):207-20. [Google Scholar]
Bender 2008
- Bender R, Bunce C, Clarke M, Gates S, Lange S, Pace NL, Thorlund K. Attention should be given to multiplicity issues in systematic reviews. Journal of Clinical Epidemiology 2008;61:857-65. [Google Scholar]
Bomprezzi 2015
- Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Therapeutic Advances in Neurological Disorders 2015;8(1):20-30. [Google Scholar]
Bourdette 2016
- Bourdette D. Rituximab for treating multiple sclerosis: off-label but on target. Neurology 2016;87:2070-1. [Google Scholar]
Brignardello‐Petersen 2021
- Brignardello-Petersen R, Guyatt GH, Mustafa RA, Chu DK, Hultcrantz M, Schünemann HJ, Tomlinson G. GRADE guidelines 33: Addressing imprecision in a network meta-analysis. J Clin Epidemiol 2021;139:49-56. [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta-analysis to evaluate the existence of small-study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161-76. [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta-analysis in Stata. PLoS One 2013;8(10):e76654. [Google Scholar]
Chen 2005
- Chen T, Hoppe FM. Simultaneous confidence intervals. In: In: Armitage P, Colton T (editors). Encyclopedia of Biostatistics (2nd edition). Chichester (UK): John Wiley & Sons, 2005, 2005. [Google Scholar]
Chouhfeh 2015
- Chouhfeh L, Kavak KS, Teter BE, Weinstock-Guttman B. Disease modifying therapies use associated with comorbid autoimmune diseases in multiple sclerosis patients. Multiple Sclerosis and Related Disorders 2015;4(3):228-33. [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Controlled Clinical Trials 1986;7:177-88. [Google Scholar]
Dias 2010
- Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta-analysis. Statistics in Medicine 08 March 2010;Volume 29(Issue 7-8):932-44. [Google Scholar]
Farber 2015
- Farber RS, Sand IK. Optimizing the initial choice and timing of therapy in relapsing-remitting multiple sclerosis. Therapeutic Advances in Neurological Disorders 2015;8(5):212-32. [Google Scholar]
FDA 2020
- CFR - Code of Federal Regulations Title 21. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=312.32 (accessed 27 October 2020).
Federal Register 2015
- Content and format of labeling for human prescription drug and biological products; requirements for pregnancy and lactation labeling. https://www.federalregister.gov/articles/2014/12/04/2014-28241/content-and-format-of-labeling-for-human-prescription-drug-and-biological-products-requirements-for (accessed 18 April 2016).
Filippini 2013
- Filippini G, Del Giovane C, Vacchi L, D'Amico R, Di Pietrantonj C, Beecher D, Salanti G. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta-analysis. Cochrane Database of Systematic Reviews 2013, Issue 6. Art. No: CD008933. [DOI: 10.1002/14651858.CD008933.pub2] [DOI] [Google Scholar]
Garattini 2013
- Garattini S, Bertelé V, Banzi R. Placebo? no thanks, it might be bad for me! European Journal of Clininical Pharmacology 2013;69(3):711-4. [Google Scholar]
Glanville 2019
- Glanville Julie, Foxlee Ruth, Wisniewski Susi, Noel-Storr Anna, Edwards Mary, Dooley Gordon. Translating the Cochrane EMBASE RCT filter from the Ovid interface to Embase.com: a case study. Health Information & Libraries Journal 2019;36(3):264-277. [Google Scholar]
Glanville 2019a
- Glanville Julie, Dooley Gordon, Wisniewski Susi, Foxlee Ruth, Noel-Storr Anna. Development of a search filter to identify reports of controlled clinical trials within CINAHL Plus. Health Information & Libraries Journal 2019;36(1):73-90. [Google Scholar]
Goodman 2006
- Goodman LS, Gilman A, Brunton LL, Lazo JS, Parker KL. Goodman & Gilman's the pharmacological basis of therapeutics. New York: McGraw-Hill, 2006. [Google Scholar]
GRADE Working Group 2004
- GRADE Working Group. Grading quality of evidence and strength of recommendations. British Medical Journal 2004;328:1490-4. [Google Scholar]
Hamidi 2018
- Hamidi V, Couto E, Ringerike T, Klemp M. A multiple treatment comparison of eleven disease-modifying drugs used for multiple sclerosis. Journal of Clinical Medicine Research 2918;10(2):88-105. [Google Scholar]
Hartung 2021
- Hartung HP, Meuth SG, Thompson AJ. Paradigm shifts: Early initiation of high-efficacy disease-modifying treatment in multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2021;27(10):1473-6. [Google Scholar]
Higgins 2011
- Higgins J, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane-handbook.org.
ICH 2015
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Introductory guide MedDRA version 18.0. http://www.meddra.org/sites/default/files/guidance/file/intguide_18_0_english.pdf (accessed 18 April 2016).
Ioannidis 2004
- Ioannidis JP, Evans SJ, Gøtzsche PC, O'Neill RT, Altman DG, Schulz K, et al, CONSORT Group. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Annals of Internal Medicine 2004;141(10):781-8. [Google Scholar]
Jackson 2014
- Jackson D, Barrett JK, Rice S, White IR, Higgins JP. A design-by-treatment interaction model for network meta-analysis with random inconsistency effects. Statistics in Medicine 2014;33(21):3639-54. [Google Scholar]
Kanters 2016
- Kanters S, Ford N, Druyts E, Thorlund K, Mills EJ, Bansback N. Use of networkmeta-analysis in clinical guidelines. Bulletin of the World Health Organization 2016;94(10):782-4. [Google Scholar]
Lebrun 2018
- Lebrun C, Rocher F. Cancer risk in patients with multiple sclerosis: potential impact of disease-modifying drugs. CNS Drugs 2018;32(10):939-49. [Google Scholar]
Lefebvre 2022
- Lefebvre C, Glanville J, Briscoe S, Featherstone R, Littlewood A, Marshall C, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). The Cochrane Collaboration, 2022. Available from training.cochrane.org/handbook.
Li 2020
- Li H, Hu F, Zhang Y, Li K. Comparative efficacy and acceptability of disease-modifying therapies in patients with relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. Journal of Neurology 2020;267(12):3489-98. [Google Scholar]
Lopez‐Leon 2020
- Lopez-Leon S, Geissbühler Y, Sabidó M, Turkson M, Wahlich C, Morris JK. A systematic review and meta-analyses of pregnancy and fetal outcomes in women with multiple sclerosis: a contribution from the IMI2 ConcePTION project. Journal of Neurology 2020;267(8):2721-31. [Google Scholar]
Lublin 1996
- Lublin F, Reingold S. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996;46:907-11. [Google Scholar]
Lublin 2014
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83(3):278-86. [Google Scholar]
Lucchetta 2019
- Lucchetta RC, Leonart LP, Becker J, Pontarolo R, Fernandez-Llimós F, Wiens A. Safety outcomes of disease-modifying therapies for relapsing-remitting multiple sclerosis: A network meta-analysis. Multiple Sclerosis and Related Disorders 2019;35:7-15. [Google Scholar]
Lycke 2015
- Lycke J. Monoclonal antibody therapies for the treatment of relapsing-remitting multiple sclerosis: differentiating mechanisms and clinical outcomes. Therapeutic Advances in Neurological Disorders 2015;8(6):274-93. [Google Scholar]
Manríquez 2008
- Manríquez Juan J. A highly sensitive search strategy for clinical trials in Literatura Latino Americana e do Caribe em Ciências da Saúde (LILACS) was developed. Journal of Clinical Epidemiology 2008;61(4):407-11. [Google Scholar]
Marshall 2018
- Marshall Iain J, Noel-Storr Anna, Kuiper Joël, Thomas James, Wallace Byron C. Machine learning for identifying Randomized Controlled Trials: An evaluation and practitioner's guide. Research Synthesis Methods 2018;9(4):602-14. [Google Scholar]
Martìnez‐Càceres 1998
- Martínez-Cáceres EM, Río J, Barrau M, Durán I, Borrás C, Tintoré M, et al. Amelioration of flulike symptoms at the onset of interferon beta-1b therapy in multiple sclerosis by low-dose oral steroids is related to a decrease in interleukin-6 induction. Annals of Neurology 1998;44(4):682-5. [Google Scholar]
McCool 2019
- McCool R, Wilson K, Arber M, Fleetwood K, Toupin S, Thom H, et al. Systematic review and network meta-analysis comparing ocrelizumab with other treatments for relapsing multiple sclerosis. Multiple Sclerosis and Related Disorders 2019;29:55-61. [Google Scholar]
McDonald 2001
- McDonald W, Compston A, Edan G, Goodkin D, Hartung H, Lublin F. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Annals of Neurology 2001;50:121-7. [Google Scholar]
McDonald 2017
- McDonald S, Noel-Storr AH, Thomas J. Harnessing the efficiencies of machine learning and Cochrane Crowd to identify randomised trials for individual Cochrane reviews. In: Global Evidence Summit; 2017 September 13-16; Cape Town, South Africa. 2017.
Miladinovic 2014
- Miladinovic B, Hozo I, Chaimani A, Djulbegovic B. Indirect treatment comparison. Stata Journal 2014;14(1):76–86. [Google Scholar]
Montalban 2018
- Montalban X, Gold R, Thompson AJ, Otero-Romero S, Amato MP, Chandraratna D, et al. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Multiple Sclerosis 2018;24(2):96-120. [Google Scholar]
Multiple‐Treatments Meta‐analysis (MTM)
- Multiple-Treatments Meta-analysis (MTM). A framework for evaluating and ranking multiple healthcare technologies. http://www.mtm.uoi.gr/ (accessed 18 April 2016).
Nikolakopoulou 2018
- Nikolakopoulou A, Mavridis D, Furukawa TA, Cipriani A, Tricco AC, Straus SE, et al. Living network meta-analysis compared withpairwise meta-analysis in comparative effectiveness research: empirical study. British Medical Journal 2018;360:k585. [Google Scholar]
Nikolakopoulou 2020
- Nikolakopoulou A, Higgins JPT, Papakonstantinou T, Chaimani A, Del Giovane C, Egger M, Salanti G. CINeMA: An approach for assessing confidence in the results of a network meta-analysis. PLOS Medicine 2020;3:17. [Google Scholar]
Noel‐Storr 2018
- Noel-Storr AH and the Project Transform Team. Cochrane Crowd: new ways of working together to produce health evidence. In: Evidence Live; 2018 June 18-20; Oxford, UK. 2018.
Peryer 2020
- Peryer G, Golder S, Junqueira D, Vohra S, Loke YK. Chapter 19: Adverse effects. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020.. Available from www.training.cochrane.org/handbook.
Peters 2008
- Peters J, Sutton A, Jones D, Abrams K, Rushton L. Contour-enhanced meta-analysis funnel plots help distinguish publication bias from other causes of asymmetry. Journal of Clinical Epidemiology 2008;61(10):991-6. [Google Scholar]
Polman 2005
- Polman C, Reingold S, Edan G, Filippi M, Hartung H, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the 'McDonald Criteria'. Annals of Neurology 2005;58:840-6. [Google Scholar]
Polman 2011
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of Neurology 2011;69:292–302. [Google Scholar]
Poser 1983
- Poser C, Paty D, Scheinberg L, McDonald W, Davis F, Ebers G, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Annals of Neurology 1983;13:227-31. [Google Scholar]
Prosperini 2020
- Prosperini L, Mancinelli CR, Solaro CM, Nociti V, Haggiag S, Cordioli C, et al. Induction versus escalation in multiple sclerosis: A 10-year real world study. Neurotherapeutics 2020;17(3):994-1004. [Google Scholar]
Rae‐Grant 2018
- Rae-Grant A, Day GS, Marrie RA, Rabinstein A, Cree BAC, Gronseth GS, et al. Practice guideline recommendations summary: Disease-modifying therapies for adults with multiple sclerosis. Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018;90(17):777-88. [Google Scholar]
Rucker 2012
- Rücker G. Network meta-analysis, electrical networks and graph theory. Research Synthesis Methods 2012;3:312-24. [Google Scholar]
Rucker 2015
- Rücker G, Schwarzer G. Ranking treatments in frequentist network meta-analysis works without resampling methods. BMC Medical Research Methodology 2015;15:58. [Google Scholar]
Salanti 2011
- Salanti G, Ades A, Ioannidis J. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64:163-71. [Google Scholar]
Salanti 2012
- Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Research Synthesis Methods 2012;3(2):80–97. [Google Scholar]
Samjoo 2020
- Samjoo IA, Worthington E, Drudge C, Zhao M, Cameron C, Häring DA, et al. Comparison of ofatumumab and other disease-modifying therapies for relapsing multiple sclerosis: a network meta-analysis. Journal of Comparative Effectiveness Research 2020;9(18):1255-74. [Google Scholar]
Schwrtzer 2015
- Schwarzer G, Carpenter GR, Rücker G. Meta-Analysis with R. In: Meta-Analysis with R. Springer, 2015. [Google Scholar]
Schünemann 2011
- Schünemann H, Oxman A, Higgins J, Vist G, Glasziou P, Guyatt G. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane-handbook.org.
Siddiqui 2018
- Siddiqui MK, Khurana IS, Budhia S, Hettle R, Harty G, Wong SL. Systematic literature review and network meta-analysis of cladribine tablets versus alternative disease-modifying treatments for relapsing-remitting multiple sclerosis. Current Medical Research and Opinion 2018;34(8):1361-71. [Google Scholar]
Silva 2022
- Silva GD, Castrillo BB, Apóstolos-Pereira SL, Callegaro D. Is there a role for off-label high-efficacy disease-modifying drugs in progressive multiple sclerosis? A network meta-analysis. Acta Neurol Scandinavica 2022;146(5):403-9. [Google Scholar]
Simpson 2021
- Simpson A, Mowry EM, Newsome SD. Early aggressive treatment approaches for multiple sclerosis. Current Treatment Options in Neurology 2021;23(7):19. [Google Scholar]
Thomas 2017
- Thomas J, Noel-Storr A, Marshall I, Wallace B, McDonald S, Mavergames C, et al. Living systematic reviews: 2. Combining human and machine effort. Journal of Clinical Epidemiology 2017;91:31-7. [Google Scholar]
Tramacere 2015
- Tramacere I, Del Giovane C, Salanti G, D'Amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: a network meta-analysis. Cochrane Database of Systematic Reviews 2015, Issue 9. Art. No: CD011381. [DOI: 10.1002/14651858.CD011381.pub2] [DOI] [Google Scholar]
Van Assche 2005
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. New England Journal of Medicine 2005;353(4):362-8. [Google Scholar]
Walton 2020
- Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Multiple Sclerosis 2020;26(14):1816-21. [Google Scholar]
White 2011
- White IR. Multivariate random-effects meta-regression: updates to mvmeta. The STATA Journal 2011;11:255-70. [Google Scholar]
White 2012
- White IR, Barrett JK, Jackson D, Higgins JPT. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Research Synthesis Methods 2012;3(2):111-25. [Google Scholar]
Williamson 2015
- Williamson EM, Berger JR. Central nervous system infections with immunomodulatory therapies. Continuum (Minneapolis Minn.) 2015;21(6 Neuroinfectious Disease):1577-98. [Google Scholar]
Xu 2018
- Xu X, Chi S, Wang Q, Li C, Xu B, Zhang J, et al. Efficacy and safety of monoclonal antibody therapies for relapsing remitting multiple sclerosis: A network meta-analysis. Multiple Sclerosis and Related Disorders 2018;25:322-8. [Google Scholar]
Yepes‐Nuñez 2019
- Yepes-Nuñez JJ, Li SA, Guyatt G, Jack SM, Brozek JL, Beyene J, et al. Development of the summary of findings table for network meta-analysis. Journal of Clinical Epidemiology 2019;115:1-13. [Google Scholar]
Zhang 2019
- Zhang Y, Salter A, Wallström E, Cutter G, Stüve O. Evolution of clinical trials in multiple sclerosis. Therapeutic advances in neurological disorders 2019;12:1756286419826547. [Google Scholar]
Zhang 2021
- Zhang Y, Salter A, Jin S, Culpepper WJ 2nd, Cutter GR, Wallin M, et al. Disease-modifying therapy prescription patterns in people with multiple sclerosis by age. Therapeutic Advances In Neurological Disorders 2021;14:17562864211006499. [Google Scholar]
Zorzela 2016
- Zorzela L, Loke YK, Ioannidis JP, Golder S, Santaguida P, Altman DG, et al. PRISMA harms checklist: improving harms reporting in systematic reviews. BMJ 2016;352:i157. [Google Scholar]
Śladowska 2022
- Śladowska K, Kawalec P, Holko P, Osiecka O. Comparative safety of high-efficacy disease-modifying therapies in relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. Neurological sciences 2022;43(9):5479-500. [Google Scholar]
References to other published versions of this review
Tramacere 2016
- Tramacere I, Benedetti MD, Capobussi M, Castellini G, Citterio A, Del Giovane C, et al. Adverse effects of immunotherapies for multiple sclerosis: a network meta-analysis. Cochrane Database of Systematic Reviews 2016, Issue 5. Art. No: CD012186. [DOI: 10.1002/14651858.CD012186] [DOI] [Google Scholar]