

Abstract
The transcription factor NF-κB is a central mediator of immune and inflammatory responses. To understand the regulation of NF-κB, it is important to probe the underlying thermodynamics, kinetics, and conformational dynamics of the NF-κB/IκBα/DNA interaction network. The development of genetic incorporation of non-canonical amino acids (ncAA) has enabled the installation of biophysical probes into proteins with site specificity. Recent single-molecule FRET (smFRET) studies of NF-κB with site-specific labeling via ncAA incorporation revealed the conformational dynamics for kinetic control of DNA-binding mediated by IκBα. Here we report the design and protocols for incorporating the ncAA p-azidophenylalanine (pAzF) into NF-κB and site-specific fluorophore labeling with copper-free click chemistry for smFRET. We also expanded the ncAA toolbox of NF-κB to include p-benzoylphenylalanine (pBpa) for UV cross-linking mass spectrometry (XL-MS) and incorporated both pAzF and pBpa into the full-length NF-κB RelA subunit which includes the intrinsically disordered transactivation domain.
Keywords: site-specific labeling, NF-κB, single-molecule FRET, cross-linking mass spectrometry, non-canonical amino acids, click chemistry
1. Introduction
The NF-κB family of transcription factors responds to a large number of extracellular stimuli and regulates the expression of hundreds of genes [1]. As a critical regulator, NF-κB is found in almost all types of animal cells and is implicated in various cellular functions including cell growth, proliferation, apoptosis, development, and stress responses. Dysregulation of NF-κB is involved in numerous diseases, especially chronic inflammation and cancers [2]. In the absence of stimuli, NF-κB is sequestered in the cytoplasm by the inhibitory protein IκBα. When a cell receives signals such as cytokines or viral infections, the IκB kinase (IKK) is activated to phosphorylate NF-κB-bound IκBα, which is then ubiquitylated and degraded by the proteasome [3]. Degradation of IκBα unmasks the nuclear localization signal (NLS) of NF-κB, which translocates from the cytoplasm to the nucleus and recognizes DNA with the consensus κB sequence. There are over 104 κB sites in the human genome, primarily in promoter and enhancer regions [4]. NF-κB activates transcription of hundreds of genes upon stimulation, including the one encoding its own inhibitor, IκBα. Activation of IκBα synthesis creates a negative feedback loop, in which the newly synthesized IκBα relocates to the nucleus, binds to NF-κB, and removes it from DNA [1]. The NF-κB:IκBα complex is then rapidly exported from the nucleus [5]. Previous kinetic experiments revealed IκBα actively removes NF-κB from DNA via the formation of a transient ternary complex—a process also known as facilitated dissociation or molecular stripping [6–8].
Over the past decades, we have characterized the thermodynamics, kinetics, and conformational dynamics of the NF-κB/IκBα/DNA interaction network with a wide variety of biophysical experiments including hydrogen-deuterium exchange mass spectrometry (HDX-MS), stopped-flow fluorescence, surface plasma resonance (SPR), fluorescence anisotropy, nuclear magnetic resonance spectroscopy (NMR), and single-molecule FRET (smFRET). Particularly, the use of small-molecule probes that are covalently attached to proteins has allowed visualization of the internal dynamics of NF-κB and IκBα with smFRET [9–12] and measurements of dissociation constants of NF-κB dimers and IκB proteins binding to NF-κB with fluorescence anisotropy [13].
The most common way to install molecular probes is to utilize reactive native amino acids such as cysteines and lysines. Cysteines are the most widely used amino acid for site-specific labeling owing to their rapid reaction with maleimide and their low native abundance in proteins. However, when proteins of interest increase in size, the probability of containing multiple native cysteines increases, complicating the usage for site-specific labeling. For a 200-residue protein, the average cysteine contents is 1.5% [14]. A medium-sized eukaryotic protein contains about 400 residues [15], meaning that there will be roughly six native cysteines on average, which have to be mutated to avoid non-specific labeling. Using cysteines for labeling is also problematic for proteins in which cysteines or disulfide bonds play important functional roles. In previous smFRET studies of IκBα we created a cysteine-free version of the protein and then introduced two cysteines which we then labeled with a mixture of donor and acceptor fluorophores. Using TIRF-based smFRET, we were able to distinguish those single molecules bearing a single donor-acceptor pair of fluorophores [10] and to demonstrate similar results with both confocal-based smFRET and TIRF-based smFRET [12]. However, more than half of the labeled molecules had to be discarded because they were labeled with two donor or two acceptor fluorophores.
The development of genetic incorporation of non-canonical amino acids (ncAAs) through an engineered amber suppressor tRNA/aminoacyl-tRNA synthetase (tRNA/aaRS) system provides a powerful solution to site-specific labeling for biophysical studies. In amber suppression, the amber stop codon, UAG, is repurposed to code for ncAAs. A large number of tRNA/aaRS pairs that read the amber codon have been engineered to incorporate a variety of ncAAs in E. coli and mammalian cells [16, 17]. Incorporation of ncAAs with a unique functional handle for biophysical probe labeling allows site-specific labeling without having to create cysteine-free proteins that can be labor intensive or alter protein functions. The usage of ncAA particularly benefits smFRET studies because smFRET often requires labeling sites other than the termini and does not tolerate non-specific labeling. Precise dual-labeling of the same protein with a FRET pair has also been achieved through the incorporation of two different ncAAs or in combination with a native cysteine [18–23].
We have incorporated the ncAA p-azidophenylalanine (pAzF) for site-specific labeling in the NF-κB dimer RelA1–325-p5039–363 with a FRET pair on its two N-terminal DNA-binding domains (NTDs). In this article, we document the detailed procedures of ncAA selection, incorporation, and site-specific labeling with click chemistry for NF-κB that lead to the discovery of slow, heterogeneous interdomain motions in NF-κB probed by smFRET and how the dynamics are regulated by IκBα to affect DNA binding. We also expanded our ncAA toolbox for NF-κB to include p-benzoylphenylalanine (pBpa) for cross-linking mass spectrometry (XL-MS) to investigate the transient protein-protein interactions within the NF-κB dimer. Both ncAAs pAzF and pBpa were incorporated into the full-length NF-κB including the 230-residue disordered transactivation domain. These protocols can be transferred and adapted for other protein complexes for biophysical experiments, especially smFRET and XL-MS.
2. The strategy of site-specific labeling NF-κB
2.1. Challenges – NF-κB contains reactive cysteines limiting the use of thiol chemistry
Installing molecular probes through conventional thiol-maleimide chemistry is challenging for NF-κB due to the large number of native cysteines it contains (8 cysteines in RelA1–325 and 7 cysteines in p5039–363). To utilize cysteines for site-specific labeling, it is often necessary to create a cysteine-free construct or selectively label the target cysteine if it is predominantly solvent exposed relative to other cysteines. Selective cysteine labeling is possible when the target is the only cysteine located at the terminus of the polypeptide chain or in an intrinsically disordered region. For example, in our previous fluorescence anisotropy experiments, we selectively labeled an external cysteine at the N-terminus of the NF-κB RelA with a 1:1 protein:dye stoichiometry by optimizing the incubation time to minimize off-target reactivity [13]. However, when the target cysteine is not predominantly exposed, it can be difficult to achieve high labeling efficiency without also labeling other off-target native cysteines. Limiting off-target labeling is especially important for experiments such as smFRET, which requires that the exact same positions are fluorescently labeled in each molecule, such that the FRET efficiency reflects the distance between two known positions. To demonstrate the challenge of selective cysteine labeling, we used the NF-κB RelA as an example and probed the reactivity of native cysteines and target cysteine with mass spectrometry. We expressed RelA1–325 with the target site S131 mutated to a cysteine and carried out thiol-maleimide reaction with N-ethylmaleimide (NEM) (Fig. 1a). The targe site is located in the helix in the NTD and was chosen because it could be used as a labeling site for FRET experiments to probe the dynamics between the two NTDs in the NF-κB dimer. Native C38 in the RelA NTD was known to be exposed and was mutated to a serine [24].
Fig. 1. Reactivity of cysteines in NF-κB probed with N-ethylmaleimide.

a. The reactivity of all cysteines including native and target in the NF-κB subunit RelA1–325 was probed with N-ethylmaleimide (NEM). b. The NF-κB RelA-p50 dimer contains multiple native cysteines (spheres). NEM-treated RelA1–325 was digested with GluC. LC-MS/MS showed that multiple native cysteines were modified by NEM (*) while the target S131C remained unmodified.
We incubated RelA1–325 with NEM at a molar ratio of 2:1 at room temperature for 15 minutes. Remaining free NEM was removed by a PD-10 desalting column. Proteolytic digestions with either pepsin or GluC were performed and analyzed by LC-MS/MS. Both pepsin and GluC digestions lead to 100% sequence coverage. Modification of cysteines by NEM is indicated by a mass shift of +125. Pepsin digest showed four out of seven native cysteines were modified by NEM and GluC digest showed five native cysteines were modified. In either pepsin or GluC digest, the target S131C was found not modified (Table 1, Fig 1b).
Table 1.
Noncanonical amino acids for single-molecule FRET applications
| ncAA | ncAA reactive handle | Fluorophore conjugate | smFRET application | Plasmid source | ncAA availability | Restrictions |
|---|---|---|---|---|---|---|
| p-acetylphenylalanine (pAcF) | Ketone | Hydroxylamine (Alexa Fluor, Invitrogen) | T4 lysozyme [18], p23 [26], amyloid β [23], N-methyl-D-aspartate receptor [21], Nucleoporin [19, 27], calmodulin [20], etc. | pEVOL-pAcF [33] | Sigma-Aldrich 857459; Chem-Impex 24756 (CAS 204856–735) | Reaction is optimized at low pH around 4. |
| N-propargyllysine (PrK) | Alkyne | Azide (cyanine dyes, Sigma-Aldrich; Alexa Fluor, Invitrogen, etc.) | Nucleoporin [27], calmodulin [20], protein disulfide isomerase [34], etc. | pEVOL-PylRS [27] | Chemical synthesis [27] | Reaction is catalyzed by Cu2+. |
| p-(propargyloxy)-phenylalanine (PrF) | Alkyne | Azide (cyanine dyes, Sigma-Aldrich; Alexa Fluor, Invitrogen, etc.) | Nucleoporin [27], NADPH-cytochrome P450 reductase [35], etc. | pEVOL-PrF [27, 36] | Chemical synthesis [27] | Reaction is catalyzed by Cu2+. |
| p-azidophenylalanine (pAzF) | Azide | Ring strained alkyne (DIBO or sDIBO Alexa fluor, Invitrogen) | Argonaute [37], NF-кB [9], etc. | pEVOL-pAzF (Addgene #31186) | Chem-Impex 06162 (CAS 33173–53-4) | ncAA does not tolerate reducing agent or light. |
| p-azidomethylphenylalanine (pN3CH2Phe) | Azide | Ring strained alkyne (DIBO or sDIBO Alexa fluor, Invitrogen) | p97 [30], etc. | pDule-pN3CH2Ph e [32] | Advanced ChemBlocks P41226 or chemical synthesis [32] | Synthesis of ncAA is required. |
| Nε-(bicyclo[6.1.0]non-4-yn-9-yl-methoxy)carbonyl-L-lysine (BCNK) | Ring strained alkyne | Tetrazine [38] | Fibroblast Growth Factor Receptor [22] | pBKBCNRS [22] | [22] | Synthesis of ncAA is reauired. |
Results from both pepsin and GluC digestions demonstrated that the majority of native cysteines in RelA were more reactive toward maleimide than the target site, and that selective cysteine labeling is not feasible for RelA when the target site is not predominately solvent exposed. It is also important to note that, even if selective cysteine labeling can be achieved, it should only be applied for ensemble experiments for which a small population of non-specifically labeled cysteines does not make significant contribution to bulk measurements or experiments that are not sensitive to labeling positions. For experiments such as smFRET, for which absolute accuracy of labeling positions is required and every single molecule is resolved, it is necessary to use a unique amino acid for site-specific labeling by either generating a cysteine-free construct or incorporating a ncAA.
2.2. Selection of noncanonical amino acids for fluorophore labeling via click chemistry
To achieve site-specific labeling of NF-κB, we first focused on the application to smFRET and chose to incorporate ncAAs that carry a handle for click chemistry under physiological conditions. Although direct incorporation of a fluorescent ncAA has been used for single-molecule imaging in vivo [25], the incorporated fluorophore can experience significant photobleaching during lysis of E. coli cells and purification of recombinant proteins, leading to poor yield. Instead, genetically incorporating a specific handle and clicking it with fluorophore conjugates after protein purification minimizes photobleaching prior to smFRET experiments.
Several ncAAs have been used for smFRET fluorophore labeling (Table 1). p-acetylphenylalanine (pAcF) contains a ketone handle that undergoes a click-type oxime ligation with hydroxylamine derived fluorophores, and has been incorporated into many proteins including T4 lysozyme [18], p23 [26], amyloid β [23], N-methyl-D-aspartate receptor [21], nucleoporins [19, 27], and calmodulin [20] for smFRET. pAcF and hydroxylamine fluorophore conjugates for smFRET are commercially available. However, the limitation of pAcF is that the oxime ligation reaction is optimally carried out at low pH around 4 and is only suitable for proteins that tolerate acidic conditions.
N-propargyllysine (PrK) is a lysine-derived ncAA with an alkyne handle that undergoes copper-catalyzed azide-alkyne cycloaddition with commercially available azide-conjugated dyes. PrK has been incorporated into T4 lysozyme, nucleoporins and RanBP3 for smFRET [27]. A systematic study of several clickable ncAAs on the protein expression, labeling, and photophysical properties suggested that PrK is the best performer for smFRET. PrK can be synthesized with a few steps and is thus relatively accessible among ncAAs. A limitation of PrK is that some proteins do not tolerate the toxicity of copper, which is required to catalyze the click reaction.
Copper-free click chemistry developed by Bertozzi and coworkers [28] has circumvented copper toxicity by using ring-strained alkynes. ncAAs with strained alkynes have been developed [29], but their synthesis and genetic incorporation can be difficult for nonexperts. Alternatively, strained alkynes such as dibenzocyclooctyne (DIBO) can be placed on the dyes to react with ncAAs with azide handles. P-azidophenylalanine (pAzF) and p-azidomethylphenylalanine (pN3CH2Phe) [30] are two examples that have been used for smFRET fluorophore labeling with copper-free click chemistry. pAzF, pN3CH2Phe, and strained alkyne conjugated dyes are all commercially available. Copper-free click chemistry has the advantage of being bioorthogonal and reacts rapidly under physiological conditions. However, the limitation of pAzF is that it is sensitive to reducing agents and light, and can be reduced to p-aminophenlylalanine (pAmF) by common reducing agents used in recombinant protein purification including DTT, βME, and TCEP [31]. pN3CH2Phe is an improved ncAA with enhanced photostability and is not reduced in E. coli cytosol or by reducing agents [32].
Other ncAA examples that have been used for smFRET include p-(propargyloxy)-phenylalanine (PrF), Nε-(bicyclo[6.1.0]non-4-yn-9-yl-methoxy)carbonyl-L-lysine (BCNK). PrF undergoes copper-catalyzed click chemistry with azides [19], and BCNK undergoes bioorthogonal Diels-Alder cycloaddition reactions with tetrazines. However, these ncAA and their clickable partners are not commercially available and can be difficult to synthesize for nonexperts.
We chose to incorporate pAzF into NF-κB because of the physiological reaction condition of copper-free click chemistry and the availability of the pEVOL-pAzF plasmid (Plasmid #31186, Addgene), pAzF (06162, Chem-Impex) and the ring strained alkyne conjugated fluorophores (Alexa Fluor DIBO/sDIBO alkyne, Invitrogen). Using this system, we were able to specifically label sites in the N-terminal domains of the NF-κB RelA and p50 subunits to monitor their conformational dynamics using smFRET. In the next section, we document the protocols for expressing and purifying the NF-κB RelA-p50 dimer with pAzF in the absence of reducing agents, and site-specific fluorophore labeling with click chemistry for the application of smFRET.
3. Protocols for pAzF incorporation and site-specific labeling for NF-κB
To probe the interdomain motions between NTDs of the NF-κB RelA-p50 dimer, we genetically incorporated the ncAA pAzF that undergoes copper-free azide-alkyne cycloaddition with ring strained alkyne dye conjugates (Fig. 2a). RelA1–325 and 5039–363 were expressed, purified, and labeled separately, and combined to form the dual-labeled NF-κB dimer (Fig. 2b).
Fig. 2. Incorporation of pAzF into NF-κB for site-specific labeling with copper-free click chemistry.

a. p-azidophenylalanine (pAzF) reacts with ring strained alkyne dibenzocyclooctyne (DIBO) under physiological conditions. b. RelA Q128pAzF and p50 F148pAzF dimers were expressed, purified, and labeled separately and combined to form the dual-labeled RelA-p50 dimer.
3.1. About the plasmids
Murine RelA1–325 (UniProt entry Q04207) and p5039–363 (UniProt entry P25799) are encoded in separate pET11a vectors with ampicillin resistance. A 6xHis-tag was introduced to the C-terminus of RelA by site-directed mutagenesis for immobilization on microscope slides through anti-His antibody. Codons for Q128 of RelA and F148 of p50 were mutated into the Amber stop codon, TAG, by site-directed mutagenesis.
The plasmid pEVOL-pAzF encodes the tRNA/aaRS for incorporating pAzF into proteins in E. coli in response to the Amber stop codon and carries chloramphenicol resistance.
3.2. Expression of NF-κB incorporating pAzF
Co-transform 100 μL BL21 (DE3) E. coli strain (New England Biolabs) with 50 ng of the expression plasmid (RelA or p50) and 50 ng of pEVOL-pAzF.
Spread cells on an LB-Agar plate with 200 μg/L ampicillin and 34 μg/mL chloramphenicol and incubate plate at 37°C overnight.
Pick a single colony and grow a 10 mL starter culture (*Zn media, **1.5x M9 media, 0.8% Dextrose, 1 mM MgSO4, 0.2 mM CaCl2, 200 ug/mL ampicillin, 34 μg/mL chloramphenicol) at 37°C, 180 rpm for 16 hours.
Inoculate 1 L M9 minimal media culture (1.5x M9 media, 0.8% Dextrose, 1 mM MgSO4, 0.2 mM CaCl2, 50 μL 20% Thiamine, 200 μg/L ampicillin, 34 μg/mL chloramphenicol) with 10 mL starter culture.
Grow cells at 37°C, 180 rpm until OD600 reaches 0.6–0.7. It usually takes 6–8 hours.
Place the flask on ice for 20 minutes to cool down.
Add 200 mg pAzF (CAS 33173–53-4, Chem-Impex) to the culture.
Add IPTG to a final concentration of 0.2 mM to induce protein expression. Add L-arabinose to a final concentration of 0.02% to induce tRNA/aaRS expression.
Transfer culture to an 18°C shaker and shake at 180 rpm for 16 hours.
Pellet cells by centrifugation at 5,000× g for 15 minutes.
*Zn media: 5 g NaCl, 10 g N-Z-Amine AS Casein enzymatic hydrolysate in 850 mL DI water
*15x M9 media: 90 g Na2HPO4, 45 g KH2PO4, 7.5 g NaCl, 30 g NH4Cl in 1L DI water, pH to 7.4 with KOH pellets.
3.3. Purification of NF-κB with pAzF
Purification of RelA with a 6xHis-tag
Resuspend the cell pellet in 35 mL of 50 mM sodium phosphate pH 8, 150 mM NaCl, 10 mM imidazole, 0.5 mM PMSF, and 1x protease inhibitor cocktail (Sigama-Aldrich).
Sonicate cells on ice to lyse.
Centrifuge cell lysate at 17,000× g for 30 minutes.
Equilibrate 5 mL Ni-NTA resin (Thermo Fisher Scientific) in a gravity column with > 50 mL of sodium phosphate pH 8, 150 mM NaCl, 10 mM imidazole pH 8.
Load the supernatant from step 3 onto the Ni-NTA column and collect flow-through for SDS-PAGE.
Wash column with > 50 mL of 50 mM sodium phosphate pH 8, 150 mM NaCl, 20 mM imidazole.
Elute protein with 10–20 mL of 50 mM sodium phosphate pH 8, 150 mM NaCl, 200 mM imidazole.
Equilibrate a PD-10 desalting column (GE Healthcare) that will be used for buffer exchange into a low-imidazole buffer with > 25 mL of PD-10 buffer (25 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA). Note: avoid buffer exchange with overnight dialysis because NF-κB aggregates in the absence of reducing agents.
Load 2.5 mL Ni column eluent to the PD-10 column. Discard flow-through.
Elute protein with 3.5 mL PD-10 buffer.
Repeat steps 8–10 for the remaining Ni-NTA column eluent.
Add protease inhibitor cocktail to prevent proteolytic digestion of the NLS of RelA.
Add glycerol to a 5% final concentration.
Estimate protein concentration by A280 absorption. The A260/A280 ratio is higher than wildtype protein due to pAzF. A more accurate concentration can be determined by BCA assay but an estimation by UV-Vis is good enough for labeling.
Separate protein into aliquots containing about 50 nmol (~0.6 mL, ~80 μM) and store at −80 °C.
Purification of p50 without a 6×His tag
Resuspend the cell pellet in 35 mL of 25 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5 mM PMSF, and 1x protease inhibitor cocktail.
Sonicate cells on ice to lyse.
Centrifuge cell lysate at 17,000× g for 30 minutes.
Equilibrate 7 mL SP Sepharose Fast Flow (GE Healthcare) resin in a gravity column with Buffer A (25mM Tris pH 7.5, 0.5mM EDTA).
Load the supernatant from step 3 onto the SP Sepharose column and collect flow-through for SDS-PAGE.
Use a gradient former (Bio-Rad) with 100 mL Buffer A in chamber 1 and 100 Buffer B (25mM Tris pH 7.5, 700mM NaCl, 0.5mM EDTA) in chamber 2. Elute protein with a 0–700 mM NaCl gradient over 200 mL at a rate of 5 mL/min and collect 10 mL fractions. Note: It is also possible to manually perform step-wise elution with buffers with 50 mM NaCl increments.
Determine fractions that contain the protein with SDS-PAGE. Combine fractions containing p50. 16. Equilibrate a PD-10 desalting column (GE Healthcare) that will be used for buffer exchange into a low-imidazole buffer with > 25 mL of PD-10 buffer (25 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA). Note: avoid buffer exchange with overnight dialysis because NF-κB aggregates in the absence of reducing agents.
Load 2.5 mL SP Sepharose eluent containing p50 to the PD-10 column. Discard flow-through.
Elute protein with 3.5 mL PD-10 buffer.
Repeat steps 8–10 for the remaining eluent.
Add glycerol to a 5% final concentration.
Estimate protein concentration by A280 absorption. The A260/A280 ratio is higher than wildtype protein due to pAzF. A more accurate concentration can be determined by BCA assay but an estimation for UV-Vis is good enough for labeling.
Separate protein into aliquots containing about 50 nmol (~0.6 mL, ~80 μM) and store at −80 °C.
3.4. Labeling NF-κB via copper-free click chemistry
Proteins should be labeled right before the smFRET experiment.
Thaw an aliquot of each NF-κB subunit (50 nmol) and add 1 μL 50 mM Alexa Fluor sDIBO alkyne (Alexa 555 or Alexa 647, in DMSO, stored at −80 °C). Keep the reaction in dark, at 4°C, overnight. Incubation > 2 days gives the best labeling efficiency.
To remove free Alexa Fluor, equilibrate a PD-10 column with SEC buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT). Wrap column with aluminum foil or do this in the dark.
Adjust sample volume to 2.5 mL and load onto PD-10. Discard flow-through.
Elute labeled protein with 3.5 mL SEC buffer.
Measure absorption at 280 nm, 555 nm, and 650 nm. Calculate labeling efficiency using the extinction coefficients , and the correction factors accounting for A280 from the dyes (, ).
3.5. Assembling NF-κB heterodimers for smFRET
Mix labeled RelA and p50 with a 1:5 donor:acceptor molar ratio to form RelA-p50 heterodimer. Excess of acceptor will minimize the chance of observing donor-only homodimers.
Purify the dual-labeled heterodimer by cation exchange chromatography (Mono S 10/10, GE Healthcare) in 25 mM Tris pH 7.5, 0.5 mM EDTA, 1mM DTT with a NaCl gradient from 100 to 700 mM to remove DNA.
(Optional) To further remove protein aggregates, purify protein with size exclusion chromatography (Superdex 200, GE Healthcare) in Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1mM DTT.
4. Results
4.1. Confirmation of pAzF incorporation
To examine the incorporation of pAzF into NF-κB, we first did a side-by-side growth comparison for p50 expression with and without the addition of pAzF. For the growth with pAzF, full-length p50 is expressed and present as a clear band when analyzed via SDS-PAGE. For the growth without pAzF, full-length p50 was not detected via SDS-PAGE (Fig. 3a). This result suggested that the tRNA/aaRS specifically incorporated pAzF at the target site and does not incorporate other amino acids into NF-κB.
Fig. 3. Expression, purification, and labeling of NF-κB.
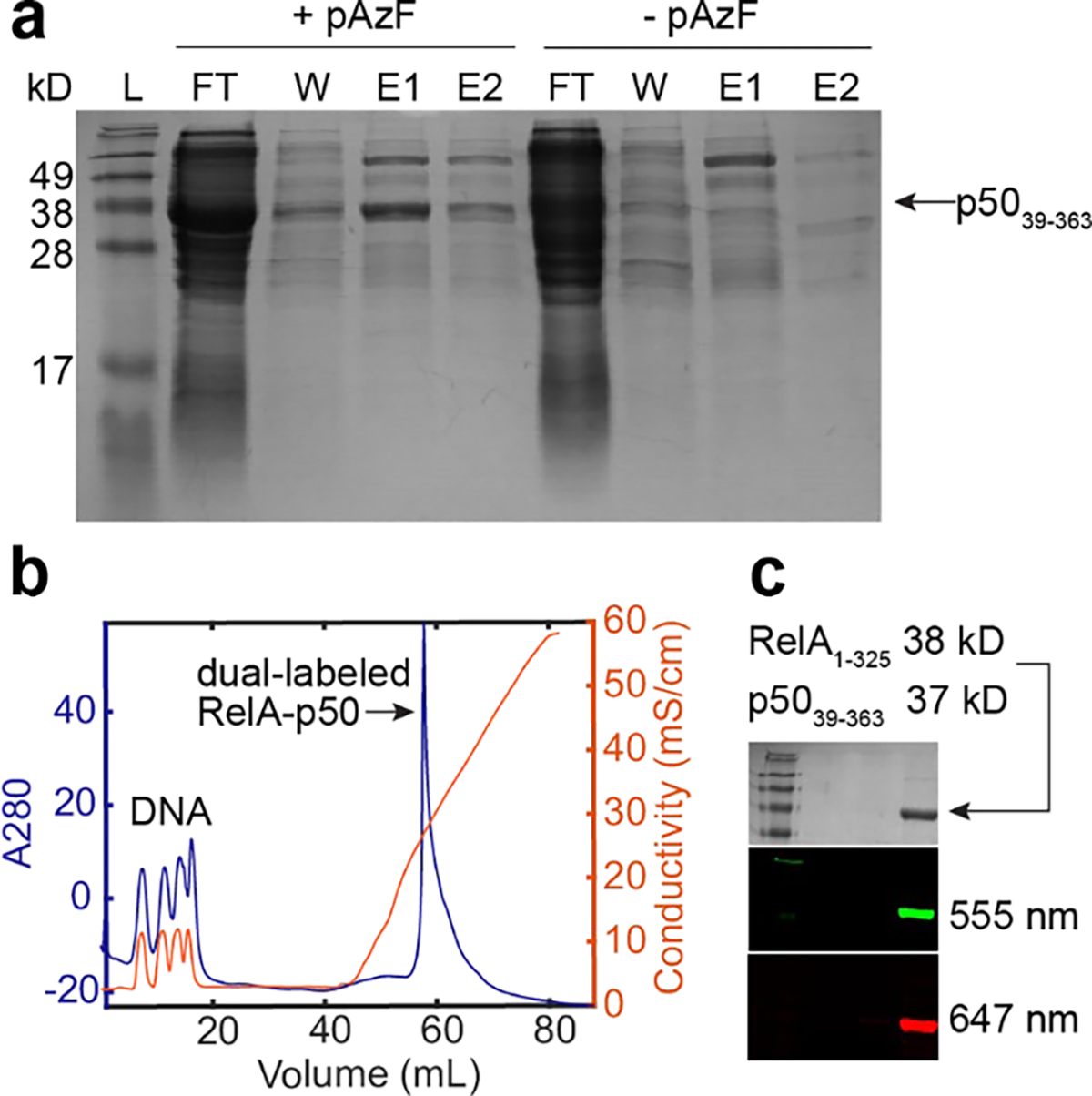
a. A side-by-side growth comparison for p5036–393 with and without the addition of pAzF showing expression of full-length p5036–393 with pAzF. b. Purification of the dual-labeled RelA-p50 with cation exchange chromatography with the Mono S column to remove DNA. c. SDS-PAGE gel stained with Coomassie and imaged with fluorescence confirmed the formation of the dual-labeled RelA-p50 dimer.
To validate the incorporation of pAzF at the desired sites, we performed pepsin digestion for Q128pAzF RelA and F148pAzF p50 and performed LC-MS/MS. The digestion was carried out in the presence of DTT and therefore a portion of pAzF was reduced to pAmF. We found the peptide containing residues 126–133 of RelA R.DLEQ[+34]AISQR.I (M+H 1093.567) with a +34 mass shift for Q128 indicating the presence of pAmF at this position, the peptide containing residues 138–161 of p50 GILHVTKKKVF[+42]ETL (M+H 1653.984) with a + 42 mass shift for F148 indicating the incorporation of pAzF.
4.2. Quantification of pAzF labeling efficiency
We quantified the labeling efficiency by UV-Vis absorption. The labeling efficiency was determined with the extinction coefficients and the correction factors accounting for from the dyes (, ). The labeling efficiency ranged from 29% to 56 % for Alexa Fluor 555 conjugated to RelA, from 20% to 35% for Alexa Fluor 647 conjugated to RelA, from 52% to 83% for Alexa Fluor 647 conjugated to p50, and from 18% to 50% for Alexa Fluor 647 conjugated to p50.
The individually labeled subunits were then combined and purified with strong cation exchange chromatography to remove bound DNA (Fig. 3b) To examine the formation of dual-labeled NF-κB dimer from cation exchange elution, we performed SDS-PAGE and imaged the gel with a Typhoon laser scanner that detects fluorescence of Alexa Fluor 555 and Alexa Fluor 647. The molecular weight for RelA1–325 and p5039–363 are 38 kD and 37 kD respectively, and therefore their bands overlap on the gel. Detection of donor or acceptor fluorescence showed the presence of both fluorophores and that the dual-labeled RelA-p50 was successfully formed (Fig. 3c)
4.3. Interdomain motions in NF-κB observed with smFRET
The double-labeled NF-κB RelA-p50 dimers were immobilized on a passivated surface for TIRF-based single-molecule FRET experiments via the interaction between the C-terminal His-tag in RelA and an anti-His antibody. Detailed procedures of TIRF-based smFRET setup have been documented in several comprehensive reviews and protocols [39, 40] and details of smFRET experiments for NF-κB can be found in our previous paper [9].
We observed anticorrelated donor and acceptor emission, indicating changing FRET between the donor-acceptor pair caused by the interdomain motions between the NTDs (Fig. 4). Particularly, we observed a continuum of conformations of NF-κB interconverting on the sub-seconds to minutes timescale in both free and DNA-bound states, and that these interdomain motions were locked by wild-type IκBα but not by the loss-of-function mutant [9]. These detailed characterizations of conformational dynamics, which were made possible by the incorporation of ncAAs, suggest how IκBα removes NF-κB from DNA and prevents rebinding by altering the interdomain motions of the NF-κB NTDs.
Fig. 4. Interdomain motions in NF-κB visualized by single-molecule FRET.

The anticorrelated fluorescence (Fl.) of donor (light green) and acceptor (dark red) of a signal RelA-p50 dimer showed changing FRET, reflecting distance change between the two NTDs over time.
4.4. Expanding the ncAA toolbox for NF-κB: Studies of the RelA transcription activation domain
Incorporation of ncAAs is a promising tool for the biophysical investigations of intrinsically disordered regions, which can be challenging to study by traditional structural and mechanistic approaches. The RelA subunit contains a 230-residue transcription activation domain (TAD), which is intrinsically disordered and whose function is not fully understood. We recently found that this domain alters the DNA binding affinity and specificity of the RelA-p50 heterodimer, highlighting the importance of understanding the crosstalk between the TAD and the DNA-binding domains of NF-κB [41]. Incorporation of ncAAs into full-length RelA enables two complementary biophysical approaches to study the effects of the RelA TAD on the structure and dynamics of the RelA-p50 dimer: smFRET probes the interdomain motions and cross-linking mass spectrometry (XL-MS) probes the transient protein-protein interactions within NF-κB.
We were able to successfully incorporate pAzF into full-length RelA including the TAD (residues 18–549) for smFRET studies using a similar approach to that described for RelA1–325. The codon for Q128 was mutated to TAG, and a 6xHis tag was included at the C-terminal end of the protein. All expression and purification steps were conducted using the protocol described above. Although the yield was lower for full-length RelA, incorporation of the 6xHis tag on the C-terminal end enabled separation of full-length RelA from truncated products via Ni-NTA chromatography and successful purification of the intact protein (Fig. 5a).
Fig. 5. Incorporation of ncAAs into full-length RelA.

a. Incorporation of pAzF into full-length RelA. b. Incorporation of p-benzoylphenylalanine (pBpa) into full-length RelA-p50 heterodimer. c. pBpa forms covalent cross-links with C-H bonds upon exposure to UV light.
p-benzoylphenylalanine (pBpa), a ncAA and a UV-inducible crosslinking agent, provides a promising tool for the investigation of intrinsically disordered regions like NF-κB’s TAD with its ability to covalently ‘lock in’ transient interactions [42, 43]. We were able to incorporate pBpa into full-length RelA in the loop region of the NTD using a similar approach to that described for pAzF. The plasmid pEVOL-pBpF (Addgene plasmid #31190) is used in place of pEVOL-pAzF to produce the appropriate tRNA/aaRS pair for pBpa incorporation at the Amber stop codon [43]. BL21(DE3) E. coli are co-transformed with pEVOL-pBpF and a plasmid containing full-length Murine RelA19–549 and N-terminally tagged 6xHis-p5039–350 in a pET11a vector. Due to the low solubility of pBpa in neutral aqueous solutions, 270 mg pBpa is dissolved in 1 mL 1M NaOH and added to the expression culture at the same time as the 10 mL starter culture instead of immediately prior to induction. As was the case for pAzF, protein expression and tRNA/aaRS expression are induced with IPTG and arabinose, respectively. Using this approach, we were able to incorporate pBpa at position 36 of full-length RelA and obtain pure RelA-p50 dimer (Fig. 5b).
The ncAA pBpa forms covalent cross-links with C-H bonds upon exposure to UV light, which can be detected using mass spectrometry (Fig. 5C) [44]. This enables detection of transient interactions between dynamic protein regions such as possible interactions between the RelA TAD and the NTD, which may not be captured by other biophysical approaches. The use of pBpa has significant advantages over traditional XL-MS approaches. Many traditional cross-linking reagents such as disuccinimidyl suberate (DSS) contain fairly long linker regions (11 Å), and therefore are able to crosslink protein regions which may not interact directly [44]. By contrast pBpa is a so-called “zero length” linker, in that it will only crosslink with a C-H bond that comes into direct contact with the ncAA. Additionally, one concern about traditional cross-linking experiments is that formation of the first covalent crosslink can bias the dynamics of the protein, such that additional crosslinks that form over the course of the experiment do not reflect contacts that occur within the native protein. Because pBpa can be specifically incorporated into a single position within a protein and can only react to form a single crosslink, that crosslink must represent a contact that occurred within the un-crosslinked protein. Despite challenges associated with ncAA incorporation, it represents a powerful tool for unbiased XL-MS experiments on dynamic proteins.
5. Conclusion
Genetic incorporation of ncAAs provides a powerful tool for site-specific installation of biophysical probes into proteins. We demonstrated and reported the protocols for incorporating the ncAAs pAzF and pBpa into NF-κB for the application of smFRET and XL-MS experiments. The ncAA toolbox for NF-κB will enable the investigation of the conformational dynamics and the communications between the intrinsically disordered TAD and DNA-binding domains.
Acknowledgments
We thank Majid Ghassemian at the Biomolecular and Proteomics Mass Spectrometry Facility of UCSD for carrying out the mass spectrometry. pEVOL-pAzF was a gift from Peter Schultz (Addgene plasmid # 31186 ; http://n2t.net/addgene:31186 ; RRID:Addgene_31186). pEVOL-pBpF was a gift from Peter Schultz (Addgene plasmid # 31190 ; http://n2t.net/addgene:31190 ; RRID:Addgene_31190).
References
- [1].Hoffmann A, Levchenko A, Scott ML, Baltimore D, The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation, Science 298(5596) (2002) 1241–5. [DOI] [PubMed] [Google Scholar]
- [2].Kumar A, Takada Y, Boriek AM, Aggarwal BB, Nuclear factor-kappaB: its role in health and disease, J Mol Med (Berl) 82(7) (2004) 434–48. [DOI] [PubMed] [Google Scholar]
- [3].Traenckner EB, Baeuerle PA, Appearance of apparently ubiquitin-conjugated I kappa B-alpha during its phosphorylation-induced degradation in intact cells, J Cell Sci Suppl 19 (1995) 79–84. [DOI] [PubMed] [Google Scholar]
- [4].Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M, Distribution of NF-kappaB-binding sites across human chromosome 22, Proc Natl Acad Sci U S A 100(21) (2003) 12247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C, Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm, J Cell Sci 110 ( Pt 3) (1997) 369–78. [DOI] [PubMed] [Google Scholar]
- [6].Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA, Kinetic enhancement of NF-kappaBxDNA dissociation by IkappaBalpha, Proc Natl Acad Sci U S A 106(46) (2009) 19328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Alverdi V, Hetrick B, Joseph S, Komives EA, Direct observation of a transient ternary complex during IkappaBalpha-mediated dissociation of NF-kappaB from DNA, Proc Natl Acad Sci U S A 111(1) (2014) 225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Potoyan DA, Zheng W, Komives EA, Wolynes PG, Molecular stripping in the NF-kappaB/IkappaB/DNA genetic regulatory network, Proc Natl Acad Sci U S A 113(1) (2016) 110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen W, Lu W, Wolynes PG, Komives EA, Single-molecule conformational dynamics of a transcription factor reveals a continuum of binding modes controlling association and dissociation, Nucleic Acids Res 49(19) (2021) 11211–11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lamboy JA, Kim H, Lee KS, Ha T, Komives EA, Visualization of the nanospring dynamics of the IkappaBalpha ankyrin repeat domain in real time, Proc Natl Acad Sci U S A 108(25) (2011) 10178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lamboy JA, Kim H, Dembinski H, Ha T, Komives EA, Single-molecule FRET reveals the native-state dynamics of the IkappaBalpha ankyrin repeat domain, J Mol Biol 425(14) (2013) 2578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Trelle MB, Ramsey KM, Lee TC, Zheng W, Lamboy J, Wolynes PG, Deniz A, Komives EA, Binding of NFkappaB Appears to Twist the Ankyrin Repeat Domain of IkappaBalpha, Biophys J 110(4) (2016) 887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ramsey KM, Chen W, Marion JD, Bergqvist S, Komives EA, Exclusivity and Compensation in NFkappaB Dimer Distributions and IkappaB Inhibition, Biochemistry 58(21) (2019) 2555–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Carugo O, Amino acid composition and protein dimension, Protein Sci 17(12) (2008) 2187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brocchieri L, Karlin S, Protein length in eukaryotic and prokaryotic proteomes, Nucleic Acids Res 33(10) (2005) 3390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu CC, Schultz PG, Adding new chemistries to the genetic code, Annu Rev Biochem 79 (2010) 413–44. [DOI] [PubMed] [Google Scholar]
- [17].Lang K, Chin JW, Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins, Chem Rev 114(9) (2014) 4764–806. [DOI] [PubMed] [Google Scholar]
- [18].Brustad EM, Lemke EA, Schultz PG, Deniz AA, A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer, J Am Chem Soc 130(52) (2008) 17664–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Milles S, Lemke EA, Single molecule study of the intrinsically disordered FG-repeat nucleoporin 153, Biophys J 101(7) (2011) 1710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim J, Seo MH, Lee S, Cho K, Yang A, Woo K, Kim HS, Park HS, Simple and efficient strategy for site-specific dual labeling of proteins for single-molecule fluorescence resonance energy transfer analysis, Anal Chem 85(3) (2013) 1468–74. [DOI] [PubMed] [Google Scholar]
- [21].Cooper DR, Dolino DM, Jaurich H, Shuang B, Ramaswamy S, Nurik CE, Chen J, Jayaraman V, Landes CF, Conformational transitions in the glycine-bound GluN1 NMDA receptor LBD via single-molecule FRET, Biophys J 109(1) (2015) 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Perdios L, Lowe AR, Saladino G, Bunney TD, Thiyagarajan N, Alexandrov Y, Dunsby C, French PM, Chin JW, Gervasio FL, Tate EW, Katan M, Conformational transition of FGFR kinase activation revealed by site-specific unnatural amino acid reporter and single molecule FRET, Sci Rep 7 (2017) 39841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Meng F, Bellaiche MMJ, Kim JY, Zerze GH, Best RB, Chung HS, Highly Disordered Amyloid-beta Monomer Probed by Single-Molecule FRET and MD Simulation, Biophys J 114(4) (2018) 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bergqvist S, Croy CH, Kjaergaard M, Huxford T, Ghosh G, Komives EA, Thermodynamics reveal that helix four in the NLS of NF-kappaB p65 anchors IkappaBalpha, forming a very stable complex, J Mol Biol 360(2) (2006) 421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pantoja R, Rodriguez EA, Dibas MI, Dougherty DA, Lester HA, Single-molecule imaging of a fluorescent unnatural amino acid incorporated into nicotinic receptors, Biophys J 96(1) (2009) 226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ratzke C, Hellenkamp B, Hugel T, Four-colour FRET reveals directionality in the Hsp90 multicomponent machinery, Nat Commun 5 (2014) 4192. [DOI] [PubMed] [Google Scholar]
- [27].Milles S, Tyagi S, Banterle N, Koehler C, VanDelinder V, Plass T, Neal AP, Lemke EA, Click strategies for single-molecule protein fluorescence, J Am Chem Soc 134(11) (2012) 5187–95. [DOI] [PubMed] [Google Scholar]
- [28].Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR, Copper-free click chemistry for dynamic in vivo imaging, Proc Natl Acad Sci U S A 104(43) (2007) 16793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Plass T, Milles S, Koehler C, Schultz C, Lemke EA, Genetically encoded copper-free click chemistry, Angew Chem Int Ed Engl 50(17) (2011) 3878–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee TC, Kang M, Kim CH, Schultz PG, Chapman E, Deniz AA, Dual Unnatural Amino Acid Incorporation and Click-Chemistry Labeling to Enable Single-Molecule FRET Studies of p97 Folding, Chembiochem 17(11) (2016) 981–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lopez CJ, Fleissner MR, Brooks EK, Hubbell WL, Stationary-phase EPR for exploring protein structure, conformation, and dynamics in spin-labeled proteins, Biochemistry 53(45) (2014) 7067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bazewicz CG, Liskov MT, Hines KJ, Brewer SH, Sensitive, site-specific, and stable vibrational probe of local protein environments: 4-azidomethyl-L-phenylalanine, J Phys Chem B 117(30) (2013) 8987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG, A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli, Biochemistry 52(10) (2013) 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chinnaraj M, Barrios DA, Frieden C, Heyduk T, Flaumenhaft R, Pozzi N, Bioorthogonal Chemistry Enables Single-Molecule FRET Measurements of Catalytically Active Protein Disulfide Isomerase, Chembiochem 22(1) (2021) 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Quast RB, Fatemi F, Kranendonk M, Margeat E, Truan G, Accurate Determination of Human CPR Conformational Equilibrium by smFRET Using Dual Orthogonal Noncanonical Amino Acid Labeling, Chembiochem 20(5) (2019) 659–666. [DOI] [PubMed] [Google Scholar]
- [36].Young TS, Ahmad I, Yin JA, Schultz PG, An enhanced system for unnatural amino acid mutagenesis in E. coli, J Mol Biol 395(2) (2010) 361–74. [DOI] [PubMed] [Google Scholar]
- [37].Zander A, Holzmeister P, Klose D, Tinnefeld P, Grohmann D, Single-molecule FRET supports the two-state model of Argonaute action, RNA Biol 11(1) (2014) 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW, Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction, Nat Chem 4(4) (2012) 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roy R, Hohng S, Ha T, A practical guide to single-molecule FRET, Nat Methods 5(6) (2008) 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Selvin PR, Ha T, Single-molecule techniques, Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- [41].Baughman HER, Narang D, Chen W, Villagran Suarez AC, Lee J, Bachochin MJ, Gunther TR, Wolynes PG, Komives EA, An intrinsically disordered transcription activation domain increases the DNA binding affinity and reduces the specificity of NFkappaB p50/RelA, J Biol Chem 298(9) (2022) 102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kauer JC, Erickson-Viitanen S, Wolfe HR Jr., DeGrado WF, p-Benzoyl-L-phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin-binding peptide, J Biol Chem 261(23) (1986) 10695–700. [PubMed] [Google Scholar]
- [43].Chin JW, Martin AB, King DS, Wang L, Schultz PG, Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli, Proc Natl Acad Sci U S A 99(17) (2002) 11020–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Holding AN, XL-MS: Protein cross-linking coupled with mass spectrometry, Methods 89 (2015) 54–63. [DOI] [PubMed] [Google Scholar]