

Abstract

Click chemistry is a set of easy, atom-economical reactions that are often utilized to combine two desired chemical entities. Click chemistry accelerates lead identification and optimization, reduces the complexity of chemical synthesis, and delivers extremely high yields without undesirable byproducts. The most well-known click chemistry reaction is the 1,3-dipolar cycloaddition of azides and alkynes to form 1,2,3-triazoles. The resulting 1,2,3-triazoles can serve as both bioisosteres and linkers, leading to an increase in their use in the field of drug discovery. The current Review focuses on the use of click chemistry to identify new molecules for treating neurodegenerative diseases and in other areas such as peptide targeting and the quantification of biomolecules.
1. Introduction
1.1. Click Reactions
Click chemistry (CC) refers to a collection of potent, incredibly dependable, and specific reactions that can produce molecules instantly by fusing together small units through heteroatom connections.1 CC was devised by Sharpless in 2001,2 and for his tremendous contributions to the field, the 2022 Nobel Prize was jointly awarded to Sharpless, along with Meldal and Bertozzi.3 CC encompasses almost all branches of chemistry, including organic chemistry,4 drug discovery,5 polymers,6,7 bioconjugation,8 and radiochemistry,9 with a broad spectrum of applications and advantages.10 “CC” originated as a way to describe reactions that produce products with high yields by the generation of carbon–heteroatom bonds. The term “click” described the simple combining of molecular building blocks, similar to how a seatbelt buckles two pieces that click together.11 CC reactions are quick, extremely specific, easy to carry out, and insensitive to oxygen and water.12−14 They can also produce a wide range of structures in large quantities. Additionally, these reactions require barely any chromatographic purification during their workup, which makes it very simple.14 These reactions are commonly referred to as spring-loaded reactions, i.e., thermodynamically driven reactions that are rapid and nonreversible to produce the reaction product with high reaction selectivity.15 The byproduct of the process could be readily eliminated without needing any chromatography techniques.16 The needed attributes of the reaction comprise facile reaction conditions, easily accessible starting components and reagents, and the utilization of no solvents or a solvent like water or one that is quickly removed, along with easy product isolation.17 Based on all of the criteria, click reactions were categorized into 4 types: (i) cycloaddition reaction, including specifically hetero-Diels–Alder reactions, and the Huisgen 1,3-cycloaddition reaction; (ii) nucleophilic ring-opening reactions (epoxides, aziridines); (iii) reactions in carbonyl chemistry ranging from the formation of oxime ethers, hydrazones, and aromatic heterocycles; and (iv) addition reactions to C–C multiple bonds including the epoxidation and dihydroxylation, as well as Staudinger ligation, a type of azide–phosphine coupling reaction10,18−21 (Figure 1). In addition to these reactions, numerous photochemical reactions also meet these strict criteria when favored substrates are utilized. A novel class of light-triggered click reactions, which is referred to as photoclick chemistry, has been created as a result of the fusion of click chemistry with harmless photochemical techniques.
Figure 1.

Different types of click reactions.
Among the several types of click reactions, the Huisgen 1,3-dipolar cycloaddition reaction between an azide and a terminal alkyne, which yields the 1,4-disubstituted 1,2,3-triazoles, has been widely explored as an effective synthetic technique for creating compound libraries. The synthetic production of 1,2,3-triazoles using azides and alkynes can also be catalyzed by Cu(0) turnings in addition to CuSO4/sodium ascorbate. The active Cu(I) catalyst entity appears to be produced by either oxidizing or reducing the Cu(0) and Cu(II) antecedents in both cases. Thus, the reaction is also referred to as the Cu(I)-assisted azide alkyne cycloaddition (Figure 2), and it is one of the most popular reactions among the other four types. Azides and alkynes are simple to work with as well as being less reactive functional units in organic chemistry. The 1,2,3-triazole ring is a desirable pharmacophore in the field of medicinal chemistry, and molecules with this structure have been discovered to exhibit significant biological properties, making them particularly ideal for drug discovery. This is brought on by the 1,2,3-triazole’s capacity to establish hydrogen bonding along with additional noncovalent links with several biological targets, including DNA, as well as its improved solubility and stability in metabolism.17,22−26 The click chemistry reactions simplify the steps needed for the successive synthesis of diverse pharmaceutical compounds, resulting in more efficient lead discovery and optimization processes and enabling medicinal chemists to tackle the shortcomings of traditional chemical synthesis.27 The biocompatibility of 1,2,3-triazoles and the cytotoxic copper catalyst are two major limitations of click reactions that must be taken into account. Alkyne homocoupling and Cu(I) saturation are two additional less severe concerns. The complexity mentioned above has also encouraged the implementation of alternate synthetic methods, which are copper-free cycloadditions, including strain-promoted azide–alkyne cycloadditions (SPAAC) and inverse-electron-demand Diels–Alder cycloadditions (IEDDA). Even though click chemistry has a few limitations, there is no disputing its significance in medical chemistry.1,28,29
Figure 2.

Representation of the copper-catalyzed azide–alkyne cycloaddition reaction.
Medicinal chemists have largely benefited from the ease of synthesizing an array of triazole derivatives for drug discovery programs.30 Numerous biological actions, including antituberculosis, antifungal and antibacterial, anticancer and antiviral, are demonstrated by the 1,2,3-triazoles.1 CC is now widely used in the era of modern drug discovery, and its focus on neurodegenerative disorders (NDDs) is of interest.31
1.2. Major Targets for the Treatment of Neurodegenerative Disorders
Neurodegenerative disorders (NDDs) are characterized by particular protein accumulations known as amyloid beta (Aβ) plaques and cerebral amyloid angiopathy (CAA), tau-protein neurofibrillary tangles (NFTs), and neuropil threads (NTs) in the brains of the patients, causing neuronal dysfunction and leading to the progression of neurological impairment and mortality.31 The most common types of NDDs are Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis, and amyotrophic lateral sclerosis.32 Neuronal dysfunction occurs in many ways, including oxidative overload, free radical formation, collapse of mitochondrial function, compromised biological energy, neuronal malfunction, neuroinflammatory processes, and neuronal structural and functional errors.33 Other mechanisms have been identified, such as mutations in leucine-rich repeat kinase 2 (LRRK2) and aggregation of α-synuclein in PD, and mutations in amyloid precursor protein (APP), PS1, and PS2 (presenilin 1 and 2) in AD.34 The major targets that can be used to treat neuronal disorders are monoamine oxidases (MAOs), acetylcholinesterase (AChE), acetylcholinesterase (AChE), butyrylcholinesterase (BChE), and β-site amyloid precursor protein cleaving enzyme 1 (BACE1). MAOs are important biological targets in neurodegeneration as they cause the deregulation of monoamine neurotransmitters.35,36 MAOIs (monoamine oxidase inhibitors) were among the initial antidepressants to be supplied in the market, originating in the latter part of the 1950s with phenelzine, tranylcypromine, isocarboxazid, and selegiline.37 The MAO-A specific inhibitors tend to be developed as potential therapies for the management of anxiety and depression, while MAO-B specific inhibitors have proved to be highly efficient in the therapy of PD and AD.38 The irreversibility of certain MAOIs developed several adverse effects compared to those of the reversible MAOIs. The sole remedy to this situation is the development of more specific, and reversible, MAOIs.39 AChE is predominantly associated with AD and PD and plays an important role in tau protein deposition, Lewy body buildup, and neuronal injury in the substantia nigra.40 Reversible AChE inhibitors, which have an indirect parasympathetic mimicking effect, are used therapeutically to treat the cholinergic deficiency in AD. Such inhibitors, by preventing the enzyme’s action, preserve the cerebral availability of AChE and can therefore mitigate but not prevent the disease’s effect on cell death. Galantamine, donepezil, and rivastigmine are traditional AChE inhibitors that are still in use.41 BChE has been shown to contribute to a key function in the development of neuritic plaques, senile plaques, and Aβ accumulation, which are the pathways involved in the pathogenesis of AD.42 The targeted BChE suppression improved learning and persistence in rats by raising ACh levels. Similar results were obtained in a mouse model of cholinergic deficiency, where in vivo BChE inhibition improved learning, memory, and cognitive performance.43 Tacrine, which was the first commercially available AChE/BChE inhibitor that had a noncompetitive, readily reversible inhibitory mechanism, was removed from the market in 2013 due to significant hepatotoxicity and is utilized solely as a reference today.41 The BACE1 constitutes the buildup of amyloid proteins within the brain cells, which results in plaque formation.44 Since BACE1 is highly abundant in the brain, feasible BACE1 inhibitors ought to be small and quickly cross the blood–brain barrier (BBB). BACE1 inhibitors can effectively decrease the amount of Aβ in neurons and throughout the brain by crossing the BBB. Verubecestat was the initial small-molecule BACE1 antagonist with BBB penetration; nonetheless, the phase 3 study of verubecestat in mild to moderate AD patients was discontinued due to insignificance.45,46
In every case, the insignificance and undesirable side effects caused certain drugs to have limitations on their clinical use. Also in spite of longer life expectancies, NDDs pose an increasing challenge and are becoming a serious threat to society. The need for patients to receive targeted, individualized care is already anticipated as a result of both genetics and environmental vulnerabilities. As a result, it is crucial to discover new pharmaceuticals that reveal the specific pathways underlying NDDs in order to gain insight into the challenges and improve the therapy.47 Using click chemistry, a wide variety of chemical compounds can be synthesized, since the reactions are rapid, incredibly precise, and simple to perform, produce products in high yields, and may easily eliminate byproducts without the use of chromatographic techniques. Among the click reactions, CuCA offers a fast and accurate synthetic approach for producing triazole motifs that are typically present in pharmaceuticals and biologically active substances. Due to its large dipole moments, this motif serves as an excellent amide substitute in bioactive compounds. The 1,2,3-triazole is resistant to both oxidative and reductive reactions as well as the process of hydrolysis (both acidic and basic), demonstrating an extremely high aromatic stabilization and a relative degree of resistance to metabolic breakdown. Triazoles are further utilized as linkers and exhibit bioisosteric impacts on aromatic rings, double bonds, peptide linkages, and an imidazole ring despite protecting essential hydrogen bonds and hydrophobic relationships. This might widen the structural variety of chemical libraries and enhance the physicochemical and pharmacokinetic attributes as well as the effectiveness of lead compounds.27,48,49
Herein, we present a detailed review of the applications of click chemistry in numerous types of NDDs, with a particular focus on drug discovery and diagnostic applications in AD.
2. Development of Monoanime Oxidase (MAO) Inhibitors Using Click Chemistry Reactions
Jia et al. (2010) demonstrated the utilization of click chemistry in high-throughput screening of inhibitory effects of 1,4-disubstituted-1H-1,2,3-triazoles against MAO-A and MAO-B. The synthesis of the titled triazoles was achieved by a copper(I)-catalyzed 1,3-dipolar cycloaddition reaction (CuCA) of alkynes and alkyl azides using CuSO4 and sodium ascorbate (i) in tert-butanol/H2O (Scheme 1). In total, 108 compounds were tested for their ability to inhibit MAO-A/B using a fluorescence-based microplate assay. Among them, compounds 1 and 2 were the most effective inhibitors of MAO-A, with half-maximal inhibitory concentration (IC50) of 0.83 and 0.97 μM, respectively. Moreover, 1 showed the highest selectivity index (SI) toward MAO-A (SI = 526.63). This study showed that compounds with an amino substituent had strong inhibitory effects on MAO-A, suggesting that prop-2-yn-1-amine is a crucial component of the inhibitory activity. Furthermore, upon comparison of the substituents at R1, unsubstituted benzene (compound 1) and pyridine (compound 2) had a more robust inhibitory action against MAO-A. A docking study showed that compound 1 binds very well with MAO-A through π–π stacking interactions with Tyr444, Tyr407, and FAD residues instead of MAO-B.50
Scheme 1. Reagents and Conditions: (I) CuSO4, Sodium Ascorbate, tert-Butanol/H2O, 24 h, Room Temperature (rt).

The click synthesis of 1,4-disubstituted-1H-1,2,3-triazoles (c1) is illustrated in Scheme 1. The reaction was carried out using CuCA, in which alkyl or phenyl azides (a1) and alkynes (b1) were reacted in the presence of CuSO4 and sodium leads to the generation of cycloaddition compounds.
The structure–activity relationship (SAR) of the 1,2,3-triazoles is shown in Figure 3. The inhibitory effect against MAO-A was stronger with an unsubstituted phenyl ring (compound 1) on R1. Pyridine substitution on R1 resulted in increased MAO-A inhibition (compound 2) compared to that with the unsubstituted phenyl ring. The amino group on R2 (propargylamine) resulted in an increased inhibitory activity.
Figure 3.

Structure–activity relationship (SAR) of 1,2,3-triazoles toward MAO inhibition.
Di Pietro et al. (2016) focused on CuCA for the synthesis of several series of 1,2,3-triazole derivatives with aminoacetylenic side chains at the 4-position of the triazole through an alkyl/aryl bridging unit for their MAO inhibition activity (Scheme 2). They explained the design and click synthesis of 76 derivatives by substituting an essential structural feature of typical irreversible MAO-B inhibitors, a propargyl amino group, at the fourth position of the 1,2,3-triazole ring and by incorporating some lipophilic groups at the 1 and 5 positions to improve hydrophobicity. All of the synthesized compounds were tested in vitro for their human MAO-A and MAO-B inhibitory properties using selegiline and clorgyline as references. The results demonstrated that compounds 3, 4, 5, and 6 exhibited a strong MAO-B inhibitory effect (IC50 values for MAO-B are 3.5 ± 0.4, 11.2 ± 2.1, 0.6 ± 0.1, and 5.2 ± 0.5 μM, respectively). Compound 5 stood out as the most effective MAO inhibitor, with submicromolar potencies against both MAO-A and MAO-B (with IC50 values of 0.5 ± 0.1 and 0.6 ± 0.1 μM, respectively), and compound 6 had very high selectivity toward MAO-B (SI value = 27.7). Reversibility studies demonstrated that compound 3 irreversibly inhibited MAO-B, because the inhibition persisted even after numerous repeated centrifugations and buffer washes. The enzyme was suppressed by 10 μM of 3 for various preincubation intervals (0–360 min) in order to evaluate the time-dependent inhibition of hMAO-B. Enzymatic efficiency revealed a sluggish time-dependent suppression of MAO-B. A parallel artificial membrane permeation assay (PAMPA), employing a combination of PBS/EtOH 70:30, was used to measure the in vitro permeability (Pe) of the innovative 1,2,3-triazole-based compounds via a lipid extract of the pig brain membrane. All compounds from the second generation showed good brain–blood barrier (BBB) permeation compared to those from the third generation.51
Scheme 2. Reagents and Conditions: (I) CuSO4, Ascorbic Acid, Na2CO3, H2O/DMF, Overnight, rt; (II) Methyl Iodide (MeI), Sodium Hydride (NaH), Tetrahydrofuran (THF), 1.5–15 h, rt; (III) Phosphoric Acid (H3PO4), Methyl Chloride, 1.5–4 h, rt; (IV) Propargyl Bromide, Cesium Carbonate (Cs2CO3) Acetone, 2–15 h, rt; or 0 °C, 2–2.5 h; or 0 °C for 30 min and Then rt for 3 h.
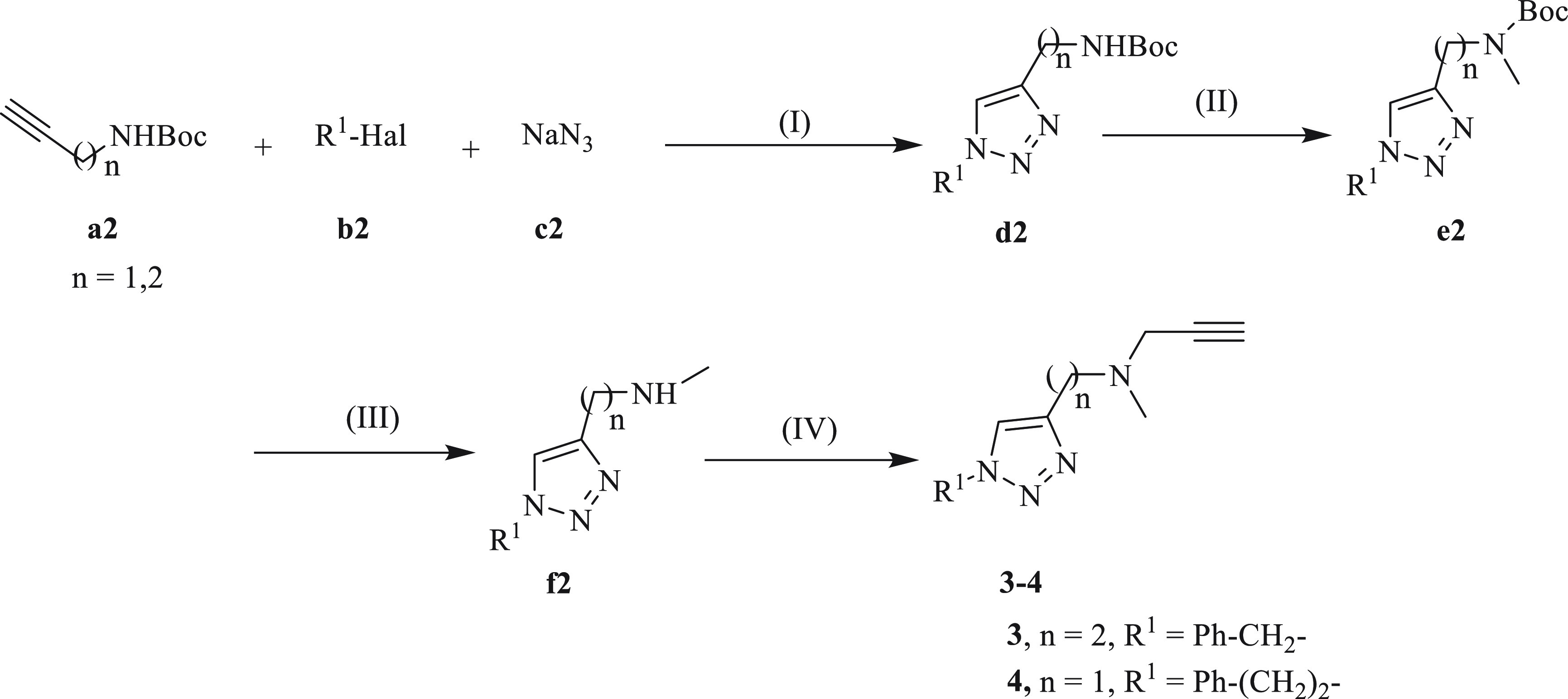
The preparation of compounds 3 and 4 is shown in Scheme 2. CuCA is the reaction that occurs in the first step using Boc-protected propargylamine (a2), alkyl halide (b2) and sodium azide (c2 to form 1-substituted triazole derivatives (d2). Followed by methylation to produce e2, which, upon acidic deprotection gave rise to secondary amines (f2). Finally, the secondary amines were propargylated to obtain the desired compounds (3, 4).
Compounds 5 and 6 were synthesized in seven to eight steps, as shown in Scheme 3. The alkynes were initially produced via the Negishi coupling of a3 and trimethylsilylacetylene, followed by desilylation of the protected alkyne derivative (b3). Subsequently, alkyne (c3) reacts with methyl azide to generate the necessary triazole (d3). The subsequent step involved reduction to produce the corresponding amines (e3). The desired compound 5 was produced by using N-methylation of f3 and acidic deprotection of g3 and propargylation. When the methylation of an N-Boc-protected amine is challenging, desired compound 6 can be obtained by converting the N-Boc-protected amine to a secondary amine (h3). This secondary amine then underwent reduction, followed by propargylation to produce compound 6.
Scheme 3. Reagents and Conditions: (V) Trimethylsilylacetylene, THF, n-BuLi, −78 °C, 30 min, and Then ZnBr2, THF, −78 to 0 C°; (VI) Silver Trifluoromethanesulfonate (AgOTf) and Methylchloride/MeOH/H2O Overnight, rt; (I) Methyl Azide, CuSO4·5H2O, Ascorbic Acid, Na2CO3, H2O/Dimethylformamide (DMF), Overnight, rt; (VII) LiAlH4, THF, Reflux, 2 h; (VIII) (Boc)2O, THF, rt; (II) MeI, NaH, THF, 60 °C, 4 h; (VII) LiAlH4, THF, 0 °C, Reflux, 72 h; (III) H3PO4, CH2Cl2, rt, 1.5–3 h; (IV) Propargyl Bromide, Cs2CO3, Acetone, 0 °C for 3.5 h or 0 °C for 30 min and Then rt for 3 h.

The SAR of N-methyl-N-[(1,2,3-triazol-4-yl)alkyl]propargylamines is shown in Figure 4. Compounds with a benzyl substitution at the R1 position of the triazole ring may have enhanced the potency toward MAO-B. The substitution of a benzyl group at the R1 position and a phenylmethyl group at the R5 position of the triazole ring, with a chain length of two carbons, produced a potent MAO-B inhibitor (compound 3). Compounds with enhanced MAO-B inhibitory properties were established by incorporating a benzyl group between the triazole and alkylamino groups (compounds 5 and 6). Substituting the alkyl amino group at the para position of the benzyl aromatic ring produced a potent derivative (compound 5), which showed high effectiveness toward both isoforms of MAO.
Figure 4.

SAR of N-methyl-N-[(1,2,3-triazol-4-yl)alkyl]propargylamines.
Haider et al. (2018) focused on creating an array of 26 harmine scaffolds combined with triazole using click chemistry to evaluate their potential to inhibit MAO. In vitro tests were performed on the newly synthesized harmine-conjugated 1,2,3-triazole derivatives to determine their inhibitory effect on the hMAO isoenzymes (MAO-A and MAO-B). The enzymatic functions of MAO-A and MAO-B were assessed by using a kynuramine-based fluorescence test. Six compounds with significant activity against MAO were identified from the inhibition assay (Scheme 4). Compounds 7 and 10 showed greater selectivity against MAO-A compared to the other compounds, with SI (MAO-A/B) values of 0.07 and 0.05, respectively. In contrast to harmine, the parent alkaloid, compounds 8 (1.21 ± 0.14 μM), 9 (0.26 ± 0.04 μM), 11 (0.36 ± 0.001 μM), and 12 (1.08 ± 0.02 μM) exhibited a more powerful suppression of MAO-B. In silico analysis incorporating the crystal structures of MAO-A and MAO-B and docking studies helped to better explain the MAO inhibition findings. The compounds were docked against the crystal structures of MAO [MAO-A (PDB ID: 2Z5X) and MAO-B (PDB ID: 2V61)]. The synthesized compounds displayed π-interactions and H2 bonding with amino acid residues such as Phe343, Asn181, His90, Tyr398, His90, and Tyr435 in the binding domain.52
Scheme 4. Reagents and Conditions: (IX) R1–Br, NaH, DMF, Reflux; (X) HBr, Acetic Acid, 115 °C, 15 h; (IV) Cs2CO3, DMF, Propargyl Bromide, 60 °C, 3 h; (I) ArCH2N3 or ArN3, CuSO4·5H2O, Sodium Ascorbate, THF–H2O (1:1), rt.

The syntheses of compounds 7–12 are explained in Scheme 4. In the first step, harmine (a4) was N-alkylated. The alkylated harmine (b4) underwent selective O-demethylation to produce c4; in the third step, it was O-propargylated to produce d4. Finally, in the fourth step, d4 was allowed to react with various easily accessible azides to produce the desired molecules.
The SAR of harmine-conjugated 1,2,3-triazoles is shown in Figure 5. MAO-A inhibition increased when an aromatic ring possessing halogens at the para position was placed at the R2 position of the triazole ring. This inhibitory effect decreased when the halogen was substituted with a cyano group. In contrast to compounds with smaller n-butyl, n-propyl, or isobutyl groups, except for compound 10, those with a large 3-phenylpropyl group at R1 did not exhibit MAO inhibition. When the R2 substitutions included an unsubstituted aromatic ring, an electron-withdrawing group (EWG), or an electron-donating group (EDG), MAO inhibition decreased in the following order: EDG > unsubstituted aromatic ring > EWG.
Figure 5.

SAR of harmine-conjugated 1,2,3-triazoles.
Mi et al. (2019) focused on creating a number of dual-target compounds using click chemistry to treat AD. The structures of hydroxypyridinones that chelate iron (known to have potential as nontoxic medicinal agents in the management of AD) and coumarin analogues (known to have MAO-B inhibitory action) were coupled using click chemistry to produce dual-target anti-AD therapeutics. Eleven compounds were synthesized; their iron-chelating activities were measured using HySS software, and their MAO-B inhibitory activities were evaluated (Scheme 5). All the compounds showed a significant chelating effect. The most effective MAO-B inhibitory activity was shown by compounds 13 (0.68 μM) and 14 (0.86 μM).53
Scheme 5. Reagents and Conditions: (I) CuSO4·5H2O, Sodium Ascorbate, CH3OH/H2O 1/4 1:1, 25 °C, 2 h; (XI) CH2Cl2, BBr3, CH3OH, 0 °C–rt.

The synthesis of the targeted compounds was carried out when propylazide-tagged hydroxypyridinones (b5 and d5) with propargyl ethers (a5 and c5) were installed on either side of the coumarin rings, that is, benzene (13) and the heterocyclic ring (14), which led to two structurally diversified triazole sandwiches. The reaction steps include the preparation of the triazole ring and subsequent demethylation of the ether groups.
The SAR showed that the hydroxymethyl group at the second position of the hydroxypyridinone ring was essential for iron-chelating activity (Figure 6). The triazole ring, which serves as a linker between the coumarin and hydroxypyridinone, preserves the ability of the compounds to inhibit MAO-B. The number of carbons in the alkyl chain connected to triazole and hydroxypyridinone had a marginal influence on their anti-MAO-B activities, and there was no large disparity in the activities of the ethyl or propyl groups. Substitution at the third or seventh positions of the coumarin ring showed a noticeably greater MAO-B inhibitory effect than that of the 4-substituted coumarin derivatives.
Figure 6.

SAR of the hybrids of coumarin derivatives and hydroxypyridinones.
Jia et al. (2017) synthesized a library of 46 azide-substituted flavones using click chemistry to evaluate their potential inhibitory activity toward MAOs. The newly synthesized flavone derivatives were tested in vitro to determine whether they had an inhibitory effect on the hMAO isoenzymes (MAO-A and MAO-B). The enzymatic activities of MAO-A and MAO-B were assessed by using a fluorescent probe method. Three compounds emerged from the inhibition assay with significant action against MAO. Compound 17 showed greater potency against MAO-A (1.6 ± 0.2 μM) and MAO-B (2.1 ± 0.7 μM). Compounds 16 (71.4 ± 7.6 μM) and 15 (75.8 ± 9.4 μM) displayed inhibition toward MAO-B. In silico analysis incorporating the crystal structures of MAO-A and MAO-B and docking studies helped to better explain the MAO inhibition findings. The active site of MAO has three functional domains (the substrate cavity, entrance cavity, and “aromatic cage”). Docking studies revealed that these inhibitors are probably located in both the substrate cavity and the aromatic cage. Their results suggest that the development of new MAO inhibitors from the C6 substitution of flavone derivatives is significant and that these compounds also have potential for the treatment of diseases associated with MAOs.54
The preparation of compounds 15, 16, and 17 is shown in Scheme 6. Here, the azides (a6 and c6) were reacted with their respective alkynes (b6 and d6) to produce the target compounds.
Scheme 6. Reagents and Conditions: (I) CuSO4, Sodium Ascorbate, THF/H2O = 1:1, rt.

The SAR of the azide-substituted flavones (Figure 7) showed that the inhibitory effect against MAO-A was stronger when X was a chlorine substituent (compounds 16 and 17). When the chain length n was equal to one, the para-chlorophenyl substituent had greater potency; that is, when R1= Cl, the para-fluorophenyl substituent was more potent when n was 2 or 4 (R1 = F). The replacement of fluorine at R1 with chlorine also diminished the efficacy toward the enzyme. Figure 7 indicates that adding a secondary amine substituent at the C6 position of the flavone is advantageous for MAO-inhibitory activity. Compounds substituted with secondary amines significantly inhibit MAO (17). Overall, the compounds with amide bonds and amine functional groups at the R-position were more effectively inhibited than those with an ether group. In contrast, the compounds containing an ether group at R demonstrated high selectivity. Co-MFA analysis of the synthesized compounds revealed that bulky groups at the R1 position boosted the inhibitory action against MAO-A and MAO-B. This was consistent with the experimental results for these compounds.
Figure 7.

SAR of azide-substituted flavones.
3. Development of Acetylcholinesterase (AChE) and Butyrylcholinesterase (BChE) Inhibitors Using Click Chemistry Reactions
de Andrade et al. (2019) used click chemistry to create two series of N-piperidine-based compounds as novel, potent, and effective human BChE (hBChE) inhibitors. Two series of molecules were designed based on donepezil, a selective human AChE (hAChE) inhibitor. The main goal of the design phase was to preserve the N-benzylpiperidine moiety and remove the 5,6-dimethoxy-1-indanone moiety from donepezil. In the first series (Scheme 7), a 1,4-disubstituted 1,2,3-triazole ring was used to replace it, whereas in the second series (Scheme 8), 1-substituted 4-phenyl-1,2,3-triazoles were used.
Scheme 7. Reagents and Conditions: (XIII) Imidazole-1-sulfonyl Azide Hydrogen Sulfate, CuSO4, NaHCO3, MeOH/H2O; or BnCl, K2CO3, Acetone; (I) Terminal Alkyne (RC≡CH), CuSO4, Sodium Ascorbate, DMF, 100 °C under Microwave (MW).

Scheme 8. Reagents and Conditions: (XIV) BnBr, K2CO3, EtOH, 90 °C; LiAlH4, DCM/THF, 70 °C, N2 (g); (XV) Azido Amino Acid, HBTU, DIPEA, DMF; (I) Phenylacetylene, CuSO4, Sodium Ascorbate, t-BuOH/H2O/DCM (1:1:1); (XVI) Morpholine/DCM (1:1); (XVII) Ac2O, Pyridine.
In contrast to the second series, which has an indirect coupling to the N-benzylpiperidine moiety through an amide bond, the first series of compounds has direct coupling to the N-benzylpiperidine moiety. All the compounds in the two series were evaluated for their inhibitory activity and selectivity toward hBChE and hAChE. Compound 18 (0.0065 ± 0.002 μM) in the first series shows good potency and selectivity toward hBChE, while the other compounds showed low inhibitory action toward hAChE and hBChE. Compounds 19 (0.0099 ± 0.0043 μM) and 20 (0.00017 ± 0.000021 μM) from the second series showed highly effective and selective inhibition of hBChE. Cell viability was evaluated to determine the cytotoxic effects of the compounds in the human neuroblastoma SH-SY5Y cell line. The mechanism of hBChE suppression by compounds 19 and 20 was further evaluated by using a kinetic study that considered the interaction between two binding sites (catalytic and peripheral). Molecular docking and molecular dynamics investigations were also performed to learn more about the binding interactions and stability of compounds 19 and 20. All the data showed that lead compounds 19 and 20 could be beneficial in the study strategies to learn more about the fascinating function of hBChE in both physiological and pathological states, as well as in the molecular mechanisms of disorders such as AD. Furthermore, compound 20 emerged as one of the most influential and specific hBChE inhibitors ever reported.55
The designed compound was synthesized in two steps (Scheme 7). The first step involved the benzylation of piperidin-4-ylmethanamine (a7) to give b7. Next, the intermediate (b7) was converted to target compound 18 either by reacting with a terminal alkyne.
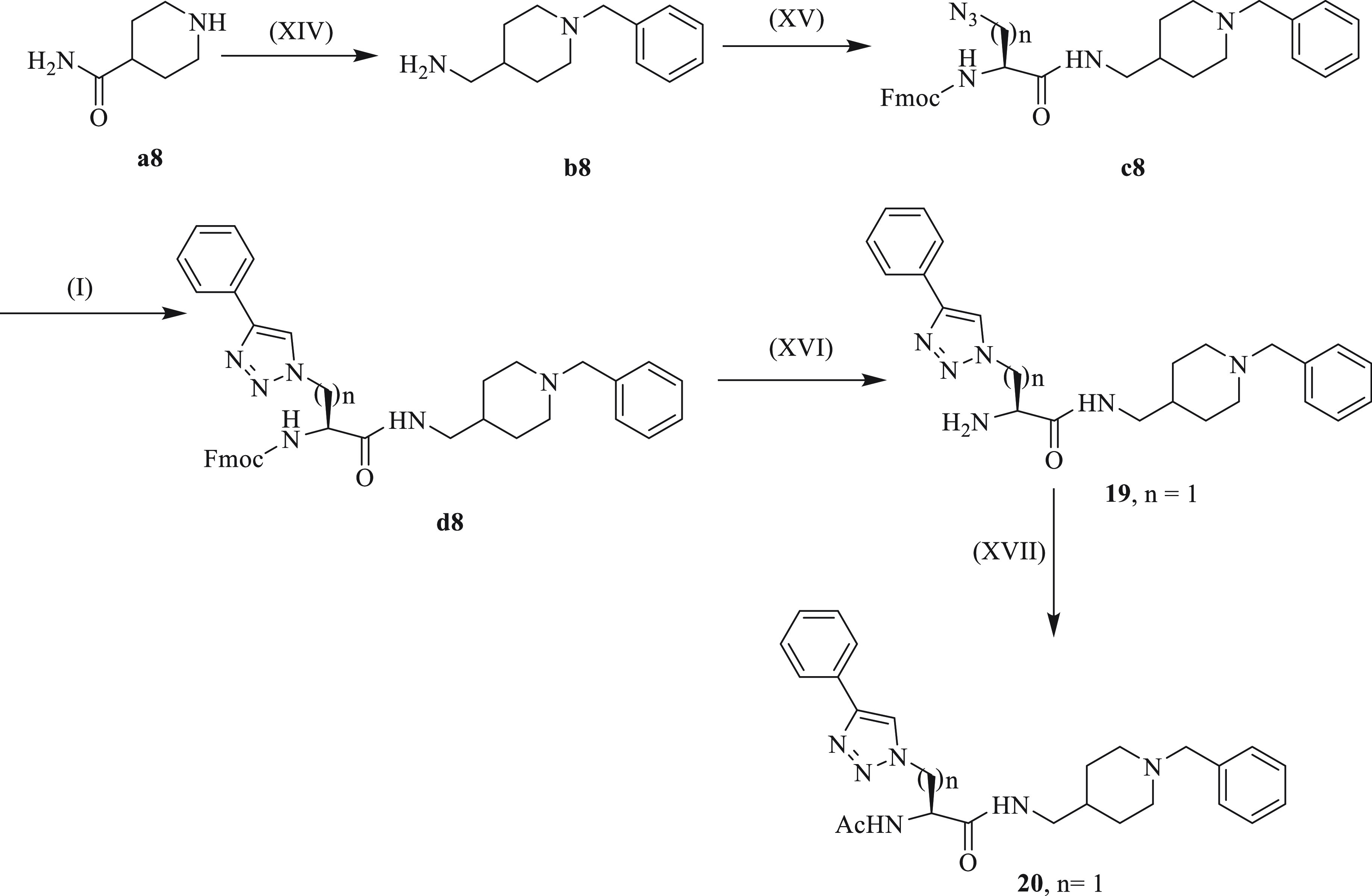
The synthesis of compounds 19 and 20 (Scheme 8) began with the benzylation of isonipecotamide (a8), followed by reduction to yield the key intermediate (b8). In the second step, amide coupling occurred between the intermediate and azido-amino acids to produce the Fmoc-protected azido-building block (c8). In the third step, phenylacetylene was reacted with the azide to form a 1,2,3-triazole-1,4-disubstituted ring (d8). The reaction with morpholine/DCM produced compound 19, which was treated with acetic anhydride and pyridine in the final step to produce compound 20.
The SAR (Figure 8) of the synthesized aryl-1,2,3-triazolyl benzylpiperidines of the first series showed a decreased inhibitory effect because of the lack of flexibility of the binding site owing to the direct coupling of benzylpiperidine with the R group through the 1,2,3-triazole ring. In the second series, compounds with shorter side chains showed increased inhibitory activity compared with those of compounds with longer side chains. In addition, acetylation of the primary amine group increased the potency and selectivity toward hBChE.
Figure 8.

SAR of aryl-1,2,3-triazolyl benzylpiperidine.
To generate novel AChE inhibitor candidates, Le-Nhat-Thuy et al. (2020) designed and synthesized several quinazoline–triazole hybrid compounds (Scheme 9). Twelve compounds were synthesized through the click reaction and split into three groups depending on the various amine moieties attached to the quinazoline scaffold at the C-4 position, specifically the 3-nitrophenylamine, benzylamine, and N-methylpiperazyl groups. All of the synthesized compounds were evaluated for their AChE inhibitory and anticancer activities. Compounds 21 (first group) and 22 and 23 (second group) showed good inhibitory activity against AChE with IC50 values of 2.06 ± 0.19 μM, 0.23 ± 0.15 μM, and 1.10 ± 0.27 μM, respectively. Compound 22 showed increased potency, and compounds 24 (37.38 ± 2.01 μM) and 25 (15.79 ± 0.18 μM) (from the third group) also exhibited the inhibitory activity but less than the other three compounds. Using the MTT assay, these compounds were tested for their cytotoxicity against the human cancer cell lines KBCCL-17 (epidermoid carcinoma), HepG2-HB-8065 (hepatocellular carcinoma), and SK-Lu-1 (nonsmall cell lung cancer); however, they had no discernible cytotoxic effects on these cell lines. Using ICM-Pro MolSoft 3.8-7, a molecular docking study was performed to examine the interactions between AChE and these compounds. The study showed that in the active site of AChE, compounds 21, 22, and 23 bind to both the peripheral anionic site (PAS) and the catalytic anionic site (CAS). This suggests that these compounds may function as dual-binding domain blockers. They also demonstrated suitable physicochemical and pharmacokinetic properties for use as potential medication candidates for AD treatment.56
Scheme 9. Reagents and Conditions: (IV) Propargyl Bromide, Cs2CO3, Acetonitrile, rt, 12 h; (XVIII) NH2OH·HCl, NaOH, MeOH, H2O, rt, 30 min; (XIX) Ac2O, Reflux, 8 h; (XX) Na2S2O4, H2O, 50–65 °C, 4 h; (XXI) DMF-DMA, Acetic Acid, Toluene, Reflux, 4 h; (XXII) Amine, Acetic Acid, Toluene, 60–110 °C, 4 h; (XXIII) Azide, DIPEA, CuI, THF, rt, 1–2 days.

The synthesis of quinazoline–triazole hybrid compounds (Scheme 9) began by the reaction of 2-nitro-5-hydroxybenzaldehyde (a9) to form 2-nitro-5-(prop-2-yn-1-yloxy)benzaldehyde (b9). In the second step, it was converted to an aldoxime (c9). In the third step, aldoxime underwent intramolecular dehydration to produce benzonitrile (d9). Subsequently, the reduction occurred to produce the corresponding amine (e9). Later it was converted into formamidine (f9). Cyclization of formamidine produced 6-(prop-2-yn-1-yloxy)quinazolin-4-amines (g9). In the last step, the hybridization of g9 with different aryl azides yielded quinazoline–triazole hybrids.
The SAR of quinazoline–triazole hybrids showed that the presence of a benzylamine moiety at the C-4 position of the quinazoline displayed increased AChE inhibition (Figure 9). The replacement of benzylamine with a N-methylpiperazyl moiety resulted in decreased inhibitory activity. Higher anti-AChE activity was produced by the nitro group attached to the aryl ring connected to the 1,2,3-triazole. However, shifting the ortho-nitrophenyl group to the meta-/para-nitrophenyl group briefly decreased the AChE inhibitory activity. The structure–activity correlations indicate that the type of amine linked to the C-4 position of the quinazoline scaffold and the type of aryl group attached to the triazole had a significant influence on the anti-AChE efficacy.
Figure 9.

SAR of the quinazoline–triazole hybrids.
4. Development of Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitor Using the Click Chemistry Approach
Nozal et al. (2021) hypothesized on the use of different linkers to produce a BACE1 inhibitor from the kinase inhibitor as the starting fragment, thereby incorporating both activities in one compound to obtain multitarget-directed ligands (MTDLs) to tackle the hallmarks of AD. To test this hypothesis, compounds 26 (Scheme 10) and 27 (Scheme 11) were synthesized using a benzothiazole-based LRRK2 inhibitor as the starting fragment. An aliphatic thioether chain and a heterocyclic 1,2,3-triazole were used to link these kinase inhibitors. Compounds 26 and 27 showed excellent inhibitory activity (IC50 values of 7.14 and 3.72 μM, respectively) against BACE1 and protein kinase. Compound 27 was the leading compound with both BACE1 and protein kinase inhibitory activities. Hence, the hypothesis was supported, and CuAAc was chosen as the best way to connect the different heterocyclic fragments. The ability of compound 27 to reduce the toxicity of Aβ40 and Aβ42 was also checked using human neuroblastoma SH-SY5Y cell lines (SK-APP cells). The excellent results prompted researchers to focus on the synthesis of further compounds.57
Scheme 10. Reagents and Conditions: (XXIV) SOCl2, 80 °C; (XXV) 2-Aminobenzothiazole, 100 °C, MW, THF; (XXVI) K2CO3, Propane-1,3-dithiol, THF, 80 °C.

Scheme 11. Reagents and Conditions: (XXVII) EDCI, DMAP, DMF, rt, 5 min, then 2-Aminobenzothiazole, rt; (XXVIII) 3-Bromopropyne, K2CO3, MeCN, 90 °C; (XXIX) NaOH, MeOH/THF (1:1 v/v), rt; (XXVII) EDCI, DMAP, DMF, rt, 5 min, then 2-aminobenzothiazol; (I) CuSO4·5H2O, Sodium Ascorbate, DMF, rt.

The synthesis of 26 (Scheme 10) began with the reaction of 4-(bromomethyl)benzoic acid (a10) to form 4-(bromomethyl)benzoyl chloride (b10). The compound was then converted to benzamide (c10). Then, coupling with propane-1,3-dithiol resulted in compound 26 (thioether derivative).
The synthesis of compound 27 is explained in Scheme 11. Azide (b11) was produced by reacting 4-(azidomethyl)benzoic acid (a11) with 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI), 4-dimethylaminopyridine (DMA), and DMF, followed by treatment with 2-aminobenzothiazole. The alkynes for CuCA were produced in three steps. The first step involved a reaction of methyl 4-hydroxybenzoate (c11) to produce the intermediate (d11). Later d11 was converted to the corresponding product (e11), which then underwent a reaction to form an adequate alkyne (f11) for CuCA. Finally, the azide (b11) and alkyne (f11) reacted to form compound 27.
Furthermore, two azides from the LRRK2 and CK1d inhibitors and three alkynes from the LRRK2, GSK3b, and CK1d inhibitors were used to synthesize the next set of compounds (Scheme 12). Four compounds were synthesized using CuAAC. The inhibitory activity of the compounds was analyzed, and compound 28 showed good inhibition against BACE1, LRRK2, and GSK3b (3.31 ± 0.27, 0.82 ± 0.14, and 2.36 ± 0.27, respectively). Among the four compounds, compounds 28 and 29 showed a considerable reduction in cytotoxicity. In addition, these two substances prevented tau phosphorylation. Okadaic acid, which induces tau phosphorylation, was used in the SH-SY5Y cell line model to assess the beneficial effects of 28 and 29 on tau phosphorylation. After receiving therapy with the novel MTDLs, relief from the toxic effects of okadaic acid was observed.
Scheme 12. Reagents and Conditions: (I) CuSO4·5H2O, Sodium Ascorbate, DMF, rt.

Scheme 12 illustrates the synthesis of compounds 28 and 29 through the click reaction.
In this study, we also showed how BACE1 could be used as a template to identify fragments to obtain promising MTDLs. Through this approach, it may be easier to recognize BACE1 binders, beginning with kinase inhibitor fragments, before synthesizing them on a larger scale, thereby preserving resources. The in situ click reaction with BACE1 was tested using azides and alkynes. Both azide and alkyne were combined in an aqueous buffer with or without BACE1, and the resulting mixtures were analyzed by LC/MS-SIM. The results revealed that the peak with the anticipated molecular weight of triazole was found only in the presence of BACE1. Six more compounds, 30–35 (Scheme 13) were prepared (using CuCA) to verify this approach, two of which served as negative controls. The inhibitory efficacies of the six compounds against BACE1 and other protein kinases were evaluated. The results demonstrated that the compounds chosen using BACE1 templated synthesis exhibited inhibitory effects on this protease, whereas negative controls did not. Finally, the BBB permeability of all the synthesized compounds was assessed using PAMPA, which identifies the compounds that are suitable candidates for the treatment of illnesses.
Scheme 13. Reagents and Conditions: (I) CuSO4·5H2O, Sodium Ascorbate, DMF, rt.

Scheme 13 shows the synthesis of compounds 30–35. Here, the azide and alkynes react to form the corresponding products via a click reaction.
5. Click Chemistry in Bioorthogonal Coupling
Du et al. (2019) reported that a self-triggered click reaction in an AD model makes neurotoxic copper a useful AD management tool. The Cu(I)-catalyzed azide–alkyne cycloaddition (CuCA) was exploited to synthesize drugs in living cells and to activate fluorophores using an endogenous Cu catalyst (Aβ40-Cu aggregates). The transformation of coumarin from a nonfluorescent state to a fluorescent state was chosen as the model reaction (Scheme 14). The transformation efficacy was 96% based on fluorescence intensity calculations. In living cells (PC12), a Caenorhabditis elegans CL2006 transgenic AD model, and brain slices from triple transgenic AD mice, the efficiency of Aβ40-Cu for catalyzing the fluorogenic click reaction was evaluated. The findings indicated a beneficial outcome.
Scheme 14. Click Reaction for the Synthesis of a Fluorescent Probe.

The authors also discussed the in situ drug development using the CuCA reaction to reduce the cytotoxicity brought on by the Aβ-Cu aggregates. To effectively disintegrate Aβ-Cu aggregates, compound 36 was developed and synthesized with Aβ-oxygenating and Cu-chelating capabilities (Scheme 15). The chelating impact of compound 36 toward Cu, the photo-oxygenating effect of suppressing Aβ aggregation, the neuroprotective ability on cytotoxicity caused by the Aβ40-Cu aggregates, and the capacity to disintegrate the Aβ-Cu aggregates by compound 36 were all evaluated. Finally, the researchers examined the possibility that Aβ40-Cu aggregates could catalyze the in-cell production of bifunctional drug 36. To deliver the prodrugs to the targeted areas, mesoporous silica nanoparticles treated with phenylboronic acid were used as controlled-release nanocarriers. The in situ-produced drug 36 reduced the burden of Aβ and dispersed Aβ-Cu aggregates, inhibited Aβ-mediated immobility, and reduced the locomotor abnormalities of the AD model CL2006 strain. This study provides new insights into the in situ multifunctional drug synthesis for the management of neurological illnesses by exploiting endogenous neurotoxic metal ions.58
Scheme 15. Synthesis of Compound 36.

Scheme 15 illustrates the rational synthesis of compound 36, in which the compound forms a complex with Cu, indicating the Cu-chelating properties of compound 36. Later, the researchers performed the in situ synthesis of 36, where the Aβ40-Cu aggregates act as the catalyst. Compound 36 dissociates into aggregates and forms a complex with Cu. Therefore, reducing the neurotoxicity caused by the Aβ40-Cu aggregation provides a new approach to the in situ synthesis of drugs against AD.
6. Role of Click Reactions in Targeting Amyloid Beta Protein in Alzheimer’s Disease
Sohma et al. (2006) developed phototriggered click peptide isoform precursors of Aβ1-42 (Aβ peptides) based on the O-acyl isopeptide method (Scheme 16). This O-acyl isopeptide method is a unique synthetic approach in the domain of peptide chemistry because it destroys the undesirable 2° structures of native peptides to enable the synthesis of peptides with challenging sequences. Using an O-acyl isopeptide protected by a photocleavable group, the researchers developed a phototriggered counterpart of Aβ1-42 which lacked the ability to self-aggregate. It was anticipated that the O-acyl isopeptide would be nonaggregative and capable of converting into the intrinsic aggregative peptides by light-irradiation “click” without the need for an extra fibril-inhibitory entity. They used this method to create 26-N-Nvoc-26-AIAβ42, a phototriggered click peptide of Aβ1-42, in which a photocleavable 6-nitroveratryloxycarbonyl (Nvoc) group was introduced to produce a new biological review system that would make it simple to manage the stimulation of the self-assembly process. Under physiological conditions, these precursors do not demonstrate any self-assembly characteristics, owing to a single ester bond. However, they were susceptible to porting to give the target Aβ1-42 in a quick and straightforward one-way (hence the name “click”) conversion process. In conclusion, these click peptides may be a key method for advancing investigation into the bioactivity of Aβ1-42 in AD by initiating Aβ1-42 self-assembly in an inducible manner.59
Scheme 16. Reaction of the Phototriggered Click Peptide.

Scheme 16 explains the reaction of phototriggered click peptide a16 (26-N-Nvoc-26-AIAβ42) in forming the target Aβ1-42 (37) in a quick and straightforward one-way conversion process. Here, the click peptide underwent photoirradiation, and the photocleavable Nvoc group was removed to form intermediate 26-AIAβ42 (b16). Then the intramolecular acyl migration of b16 gives the target Aβ1-42 (37).
Jones et al. (2017) synthesized three multitarget phenol–triazoles that could be used as AD therapeutics. The Huisgen 1,3-dipolar cycloaddition reaction was used to synthesize this series (click chemistry). The Lipinski rule, log BB for drug likeness, and BBB penetration were among the physiochemical parameters of all of the tested series, and all compounds displayed positive physiochemical traits. The phenol–triazole series was also assessed for its metal binding affinity (Cu) and antioxidant properties and checked for interaction with the Aβ peptide through 2D-NMR studies. Compound 38 showed a greater number of interactions throughout the length of the Aβ peptide, while compounds 39 and 40 exhibited close interactions with particular residues of the peptides. The Aβ aggregation profiles of this series were determined. Finally the researchers investigated whether these substances could reduce the neurotoxicity caused by Aβ in human neuronal cultures and found that 38 and 40 showed neuroprotective effects. These findings implied that the phenol–triazole ligand scaffold can target various AD-related factors, thereby supporting further therapeutic research.60
The click synthesis of compounds 38–40 is explained in Scheme 17. Alkyne-substituted phenol 2-((trimethylsilyl)ethynyl)phenol (a17) and various azides (b17) were reacted to form the final compounds.
Scheme 17. Reagents and Conditions: (I) CuSO4, Sodium Ascorbate.

The SAR of the phenol–triazole ligand scaffold is demonstrated in Figure 10. The triazole ring plays a significant role in ligand peptide interactions since the triazoles are able to mimic and rigidify the conformation of the amide backbone (peptide bond).61 The R group on the triazole ring influences ligand-Aβ peptide interactions as well as neuroprotection. When R was substituted with a propanol group, 38 exhibited interactions across the entire peptide length. The propanol group was replaced by morpholine (compound 39) and thiomorpholine (compound 40), and the substitutions displayed a superior specificity for the Aβ peptide residues. In addition, these substitutions play a crucial role in Aβ aggregation.
Figure 10.

SAR of phenol–triazole ligand scaffold.
7. Click Chemistry in Fluorescence Imaging
Ahmed et al. (2019) reported dansyl azide (DNS-AZ) as a fluorescent marking tool based on click chemistry for biomolecular characterization. The fluorescence marking and tag of biomolecules is a powerful method for examining various biological activities. DNS-AZ has previously been reported as a redox-based fluorescence marker. Click chemistry has also been shown as an effective technique for bioorthogonal labeling inside biological systems because of the use of aqueous solutions and moderate reaction conditions. Rasagiline mesylate (RSM) is an MAO-B-specific irreversible antagonist authorized for use as either primary or adjunctive therapy for PD. Only a limited number of bioanalytical techniques are available to determine RSM in plasma samples, and because of the challenges encountered in quantifying RSM using inadequate analytical processes, this study aimed to create a new HPLC fluorescence approach for the selective assessment of RSM in rat plasma using this fluorescent marking tool.
By combining RSM and DNS-AZ through a click reaction (Scheme 18), the first step of the study was the production of an inert fluorescent 1,2,3-triazole analogue. To generate a highly fluorescent final product, all reagents, reaction temperatures, and durations were optimized. Selegiline was utilized as an internal standard because it shares the same physicochemical properties. The structure of the resulting compound 41 was verified by tandem mass ultraperformance liquid chromatography. To ensure that the created method was appropriate for the intended use, it was evaluated in accordance with the U.S. FDA criteria. The linearity of the devised method was assessed in rat plasma with concentrations between 0.5 and 100 ng/mL (n = 5), and it was found to be linear with adequate precision and accuracy values. After administering one oral dose of the RSM pill to rats, the devised method was used to analyze RSM in pharmacokinetic studies to establish its usability in real sample evaluations. The proposed approach demonstrates the ability to measure the RSM in actual samples and can be used as a tool in health management.62
Scheme 18. Synthesis of Compound 41.

Scheme 18 shows the synthesis of the fluorescent derivatization product. A reaction occurred between RSM and DNS-AZ with the optimized reagents. A click reaction occurred between the alkyne and azide to give the product.
8. Future Perspectives
Research and development of drugs are especially challenging when it comes to neurodegenerative diseases. The onset of neurodegenerative diseases is mostly attributable to a combination of genetic anomalies, environmental variables, and aging. There is always a need for producing more therapeutics in this field due to the increasing demands. In comparison to the conventional method, click chemistry allows for the rapid synthesis of a large number of chemical libraries. Along with positive aspects like modularity, excellent yield and high chemo- and regioselectivity facilitate lead optimization and drug discovery. The 1,2,3-triazole scaffold is frequently used in drug development processes, and various triazole-based compounds have effectively evolved into marketed drugs. They are beneficial not only for NDDs but also for many other targets. For example: rufinamide (anticonvulsant), carboxyamidotriazole (anticancer), tazobactam (antibacterial agent), and so on.63 The click reaction has several benefits as mentioned and applications like bioconjugation, biomedical imaging, proteomics (protein profiling, protein labeling, and enzyme–inhibitor screening), and material science. Also, the bioorthogonal properties of click reactions can be utilized as tools for the selective tagging and characterization of biological molecules in cellular extracts. All of these uses have sparked a great deal of study and advancement in this area. Consequently, it will satisfy the preclinical and clinical requirements of many systemic and targeted diseases in the near future.
9. Conclusion
CC reactions are quick, extremely specific, easy to carry out, and intolerant of oxygen and water. In this Review, we discuss the role of click reactions in major targets of the NDDs. Among the several types of click reactions, the Huisgen 1,3-dipolar cycloaddition reaction between an azide and a terminal alkyne that yields the 1,4-disubstituted 1,2,3-triazoles has been given more emphasis in this Review. From the reports reviewed, it is clear that the click reactions induce a fast construction of numerous compounds under facile reaction conditions with high reaction selectivity along with easy product isolation. This Review also highlights the SARs of compounds joined by the 1,2,3-triazole linker, providing potential interactions with biological targets and being resistant to metabolic depletion and thereby providing a neuroprotective effect. The dual-target hybrids, i.e., with both iron ion-chelating and MAO-B-inhibitory activities, can also be synthesized through click reactions. Also, the self-triggered click reaction in an AD model makes the neurotoxic copper a useful tool as a catalyst in the Cu(I)-catalyzed azide–alkyne cycloaddition to synthesize drugs in living cells for the management of AD. From the merits discussed in the area of neuroprotection, the click reactions, mainly the copper-catalyzed azide–alkyne cycloaddition reactions, can potentially accelerate the exploration for designing new therapeutics for the treatment of neurological disorders.
Acknowledgments
The authors are thankful to AlMaarefa University for their support. Prof. Dr. Bijo Mathew thanks the Indian Council Social Science Research for the financial support (No. 02/90/2022-23/RP/MN).
The authors declare no competing financial interest.
References
- Hou J.; Liu X.; Shen J.; Zhao G.; Wang P. G. The Impact of Click Chemistry in Medicinal Chemistry. Expert Opin Drug Discov 2012, 7 (6), 489–501. 10.1517/17460441.2012.682725. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P.; Chaturvedi N.; Gupta S.; Mishra A.; Singh M.; Siddhartha T. Click Chemistry: A New Approach for Drug Discovery. Int. J. Pharm. Sci. Rev. Res. 2011, 10, 111–117. [Google Scholar]
- The Editors of Encyclopaedia Britannica . K. Barry Sharpless. In Encyclopaedia Britannica; Encyclopaedia Britannica, Inc.: 2023. [Google Scholar]
- Rostovtsev V. V; Green L. G.; Fokin V. V.; Sharpless K. B.; Coolen H. K.; van Leeuwen P. W.; Nolte R. J.; Harrowfield J. M. In Calixarenes; Kluwer Academic Publishers: 2002; Vol. 114. [Google Scholar]
- Manetsch R.; Krasiński A.; Radić Z.; Raushel J.; Taylor P.; Sharpless K. B.; Kolb H. C. In Situ Click Chemistry: Enzyme Inhibitors Made to Their Own Specifications. J. Am. Chem. Soc. 2004, 126 (40), 12809–12818. 10.1021/ja046382g. [DOI] [PubMed] [Google Scholar]
- Helms B.; Mynar J. L.; Hawker C. J.; Fréchet J. M. J. Dendronized Linear Polymers via “Click Chemistry. J. Am. Chem. Soc. 2004, 126 (46), 15020–15021. 10.1021/ja044744e. [DOI] [PubMed] [Google Scholar]
- Karim Md. A.; Cho Y.-R.; Park J. S.; Ryu T. I.; Lee M. J.; Song M.; Jin S.-H.; Lee J. W.; Gal Y.-S. Comparison of Three Different Click Reaction Methods for the Synthesis of Fluorene-Based Polymers and Performance in Quasi-Solid-State DSSCs. Macromol. Chem. Phys. 2008, 209 (19), 1967–1975. 10.1002/macp.200800329. [DOI] [Google Scholar]
- Wang Q.; Chan T. R.; Hilgraf R.; Fokin V. V.; Sharpless K. B.; Finn M. G. Bioconjugation by Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2003, 125 (11), 3192–3193. 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- Schirrmacher R.; Wangler C.; Schirrmacher E. Recent Developments and Trends in 18F-Radiochemistry: Syntheses and Applications. Mini Rev. Org. Chem. 2007, 4 (4), 317–329. 10.2174/157019307782411699. [DOI] [Google Scholar]
- Nwe K.; Brechbiel M. W. Growing Applications of “Click Chemistry” for Bioconjugation in Contemporary Biomedical Research. Cancer Biother Radiopharm 2009, 24 (3), 289–302. 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tressaud A.Fluorine, a Key Element for the 21st Century. In Fluorine; Elsevier: 2019; pp 77–150. 10.1016/B978-0-12-812990-6.00002-7. [DOI] [Google Scholar]
- Zhang Z.-B.; Wu J.-J.; Su Y.; Zhou J.; Gao Y.; Yu H.-Y.; Gu J.-S. Layer-by-Layer Assembly of Graphene Oxide on Polypropylene Macroporous Membranes via Click Chemistry to Improve Antibacterial and Antifouling Performance. Appl. Surf. Sci. 2015, 332, 300–307. 10.1016/j.apsusc.2015.01.193. [DOI] [Google Scholar]
- Pachón L. D.; van Maarseveen J. H.; Rothenberg G. Click Chemistry: Copper Clusters Catalyse the Cycloaddition of Azides with Terminal Alkynes. Adv. Synth Catal 2005, 347 (6), 811–815. 10.1002/adsc.200404383. [DOI] [Google Scholar]
- Aragão-Leoneti V.; Campo V. L.; Gomes A. S.; Field R. A.; Carvalho I. Application of Copper(I)-Catalysed Azide/Alkyne Cycloaddition (CuAAC) ‘Click Chemistry’ in Carbohydrate Drug and Neoglycopolymer Synthesis. Tetrahedron 2010, 66 (49), 9475–9492. 10.1016/j.tet.2010.10.001. [DOI] [Google Scholar]
- Shankaraiah N.; Sakla A. P.; Laxmikeshav K.; Tokala R. Reliability of Click Chemistry on Drug Discovery: A Personal Account. Chem. Rec. 2020, 20 (4), 253–272. 10.1002/tcr.201900027. [DOI] [PubMed] [Google Scholar]
- Hou J.; Liu X.; Shen J.; Zhao G.; Wang P. G. The Impact of Click Chemistry in Medicinal Chemistry. Expert Opin Drug Discov 2012, 7 (6), 489–501. 10.1517/17460441.2012.682725. [DOI] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Billington R. A.; Canonico P. L.; Sorba G.; Genazzani A. A. Click Chemistry Reactions in Medicinal Chemistry: Applications of the 1,3-Dipolar Cycloaddition between Azides and Alkynes. Med. Res. Rev. 2008, 28 (2), 278–308. 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- Jørgensen K. A. Catalytic Asymmetric Hetero-Diels–Alder Reactions of Carbonyl Compounds and Imines. Angew. Chem. 2000, 39 (20), 3558–3588. . [DOI] [PubMed] [Google Scholar]
- Adolfsson H.; Converso A.; Sharpless K. B. Comparison of Amine Additives Most Effective in the New Methyltrioxorhenium-Catalyzed Epoxidation Process. Tetrahedron Lett. 1999, 40 (21), 3991–3994. 10.1016/S0040-4039(99)00661-9. [DOI] [Google Scholar]
- Kolb H. C.; VanNieuwenhze M. S.; Sharpless K. B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94 (8), 2483–2547. 10.1021/cr00032a009. [DOI] [Google Scholar]
- Gololobov Yu. G.; Zhmurova I. N.; Kasukhin L. F. Sixty Years of Staudinger Reaction. Tetrahedron 1981, 37 (3), 437–472. 10.1016/S0040-4020(01)92417-2. [DOI] [Google Scholar]
- Shankaraiah N.; Sakla A. P.; Laxmikeshav K.; Tokala R. Reliability of Click Chemistry on Drug Discovery: A Personal Account. Chem. Rec. 2020, 20 (4), 253–272. 10.1002/tcr.201900027. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Sharpless K. B. The Growing Impact of Click Chemistry on Drug Discovery. Drug Discov Today 2003, 8 (24), 1128–1137. 10.1016/S1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- Pachón L. D.; van Maarseveen J. H.; Rothenberg G. Click Chemistry: Copper Clusters Catalyse the Cycloaddition of Azides with Terminal Alkynes. Adv. Synth Catal 2005, 347 (6), 811–815. 10.1002/adsc.200404383. [DOI] [Google Scholar]
- Kumar G. S.; Lin Q. Light-Triggered Click Chemistry. Chem. Rev. 2021, 121 (12), 6991–7031. 10.1021/acs.chemrev.0c00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani A.; Singh G.; Singh A.; Maqbool U.; Kaur G.; Singh J. CuAAC-Ensembled 1,2,3-Triazole-Linked Isosteres as Pharmacophores in Drug Discovery: Review. RSC Adv. 2020, 10 (10), 5610–5635. 10.1039/C9RA09510A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan B.; Balasubramanian K. K.. Applications of Click Chemistry in Drug Discovery and Development. In Click Reactions in Organic Synthesis; Wiley: 2016; pp 25–76. 10.1002/9783527694174.ch2. [DOI] [Google Scholar]
- Kim E.; Koo H. Biomedical Applications of Copper-Free Click Chemistry: In Vitro, in Vivo, and Ex Vivo. Chem. Sci. 2019, 10 (34), 7835–7851. 10.1039/C9SC03368H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Contreras M.; He H.; Little K. N.; Lee J. P.; Campbell R. P.; Royzen M.; Zhu L. SNAP/CLIP-Tags and Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC)/Inverse Electron Demand Diels–Alder (IEDDA) for Intracellular Orthogonal/Bioorthogonal Labeling. Bioconjug Chem. 2020, 31 (5), 1370–1381. 10.1021/acs.bioconjchem.0c00107. [DOI] [PubMed] [Google Scholar]
- Thirumurugan P.; Matosiuk D.; Jozwiak K. Click Chemistry for Drug Development and Diverse Chemical–Biology Applications. Chem. Rev. 2013, 113 (7), 4905–4979. 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- Dugger B. N.; Dickson D. W. Pathology of Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2017, 9 (7), a028035 10.1101/cshperspect.a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayan J.; Roshi H.; Ashraf F. F. P.; Nair P. G.; Vijayakumar A.; Nair A. S.; Pappachen L. K.; Abdelgawad M. A.; Parambi D. G. T.; Aleya L.; Mathew B. Effects of Radiation Exposure on Brain Health: A State of the Art and New Challenges. Environmental Science and Pollution Research 2022, 29 (58), 87068–87081. 10.1007/s11356-022-23703-4. [DOI] [PubMed] [Google Scholar]
- Jellinger K. A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell Mol. Med. 2010, 14, 457. 10.1111/j.1582-4934.2010.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L.; Cookson M. R.; Petrucelli L.; La Spada A. R. Converging Pathways in Neurodegeneration, from Genetics to Mechanisms. Nat. Neurosci 2018, 21 (10), 1300–1309. 10.1038/s41593-018-0237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung A. W. K.; Georgieva M. G.; Atanasov A. G.; Tzvetkov N. T. Monoamine Oxidases (MAOs) as Privileged Molecular Targets in Neuroscience: Research Literature Analysis. Front Mol. Neurosci 2019, 12, 143. 10.3389/fnmol.2019.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi M.; Maruyama W.; Inaba-Hasegawa K.; Akao Y. Type A Monoamine Oxidase Regulates Life and Death of Neurons in Neurodegeneration and Neuroprotection 2011, 100, 85–106. 10.1016/B978-0-12-386467-3.00005-4. [DOI] [PubMed] [Google Scholar]
- Reinfeld S. Should Residents Be Taught How to Prescribe Monoamine Oxidase Inhibitors?. Curr. Psychiatr 2022, 21 (12), e1–e2. 10.12788/cp.0316. [DOI] [Google Scholar]
- Kumar B.; Sheetal S.; Mantha A. K.; Kumar V. Recent Developments on the Structure–Activity Relationship Studies of MAO Inhibitors and Their Role in Different Neurological Disorders. RSC Adv. 2016, 6 (48), 42660–42683. 10.1039/C6RA00302H. [DOI] [Google Scholar]
- Bhawna; Kumar A.; Bhatia M.; Kapoor A.; Kumar P.; Kumar S. Monoamine Oxidase Inhibitors: A Concise Review with Special Emphasis on Structure Activity Relationship Studies. Eur. J. Med. Chem. 2022, 242, 114655 10.1016/j.ejmech.2022.114655. [DOI] [PubMed] [Google Scholar]
- Walczak-Nowicka Ł. J.; Herbet M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in Their Pathogenesis. Int. J. Mol. Sci. 2021, 22 (17), 9290. 10.3390/ijms22179290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchio I.; Sorrentino L.; Paoletti A.; Marra R.; Arbitrio M. The State of The Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 117957352110291 10.1177/11795735211029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig N. H.; Utsuki T.; Yu Q.; Zhu X.; Holloway H. W.; Perry T.; Lee B.; Ingram D. K.; Lahiri D. K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin 2001, 17 (3), 159–165. 10.1185/0300799039117057. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Manoharan A.; J J.; Abdelgawad M. A.; Mahdi W. A.; Alshehri S.; Ghoneim M. M.; Pappachen L. K.; Zachariah S. M.; Aneesh T. P.; Mathew B. Exploiting Butyrylcholinesterase Inhibitors through a Combined 3-D Pharmacophore Modeling, QSAR, Molecular Docking, and Molecular Dynamics Investigation. RSC Adv. 2023, 13 (14), 9513–9529. 10.1039/D3RA00526G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evin G.; Hince C. BACE1 as a Therapeutic Target in Alzheimer’s Disease: Rationale and Current Status. Drugs Aging 2013, 30 (10), 755–764. 10.1007/s40266-013-0099-3. [DOI] [PubMed] [Google Scholar]
- Das B.; Yan R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33 (3), 251–263. 10.1007/s40263-019-00613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H.; Vassar R.; De Strooper B.; Hardy J.; Willem M.; Singh N.; Zhou J.; Yan R.; Vanmechelen E.; De Vos A.; Nisticò R.; Corbo M.; Imbimbo B. P.; Streffer J.; Voytyuk I.; Timmers M.; Tahami Monfared A. A.; Irizarry M.; Albala B.; Koyama A.; Watanabe N.; Kimura T.; Yarenis L.; Lista S.; Kramer L.; Vergallo A. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89 (8), 745–756. 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kins S.; Schäfer K.-H.; Endres K. Drug Development for Neurodegenerative Diseases. Biol. Chem. 2022, 403 (1), 1–1. 10.1515/hsz-2021-0413. [DOI] [PubMed] [Google Scholar]
- Dheer D.; Singh V.; Shankar R. Medicinal Attributes of 1,2,3-Triazoles: Current Developments. Bioorg Chem. 2017, 71, 30–54. 10.1016/j.bioorg.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Hao X.; Jing L.; Wu G.; Kang D.; Liu X.; Zhan P. Recent Applications of Click Chemistry in Drug Discovery. Expert Opin Drug Discov 2019, 14 (8), 779–789. 10.1080/17460441.2019.1614910. [DOI] [PubMed] [Google Scholar]
- Jia Z.; Zhu Q. Click’ Assembly of Selective Inhibitors for MAO-A. Bioorg. Med. Chem. Lett. 2010, 20 (21), 6222–6225. 10.1016/j.bmcl.2010.08.104. [DOI] [PubMed] [Google Scholar]
- Di Pietro O.; Alencar N.; Esteban G.; Viayna E.; Szałaj N.; Vázquez J.; Juárez-Jiménez J.; Sola I.; Pérez B.; Solé M.; Unzeta M.; Muñoz-Torrero D.; Luque F. J. Design, Synthesis and Biological Evaluation of N-Methyl-N-[(1,2,3-Triazol-4-Yl)Alkyl]Propargylamines as Novel Monoamine Oxidase B Inhibitors. Bioorg. Med. Chem. 2016, 24 (20), 4835–4854. 10.1016/j.bmc.2016.06.045. [DOI] [PubMed] [Google Scholar]
- Haider S.; Alhusban M.; Chaurasiya N. D.; Tekwani B. L.; Chittiboyina A. G.; Khan I. A. Isoform Selectivity of Harmine-Conjugated 1,2,3-Triazoles against Human Monoamine Oxidase. Future Med. Chem. 2018, 10 (12), 1435–1448. 10.4155/fmc-2018-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Z.; Gan B.; Yu S.; Guo J.; Zhang C.; Jiang X.; Zhou T.; Su J.; Bai R.; Xie Y. Dual-Target Anti-Alzheimer’s Disease Agents with Both Iron Ion Chelating and Monoamine Oxidase-B Inhibitory Activity. J. Enzyme Inhib Med. Chem. 2019, 34 (1), 1489–1497. 10.1080/14756366.2019.1634703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W. Z.; Cheng F.; Zhang Y. J.; Ge J. Y.; Yao S. Q.; Zhu Q. Rapid Synthesis of Flavone-Based Monoamine Oxidase (MAO) Inhibitors Targeting Two Active Sites Using Click Chemistry. Chem. Biol. Drug Des 2017, 89 (1), 141–151. 10.1111/cbdd.12841. [DOI] [PubMed] [Google Scholar]
- de Andrade P.; Mantoani S. P.; Gonçalves Nunes P. S.; Magadán C. R.; Pérez C.; Xavier D. J.; Hojo E. T. S.; Campillo N. E.; Martínez A.; Carvalho I. Highly Potent and Selective Aryl-1,2,3-Triazolyl Benzylpiperidine Inhibitors toward Butyrylcholinesterase in Alzheimer’s Disease. Bioorg. Med. Chem. 2019, 27 (6), 931–943. 10.1016/j.bmc.2018.12.030. [DOI] [PubMed] [Google Scholar]
- Le-Nhat-Thuy G.; Nguyen Thi N.; Pham-The H.; Dang Thi T. A.; Nguyen Thi H.; Nguyen Thi T. H.; Nguyen Hoang S.; Nguyen T. V. Synthesis and Biological Evaluation of Novel Quinazoline-Triazole Hybrid Compounds with Potential Use in Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2020, 30 (18), 127404 10.1016/j.bmcl.2020.127404. [DOI] [PubMed] [Google Scholar]
- Nozal V.; García-Rubia A.; Cuevas E. P.; Pérez C.; Tosat-Bitrián C.; Bartolomé F.; Carro E.; Ramírez D.; Palomo V.; Martínez A. From Kinase Inhibitors to Multitarget Ligands as Powerful Drug Leads for Alzheimer’s Disease Using Protein-Templated Synthesis. Angew. Chem. 2021, 133 (35), 19493–19503. 10.1002/ange.202106295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Yu D.; Du X.; Scott P.; Ren J.; Qu X. Self-Triggered Click Reaction in an Alzheimer’s Disease Model: In Situ Bifunctional Drug Synthesis Catalyzed by Neurotoxic Copper Accumulated in Amyloid-β Plaques. Chem. Sci. 2019, 10 (44), 10343–10350. 10.1039/C9SC04387J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohma Y.; Kiso Y. Click Peptides” — Chemical Biology-Oriented Synthesis of Alzheimer’s Disease-Related Amyloid β Peptide (Aβ) Analogues Based on the “O-Acyl Isopeptide Method”. ChemBioChem 2006, 7 (10), 1549–1557. 10.1002/cbic.200600112. [DOI] [PubMed] [Google Scholar]
- Jones M. R.; Mathieu E.; Dyrager C.; Faissner S.; Vaillancourt Z.; Korshavn K. J.; Lim M. H.; Ramamoorthy A.; Wee Yong V.; Tsutsui S.; Stys P. K.; Storr T. Multi-Target-Directed Phenol–Triazole Ligands as Therapeutic Agents for Alzheimer’s Disease. Chem. Sci. 2017, 8 (8), 5636–5643. 10.1039/C7SC01269A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agouram N.; El Hadrami E. M.; Bentama A. 1,2,3-Triazoles as Biomimetics in Peptide Science. Molecules 2021, 26 (10), 2937. 10.3390/molecules26102937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S.; Abdallah N. A. Dansyl Azide as a Selective Fluorescence Tagging Probe for Click Chemistry Reactions and Its Application to Monitor Rasagiline in Pharmacokinetic Studies. J. Pharm. Biomed Anal 2019, 165, 357–365. 10.1016/j.jpba.2018.12.026. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Saxena M.; Rishi N. An Overview of Recent Advances in Biomedical Applications of Click Chemistry. Bioconjug Chem. 2021, 32 (8), 1455–1471. 10.1021/acs.bioconjchem.1c00247. [DOI] [PubMed] [Google Scholar]