

Abstract
The cystine/glutamate antiporter system xc− (Sxc−) mediates the exchange of intracellular L-glutamate (L-Glu) with extracellular L-cystine (L-Cys2). Both the import of L-Cys2 and the export of L-Glu take on added significance in CNS cells, especially astrocytes. When the relative activity of Sxc− overwhelms the regulatory capacity of the EAATs, the efflux of L-Glu through the antiporter can be significant enough to trigger excitotoxic pathology, as is thought to occur in glioblastoma. This has prompted considerable interest in the pharmacological specificity of Sxc− and the development of inhibitors. The present study explores a series of analogues that are structurally related to sulfasalazine, a widely employed inhibitor of Sxc−. We identify a number of novel aryl-substituted amino-naphthylsulfonate analogues that inhibit Sxc− more potently than sulfasalazine. Interestingly, the inhibitors switch from a competitive to noncompetitive mechanism with increased length and lipophilic substitutions, a structure-activity relationship that was previously observed with aryl-substituted isoxazole. These results suggest that the two classes of inhibitors may interact with some of the same domains on the antiporter protein and that the substrate and inhibitor binding sites may be in close proximity to one another. Molecular modeling is used to explore this possibility.
Introduction
The cystine/glutamate antiporter system xc− (Sxc−) mediates the exchange of intracellular L-glutamate (L-Glu) with extracellular L-cystine (L-Cys2) in a variety of CNS cells, including: astrocytes, oligodendrocytes, retinal Muller cells, and immature cortical neurons (for review see: [1]). The import of L-Cys2 and the export of L-Glu both take on added significance within the CNS given the reliance of astrocytes on L-Cys2 as a precursor for the antioxidant glutathione (GSH) and the ability of L-Glu to contribute to excitatory signaling and excitotoxic pathology [2]. The entry of L-Cys2 into astrocytes not only appears to be a rate limiting step in maintaining the intracellular pools of L-cysteine (L-CysH) and GSH, their subsequent export serves as an extracellular source of L-CysH for the synthesis of GHS in neurons, which appear more efficient at transporting L-CysH than L-Cys2 [3]. From a translational perspective, inhibiting Sxc− and reducing intracellular GSH levels also provides a therapeutic strategy to increase chemosensitivity and reduce chemoresistance in cancer cells [4–6]. More recently, attention has focused on the Sxc−-mediated efflux of L-Glu into extrasynaptic spaces, where it holds the potential to act on excitatory amino acid (EAA) receptors and contribute to signaling, neurotransmitter release and synaptic organization [2,7–9]. This efflux of L-Glu is typically countered by its uptake via Na+-dependent excitatory amino acid transporters (EAATs), which are also highly expressed in astrocytes. When the relative activity of Sxc− overwhelms the regulatory capacity of the EAATs, the efflux of L-Glu through the antiporter can be significant enough to trigger excitotoxic pathology [2,10]. This especially appears to be the case in patients with glioblastoma, where the overexpression of Sxc− contributes to tumor growth and tumor-associated seizures [11–15].
Classified within the Amino acid, Polyamine, and organic Cation (APC) transporter super-family and L-Amino acid Transporter (LAT) family [16], Sxc− is a eukaryotic Heteromeric Amino acid Transporter (HAT) (aka glycoprotein-associated amino acid exchangers). In the instance of Sxc−, the heterodimer is comprised of xCT (SLC7A11), a non-glycosylated light chain subunit that mediates the exchange of the substrates, and 4F2hc, a type II N-glycosylated heavy chain subunit that is covalently linked to xCT via a disulfide bond and acts in trafficking the antiporter to the cell surface. Sxc− functions in a Na-independent, Cl-dependent, and electroneutral manner that readily distinguishes it from the Na+-dependent EAATs. Activity can be quantified via radiolabeled flux assays with either L-Glu or L-Cys2 as substrates [17], with each acting as a competitive inhibitor of the other, or by fluorometrically measuring the substrate-induced efflux of L-Glu through Sxc− with an enzyme-coupled metabolic assay [18–20].
The pharmacology of Sxc− has been well studied, with a variety of alternate substrates and non-substrate inhibitors having been identified [1]. Not surprisingly, substrates share considerable structural homology with L-Glu and L-Cys2 and include α-aminoadipic acid, α-aminosuberic acid, and cystathionine, as well as more novel analogues, such as ibotenate , quisqualate, L−β-N-oxalyl-L-α.β-diaminopropionate (L−β-ODAP), L-alanosine, and β-N-methylamino-L-alanine (BMAA) [1,21–25]. Non-substrate inhibitors (compounds that bind to and inhibit the transporter, but are not translocated) include a number of carboxyphenylglycine (CPG) analogues (e.g. S-4-carboxy-phenylglycine, S-4-carboxy,3-hydroxy-phenylglycine, R,S-sulphothienylglycine), aryl-substituted isoxazoles (e.g., 5-benzyl-4-bis-TFM-HMICA, 5-naphthyl-4-bis-TFM-HMICA, 5–4-TFM-benzyl-4-bis-TFM-HMICA), the ferroptosis activator erastin, the anticancer drug sorafenib, and sulfasalazine and its derivatives [1,17–20,26–30].
Of these inhibitors, sulfasalazine (SAS) has proved to be one of the most useful, owing in part to its FDA-approval as a drug (Azulfidine) to treat ulcerative colitis, Crohn’s disease and rheumatoid arthritis. Its effectiveness in these disorders arise from SAS being a pro-drug that is rapidly metabolized to 5-aminosalicylic acid and sulfapyridine in vivo, both of which exhibit anti-inflammatory activity. Interestingly, this has limited the use of SAS as a drug targeting Sxc− in vivo, as it is only the intact molecule that inhibits the antiporter. Nonetheless, SAS has been useful in a number of “proof of concept” studies to explore the role Sxc− in various disease models, including: reducing the acute release of L-glutamate and epileptiform activity in mice with implanted U251GFP glioma cells, decreasing hyperexcitability in mouse cortical slices under epileptogenic conditions, and sensitizing cancer cells to radiation [31–33]. The present study explores a series of SAS analogues containing one or more hydroxy-benzoic acid or amino-naphthyl sulfonic acid moieties linked together by diazo bonds as potential inhibitors of the Sxc−-mediated uptake of L-Glu into SNB19 human glioblastoma cells. We identify a number of novel inhibitors of Sxc− that are more potent than SAS and add to a growing understanding of the structure-activity relationships (SARs) that dictate the pharmacological specificity of Sxc−. Interestingly, kinetic characterization of the inhibitors yield both competitive and noncompetitive inhibitors, similar to the SAR observed in previous studies with aryl-substituted isoxazole that switched from a competitive to a noncompetitive mechanism of inhibition with increasing length and lipophilic substitution [17]. Molecular docking studies using an xCT homology model based upon the bacterial arginine/agmatine exchanger AdiC [34], are also used to identify residues that likely participate in substrate binding, as well as adjacent domains that may accommodate the noncompetitive inhibitors and represent an allosteric regulatory site on the antiporter.
Materials and Methods:
The azo-containing dyes( Acid Black 1, Pontacyl Violet 6R, Pontacyl Carmine 2B, Acid Red 1, Gallion, Chromotrope 2B and 2R, Chrome Yellow, Mordant Orange 1, and Alizarin Yellow GG) were obtained from TCI America (Portland, OR). All other chemical were obtained from Sigma-Aldrich (St. Louis, Mo). L-[3,4-3H]-Glutamate was obtained from PerkinElmer (Waltham, MA). All other materials were prepared or obtained as detailed below.
Synthesis of mono- and bis-azo-amino-naphthyl sulfonates:
The azo amino-naphthylene sulfonic acid (AANS) analogues reported in this study were prepared using known azo-coupling procedures as previously reported [35,36] in which an analine or symmetrical dianilino structure is diazotized with a near stoichiometric amount of HNO2/HCl and reacted with one or two equivalents of 4-amino-1-naphthylene sulfonic acid. The crude products typically precipitated or precipitated with the addition of solid NaCl. The compounds were then collected by filtration, and products were chromatographed on silica using CH2Cl2/MeOH (9:1) to a purity greater than 95%. Yields were generally poor ranging from 5–25%. The regiochemistry of the diazo coupling occurred at the 2-position as determined by NMR, and no NMR evidence of other isomers were observed. Experiments conducted to produce bis-diazotization were accompanied by the mono-azo product that was removed chromatographically. The 1H and 13C NMR, UV-Vis, mass spec characteristics and combustion analyses were consistent with the assigned structures.
Cell culture
SNB-19 glioma cells, purchased from American Type Culture Collection (Manassas, VA), were grown in DMEM/F-12 medium (pH 7.4) containing 1 mM pyruvate and 16 mM NaHCO3 and supplemented with 10% fetal calf serum. The cells were cultured in 150 cm2 flasks (Corning or Biologix) and maintained at 37°C in a humidified atmosphere of 5% CO2. In the 3H-L-Glu uptake experiments, cells were seeded in 12 well culture plates (Greiner Bio-one) at a density of 3×104 cells/well and maintained for 3 days until 80–90% confluent.
Glutamate uptake assay
Uptake of 3H-L-Glu into cultured cells was quantified using a modification of the procedure of Martin and Shane as previously described by [19]. Briefly, after removal of culture media, wells were rinsed three times and pre-incubated in 1 ml Na+-free HEPES buffered (pH 7.4) Hank’s balanced salt solution (HBHS) at 30°C for 5 min. The Na+-free buffer contained: 137.5 mM choline-Cl, 5.36 mM KCl, 0.77 mM KH2PO4, 0.71 mM MgSO4•7H2O, 1.1 mM CaCl2, 11 mM D-glucose, and 10 mM HEPES. Uptake was initiated by aspiration of the pre-incubation buffer and the addition of a 500 μl aliquot of Na+-free transport buffer containing 3H-L-Glu (4–16 mCi/ml) mixed with L-Glu (10 μM-500 μM, final concentration). In those assays that evaluated inhibitor activity, the 500 μl aliquot of transport buffer contained both the 3H-L-Glu and potential inhibitors to ensure simultaneous addition. Following a 5 min incubation at 30°C, the assays were terminated by three sequential 1 ml washes with ice cold buffer after which the cells were dissolved in 1 ml of 0.4 M NaOH for 24 h. Aliquots (200 μl each) were then transferred into two 5 ml glass scintillation vial and neutralized with the addition of 5 μl glacial acetic acid followed by 3 ml Liquiscint© scintillation fluid (National Diagnostics) to each sample. Incorporation of radioactivity was quantified by liquid scintillation counting (LSC, Beckman LS 6500) and the two aliquots averaged to yield a value for that well. Within each experiment assays were performed in duplicate wells and averaged for an n=1. Protein concentrations were determined by the bicinchoninic acid (BCA) method (Pierce) and used to normalize the of 3H-L-Glu accumulation into the cells. Values are reported as mean ± S.E.M. (n ≥ 3 experiments) and have been corrected for non-specific uptake (e.g., leakage and binding) by subtracting the amount of 3H-L-Glu accumulation at 4°C.
Kinetic analyses
Michaelis–Menten and Lineweaver–Burk (LWB) plots and associated kinetic parameters (Km and Vmax) for transport inhibitors were estimated using a non-linear curve fitting analysis (Kaleida-Graph 4.1.3). Ki determinations from LWB replots were calculated using linear-regression analysis (KaleidaGraph 4.1.3).
Molecular modeling
A Sxc− comparative model was constructed using a published sequence alignment [37] to map the sequence of xCT (the transporter subunit of Sxc−) onto a crystal structure of the homologous bacterial arginine transporter AdiC (Protein Data Bank accession # 3OB6). The 3OB6 structure was chosen as the modeling template because it was crystallized in an open-outward conformation with electron densities for oxygen indicating the presence of arginine in the substrate binding site. Small molecule computational docking of substrate and inhibitors into the xCT model was done at physiological pH with flexible side chains using AutoDock Vina included with YASARA molecular modeling software (v. 19.5.23, yasara.org). Docked poses for glutamate and competitive inhibitors were screened for similarities between favored poses, with the positioning of the α-carboxylate oxygens of the substrate L-arginine (L-Arg) captured in the 3OB6 crystal structure used as a reference point. Ghasemitarei et al. [38] also reported an orientation of L-Cys2 in a Sxc− comparative model that is consistent with the pose of glutamate reported below (Figure 2A). Minimizations of selected docking poses were done using the YAMBER3 force field included with YASARA. Discovery Studio 2018 visualizer software (3dsbiovia.com) was used to determine contacts and hydrogen bonding between docked poses and xCT, and PyMOL (v. 2.1, pymol.org) was used for visualization and figure rendering.
Figure 2. Molecular modeling of Sxc- transporter site on subunit xCT.
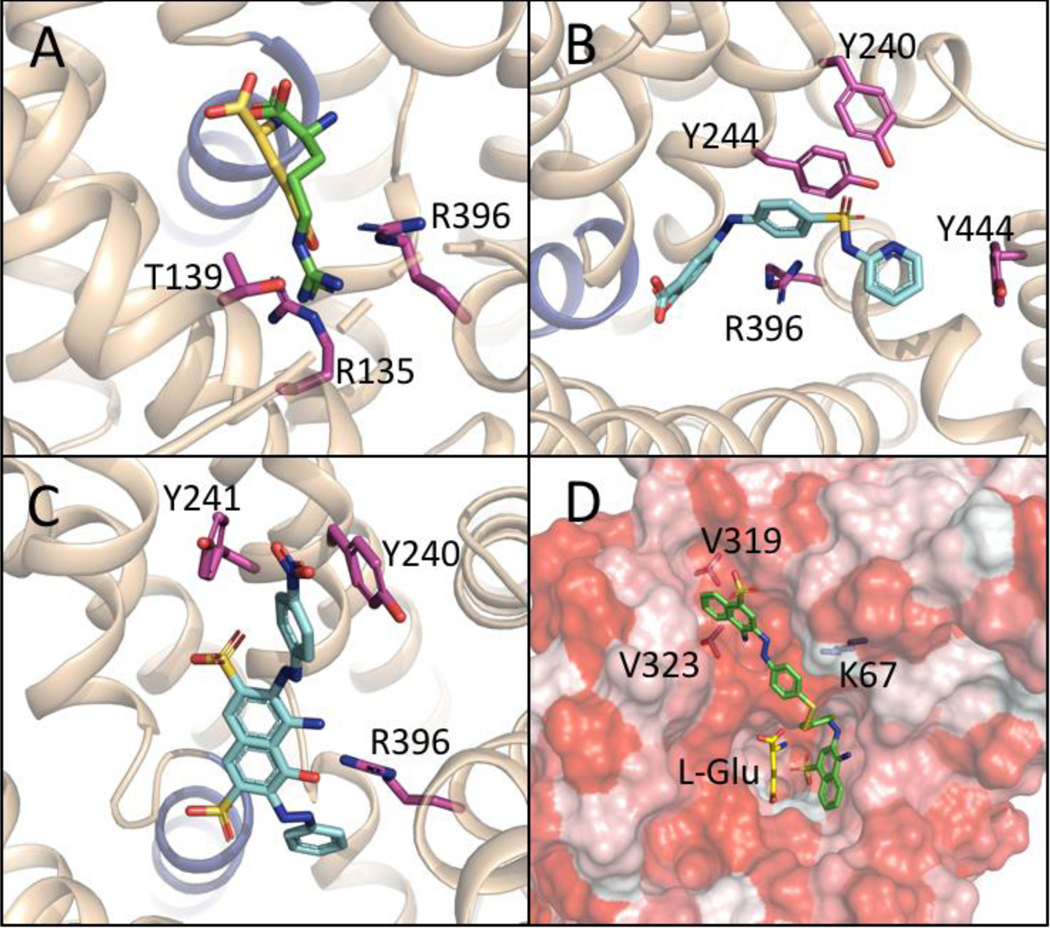
Figure. 2 Docking studies of L-Glu, SAS, Acid Black, and AANS #7 at a comparative model of xCT created from an outward facing, substrate-bound Arg/Agmantine antiporter (AdiC, PDB accession code 3OB6). A: L-Glu (yellow) overlaid with the position of bound Arginine (green) from the crystal structure 3OB6; displaying interactions with R135, R396, T139 (purple) and TM1a (blue ribbon). B: Competitive inhibitor SAS (blue) interacting with the backbone of TM1a and R396, as well as Y240, Y244, and Y444 (purple). C: Competitive inhibitor Acid Black (blue) similarly interacting with the backbone (blue ribbon) of TM1a and R396 as well as Y240 and Y241 (purple). D: Noncompetitive inhibitor AANS #7 (green) binding in a hypothetical allosteric site (hydrophobic surfaces indicated in red), and interacting with residues V319, V323, and K67, while leaving space for the substrate L-Glu (yellow) to bind.
Results and Discussion
The similarity in structure between SAS and commercially available dyes containing analogous azo-linked salicylate groups (e.g., Mordant Orange, Alizarin Yellow GG, Chrome Yellow) prompted the assay of these and a series of synthetically prepared mono- and bis-azo-linked amino-naphthly-sulfonates (AANSs) for the ability to block the Sxc−-mediated uptake of 3H-L-Glu into SNB19 cells (Tables 1 and 2). The structures and concentration-dependence with which the compounds exhibiting no, little or moderate inhibitory activity, compared to SAS, were determined against a single concentration of L-Glu (100 μM) and are reported as IC50 values (mean ± SEM) in Table 1. Of the commercially available azo-containing dyes that were assayed as Sxc− inhibitors, only Acid Black proved more potent than SAS. The salicylic acid-containing dyes Mordant Orange, Alizarin Yellow GG, and Chrome Yellow exhibited a moderate level of inhibition with IC50 values between 25 and 40 μM, yet still about 5-fold less effective than SAS. Given the high degree of structural similarity, this would suggest that the azo-salicylic acid scaffold is a key binding component, but that the nitrophenyl group present in Mordant Orange and Alizarin , as well as the sulfonaphthylene in Chrome Yellow, do not form as favorable binding interactions with Sxc− as the pyridyl-sulfonamide of SAS. The other commercially available azo-containing dyes incorporate a naphthylene sulfonic acid scaffold variously substituted with amino and hydroxy groups. Among these, Acid Black proved to be about 2–3 fold more potent than SAS, exhibiting a Ki of about 3 μM (Table 2) and confirmed the usefulness of the azo-linked amino-naphthylene sulfonic acid (AANS) structure as a core template. More detailed kinetic characterization revealed a competitive mechanism, indicating that the binding of Acid Black and L-Glu to xCT (the transporter subunit of Sxc−) are mutually exclusive and likely overlap with respect to their interactions within the xCT domains that participate in substrate binding. Comparisons between Acid Black and the less active sulfonate dye analogues suggest a number of important SAR features: i) the addition of at least one appended aryl substituent is critical, as 4-amino-5-hydroxy-2,7-naphthylene-disulfonate lacks this element and is inactive, ii) derivatization of the amino group in the 4 position (e.g., an N-acetamide on Acid Red) results in a marked loss of activity, but will tolerate replacement with a hydroxy group, albeit with a decrease in potency (e.g., compare Acid Red with Chromotrope 2R), iii) inhibitory activity appears to be increased when the azo-linked analine group is unsubstituted (e.g., compare Gallion and Chromotrope 2B with Chromotrope 2R), and iv) having two aryl groups appended to the aromatic core, as found in Acid Black and SAS markedly increases inhibitory activity (e.g., Chromotrope 2R is less active).
Table 1:
IC50 values for the inhibition of Sxc−-mediated uptake of 3H-L-Glu uptake into SNB19 cell by inactive, low and moderate inhibitors.
| Compound | Structure | IC50 μM Mean ± SEM (n ≥ 3) |
|---|---|---|
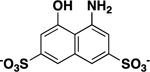
| 4-Amino-5-OH-2,7-naphthylene-disulfonate |
|
> 500 |
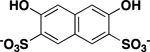
| 3,6-dihydroxy-2,7-naphthylene-disulfonate |
|
> 500 |
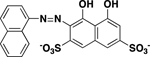
| Pontacyl Violet 6R |
|
>250 |
| Pontacyl Carmine 2B |

|
>250 |
| Acid Red 1 |
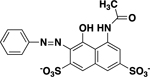
|
191 ± 8 |
| Gallion |

|
155 ± 11 |
| Chromotrope 2B |
|
130 ± 23 |
| AANS #1 (RB606) |

|
53 ± 12 |
| Mordant Orange 1 |
|
38 ± 3 |
| Alizarin Yellow GG |
|
30 ± 7 |
| Chrome Yellow |
|
27 ± 4 |
| Chromotrope 2R |
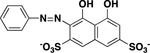
|
21 ± 5 |
| AANS #2 (RB 624) |
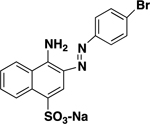
|
25 ± 4 |
| AANS #3 (RB 629 |

|
14 ± 2 |
| AANS #4 (RB 617) |

|
14 ± 2 |
Table 2:
Ki values and mechanisms for the inhibition of Sxc−-mediated uptake of 3H-L-Glu uptake into SNB19 cell by SAS and select AANSs.
| Compound | Structure | Ki (μM) Mean ± SEM (n) | Inhibitory Mechanism |
|---|---|---|---|
| Sulfasalazine |
|
10.4 ± 1.3 (7) | Competitive |
| Acid Black 1 |
|
3.4 ± 0.9 (5) | Competitive |
| AANS #5 (RB594) |
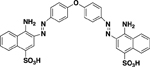
|
11 ± 1.3 (5) | Noncompetitive (Mixed) |
| AANS #6 (RB 626) |
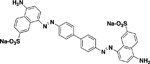
|
4.3 ± 0.8 (5) | Noncompetitive (Mixed) |
| AANS #7 (RB 605) |

|
4.3 ± 1.5 (3) | Noncompetitive |
Although the available azo-dyes possess sufficient structural diversity, more customized inhibitors were needed to probe specific interactions and provide deeper insights into the binding characteristics and pharmacology of the transporter. One chemical feature that distinguishes the commercially available dye structures used in this study is the presence of one (e.g., Chromotrope 2B & 2R) or two (e.g., Acid Black) azo-linked aryl substituents on the amino-naphthylene sulfonic acid (AANS) core. A second important element is the position of the AANS group(s) and whether or not the amino-napththyl sulfonate serves to mimic the functional groups of L-Glu that participate in substrate binding. To address this, the first set of custom inhibitors included a lipophilic group at the 2-position of an amino naphthylene sulfonic acid via an azo linkage. In the second set, two amino naphthylene sulfonic acid groups were connected via central azo lipophilic linkers to produce “dual” head groups (potentially) representing glutamate isosteres. Although previous work showed that Congo Red was a relatively poor blocker of glutamate uptake by Sxc− [39], it was chosen to develop analogs because the addition of one or two AANS moieties could be conducted regioselectively to various lipophilic linkers [36]. Congo Red-based analogs also presented certain pharmacologic advantages as an inhibitor set: i) there is one sulfonic acid per naphthylene ring which may provide some insight to the importance of the placement of this functional group in the molecule, and ii) the regiochemistry of the azo coupling and reduced number of synthetic operations improves accessibility to analogs and future development.
Consistent with the SARs of Acid Black and SAS, the mono-AANSs exhibited less inhibitory activity than the bis-AANS analogues as exemplified by AANSs #1–4 (see Table 2). That being said, AANSs #2–4, were effective inhibitors, exhibiting IC50 values of 15–25μM, which are comparable to or lower than the potencies reported for several well-known Sxc− inhibitors assayed under similar conditions, including: L-Cys2, L-serine-O-sulfate, ibotenate, L-homocysteate, and aryl-substituted isoxazoles (4-bis-TFM-HMICA and5-benzyly-4-bis-TFM-HMICA) [17,19]. Examination of AANSs #1–4 (and Acid Black) suggest that greater diversity in R-group substitutions is allowable with respect to the azo-linked aryl groups at position 3 on the naphthylene core compared to substitutions at the 6 position, which is a simple analine or pyridine group in Acid Black and SAS, respectively. Interestingly, the amino-naphthyl-sulfonate moieties in the symmetrical bis-azo analogues (AANSs #5–7, Table2), which were among the most potent inhibitors characterized, have dual AANS substituents positioned at the distal ends of the molecule rather than as a central core, as is the case in Acid Black. Also, AANS #6 stands apart from the other AANSs and Acid Black with respect to linkage point to the naphthylene ring (4 vs 3), as well as the relative positioning of the amino and sulfonate groups.
The most potent inhibitors (Table 2) were kinetically characterized in greater detail using a standard Michaelis-Menten analysis in which the concentrations of both the inhibitor and substrate (L-Glu) were systematically varied rather than the single concentration of L-Glu (100 μM) employed in the IC50 determinations. Both SAS and Acid Black yielded V vs [S] and Lineweaver-Burk (LWB) plots consistent with competitive inhibition (i.e., increase in Km with little or no change in Vmax and a pattern on the LWB plot of intersecting lines on the X axis). Representative plots are shown in Figures 1A-D. A replot (inset) of the slopes from the LWB vs [I] was linear and yielded Ki values of about 10 μM and 3 μM (means from n ≥ 3 assays, Table 2), respectively. The competitive mechanism dictates that the binding of these inhibitors to Sxc− are mutually exclusive with respect to the binding of the substrate L-Glu. This would be consistent with overlapping binding sites and raises the possibility that functional groups on the inhibitors may be interacting with some of the same xCT resides that participate in substrate binding. It must be noted, however, that to be competitive, the binding of the inhibitor and substrate need only be mutually exclusive. In contrast to the competitive mechanism, AANS #7 yielded kinetic plots indicative of noncompetitive binding (i.e., decrease in Vmax with little or no change in Km and a pattern on the LWB plot of intersecting lines on the Y axis). Representative plots are shown in Figures 1E-F. A replot (inset) of the slopes from the LWB vs [I] was linear and yielded Ki values of about 4 μM (mean from n ≥ 3 assays, Table 2). The noncompetitive mechanism indicates that the inhibitor can bind to the antiporter, whether or not the substrate (L-Glu) is bound, to produce a “dead-end” complex that cannot translocate substrate. AANS #5 and #6 also proved to be potent noncompetitive inhibitors, but yielded LWB plots consistent with mixed inhibition (intersecting lines above the X axis in the second quadrant, data not shown). Plots indicative of mixed inhibition can arise from multiple mechanisms, the two most common of which are observed when: i) an inhibitor binds in two different configurations, one competitive and one noncompetitive or ii) an inhibitor binds to a noncompetitive (i.e., allosteric) site where, unlike a “pure” noncompetitive inhibitor (e.g., AANS #7), it also influences the binding properties of the substrate [40]. In the instance of the former, it can be very difficult to kinetically resolve the action of an inhibitor at two distinct sites if the respective Ki values are close to one another. Similar results were previously observed with a series of aryl-substituted isoxazoles, where the addition of a single aryl group to an isoxazole core at either the 4 or 5 position (e.g., 4-bis-TFM-HMICA) yielded competitive inhibitors, while di-aryl-isoxazole substituted at both positions produced (e.g., 5-benzyl-4-bis-TFM-HMICA, 5-naphthyl-4-bis-TFM-HMICA, 5–4-TFM-benzyl-4-bis-TFM-HMICA) produced noncompetitive inhibitors [17]. Collectively, the switch from competitive to noncompetitive binding through structural modifications of shared core structures raises the likelihood that specific fragments of the molecules, whether present in a competitive or noncompetitive inhibitors, may be interacting with similar regions of the transporter. In turn, this suggests that the substrate binding site, which could be blocked by competitive inhibitors, may be in close proximity to the site at which the noncompetitive inhibitors are binding. Such a possibility is consistent with the identification of inhibitors that act at a “vestibule” or allosteric regulatory site adjacent to the substrate binding domains on LeuT and the serotonin transporter [41,42]. When these sites are occupied (with or without the substrate bound) the transporter is prevented from accessing the conformational states necessary for substrate translocation via the alternate access mechanism. Interestingly, the overall structural character of bis-AANS (#5–7) and the di-aryl-substituted isoxazoles is similar to that of two other potent inhibitors of Sxc− that do not possess typical isosteric elements of either L-Glu or L-Cys2, erastin and sorafenib [20]. While the mechanism of inhibition for these compounds has yet to be reported, the structural similarities make it tempting to speculate that they may also act as noncompetitive inhibitors and may be binding to Sxc− in manner analogous to the bis-AANSs.
Figure 1. Kinetic analyses of competitive and noncompetitive inhibitors of Sxc-Figure 1.

Representative kinetic analyses for the inhibition of Sxc−-mediated (Na+-independent, Cl−-dependent) uptake of 3H-L-Glu into SNB19 cells by SAS (A-B), Acid Black (C-D) and AANS #7 (E-F). Replots of the slopes generated from the LWB plots (inset) were used to determine Ki values. Variation in uptake rates were attributable to differences in Sxc− expression levels. Data from n ≥ 3 such analyses were averaged to yield values reported in Table 2. V vs [S] and LWB plots for SAS and Acid Black are consistent with competitive inhibition, while those for AANS #7 are consistent with noncompetitive inhibition.
In order to visualize possible structural implications for the binding of substrates, competitive inhibitors, and noncompetitive inhibitors, comparative models of the xCT subunit of the Sxc− transporter were constructed using crystal structures of homologous bacterial transporters/antiporters. Because the competitive and noncompetitive inhibitors were applied extracellularly and kinetically assessed for the ability to reduce the Sxc−-mediated uptake of L-Glu into the SNB19 cell, an outward-facing, substrate-bound Arginine/Agmatine antiporter AdiC (PDB accession code 3OB6) was selected as the most relevant modeling template [34]. Advantageously, the 30B6 crystal structure includes a bound L-arginine (L-Arg) which defines the substrate binding cavity and served as a key reference point for evaluating the multiple, favored poses resulting from docking L-Glu into the substrate cavity with AutoDock Vina in the YASARA molecular modeling software. Figure 2A depicts a resulting favored pose of L-Glu (yellow) docked into the xCT substrate binding site overlaid with the L-Arg (green) that was crystalized within AdiC substrate site. Consistent with the 3OB6 structure, the α-amine and α-carboxylate groups of L-Glu are positioned similarly to those of L-Arg, which are oriented toward the dipole resulting from a break in helix 1A (shown in blue in Figures 2A-C) [34]. This provided a sequence-independent reference point in the xCT homology model that was taken into consideration when selecting among favored poses of docked L-Glu. The resulting position of L-Glu also orients its distal carboxylate towards two conspicuous arginine residues in TM3 and TM10 (R135 and R396, highlighted in purple in Figure 2A and not to be confused with the L-Arg that is a substrate of AdiC). These arginine residues, which have been previously implicated as key components in the xCT substrate binding site [18,37], likely act together to coordinate the distal carboxylate of L-Glu. Other predicted points of interaction between L-Glu and xCT include a stabilizing hydrogen bond between the distal carboxylate and T139, which is near the interacting R135 and R396 residues, as well as multiple hydrogen bonds with T56, I57, A60, and G61 residues along the backbone of helix TM1 that may stabilize the α-amino group. Lastly, the positioning of L-Glu is also consistent with the proposed location of L-Cys2 in the substrate site of xCT in a comparable modeling study [38].
A representative favored pose of SAS (blue) docked with xCT is depicted in Figure 2B. The carboxylate moiety of the SAS is coordinated by the TM1a dipole, similar to both the L-Glu pose in Figure 1A and to the bound L-Arg included in the 3OB6 crystal structure. This differs somewhat from the position of SAS proposed by Patel and colleagues in their xCT modeling studies (also based on a 30B6 homology model) which depict the carboxylate moiety of SAS intermittently interacting with R135 and R396 [18]. Their conclusion may not have taken the potential helix dipole interactions into account. Further, inspection of 2D ligand maps (not shown) suggest the aromatic rings of SAS in close contact with R396, Y240, Y244, and Y444, forming pi-pi and pi-cation interactions. Importantly, the region occupied by the bound SAS overlaps with the region occupied by bound L-Glu, a relationship that is consistent with the mutually exclusivity of substrate and inhibitor binding in competitive mechanisms. Similarly, the docked pose of competitive inhibitor Acid Black (blue), depicted in Figure 2C, also overlaps with the substrate site, and shows similar pi-mediated interactions with R396, Y240 and Y241, while the negatively charged sulfoxide group is coordinated by the TM1a dipole.
As discussed above, we hypothesize that the xCT domains with which the newly described noncompetitive inhibitors are interacting are in close proximity to the substrate binding site, owing to the SAR switch from competitive to noncompetitive inhibition observed as a consequence of sequentially appending aryl groups to similar core templates in both aryl-substituted isoxazoles [17] and the newly described bis-AANSs (#5–7). By definition, however, these noncompetitive inhibitors would have to bind to xCT in a manner that would still allow L-Glu to bind. One of the favored poses obtained when docking the “pure” noncompetitive inhibitor AANS #7 (Figure 2D, green), placed the inhibitor into a hydrophobic groove formed by TMs 3 and 8 (Figure 2D). Other key residues of this hypothetical allosteric site include V319 and V323 in TM8, and K67 in the extracellular loop spanning TMs 1 and 2. Importantly, the region occupied by AANS #7 when bound in this manner also appears to accommodate L-Glu (yellow). Consistent with its action as a noncompetitive inhibitor and the ability to form a “dead end” complex, AANS #7 is positioned such that it could impede the movement of TM8, which has been implicated as critical for the transport cycle [34]. One consequence of identifying a distinct site at which the noncompetitive inhibitors are acting is that it will very likely exhibit a SAR profile that is different from the substrate binding site, which carries with it significant implications for drug design. For example, the SAS analogues characterized here are of limited therapeutic value for inhibiting Sxc− in glioblastoma cells, owing to the presence of functional groups that are necessary to interact with the substrate binding domains, but generally render the compounds unable to cross the blood brain barrier [13]. However, these functional groups may be of less importance in binding to the noncompetitive site and leave open the possibility of developing Sxc− inhibitors with improved pharmacokinetic/pharmacodynamic properties. Future studies employing molecular dynamics and homology models of other transporter states in the alternate access mechanism (e.g., occluded, substrate bound) should shed greater insight into the mechanism by which the competitive and noncompetitive inhibitors block substrate translocation. The current findings help further define the pharmacology of Sxc− inhibitors and identify putative binding domains on the xCT protein, both of which should prove of value in developing more effective drugs to regulate Sxc− activity in vivo.
Acknowledgment
This work was supported in part by NIH NINDS Grant R15 NS088899 (RB) and NIGMS Grant P20GM103546 (RB, DH, M.C). Modeling studies were carried out in the Molecular Modeling Core Facility supported by the UM’s NIH COBRE Center for Biomolecular Structure and Dynamics. DS, HL, and AD were supported by undergraduate research fellowships from UM’s Center for Structural & Functional Neuroscience. The author’s also acknowledge many years of insightful discussion of glutamate transporters with M.B. Robinson.
Abbreviations
- AANS
Amino-naphthylsulfonate
- L-Arg
L-Arginine
- 5-Benzyl-4-bis-TFM-HMICA
(Z)-5-benzyl-4-(1-(2-(3,5-bis(trifluoromethyl)phenyl)hydrazono)ethyl)isoxazole-3-carboxylic acid
- L-Cys2
L-Cystine
- L-CysH
L-Cysteine
- EAAT
Excitatory amino acid transporter
- L-Glu
L-Glutamate
- GSH
Glutathione
- 5-Naphthyl-4-bis-TFM-HMICA
(Z)-4-(1-(2-(3,5-bis(trifluoromethyl)phenyl)hydrazono)ethyl)-5-(naphthalen-2-ylmethyl)isoxazole-3-carboxylic acid
- SAR
Structure-activity relationship
- SAS
Sulfasalazine
- 4-bis-TFM-HMICA
(Z)-4-(1-(2-(3,5-bis(trifluoromethyl)phenyl)hydrazono)ethyl)-5-methylisoxazole-3-carboxylic acid
- 5–4-TFM-Benzyl-4-bis-TFM-HMICA
Z)-4-(1-(2-(3,5-bis(trifluoromethyl)phenyl)hydrazono)ethyl)-5-(4-(trifluoromethyl)benzyl)isoxazole-3-carboxylic acid
References
- 1.Bridges RJ, Natale NR, and Patel S, (2012) System xc- cystine/glutamate antiporter, an update on molecular pharmacology and roles within the CNS. Br J Pharmacology, 165(1): p. 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridges R, Lutgen V, Lobner D, and Baker DA, (2012) Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev, 64(3): p. 780–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dringen R, Gutterer JM, and Hirrlinger J, (2000) Glutathione metabolism in brain: metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem, 267: p. 4912–4916. [DOI] [PubMed] [Google Scholar]
- 4.Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Cassady K, and Aboody KS, (2016) Increased Expression of System xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol Cancer Res, 14(12): p. 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, Lam S, Gout PW, and Wang YZ, (2009) The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol, 64(3): p. 463–72. [DOI] [PubMed] [Google Scholar]
- 6.Cobler L, Zhang H, Suri P, Park C, and Timmerman LA, (2018) xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget, 9(64): p. 32280–32297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DA, Xi ZX, Hui S, Swanson CJ, and Kalivas PW, (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. The journal of Neuroscience, 22(20): p. 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augustin H, Grosjean Y, Chen K, Sheng Q, and Featherstone D, (2007) Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J. Neurosci, 27: p. 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massie A, Boillee S, Hewett S, Knackstedt L, and Lewerenz J, (2015) Main path and byways: non-vesicular glutamate release by system xc(−) as an important modifier of glutamatergic neurotransmission. J Neurochem, 135(6): p. 1062–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheldon AL and Robinson MB, (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int, 51(6–7): p. 333–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, and Sontheimer H, (2007) Autocrine glutamate signaling promotes glima cell invasion. Cancer Res, 67: p. 9463–9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, and Sontheimer H, (2005) Inhibition of cystine uptake disrupts the growth of primary brain tumors. The Journal of Neuroscience, 25(31): p. 7101–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sontheimer H and Bridges RJ, (2012) Sulfasalazine for brain cancer fits. Expert Opin Investig Drugs, 21(5): p. 575–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahlmanns M, Yakubov E, Chen D, Sehm T, Rauh M, Savaskan N, and Wrosch JK, (2017) Chemotherapeutic xCT inhibitors sorafenib and erastin unraveled with the synaptic optogenetic function analysis tool. Cell Death Discov, 3: p. 17030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert SM, et al. , (2015) SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med, 7(289): p. 289ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, and Kanai Y, (2003) CATs and HATs : the SLC7 family of amino acid transporters. Eur J Physiol, 447(5): p. 532–542. [DOI] [PubMed] [Google Scholar]
- 17.Newell JL, Keyari CM, McDaniel SW, Diaz PJ, Natale NR, Patel SA, and Bridges RJ, (2014) Novel di-aryl-substituted isoxazoles act as noncompetitive inhibitors of the system Xc(−) cystine/glutamate exchanger. Neurochem Int, 73: p. 132–8. [DOI] [PubMed] [Google Scholar]
- 18.Patel DN, Kharkar PS, Gandhi NS, Kaur E, Dutt S, and Nandave M, (2019) Novel analogs of sulfasalazine as system xc - antiporter inhibitors: Insights from the molecular modeling studies. Drug Dev. Res, epub. [DOI] [PubMed] [Google Scholar]
- 19.Patel SA, Warren BA, Rhoderick JF, and Bridges RJ, (2004) Differentiation of substrate and non-substrate inhibitors of transport system x(c)(−): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacol., 46: p. 273–284. [DOI] [PubMed] [Google Scholar]
- 20.Dixon SJ, et al. , (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife, 3: p. e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi S, et al. , (2015) Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. J Biol Chem, 290(14): p. 8778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H, et al. , (2014) A simple route to [11C]N-Me labeling of aminosuberic acid for proof of feasibility imaging of the x(C)(−) transporter. Bioorg Med Chem Lett, 24(23): p. 5512–5. [DOI] [PubMed] [Google Scholar]
- 23.Warren BA, Patel SA, Nunn PB, and Bridges RJ, (2004) The Lathyrus excitotoxin B-N-oxalyl-L-a, B-diaminopropionic acid is a substrate of the L-cystine/L-glutamate exchanger system xc-. Toxicology and Applied Pharmacology, 200: p. 83–92. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Dai Z, Barbacioru C, and Sadée W, (2005) Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res, 65: p. 7446–7454. [DOI] [PubMed] [Google Scholar]
- 25.Albano R and Lobner D, (2018) Transport of BMAA into Neurons and Astrocytes by System xc. Neurotox Res, 33(1): p. 1–5. [DOI] [PubMed] [Google Scholar]
- 26.Ye ZC and Sontheimer H, (1999) Glioma cells release excitotoxic concentrations of glutamate. Cancer Res, 59: p. 4383–4391. [PubMed] [Google Scholar]
- 27.Dixon SJ, et al. , (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 149(5): p. 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shukla K, Thomas AG, Ferraris DV, Hin N, Sattler R, Alt J, Rojas C, Slusher BS, and Tsukamoto T, (2011) Inhibition of xc(−) transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett, 21(20): p. 6184–7. [DOI] [PubMed] [Google Scholar]
- 29.Matti AA, Mirzaei J, Rudolph J, Smith SA, Newell JL, Patel SA, Braden MR, Bridges RJ, and Natale NR, (2013) Microwave accelerated synthesis of isoxazole hydrazide inhibitors of the system [Formula: see text] transporter: Initial homology model. Bioorg Med Chem Lett. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gout PW, Buckley AR, Simms CR, and Bruchovsky N, (2001) Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia, 15: p. 1633–1640. [DOI] [PubMed] [Google Scholar]
- 31.Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, and Sontheimer H, (2011) Glutamate release by primary brain tumors induces epileptic activity. Nat Med, 17(10): p. 1269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, and Yamashita T, (2018) Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS One, 13(4): p. e0195151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sleire L, et al. , (2015) Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene, 34(49): p. 5951–9. [DOI] [PubMed] [Google Scholar]
- 34.Kowalczyk L, et al. , (2011) Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc Natl Acad Sci U S A, 108(10): p. 3935–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartwell JL and Fieser LF, (1936) Coupling of O-tolidine and Chicago Acid. Org. Synth, 16: p. 12–17. [Google Scholar]
- 36.Song DH, Yoo HY, and Kim JP, (2007) Synthesis of stilbene-based azo dyes and application for dichroic materials in poly(vinyl alcohol) polarizing films. Dyes Pigments, 75: p. 727–731. [Google Scholar]
- 37.Shaffer PL, Goehring A, Shankaranarayanan A, and Gouaux E, (2009) Structure and mechanism of a Na+-independent amino acid transporter. Science, 325(5943): p. 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghasemitarei M, Yusupov M, Razzokov J, Shokri B, and Bogaerts A, (2019) Transport of cystine across xC(−) antiporter. Arch Biochem Biophys, 664: p. 117–126. [DOI] [PubMed] [Google Scholar]
- 39.Ahmed SK, Etoga JL, Patel S, Bridges RJ, and Thompson CM, (2011) Use of the hydantoin isostere to produce inhibitors showing selectivity toward the vesicular glutamate transporter versus the obligate exchange transporter System Xc-. Bioorg Med Chem Lett, 21(14): p. 4358–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segel I, Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. 1993: Wiley. [Google Scholar]
- 41.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, and Wang DN, (2009) Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol, 16(6): p. 652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh SK, Piscitelli CL, Yamashita A, and Gouaux E, (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science, 322(5908): p. 1655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]