

Abstract
The macromolecular composition and a number of parameters affecting chromosome replication were examined over a range of exponential growth rates in two common Escherichia coli strains, B/r and K-12 AB1157. Based on improved measurements of DNA after treatment of exponential cultures with rifampin, the cell mass per chromosomal replication origin (initiation mass) and the time required to replicate the chromosome from origin to terminus (C period) were determined. For these two strains, the initiation mass approached values of 8 × 10−10 and 10 × 10−10 units of optical density (at 460 nm) of culture mass per oriC, respectively, at growth rates above 1 doubling/h (at 37°C). The amount of protein per oriC decreased with increasing growth rate for AB1157 and remained nearly constant for the B/r strain. The C period decreased for both strains in an essentially identical manner from about 70 min at 0.6 doublings/h to about 33 min at 3 doublings/h. From the initiation mass and C period, relative or absolute copy numbers for genes with known map locations can be accurately determined at different growth rates. At growth rates above 2 doublings/h, when chromosomes are highly branched, genes near the origin are about threefold more prevalent than genes near the terminus. At a growth rate of 0.6 doubling/h, this ratio is only about 1.7, which reflects the lower degree of chromosome branching.
During exponential growth of a bacterium such as Escherichia coli, chromosome replication and other aspects of macromolecular synthesis are maintained in precise balance. At a given growth rate, the activity of a particular gene, measured either as RNA transcripts or as protein product, depends both on genetic control of the gene in question and on the copy number. The average copy number of a gene per unit of cell mass depends on (i) the location of the gene relative to the replication origin and terminus, (ii) the velocity of replication fork movement, and (iii) the timing of initiation of replication at the origin. The location or map position of a gene is determined by standard genetic or molecular techniques. The replication velocity is given by the time required to replicate the chromosome (C period) and was first measured by Helmstetter and Cooper (20) for E. coli B/r. The timing of replication initiation is characterized by the cell mass or by the amount of protein per replication origin at the time of initiation (initiation mass, Mi [16]). More conveniently, a related parameter (M0) is used; this is the cell mass or protein in a given volume of exponential culture, divided by the total number of oriC copies in that volume. The two parameters are related by the simple relationship M0 = ln 2 · Mi (10). Of the two parameters, C and M0, the former determines the extent of chromosome branching and thereby affects the copy number of particular genes depending on their map location; the latter determines the overall DNA concentration and thereby affects the transcription of all genes by influencing the concentration of free RNA polymerase.
Few attempts have been made to measure the initiation mass, particularly for E. coli K-12 strains. Because of this, uncertainty exists as to whether the initiation mass is constant (16), increases (15), or decreases (32) with increasing growth rate. Moreover, there is also uncertainty about whether the C period is constant at growth rates above 1 doubling per h (20) or whether it continues to decrease with increasing growth rate (14). In this work, we have attempted to carefully measure the initiation mass (M0) and replication velocity (C period), in addition to DNA, RNA, and protein content, for E. coli B/r and the K-12 strain AB1157 at different growth rates. The results supplement and extend our previously reported values for the macromolecular composition of E. coli B/r (11). In the accompanying Appendix, we have used the measurements of M0 and C to determine the copy number of various loci located around the bacterial chromosome. This provides a basis for distinguishing between the contributions of gene dosage and the effects of genetic regulation on the level of expression of a gene.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. coli K-12 strain AB1157 (2) requires arginine, histidine, proline, leucine, threonine, and thiamine. The E. coli B/r A strain HB123 requires phenylalanine and threonine. Cultures were grown at 37°C in a shaker water bath, with medium C (18), supplemented with 0.2% (wt/vol) carbon source (succinate, glycerol, or glucose) and required amino acids at 50 μg/ml and thiamine (for AB1157) at 2 μg/ml. To obtain faster growth, 0.2% Difco Casamino Acids plus 50 μg of tryptophan per ml and 500 μg of serine per ml or Luria-Bertani (LB) medium (27) with 0.2% glucose were used.
For consistency, the succinate minimal medium for both strains was supplemented with the same five amino acids (arginine, histidine, proline, leucine, and threonine required by AB1157). In our hands, strain AB1157 grew at almost the same rate in succinate minimal medium as it did in glycerol minimal medium. This was surprising since glycerol, a glycolytic intermediate, is considered to be more energy rich than succinate, a Krebs cycle intermediate. With five required amino acids, these media used are not typical minimal media; it is likely that some of the amino acids were also metabolized as a carbon source.
For both E. coli B/r and AB1157, threonine, when present at low concentrations, is depleted from the medium in overnight cultures. The resulting amino acid starvation produced a growth lag and perturbed the physiology of experimental cultures started from such overnight cultures. To avoid such starvation effects, the threonine concentration in overnight cultures was increased to 200 μg/ml. Experimental cultures of bacteria were started by 1:500 (poor media) to 1:2,000 (rich media) dilution of an overnight culture in a medium equal to or poorer than the medium to be used, in order to avoid a nutritional shift-down associated with a long growth lag (24). Except for cultures in LB medium, growth was monitored by measuring the turbidity, or optical density (OD) at a 460-nm wavelength (OD460), with a 1-cm light path. In the case of LB medium, the OD600 was measured. By monitoring both the OD460 and the OD600 of cultures growing in different media, parallel growth curves were obtained, differing by a factor of 1.60 ± 0.03 (OD460/OD600), which was used to convert OD600 units into OD460 units for cultures in LB medium. Rifampin at 300 μg/ml (final concentration) was added at an OD460 of about 0.7 for minimal medium or at an OD600 of about 0.43 for LB medium.
Collection of bacteria on glass fiber filters.
Bacteria in culture samples (2.5 ml for DNA; 4.0 ml for RNA and protein) were collected on 24-mm glass fiber filters. Whatman GF/F filters with 0.7-μm maximum pore size retained 99% of the bacterial mass under all conditions. For samples used for DNA determinations, collection filters were presoaked in 1 mM perchloric acid (PCA) to reduce the background in the diphenylamine assay. To remove small metabolites, the bacteria were treated with 0.45 M PCA at 0°C before filtration. The bacteria were washed on the filters with 1 mM PCA, and the filters were placed into vials (scintillation vials) for further processing (see below). Since LB medium contains significant amounts of acid-precipitable oligonucleotides and polypeptides, bacteria in LB medium were immediately collected on glass fiber filters without acid precipitation and washed with 1 mM MgSO4 at 0°C. After this washing, about 3 ml of 0.5 M PCA was slowly filtered through the same filter with reduced vacuum. When samples contained rifampin, filters were further washed with acetone to remove all visible rifampin.
Determination of DNA in samples of bacterial culture.
DNA was determined by the diphenylamine assay (13) as follows. To each vial with a dry filter, 0.5 ml of 1.6 M PCA was added, and the closed vials were incubated at 70°C for 30 min before the addition of 1 ml of diphenylamine reagent (1 g of diphenylamine, reagent grade [Kodak], and 125 μl of aqueous solution of 32-mg/ml acetaldehyde per 50 ml of glacial acetic acid). After incubation at 30°C for 14 to 16 h overnight, reaction mixtures were clarified by filtration through glass fiber filters before the A600 was read. For filtration, a special apparatus designed to facilitate the collection of small volumes of filtrate was used. Samples obtained with medium without bacteria and processed in an identical manner served as medium blanks. Assays of known amounts of deoxyadenosine (AdR) or calf thymus DNA (CT-DNA) for calibration were incubated at the same time; for these assays, acid precipitation and filtration steps were omitted (see Appendix for further details).
Concentrations of DNA or AdR used for calibration were measured by their UV absorbance at pH 12 (0.01 M NaOH), with molar extinction coefficients (ɛ260 = A260 of 1 mM solution at pH 12) of 15.4 for AdR and 10.61 for CT-DNA (calculated from GC content = 43% [23]). Assay values increased linearly with the amount of DNA up to A600 values of at least 1.0. The E. coli genome was assumed to be 4.7 Mbp (3).
RNA and protein determinations.
Four to five 4-ml samples were taken from exponential cultures over a period of one to two generations and added to 0.16 ml of concentrated PCA at 0°C. Bacteria were collected on glass fiber filters as described above. For cultures in LB medium, the medium was again first removed by filtration and washed out before acid treatment of the filter as described above. In addition, three samples of medium without bacteria were taken as medium blanks. The dried filters were placed in scintillation vials, and 2.0 ml of freshly prepared 0.2 M NaOH was added to dissolve proteins and hydrolyze RNA. After overnight incubation at 30°C, 0.5 ml of the alkaline solution was removed for protein determination, by the Lowry assay (26) as described elsewhere (12). To the remaining 1.5 ml of hydrolysate, an equal volume of 0.5 M PCA was added to precipitate protein and DNA at 0°C. The precipitate and debris were removed from the samples by filtration through glass fiber filters. The UV absorption of the clear filtrates (RNA hydrolysates) was measured at 260 nm. The absorption spectrum suggests that the samples were not contaminated to a significant extent by UV-absorbing nonnucleotide materials. The A260 values were converted into RNA nucleotides by assuming that one A260 unit (at acid pH) corresponds to 93 nmol of RNA nucleotides, calculated from the base composition of E. coli RNA (28).
For calibration of the Lowry assay, a concentrated stock solution of bovine serum albumine (BSA) was first diluted into 0.2 M NaOH to a final concentration of approximately 1 mg/ml. From this solution, a series of further dilutions into 0.2 M NaOH were made to obtain concentrations between 0 and 1 mg/ml in steps of 0.2 mg/ml; duplicate 0.5-ml aliquots from each dilution were assayed by the addition to 2.5 ml of alkaline copper reagent (0.02% [wt/vol] sodium tartrate, 0.01% [wt/vol] CuSO4, 0.2 M Na2CO3, 0.1 M NaOH) and 0.5 ml of 1 N Folin–Ciocalteu’s phenol reagent (Sigma Chemical). The A750 of the assay was read after 3 to 4 h.
BSA concentrations were determined from their A280. After a sample of BSA was dried for 24 h at 50°C under vacuum in a tube of known weight, the BSA was weighed with an analytical balance and dissolved in a given volume of water. The ɛ280 of the aqueous solution was found to be 0.621/mg. In previous work, this value was determined to be 0.695 (30). The difference presumably reflects differences in the particular commercial BSA preparation.
The A750 of the protein assay increases nonlinearly with the protein concentration; the calibration curve could be approximated by a parabola, similar to that described below for the mass density (equation 9 [Appendix]). The parameters defining this parabola were obtained by a best-fit method (see Fig. A1 [Appendix] and Table 1) and used to convert the observed A750 values into protein concentrations.
TABLE 1.
Activity of the gene rplN for r-protein L14 in E. coli B/r growing at different rates at 37°C
| τ (min) | μ (doublings/h) | 1012 L14 molecules/OD460 U of cell massa | 103 L14 molecules/oriCb | No. of rplN genes/oriCc | 103 L14 molecules/rplN genesd | No. of L14 molecules synthesized/min/ rplN genee |
|---|---|---|---|---|---|---|
| 100 | 0.6 | 6 | 3 | 0.90 | 4 | 26 |
| 60 | 1.0 | 7 | 4 | 0.85 | 5 | 58 |
| 40 | 1.5 | 8 | 8 | 0.82 | 9 | 158 |
| 30 | 2.0 | 10 | 9 | 0.80 | 12 | 270 |
| 24 | 2.5 | 11 | 10 | 0.79 | 13 | 369 |
| 20 | 3.0 | 13 | 11 | 0.77 | 14 | 474 |
The number of L14 molecules in a culture was assumed to be equal to the number of ribosomes present. The number of ribosomes per cell mass was obtained from the total amount of RNA per mass (in RNA nucleotides per OD460 unit [Fig. 4b]) by multiplication with 0.84 (84% of total RNA is rRNA) and division by the number of rRNA nucleotides in a 70S ribosome (4,566 [11]).
The number of L14 molecules per oriC was found from the number of L14 molecules per cell mass above by multiplication with mass per oriC (initiation mass [Fig. 5a]).
The number of rplN genes per oriC was found from the map location of rplN (at 73 min [1]) and the C period (Fig. 3a), by equations 4 and 6 (Appendix).
The number of L14 molecules per rplN gene was found by dividing the number of L14 molecules per oriC by the number of rplN genes per oriC.
The synthesis rate of L14 per rplN gene (in L14 molecules per minute per gene) equals the product of the number of L14 molecules per gene multiplied by the growth rate (ln 2/τ).
RESULTS
Principle of the method.
Figure 1 illustrates an idealized plot of the accumulation of DNA (measured in genome equivalents) in an exponential culture with a doubling time of 45 min. Since most chromosomes in the culture are in the process of replication, they will have multiple origins (in this case, two) and a single terminus and will contain more than one genome equivalent of DNA. The numbers of replication origins, termini, and DNA genome equivalents (NoriC, NterC, and G, respectively) increase exponentially in a parallel fashion with the same doubling time (τ), such that, in a semilog plot of these parameters versus growth time, the horizontal distance between the origin and terminus curve represents the time required to replicate the chromosome from origin to terminus (C period) and the vertical distance between the origin and DNA curve represents the number of origins per genome equivalent of DNA (NoriC/G, or ΔG; Fig. 1).
FIG. 1.

Theoretical accumulation of DNA in an exponential culture of E. coli treated with rifampin. The method used to find the number of replication origins and the C period from the accumulation of DNA after stopping initiations with rifampin (Rif) is illustrated. Solid line, DNA in genome equivalents (Geq) in a given volume of culture, normalized to 1.0 at t = 0 when rifampin is added to the culture; stippled line, number of replication origins (Ori); dotted line, number of replication termini (Ter). The horizontal distance between the origin and terminus curve corresponds to C, and the vertical distance between the origin and DNA curve (ΔG) represents the number of origins per genome equivalent of DNA. Relat., relative.
Rifampin inhibits initiation of transcription, and its action indirectly blocks initiation of new rounds of replication without affecting elongation and completion of replications already in progress (31). As a consequence, the number of origins in a culture remains constant at a value equal to the number of origins present at the time of rifampin addition. At the same time, the rate of accumulation of DNA begins to diminish because there are no new replication forks generated by initiation and existing forks gradually reach the replication terminus and cease to function. The number of termini continues to increase at an exponential rate until all forks have reached the terminus. At this point, the numbers of origins, genome equivalents of DNA, and termini are all equal; that is, the chromosomes are fully replicated, contain a single genome equivalent of DNA, and possess a single origin and a single terminus.
The experiments to be described below measure the accumulation of DNA after the addition of rifampin to exponential cultures. The factor increase in DNA after rifampin addition (ΔG) is a measure of the number of origins per genome at time zero (Fig. 1). Similarly, the time required for the DNA accumulation curve to reach the plateau is a measure of the C period.
Determination of chromosomal replication origins and replication velocity.
The number of replication origins, oriC, and the replication velocity (C period; the time to replicate the chromosome from origin to terminus) were determined for the E. coli B/r A strain HB123 and the K-12 strain AB1157 by monitoring the accumulation of DNA after the addition of rifampin (see above and Appendix, first section). The nonradioactive method used here gives absolute values for DNA genome equivalents and origins. Figure 2 shows an example for strain B/r grown in LB medium. During exponential growth before the addition of rifampin, both mass density and DNA increased with the same 21-min doubling time. From the data in Fig. 2a and b, the DNA/mass ratio (G/M) was determined to be 7.3 × 108 genome equivalents of DNA per OD460 unit of culture mass.
FIG. 2.

Accumulation of DNA in E. coli B/r grown in LB medium after treatment with rifampin (Rif) to inhibit initiation of rounds of replication. (a) Growth curve (OD600); (b) DNA accumulation (A600 of diphenylamine assay); (c) replot of the DNA curve in panel b after normalization to 1.0 at t = 0, the time of rifampin addition. The parameter γ is defined as the difference of the exponential curve (dashed) minus observed values. The solid curve was generated as a best fit by equation 3 (Appendix). (d) The 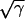 curve extrapolates to d = 2.5 min on the abscissa. This value is an estimate of the delay in the action of rifampin on initiation of replication (see text and Appendix). Relat., relative; Theor., theoretical.
curve extrapolates to d = 2.5 min on the abscissa. This value is an estimate of the delay in the action of rifampin on initiation of replication (see text and Appendix). Relat., relative; Theor., theoretical.
To find the number of replication origins and the C period from the DNA curve, the observed DNA values (A600 in Fig. 2b) were normalized to 1.0 at the time of rifampin addition (t = 0; Fig. 2c), and these normalized DNA values were subtracted from the exponential term 2t/τ (dashed curve in Fig. 2b and c). The square root of this difference was plotted as a function of the sampling time (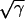 in Fig. 2d). Extrapolation of the
in Fig. 2d). Extrapolation of the 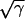 curve to the time axis gives a value that corresponds to the delay (d) in the action of rifampin on initiation of chromosome replication (8). In the experiment whose results are shown in Fig. 2, d was estimated to be 2.5 min (Fig. 2d). In other experiments, the extent of the delay varied both as a function of the growth medium and from culture to culture in a given medium. In minimal media, delays of up to 12 min have been observed (see Discussion). From the delay (d) and the culture doubling time (τ), an expectation for the DNA accumulation after rifampin addition was calculated (with equation 3 [Appendix]) with an assumed value for C. By reiterative calculations in which the value of C was varied, the best fit (solid curve in Fig. 2c) with observed data points was obtained; this happened when C was set at 33 min (Fig. 2c). Thus, a C period of 33 min most closely approximates the kinetics of DNA accumulation after rifampin treatment of the culture. In these curves, points taken between 10 and 30 min after rifampin addition often fell slightly below the theoretical best-fit curve. This suggests that the replication velocity may be somewhat reduced in the presence of rifampin.
curve to the time axis gives a value that corresponds to the delay (d) in the action of rifampin on initiation of chromosome replication (8). In the experiment whose results are shown in Fig. 2, d was estimated to be 2.5 min (Fig. 2d). In other experiments, the extent of the delay varied both as a function of the growth medium and from culture to culture in a given medium. In minimal media, delays of up to 12 min have been observed (see Discussion). From the delay (d) and the culture doubling time (τ), an expectation for the DNA accumulation after rifampin addition was calculated (with equation 3 [Appendix]) with an assumed value for C. By reiterative calculations in which the value of C was varied, the best fit (solid curve in Fig. 2c) with observed data points was obtained; this happened when C was set at 33 min (Fig. 2c). Thus, a C period of 33 min most closely approximates the kinetics of DNA accumulation after rifampin treatment of the culture. In these curves, points taken between 10 and 30 min after rifampin addition often fell slightly below the theoretical best-fit curve. This suggests that the replication velocity may be somewhat reduced in the presence of rifampin.
Similar experiments were carried out with E. coli B/r and the K-12 strain AB1157 growing in a variety of media that gave exponential growth rates of between 0.6 and 3.0 doublings/h. For both strains, the estimate for the length of C in these experiments was not constant; instead, C decreased from about 70 min in duration at a growth rate of 0.6 doublings/h to about 33 min at a growth rate of 3.0 doublings/h (Fig. 3a). The estimates of C for B/r and K-12 strains appear to fall on the same decreasing curve that extrapolates to C = 80 min for μ → 0 (μ is the bacterial growth rate in doublings per hour, equal to 60/τ).
FIG. 3.

Growth rate variation in the velocity of chromosome replication. (a and b) C period (in minutes in panel a; period as a fraction of culture doubling time, τ, in panel b) and (c) number of origins per genome equivalent of DNA are plotted as functions of growth rate for E. coli B/r (circles) and the K-12 strain AB1157 (triangles). The media used were (in the order of increasing growth rate) succinate minimal (open symbols), glycerol minimal, glucose minimal, glucose-amino acids, and LB (solid symbols). Genome equ., genome equivalents.
The velocity of the replication forks relative to the culture growth rate (i.e., the ratio of the C period and doubling time, C/τ) determines the extent of chromosome branching (see Appendix). For example, when C is less than τ (i.e., C/τ < 1) there are either zero or two forks per chromosome, and when C is greater than τ (C/τ > 1), there are either two or six forks per chromosome. As the ratio C/τ increases, the proportion of chromosomes with the higher number of forks increases. From the C-period data in Fig. 3a, the curve describing C/τ as a function of growth rate has been calculated (Fig. 3b). At high growth rates above 1.5 doublings/h, C/τ was found to approach a constant value (1.6 [Fig. 3b]), which means that the replication velocity changes to about the same extent as the exponential growth rate. At growth rates below 1.5 doublings/h, C/τ appears to extrapolate to a value of zero or near zero for very slowly growing cultures. If this extrapolation is valid, it implies that in poor medium, exponential mass doubling times increase more rapidly than the C period. Consequently, in very slowly growing cultures, only a small proportion of cells will be in the process of replicating their chromosomes, so that most cells will have either a single unreplicated chromosome or two fully replicated chromosomes.
The number of oriC copies per genome equivalent of DNA is equal to the value of ΔG after rifampin addition (Fig. 1). ΔG depends on C/τ and was calculated from the data in Fig. 3b (with equation 1 [Appendix]). In our experiments, ΔG had a maximum value of about 1.65 at the highest growth rates (Fig. 3c). Values of ΔG > 1.0 indicate branched chromosome structures, i.e., structures with a higher proportion of origins. The apparent plateau in the ΔG curve at growth rates above 2.0 doublings/h indicates that the extent of chromosome branching does not further increase during fast growth and is reflected in a constant C/τ value (Fig. 3b). During slow growth, the ΔG curve extrapolates to 1.0, i.e., one origin per genome equivalent, corresponding to unreplicated (or fully replicated) chromosomes. This would be expected if the culture doubling time became much longer than the chromosome replication time (C/τ → 0 for μ → 0 [Fig. 3b]).
Protein and RNA determinations.
In addition to DNA, protein and RNA were measured in exponential cultures of E. coli B/r and AB1157 (Fig. 4). The two strains were similar in their macromolecular composition, and their compositions changed nearly in parallel as the exponential growth rate was changed. The values for the B/r strain are similar to, and, because of the refined methodologies employed here, presumably more accurate than, our previously reported values (11). For E. coli K-12 strains, no systematic studies of this kind have been reported.
FIG. 4.

Protein (a), RNA (b), and DNA (c) per mass unit of culture (OD460) were measured as functions of growth rate for E. coli B/r and the K-12 strain AB1157. The media and symbols used are as described in the legend to Fig. 3. aa, amino acids; nuc, nucleotides; Geq, genome equivalents.
Initiation mass for B/r and K-12 strains.
The cell mass per origin (initiation mass, M0) was obtained from the number of origins per genome equivalent of DNA (ΔG [Fig. 3b]) and the number of genome equivalents of DNA per OD460 unit of cell mass (Fig. 4c). For E. coli B/r, the initiation mass reached a nearly constant value at growth rates above 1.3 doublings/h, equal to 8 × 10−10 OD460 units per oriC (Fig. 5a). This is in good agreement with a previous report (15), in which the same plateau was reached at 1.6 doublings/h. The initiation mass for the K-12 strain was about 25% higher than the B/r initiation mass and was equal to 1.0 × 10−9 OD460 units per oriC.
FIG. 5.

Relationship between initiation mass and exponential growth rate. Initiation mass expressed as OD460 units of culture mass per oriC (a) and as total protein in amino acid residues per oriC (b) as a function of the growth rate is illustrated. The media and symbols used are as described in the legend to Fig. 3. Open symbols, succinate medium; solid symbols, other media. aa, amino acids; Prot., protein.
The initiation of chromosome replication has often been linked to protein accumulation (reviewed in reference 9). The amount of protein per origin was therefore calculated by multiplying the average mass-per-origin values shown in Fig. 5a by the average protein-per-mass values shown in Fig. 4a. The calculation indicates that for strain B/r, protein per origin was nearly constant, ranging between 3 × 108 and 4 × 108 amino acid residues per oriC, in agreement with previous reports (11). In LB medium, the amount of protein per origin may be slightly lower. For the K-12 strain, the amount of protein per oriC decreased from about 6 × 108 to 4 × 108 amino acid residues in the range from 0.75 to 2.5 doublings/h.
DISCUSSION
Deviations of observed parameters from apparent growth rate functions.
Schaechter et al. (29) found that the composition of bacteria grown in different media is uniquely correlated with the growth rate. Since then, physiological parameters have often been plotted as functions of growth rate. However, the growth rate is not an independent variable; like all other parameters, it depends on the composition of the growth medium, which does not vary in a continuous manner. Therefore, data points obtained with different growth media may not represent a defined function of growth rate, and a curve connecting different observed points should not necessarily imply that actual points exist on this curve between and beyond observed points.
For AB1157 grown in glucose-amino acids medium, the values for the C period, and thus also the values for C/τ and oriC per genome (Fig. 3, triangles at 2 doublings/h), representing four repeats of the same experiment done on different days, are on the average significantly above the curves drawn. Because every observed point shown in Fig. 3 was obtained as an average from many samples taken from a single exponential culture (as in Fig. 2), the fluctuations of the points in the plot of Fig. 3 are assumed to represent mainly variations from culture to culture, rather than inaccuracies of the measuring data. The exponentially decreasing curve in Fig. 3a and the monotonously increasing curves in Fig. 3b and c appear to be reasonable interpretations of the trend. If the curves had been drawn through the averages of the measuring points, the C/τ and oriC-per-genome curves for AB1157 would show a maximum, and the C-period curve would show a bump, at 2 doublings/h.
Growth rate dependence of the initiation mass.
For both E. coli strains studied here, the cell mass per replication origin (M0) approached nearly constant levels with increasing growth rate; the values for the K-12 strain were about 25% higher than the values for E. coli B/r (Fig. 5a). Observations obtained with a flow cytometer have suggested that the initiation mass for E. coli K-12 decreases with increasing growth rate (32). The discrepancy might be due to differences in measurement of cell mass. The flow cytometer measures the light scattering of individual cells, whereas we have used culture turbidity as a measure for mass density. When the initiation mass was expressed in terms of protein per oriC, it decreased with increasing growth rate for AB1157 (Fig. 5b). This result agrees with the work of Wold et al. (32), who used a fluorescent protein dye in connection with the flow cytometer to estimate protein per oriC. For the control of replication, as well as for the calculation of gene dosages and absolute gene activities, protein rather than culture turbidity per origin is assumed to be the more relevant parameter.
The constancy or nonconstancy of the initiation mass has been frequently discussed in the literature (e.g., see references 9, 16, and 22). In consecutive cell cycles within a given culture, replication is not initiated at the same exact time (cell age) during the cell cycle. Instead, replication is initiated when the cell has reached a particular mass, or amount of protein. The cell age at which this happens varies from one division cycle to the next because the timing of the division event is imprecise. For this reason, the mass of newborn daughter cells shows a stochastic variation. This has been shown elsewhere for synchronous cultures (6) and more recently for exponential cultures growing very slowly with nonoverlapping rounds of replication (5). This indicates that replication is linked to the accumulation of cell mass or, more likely, to the accumulation of some protein. The values for the cell mass or protein at the time of initiation vary discontinuously with the growth rate, showing twofold steps, but the number of replication origins per cell in which initiation occurs also varies with twofold steps, so that the cell mass or protein per replication origin becomes a smooth function of the growth rate (16). This suggests that a certain structural protein which is made as a constant proportion of total protein is required in stoichiometric amounts per oriC to trigger initiation of replication; this protein is assumed to accumulate during the cell cycle either at oriC (17) or at a structure that also doubles at the time of replication initiation (9). However, the constancy of the initiation mass from initiation to initiation within a given culture must be distinguished from potential changes in the initiation mass at different growth rates. At different growth rates, the protein (or mass) per origin would be constant only if the initiation protein were synthesized as a constant fraction of total protein (or mass), independent of the growth rate. There is no compelling reason to believe that this is or should be the case.
Growth rate dependence of the replication velocity.
Available literature values for the C period have been compiled and plotted as a function of growth rate by Helmstetter (19). Those data suggest a decreasing C period when the growth rate increases from about 0.1 to 1.0 doublings/h and a nearly constant C period of about 43 min at growth rates between 1.0 and 2.5 doublings/h. Our data (Fig. 3a) showed a gradual decrease in the C period with increasing growth rate, in agreement with previously determined values of C from this laboratory (Fig. 4 in reference 14). By inclusion of cultures grown in LB medium, the lowest values observed for C were 33 to 34 min at 3.0 doublings/h (Fig. 3b). The discrepancies between our new values and some of the previously reported values might result from the more precise determination in this work of the delay in the cessation of replication initiation after the addition of rifampin (as in Fig. 2d).
Our method for determination of C depends on the assumption that rounds of replication go essentially to completion in the presence of rifampin. That this is probably the case has been shown elsewhere by flow cytometry (31), which shows the accumulation of discrete bands of DNA during rifampin treatment. These bands represent integer multiples of genome equivalents; i.e., the cells have one, two, four, or eight fully replicated chromosomes, depending on the exponential growth rate before the drug treatment. This observation does not rule out the possibility that some last sequences near the replication terminus are not replicated in the presence of rifampin. However, this would not significantly affect our calculation of C.
Delay in the action of rifampin on initiation of replication.
The analysis of DNA accumulation curves after rifampin addition indicated a few minutes’ delay in the action of rifampin on initiation. This delay was 1 to 3 min in rich media and 5 to 12 min in minimal media (Fig. 2d and other data not shown). During this delay, DNA continues to accumulate exponentially. An alternative interpretation of this apparent delay would be that rifampin stops initiation immediately but that replication forks pause for a few minutes at a second control site shortly after initiation. Such temporary stalling of replication forks close to oriC has been shown elsewhere for Bacillus subtilis (25). If that were also true for E. coli, then the C periods estimated here (Fig. 2b) would represent the times for replication of the chromosome from this putative pause site, rather than from oriC, to the terminus. For estimating gene dosages, it does not matter how the delay is interpreted as long as the pause site is close to oriC. For the mechanism of replication control, the distinction would be important, however, since a pause site would suggest that the timing of replication is controlled at two levels, at oriC and at a downstream site.
Use of replication data to determine gene dosages.
As was pointed out above (in the introduction), to evaluate the transcriptional control of bacterial genes, especially when experiments involve different growth rates, it is necessary to relate the observed gene expression to the number of gene copies present. For genes with known map locations on the bacterial chromosome, accurate gene dosages can be found from the values of the initiation mass and the C period presented above (Fig. 5 and 3, respectively), by using the mathematical relationships provided below (Appendix). As an example, Table 1 shows how the activity of the first gene in the r-protein spc operon, rplN (r-protein L14), may be found from observed data in Fig. 3, 4, and 5 above and from genetic data on the map locations of rplN and oriC. In this case, the gene activity was calculated as number of L14 molecules produced per minute per gene. The increase in the synthesis rate per rplN gene with increasing growth rate reflects the genetic (transcriptional and translational) control of this locus.
ACKNOWLEDGMENTS
This work was supported by grants from NIH and MRC.
We thank E. Boye for advice in the preparation of the manuscript.
Appendix
This Appendix has four parts. In the first part, the principle of the method to determine the oriC copy numbers and the C period is explained in detail. The relationships and equations in this part have been derived previously in different contexts (e.g., see references 7, 8, 10, and 21). The second part describes how the initiation mass and C period can be used to find exact copy numbers of genes with known map locations on the bacterial chromosome. The third part describes the relationship between culture turbidity and cell mass density. This relationship has been used for accurate determinations of culture mass and generation time. The last part describes controls for the colorimetric DNA assay and for the conversion of assay values into absolute units in E. coli genome equivalents.
(i) Principle of method to determine the initiation mass and C period. After the addition of the antibiotic rifampin to a bacterial culture, initiation of new rounds of replication stops, but ongoing rounds of replication are completed. This has been most directly shown with the aid of a flow cytometer that measures the amount of DNA in individual cells: 90 min after the addition of rifampin, all cells have only fully replicated chromosomes (31). When initiation of replication stops, the number of replication origins becomes constant, and at essentially undiminished speed of the replication forks, the numbers of replication termini and of DNA genome equivalents (G) become equal to this number of origins after C minutes (Fig. 1). Thus, the number of oriC copies (NoriC) present at the time of rifampin addition equals the number of DNA genome equivalents reached at the plateau. Figure 1 also shows that the increase factor in DNA (ΔG) is equal to NoriC/G. Since NoriC/G is a function of the replication velocity relative to the bacterial growth rate (ratio C/τ), ΔG can be used to find C, by using the relationship (8)
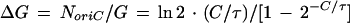 |
1 |
Together with the amount of DNA per mass present during exponential growth (G/M), ΔG is then used to determine the number of copies of oriC per culture mass (1/M0), i.e., the reciprocal of the initiation mass:
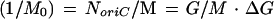 |
2 |
The curve describing the residual accumulation of DNA after stopping initiation (solid curve in Fig. 1) is given by the relationship (8)
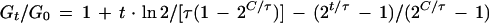 |
3 |
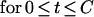 |
Gt and G0 are the amounts of DNA present at the times t and 0, respectively. Equation 3 is based on the two assumptions that rifampin immediately stops initiation and that it does not affect the velocity of the replication forks. A comparison of the observed DNA curve with this theoretical expectation allows one to determine a potential delay (d) in the action of rifampin on initiation of replication, which must be taken into account for determination of M0 and C. Mathematically, this is done by replacing t in equation 3 with the difference (t − d) and the first term on the right side, i.e., 1, with 2d/τ, which represents the exponential growth during the delay. Equation 3 then describes the accumulation of DNA in the time range between d and C + d. Before t = d, DNA accumulation is described by the exponential growth function, 2t/τ, and after t = (C + d), the amount of DNA remains constant, equal to the plateau value given by equation 3 for t = (C + d).
(ii) Determination of gene copy numbers from initiation mass and C period. The average copy number of a gene per cell is to be distinguished from its copy number per cell mass. Only the latter is relevant for studies on gene control. The copy number per cell mass depends on the control of replication characterized by the initiation mass, whereas the copy number per cell depends on the control of cell division and not on the control of chromosome replication.
The copy number (NX) of any gene X per cell mass (M) can be found if the map location (m) of that gene on the E. coli chromosome relative to the replication origin is known. The ratio of copy numbers, NX/NoriC, is given by the relationship (7)
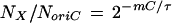 |
4 |
Combining equations 2 and 4, we find the copy number of gene X per unit of culture mass:
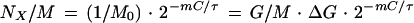 |
5 |
For example, for m = 0 (oriC), equation 5 gives NoriC/M = 1/M0, and for m = 1, i.e., for a gene near the replication terminus (terC), we find NterC/M = 2−C/τ/M0. This shows that genes near the replication origin have the highest copy number and genes near the replication terminus have the lowest copy number; the maximum difference in an exponential culture is given by the factor 2C/τ, corresponding to the ratio of origins over termini. Moreover, the faster a culture grows (small τ), the lower the relative frequency of genes at some distance from oriC is. During growth in rich medium (assuming τ = 20 min and C = 33 min [Fig. 3a]), the greatest differential is expected to be 233/20 = 3.1. Thus, during growth in rich medium, a gene located near oriC has a 3.1-fold higher copy number than a gene located near terC.
The location of a gene on the circular chromosome map is given in map units (MU) between 0 and 100 min (representing the chromosome transfer time in a particular type of mating experiment). With oriC located at 84 min on the E. coli chromosome map (1) and bidirectional chromosome replication (4), the location in MU can be converted into the location relative to oriC (m) by using one of the following relationships (11):
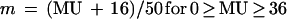 |
6a |
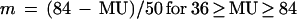 |
6b |
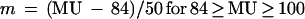 |
6c |
(iii) Determination of cell mass density and growth. The turbidity (OD) of a culture does not increase exactly in proportion to the mass density (M), as can be seen by making a number of dilutions from a concentrated culture and plotting the observed OD against the relative concentration (Fig. A1). Rather than diluting every sample of culture before reading the OD, it is more accurate to read the OD directly and correct for the nonlinearity. The observed OD curve in Fig. A1 can be approximated by the parabola
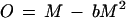 |
7 |
where O is the observed OD, b is an empirically determined factor (see below), and M is the (wanted) true mass density, defined by the initial slope (a) of the curve and the relative concentration (C) in the diluted sample (setting the concentration of the undiluted culture equal to 1.0 [Fig. A1]) as follows:
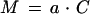 |
8 |
The quadratic term in equation 7 can be understood as the probability that two bacteria are in one line of sight between the light source and detector of the spectrophotometer, so that the second bacterium is in the shade of another one and does not contribute to the light scattering observed. The parameters a and b in equations 7 and 8 have been determined by a best-fit method from the observed data in Fig. A1 (Table A1). To find the true mass density from the observed OD, only b is needed (resolving equation 7 for M):
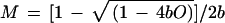 |
9 |
For E. coli B/r and K-12 strains, we found that b = 0.15 if the OD460 is measured (example in Table A1) and b = 0.17 if the OD600 is measured. Different growth media did not seem to make a significant difference.
FIG. A1.

Relationship between culture mass density and OD. A culture with an OD460 = 0.812 was diluted to 0.2, 0.4, etc. (relative concentrations on abscissa), and the OD460 of these dilutions was determined (points on the curve). The curve shown is a parabola that has been calculated (equation 7, with b = 0.15). The parabolic relationship can be used to find exact mass densities (triangles, curve labeled M) of cultures from the observed OD (see text for details). obs., observed; calc., calculated; Relat., relative.
TABLE A1.
Determination of culture mass densitya
| Relative concnb (C) | Observed ODc (Oo) | Calculated ODd (Oc) | Deviation (%)e [(Oc − Oo)/Oo] | Mass densityf (M) |
|---|---|---|---|---|
| 0.0 | 0.000 | 0.000 | 0.0 | 0.000 |
| 0.2 | 0.184 | 0.186 | 0.9 | 0.191 |
| 0.4 | 0.361 | 0.360 | −0.1 | 0.382 |
| 0.6 | 0.530 | 0.524 | −1.1 | 0.574 |
| 0.8 | 0.681 | 0.677 | −0.6 | 0.765 |
| 1.0 | 0.812 | 0.819 | 0.9 | 0.956 |
A culture of E. coli B/r was grown in glucose minimal medium to an OD460 of 0.812, when 200 μg of chloramphenicol per ml was added and the culture was chilled to stop growth.
Different mass densities were obtained by adding 1, 2, 3, or 4 ml of culture to 4, 3, 2, or 1 ml, respectively, of growth medium for relative concentrations (C) of 0.2 to 0.8.
The OD460 of the culture dilutions was determined (1-cm light path; observed OD, Oo).
The OD460 of the culture was calculated (Oc) from C, by equations 7 and 8 with a = 0.956 and b = 0.15 (best-fit method).
The difference calculated minus observed OD, expressed as a percentage. With a = 0.956 and b = 0.15, both the individual deviations and their sum were minimized (sum = −0.1%).
The mass density (M = corrected OD) was calculated from the observed OD for the different dilutions, by equation 9 with b = 0.15. The calculated values fall on the straight line labeled M in Fig. A1.
The OD of the medium without bacteria (medium blank) is not exactly zero, especially for LB medium. The observed medium blank value (B) was subtracted in the calculation of the culture mass density (M) by substituting the difference (O − B) for O in equation 9.
Equation 9 is valid for OD readings up to about 1.3. For mass densities > 1.3, as in stationary cultures, dilutions have to be made. Generally, the observed mass densities, after correction by equation 9, deviated by less than 1% from the best-fit exponential curve.
(iv) Determination of DNA by the diphenylamine assay. The diphenylamine assay is specific for deoxyribose. In the case of DNA, nucleotide bases have to be removed to make deoxyribose residues in the DNA accessible to the diphenylamine reagent. This is done by a 30-min treatment of the DNA with PCA at 70°C, which generates apurinic acid through efficient depurination. The diphenylamine reaction is enhanced about threefold by acetaldehyde and produces a blue compound measured by its light absorption at 600 nm (A600). The exact chemistry of the reaction is not known (13).
Acetaldehyde was also found to give a deoxyribose-independent diphenylamine reaction (Fig. A2a; blanks) in addition to enhancing the deoxyribose-dependent reaction (Fig. A2b). The deoxyribose-dependent reaction was complete after about 10 h and was independent of the acetaldehyde concentration above 0.08 mg/ml (Fig. A2b), whereas the deoxyribose-independent reaction continued for longer periods and increased with the acetaldehyde concentration (Fig. A2a). Acetaldehyde concentrations producing the lowest blank value without significantly reducing the deoxyribose-dependent reaction give the highest signal-to-noise ratio and therefore the greatest accuracy. Since stock solutions of acetaldehyde have a tendency to decompose during storage at 4°C, the optimal acetaldehyde concentration was determined (as illustrated in Fig. A2) before determining DNA in bacterial cultures.
FIG. A2.

Effect of different acetaldehyde concentrations on the colorimetric diphenylamine assay for DNA. (a) Assays with DNA (filled symbols) and without DNA (open symbols; blanks). (b) Differences of assay values, with DNA minus blank. The graph shows that acetaldehyde alone reacts with the diphenylamine without reaching a plateau, whereas the reaction with DNA is complete in 5 to 10 h and independent of the acetaldehyde concentration in the range tested. Blk., blank; nucleot., nucleotides; acetald., acetaldehyde.
Glass fiber filters used to collect bacteria for DNA determinations, but without any bacteria or DNA, were found to react weakly in the diphenylamine assay. The reaction was proportional to the number of filters in the assay. This increased the blank values in the DNA assays and led to extra scatter in the data. Repeated washes of the filters with salt (1 M NaCl) or alkali (1 M NaOH) did not remove the substrate for the reaction but rather increased the readings obtained with filters only, suggesting that sodium or other metal ions bound to the glass surface might be involved. Acid washes, however, decreased the reaction (although not to zero) and reduced the scatter. Thus, for improved accuracy, filters for measuring DNA in bacteria were soaked overnight in 1 mM PCA before use.
The DNA assay may be calibrated with known amounts of deoxyribose, deoxyribonucleosides, or DNA. For this purpose, we have routinely used either AdR or CT-DNA. Per mole of purine nucleotides, CT-DNA gave 23% higher assay values than did AdR (Fig. A3b). Since the 23% difference was found to be independent of the source of DNA (calf thymus, salmon sperm, and phage λ, all highly purified) or of the nucleoside (AdR and deoxyguanosine, different lot numbers and brands), we have assumed that the calibration with known amounts of DNA gives correct values for assaying unknown DNA. For convenience, however, DNA assays were generally calibrated with AdR, in which case an empirical factor (1.23 ± 0.03, as in Fig. A3b) was applied. The 23% difference in the final plateau and the observation that the reaction kinetics differ for DNA and nucleosides (Fig. A3b; ratio of assay values, CT-DNA to AdR, decreases with reaction time) suggest that the deoxyribose residues in apurinic acid, i.e., with two phosphate ester bonds, react differently with diphenylamine than does the deoxyribose in nucleosides like AdR or deoxyguanosine, i.e., without phosphate bonds.
FIG. A3.

Calibration of diphenylamine assay for DNA, with either CT-DNA or AdR. (a) Reaction kinetics observed with equal amounts of CT-DNA or AdR. In DNA, only deoxyribose residues of purine nucleotides are assumed to react. (b) Ratio of the two curves in panel a, CT-DNA/AdR, after division by 0.5 (50% purines in DNA). The curve shows that the reaction with DNA (apurinic acid) is faster than the reaction with AdR and reaches a 23% higher value than expected from the assumption that the contribution of purine nucleotides in DNA to the diphenylamine reaction is equivalent to that of AdR. Blk., blank; nucleot., nucleotides.
REFERENCES
- 1.Bachmann B J. Linkage map of Escherichia coli K-12, edition 8. Microbiol Rev. 1990;54:130–197. doi: 10.1128/mr.54.2.130-197.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachmann B J. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: ASM Press; 1996. pp. 2460–2488. [Google Scholar]
- 3.Berlyn M K B, Low K B, Rudd K E. Linkage map of Escherichia coli K-12, edition 9. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: ASM Press; 1996. pp. 1715–1902. [Google Scholar]
- 4.Bird R E, Louarn J, Martuscelli J, Caro L. Origin and sequence of chromosome replication in Escherichia coli. J Mol Biol. 1972;70:549–566. doi: 10.1016/0022-2836(72)90559-1. [DOI] [PubMed] [Google Scholar]
- 5.Boye E, Stokke T, Kleckner N, Skarstad K. Coordinating DNA replication initiation with cell growth: differential roles for DnaA and SeqA proteins. Proc Natl Acad Sci USA. 1996;93:12206–12211. doi: 10.1073/pnas.93.22.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bremer H, Chuang L. The cell cycle in Escherichia coli B/r. J Theor Biol. 1981;88:47–81. doi: 10.1016/0022-5193(81)90328-3. [DOI] [PubMed] [Google Scholar]
- 7.Bremer H, Churchward G. An examination of the Cooper-Helmstetter theory of DNA replication in bacteria and its underlying assumptions. J Theor Biol. 1977;69:645–654. doi: 10.1016/0022-5193(77)90373-3. [DOI] [PubMed] [Google Scholar]
- 8.Bremer H, Churchward G. Deoxyribonucleic acid synthesis after inhibition of initiation of rounds of replication in Escherichia coli B/r. J Bacteriol. 1977;130:692–697. doi: 10.1128/jb.130.2.692-697.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bremer H, Churchward G. Control of cyclic chromosome replication in Escherichia coli. Microbiol Rev. 1990;55:459–475. doi: 10.1128/mr.55.3.459-475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bremer H, Churchward G, Young R. Relation between growth and replication in bacteria. J Theor Biol. 1979;81:533–545. doi: 10.1016/0022-5193(79)90051-1. [DOI] [PubMed] [Google Scholar]
- 11.Bremer H, Dennis P P. Modulation of chemical composition and other parameters of the cell by growth rate. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: ASM Press; 1996. pp. 1553–1569. [Google Scholar]
- 12.Brunschede H, Dove T, Bremer H. Establishment of exponential growth after a nutritional shift-up in Escherichia coli B/r: accumulation of deoxyribonucleic acid, ribonucleic acid, and protein. J Bacteriol. 1977;129:1020–1033. doi: 10.1128/jb.129.2.1020-1033.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burton K. A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric determination of DNA. Biochem J. 1956;62:315–323. doi: 10.1042/bj0620315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Churchward G, Bremer H. Determination of the DNA replication time in exponentially growing Escherichia coli B/r. J Bacteriol. 1977;130:1206–1213. doi: 10.1128/jb.130.3.1206-1213.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churchward G, Estiva E, Bremer H. Growth rate-dependent control of chromosome replication initiation in Escherichia coli. J Bacteriol. 1981;145:1232–1238. doi: 10.1128/jb.145.3.1232-1238.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donachie W. Relationships between cell size and time of initiation of DNA replication. Nature. 1968;219:1077–1079. doi: 10.1038/2191077a0. [DOI] [PubMed] [Google Scholar]
- 17.Hansen F G, Christensen B B, Atlung T. The initiator titration model: computer simulation of chromosome and minichromosome control. Res Microbiol. 1991;142:161–167. doi: 10.1016/0923-2508(91)90025-6. [DOI] [PubMed] [Google Scholar]
- 18.Helmstetter C. Rate of DNA synthesis during the division cycle of Escherichia coli B/r. J Mol Biol. 1967;24:417–427. [Google Scholar]
- 19.Helmstetter C E. Timing of synthetic activities in the cell cycle. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: ASM Press; 1996. pp. 1627–1639. [Google Scholar]
- 20.Helmstetter C E, Cooper S. DNA synthesis during the division cycle of rapidly growing E. coli B/r. J Mol Biol. 1968;31:507–518. doi: 10.1016/0022-2836(68)90424-5. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez V J, Bremer H. Characterization of RNA and DNA synthesis in Escherichia coli strains devoid of ppGpp. J Biol Chem. 1993;268:10851–10862. [PubMed] [Google Scholar]
- 22.Herrick J, Kohiyama M, Atlung T, Hansen F G. The initiation mess? Mol Microbiol. 1996;19:659–666. doi: 10.1046/j.1365-2958.1996.432956.x. [DOI] [PubMed] [Google Scholar]
- 23.Hurst R O, Marko A M, Butler G C. The mononucleotide content of some deoxyribonucleic acids. J Biol Chem. 1953;204:847–856. [PubMed] [Google Scholar]
- 24.Kjeldgaard N O, Maaloe O, Schaechter M. The transition between different physiological states during balanced growth of Salmonella typhimurium. J Gen Microbiol. 1958;19:607–616. doi: 10.1099/00221287-19-3-607. [DOI] [PubMed] [Google Scholar]
- 25.Levine A, Vannier F, Dehbi M, Henckes G, Seror S J. The stringent response blocks DNA replication outside the ori region in Bacillus subtilis and at the origin in Escherichia coli. J Mol Biol. 1991;219:605–613. doi: 10.1016/0022-2836(91)90657-r. [DOI] [PubMed] [Google Scholar]
- 26.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 28.Nierlich D. Regulation of ribonucleic acid synthesis in growing bacterial cells. II. Control over the composition of newly made RNA. J Mol Biol. 1972;72:765–777. doi: 10.1016/0022-2836(72)90190-8. [DOI] [PubMed] [Google Scholar]
- 29.Schaechter M, Maaloe O, Kjeldgaard N. Dependency on medium and temperature of cell size and chemical composition during balanced growth of Salmonella typhimurium. J Gen Microbiol. 1958;19:592–606. doi: 10.1099/00221287-19-3-592. [DOI] [PubMed] [Google Scholar]
- 30.Shepherd N S, Churchward G, Bremer H. Synthesis and activity of ribonucleic acid polymerase in Escherichia coli. J Bacteriol. 1980;141:1098–1108. doi: 10.1128/jb.141.3.1098-1108.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skarstad K, Boye E, Steen H B. Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J. 1986;5:1711–1717. doi: 10.1002/j.1460-2075.1986.tb04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wold S, Skarstad K, Steen H B, Stocke T, Boye E. The initiation mass for DNA replication in Escherichia coli K-12 is dependent on growth rate. EMBO J. 1994;13:2097–2102. doi: 10.1002/j.1460-2075.1994.tb06485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]