

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection can lead to fatal outcomes for subgroups of patients with pre-existing co-morbidities. We previously reported a significant association between high expression levels of a cancer stem cell protein, doublecortin-like kinase 1 (DCLK1), in the lungs and macrophages of SARS-CoV-2-infected patients and the severity of coronavirus disease 2019 (COVID-19). Herein, we demonstrate a pivotal role of DCLK1 in the viral replication cycle and the dysregulation of cell signaling that contributes to SARS-CoV-2 pathology. Through CRISPR/Cas9-mediated DCLK1 knockout and inhibition of its kinase using a small molecule kinase inhibitor of DCLK1 (DCLK1-IN-1), we effectively blocked the viral replication-transcription processes. Furthermore, DCLK1 inhibition reversed the virus-induced positive and/or negative modulation of the cellular interactome and signaling pathways. We observed a decrease in the phosphorylation of a serine/arginine-rich region in the nucleocapsid protein, which regulates viral replication and packaging, upon treatment with DCLK1-IN-1. In a murine model of COVID-19, intranasal inoculation of SARS-CoV-2 induced severe lung pathology accompanied by increased DCLK1 expression, high titers of viral genomic and subgenomic RNAs, and elevated levels of inflammatory cytokines (interleukin-6 and tumor necrosis factor alpha). Remarkably, treatment of infected mice with DCLK1-IN-1 reduced viral RNAs, downregulated inflammatory cytokines, restored normal cell signaling pathways, and improved lung pathology. In conclusion, our findings underscore the crucial role of DCLK1 in SARS-CoV-2 pathology and suggest it as a promising target for therapeutic intervention.
IMPORTANCE
Severe COVID-19 and post-acute sequelae often afflict patients with underlying co-morbidities. There is a pressing need for highly effective treatment, particularly in light of the emergence of SARS-CoV-2 variants. In a previous study, we demonstrated that DCLK1, a protein associated with cancer stem cells, is highly expressed in the lungs of COVID-19 patients and enhances viral production and hyperinflammatory responses. In this study, we report the pivotal role of DCLK1-regulated mechanisms in driving SARS-CoV-2 replication-transcription processes and pathogenic signaling. Notably, pharmacological inhibition of DCLK1 kinase during SARS-CoV-2 effectively impedes these processes and counteracts virus-induced alternations in global cell signaling. These findings hold significant potential for immediate application in treating COVID-19.
KEYWORDS: SARS-CoV-2, COVID-19, DCLK1, interactome, DCLK1-IN-1, replication-transcription, pathology, host-virus interaction, cell signaling
INTRODUCTION
The clinical outcomes of coronavirus disease 2019 (COVID-19) are largely dictated by the host’s responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (1 – 3). In severe cases, multiple organs can be affected, leading to long-lasting health complications even after recovery (4 – 6). Advanced age and pre-existing co-morbidities, such as diabetes, hypertension, obesity, and liver cirrhosis, significantly increase the risk of developing severe COVID-19 (7, 8). Additionally, high viral loads in plasma have been associated with elevated disease severity and mortality rates. The emergence of SAR-CoV-2 variants and limited efficacy of existing anti-viral drugs highlight the need for a deeper understanding of viral replication mechanisms and pathology to develop more effective therapeutics.
The impact of SARS-CoV-2 infection on cellular signaling networks in COVID-19 has been explored through transcriptome and proteome analyses (9). For instance, in Vero-E6 cells, SARS-CoV-2 infection triggers the activation p38 MAPK, facilitating the formation of viral particle-containing filopodial protrusions with the involvement of casein kinase II. Additionally, infection induces phosphorylation changes in mitotic kinases, leading to cell cycle arrest (10). In hospitalized patients with COVID-19, single-cell RNA sequencing of peripheral blood mononuclear cells and infected lung tissue has revealed highly variable host immune responses. These responses include disrupted interferon responses, dysregulation of myeloid cells, and profound immune exhaustion, characterized by skewed B- and T-cell repertoires (1, 3, 11, 12). Furthermore, patients with severe COVID-19 exhibit elevated secretion of inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6, along with increased levels of alarmins (e.g., S100A8/S100A9). Moreover, enhanced fibrotic responses and impaired regeneration of type 2 pneumocytes contribute to the pathogenesis of severe disease (2, 3, 9).
Our recent study reported that SARS-CoV-2 infection induces the expression of doublecortin-like kinase 1 (DCLK1), a regulator of microtubule dynamics, within the lung. DCLK1, in turn, amplifies viral production and the release of inflammatory cytokines (13). Typically, in a majority of cells, DCLK1 is only expressed in response to cellular injury or stress as a multifunctional kinase. The N-terminal domain (NTD) of DCLK1 contains conserved microtubule-binding doublecortin (DC) motifs (DC1 and DC2). The microtubule-binding functions of DCLK1 are partially redundant with doublecortin proteins that lack a kinase domain (14). The CTD of DCLK1 comprises a serine/threonine kinase domain and distal regulatory tail. DCLK1 undergoes autophosphorylation at the T688 residue, which inhibits kinase activity and autophosphorylation of DC domains (15). Unphosphorylated DC domains strongly bind to microtubules and play a role in their regulation and stability. Therefore, DCLK1 functions as a self-regulating molecule with its kinase activities being pivotal in various biological processes (15 – 17). In this study, we investigate the role of DCLK1 in regulating SAR-CoV-2 replication and infection-induced pathology. Additionally, we describe a selective small molecule kinase inhibitor of DCLK1 (DCLK1-IN-1) (18, 19) that effectively impedes viral replication and restores dysregulated signaling pathways. These findings further underscore the significance of DCLK1 as a potential therapeutic target for mitigating severe COVID-19.
RESULTS
DCLK1 is a key host factor required for SARS-CoV-2 replication
We previously reported that high-level expression of a cancer stem cell protein, DCLK1, in the lungs and macrophages of SARS-CoV-2-infected patients correlated with COVID-19 severity (13). Herein, we determined underlying mechanisms of these observations. To begin with, we targeted DCLK1 in lung Calu-3 cells using the CRISPR/Cas9 approach and determined its role in the SARS-COV-2 replication cycle. Calu-3 lung cells were transduced with lenti-CRISPR-v2 vectors expressing three custom-designed DCLK1-targeting sgRNAs (Fig. 1A, lanes 3 through 5) or negative control sgRNA (lane 2) and the transduced cells were selected in the presence of puromycin. The total lysates of resistant cells for sgRNA #2 (lane 4) showed 70% knockout of DCLK1 (termed Calu-3-DKO). We did not observe any deleterious effects on cell growth due to DCLK1 knockdown. SARS-CoV-2-infected Calu-3 cells (wild type) showed characteristic Spike-rich multinucleated syncytia (Fig. 1B). In contrast, Calu-3-DKO cells did not exhibit syncytia and showed little to no S protein staining. By Western blot, DCLK1 knockdown reduced the expression of S and N proteins by 70%–80% (Fig. 1C). Infected Calu-3-DKO cells also showed a 10-fold decrease in viral gRNA and a 3-fold decrease of N protein mRNA compared to control infected cells (Fig. 1D). These results provide direct evidence that DCLK1 regulates the SARS-CoV-2 replicative cycle.
Fig 1.
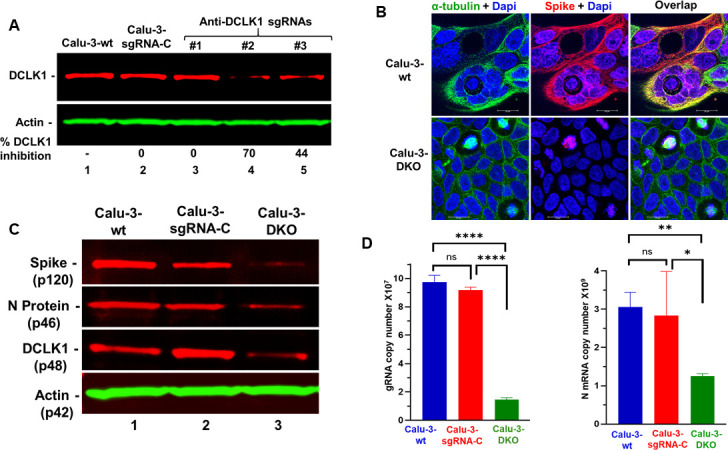
CRISPR/Cas9-mediated knockdown of DCLK1 blocks SARS-CoV-2 replication-transcription processes. (A) LentiCRISPR-v2 vectors expressing anti-DCLK1 synthetic guide RNAs (sgRNAs #1, #2, and #3; lanes 3–5) were used to target DCLK1, while control synthetic guide RNA was used as a control (sgRNA-C, lane 2). Additional controls included untreated cells (Calu-3-wt, lane 1). Puromycin-resistant cells were pooled to determine the relative expression of DCLK1. sgRNA #2-treated cells showed 70% inhibition of DCLK1 (Calu-3-DKO cells) and were selected for further study. (B) Calu-3 cells were infected with SARS-CoV-2 for 48 h and imaged by confocal microscope after immunofluorescence staining for microtubules (green), Spike protein (red), and nuclei (blue). (C) Western blots of total lysates of infected cell lines for Spike protein, N protein, and actin. (D) Reverse transcription-quantitative PCR for average copy number of viral gRNA (left) and N protein mRNA (right) in infected cell lines. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, ns (not significant) P > 0.05.
SARS-CoV-2-induced dysregulation of cellular proteome is normalized by DCLK1-IN-1
To further investigate potential mechanisms for DCLK1 regulation of SARS-CoV-2 replication and its global impacts on the interactome, we used DCLK1-IN-1 to inhibit DCLK1 kinase. This drug has been characterized for its specific binding to the DCLK1 kinase domain and compared to similar kinases (16, 18, 19). We found that in infected lung cells, DCLK1-IN-1 exhibited an IC50 of 2.3 µM for SARS-CoV-2 production and 2.8 µM for S protein expression (Fig. S1). Therefore, we selected 5.0 µM as a concentration of DCLK1-IN-1 to use in subsequent experiments (Fig. S2) (20). Total lysates of infected Calu-3 cells (A2) and infected cells treated with dimethyl sulfoxide (DMSO as vehicle control, A3) or DCLK1-IN-1 (A4) were used for label-free quantitative proteomic analysis. Uninfected cells (A1) were the baseline control. A total of 18,940 unique peptides corresponding to 3,257 proteins were identified and quantitated (a set of these proteins are shown in Fig. S3A and B). Principal component analysis of total protein abundance showed high reproducibility among biological replicates for each condition (Fig. 2A). However, significant differences were observed across these conditions. Similar differences were observed in heat maps that clustered protein groups based on abundances across samples (Fig. 2B and C). Similarly, several proteins were significantly increased or decreased in abundance in response to DCLK1-IN-1 (Fig. 2D). Comparative analysis of increased proteins among uninfected, infected, and DCLK1-IN-1-treated cells revealed a subset of eight common host proteins: G1 to S phase transition 2 (GSPT2, a polypeptide chain release factor also known as ERF3B), galectin-1 (LGALS1, immune tolerance), enolase 2 (promotes cell survival), H3 clustered histone 15 (involved in nucleosome structure), latexin (putative tumor suppressor), sterile alpha motif domain containing 4B (SAMD4B, putative transcriptional repressor), spectrin alpha, erythrocytic 1 (involved in cell shape, arrangement of transmembrane proteins, and organization of organelles), and WD repeat domain 75 (WDR75, involved in ribosome biogenesis and premRNA processing). This subset was part of a larger set of 77 upregulated proteins that were identified in infected cells and normalized following DCLK1-IN-1 treatment (Fig. 2E). We were able to validate normalization of protein abundance for four of these potential protein targets (Fig. 2F).
Fig 2.
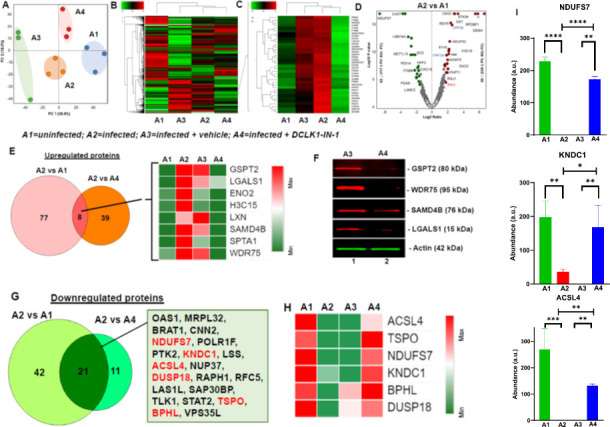
SARS-CoV-2-mediated alteration in proteomic profile of lung epithelial cells are restored following DCLK1 inhibition. Calu-3 cell lysates from uninfected (A1), SARS-CoV-2-infected (A2), infected cells treated with vehicle (DMSO) (A3), or DCLK1-IN-1 (A4) underwent proteomic analysis (experiments performed in triplicate). (A) Principal component analysis of total protein abundance for each sample shows close clustering of total normalized protein abundance (peak area) for A1 and A4 compared to A2 and A3. (B) Heat map clustering for differential protein abundance for each experimental condition. (C) Heat maps show multiple proteins induced by SARS-CoV-2 (A2 and A3, red) and normalization toward levels in uninfected cells (A1, green) following treatment with DCLK1-IN-1 (A4, green). (D) Volcano plots show significantly increased (red circles) and decreased (green circles) protein levels in infected (A2) compared to uninfected (A1) cells. (E) Eight proteins are identified that were induced by infection and normalized by DCLK1-IN-1. (F) Western blots validate proteomic data for a subset of proteins normalized by DCLK1-IN-1 as indicated. (G) Twenty-one genes were downregulated in SARS-CoV-2-infected cells, and expression was restored by DCLK1-IN-1 (see box; the six most downregulated genes are shown in red). (H) Heat map showing six proteins most downregulated by infection (A2) compared to uninfected control (A1). Downregulated protein expression is restored by DCLK1-IN-1 (A4) and not DMSO (A3). (I) Examples of protein levels in infected cells that were restored by DCLK1-IN-1. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns (not significant) P > 0.05.
Proteomic analysis also identified 42 downregulated host proteins in SARS-CoV-2-infected Calu-3 cells compared to uninfected cells (Fig. 2G). Among these, the expression of acyl-CoA synthetase (CSL4), translocator protein for mitochondria (SPO), NADH-ubiquinone oxidoreductase 20-kDa subunit, complex I-20 kD (NDUFS7), kinase non-catalytic C-lobe domain containing 1, biphenyl hydrolase like, and dual specificity protein phosphatase 18 were restored by DCLK1-IN-1 (Fig. 2H and I). As anticipated, SARS-CoV-2 infection significantly altered the expression of the host proteome. However, a number of proteins were restored by DCLK1 kinase inhibitor.
DCLK1 kinase inhibitor downregulates abundance and phosphorylation of viral proteins required for SARS-CoV-2 replication-transcription
Proteomic analysis also confirmed an inhibition of SARS-CoV-2 proteins by DCLK1-IN-1 treatment. Specifically, M and S proteins (Fig. 3A through C) and ORF7a, ORF8, and ORF9b regulatory proteins (Fig. 3D through F) were decreased by DCLK1-IN-1. Since ORF8 and ORF9b are known to inhibit type I/II interferon responses and classical nuclear factor kappa B (NF-κB) signaling (21, 22), the downregulation of these proteins by the DCLK1 kinase inhibitor may help relieve viral-mediated immune invasion and inflammatory responses. In addition, viral particle assembly would also be expected to be hampered by decreased M and S protein expression (23). We noted that a low-level reduction of Spike (Fig. 3A) and ORF8 (Fig. 3E) following DMSO treatment was observed. It could be the result of non-specific effects on intracellular transport, extracellular signaling, and/or cell permeability during infection. However, similar changes in protein abundance were not observed for other viral proteins.
Fig 3.
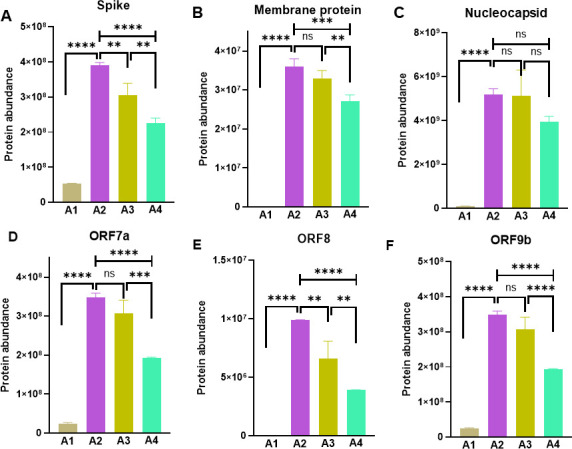
SARS-CoV-2 encoded structural and non-structural protein expression is blocked by DCLK1 inhibition. Viral structural (A–C) and regulatory proteins (D–F) of SARS-CoV-2 are significantly downregulated by DCLK1-IN-1 treatment of infected Calu3 cells when compared to untreated infected controls. Analyses were carried out using quantitative proteomics. A1, uninfected; A2, infected; A3, infected and treated with DMSO as vehicle; and A4, infected and treated with DCLK1-IN-1. **P ≤ 0.01, ***P ≤ 0.001, ***P ≤ 0.0001, ns (not significant) P > 0.05.
Since DCLK1 triggers significant changes in cell signaling during SARS-CoV-2 infection, we carried out an in-depth phosphoproteomic analysis of drug-treated infected cells to determine changes in the global phosphorylation landscape of viral and cellular proteins (Fig. S4A and B). In SARS-CoV-2-infected cells, ~120 phosphorylated sites were detected in viral proteins (N, M, Spike, and non-structural 3a proteins). Among 11 phosphorylated sites in N, eight amino acids were serines (176, 180, 183, 186, 187, 188, 201, and 202), and all were located in the serine/arginine (SR)-rich domain of the protein (Fig. 4A and B). Phosphorylation of this domain in N protein has been shown to modulate RNA packaging and replication/transcription processes (10, 24). Additional phosphorylation of serine residues was also detected in the NTD of N protein (Ser2, Ser23, and Ser79). Treatment of infected cells with DCLK1-IN-1 significantly decreased the phosphorylation of five serines in N protein (Ser2/Ser176/Ser180/Ser201/Ser202 (red stars in Fig. 4C). However, DCLK1-IN-1 had no effect on the phosphorylation of other serines in this protein (i.e., Ser183/Ser186/Ser187/Ser188. Interestingly, except for Ser2, the phosphorylation sites in N protein that were regulated by DCLK1-IN-1 were all located in the S-R-rich region. Amino acids in the S-R-rich domain are highly conserved in all current Omicron lineages (Fig. 4C); we tested whether DCLK1-IN-1 could also inhibit the replication of SARS-CoV-2 Omicron variants (B.1.1.529) using the 50% tissue culture infectious dose (TCID50) assay. Indeed, DCLK1-IN-1 significantly blocked viral production (Fig. 4D), similar to the Wuhan alpha lineage (Fig. S1) (13). Phosphorylation of M protein at Ser212, Ser213, and Ser214 during SARS-CoV-2 infection was also reduced by DCLK1-IN-1 (data not shown).
Fig 4.

DCLK1 regulates phosphorylation of nucleocapsid (N) protein. (A) Quantitative phosphoproteomic analysis of SARS-CoV-2-infected Calu-3 cells after treatment with DCLK1-IN-1 (blue bars, A4P) or DMSO (orange bars, A3P). These data are compared with the abundance of phosphopeptides in infected cells (red bars, A2P). Lysates from uninfected/untreated Calu-3 cells are also shown (negative controls, A1P). (B) Amino acid sequence in the SR-rich region of N protein. Phosphorylated serines are shown in red with stars indicating sites that had a reduction in phosphorylation after treatment with DCLK1-IN-1. Reduction in serine phosphorylation by DMSO is indicated by hashtags. (C) DCLK1-IN-1-mediated inhibition of phosphorylation is directed primarily toward conserved serines (red) in the highly conserved SR-rich region of N protein that is found in all omicron variants (BA.1 through BA.5) and the original Wuhan strain (top). (D) DCLK1-IN-1 inhibits Omicron strain viral production in infected Calu-3 cells; TCID50 for DMSO (vehicle) set at 100% and compared with DCLK1-IN-1. (E) Enhanced phosphorylation of eight host proteins due to SARS-CoV-2 infection is repressed by DCLK1-IN-1 (sample designation as in panel A). (F) Examples of host proteins (MAD1L1 and SYMPK) where phosphorylation is restored by DCLK1-IN-1 (blue bars) compared with uninfected Calu-3 cells (green bars). Reduction of protein phosphorylation due to infection (A2P) is not restored by DMSO (A3P). CTD, C-terminal domain; NTD, N-terminal domain.*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns (not significant) P > 0.05.
Next, we determined the effects of DCLK1 kinase inhibitor on the phosphorylation of host proteins. As expected, the phosphorylation landscape of Calu-3 cells was dramatically changed by SARS-CoV-2 infection (Fig. S4D and E). We found that infection increased the phosphorylation of eight host proteins (Fig. 4E). Increased phosphorylation was identified in five proteins: p-HMGCS1-Ser293, p-LRBA-Ser1261, p-USP42-Ser754, p-IFIT3/IRF3-Ser478, p-AP3D1-Ser1261, and p-DNMT1-Ser143 (25 – 28). Phosphorylation of these proteins was significantly reversed by DCLK1-IN-1. These findings show that infection alters multiple regulatory proteins and that these changes can be normalized by DCLK1-IN-1. In addition, DCLK1-IN-1 also partially restored the phosphorylation of p-MAD1L1-Ser16 and p-SyMPK-S520 proteins that were otherwise suppressed by SARS-CoV-2 infection (Fig. 4F). MAD1L1 is a component of the spindle-assembly checkpoint that blocks the onset of anaphase and is highly expressed in late S and G2/M phase. Symplekin is a scaffold protein that functions as a component of a multimolecular complex involved in histone mRNA 3′-end processing.
Signaling dysregulation and EGFR phosphorylation by SARS-CoV-2 are partially normalized by DCLK1-IN-1
We next determined whether disturbances in cellular signaling due to SARS-CoV-2 infection could be normalized by DCLK1-IN-1. Levels of signaling proteins and phosphorylation of kinases were compared for SARS-CoV-2-infected Calu-3 cells (A2) to uninfected cells (A1), infected controls treated with DMSO as a vehicle (A3), and infected cells treated with DCLK1-IN-1 (A4). Reactome and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses showed enrichment of an NF-κB activated pathway, interferon and cytokine signaling, and TRAF3-dependent IRF activation (Fig. S5A). DCLK1-IN-1 treatment led to an increased abundance of proteins involved in the activation of anti-viral responses, interferon signaling, cell cycle checkpoints, cytokine signaling, Rho GTPase signaling and formin enrichment, and epigenetic regulation of gene expression (Fig. S5B, A4 vs A2). The activation of GTPase-formins by DCLK1-IN-1 could help cells recover cytoskeleton organization, although this remains to be tested.
Kinases play key roles in SARS-CoV-2 replication and pathology (10). We found that SARS-CoV-2 infection of Calu-3 cells differentially regulated the expression of 21 kinases (Fig. 5A). Among these kinases, protein levels for MTOR, TK1, PIK3R1, PTK2, FASTKD3, EPS8L1, EPS8L2, and MAP2K1 were normalized by DCLK1-IN-1. Interestingly, distinct phosphorylation signatures of EGFR family members that were induced by infection were normalized by DCLK1-IN-1. Notably, a significant decrease was observed in the phosphorylation of p-EGFR (T693), p-FGFR3 (S440 and S441), p-ERBB2/p-HER2 (S108, S1073, and T701), and p-PRPF4B (Y849), while increased phosphorylation was observed for p-EEF2K(S445), p-TNIK(S640), p-BMP2K(S1029), and p-EGFR (S1071) (Fig. 5B). DCLK1-IN-1 restored the phosphorylation of these proteins to levels similar to that observed in uninfected cells (A2 and A3 compared with A4; Fig. 5B and C). These results suggest that inhibition of DCLK1 kinase during viral infection reduces aberrant activation or deactivation of cellular kinases and their associated signaling pathways. A protein-protein interaction network analysis showed significant changes in the abundance of 28 proteins due to infection (1.5‐fold, P < 0.05). The majority of these proteins reverted back to normal upon treatment with DCLK1-IN-1 (Fig. 5D). These analyses indicate that a large number of dysregulated proteins and signaling pathways are normalized by DCLK1-IN-1.
Fig 5.

SARS-CoV-2-induced alterations in EGFR family members’ kinase activity are restored following DCLK1 inhibition. Proteomic and phospho-proteomic analyses of total cell lysates from uninfected cells (A1), SARS-CoV-2-infected Calu-3 cells (A2), and infected cells treated with DMSO (A3) or DCLK1-IN-1 (A4) identify significant changes in the abundance of cellular kinases. (A) Heat map shows an altered abundance of 21 kinases in infected cells; these changes are partly restored by DCLK1-IN-1. Red and green stars are the known interactors of the M and N proteins, respectively. (B) Phosphorylation signatures for eight kinases are altered by SARS-CoV-2 infection (A2P). The degree of phosphorylation at these sites (A1P) is significantly restored by DCLK1-IN-1 (A4P). Red and green stars represent known interactors based on BiGRID database for SARS-CoV-2 M and N proteins. (C) KEGG pathway analysis of differentially abundant kinases (see Materials and Methods for details). (D) A protein-protein interaction network for 28 proteins shows significant changes in abundance in response to SARS-CoV-2 infection (1.5‐fold, P < 0.05). Non-interacting nodes are not shown. Line colors indicate the strength of data based on experimental results (pink), co‐expression (black), gene‐fusion (red), or co‐occurrence (blue) (http://string-db.org/).
DCLK1 kinase inhibitor blocks the viral replication cycle in a murine model of SARS-CoV-2 infection
To corroborate our cell culture findings, we assessed the effects of Dclk1 on SARS-CoV-2 replication cycle and pathology in a K18-hACE2 transgenic murine model that expresses human ACE2 (29 – 31). The intranasal inoculation of SARS-CoV-2 in these mice resulted in productive viral infection as confirmed by the expression of S and N proteins in the lung (Fig. 6A). Lung cell lysates from infected mice showed significantly increased expression of both long (84 kDa) and short (48 kDa) isoforms of Dclk1 compared to uninfected controls (Fig. 6B). In this model, we also confirmed infection-associated increases in LGALS1, TSPO, and WDR75 (Fig. 6A and B) as shown in the proteomic heat map data for SARS-CoV-2-infected Calu-3 cells (Fig. 2E). HLA-A expression was also increased, suggesting heightened presentation of viral antigens to immune cells in infected mice.
Fig 6.

Expression of viral proteins and RNAs are blocked by DCLK1-IN-1 in the K18-hACE2 model of SARS-CoV-2 infection. K18-hACE2 transgenic mice are a model for SARS-CoV-2 infection. (A) Western blots of total cell lysates from necropsied lungs of uninfected mice (U1–U6) or mice infected with SARS-CoV-2 (M1–M6); lysates were prepared 5 days post-infection, and each group consisted of three male and three female mice. (B and C) Quantitative evaluation of band intensities for host proteins shown in panel A; band intensities for Spike and N proteins were not quantified since these proteins were undetectable in uninfected mice. (D) Representative H&E stained lung for each treatment group (n = 6). Histopathological outcomes for DMSO-treated or DCLK1-IN-1-treated mice were compared with infected (group 1) and uninfected (group 4) mice; the experiment was replicated once with similar results (data not shown). (E) Total RNAs from mouse lung by qRT-PCR with copy numbers of viral genomic RNA (left panel) and N protein mRNA (right panel) calculated from C t values. As controls, group 4 mice were negative for both RNAs; for group 2 mice, RNA levels were similar to those of group 1 (data not shown). (F) Western blots for Spike and N proteins from total lung cell lysates for two representative mice in each group. (G) Quantified band intensities of Spike and N proteins (repeated twice, n = 6) were quantitated using Image J software. (H) Quantitative qRT-PCR for IL-6, TNF-α, and IL-1β mRNAs in DCLK1-IN-1-treated and control mice using total RNAs isolated from mouse lung (n = 3 per group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns (not significant) P > 0.05.
SARS-CoV-2 infection in K18-hACE2 mice showed histopathological changes in the lung that were consistent with COVID-19, e.g., perivascular inflammation, edema, thrombosis, necrosis, syncytial cells, and infiltration of immune cells including lymphocytes, macrophages, and neutrophils (Fig. 6D). To assess the effect of Dclk1 on SARS-CoV-2 infection, we administered DCLK1-IN-1 to infected mice. DCLK1-IN-1 has a favorable pharmacokinetic profile in mice (half-life 2.09 h) with maximum tolerated doses of up to 100 mg/kg with no significant adverse effects (18). The histopathological features noted in the lungs of infected mice were significantly improved by DCLK1-IN-1 (Fig. 6D). In addition, substantial reductions in viral genomic and N protein mRNAs (100-fold for genomic and 8-fold for N; Fig. 6E) were observed. Lungs of infected mice treated with DCLK1-IN-1 also showed significant reductions in S and N proteins compared to controls (Fig. 6F and G). IL-6, TNF-α, and IL-1β are signature inflammatory cytokines in COVID-19 (32). The lungs of infected mice showed increased mRNA levels for IL-6, TNF-α, and IL-1β. DCLK1-IN-1 treatment significantly decreased the relative abundance of mRNAs for IL-6 and TNF-α but not IL-1β (Fig. 6H). Increases in the serum levels of these cytokines due to infection were not observed, possibly due to the short duration of this model (i.e., only 5 days). Overall, these observations were most consistent with mild-to-moderate COVID-19-like illness. Systemic treatment with DCLK1-IN-1 showed marked efficacy against viral replication-transcription processes and pathological changes due to pneumonia.
Immunofluorescence and confocal microscopy of lungs from infected K18-hACE2 mice showed abundant expression of Spike protein in type II pneumocytes that were also expressing surfactant protein C (SP-C) (Fig. 7A). This is similar to previous reports in human cells (33). Intense Dclk1 staining was also observed in infected cells. In addition, we noted sporadic CD206+ M2-like macrophages that exhibited strong staining for Dclk1 and SP-C but were otherwise negative for Spike protein (Fig. 7B). Scanned lung slides from all four groups were quantitated for staining intensities. Marked increases in co-staining for Spike protein, Dclk1, CD206, and SP-C were noted in SARS-CoV-2-infected mice treated with DMSO (as a vehicle control) compared to infected mice treated with DCLK1-IN-1 (Fig. 7C). Stark reductions in Spike protein and Dclk1 expression for DCLK1-IN-1-treated mice suggest potential benefits in targeting Dclk1 (Fig. 7D).
Fig 7.
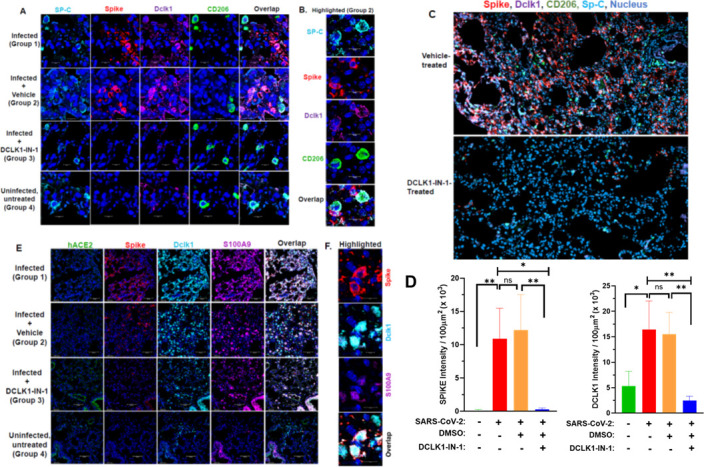
DCLK1-IN-1 shows efficacy in the treatment of SARS-CoV-2-infected K18-hACE2 mice. Lungs from K18-hACE2 transgenic mice were obtained at necropsy 5 days post-infection with SARS-CoV-2. (A) Confocal microscopy shows immunofluorescence staining for SP-C (cyan), Spike (red), Dclk1 (magenta), and CD206 (green) from infected mice and infected mice treated with DCLK1-IN-1 (blue, nucleus; ×20 magnification). (B) Spike-negative and CD206+ M2-like macrophages stain for SP-C and DCLK1 and are highlighted in close contact with infected cells (×40 magnification). (C) Quantitation of staining intensities for Spike and Dclk1 for all groups as indicated. (D) Lungs stained for Spike (red), CD206 (green), Dclk1 (magenta), SP-C (cyan), and nuclei (blue) were scanned by AxioScan (magnification ×10) and images shown for DMSO treated (control, upper panel) and DCLK1-IN-1 treated (lower panel). (E) Representative images of lungs from groups of mice stained for hACE2 (green), Spike (red), Dclk1 (cyan), S100A9 (magenta), and nuclei (blue). (F) Infected lung cells co-expressing Dclk1 and S100A9 are highlighted (magnification ×40). *P ≤ 0.05, **P ≤ 0.01, ns (not significant) P > 0.05.
We recently reported the co-expression of S100A9, a pro-inflammatory marker, and DCLK1 in infected lung cells and in alveolar CD206+ M2-like macrophages in patients with moderate-to-severe COVID-19 (13). Others have reported that the expression of the ACE2 receptor for SARS-CoV-2 declines substantially after infection. Both prior observations were reproduced in our infected K18-hACE2 mice (Fig. 7E and F for ACE2 and S100A9; Fig. 7A for M2-like macrophages). As expected, hACE2 expression was only observed in lungs from uninfected and DCLK1-IN-1-treated mice. Expression of this viral receptor was downregulated, however, in infected mice and infected mice treated only with the DMSO vehicle. DCLK1-IN-1 also led to a modest decrease in S100A9 expression. These results further confirm the K18-hACE2 murine model of SARS-CoV-2 infection as a reliable surrogate for acute viral pneumonia seen in COVID-19. DCLK1-IN-1 showed significant positive efficacy in this model with results that were consistent with in vitro findings from infected Calu-3 cells.
DISCUSSION
DCLK1 is an autoregulatory kinase that plays a crucial role in multiple biological processes including microtubule dynamics, intracellular trafficking, neuronal growth, cell signaling, cancer, and inflammation (16, 34 – 36). We recently discovered a novel function of DCLK1 in promoting SARS-CoV-2 production and exacerbation of COVID-19 (13). For in-depth examination of the DCLK1-driven mechanisms, we employed CRISPR/Cas9 to inactivate DCLK1 in Calu-3 lung cells and treated SARS-CoV-2-infected mice with a specific inhibitor of DCLK1 kinase (DCLK1-IN-1) followed by global protein analysis. Through these approaches, we demonstrated that DCLK1 enhances SARS-CoV-2 replication-transcription processes. Moreover, DCLK1-IN-1 effectively mitigated infection-induced dysregulation of the cellular proteome, phosphoproteome, and signaling pathways. Collectively, these findings suggest that DCLK1 drives SARS-CoV-2 propagation, and targeting DCLK1 inhibits viral replication cycle and restores interactome balance.
Several host kinases (e.g., p38, CK2, CDK, AXL, and SRPK1/2) have been implicated in SARS-CoV-2 replication and pathology (10, 37, 38). Inhibitors that target these kinases are currently being studied as potential anti-viral agents. However, unlike previously studied kinases that regulate crucial cellular functions and signaling pathways, DCLK1 stands out because it is predominantly induced in infected epithelial cells and polarized macrophages responding to infection. Under homeostatic conditions, pulmonary epithelial cells and cells in many other organs have minimal to undetectable levels of DCLK1 (13, 17, 39, 40). Apart from its kinase function, DCLK1 also possesses DC motifs that bind and polymerize tubulins and thereby regulate microtubule dynamics. Many viruses, including SAR-CoV-2, rely on microtubule networks for replication and intracellular transport of viral particles. Interfering with tubulin binding of DC motifs in DCLK1 may present an additional strategy to limit viral replication. Further investigation is needed to determine the extent to which these mechanisms operate independently or synergistically to limit SARS-CoV-2 replication.
During the early stages of SARS-CoV-2 infection, N protein localizes to viral replication-transcription complexes, facilitating RNA structural rearrangements for subgenomic transcription (24). Hyperphosphorylated N protein forms dynamic liquid-like condensates that colocalize with NSP3 and membrane vesicles, supporting viral subgenomic transcription (24, 41). Inhibition of phosphorylation of the SR-rich region hampers these processes (24). In this study, we found that DCLK1 facilitated the phosphorylation of at least 4 of 10 conserved serines in this region. Previous research suggests that other kinases, apart from Cdk1 and GSK3 (24), and SRPK1/2 (38), are likely involved in phosphorylating this domain. Changes in phosphorylation within the SR-rich region correspond to significant decreases in gRNAs and N protein mRNAs in the lungs of SARs-CoV-2-infected mice. Targeting phosphorylation in the SR-rich region offers a potentially crucial advantage as this region is highly conserved across all sublineages of SARS-CoV-2. Various lineages exhibit different responses to anti-viral drugs and monoclonal antibodies due to mutations in the sequences of targeted regions (e.g., S protein) (42). Developing drugs that specifically target phosphorylation within the invariant SR-rich region of N protein may lead to therapies that are less likely to lose effectiveness as new lineages emerge.
While DCLK1-IN-1 inhibits the DCLK1 kinase domain in both large and small isoforms of the protein, it is important to consider potential off-target effects of a small molecule inhibitor on other ATP-dependent kinases. To address this concern, we conducted an analysis of protein expression levels and phosphorylation status of cellular kinases in SARS-CoV-2-infected lung cells. We found that infection altered the expression of 109 kinases with notable upregulation of MTOR, PIK3R1, MAP2K1, DAPK3, and EPS8L1/2 and downregulation of PTK2, NME3, TLK1, and OXSR1 (Fig. 5). The expression level of these kinases was fully or partially normalized upon treatment with DCLK1-IN-1, indicating its specific blocking effect on kinases affected by SARS-CoV-2 infection. The restoration of kinases such as EGFR/ERBB2, in conjunction with viral clearance, could contribute to cell growth, survival, and recovery. For instance, resetting ERK-mediated phosphorylation of ERBB2/HER2 at T701 can lead to a compensatory activation of AKT and thereby improve cell survival (43). Further detailed analyses of cellular phosphorylation events will provide in-depth understanding of their roles in SARS-CoV-2 pathogenesis. The specificity, selectivity, and off-target effects of DCLK1-IN-1 have been extensively investigated (18, 19). The structural basis of DCLK1-IN-1 selectivity, KINOMEscan and activity-based proteomic profiling against 250 cellular kinases has confirmed the pronounced selectivity of DCLK1-IN-1 for its intended target, DCLK1. We performed a 48-h treatment of cells with this inhibitor when viral production was at its maximum (13). At this time point, or beyond this time, infected cells form syncytia, become deformed, swell, and die. Therefore, the restoration of all 109 kinases is not feasible under these experimental conditions. Because of this limitation, only partial reversal of kinases was seen.
Our proteomic and phosphoproteomic findings on SARS-CoV-2-infected cells provide additional insights into the effects of DCLK1-IN-1. For example, the binding of NSP1 to the 40S ribosomal subunit blocks the mRNA entry channel, leading to the destabilization of mRNAs and inhibition of host mRNA translation (44, 45). In the cell culture conditions used in this study, the elevated expression of GSPT2/eRF3b (a translation termination factor) in infected cells was restored to normal levels by DCLK1-IN-1. Similarly, the viral-induced increases in proteins known to perturb immune regulation such as LGALS1 (associated with tolerogenic signaling and immunosuppressive for CD8 T cells), SAMD4B (linked to mTOR and mitochondrial functions), and WDR75 (a regulator of Ras and ribosomal RNA synthesis) were all reversed by DCLK1-IN-1. The phosphoproteomic analyses partially confirmed changes in the phosphorylation landscape of several signaling pathways (e.g., MAP2K, mTOR, and PI3K) which had been previously mapped for SARS-CoV-2-infected Vero-E6 and A549-ACE2 cells (10). Additionally, we discovered several host proteins with increased levels of phosphorylation during SARS-CoV-2 infection. These findings suggest that DCLK1 may directly or indirectly regulate multiple phosphorylation during SARS-CoV-2 infection. However, further investigation is needed to establish a direct causal effect for DCLK1 on SARS-CoV-2 replication. One possible next step would be to determine whether DCLK1-mediated phosphorylation of N protein balances its dual function in replication and packaging and thereby acts as a “molecular switch” in the viral life cycle (Fig. 8).
Fig 8.
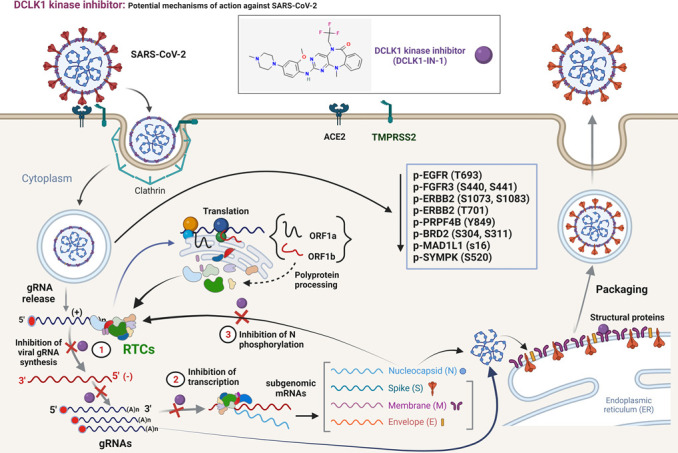
Proposed mechanism of action for efficacy of DCLK1 kinase inhibitor in SARS-CoV-2 infection. SARS-CoV-2 enters target cells through interactions of Spike protein with cellular surface protein ACE2. Subsequently, Spike protein is cleaved by transmembrane serine protease 2. After entry into the cytosol, gRNA containing 5′ cap (red circles) and a poly(A) tail is released from nucleocapsid (N) protein core particles and translated to produce polyproteins (pp1a and pp1b). These polyproteins are cleaved into 16 non-structural proteins that assemble into RTCs. RTCs synthesize new gRNAs via negative-strand RNA intermediates and also produce subgenomic mRNAs that encode structural and accessory proteins. The exact composition and involvement of cellular proteins in the regulation of RTCs are poorly defined. Viral particles are assembled by coating gRNAs with heavily phosphorylated N proteins to produce structures that bud into the endoplasmic reticulum-golgi intermediate compartment. During this process, viral particles acquire a lipid bilayer containing Spike, membrane (M), and envelope (E) proteins. Doublecortin motifs of DCLK1 can help with the movement of RTCs and transport viral particles by regulating microtubule dynamics. Thus, DCLK1-IN-1 acts on multiple steps of the viral replication cycle. gRNA, genomic RNA; RTC, replication-transcription complex.
The downregulation of DCLK1 by CRISPR/Cas9 and corresponding inhibition of gRNA, sgRNA, and viral proteins reiterated DCLK1 regulation of SARS-CoV-2 replication. DCLK1-IN-1 similarly showed anti-viral effects. We designed guide RNAs to specifically target DCLK1 during CRISPR/Cas-mediated knockdown of the gene. We saw no evidence that DCLK2 paralog complemented the negative effects of DCLK1 knockout on the replication and transcription of SARS-CoV-2 RNA. In addition, we previously showed that DCLK1 overexpression alone in Huh7 cells amplified SARS-CoV-2 production (13). Together, these data support a unique role for DCLK1 in SARS-COV-2 replication and pathology. However, potential contributions of DCLK2 or DCX (N-terminus homolog of DCLK1/2) in COVID-19 are not entirely precluded.
The administration of DCLK1-IN-1 to SARS-CoV-2-infected K18-hACE2 transgenic mice substantiated our cell-based results. We were unable to assess its efficacy for severe disease as this model of SARS-CoV-2 infection does not fully recapitulate the classic cytokine storm seen in severe COVID-19. However, a two- to three-fold infection-induced TNF-α and IL-6 mRNAs in the lungs were normalized by DCLK1-IN-1. Different models and additional study will be required to assess this manifestation of severe infection. Potential off-target effects of DCLK1-IN-1 could ultimately limit its clinical utility. Prior studies using DCLK1-IN-1 in mice did not exhibit significant adverse effects during the short-term treatment schedule (18). Similarly, we did not find any significant side effects in our murine model, although the study was of short duration. This inhibitor has yet to be evaluated in human clinical trials. The importance of DCLK1 as a potential target for inhibiting SARS-CoV-2 replication and limiting pathogenesis is magnified by its lack of expression in healthy lungs and other organs [this study and references (13, 39)]. Thus, targeting viral-induced DCLK1 instead of constitutive cellular kinases could be advantageous (46). In summary, the inhibition of DCLK1 kinase represents a novel and potentially safe and efficacious treatment strategy for COVID-19.
MATERIALS AND METHODS
Regulatory approvals
SARS-CoV-2 infection, treatment, and necropsy of transgenic mice (K18-hACE2) were performed in an animal biosafety level 3 facility.
Preparation of SARS-CoV-2 virus and TCID50 assay
SARS-CoV-2 (Wuhan and Omicron) strains were obtained from BEI Resources at the National Institutes of Allergy and Infectious Diseases in Bethesda, MD (Cat. No. NR-52281). SARS-CoV-2 was grown for up to three passages in Vero-E6 cells [Cat. No. CRL-1586, American Type Culture Collection (ATCC)] that were cultured in complete Dulbecco’s Modified Eagle’s Medium (DMEM) containing 5% FBS and antibiotics (Pen/Strep, Gibco) at 37°C and 5% CO2. To passage SARS-CoV-2, Vero-E6 cells were grown in a T-150 flask to 50% confluency (~10 million cells) and inoculated with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.001. Infection of cells was carried out in 3 mL of DMEM without fetal bovine serum (FBS) for 1 h at 37°C with gentle mixing. Cells were placed in complete media for 48 h. Virus was harvested from spent culture supernatants, centrifuged, and stored at −80°C. All experiments involving viral infection were conducted using viruses from the same stock. SARS-CoV-2 was titrated using the TCID50 method (47). For TCID50 assays, Vero-E6 cells were seeded at 10,000 cells per well in a 96-well plate, infected with serially diluted SARS-CoV-2-containing spent media, and cytopathic effects were determined after 96 h. The number of virus-positive wells was used to calculate a TCID50/mL at each dilution.
Quantitation of viral RNA copy number
Total RNAs from sera and lung tissues were isolated using an RNA isolation kit (Qiagen). A one-step real-time reverse transcription-quantitative PCR (RT-qPCR) kit and standards were used for quantitation of viral RNAs (2019-nCoV-PCR-AUS, PerkinElmer). Real-time PCR was performed on a BioRad CFX 1000 using the following cycles: reverse transcription at 50°C for 10 min, hot start at 95°C for 10 min, and 40 cycles of denaturation at 95°C for 10 s and annealing/extension at 60°C for 30 s. Results were normalized and expressed as RNA copies per microgram of total RNA.
Infection of lung adenocarcinoma cells with SARS-CoV-2, treatment with DCLK1 kinase inhibitor, and inactivation of DCLK1 gene
Lung adenocarcinoma cells (Calu-3, Cat. No. HTB-55, ATCC) were infected for 4 h at 37°C with SARS-CoV-2 at an MOI = 1. Cell supernatants were collected at 24, 48, 72, and 96 h post-infection to detect productive virus. Viral titers in spent media were measured by TCID50 (47). Infected cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher), and total lysates were prepared using Bullet Blender’s protocol (Next Advance, Inc.) for proteomic and phosphoproteomic analyses and Western blot (see below). Antibodies and fluorescence conjugated reagents (listed in Table S1) and imaging were used with a LI-COR imaging system (LI-COR Biosciences). Band intensities were calculated using Image Studio Digits. Images were produced in compliance with digital image and integrity policies.
We used a CRISPR/Cas9 approach to target DCLK1 in Calu-3 cells. Calu-3 cells were transduced with a lentiCRISPR-v2 (Addgene #52961) vector containing sgRNA against DCLK1. The cells were selected with 10 µg/mL puromycin, and total lysates of resistant cells were assessed by Western blot for DCLK1 knockdown. The sgDCLK1 #1: 5′-AGTAGAGAGCTGACTACCAG-3′, sgDCLK#2: 5′-AGTAGAGAGCTGACTACCA-3′, and sgDCLK1#3: 5′-GGAGTAGAGAGCTGACTACC-3′ were annealed and ligated into BsmB1 cut lentiCRISPRv2 vector as previously described (48). Cells were infected with SARS-CoV-2 (MOI = 1) for 48 h and immunostained for confocal microscopy. Similar cultures were used for quantification of viral RNAs. Cells transduced with lentiCRISPR-v2-sgLACZ were used as negative controls (sgRNA-C). To maintain cellular heterogeneity in the culture, we used a pooled population of puromycin-resistant cells.
Proteomic and phosphoproteomic analyses of SARS-CoV-2-infected cells
In-solution digestions with trypsin/LysC were performed for each 100-µg protein sample (Cat. No. V5071, Promega). Tryptic peptides were desalted using C18 Sep-Pak plus cartridges (Cat. No. WAT023590, Waters) and were lyophilized for 8 h to dryness. Phosphopeptides were enriched with High Select Phosphopeptide Enrichment Kits (Cat. No. A32993, Thermo Scientific). Dried peptides were reconstituted with 200-µL buffer A (0.1% formic acid). For each sample, 2 µg of peptides was injected for liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis using Dionex UltiMate 3,000 Ultra-High-Performance Liquid Chromatography (UHPLC) system connected to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific). The LC-MS/MS analysis was performed as described earlier (49). Briefly, re-suspended tryptic peptides were loaded onto an in-house trap column (150 µm × 3 cm packed with Bio-C18 3 µm resin; Sepax Technologies, Delaware) using mobile phase A (0.1% formic acid in LC-MS grade water) at a flow rate of 3 µL/min for 10 min and separated on an analytical column at a flow rate of 350 nL/min (75 µm × 30 cm in-house packed with Bio-C18 3 µm resin, Sepax Technologies). Total run time was 90 min, including column wash and re-equilibration.
The Q Exactive HF-X mass spectrometer was operated as follows: positive polarity, spray voltage 2.0 kV, funnel reflex lens (RF) lens value at 40%, and capillary temperature of 320°C. The data-dependent acquisition using the full MS-ddMS2 setup was used. The spectra were collected using a top 15 data-dependent method. Full MS resolution was set to 60,000 at 200 m/z, and full mass spectra (MS) automatic gain control (AGC) target was 3E6 with a maximum injection time of 45 ms. The ion mass range was set to 350–2,000 m/z. An AGC target value for fragment spectra was set to 1E5. The isolation width was set to 1.3 m/z units, and the first mass was fixed at 100 m/z. The normalized collision energy of higher-energy collisional dissociation (HCD) was set to 30%. Peptide match was set to preferred, and isotope exclusion was on. MS1 and MS2 spectra were acquired in profile and centroid modes, respectively. Precursors were fragmented by HCD using 30% normalized collision energy and analyzed in the Orbitrap at a resolution of 15,000 (at 200 m/z). The dynamic exclusion duration of fragmented precursor ions was set to 30 s. The ion selection abundance threshold was set at 2E5 with charge state exclusion of unassigned and z = 1 and z = 6–8 ions.
RAW files were searched against the UniProt reviewed Human protein (TaxID: UP000005640) and SARS-CoV-2 protein databases (TaxID: UP000000354) using the Sequest algorithm within Proteome Discoverer v.2.4 (Thermo Fisher Scientific). The Sequest database search was performed with the following parameters: trypsin enzyme cleavage specificity, two possible missed cleavages, 10-ppm mass tolerance for precursor ions, and 0.02-Da mass tolerance for fragment ions. Search parameters permitted dynamic modification of methionine oxidation (+15.9949 Da) and static modification of carbamidomethylation (+57.0215 Da) on cysteine. To identify the phosphopeptides, phosphorylation (+79.966 Da) of serine (S), threonine (T), and tyrosine (Y) was added as variable modifications. Peptide assignments from the database search were filtered down to a 1% false discovery rate. Label-free quantitation across samples employed the Minora algorithm and adjoining bioinformatics tools available in Proteome Discoverer.
Bioinformatic analysis of bottom-up proteomic results
Bioinformatic platforms such as ShinyGO v.0.76 (50) and STRING (string-db.org) were used for pathway and protein-protein network analysis. Morpheus (software.broadinstitute. org/morpheus) was used to generate heat maps. Venn diagrams were generated using Venn Diagram Plotter (PNNL, pnnl-comp-mass-spec.github.io/Venn-Diagram-Plotter). The protein-protein interaction map was generated using STRING (https://string-db.org/).
Treatment of SARS-CoV-2-infected transgenic mice with a DCLK1 kinase inhibitor
K18-hACE2 transgenic mice (C57BL/6J background) expressing human ACE2 under the control of the keratin 18 promoter were purchased from The Jackson Laboratory (Cat. No. 034860). All mice were observed and weighed daily. Male and female mice were used in equal numbers (4–6 weeks old, n = 12) and randomized according to the following groups: group 1: SARS-CoV-2-infected, untreated positive control; group 2: SARS-CoV-2-infected and treated only with vehicle (DMSO); group 3: SARS-CoV-2-infected and treated with DCLK1-IN-1 (10-mg/kg body weight); and group 4: wild-type negative control with no infection. Groups 1, 2, and 3 mice were intranasally infected with SARS-CoV-2 (2.8 × 104 plaque forming units in 20 µL with 10 µL per naris). The treatments for groups 2 and 3 mice were started 6 h post-infection using vehicle or DCLK1-IN-1 by intraperitoneal injection, respectively, once daily for 4 days. Group 1 and group 4 control mice were administered phosphate-buffered saline. Mice were euthanized 5 days post-infection for analysis of blood and lung tissues. Total RNAs were isolated from lungs and subjected to RT-qPCR for viral genomic RNA and N protein mRNA.
Histology, immunohistochemistry, and confocal microscopy
Murine lung tissues were subjected to immunofluorescence using an Akoya Opal 4-Color auto immunohistochemistry kit (Cat. No. NEL820001KT). Stained slides were imaged and evaluated by confocal microscopy (Leica SP8). Slides were scanned on a Zeiss AxioScan instrument and staining intensities were quantitated using the HALO image analysis software (Zeiss). Calu-3 cells were stained as previously described (13).
Cytokine analysis
Murine sera were analyzed using a custom-made human Magnetic Luminex Assay Kit for TNF-α, IL-1β, IL-6, and S100A9 (R&D Systems). Samples were processed in replicates and quantified using the BioPlex 200 System (Bio-Rad). The concentration of each analyte was expressed as picogram per milliliter of serum. Analyte levels in normal uninfected murine sera were compared with sera from SARS-CoV-2-infected mice that had been treated with DMSO or DCLK1-IN-1.
Statistical analysis
Statistical analyses were performed using Prism Graphpad software v.9.0. Comparison between groups was made using one-way analysis of variance. Multiple comparisons were done using the Šidák test with a single pooled variance. For proteomic and phosphoproteomic analyses, a 1.5-fold increase or decrease in abundance with a P value of ≤0.05 was considered as statistically significant. Cytokine/chemokine/growth factor data are presented as mean ± standard error of the mean (P values: *P < 0.05 , **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant, ≥0.05.
ACKNOWLEDGMENTS
The OUHSC Laboratory for Molecular Biology and Cytometry Research provided services for flow cytometry and confocal microscopy. Immunohistochemistry was performed by the Stephenson Cancer Center Pathology Core Laboratory (supported by the NIGMS P20GM103639). N.Ah. gratefully acknowledges the initial funding support from the OU VPRP (Norman) Office for the establishment of the Proteomics Core Facility. This work was supported by Stephenson Cancer Center CB Pilot Project, Presbyterian Health Foundation, and OK-INBRE award through the National Institute of General Medical Sciences of the National Institutes of Health (under award P20GM103447, awarded to N.Al.) and National Cancer Institute (R01-CA230641, awarded to M.M.H).
Conceptualization: N.Al. and M.M.H.; methodology: N.Al., R.B.U., N.Ah., J.L.L., N.D.R., J.P., S.D., and B.N.H.; investigation: N.Al., M.M.H., and N.Ah.; funding acquisition: N.Al. and M.M.H.; project administration: N.Al., M.M.H., C.W.H., and M.S.B.; supervision: N.Al. and M.M.H.; review and editing: N.Al., M.M.H., N.Ah., C.W.H., and M.S.B..
Contributor Information
Naushad Ali, Email: naushad-ali@ouhsc.edu.
Rebecca Ellis Dutch, University of Kentucky College of Medicine, Lexington, Kentucky, USA .
DATA AVAILABILITY
All data associated with this study are presented in the paper and supplementary materials. Materials will be available upon request. Proteomics data (RAW files and result files) have been submitted and are now available in the public database repository MassIVE database (https://massive.ucsd.edu/) under accession numbers: MSV000091149 (proteomics) and MSV000091115 (phosphoproteomics).
ETHICS APPROVAL
All experiments relating to infectious clones and cultures were performed in an animal biosafety level 3 facility after approval by the OUHSC Institutional Biosafety Committee (protocol #100492). Lung adenocarcinoma cells were used in accordance with guidelines approved by the OUHSC Institutional Biosafety Committee (protocol #200470–2440A). Animal protocols were approved by the OUHSC Institutional Animal Care and Use Committee (protocol #21–064-CHI).
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.01194-23.
Supplemental figures and table.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, Alanio C, Kuri-Cervantes L, Pampena MB, D’Andrea K, et al. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369:eabc8511. doi: 10.1126/science.abc8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, et al. 2021. A molecular single-cell lung atlas of lethal COVID-19. Nature 598:114–119. doi: 10.1038/s41586-021-03921-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Silvin A, Chapuis N, Dunsmore G, Goubet A-G, Dubuisson A, Derosa L, Almire C, Hénon C, Kosmider O, Droin N, et al. 2020. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 182:1401–1418. doi: 10.1016/j.cell.2020.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su Y, Yuan D, Chen DG, Ng RH, Wang K, Choi J, Li S, Hong S, Zhang R, Xie J, et al. 2022. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 185:881–895. doi: 10.1016/j.cell.2022.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Proal AD, VanElzakker MB. 2021. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol 12:698169. doi: 10.3389/fmicb.2021.698169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lopes-Pacheco M, Silva PL, Cruz FF, Battaglini D, Robba C, Pelosi P, Morales MM, Caruso Neves C, Rocco PRM. 2021. Pathogenesis of multiple organ injury in COVID-19 and potential therapeutic strategies. Front Physiol 12:593223. doi: 10.3389/fphys.2021.593223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaeuffer C, Le Hyaric C, Fabacher T, Mootien J, Dervieux B, Ruch Y, Hugerot A, Zhu Y-J, Pointurier V, Clere-Jehl R, Greigert V, Kassegne L, Lefebvre N, Gallais F, Meyer N, Hansmann Y, Hinschberger O, Danion F, COVID Alsace Study Group . 2020. Clinical characteristics and risk factors associated with severe COVID-19 prospective analysis of 1,045 hospitalised cases in north-eastern France, march 2020. Euro Surveill 25:2000895. doi: 10.2807/1560-7917.ES.2020.25.48.2000895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao Y, Ding M, Dong X, Zhang J, Kursat Azkur A, Azkur D, Gan H, Sun Y, Fu W, Li W, Liang H, Cao Y, Yan Q, Cao C, Gao H, Brüggen M, van de Veen W, Sokolowska M, Akdis M, Akdis CA. 2021. Risk factors for severe and critically ill COVID-19 patients: a review. Allergy 76:428–455. doi: 10.1111/all.14657 [DOI] [PubMed] [Google Scholar]
- 9. Wu M, Chen Y, Xia H, Wang C, Tan CY, Cai X, Liu Y, Ji F, Xiong P, Liu R, Guan Y, Duan Y, Kuang D, Xu S, Cai H, Xia Q, Yang D, Wang MW, Chiu IM, Cheng C, Ahern PP, Liu L, Wang G, Surana NK, Xia T, Kasper DL. 2020. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc Natl Acad Sci U S A 117:28336–28343. doi: 10.1073/pnas.2018030117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, et al. 2020. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182:685–712. doi: 10.1016/j.cell.2020.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, Li J, Liu Y, Tang F, Zhang F, et al. 2021. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 184:5838. doi: 10.1016/j.cell.2021.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, Krämer B, Krammer T, Brumhard S, Bonaguro L, et al. 2020. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182:1419–1440. doi: 10.1016/j.cell.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Undi RB, Larabee JL, Filiberti A, Ulahannan S, Aravindan S, Stroberg E, Barton LM, Duval EJ, Mukhopadhyay S, Henthorn JC, Akins D, Houchen CW, Huycke MM, Ali N. 2022. Targeting doublecortin-like kinase 1 (DCLK1)-regulated SARS-CoV-2 pathogenesis in COVID-19. J Virol 96:e0096722. doi: 10.1128/jvi.00967-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koizumi H, Tanaka T, Gleeson JG. 2006. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 49:55–66. doi: 10.1016/j.neuron.2005.10.040 [DOI] [PubMed] [Google Scholar]
- 15. Agulto RL, Rogers MM, Tan TC, Ramkumar A, Downing AM, Bodin H, Castro J, Nowakowski DW, Ori-McKenney KM. 2021. Autoregulatory control of microtubule binding in doublecortin-like kinase 1. Elife 10:e60126. doi: 10.7554/eLife.60126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Ferguson FM, Li L, Kuljanin M, Mills CE, Subramanian K, Harshbarger W, Gondi S, Wang J, Sorger PK, Mancias JD, Gray NS, Westover KD. 2020. Chemical biology toolkit for DCLK1 reveals connection to RNA processing. Cell Chem Biol 27:1229–1240. doi: 10.1016/j.chembiol.2020.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, Isomura A, Kawada K, Sakai Y, Yanagita M, Kageyama R, Kawaguchi Y, Taketo MM, Yonehara S, Chiba T. 2013. DCLK1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet 45:98–103. doi: 10.1038/ng.2481 [DOI] [PubMed] [Google Scholar]
- 18. Ferguson FM, Nabet B, Raghavan S, Liu Y, Leggett AL, Kuljanin M, Kalekar RL, Yang A, He S, Wang J, Ng RWS, Sulahian R, Li L, Poulin EJ, Huang L, Koren J, Dieguez-Martinez N, Espinosa S, Zeng Z, Corona CR, Vasta JD, Ohi R, Sim T, Kim ND, Harshbarger W, Lizcano JM, Robers MB, Muthaswamy S, Lin CY, Look AT, Haigis KM, Mancias JD, Wolpin BM, Aguirre AJ, Hahn WC, Westover KD, Gray NS. 2020. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat Chem Biol 16:635–643. doi: 10.1038/s41589-020-0506-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patel O, Roy MJ, Kropp A, Hardy JM, Dai W, Lucet IS. 2021. Structural basis for small molecule targeting of doublecortin like kinase 1 with DCLK1-IN-1. Commun Biol 4:1105. doi: 10.1038/s42003-021-02631-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahsan N, Rao RSP, Wilson RS, Punyamurtula U, Salvato F, Petersen M, Ahmed MK, Abid MR, Verburgt JC, Kihara D, Yang Z, Fornelli L, Foster SB, Ramratnam B. 2021. Mass spectrometry‐based proteomic platforms for better understanding of SARS‐CoV‐2 induced pathogenesis and potential diagnostic approaches. Proteomics 21:e2000279. doi: 10.1002/pmic.202000279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, Xia B, Ma X, Yang T, Yu F, Liu J, Liu B, Song Z, Chen J, Yan S, Wu L, Pan T, Zhang X, Li R, Huang W, He X, Xiao F, Zhang J, Zhang H. 2021. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-iota. Proc Natl Acad Sci U S A 118:e2024202118. doi: 10.1073/pnas.2024202118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, Zheng YX, Wang XN, Qi H, Zhang J, Wang PH, Han ZG, Tao SC. 2020. SARS-CoV-2 ORF9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol 17:998–1000. doi: 10.1038/s41423-020-0514-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, Lavillette D, Denolly S. 2021. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the Spike protein, allowing assembly of virus-like particles. J Biol Chem 296:100111. doi: 10.1074/jbc.RA120.016175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, Frankel AD, Morgan DO. 2020. Phosphoregulation of phase separation by the SARS-CoV-2 N protein suggests a biophysical basis for its dual functions. Mol Cell 80:1092–1103. doi: 10.1016/j.molcel.2020.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chikhalya A, Dittmann M, Zheng Y, Sohn SY, Rice CM, Hearing P. 2021. Human IFIT3 protein induces interferon signaling and inhibits adenovirus immediate early gene expression. mBio 12:e0282921. doi: 10.1128/mBio.02829-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salem S, Mosaad R, Lotfy R, Elbadry M. 2023. Altered expression of DNA methyltransferases and methylation status of the TLR4 and TNF-alpha promoters in COVID-19. Arch Virol 168:95. doi: 10.1007/s00705-023-05722-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu HY, Gu H, Qu H, Bao W, Li Y, Cai D. 2022. Aberrant cholesterol metabolic genes regulation in a negative feedback loop induced by an alphacoronavirus. Front Nutr 9:870680. doi: 10.3389/fnut.2022.870680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mishra R, Kumawat KL, Basu A, Banerjea AC. 2022. Japanese encephalitis virus infection increases USP42 to stabilize TRIM21 and OAS1 for neuroinflammatory and anti-viral response in human microglia. Virology 573:131–140. doi: 10.1016/j.virol.2022.06.012 [DOI] [PubMed] [Google Scholar]
- 29. McCray PB, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S. 2007. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus . J Virol 81:813–821. doi: 10.1128/JVI.02012-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golden JW, Cline CR, Zeng X, Garrison AR, Carey BD, Mucker EM, White LE, Shamblin JD, Brocato RL, Liu J, Babka AM, Rauch HB, Smith JM, Hollidge BS, Fitzpatrick C, Badger CV, Hooper JW. 2020. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 5:e142032. doi: 10.1172/jci.insight.142032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yinda CK, Port JR, Bushmaker T, Offei Owusu I, Purushotham JN, Avanzato VA, Fischer RJ, Schulz JE, Holbrook MG, Hebner MJ, Rosenke R, Thomas T, Marzi A, Best SM, de Wit E, Shaia C, van Doremalen N, Munster VJ, Subbarao K. 2021. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLoS Pathog 17:e1009195. doi: 10.1371/journal.ppat.1009195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes S-S, Bosurgi L, Dutzmann J, Sedding D, Frese T, Girndt M, Höll JI, Gekle M, Mikolajczyk R, Binder M. 2022. The IL-1beta, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep Med 3:100663. doi: 10.1016/j.xcrm.2022.100663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mulay A, Konda B, Garcia G Jr, Yao C, Beil S, Villalba JM, Koziol C, Sen C, Purkayastha A, Kolls JK, Pociask DA, Pessina P, de Aja JS, Garcia-de-Alba C, Kim CF, Gomperts B, Arumugaswami V, Stripp BR. 2021. SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep 35:109055. doi: 10.1016/j.celrep.2021.109055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ali N, Nguyen CB, Chandrakesan P, Wolf RF, Qu D, May R, Goretsky T, Fazili J, Barrett TA, Li M, Huycke MM, Bronze MS, Houchen CW. 2020. Doublecortin-like kinase 1 promotes hepatocyte clonogenicity and oncogenic programming via non-canonical beta-catenin-dependent mechanism. Sci Rep 10:10578. doi: 10.1038/s41598-020-67401-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lipka J, Kapitein LC, Jaworski J, Hoogenraad CC. 2016. Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J 35:302–318. doi: 10.15252/embj.201592929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo W, Jin Y, Jiang Y, Yang L, Xu H, Wu D, Zhang Y, Yin L, Khan ZA, Liang G, Wang Y. 2023. Doublecortin-like kinase 1 activates NF-kappaB to induce inflammatory responses by binding directly to IKKbeta. Cell Death Differ 30:1184–1197. doi: 10.1038/s41418-023-01147-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rayaprolu V, Fulton BO, Rafique A, Arturo E, Williams D, Hariharan C, Callaway H, Parvate A, Schendel SL, Parekh D, Hui S, Shaffer K, Pascal KE, Wloga E, Giordano S, Negron N, Ni M, Copin R, Atwal GS, Franklin M, Boytz RM, Donahue C, Davey R, Baum A, Kyratsous CA, Saphire EO. 2023. Structure of the Inmazeb cocktail and resistance to Ebola virus escape. Cell Host Microbe 31:260–272. doi: 10.1016/j.chom.2023.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yaron TM, Heaton BE, Levy TM, Johnson JL, Jordan TX, Cohen BM, Kerelsky A, Lin T-Y, Liberatore KM, Bulaon DK, et al. 2022. Host protein Kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci Signal 15:eabm0808. doi: 10.1126/scisignal.abm0808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ali N, Chandrakesan P, Nguyen CB, Husain S, Gillaspy AF, Huycke M, Berry WL, May R, Qu D, Weygant N, Sureban SM, Bronze MS, Dhanasekaran DN, Houchen CW. 2015. Inflammatory and oncogenic roles of a tumor stem cell marker doublecortin-like kinase (DCLK1) in virus-induced chronic liver diseases. Oncotarget 6:20327–20344. doi: 10.18632/oncotarget.3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westphalen CB, Takemoto Y, Macchini M, Jiang Z, Renz BW, Chen X, Ormanns S, Nagar K, Tailor Y, et al. 2017. DCLK1 defines quiescent pancreatic progenitors that promote injury-induced regeneration and tumorigenesis. Cell Stem Cell 31. doi: 10.1016/j.ccell.2016.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M. 2020. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun 11:6041. doi: 10.1038/s41467-020-19843-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takashita E, Yamayoshi S, Halfmann P, Wilson N, Ries H, Richardson A, Bobholz M, Vuyk W, Maddox R, Baker DA, Friedrich TC, O’Connor DH, Uraki R, Ito M, Sakai-Tagawa Y, Adachi E, Saito M, Koga M, Tsutsumi T, Iwatsuki-Horimoto K, Kiso M, Yotsuyanagi H, Watanabe S, Hasegawa H, Imai M, Kawaoka Y. 2022. Efficacy of antiviral agents against the omicron subvariant BA.2.75. N Engl J Med 387:2094–2097. doi: 10.1056/NEJMc2211845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen CH, Hsia TC, Yeh MH, Chen TW, Chen YJ, Chen JT, Wei YL, Tu CY, Huang WC. 2017. MEK inhibitors induce AKT activation and drug resistance by suppressing negative feedback ERK-mediated HER2 phosphorylation at Thr701. Mol Oncol 11:1273–1287. doi: 10.1002/1878-0261.12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Slesarev GG. 1966. Anterior philodesis in dropped foot using Lavsan. Ortop Travmatol Protez 27:75–76. [PubMed] [Google Scholar]
- 45. Zhang D, Zhu L, Wang Y, Li P, Gao Y. 2022. Translational control of COVID-19 and its therapeutic implication. Front Immunol 13:857490. doi: 10.3389/fimmu.2022.857490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boytz R, Słabicki M, Ramaswamy S, Patten JJ, Zou C, Meng C, Hurst BL, Wang J, Nowak RP, Yang PL, Sattler M, Stone RM, Griffin JD, Gray NS, Gummuluru S, Davey RA, Weisberg E. 2023. Anti-SARS-CoV-2 activity of targeted kinase inhibitors: repurposing clinically available drugs for COVID-19 therapy. J Med Virol 95:e28157. doi: 10.1002/jmv.28157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Hyg 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408 [DOI] [Google Scholar]
- 48. Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ahsan N, Fornelli L, Najar FZ, Gamagedara S, Hossan MR, Rao RSP, Punyamurtula U, Bauer A, Yang Z, Foster SB, Kane MA. 2023. Proteomics evaluation of five economical commercial abundant protein depletion kits for enrichment of diseases-specific biomarkers from blood serum. Proteomics:e2300150. doi: 10.1002/pmic.202300150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ge SX, Jung D, Yao R. 2020. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36:2628–2629. doi: 10.1093/bioinformatics/btz931 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figures and table.
Data Availability Statement
All data associated with this study are presented in the paper and supplementary materials. Materials will be available upon request. Proteomics data (RAW files and result files) have been submitted and are now available in the public database repository MassIVE database (https://massive.ucsd.edu/) under accession numbers: MSV000091149 (proteomics) and MSV000091115 (phosphoproteomics).