

ABSTRACT
In contrast to microbial metagenomics, there has still been only limited efforts to benchmark performance of virome analysis approaches in terms of faithfulness to community structure and of completeness of virome description. While natural communities are more readily accessible, synthetic communities assembled using well-characterized isolates allow more accurate performance evaluation. Starting from authenticated, quality-controlled reference isolates from the DSMZ Plant Virus Collection, we have assembled synthetic communities of varying complexity up to a highly complex community of 72 viral agents (115 viral molecules) comprising isolates from 21 viral families and 61 genera. These communities were then analyzed using two approaches frequently used in ecology-oriented plant virus metagenomics: a virion-associated nucleic acids (VANA)-based strategy and a highly purified double-stranded RNAs (dsRNAs)-based one. The results obtained allowed to compare diagnostic sensitivity of these two approaches for groups of viruses and satellites with different genome types and confirmed that the dsRNA-based approach provides a more complete representation of the RNA virome. However, for viromes of low to medium complexity, VANA appears a reasonable alternative and would be the preferred choiceif analysis of DNA viruses is of importance. They also allowed to identify several important parameters and to propose hypotheses to explain differences in performance, in particular, differences in the imbalance in the representation of individual viruses using each approach. Remarkably, these analyses highlight a strong direct relationship between the completeness of virome description and sample sequencing depth which should prove useful in further virome analysis efforts.
IMPORTANCE
We report here efforts to benchmark performance of two widespread approaches for virome analysis, which target either virion-associated nucleic acids (VANA) or highly purified double-stranded RNAs (dsRNAs). This was achieved using synthetic communities of varying complexity levels, up to a highly complex community of 72 viral agents (115 viral molecules) comprising isolates from 21 families and 61 genera of plant viruses. The results obtained confirm that the dsRNA-based approach provides a more complete representation of the RNA virome, in particular, for high complexity ones. However, for viromes of low to medium complexity, VANA appears a reasonable alternative and would be the preferred choice if analysis of DNA viruses is of importance. Several parameters impacting performance were identified as well as a direct relationship between the completeness of virome description and sample sequencing depth. The strategy, results, and tools used here should prove useful in a range of virome analysis efforts.
KEYWORDS: virome, VANA, dsRNA, synthetic community, metagenome, double-stranded RNA, high-thropughput sequencing
INTRODUCTION
Significant advances in the development of molecular methods have been made in the last decades, including innovative sequencing technologies based on DNA/RNA approaches such as targeted reverse-transcription (RT) PCR or non-targeted high-throughput sequencing (HTS). HTS, also known as next-generation sequencing, enables high-speed, high-throughput sequencing of native DNA/RNA or amplified DNA, generating enormous amounts of sequencing data. These developments led to major advances in the field of metagenomics, i.e., the sequencing of the entire genetic material of a sample, and to a new understanding of microbial diversity (1, 2). Viral metagenomics has revealed the immense diversity and ubiquity of viruses in nature and thus revolutionized our vision of these biological agents (1, 3 – 8). Specifically, these metagenomic studies have revealed that virus sequence data available in public databases are biased toward human viruses or viruses of anthropological significance, with, e.g., influenza-like viruses found in fish and amphibian hosts (9) or more than 75% of the plant virus species characterized up to 2006 having been isolated from crops (10). These findings, together with reports on viruses associated with hosts different from those known for the vast majority of their relatives, such as flavi-like viruses found in plants (11, 12), have raised novel questions about virus-hosts co-divergence or host switching.
In plant virology in particular, advances in the development of viral metagenome analyses have been of great importance in terms of early detection of known viruses and discovery of novel plant viruses (4, 7, 13, 14), as more than half of emerging diseases in plants are thought to be caused by viruses (15). HTS has a huge potential in plant virus diagnostics because it allows to picture the complete phytosanitary status of a plant and to differentiate between virus variants that may contribute differentially to disease etiology (14). For example, in a metagenomic analysis of sour cherry showing symptoms of Shirofugen stunt disease (SSD), a divergent isolate of little cherry virus 1 (LChV1) was identified in the absence of any other viral agent, suggesting that LChV1 could be responsible for the SSD disease (16). However, metagenomic approaches have also revealed that plants are often infected by more than one virus (17), complicating the unraveling of the etiology of plant viral diseases.
HTS has also renewed the link between classical plant virology and ecology (4, 18). Viromes identified from both cultivated and uncultivated plant populations enabled the study of ecological processes such as the movement of viruses between different host reservoirs, the effects of management practices or of the anthropological simplification of ecosystems (19 – 23).
For the efficient characterization of complex plant-associated viromes, there is generally a need to enrich viral sequences and conversely reduce the amount of host plant sequences that are generated. Different target nucleic acid populations have been used for virome studies but, coupled with the virus enrichment constraint, the most widely used approaches have targeted virion-associated nucleic acids (VANA) or double-stranded RNAs (dsRNAs) (4, 7). For single plant samples or low-complexity samples, the use of total RNA or small interfering RNA (siRNA) sequencing are considered the most universal and straightforward options (24, 25), but when the viromes of entire plant communities are analyzed from complex plant pools, VANA or dsRNA enrichment methods are generally preferred (4, 7, 19, 21, 26). A huge number of bioinformatic tools are available for HTS data analysis and have been, together with nucleic acid preparation strategies, extensively reviewed (13, 27, 28). The choice of a specific viral enrichment method or bioinformatic pipeline depends on the experimental objectives. Even though there have been some efforts toward performance comparisons of different virome analysis approaches (29, 30), there is a need to better benchmark them and assess their respective efficiency at providing a faithful and comprehensive description of complex viromes, without introducing biases. In a virus discovery study on single quarantine plants, VANA was shown to assemble longer contigs compared to siRNA for a novel DNA Mastrevirus (31), while in a study investigating the virome of native plants in Oklahoma, more viral operational taxonomy units (OTUs) could be detected with dsRNA compared to VANA (26). Ma et al. (32) provided a more comprehensive comparison of these two approaches using the natural viral communities present in complex plant pools from managed and unmanaged sites. The authors found significant differences with more viral contigs and, on average, longer contigs assembled from libraries prepared from dsRNA. With regard to viral richness, more OTUs were detected by the dsRNA approach compared to the VANA one. However, most DNA viruses were only detected using VANA.
Standardization is fundamental for the reliable representation of microbiome/virome in metagenomic studies and is challenged by the rapid development of sequencing platforms, protocols, and bioinformatic pipelines (33). Benchmarking is a powerful tool to provide standards that can be used to compare and evaluate the performance of the different steps required in metagenomic studies, including target nucleic acids population extraction, library preparation, sequencing (and sequencing platform), and, finally, bioinformatics sequence analysis. In this context, benchmarking studies in metagenomics are often based on mock communities that are microbial assemblages of known composition which can be used to compare the actual vs the expected performance of a process. Besides the use of actual empirical phytoviromes (32), the use of synthetic communities could therefore provide a more precise and detailed benchmarking of HTS-based virome description strategies. Bacterial and fungal mock communities have thus been developed and used to compare the performance of different sequencing platforms, e.g., short-read Illumina or long-read PacBio SMRT sequencing (34 – 36). In recent years, viral mock communities have also been developed, especially in the medical and clinical field, to benchmark protocols in human virome studies. For example, the nucleic acid preparation step for the virome analysis of fecal samples was optimized using a combination of both viral and bacterial mock communities (37). In another study, the bias introduced by viral enrichment or random amplification was assessed using a DNA virus mock community (38). Viral synthetic communities have also been used to benchmark library preparation approaches in environmental (39) and insect (40) virome studies. However, the use of synthetic communities in plant virome studies is lagging behind. So far, the only study using a defined mix of plant viruses to assess different nucleic acid preparation protocols was performed by Gafaar and Ziebell (30). This study revealed a better performance of enriched dsRNAs as compared to ribodepleted total RNA or siRNAs for virus detection. However, only low-complexity synthetic communities have been used so far, whereas most of the viral metagenomes associated with natural plant communities are composed of a complex and diverse mixture of DNA and RNA viruses that are studied from pooled plant samples. In the present work, we used a total of 22 synthetic plant virus communities of varying degrees of complexity to compare the diagnostic performance of VANA- and dsRNA-based approaches for virome description and analyzed how this performance is affected by sequencing depth and other parameters. In parallel, a first attempt at contrasting the performance of VANA and dsRNA approaches with those of RNASeq was conducted, using synthetic data sets assembled in silico from single-isolate RNASeq data.
MATERIALS AND METHODS
Mock viral communities design
A list of 61 different viruses (assigned to 59 different genera from 18 different families plus one unassigned virus) was selected among those kept in collection and available at the Leibniz-Institute DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany), taking into consideration three main criteria: (i) maximizing viral diversity by including viruses with all genome types (ssDNA, dsDNA-RT, dsRNA, +ssRNA, −ssRNA), (ii) including (with one exception) only a single representative virus per viral genus, and (iii) selecting viruses/isolates for which a complete or near-complete genomic sequence is available. In some cases, these genomic sequences had been determined previously, while in other cases, they were developed specifically in the frame of efforts to further improve the characterization of isolates distributed by the DSMZ through the European Union-funded EVA-Global initiative (https://www.european-virus-archive.com/). Quality-controlled samples were obtained from the DSMZ in the form of infected, lyophilized plant material in vacuum-sealed vials. The complete list of the isolates used, together with their properties and the propagation host in which they were provided, is given in Table 1.
TABLE 1.
Viral isolates used to construct mock viral communities of varying complexity d
| Family | Genus | Virus | Acronym | Genome | Code a | Host b | Sequence accession number(s) |
|---|---|---|---|---|---|---|---|
| Alphaflexiviridae | Allexivirus | Shallot virus X | ShVX | ssRNA(+) | PV-0622 | Chenopodium murale | MW854280 |
| Alphaflexiviridae | Potexvirus | Lettuce virus X | LeVX | ssRNA(+) | PV-0904 | Nicotiana benthamiana | MW248356 |
| Benyviridae | Benyvirus | Beet necrotic yellow vein virus | BNYVV | ssRNA(+) | PV-0467 | Chenopodium quinoa | OK181765-67; M36896 |
| Betaflexiviridae | Capillovirus | Apple stem grooving virus | ASGV | ssRNA(+) | PV-0199 | Chenopodium quinoa | MW582790 |
| Betaflexiviridae | Carlavirus | Poplar mosaic virus | PopMV | ssRNA(+) | PV-0341 | Nicotiana benthamiana | ON924213 |
| Betaflexiviridae | Trichovirus | Apple chlorotic leaf spot virus | ACLSV | ssRNA(+) | PV-0998 | Chenopodium quinoa | OK340218-19 c |
| Betaflexiviridae | Tepovirus | Potato virus T | PVT | ssRNA(+) | PV-1145 | Nicotiana hesperis | MZ405665 |
| Bromoviridae | Alfamovirus | Alfalfa mosaic virus | AMV | ssRNA(+) | PV-0779 | Nicotiana tabacum “Samsun nn” | MZ405653-55 |
| Bromoviridae | Anulavirus | Pelargonium zonate spot virus | PZSV | ssRNA(+) | PV-0259 | Nicotiana glutinosa “24A” | ON398493-95 |
| Bromoviridae | Bromovirus | Brome mosaic virus | BMV | ssRNA(+) | PV-0194 | Hordeum vulgare | MW582787-89 |
| Bromoviridae | Cucumovirus | Peanut stunt virus | PSV | ssRNA(+) | PV-0190 | Nicotiana benthamiana | MW307259-61 |
| Bromoviridae | Ilarvirus | Parietaria mottle virus | PMoV | ssRNA(+) | PV-0400 | Chenopodium quinoa | MZ405646-48 |
| Closteroviridae | Closterovirus | Beet yellows virus | BYV | ssRNA(+) | PV-1260 | Beta macrocarpa | MT815988 |
| Closteroviridae | Crinivirus | Tomato chlorosis virus | ToCV | ssRNA(+) | PV-1242 | Solanum lycopersicum | ON398512-13 |
| Potyviridae | Bymovirus | Barley yellow mosaic virus | BaYMV | ssRNA(+) | PV-0634 | Hordeum vulgare | OL311692-93 |
| Potyviridae | Ipomovirus | Cucumber vein yellowing virus | CVYV | ssRNA(+) | PV-0776 | Cucumis sativus | OK181771 |
| Potyviridae | Potyvirus | Bidens mottle virus | BiMoV | ssRNA(+) | PV-0752 | Nicotiana benthamiana | ON398504 |
| Potyviridae | Rymovirus | Agropyron mosaic virus | AgMV | ssRNA(+) | PV-0729 | Triticum aestivum | OM471970 |
| Potyviridae | Tritimovirus | Brome streak mosaic virus | BrSMV | ssRNA(+) | PV-0431 | Hordeum vulgare | OP357935 |
| Potyviridae | Unassigned | Spartina mottle virus | SpMV | ssRNA(+) | PV-0970 | Spartina sp. | MN788417 |
| Secoviridae | Cheravirus | Arracacha virus B | AVB | ssRNA(+) | PV-0082 | Chenopodium murale | MW582785-86 |
| Secoviridae | Comovirus | Squash mosaic virus | SqMV | ssRNA(+) | PV-0581 | Cucurbita pepo | ON398498-99 |
| Secoviridae | Fabavirus | Broad been wilt virus 1 | BBWV-1 | ssRNA(+) | PV-0067 | Chenopodium quinoa | MT663310-11 |
| Secoviridae | Nepovirus | Tomato black ring virus | TBRV | ssRNA(+) | PV-0191 | Nicotiana clevelandii | MW057704-05 |
| Secoviridae | Sequivirus | Carrot necrotic dieback virus | CNDV | ssRNA(+) | PV-0976 | Nicotiana benthamiana | MW080951 |
| Secoviridae | Stralirivirus | Strawberry latent ringspot virus | SLRSV | ssRNA(+) | PV-0247 | Chenopodium quinoa | MZ405640-41 |
| Solemoviridae | Sobemovirus | Rice yellow mottle virus | RYMV | ssRNA(+) | PV-0732 | Oryza sativa | MT701719 |
| Solemoviridae | Enamovirus | Pea enation mosaic virus 1 | PEMV1 | ssRNA(+) | PV-0088 | Pisum sativum | MW961146 |
| Solemoviridae | Polerovirus | Cucurbit aphid-borne yellows virus | CABYV | ssRNA(+) | PV-1017 | Physalis floridana | MZ202344 |
| Tombusviridae | Alphacarmovirus | Calibrachoa mottle virus | CbMV | ssRNA(+) | PV-0611 | Chenopodium quinoa | OK181769 |
| Tombusviridae | Alphanecrovirus | Tobacco necrosis virus A | TNV-A | ssRNA(+) | PV-0186 | Chenopodium quinoa | MT675968 |
| Tombusviridae | Aureusvirus | Johnsongrass chlorotic stripe mosaic virus | JCSMV | ssRNA(+) | PV-0605 | Zea mays | MT682309 |
| Tombusviridae | Betacarmovirus | Turnip crinkle virus | TCV | ssRNA(+) | PV-0293 | Nicotiana benthamiana | OK181761 |
| Tombusviridae | Betanecrovirus | Beet black scorch virus | BBSV | ssRNA(+) | PV-0951 | Chenopodium quinoa | OK058516 |
| Tombusviridae | Dianthovirus | Carnation ringspot virus | CRSV | ssRNA(+) | PV-0097 | Nicotiana clevelandii | MT682300-01 |
| Tombusviridae | Gammacarmovirus | Melon necrotic spot virus | MNSV | ssRNA(+) | PV-0378 | Cucumis sativus | ON398496 |
| Tombusviridae | Machlomovirus | Maize chlorotic mottle virus | MCMV | ssRNA(+) | PV-1087 | Zea mays | OK181780 |
| Tombusviridae | Pelarspovirus | Pelargonium line pattern virus | PLPV | ssRNA(+) | PV-0193 | Chenopodium quinoa | MW854266 |
| Tombusviridae | Tombusvirus | Tomato bushy stunt virus | TBSV | ssRNA(+) | PV-0268 | Nicotiana clevelandii | MW582792 |
| Tombusviridae | Umbravirus | Carrot mottle virus | CMoV | ssRNA(+) | PV-0968 | Nicotiana benthamiana | OK058520 |
| Tombusviridae | Umbravirus | Pea enation mosaic virus 2 | PEMV2 | ssRNA(+) | PV-0088 | Pisum sativum | MW961147; MW961148 c |
| Tospoviridae | Orthotospovirus | Impatiens necrotic spot virus | INSV | ssRNA(+/-) | PV-0280 | Nicotiana benthamiana | MW582795-97 |
| Tymoviridae | Tymovirus | Turnip yellow mosaic virus | TYMV | ssRNA(+) | PV-0299 | Brassica rapa | ON924209 |
| Virgaviridae | Furovirus | Soil-borne wheat mosaic virus | SBWMV | ssRNA(+) | PV-0748 | Triticum aestivum | MZ405651-52 |
| Virgaviridae | Hordeivirus | Barley stripe mosaic virus | BSMV | ssRNA(+) | PV-0330 | Hordeum vulgare | ON924210-12 |
| Virgaviridae | Pecluvirus | Peanut clump virus | PCV | ssRNA(+) | PV-0291 | Nicotiana benthamiana | MW961156-57 |
| Virgaviridae | Pomovirus | Potato mop-top virus | PMTV | ssRNA(+) | PV-0582 | Nicotiana benthamiana | ON398500-02 |
| Virgaviridae | Tobamovirus | Paprika mild mottle virus | PaMMV | ssRNA(+) | PV-0606 | Nicotiana benthamiana | OK181768 |
| Virgaviridae | Tobravirus | Pea early-browning virus | PEBV | ssRNA(+) | PV-0298 | Chenopodium quinoa | MW854268-69 |
| Not assigned | Idaeovirus | Raspberry bushy dwarf virus | RBDV | ssRNA(+) | PV-0053 | Chenopodium quinoa | MW582777-78 |
| Rhabdoviridae | Cytorhabdovirus | Lettuce necrotic yellows virus | LNYV | ssRNA(−) | PV-0085 | Nicotiana glutinosa “24A” | MZ202327 |
| Rhabdoviridae | Varicosavirus | Beet oak leaf virus | BOLV | ssRNA(−) | PV-1034 | Spinacia oleracea | OQ975887-88 |
| Rhabdoviridae | Alphanucleorhabdovirus | Physostegia chlorotic mottle virus | PhCMoV | ssRNA(−) | PV-1182 | Nicotiana occidentalis “37B” | KX636164 |
| Rhabdoviridae | Betanucleorhabdovirus | Sonchus yellow net virus | SYNV | ssRNA(−) | PV-0052 | Nicotiana clevelandii | MT613317 |
| Aspiviridae | Ophiovirus | Lettuce ring necrosis virus | LRNV | ssRNA(−) | PV-0983 | Nicotiana occidentalis “P1” | ON398506-09 |
| Partitiviridae | Alphacryptovirus | Poinsettia latent virus | PnLV | dsRNA | PV-0629 | Euphorbia pulcherrima | ON398503 |
| Caulimoviridae | Badnavirus | Banana streak OL virus | BSOLV | dsDNA-RT | PV-0492 | Musa sp. | OQ102041 |
| Caulimoviridae | Caulimovirus | Cauliflower mosaic virus | CaMV | dsDNA-RT | PV-0229 | Brassica rapa | OP947586 |
| Geminiviridae | Begomovirus | Squash leaf curl virus | SLCV | ssDNA | PV-1299 | Cucurbita pepo | MW582809-10 |
| Geminiviridae | Mastrevirus | Maize streak virus | MSV | ssDNA | PV-1103 | Zea mays | OQ102042-44 |
| Nanoviridae | Babuvirus | Banana bunchy top virus | BBTV | ssDNA | PV-1166 | Musa sp. | OQ102052-57 |
DSMZ catalog code.
Host in which the virus isolate was propagated and lyophilized.
Several variants are present in the propagated sample and accession numbers for the variants are provided.
The taxonomic status of the various viruses is indicated, together with their DSMZ catalog code, their propagation host, and the GenBank accession number(s) of their genomic sequence(s).
Initial low-complexity pools were generated by assembling 30 mg of virus-infected samples into 12 viral communities comprising five viruses each (150 mg of plant material each) and containing at least one virus with a genome type different from +ssRNA (Table S1). Pea enation mosaic virus was counted as one virus, when it is in fact a co-infection of pea enation mosaic virus 1 (Enamovirus) and pea enation mosaic virus 2 (Umbravirus). Stepwise combinations of these five-viruses mock communities were then assembled to create communities of increasing degrees of complexity (Fig. S1), yielding a total of 22 communities with complexity ranging from 5 to 60 viruses.
Double-stranded RNA extraction
Double-stranded RNAs were purified from pooled samples according to reference (41) with some minor modifications. Briefly, instead of 75 mg, 150 mg dried plant material (representing a pool of five plants, Table S1) was used as starting material and buffer volumes increased proportionally. Plants were ground in liquid nitrogen until a fine powder was obtained which was then mixed with the phenol-extraction buffer. Following gentle agitation for 30 min and centrifugation, the supernatant was decanted and half of it was directly further processed, while the other half was used for the stepwise gradual assembly of pairs of communities used to generate more complex viral communities. In this way, six communities of 10 viruses each, then three communities of 20 viruses, and, finally, a single community of 60 viruses could be assembled. Between each step, assembled samples were vortexed for at least 30 s for optimal homogenization. A detailed scheme of the pooling strategy to form communities of different complexities is shown in Fig. S1. Irrespective of its complexity, a supernatant volume corresponding to an initial input of 75 mg of plant sample was thus obtained and further processed as per the protocol of Marais et al. (41) which involves two rounds of CC41 cellulose (Whatman) chromatography followed by a nuclease treatment (DNase RQ1 plus RNaseA under high salt conditions) to remove any remaining host DNA and single-stranded RNA. A negative extraction control using only buffer was systematically included. Purified dsRNAs were finally converted to cDNA and randomly amplified while simultaneously adding MID (multiplex identifier) tags (41, 42).
VANA extraction
VANA extractions were performed on pools of five viruses similarly prepared as for dsRNA, using the protocol of François et al. (42) with minor modifications. Briefly, 150 mg of lyophilized plant material (representing a pool of five plants, Table S1) was ground in Hank’s buffered salt solution (1:10) with four metal beads within a grinding machine (Fastprep 24, MP Biomedicals). Following two centrifugation steps (4,000 g for 5 min at 4°C and 8,000 g at 4°C for 3 min), the supernatants were split and used in the same stepwise assembly of more complex communities as for the dsRNA approach (Fig. S1). A negative, buffer-only, extraction control was systematically included. Each of the thus generated samples, representing different degrees of community complexity, was filtered through a 0.45 µm filter and centrifuged at 148,000 g for 2.5 hours at 4°C to concentrate the virus particles. Unprotected nucleic acids were eliminated by DNase and RNase treatment at 37°C for 1.5 hours. Viral RNA and DNA were then isolated using the NucleoSpin Virus kit (Macherey Nagel, Hoerdt, France), using only 80 µL of sample in the first lysis step and omitting the addition of proteinase K. Extracted RNAs were transformed to cDNA using Superscript III reverse transcriptase (ThermoFisher Scientific/Invitrogen), cDNAs were further purified with the QIAquick PCR purification Kit (Qiagen, Courtaboeuf, France), and a complementary strand was synthesized using the Klenow fragment of DNA polymerase I. Finally, a random PCR amplification adding barcoded dodeca linkers and corresponding MID primers during reverse transcription and PCR, respectively, was performed (42).
Illumina sequencing
PCR products from all communities analyzed using the dsRNA and VANA procedures were finally purified using the MinElute PCR purification kit (Qiagen) and equimolar quantities of amplification products were sent to Illumina sequencing in multiplexed format (2 × 150 bp) on two lanes (one for VANA and one for dsRNA, respectively) on a NovaSeq 6000 system at the GetPlaGe platform (GenoToul INRAE Toulouse, France).
Generation of synthetic datasets for viral communities using single-isolate RNASeq data
For all but one of the viral isolates used to build the synthetic communities, available single-isolate ribodepleted RNASeq data sets (Leibniz-Institute DSMZ) were used to reconstruct in silico data sets corresponding to the different communities with read number and average reads length paralleling those from the VANA and dsRNA datasets. These reconstructed datasets, mimicking the analysis of the various communities by RNASeq, were analyzed in parallel to those generated by the VANA and dsRNA approaches.
HTS data analysis
Sequencing reads were imported into CLC Genomics Workbench v. 21.0.3. (CLC-GW, Qiagen) and adapters were removed from reads followed by trimming on quality and length using default settings and a minimum read length of 60 nucleotides (nt). Final trimmed reads were on average 111–113 nt long for the various datasets. Datasets were normalized by resampling at varying depth as needed, using the random reads sampling tool in CLC-GW.
To analyze virus detection performance as a function of contig size, de novo assembly was performed with CLC-GW (word size, 50; bubble size, 300) using various minimum contig lengths (125, 175, 250, 350, 500, 1000 nt). In order to identify viruses possibly present in the samples used, in addition to the expected reference viruses, contigs were annotated by a BlastX analysis (43) against the viral RefSeq portion of the non-redundant (nr/nt) National Center for Biotechnology Information (NCBI) GenBank database. For the additional viruses thus identified, a genomic scaffold was reconstructed and extended by repeated rounds of residual reads mapping using CLC-GW, thus yielding near-complete genome sequences that were used as reference for the relevant virus (Table 2). In a few cases, these assemblies were considered too incomplete and the closest complete genomic sequence in GenBank was selected as reference sequence (Table 2).
TABLE 2.
Additional viruses identified by analysis of the HTS data in the samples used to assemble the synthetic mock communities of varying complexity
| Family | Genus | Virus | Acronym | Genome type | Reference sequence accession number a |
|---|---|---|---|---|---|
| Virgaviridae | Tobamovirus | Tobacco mosaic virus | TMV | ssRNA(+) | OQ953825 |
| Tymoviridae | Unassigned | Poinsettia mosaic virus | PnMV | ssRNA(+) | OQ953828 |
| Endornaviridae | Alphaendornavirus | Hordeum vulgare endornavirus | HvEV | ssRNA(+) | OQ953829 |
| Solemoviridae | Polerovirus | Turnip yellows virus | TuYV | ssRNA(+) | JQ862472 |
| Geminiviridae | Mastrevirus | Maize streak Réunion virus | MSRV | ssDNA | OQ953826 |
| Totiviridae | Unassigned | Maize-associated totivirus | MATV | dsRNA | OQ953827 |
| Totiviridae | Unassigned | Maize-associated totivirus 2 | MTV-2 | dsRNA | MN428829 |
| Mitoviridae | Duamitovirus | Chenopodium quinoa mitovirus 1 | CqMV1 | ssRNA(+) | MT089917 |
| Small linear ssRNA satellite | Turnip crinkle satellite RNA F | TCVsatRNA F | ssRNA | X12749 | |
| Small linear ssRNA satellite | Pea enation mosaic virus satellite RNA | PEMVsatRNA | ssRNA | OQ953831 | |
| Small linear ssRNA satellite | Strawberry latent ringspot virus satellite RNA | SLRSVsatRNA | ssRNA | OQ953830 | |
Accession number of the closest sequence in GenBank that was used as reference for reads mapping.
In order to determine virus detection performance, unassembled reads or de novo assembled contigs were mapped against the reference genome segment(s) for each virus (Tables 1 and 2) using very stringent mapping parameters (length fraction 100%, minimal similarity fraction 90%) in CLC-GW. In order to take into account inter-sample crosstalk due to index hopping (44, 45), a threshold of positive detection was computed for each viral molecule by calculating the average plus 3 standard deviations (SD) of background virus reads observed in libraries generated from communities that did not contain the corresponding virus. Assuming a normal distribution of background reads, the use of such a positivity threshold would provide a <1% risk of reporting a false-positive detection (https://en.wikipedia.org/wiki/68%E2%80%9395%E2%80%9399.7_rule).
Comparison of parameters (number, average length) for de novo assembled viral contigs obtained from VANA and dsRNA data sets normalized at different sequencing depths was performed with five resampling repeats at each depth. Statistically significant differences were identified using a two-sample t-test.
RESULTS
Viruses or virus-like agents identified from viral communities HTS data
The analysis of reads from both the VANA and dsRNA approaches for all communities revealed the presence of all expected viruses, although a few viruses were only represented by a limited number of reads or were only detected using one of the two approaches. Overall, only lettuce ring necrosis virus turned out to be fully absent from VANA reads, while banana bunchy top virus was only represented by a single dsRNA read. It should also be noted that not all viruses could be detected in all the communities of different complexity in which they were expected.
In addition to the expected 61 viruses, evidence for the presence in some communities of additional viruses or virus-like agents was obtained through the BlastX indexing of de novo assembled contigs from the low-complexity, five-viruses communities. A total of 11 unexpected agents were thus identified (Table 2). These include three linear ssRNA satellites associated with the helper virus isolates included in the communities (turnip crinkle satellite F, pea enation mosaic satellite RNA, and strawberry latent ringspot virus satellite RNA), latent viruses associated with the propagation hosts used (Hordeum vulgare endornavirus, maize-associated totivirus, maize-associated totivirus2, and Chenopodium quinoa mitovirus 1), as well as viruses in co-infection with some of the viral isolates used (poinsettia mosaic virus, tobacco mosaic virus, turnip yellows virus, and maize streak Réunion virus) (Table 2). Taken together, these agents represent three additional viral families, for a total of 21 viral families (plus satellites) used for the assembly of communities. For these additional agents, either a nearly complete genome was reconstructed from sequencing reads and used as the mapping reference or the closest full genome sequence in GenBank was used for further mapping analyses (Table 2). For all other viral isolates included in the communities, complete or nearly complete genomic sequences were available (Table 1).
While the communities of varying complexities analyzed here will be referred to as 5-viruses, 10-viruses, 20-viruses, and 60-viruses, it should be kept in mind that the real number of viruses present in a given community might be slightly different because of (i) the presence of one or more of the additional viruses and (ii) the counting of pea enation mosaic virus as one virus when it is in fact a co-infection of pea enation mosaic virus 1 (Enamovirus) and pea enation mosaic virus 2 (Umbravirus).
Read mapping analysis of VANA and dsRNA datasets for the communities of various complexities
To be able to compare results between low- and high-complexity communities, all datasets were normalized by randomly subsampling 120K cleaned reads, the depth of the five-viruses community with the lowest number of reads. To address the issue of inter-sample crosstalk caused by index jumping (44, 45), a threshold of positive detection was computed for each viral molecule by calculating the average + 3 standard deviations (SDs) of background reads in libraries generated from communities that did not contain the corresponding virus. Assuming a normal distribution of crosstalk read numbers, this strategy ensures that the probability of having a mapped read number higher than the threshold by chance (false-positive detection) is lower than 1%.
In general, the proportion of viral reads in both VANA and dsRNA datasets was high (64%−89%) and was slightly affected by community complexity, with a general trend to reach higher values when analyzing more complex communities (Fig. 1A). The proportion of viral reads in the dsRNA datasets was slightly higher than in the corresponding VANA datasets, with the strongest differential observed for lower-complexity communities of 5 and 10 viruses (64%−65% viral reads as compared to 79%−82%, Fig. 1A). In contrast, the average proportion of viral reads in RNASeq datasets for individual virus isolates following ribodepletion was 19.6% but with a very large standard deviation of 26.1%.
Fig 1.

Average proportion of viral reads (DNA and RNA viruses) in VANA (gray) and dsRNA (blue) data sets from viral communities of different complexities (A) and number of viruses detected at an even 120K reads depth for communities of different complexities (B). In Fig. 1B, RNA viruses are indicated by solid bars while DNA viruses are indicated by dashed bars.
Using the 12 communities of five viruses and a sequencing depth of 120K reads, 67 viruses were detected with both VANA and dsRNA approaches (with detection of reads for at least one genomic molecule considered as positive detection for a virus with a multipartite genome), out of the total of 72 viruses or virus-like agents present in the 12 communities analyzed (93.1%). However, VANA yielded reads for all six DNA viruses used (100%), while dsRNA yielded reads for only three of them (50%). Conversely, VANA yielded reads for 61 of the 66 RNA viruses or satellites (92.4%), when dsRNA yielded reads for 64 of them (97.0%) (Fig. 1B). As expected, and previously reported, the performance of VANA is thus superior for DNA viruses but that of dsRNA is slightly superior for RNA viruses. Using the data sets reconstructed from single plant RNASeq data, an overall rate of detection of 97.2% of the 71 viruses was obtained (no RNASeq data were available for one of the isolates used, which was therefore excluded from all computations).
The impact of increasing community complexity is reflected by the diminishing number of viruses detected at an equal sequencing effort of 120K reads. The performance of VANA gradually deteriorated, with detection decreasing from 61 RNA viruses detected to 58 (10-viruses communities) and then to 52 (20-viruses communities) to reach only 34 RNA viruses detected (51.5%) in the most complex community (Fig. 1B). The same pattern was observed for DNA viruses, with all six DNA viruses detected in the 10- and 20-viruses communities but only one detected when analyzing the 60-viruses community. In the case of the dsRNA approach, performance was marginally reduced for the 10- and 20-viruses communities (65 and 63 RNA viruses detected, respectively) and less affected than for the VANA approach for the most complex community, with still 57 of 66 RNA viruses detected (86.4%) (Fig. 1B). Remarkably, performance was the least affected for the RNASeq approach using reconstructed communities data, with still 65 viruses (91.5%) detected for the most complex community (5/6 DNA viruses and 59/65 RNA viruses, or 90.7%).
If trying to compensate for community complexity by proportionally increasing the sequencing effort for more complex communities, the erosion in performance is less important for VANA, with still 57 of 66 RNA viruses detected for the 60-viruses community (86.4%) and five of the six DNA viruses (83.3%) at a 1.44 M reads depth (12 × 120K). The performance of dsRNA, on the other hand, is no longer impaired, as all 66 RNA viruses (100%) were detected for the most complex community (result not shown). Similarly, the performance of RNASeq was no longer substantially impacted, with all DNA viruses and all but one RNA viruses detected.
The stronger degradation of VANA performance, as community complexity increases, correlates with a more uneven distribution of read numbers between viruses and the stronger dominance of a few viruses, in particular turnip yellow mosaic virus (TYMV). In the 60-viruses community VANA dataset, TYMV represented 67% of the reads while the corresponding value for the dsRNA dataset was only 28%. As shown in Fig. 2, even if spanning a 5 to 6 logs scale, the percentage of reads for each virus in the total datasets tends to be more evenly distributed between viruses in the dsRNA dataset than in the VANA data set for the 60-viruses community. By contrast and excluding a single sample showing extremely low viral read numbers, the variation in the proportion of viral reads in individual viral isolates analyzed by RNASeq showed much less variability as it remained within a 3 logs range of variation.
Fig 2.
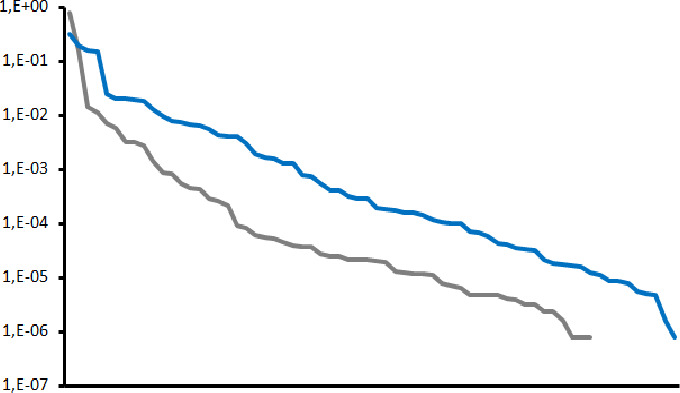
Distribution of percentage of mapped VANA (gray) and dsRNA (blue) reads for each detected virus in the 60-viruses community using a normalized 1.44 million reads sequencing depth. The percentages of mapped reads for each virus are shown on a logarithmic scale, from 1,E + 00 (100%) to 1,E − 07 (0.000001%).
Although allowing to compare the performance of the VANA and dsRNA approaches, these analyses based on the mapping of reads against cognate reference genomes do not mimic the situation in metagenomic studies, in which a high proportion of viruses are expected to be novel and for which therefore no suitable reference genome is available. We therefore analyzed the performance of these two approaches following the de novo assembly of reads into contigs, which is known to reduce the proportion of un-annotated “dark matter” (46).
Impact of minimal contig length on the number of detected viruses
We first evaluated the impact of the minimal contig length on the number of detected viruses using the most complex community of 60 viruses and deep data sets normalized at 10M reads. As expected, and shown in Fig. 3, the number of detected viruses decreased as minimal contig length increased. The pattern observed for RNA viruses is similarly observed for DNA viruses. The dsRNA approach consistently detected more RNA viruses than the VANA one, irrespective of the minimal contig length used, but the difference increased as minimal contig length increased. Using the shortest, 125 nt contig length, VANA identified 54 of the 66 RNA viruses or satellites present in the community (81.8%), while dsRNA identified 63 of them (95.5%) (Fig. 3). The corresponding values for DNA viruses are, respectively, 4/6 (66.7%) and 3/6 (50%).
Fig 3.
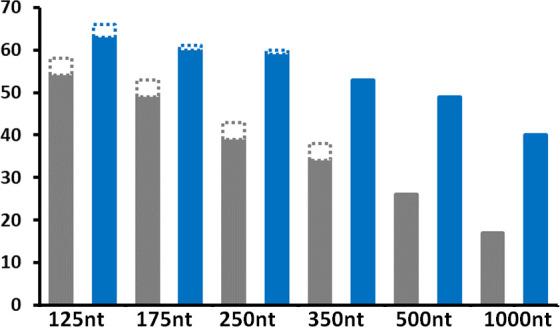
Number of detected viruses using VANA (gray) or dsRNA (blue) in the 60-viruses community (over a total of 69 viruses plus three satellites) as a function of minimal contig length at a sequencing depth of 10M reads. RNA viruses are indicated by solid bars while DNA viruses are indicated by dashed bars.
On the other hand, the coverage of the detected viruses (fraction of the target molecules represented in contigs) was much less affected by minimal contig length. While being relatively stable for the dsRNA approach, for which it varied between 66.5% and 74.9% with no clear trend, it showed a tendency to increase with contig length for the VANA approach, from 50.2% (>125 nt contigs) to 76.7% (>1,000 nt contigs) (Fig. S2).
For further analyses, an intermediate 250 nt minimal contig length was retained as it corresponds to an encoded 83 amino acid sequence that was felt sufficient for many conserved protein domain searches which are often used in virome analysis or annotation (47).
Effects of community complexity on virome description performance
We evaluated how, for a given sequencing depth, community complexity affects virome description performance following contigs assembly. For this, all data sets were normalized at a 120K reads depth. Similar to the initial analysis using reads mapping, the number of detected viruses was reduced as community complexity increased. Again, dsRNA outperformed VANA at all complexity levels, though the difference in performance remained limited for low to medium community complexities (Fig. S3). VANA performance degradation was, however, more drastic at high community complexity, dropping from 44 RNA viruses and 4 DNA viruses detected for communities of five viruses (66.7% of total viruses) to 11 RNA viruses and 1 DNA virus detected (16.7%) for the 60-viruses community. The corresponding values for dsRNA were 53 (80.3%) and 26 RNA viruses (39.4%), with no DNA virus detected (Fig. S3). Remarkably, RNASeq turned out to be the least affected, with, respectively, 57/71 (80.3%, five-viruses communities) and 34/71 viruses (47.9%, 60-viruses community) detected. These results indicate that even for limited complexity communities involving only five viruses, read numbers significantly higher than 120K are needed by the various techniques to achieve a 100% detection performance with a wide range of viruses.
If trying to compensate increased virome complexity by a parallel increase in sequencing depth, a negative impact of complexity is still seen but is much less severe. For example, for the most complex community of 60 viruses at a 1.44M depth (12 * 120K reads), VANA detected 23 RNA viruses and 2 DNA viruses (compared to 44 RNA viruses and 4 DNA viruses when analyzing individually the 12 pools of five viruses at 120K reads depth), which corresponds to a reduction in performance of 47.9%. For its part, dsRNA detected 42 RNA viruses (no DNA virus), to be compared with 53 viruses when individually analyzing the 12 pools of five viruses, corresponding to a reduction in performance of 20.7% (Fig. S4). The corresponding value for RNASeq was 55 viruses detected, corresponding to a performance equivalent to the analysis of the 12 communities of five viruses. The loss in performance resulting from high community complexity is therefore only significant for the dsRNA and VANA approaches, and strongest in the case of VANA.
Impact of sequencing depth on de novo assembly
The 60-viruses community was used to investigate the influence of sequencing depth on de novo assembly performance itself. The VANA and dsRNA data sets were therefore resampled at different depths (100K, 300K, 1M, 3M, and 10M reads, five random resampling at each depth) and assembled, and the obtained contigs mapped against the viral reference genomes to determine the average assembly parameters and viral contigs parameters. The results are shown in Table S2 and, for viral contigs alone, in Table 3.
TABLE 3.
Comparison of the number and average length of de novo assembled viral contigs obtained for VANA and dsRNA data sets normalized at different sequencing depths (100K, 300K, 1M, 3M, and 10M reads, five resampling repeats at each depth) a
| VANA average ± SD | dsRNA average ± SD | Two sample t-test | ||
|---|---|---|---|---|
| 100K reads | nb viral contigs | 33.6 ± 1.9 | 101.8 ± 2.9 | 9.2E-11 |
| Viral contigs average length | 733.4 ± 23.7 | 747.4 ± 17.1 | 0.32 | |
| 300K reads | nb viral contigs | 70.2 ± 5.4 | 129.4 ± 8.1 | 8.0E-07 |
| Viral contigs average length | 643.4 ± 27.8 | 887.8 ± 38.2 | 2.8E-06 | |
| 1M reads | nb viral contigs | 106.2 ± 6.3 | 159.2 ± 6.6 | 1.1E-06 |
| Viral contigs average length | 694.8 ± 30.3 | 1019.6 ± 40.9 | 5.7E-07 | |
| 3M reads | nb viral contigs | 129.6 ± 4.8 | 207.6 ± 3.8 | 2.5E-09 |
| Viral contigs average length | 798.4 ± 15.9 | 1067.6 ± 11.5 | 1.4E-09 | |
| 10M reads | nb viral contigs | 201.2 ± 4.1 | 268 ± 2.9 | 1.8E-09 |
| Viral contigs average length | 791.2 ± 11.1 | 1121.4 ± 10.6 | 3.9E-11 |
The SDs and the statistical differences (P-values) are also shown.
As expected, all assembly parameters (number of contigs, average contig length, N50, maximal contig length) increased with sequencing depth (Table S2). The same tends to be true for viral contigs (number and length, Table 3), while the proportion of viral contigs tended to diminish as sequencing depth increased, likely reflecting increased probability of assembly of non-viral reads (Table S2). Although at the lowest 100K reads sequencing depth, few assembly parameters were found to be statistically different, both the total number of assembled contigs and the number of viral contigs were found to be highly statistically different, with dsRNA yielding about threefold more contigs and threefold more viral contigs than VANA (Table 3 and Table S2). This trend was observed at all sequencing depth, with 1.3- to 1.8-fold more viral contigs observed for dsRNA.
At other sequencing depths, differences between the VANA and dsRNA assemblies proved systematically highly significant, with dsRNA consistently yielding more numerous and longer contigs as well as more numerous and longer viral contigs. On the other hand, the proportion of viral contigs was found consistently higher in assemblies of the VANA data sets (Table S2).
It should be noted that the better assembly performance of dsRNA is independent of minimal contig length (Table 4). In particular, using the most complex community and 10 million reads data sets, the higher performance of dsRNA over VANA was observed for all assembly parameters (number of contigs, average length, N50, maximum length) and for both viral contigs parameters (number and average length) at all minimal contigs length (from 125 to 1,000 nt) with a single exception, the number of viral contigs >125 nt long (1,852 for VANA vs 1,672 for dsRNA) (Table 4). At all other minimal contig lengths, VANA showed from 19.2% (contigs ≥ 175 nt) to 50.7% (>1 kb contigs) fewer viral contigs than dsRNA and these contigs were 23%−33% shorter on average than the dsRNA ones (Table 4).
TABLE 4.
Performance parameters of de novo assembly using different minimal contigs length of normalized, 10M reads, VANA, and dsRNA data sets for the 60-viruses synthetic community
| Minimal contig length | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 125 nt | 175 nt | 250 nt | 350 nt | 500 nt | 1,000 nt | |||||||
| VANA | dsRNA | VANA | dsRNA | VANA | dsRNA | VANA | dsRNA | VANA | dsRNA | VANA | dsRNA | |
| nb contigs | 1947 | 2212 | 416 | 784 | 220 | 437 | 144 | 276 | 86 | 182 | 37 | 88 |
| Average length | 235 | 324 | 506 | 607 | 757 | 907 | 985 | 1243 | 1355 | 1662 | 2191 | 2696 |
| N25 | 547 | 1764 | 1836 | 3642 | 2334 | 4007 | 2560 | 4060 | 3449 | 4505 | 3824 | 5671 |
| N50 | 206 | 352 | 628 | 1005 | 994 | 1521 | 1277 | 1955 | 1709 | 2775 | 2277 | 3705 |
| N75 | 156 | 191 | 313 | 352 | 481 | 558 | 618 | 773 | 888 | 1117 | 1653 | 1782 |
| Max | 6549 | 13919 | 6652 | 13919 | 6652 | 13919 | 6549 | 13919 | 6652 | 13919 | 6652 | 13919 |
| nb viral contigs | 1852 | 1672 | 378 | 468 | 204 | 269 | 137 | 181 | 84 | 131 | 37 | 75 |
| % viral contigs | 95% | 76% | 91% | 60% | 93% | 62% | 95% | 66% | 98% | 72% | 100% | 85% |
| Viral contigs average length | 235 | 327 | 525 | 741 | 783 | 1123 | 1008 | 1508 | 1368 | 1921 | 2191 | 2833 |
| Bases in viral contigs | 435421 | 547507 | 198486 | 347003 | 159827 | 302074 | 138102 | 272883 | 114951 | 251656 | 81077 | 212467 |
| % bases in viral contigs | 95.20% | 76.40% | 94.40% | 72.90% | 96.00% | 76.20% | 97.40% | 79.50% | 98.60% | 83.20% | 100% | 89.50% |
As compared to VANA and dsRNA assemblies, RNASeq assemblies generated more viral contigs at low sequencing depth (ca. 10%−30% more than dsRNA for depth of 100K to 1M reads) but ca. 15% fewer viral contigs at the 10M depth. On the other hand, a striking difference in the length of viral contigs was also observed, with RNASeq contigs increasing from an average of 1 kb (100K depth, 34% longer than dsRNA contigs on average) to 2.1 kb (10M depth, 89% longer than for dsRNA).
Impact of sequencing depth on virus identification performance
We proceeded to evaluate the performance of VANA and dsRNA in identifying the expected viruses or viral molecules as affected by sequencing depth. The contigs obtained for the various data sets resampled at different depths (five resampling per sequencing depth) were mapped on individual reference sequences. This allowed to evaluate both the proportion of detected viruses and the coverage of the detected viral molecules, together with their standard deviation (Fig. S5). Once again, at all sequencing depths and for both parameters, dsRNA outperformed VANA for RNA viruses, while VANA outperformed dsRNA for DNA viruses. In all cases, average coverage of detected segments of RNA viruses showed a high standard deviation but dsRNA contigs covered 9% to 22% more of the detected molecules than VANA contigs.
Similarly, and as expected from single reads mapping data, dsRNA outperformed VANA for the identification of RNA viruses present in the most complex, 60-viruses community. For VANA, performance ranged from 17.7% of RNA viruses identified at the 100K reads depth to 60.3% at the 10 million reads depth. The corresponding values for dsRNA are, respectively, 35.2% and 89.7% and those for RNASeq are, respectively, 46.2% and 90.8%. The performance of RNASeq therefore appears to be nearly identical to that of dsRNA for RNA viruses, and superior for DNA viruses with 5/6 viruses detected for the 3M and 10M reads depth.
A plot of the observed proportion of detected RNA viruses over a logarithmic scale of the sequencing effort is shown in Fig. 4. It shows a remarkable pattern with linear regression r² coefficients of 0.97–0.99, suggesting a very strong and monotonous relationship between sequencing depth and the proportion of the viruses present in the community that are represented by at least one assembled contig. An extension of that trend would suggest that a depth of about 30 million reads would be needed for the dsRNA approach to recover at least one contig for each of the 66 RNA viruses present in the synthetic community, while in excess of 1 billion reads would be needed to achieve a comparable performance using VANA. If taking into account also DNA viruses to calculate a proportion of detected viruses, similar linear relationships are still observed, but the performance of the dsRNA approach is slightly degraded as expected from its poor ability to detect DNA viruses (Fig. 4). Analyzed in a similar fashion, the RNASeq data showed the same linear relationship, although with a slightly lower r² value of 93.7% and a predicted detection of all 71 viruses and satellites with 16–17M reads.
Fig 4.
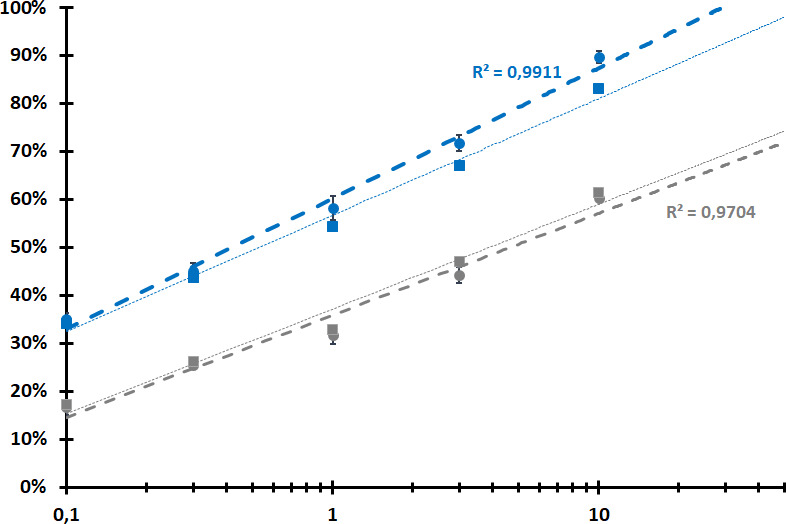
Observed percentages of detected viruses in the 60-viruses community as a function of sequencing depth expressed in million reads per sample and plotted on a logarithmic scale. VANA results are in gray, dsRNA results are in blue. Linear regression curves are shown for RNA viruses (round dots, thick lines,) as well as considering both RNA and DNA viruses (square dots, think lines). Linear r² coefficients are shown only for the RNA virus curves.
Due to a more limited number of reads available for virus communities up to the 20-viruses pools, a similar evaluation could not be as extensively performed for these lower complexity communities. However, an analysis at three sequencing depths (100K reads, 300K reads, 875K reads) of the 20-viruses communities data provided comparable results with r² correlation coefficients of 0.95–0.98, suggesting that the linear correlation between the percentage of viruses recovered and the log of the sequencing depth is independent of the complexity of the analyzed community (result not shown).
An analysis performed at the level of individual viral genomic molecules (115 viral molecules) allows to evaluate the performance of the two methods using the most complex, 60-viruses pool, for groups of viruses with different genome types. The numbers of viral molecules are, however, small for RNA satellites, dsRNA viruses, and dsDNA viruses. The results, using a 10 million reads sequencing depth, are summarized in Table 5. Considering individual molecules, VANA had at least one contig for only 50% of the viral molecules present in the most complex synthetic community, to be compared with a 76.5% value for dsRNA. But while the VANA performance was at an intermediate level for all virus groups analyzed, dsRNA showed good performance for +ssRNA viruses (89.5% of molecules), RNA satellites (100%), and dsRNA viruses (100%). The dsRNA performance was, however, poor for DNA viruses, as expected, but also for −ssRNA viruses (41.7% of detected molecules only).
TABLE 5.
Detection performance of VANA, dsRNA, and RNASeq methods at the level of individual viral genomic molecules (from a total of 115 viral molecules) using the most complex, 60-viruses pool, for groups of viruses with different genome types at 10M reads sequencing depth
| # viral molecules | VANA | dsRNA | RNASeq | ||||
|---|---|---|---|---|---|---|---|
| # detected | % detected | # detected | % detected | # detected | % detected | ||
| +ssRNA viruses | 86 | 50 | 58.1% | 77 | 89.5% | 81 | 96.,3% |
| ssRNA viruses | 12 | 1 | 8.3% | 5 | 41.7% | 12 | 100% |
| RNA satellites | 3 | 1 | 33.3% | 3 | 100% | 2 | 66.,6% |
| dsRNA viruses | 2 | 0 | 0% | 2 | 100% | 0 | 0,0% |
| ssDNA viruses | 10 | 4 | 40.0% | 0 | 0% | 4 | 40.0% |
| dsDNA viruses | 2 | 1 | 50.0% | 1 | 50.0% | 1 | 50.0% |
| Total | 115 | 57 | 49.6% | 88 | 76.5% | 100 | 87.7% |
DISCUSSION
While synthetic communities have been widely used to benchmark metagenomic processes targeting bacteria and fungi, methodological benchmarking approaches in virome studies are still limited and largely confined to clinical settings (38, 48, 49) and, to some extent, to environmental virome studies (50, 51). Such approaches are today largely lacking in plant virology. Here, we used well-authenticated and sequence-characterized plant virus isolates from a public bioresource center (Leibniz-Institute DSMZ) that allowed for the simple construction of synthetic viral communities of varying complexity. Although some of the viruses were detected by only very low read numbers, no virus was fully absent from all generated data sets, validating the approach and the samples used. The fact that some viruses were identified only by low read numbers could have a variety of reasons, such as low virus titer in some samples, competition with other viruses for reads representation in the assembled communities, or difficulties in extracting viral nucleic acids from some plant species. In addition, the fact that freeze-dried plant material was used in this study may have had a negative impact on results and the analysis of fresh plant tissues might have provided superior results. In this respect, it should be noted that the two viruses present as infected banana samples, banana streak OL virus (BSOLV) and banana bunchy top virus (BBTV), were only detected by very low read numbers using both VANA and dsRNA, despite the fact that these techniques have successfully been used in the past to analyze banana samples (52, 53). The RNASeq data on the same viral isolates shows about 0.9% of viral reads for BSOLV, but BBTV was the individual sample with the fewest reads by far in the RNASeq analysis, suggesting a low viral concentration in that particular sample.
A total of 11 additional viruses or viral agents were identified in the constructed communities. In most cases, these correspond to satellites that had not been specifically indexed in the viral isolates used or of viruses latently infecting propagation hosts, such as Hordeum vulgare endornavirus, which is present in many barley varieties, or Chenopodium quinoa mitovirus.
The communities assembled cover all known plant virus genome types, 21 viral families (plus satellites and one virus unassigned in a family), and a total of 61 genera (plus four viruses not currently assigned to a genus and three satellites). It is thus, probably to date, the largest scale effort to build synthetic viral communities and use them for the benchmarking of phytovirome analysis approaches. In some benchmarking studies, the nucleic acid proportions of the individual viruses involved in the virus community were quantified prior to extraction (36, 39). The fact that no special effort was made here to normalize or measure the concentration of the different viruses is a limitation for some comparisons. On the other hand, the samples used involved different propagation hosts and actual virus titers in those hosts, so that the communities assembled reflect actual samples from plant virome studies. The results obtained indicate that a range of parameters impact the completeness of the virome description achieved. Not surprisingly, such parameters include (i) sequencing depth, (ii) community complexity, (iii) use of de novo assembled contigs vs use of unassembled reads, and (iv) minimal contig length.
The key objective of this work was to compare the performance of the VANA and dsRNA approaches, which are the two techniques most widely used in ecology-oriented viral metagenomics experiments involving the analysis of complex pools of plants. The results provided here for RNASeq following ribodepletion should be considered with caution, since they are not fully comparable with the VANA or dsRNA data. Indeed, the RNASeq data sets for the various communities were assembled in silico, from data obtained by single-isolate sequencing. This means that any interactions between plant samples or competition between viruses for representation in the datasets were eliminated, contrary to the situation with the VANA and dsRNA experiments. Given that RNASeq is considered an unbiased approach (hence its use for transcriptome analysis), this should not be a problem but the existence of unforeseen effects affecting the results cannot be completely ruled out. As compared to dsRNA and VANA, the results obtained for RNASeq using the in silico assembled communities show (i) a much lower imbalance in the representation of the various viruses (3 logs variation as opposed to 5–6 logs), (ii) on average, significantly longer viral contigs, irrespective of sequencing depth, and (iii) an overall excellent performance with 90% of the viruses identified at 10M reads depth for the most complex, 60-viruses community. This last result favorably compares with the dsRNA performance for all viral categories with the exception of viruses with dsRNA genomes (Table 5). This performance comes as a surprise given the absence of enrichment (besides ribodepletion) in RNASeq. However, the relatively narrow range of variation in the proportion of viral reads for different viruses, possibly implying reduced competition for representation between viruses, and the even distribution of RNASeq reads along viral genomes, possibly favoring a more efficient genome assembly, could have contributed to the RNASeq performance. In any case, these results surprisingly suggest that RNASeq could have a very good potential for the analysis of complex viral communities and clearly call for direct benchmarking efforts using RNASeq and complex synthetic or natural communities in order to unambiguously validate this potential.
As previously reported using natural communities (32), the dsRNA approach provided in all comparisons a more complete description of the RNA virome than the VANA approach but performed very poorly with DNA viruses. However, the differential with VANA is more limited for the less complex communities of 5 or 10 viruses. According to our own experience, this level of complexity is most often seen when analyzing single plants or pools of 5–20 plants of the same species, with vegetatively propagated plants tending to have more complex viromes. Higher complexity levels are usually encountered when analyzing larger pools composed of plants belonging to different species. The dsRNA approach is therefore recommended whenever analyzing complex viromes or when an emphasis on RNA viruses is of importance, in particular since dsRNA allows comparable levels of completeness with a lower sequencing effort. On the other hand, for viromes of low to medium complexity, the results reported here show VANA to be a reasonable alternative. For example, at 480K reads depth, VANA detected 57.4% of all viruses for the 20-viruses communities as compared to 61.8% for dsRNA (result not shown, see also Fig. S4 for the compared rates of detection of RNA viruses only). VANA should of course be the preferred choice if analysis of DNA viruses is of importance. The reason for the better performance of the dsRNA approach for high-complexity viromes is not fully clear but might result from a lower level of competition between viral nucleic acid molecules for representation in complex pools, resulting in a somewhat less imbalanced distribution of read numbers between viruses (Fig. 2). Different human microbiome studies have shown that different steps of RNA/DNA extraction such as homogenization, centrifugation, filtration, and chloroform treatment can have a major impact on the quantitative and qualitative composition of identified viral communities, skewing viral metagenome assemblies (37, 38, 54). Another critical step is library preparation, which often involves a random amplification PCR to increase virus genetic material and to add linkers, allowing samples multiplexing during HTS sequencing and thus reducing sequencing costs. The amplification step may alter the relative abundance of viruses and can lead to uneven coverage if random primers do not anneal randomly on viral genomes. Indeed, in the case of faba bean necrotic stunt virus, the relative frequencies of the different genome segments determined by qPCR was significantly different before and after a rolling circle amplification step used prior to HTS sequencing (55). Furthermore, different library preparation techniques have been found to require different sequencing depths to achieve the same genome coverage (56). Regardless of the experiment, it is advisable to develop an estimate of the sequencing depth needed, so as to be able to answer the biological question at hand while avoiding excessive sequencing costs. Here, we identified a very robust correlation between the percentage of viruses identified in complex communities and the log of the sequencing depth. This is an interesting result, since it allows to gauge the sequencing effort needed for a particular level of virome description or, conversely, to gauge the extent of virome description that can be expected from a particular sequencing depth. Besides metagenomic studies, this finding might have practical implications for diagnostics since many plants, in particular vegetatively propagated ones, frequently display complex mixed infections involving a range of viruses.
Virus detection in metagenomic studies is constrained by the degree of complexity of the virus communities analyzed. Our results suggest that the detection efficiency of either mapping of unassembled reads or analysis of de novo assembled contigs was affected by community complexity with a general trend of detecting a lower proportion of viruses in more complex communities. However, the read mapping strategy was more efficient at all complexities (Fig. 1B and Fig. S3), confirming results obtained through performance testing of sequence analysis strategies (57). This may be due to the complexity of de novo assembly of complex communities, linked with insufficient coverage or uneven coverage of low abundance viruses within such communities. Correspondingly, we observed a lower virus detection rate when using longer minimal contig sizes in the de novo assembly, which again might be attributed to difficulties in assembling reads from more complex communities, for example, when coexisting viruses share highly similar regions in their genomes, leading to higher fragmentation and reduced contig sizes (58).
Lastly, it has been reported that the quality and completeness of virome description is also affected by the bioinformatic analysis used (58 – 61). The normalized 10M reads data sets generated in the present study with the 60-viruses community, which are available at https://doi.org/10.57745/T4UYPC, together with the community composition and the complete or near-complete reference genomic sequences used here should prove very useful tools to benchmark virome characterization pipelines.
ACKNOWLEDGMENTS
The authors wish to thank the INRAE GetPlaGe Platform (GenoToul,Toulouse, France) for Illumina sequencing.
This study was funded by the European Union through a Horizon 2020 Marie Skłodowska-Curie Actions Innovative Training Network (H2020 MSCA- 60 ITN) project “INEXTVIR” (GA 813542). The individual sequencing of some of the isolates used in this study was performed as part of the EVA-Global project which has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 871029.
O.M. is recipient of a PhD fellowship from CIRAD and the ANR (Phytovirus project number: ANR-19-CE35-0008-02).
Contributor Information
Thierry Candresse, Email: thierry.candresse@inrae.fr.
W. Allen Miller, Iowa State University, Ames, Iowa, USA .
ETHICS APPROVAL
This article does not contain any studies with human participants or animals performed by any of the authors.
DATA AVAILABILITY
Trimmed sequencing reads for all viral communities analyzed by dsRNA or VANA approaches are available from the French Recherche Data Gouv multidisciplinary repository at https://doi.org/10.57745/42WNRJ. The normalized 10M reads dsRNA or VANA data sets generated using the 60-viruses community have also been made available together with the community composition and the complete or near-complete reference genomic sequences used are also available from the same repository at https://doi.org/10.57745/T4UYPC.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.01300-23.
Fig. S1 to S5.
Tables S1 and S2.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Zhang Y-Z, Chen Y-M, Wang W, Qin X-C, Holmes EC. 2019. Expanding the RNA virosphere by unbiased metagenomics. Annu Rev Virol 6:119–139. doi: 10.1146/annurev-virology-092818-015851 [DOI] [PubMed] [Google Scholar]
- 2. Jian H, Yi Y, Wang J, Hao Y, Zhang M, Wang S, Meng C, Zhang Y, Jing H, Wang Y, Xiao X. 2021. Diversity and distribution of viruses inhabiting the deepest ocean on earth. ISME J 15:3094–3110. doi: 10.1038/s41396-021-00994-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefeuvre P, Martin DP, Elena SF, Shepherd DN, Roumagnac P, Varsani A. 2019. Evolution and ecology of plant viruses. Nat Rev Microbiol 17:632–644. doi: 10.1038/s41579-019-0232-3 [DOI] [PubMed] [Google Scholar]
- 4. Maclot F, Candresse T, Filloux D, Malmstrom CM, Roumagnac P, van der Vlugt R, Massart S. 2020. Illuminating an ecological blackbox: using high throughput sequencing to characterize the plant virome across scales. Front Microbiol 11:578064. doi: 10.3389/fmicb.2020.578064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roux S, Matthijnssens J, Dutilh BE. 2019. Metagenomics in virology. Reference module in life sciences. doi: 10.1016/B978-0-12-809633-8.20957-6:B978-0-12-809633-8.20957-6 [DOI] [Google Scholar]
- 6. Greninger AL. 2018. A decade of RNA virus metagenomics is (not) enough. Virus Res 244:218–229. doi: 10.1016/j.virusres.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moubset O, François S, Maclot F, Palanga E, Julian C, Claude L, Fernandez E, Rott P, Daugrois J-H, Antoine-Lorquin A, et al. 2022. Virion-associated nucleic acid-based metagenomics: a decade of advances in molecular characterization of plant viruses. Phytopathology 112:2253–2272. doi: 10.1094/PHYTO-03-22-0096-RVW [DOI] [PubMed] [Google Scholar]
- 8. Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, Chen IM, Ivanova N, Zeigler Allen L, Paez-Espino D, Bryant DA, Bhaya D, Krupovic M, Dolja VV, Kyrpides NC, Koonin EV, Gophna U, RNA Virus Discovery Consortium . 2022. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell 185:4023–4037. doi: 10.1016/j.cell.2022.08.023 [DOI] [PubMed] [Google Scholar]
- 9. Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, Wang W, Eden JS, Shen JJ, Liu L, Holmes EC, Zhang YZ. 2018. The evolutionary history of vertebrate RNA viruses. Nature 556:197–202. doi: 10.1038/s41586-018-0012-7 [DOI] [PubMed] [Google Scholar]
- 10. Wren JD, Roossinck MJ, Nelson RS, Scheets K, Palmer MW, Melcher U. 2006. Plant virus biodiversity and ecology. PLoS Biol 4:e80. doi: 10.1371/journal.pbio.0040080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi K, Atsumi G, Iwadate Y, Tomita R, Chiba K, Akasaka S, Nishihara M, Takahashi H, Yamaoka N, Nishiguchi M, Sekine K-T. 2013. Gentian Kobu-sho-associated virus: a tentative, novel double-stranded RNA virus that is relevant to gentian Kobu-sho syndrome. J Gen Plant Pathol 79:56–63. doi: 10.1007/s10327-012-0423-5 [DOI] [Google Scholar]
- 12. Schönegger D, Marais A, Faure C, Candresse T. 2022. A new flavi-like virus identified in populations of wild carrots. Arch Virol 167:2407–2409. doi: 10.1007/s00705-022-05544-1 [DOI] [PubMed] [Google Scholar]
- 13. Roossinck MJ, Martin DP, Roumagnac P. 2015. Plant virus metagenomics: advances in virus discovery. Phytopathology 105:716–727. doi: 10.1094/PHYTO-12-14-0356-RVW [DOI] [PubMed] [Google Scholar]
- 14. Maree HJ, Fox A, Al Rwahnih M, Boonham N, Candresse T. 2018. Application of HTS for routine plant virus diagnostics: state of the art and challenges. Front Plant Sci 9:1082. doi: 10.3389/fpls.2018.01082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 19:535–544. doi: 10.1016/j.tree.2004.07.021 [DOI] [PubMed] [Google Scholar]
- 16. Candresse T, Marais A, Faure C, Gentit P. 2013. Association of little cherry virus 1 (LChV1) with the shirofugen stunt disease and characterization of the genome of a divergent LChV1 isolate. Phytopathology 103:293–298. doi: 10.1094/PHYTO-10-12-0275-R [DOI] [PubMed] [Google Scholar]
- 17. Moreno AB, López-Moya JJ. 2020. When viruses play team sports: mixed infections in plants. Phytopathology 110:29–48. doi: 10.1094/PHYTO-07-19-0250-FI [DOI] [PubMed] [Google Scholar]
- 18. Malmstrom CM, Melcher U, Bosque-Pérez NA. 2011. The expanding field of plant virus ecology: historical foundations, knowledge gaps, and research directions. Virus Res 159:84–94. doi: 10.1016/j.virusres.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 19. Bernardo P, Charles-Dominique T, Barakat M, Ortet P, Fernandez E, Filloux D, Hartnady P, Rebelo TA, Cousins SR, Mesleard F, Cohez D, Yavercovski N, Varsani A, Harkins GW, Peterschmitt M, Malmstrom CM, Martin DP, Roumagnac P. 2018. Geometagenomics Illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J 12:173–184. doi: 10.1038/ismej.2017.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma Y, Marais A, Lefebvre M, Faure C, Candresse T. 2020. Metagenomic analysis of virome cross-talk between cultivated solanum lycopersicum and wild solanum nigrum. Virology 540:38–44. doi: 10.1016/j.virol.2019.11.009 [DOI] [PubMed] [Google Scholar]
- 21. Ma Y, Fort T, Marais A, Lefebvre M, Theil S, Vacher C, Candresse T. 2021. Leaf-associated fungal and viral communities of wild plant populations differ between cultivated and natural ecosystems. Plant Environ Interact 2:87–99. doi: 10.1002/pei3.10043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Susi H, Laine AL. 2021. Agricultural land use disrupts biodiversity mediation of virus infections in wild plant populations. New Phytol 230:2447–2458. doi: 10.1111/nph.17156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maachi A, Donaire L, Hernando Y, Aranda MA. 2022. Genetic differentiation and migration fluxes of viruses from melon crops and crop edge weeds. J Virol 96:e0042122. doi: 10.1128/jvi.00421-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R. 2009. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388:1–7. doi: 10.1016/j.virol.2009.03.024 [DOI] [PubMed] [Google Scholar]
- 25. Kashif M, Pietilä S, Artola K, Jones RAC, Tugume AK, Mäkinen V, Valkonen JPT. 2012. Detection of viruses in sweetpotato from Honduras and Guatemala augmented by deep-sequencing of small-RNAs. Plant Dis 96:1430–1437. doi: 10.1094/PDIS-03-12-0268-RE [DOI] [PubMed] [Google Scholar]
- 26. Thapa V, McGlinn DJ, Melcher U, Palmer MW, Roossinck MJ. 2015. Determinants of taxonomic composition of plant viruses at the nature conservancy’s tallgrass prairie preserve, Oklahoma. Virus Evol 1:vev007. doi: 10.1093/ve/vev007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Villamor DEV, Ho T, Al Rwahnih M, Martin RR, Tzanetakis IE. 2019. High throughput sequencing for plant virus detection and discovery. Phytopathology 109:716–725. doi: 10.1094/PHYTO-07-18-0257-RVW [DOI] [PubMed] [Google Scholar]
- 28. Kutnjak D, Tamisier L, Adams I, Boonham N, Candresse T, Chiumenti M, De Jonghe K, Kreuze JF, Lefebvre M, Silva G, Malapi-Wight M, Margaria P, Mavrič Pleško I, McGreig S, Miozzi L, Remenant B, Reynard J-S, Rollin J, Rott M, Schumpp O, Massart S, Haegeman A. 2021. A primer on the analysis of high-throughput sequencing data for detection of plant viruses. Microorganisms 9:841. doi: 10.3390/microorganisms9040841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pecman A, Kutnjak D, Gutiérrez-Aguirre I, Adams I, Fox A, Boonham N, Ravnikar M. 2017. Next generation sequencing for detection and discovery of plant viruses and viroids: comparison of two approaches. Front Microbiol 8:1998. doi: 10.3389/fmicb.2017.01998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaafar YZA, Ziebell H. 2020. Comparative study on three viral enrichment approaches based on RNA extraction for plant virus/viroid detection using high-throughput sequencing. PLoS ONE 15:e0237951. doi: 10.1371/journal.pone.0237951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Candresse T, Filloux D, Muhire B, Julian C, Galzi S, Fort G, Bernardo P, Daugrois JH, Fernandez E, Martin DP, Varsani A, Roumagnac P. 2014. Appearances can be deceptive: revealing a hidden viral infection with deep sequencing in a plant quarantine context. PLoS ONE 9:e102945. doi: 10.1371/journal.pone.0102945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma Y, Marais A, Lefebvre M, Theil S, Svanella-Dumas L, Faure C, Candresse T. 2019. Phytovirome analysis of wild plant populations: comparison of double-stranded RNA and virion-associated nucleic acid metagenomic approaches. J Virol 94:e01462-19. doi: 10.1128/JVI.01462-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Massart S, Adams I, Al Rwahnih M, Baeyen S, Bilodeau GJ, Blouin AG, Boonham N, Candresse T, Chandellier A, De Jonghe K, et al. 2022. Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests. Peer Community Journal 2:e62. doi: 10.24072/pcjournal.181 [DOI] [Google Scholar]
- 34. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egan CP, Rummel A, Kokkoris V, Klironomos J, Lekberg Y, Hart M. 2018. Using mock communities of arbuscular mycorrhizal fungi to evaluate fidelity associated with illumina sequencing. Fungal Ecology 33:52–64. doi: 10.1016/j.funeco.2018.01.004 [DOI] [Google Scholar]
- 36. Sevim V, Lee J, Egan R, Clum A, Hundley H, Lee J, Everroad RC, Detweiler AM, Bebout BM, Pett-Ridge J, Göker M, Murray AE, Lindemann SR, Klenk H-P, O’Malley R, Zane M, Cheng J-F, Copeland A, Daum C, Singer E, Woyke T. 2019. Shotgun metagenome data of a defined mock community using oxford nanopore, pacbio and Illumina technologies. Sci Data 6:285. doi: 10.1038/s41597-019-0287-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conceição-Neto N, Zeller M, Lefrère H, De Bruyn P, Beller L, Deboutte W, Yinda CK, Lavigne R, Maes P, Van Ranst M, Heylen E, Matthijnssens J. 2015. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci Rep 5:16532. doi: 10.1038/srep16532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parras-Moltó M, Rodríguez-Galet A, Suárez-Rodríguez P, López-Bueno A. 2018. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 6:119. doi: 10.1186/s40168-018-0507-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roux S, Solonenko NE, Dang VT, Poulos BT, Schwenck SM, Goldsmith DB, Coleman ML, Breitbart M, Sullivan MB. 2016. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. doi: 10.7717/peerj.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gil P, Dupuy V, Koual R, Exbrayat A, Loire E, Fall AG, Gimonneau G, Biteye B, Talla Seck M, Rakotoarivony I, Marie A, Frances B, Lambert G, Reveillaud J, Balenghien T, Garros C, Albina E, Eloit M, Gutierrez S. 2021. A library preparation optimized for metagenomics of RNA viruses. Mol Ecol Resour 21:1788–1807. doi: 10.1111/1755-0998.13378 [DOI] [PubMed] [Google Scholar]
- 41. Marais A, Faure C, Bergey B, Candresse T. 2018. Viral double-stranded RNAs (dsRNAs) from plants: alternative nucleic acid substrates for high-throughput sequencing, p 45–53. In Pantaleo V, Chiumenti M (ed), Viral metagenomics: methods in molecular biology. Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- 42. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P. 2018. Viral metagenomics approaches for high-resolution screening of multiplexed arthropod and plant viral communities, p 77–95. In Pantaleo V, Chiumenti M (ed), Viral metagenomics: methods and protocols. Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- 43. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 44. Illumina . 2017. Effects of index misassignment on multiplexing and downstream analysis. Available from: https://www.illumina.com/content/dam/illumina-marketing/documents/products/ whitepapers/index-hopping-white-paper-770-2017-004.pdf
- 45. van der Valk T, Vezzi F, Ormestad M, Dalén L, Guschanski K. 2020. Index hopping on the illumina HiseqX platform and its consequences for ancient DNA studies. Mol Ecol Resour 20:1171–1181. doi: 10.1111/1755-0998.13009 [DOI] [PubMed] [Google Scholar]
- 46. François S, Filloux D, Frayssinet M, Roumagnac P, Martin DP, Ogliastro M, Froissart R. 2018. Increase in taxonomic assignment efficiency of viral reads in metagenomic studies. Virus Res 244:230–234. doi: 10.1016/j.virusres.2017.11.011 [DOI] [PubMed] [Google Scholar]
- 47. Lefebvre M, Theil S, Ma Y, Candresse T. 2019. The virannot pipeline: a resource for automated viral diversity estimation and operational taxonomy units assignation for virome sequencing data. Phytobiomes J 3:256–259. doi: 10.1094/PBIOMES-07-19-0037-A [DOI] [Google Scholar]
- 48. Ajami NJ, Wong MC, Ross MC, Lloyd RE, Petrosino JF. 2018. Maximal viral information recovery from sequence data using VirMAP. Nat Commun 9:3205. doi: 10.1038/s41467-018-05658-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Santiago-Rodriguez TM, Hollister EB. 2020. Potential applications of human viral metagenomics and reference materials: considerations for current and future viruses. Appl Environ Microbiol 86:e01794-20. doi: 10.1128/AEM.01794-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roux S, Brum JR, Dutilh BE, Sunagawa S, Duhaime MB, Loy A, Poulos BT, Solonenko N, Lara E, Poulain J, Pesant S, Kandels-Lewis S, Dimier C, Picheral M, Searson S, Cruaud C, Alberti A, Duarte CM, Gasol JM, Vaqué D, Tara Oceans Coordinators, Bork P, Acinas SG, Wincker P, Sullivan MB. 2016. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537:689–693. doi: 10.1038/nature19366 [DOI] [PubMed] [Google Scholar]
- 51. Zablocki O, Michelsen M, Burris M, Solonenko N, Warwick-Dugdale J, Ghosh R, Pett-Ridge J, Sullivan MB, Temperton B. 2021. VirION2: a short- and long-read sequencing and informatics workflow to study the genomic diversity of viruses in nature. PeerJ 9:e11088. doi: 10.7717/peerj.11088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Filloux D, Dallot S, Delaunay A, Galzi S, Jacquot E, Roumagnac P. 2015. Metagenomics approaches based on virion-associated nucleic acids (VANA): an innovative tool for assessing without a priori viral diversity of plants. Meth Molec Biol 1302:249–257. doi: 10.1007/978-1-4939-2620-6 [DOI] [PubMed] [Google Scholar]
- 53. Teycheney PY, Bandou E, Gomez RM. 2015. Viral treasure hunt in European outermost territories: how metagenomics boosts the discovery of novel viral species in tropical and sub-tropical crops germplasm. Available from: https://agritrop.cirad.fr/575812/
- 54. Kleiner M, Hooper LV, Duerkop BA. 2015. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16:7. doi: 10.1186/s12864-014-1207-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gallet R, Fabre F, Michalakis Y, Blanc S. 2017. The number of target molecules of the amplification step limits accuracy and sensitivity in ultradeep-sequencing viral population studies. J Virol 91:e00561-17. doi: 10.1128/JVI.00561-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Visser M, Bester R, Burger JT, Maree HJ. 2016. Next-generation sequencing for virus detection: covering all the bases. Virol J 13:85. doi: 10.1186/s12985-016-0539-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Massart S, Chiumenti M, De Jonghe K, Glover R, Haegeman A, Koloniuk I, Komínek P, Kreuze J, Kutnjak D, Lotos L, et al. 2019. Virus detection by high-throughput sequencing of small RNAs: large-scale performance testing of sequence analysis strategies. Phytopathology 109:488–497. doi: 10.1094/PHYTO-02-18-0067-R [DOI] [PubMed] [Google Scholar]
- 58. Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB. 2017. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5:e3817. doi: 10.7717/peerj.3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Breitwieser FP, Lu J, Salzberg SL. 2019. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform 20:1125–1136. doi: 10.1093/bib/bbx120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rampelli S, Soverini M, Turroni S, Quercia S, Biagi E, Brigidi P, Candela M. 2016. Viromescan: a new tool for metagenomic viral community profiling. BMC Genomics 17:165. doi: 10.1186/s12864-016-2446-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sutton TDS, Clooney AG, Ryan FJ, Ross RP, Hill C. 2019. Choice of assembly software has a critical impact on virome characterisation. Microbiome 7:12. doi: 10.1186/s40168-019-0626-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S5.
Tables S1 and S2.
Data Availability Statement
Trimmed sequencing reads for all viral communities analyzed by dsRNA or VANA approaches are available from the French Recherche Data Gouv multidisciplinary repository at https://doi.org/10.57745/42WNRJ. The normalized 10M reads dsRNA or VANA data sets generated using the 60-viruses community have also been made available together with the community composition and the complete or near-complete reference genomic sequences used are also available from the same repository at https://doi.org/10.57745/T4UYPC.