

Abstract
Water caltrop (Trapa spp., Lythraceae) is a traditional but currently underutilized non-cereal crop. Here, we generated chromosome-level genome assemblies for the two diploid progenitors of allotetraploid Trapa. natans (4x, AABB), i.e., diploid T. natans (2x, AA) and Trapa incisa (2x, BB). In conjunction with four published (sub)genomes of Trapa, we used gene-based and graph-based pangenomic approaches and a pangenomic transposable element (TE) library to develop Trapa genomic resources. The pangenome displayed substantial gene-content variation with dispensable and private gene clusters occupying a large proportion (51.95%) of the total cluster sets in the six (sub)genomes. Genotyping of presence-absence variation (PAVs) identified 40 453 PAVs associated with 2570 genes specific to A- or B-lineages, of which 1428 were differentially expressed, and were enriched in organ development process, organic substance metabolic process and response to stimulus. Comparative genome analyses showed that the allotetraploid T. natans underwent asymmetric subgenome divergence, with the B-subgenome being more dominant than the A-subgenome. Multiple factors, including PAVs, asymmetrical amplification of TEs, homeologous exchanges (HEs), and homeolog expression divergence, together affected genome evolution after polyploidization. Overall, this study sheds lights on the genome architecture and evolution of Trapa, and facilitates its functional genomic studies and breeding program.
Introduction
Underutilized or ‘neglected’ crops are mostly wild or semi-domesticated species that have been used for food, medicine or cultural practices (etc.) for centuries but are no longer widely used or commercialized as part of mainstream agriculture ([1]; see also [2–4]). Nevertheless, many underutilized crops possess a high content of micronutrients for mitigating malnutrition, and are often adapted to unique climatic and environmental conditions (e.g. [3]). Thus, they are not only important to local people, but can also have the potential to improve the resilience and sustainability of food production systems [4, 5].
The annual herbaceous and aquatic genus Trapa L. (Lythraceae), has traditionally been divided into two species, i.e., Trapa natans L. with diploid (2n = 2x = 48) and tetraploid (2n = 4x = 96) cytotypes, and diploid Trapa incisa Sieb. and Zucc. (2n = 2x = 48) [6, 7]. The fruits of Trapa spp., also known as water caltrop, possess a high content of starch and were once an important food source, but are presently mostly underutilized [8]. Archaeological evidence suggests that water caltrop has been domesticated in the Yangtze River basin since the Neolithic period [9]. Based on the assembly of tetraploid T. natans and population genomics analyses of wild and cultivated accessions of Trapa in our previous study (Fig. S1) [10], we have found that tetraploid T. natans (AABB) is an allotetraploid hybrid between diploid T. natans (AA) and T. incisa (BB); the cultivated water caltrop was domesticated from diploid T. natans at c. 6300 (5600–13 900) yrs bp, and was subject to further artificial selection in historical times, during the Tang and Song Dynasties (618–1279 ad). In addition, recent whole-genome sequencing and genome analysis of diploid T. natans and T. incisa have uncovered abundant genomic variations between the two species [11].
Genomic structural variants (SVs, variants ≥ 50 bp) are important sources of functional variation, and can play important roles in domestication, adaptation and speciation [12–14]. With the exponentially rising amount of reference genomes, extensive SVs have been discovered even within species, implying that a single reference genome is not sufficient to infer the full species genetic diversity (reviewed in [15]). Thus, generating a species-representative genome or ‘pangenome’ is the method of choice for better capturing both structural and nucleotide diversity [16, 17]. Pangenomes have recently been generated for various crops, such as soybean [18], rice [19, 20], maize [21], and wheat [22]. All these studies have highlighted the essential role of presence-absence variations (PAVs) within a species in determining the genetic basis of agronomic traits. For instance, PAVs localized within MYB genes were identified as a potential cause underlying the variation of grain colour in sorghum [23]. However, until now, only a few pangenome studies have been performed on underutilized crops, such as sesame [24] or pigeon pea [25].
In this study, we generated two chromosome-level genome assemblies of diploid T. natans (2x, AA) and T. incisa (2x, BB). These two newly sequenced genomes, and three previously published genome of Trapa [10, 11], were employed to construct a gene-based pangenome of Trapa. Based on this pangenome, we defined the core/dispensable/private gene clusters. We also built a pan transposable element (TE) library, and compared the divergence of TEs among the two subgenomes of allotetraploid T. natans and the four (sub)genomes of its diploid progenitors. In addition, we generated a graph-based pangenome to genotype PAVs, and identified genes with PAVs that likely contributed to speciation and phenotypic divergence between diploid T. natans and T. incisa. Finally, we investigated the genomic variations during allopolyploidization and subgenome dominance in allotetraploid T. natans. Overall, our study will contribute to a better understanding of the genome evolution underlying the diversification and polyploidization of Trapa. This pangenome resource will facilitate further studies on the evolutionary and functional genomics of water caltrop.
Results
De novo genome assembly and annotation of diploid T. natans and T. incisa
To construct the pangenome representing the full range of genetic diversity of Trapa, we sequenced and assembled the genomes of a traditional cultivar of diploid T. natans (i.e., a cultivar called ‘Nahuling’ with no horns, hereinafter referred to as ‘TnA_NL’) and one sample of T. incisa from Heilongjiang River, China (hereinafter referred to as ‘TiB_HR’). By adopting a hybrid assembly approach, we used a combination of PacBio long reads, Illumina short reads, and a Hi-C chromatin interaction map (see details in Materials and methods). The assembled genome of the TnA_NL was 477.43 Mb with a contig N50 of 6.27 Mb, which was 89.3% of the estimated genome size (534.47 Mb) determined by k-mer analysis. The resulting genome of TiB_HR was 470.61 Mb with a contig N50 of 12.07 Mb, accounting for 93.4% of the estimated genome size (503.46 Mb) (Table 1; Figs S2 and S3, see online supplementary material). Both assembled genomes showed low heterozygosity (TnA_NL: 0.31%; TiB_HR: 0.07%). The chromosome-scale scaffolds were finally assembled based on Hi-C data. Approximately 99.7% (475.99 Mb) and 97.8% (460.48 Mb) of the assembled sequences were anchored onto the respective 24 pseudo-chromosomes of TnA_NL and TiB_HR, respectively (Table 1; Table S1 and Figs S4 and S5, see online supplementary material). Based on our analyses of Benchmarking Universal Single-Copy Orthologs (BUSCO), we identified 1614 universal single-copy genes and most of them could be fully annotated onto the genome assemblies of TnA_NL (1578; 97.77%) and TiB_HR (1571; 97.34%) (Table 1). Core Eukaryotic Genes Mapping Approach (CEGMA) analyses revealed that 236 (95.16%) and 235 (94.76%) of the 248 core eukaryotic genes were present in complete length in the respective genomes (Table S2, see online supplementary material). In addition, a very high proportion of Illumina short reads could be remapped to each assembled genome (TnA_NL: 98.70%; TiB_HR: 97.41%) (Table S3, see online supplementary material). In total, 32 457 and 34 940 protein-coding genes were predicted for TnA_NL and TiB_HR, with an average of 5.27 and 5.09 exons per gene, respectively (Table 1; Tables S4 and S5, see online supplementary material).
Table 1.
Summary of genome assembly and annotation of Trapa
| Genomic features | TiB_HR | TiB_YR a | TnA_NL | TnA_WL a | Tn_tetra b |
|---|---|---|---|---|---|
| Species | T. incisa | T. incisa | T. natans | T. natans | T. natans |
| Genome composition | BB | BB | AA | AA | AABB |
| Location/Cultivar | Heilongjiang River | Yangtze River | Nahuling | Wuling | |
| Assembly size (Mb) | 470.61 | 463.97 | 477.43 | 479.9 | 1056.98 |
| Number of scaffolds | 298 | 208 | 33 | 194 | 2873 |
| Number of contigs | 298 | 262 | 249 | 325 | 3854 |
| Contig N50 (Mb) | 12.07 | 13.77 | 6.27 | 13.52 | 3.19 |
| Scaffold N50 (Mb) | 12.07 | 13.77 | 21.23 | 13.52 | 20.8 |
| Genome in chromosomes | 97.84% | 98.14% | 99.70% | 98.01% | 89.30% |
| Number of annotated genes | 34 940 | 33 315 | 32 457 | 33 306 | 68 946 |
| Average number of exons per gene | 5.09 | 5.45 | 5.27 | 5.48 | 5.09 |
| Complete BUSCO | 98.80% | 97.60% | 98.00% | 97.70% | 97.80% |
Construction of the gene-based pangenome of Trapa
Following the protocol of Lu et al. [10], we distinguished two subgenomes, termed ‘A-subgenome’ vs. ‘B-subgenome’, within the published genome of tetraploid T. natans [10]. In total, six assembled (sub)genomes, including two genomes of diploid T. natans (TnA_NL and TnA_WL) (this study), two genomes of T. incisa (TiB_HR and TiB_YR) [11], and two subgenomes of allotetraploid T. natans (TnAt and TnBt) [10], were used to construct a gene-based pangenome (Table 1). Sequence similarity analysis classified the predicted genes from the six (sub)genomes into 39 252 non-redundant gene clusters. Of these gene clusters, 18 859 (48.05%), 11 352 (28.92%) and 9041 (23.03%) were defined as core gene clusters, dispensable gene clusters, and private gene clusters, respectively (Fig. 1a and c). As shown in Fig. 1b, the number of core genes found in all genomes decreases as the number of genomes increases. The dispensable and private gene clusters occupied a large proportion (51.95%) of the total cluster sets in the six accessions. However, in individual accessions, the genes from the dispensable and private gene clusters averaged only 26.38% of the total genes (Fig. 1d). In addition, all six (sub)genomes contained a similar proportion of core (72.42–74.41%), dispensable (20.27–24.32%), and private (3.09–5.32%) genes (Fig. 1d).
Figure 1.
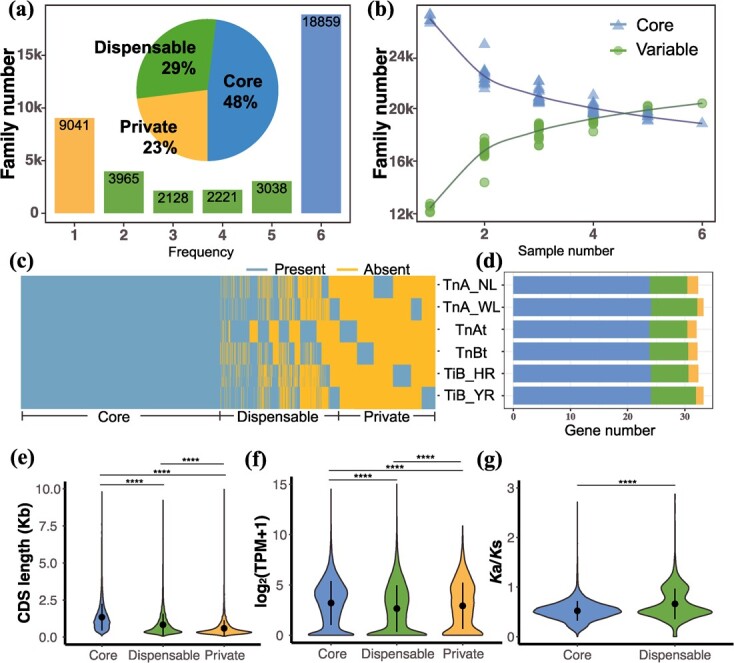
Composition and characteristics of the gene-based pangenome of Trapa. (a) Compositions of the gene-based pangenomes. The histogram shows the number of gene family clusters in the six (sub)genomes (diploid T. natans: TnA_NL and TnA_WL; diploid T. incisa: TiB_HR and TiB_YR; allotetraploid T. natans: TnAt vs. TnBt) with different frequencies. The pie chart shows the proportion of core, dispensable, and private gene family clusters. (b) Variation of variable (i.e., dispesable and private gene family clusters) and core gene family clusters with the number of water caltrop genomes increasing. (c) Presence and absence information of pan-gene family clusters in the six water caltrop (sub)genomes. (d) The number of classified genes in each (sub)genome. Summary of individual gene characteristics per gene cluster in terms of (e) CDS length, (f) expression level, and (g)Ka/Ks values of each gene in core, dispensable, and private gene clusters. Asterisks denote statistical significance across three types of gene clusters.
The average lengths of the dispensable (832 bp) and private (590 bp) genes were significantly shorter than those of the core genes (1349 bp) (P < 2.2e-16, Wilcoxon rank-sum test) (Fig. 1e). Based on our RNA sequencing (RNA-seq), core genes [3.21 log2(TPM + 1)] had significantly higher expression levels than dispensable genes [2.65 log2(TPM + 1), P < 2.2e-16] and private genes [2.92 log2(TPM + 1), P = 8.3e-13, Wilcoxon rank-sum test] (Fig. 1f). Interestingly, a significantly higher ratio of non-synonymous/synonymous nucleotide substitutions (Ka/Ks) was observed in dispensable as compared to core genes (1.71 vs. 0.54, P < 2.2e-16, Wilcoxon rank-sum test) (Fig. 1g). Gene Ontology (GO) enrichment analysis indicated that core genes were significantly enriched for essential functions, including photoperiodism (e.g., GO:0048573; GO:0009658), meristem (e.g., GO:0000725; GO:0061982), and several terms related to chromosome organization (e.g., GO:0051276; GO:0098813) (Fig. S6 and Table S6, see online supplementary material). In contrast, dispensable genes were significantly enriched in biological processes related to root development (e.g., GO:0048527; GO:0080022), biosynthesis of secondary metabolites (e.g., GO:0009787; GO:0009699), and response to abiotic stress (GO:0071456; GO:0009416) (Fig. S7 and Table S7, see online supplementary material).
Comparison of TEs between allotetraploid T. natans, diploid T. natans, and T. incisa
TEs are among the most variable parts of the genome and may be important drivers of rapid adaptation and species divergence (e.g. [26, 27]). To examine the differences in TE distribution between species, we constructed a pan-TE library of the water caltrop pangenome, including a total of 1616 TE families, and re-annotated the six (sub)genomes (Fig. 2a; Table S8, see online supplementary material). As a result, TE and non-TE repeats accounted for 55.05% to 56.44% of the (sub)genomes (average: 55.77%), whereby long terminal repeat retrotransposons (LTR-RTs) were predominant (average: 40.16%; Fig. 2b; Table S9, see online supplementary material). The two main classes of of LTR-RTs were Gypsy (RLG) and Copia (RLC) elements, making up 37.20% and 1.17% of the (sub)genomes, respectively. For each (sub)genome, the size of TE families varied from a single element to 30 665 elements, with the 10 largest families accounting for an average of 52.39% of all TEs (Fig. 2a; Table S8, see online supplementary material).
Figure 2.
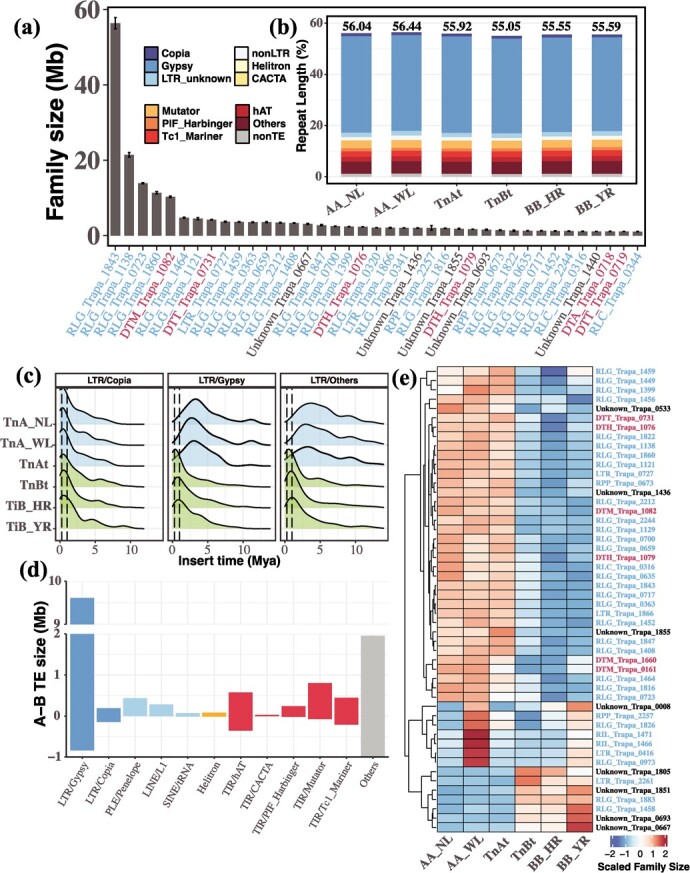
The landscape and insertion times of transposable elements (TEs) of Trapa. (a) Mean size of the 39 largest TE families (>1 Mb) across the six (sub)genomes (diploid T. natans: TnA_NL and TnA_WL; diploid T. incisa: TiB_HR and TiB_YR; allotetraploid T. natans: TnAt snd TnBt). The x-axis indicates the names of the TE families (blue: retrotransposons; red: DNA transposons). The error bars denote the standard deviation among the six (sub)genomes of Trapa. (b) Length (%) of repetitive elements per (sub)genome, as inferred by panEDTA annotation. (c) Estimated insertion times (in million years ago, Mya) of full-length long terminal repeat (FL-LTR) retrotransposons (Copia, Gypsy, and others) for each (sub)genome. Density distributions represent the A- (TnA_NL, TnA_WL, and TnAt) and B- (TiB_HR, TiB_YR, and TnBt) lineages, respectively. The two dashed lines represent the inferred times of the allotetraploidization (left, c. 0.27 Mya) and the divergence between diploid T. incisa and T. natans (right, c. 1 Mya). (d) Size differences in major TE families between the A- and B-lineages. Positive values represent families that are larger in the A-lineage than in the B-lineage, while negative values represent those that are larger in the B-lineage than in the A-lineage. (e) Heatmap of the scaled sizes of the 50 most different TE families between A- and B-lineages. Column names in blue vs. red indicate retrotransposons vs. DNA transposons.
The vast majority of TE families (1564 out of 1616, 96.78%) were consistently observed across the six (sub)genomes, but their sizes varied widely. For the A (i.e., TnA_NL, TnA_WL and TnAt) and B (i.e., TiB_HR, TiB_YR and TnBt) lineages, 164 TE families differed by at least 10 kb in size, of which 127 families (77.44%, including 57 Gypsy families) were larger in A-lineage as compared to B-lineage (Fig. 2d and e; Table S8, see online supplementary material). Burst time analysis of intact LTR-RTs revealed that Copia families in both A- and B-lineages experienced a recent burst (or expansion) at c. 0.29 million years ago (Mya) (Fig. 2c), possibly coinciding with a recent allopolyploidization event (c. 0.27 Mya) [10]. However, the expansion of the Gypsy families occurred much earlier in the A-lineage (c. 3.04 Mya) than in the B-lineage (c. 0.48 Mya) (Fig. 2c).
Construction of the graph-based pangenome
We constructed a graph-based pangenome for Trapa using the assembled (sub)genomes. To this aim, we chose the genome of diploid T. natans (TnA_NL) as the ‘backbone’, and identified insertions and deletions (≥ 50 bp) from the six (sub)genomes with long reads. Subsequently, 211 598 non-redundant PAVs were integrated into a variation graph. The resulting graph-based pangenome of Trapa spanned 558.12 Mb, of which about 80.69 Mb were absent from the TnA genome. To validate the quality of the graph-based pangenome, the paired-end short reads of 57 individuals, including allotetraploid T. natans and diploid T. natans/T. incisa from our previous study [10], were mapped on the graph and linear genomes, respectively. On average, 92.42% of the reads were properly mapped on the graph, which was much higher than that obtained from linear-genome mapping (79.18–90.52%; Fig. 3a). Moreover, our simulation studies showed that the graph-based mapping had either higher precision or higher recall relative to the linear mapping (Fig. 3b; Fig. S8, see online supplementary material).
Figure 3.
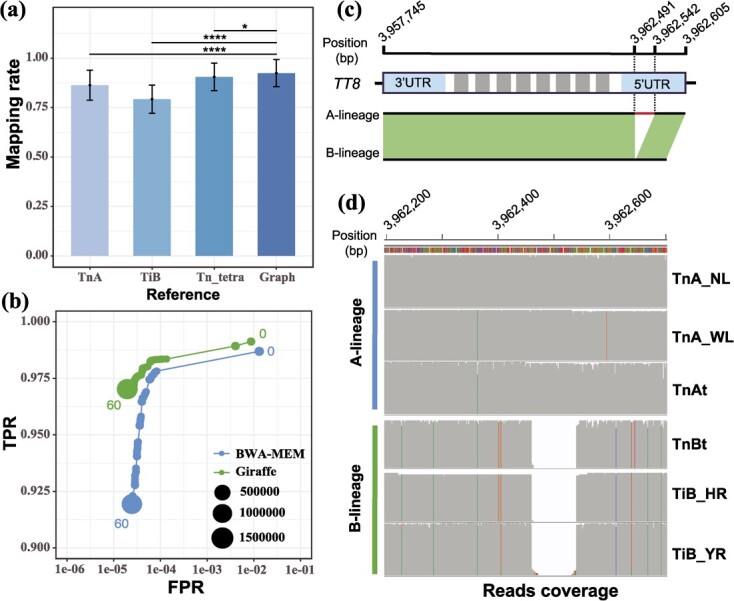
Comparison of mapping accuracy between graph and linear genomes. (a) The genome mapping rate of re-sequenced individuals with respect to three reference genomes of diploid T. natans (TnA), diploid T. incisa (TiB), and allotetraploid T. natans (Tn_tetra) and the graph-based pangenome of Trapa, respectively. (b) Receiver operating characteristic (ROC) graph illustrating true-positive vs. false-positive rates (TPRs vs. FPRs) at different mapping quality thresholds (i = 0–60) for graph-based and linear genome mapping approaches, respectively. The size of each circle is proportional to the log-scaled number of reads with the respective mapping quality. (c) An 80-bp deletion is located within the 5’UTR of the TT8 gene of B lineage. The blue and grey boxs represent UTRs and exons, respectively. (d) Validation of the deletion based on the mapping of Pac Bio long reads onto the genome assembly of TnA_NL.
As PAVs can largely contribute to the observed phenotypic variations (reviewed in [28]), we further genotyped the PAVs among six (sub)genomes based on the water caltrop graph-based pangenome. After filtering, 156 616 PAVs were retained, of which 40 453 (25.90%) were differentiated between A- and B-lineages. A total of 2570 genes were found to contain inter-lineage PAVs. These genes were mainly enriched in GO terms involving three biological processes/physiological pathways, i.e., organ development process, organic substance metabolic process and response to stimulus (Table S10, see online supplementary material). Not unexpectedly, a subset of these genes with PAVs were found to play a crucial role in phenotypic divergence and reproductive isolation between two diploids TnA and TiB (Fig. 3c and d; Fig. S1 and Table S11, see online supplementary material). For example, 102 genes (e.g., TT8, CRY2, GED1, AGL61) were involved in the developmental processes of flower, seed, and fruit, of which PID and CRA1 play a role in asymmetric cotyledon development; 21 genes (e.g., ECT2, PSY1R, SIZ1, ORE15) were identified as potentially related to cell proliferation and expansion during plant organogenesis, which could contribute to the difference in plant size between TnA and TiB. In addition, 12 genes (e.g., SS3, SS4, SPS2F, SBE2.1) were found to be associated with the biosynthesis of sucrose/starch (Table S11, see online supplementary material). In addition, based on gene expression profile differences in four tissues of TnA_NL and TiB_HR, including flower bud (FB), fertilized flower (FF), juvenile fruit (JF), and leaf (L), 1428 genes were found to differentially express in at least one tissue (Table S12, see online supplementary material). Permutation test indicated that the number of differentially expressed genes (DEGs) was significantly higher than expected by chance (P < 2e-16).
Evolutionary trajectory of genes affected by PAVs during polyploidization
To estimate the genomic variation during allopolyploidization in Trapa, we performed pairwise whole-genome alignments of each subgenome and genomes of its presumed diploid progenitors. On average 1 569 241 SNPs, 271 920 small InDels (<50 bp), 6260 deletions (≥50 bp), and 7094 insertions (≥50 bp) were identified in the A-subgenome (TnAt); 3 327 898 SNPs, 433 964 InDels, 12 300 deletions, and 14 213 insertions were detected in the B-subgenome (TnBt) (Fig. 4a; Table S13, see online supplementary material). Besides, we found much more variations between TiB_YR vs. TnBt as compared to TiB_HR vs. TnBt, consistent with the phylogenetic tree of Trapa (Fig. 4b; Table S13, see online supplementary material). Thus, we chose the reference genome of TnA_NL and TiB_HR as representatives for the following comparative analyses of subgenomes.
Figure 4.
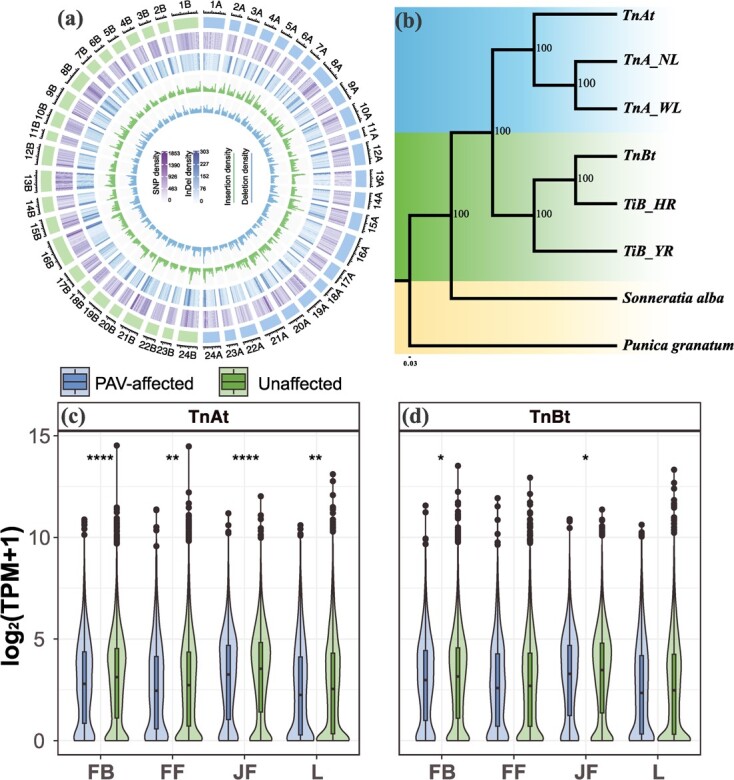
Phylogenetic relationships of Trapa accessions and distribution of genomic variations between subgenomes of allotetraploid T. natans and its progenitor diploid genomes. (a) Circos plot of the 24 chromosomes of TnAt (CHR_1At–24At) and TnBt (CHR_1Bt–24Bt), respectively, shows the density distributions of genomic variations compared to their presumed progenitor diploid genomes (TnA_NL and TiB_HR). SNPs, InDels, PAVs are included in this plot. (b) The maximum likelihood tree of Trapa based on 4150 single-copy orthologous genes. Sonneratia alba and Punica granatum were used as outgroups [29, 30]. (c) and (d) Violin plot of expression levels of PAV-affected genes in (c) the TnAt and (d)TnBt subgenomes for each of four tissues (FB: flower bud; FF: fertilized flower; JF: juvenile fruit; and L:leaf).
We further defined homeologous genes with PAVs occurring within 2 kb upstream and downstream of protein-coding genes as potentially cis-regulated genes. Of the 14 212 single-copy orthologous genes shared among the four (sub)genomes, we identified 3234 homeologous pairs with PAVs. Of them, 1623 genes were shared between the two subgenomes of allotetraploid T. natans (TnAt and TnBt), while 785 and 826 were specific to TnAt and TnBt, respectively. Those potentially PAV-affected genes in both subgenomes related to allopolyploidization had significantly higher Ka/Ks ratios than those for unaffected genes (both Ka/Ks values <0.5; P = 0.001 for TnAt, P = 3.32-6e for TnBt, Wilcoxon rank-sum test) (Fig. S9, see online supplementary material), indicating that these PAV-affected genes underwent relaxed purifying selection. We further evaluated the effects of PAVs on levels of gene expression in four tissues, i.e., flower bud (FB), fertilized flower (FF), juvenile fruit (JF), and leaf (L). The average expression levels of PAV-affected genes were significantly lower than those of the remaining genes in all four tissues of TnAt, especially for flower bud and juvenile fruit (Fig. 4c and d). However, when compared with the unaffected genes, the PAV-affected genes had a significantly lower expression level only in the flower bud and juvenile fruit of TnBt. This suggests that PAVs could have a greater impact on homologous gene expression in TnAt than in TnBt.
Homeologous exchanges and homeolog expression pattern
Homeologous exchange (HE) between the constituent subgenomes is a frequent type of structural variant in polyploids [31]. In this study, we identified 61 HEs in allotetraploid T. natans, including 35 from TnBt to TnAt (5.08 Mb) and 26 from TnAt to TnBt (3.67 Mb) (Fig. 5a and b; Tables S14–S16, see online supplementary material). Functional annotation of genes within HEs revealed that these genes were enriched in organ development [e.g., trichome morphogenesis (GO:0010090), leaf senescence (GO:0010150), and leaf development (GO:0048366)] and stress response processes [e.g., response to light intensity (GO:0009642), cellular response to oxygen-containing compound (GO:1901701), and cellular response to endogenous stimulus (GO:0071495)] (P < 0.05) (for details, Tables S14–S16, see online supplementary material), suggesting a role of asymmetric HEs in biological function.
Figure 5.
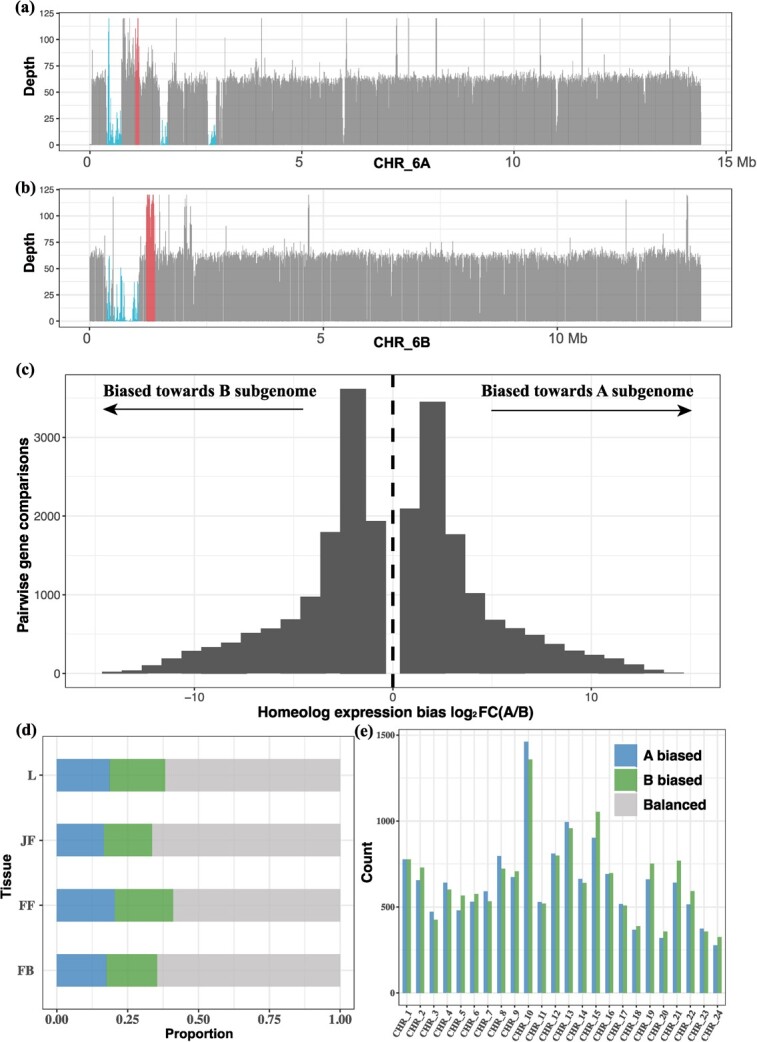
Homeologous exchange (HE) regions and homeologous expression bias (HBE) in the two subgenomes (TnAt vs. TnBt) of allotetraploid Trapa natans (2n = 4x = 96). (a) and (b) HE regions, as exemplified for one chromosome each of the TnAt and TnBt subgenomes. Coverage depth was obtained for CHR_6At (a) and CHR_6Bt chromosomes (b) after mapping Illumina sequence reads from allotetraploid T. natans on the diploid T. natans and T. incisa genome assemblies concatenated together. The regions with red lines represent windows whose coverage depth was 1.5 times greater than the whole-genome average depth, while the blue lines show deletions with low or no coverage. (c) Histograms of genome-wide expression of syntenic homeologous genes among the TnAt and TnBt subgenomes. (d) and (e) Pairwise comparisons between syntenic gene pairs among four tissues (FB: flower bud; FF,: fertilized flower; JF: juvenile fruit; L: leaf) (d) and among the 24 chromosome pairs (e).
Based on the expression profiles of the four tissues described above (flower bud, fertilized flower, juvenile fruit, and leaf), we tested for signatures of homeolog expression bias (HEB) in allotetraploid T. natans. Of the 24 966 syntenic gene pairs between the TnAt and TnBt subgenomes, 10 626 and 2241 displayed HEB in at least one tissue and all sampled tissues, respectively (Table S17, see online supplementary material). Based on pairwise comparisons between syntenic gene pairs, there was a slight bias in expression level towards the B-subgenome across all four tissues and most chromosome pairs (P = 0.022, Wilcoxon rank-sum test) (Fig. 5c–e; Fig. S10, see online supplementary material). At the chromosome level, a total of 13 pairs of chromosomes showed significant HEB (P < 0.05, Wilcoxon rank-sum test), and eight of those showed a bias in expression level towards the B-subgenome (Fig. 5e).
Discussion
Considerable efforts are essential in the exploitation of sustainable food supply to meet the upcoming food production challenge [4, 21]. The cultivated diploid Trapa natans, containing about 67.5% high-quality starch of the fruit’s dry weight [32, 33], is a potential secure food source, especially in wasteland regions [8]. Even though the previously published reference genomes of allopolyploid T. natans, diploid T. natans and T. incisa provided strong support for its allotetraploid origin and domestication of diploid T. natans [10, 11], it does not capture fully the genetic variability within water caltrop.
In this study, we have constructed a gene-based pangenome dataset for Trapa. Based on this, the proportion of core gene clusters predicted in Trapa (48.05%) proved to be higher than those of soybean (35.87%, [18]), rice (30.58%, [19]), and sorghum (36%, [23]). This might be due to a combination of its relatively recent speciation and the limited size of genome assemblies. Nevertheless, the constructed Trapa pangenome exhibited extensive variation in gene content, as indicated by the prediction of 28.92% dispensable and 23.03% private gene clusters within the six (sub)genomes. Based on GO enrichment analyses, the core genes were mainly comprised of conserved and housekeeping genes (Fig. 1c–e; Fig. S6, see online supplementary material), while these dispensable genes were primarily related to biological processes related to organ development, metabolism, and biotic and abiotic stress response (Fig. S7, see online supplementary material). This implies that dispensable genes might have a crucial role in phenotypic variation and adaptation to abiotic/biotic stresses of water caltrop. Similar patterns are also found in the pangenomes of other crops, including rice [19], Brassica species [34, 35] sesame [24], soybean [18], and pigeon pea [25]. Interestingly, although we observed that dispensable genes had relatively lower expression level compared with core genes (Fig. 1f), their significantly higher Ka/Ks values (Fig. 1g) suggest that dispensable genes are evolving faster due to relaxed functional constraints, likely promoting speciation and phenotypic divergence (Fig. S7, see online supplementary material).
By not only focusing on SNPs and small InDels, many recent studies have also found that structural variations (SVs) play a major role in species diversification and phenotype variation in both plants and animals [14, 36, 37]. In our study, when compared to T. incisa (BB), diploid T. natans (AA) has stouter stems with larger fruits (seeds), leaves, and flowers and more vigorous roots (Fig. S1, see online supplementary material) [38]. To precisely identify the potential PAVs that may contribute to reproductive isolation and phenotypic divergence between T. incisa and diploid T. natans, we have constructed a graph-based pangenome based on 211 598 non-redundant PAVs from the six (sub)genomes. Consistent with patterns observed in recent studies on humans [39], yeasts [39], cows [40], and tomatoes [41], both the mapping rate and mapping accuracy of our graph-based pangenome of Trapa are much higher than that of the corresponding linear genome (Fig. 3a and b; Fig. S8, see online supplementary material). As expected, 2570 genes with essential functions were associated with PAVs specific to A- or B-lineages. For example, an 80-bp deletion within 5’UTR of TT8 was found to be specific to B-lineage (Fig. 3c and d). TT8, a bHLH transcription factor, has been proven to affect post-zygotic reproductive barrier in Arabidopsis thaliana [42]. It was also differentially expressed between the two lineages among four tissues of Trapa (Table S12, see online supplementary material). Besides, some genes under positive selection in cultivated water caltrop (such as PID and CRA1 [10]) were also found to be associated with PAVs (Table S11, see online supplementary material). This suggests that these genes are likely related to speciation and domestication. Clearly, however, in addition to the PAV information provided herein, further research is required to complement the graph-based pangenome of Trapa with other types of genomic SVs (e.g., copy number variations, inversions, and chromosomal rearrangements) as well as single nucleotide polymorphisms (SNPs) and small InDels, etc. [39].
Our findings have also revealed that the B-subgenome (TnBt) is the dominant one in allotetraploid T. natans based on inferences in four important respects. Firstly, when compared to TnBt, TnAt subgenome lost more genes but retained more TEs during polyploidization (Fig. 2a; Tables S9 and S18, see online supplementary material), suggesting this TE-rich subgenome may experience more gene losses, and eventually become a recessive subgenome due to ongoing gene loss [43]. Similar patterns have been observed in the genome of tetraploid broomcorn millet (Panicum miliaceum), one of the earliest domesticated crops [44]. Besides, PAVs were found to have a greater impact on homologous gene expression in TnAt than in TnBt (Fig. 4c and d). Further, partitioning of homeolog gene expression is largely established in allotetraploid T. natans with the presence of slight bias towards the B-subgenome across four tissues and most chromosome pairs (Fig. 5 c–e). Such gene expression variation between the two subgenomes may contribute to the increased complexity of regulatory networks after allopolyploidization (see also [43]). Finally, homeologous exchanges from TnBt to TnAt were found to be more frequent than the reverse (Table S14, see online supplementary material). These findings indicate that multiple factors, including PAVs, asymmetrical amplification of Tes, Hes, and homeolog expression divergence, together affect a route for genome evolution after polyploidization. PAV-affected genes in both subgenomes were found to be under natural selection, and some genes related to Hes were enriched in organ development and stress response processes. These genes might have contributed to the vigour and broad adaptation of allotetraploid T. natans (Tables S14–S16, see online supplementary material). In summary, the pangenome of Trapa affords a platform for a thorough exploration of genomic variation of Trapa species, thereby promoting a better understanding of the evolutionary and functional genomics of this currently underutilized crop variation.
Materials and methods
Plant materials
For genome sequencing, the plant sample of diploid T. incisa was collected from Xingkai Lake National Nature Reserve (132.32°E, 45.37°N), and that of diploid T. natans was collected at Jiaxing Academy of Agricultural Science (120.69°E, 30.86°N). For genome annotation of T. incisa, both Illumina short-read and PacBio long-read RNA sequencing were performed. The long-read transcriptome data were derived from the evenly mixed sample of six tissues, while short-read transcriptome data were generated from different tissues separately (Table S19, see online supplementary material). For genome annotation of T. natans, previously released short-read transcriptome sequencing data (SRR14597430–SRR14597415, [10]) separately derived from different tissues were used (Table S19, see online supplementary material).
Genome assembly and quality assessment
Both genomes were assembled using a hybrid strategy that combined PacBio long reads, Illumina short reads, and a Hi-C chromatin interaction map. After quality controlling, 23.57 Gb of PacBio HiFi data, 55.73 Gb of Illumina data, and 40 Gb of Hi-C data (Table S20, see online supplementary material) were used for de novo genome assembly of T. incisa.Firstly, basing on k-mer frequency distribution analysis, Illumina short reads were used to estimate genome size and heterozygosity of this individual with jellyfish v.2.3.0 [45] and genomescope2 [46]. Then, PacBio HiFi reads were subjected to draft assembly with default parameters using hifiasm v.0.16.0 [47]. Finally, the Hi-C clean reads were aligned to the draft assembly with bwa-mem [48]. Allhic v.0.9.8 [49] was applied to perform genome assembly at the chromosome level using the corrected contigs. Juicebox tool v.2.12 [50] was applied to adjust chromosome construction manually. Benchmarking Universal Single-Copy Orthologs (busco v.4.0.5) [51] and Core Eukaryotic Genes Mapping Approach (cegma v.2.5) [52] were applied to evaluate the completeness of the genome assembly. In addition, Illumina reads were aligned to the reference genome to assess the mapping rate.
For diploid T. natans, 127 Gb of PacBio Continuous Long Reads (CLR) data, 33.7 Gb of Illumina data, and 40 Gb of Hi-C data were used for de novo genome assembly (Table S20, see online supplementary material). The clean reads were subjected to self-correction, trimming, and assembly using canu v.2.2 [53]. Afterwards, error-corrected contigs were assessed and anchored onto chromosomes, using the same pipeline as mentioned above.
Annotations of transposable elements (TEs) and gene models
To predict the TEs in Trapa genomes, we first constructed a de novo TE library for each of the six (sub)genomes using the exstensivede-novote annotator (edta v.2.1 [27, 54] with parameters ‘--overwrite 1 --sensitive 1 --anno 1 --evaluate 0’. Classifications of these TE libraries were refined using the script deepte.py with the predefined plant model [55]. Then, the script panedta.sh [27] was performed to generate the pan-TE library by eliminating low-copy, incomplete, and redundant sequences across individual TE libraries. Finally, this filtered pan-TE library was used to re-annotate all genomes with consistent TE family IDs, including both structurally intact and fragmented TEs.
Prediction of protein-coding genes of diploid T. natans and T. incisa was performed using the repeat-masked genome with three distinct approaches, i.e., ab initio gene prediction, homology-based prediction, and transcriptome-assisted annotation. For the ab initio gene prediction, augustus v.3.2.3 [56], glimmerhmm v.3.0.4 [57], genscan v.1.0 [58], snap v.2013.11.29 [59], and geneid v.1.4.4 [60] were applied. For the homology-based gene prediction, the protein-coding sequences from allotetraploid T. natans (PRJNA725399 [10]), Corymbia citriodora (PRJNA234431 [61]), Eucalyptus grandis (AUSX00000000 [62]), and Punica granatum (PRJNA355913 [63]) were mapped to the assembled genomes using tblastn v.2.2.26 (E-value ≤1e-5 [64]) to obtain high-quality protein structures. To perform transcripts-assistant annotation, PacBio ISO-seq and Illumina short-read RNA-seq data were used for T. incisa (see details in Table S19, see online supplementary material). Smrtlink v.11.1 (https://www.pacb.com/support/software-downloads/) and isoseq v.3 (https://github.com/PacificBiosciences/IsoSeq) were used to extract isoform sequences and process the polished consensus sequences. A total of 54.97 Gb PacBio ISO-Seq reads were directly mapped to the genome of T. incisa by gmap v.2021-12-17 [65]. The short-read RNA-seq data were aligned to the genome sequence with tophat v.2.0.11 [66]. Subsequently, the mapped reads were assembled into longer transcripts using cufflinks v.2.2.1 [67]. The transcripts from all tissues were merged and subjected to transdecoder in pasa v2.4.1 [68] to predict and filter protein-coding sequencing. Only complete transcripts were retained for further analysis. For the diploid T. natans, only previously released short-read transcriptome data were used to perform transcriptome-assisted annotation with the same pipeline as mentioned above (Table S19, see online supplementary material). All genes predicted by the above methods were integrated into a non-redundant gene set using evidencemodeler (evm) v.1.1.1 [68]. The EVM-predicted genes were further updated with pasa v.2.4.1 [68] to predict the untranslated regions and alternative splicings. The resulting protein models were functionally annotated according to the best matches with proteins deposited in go, kegg, swiss-prot, trembl and a non-redundant protein database using blastp (E-value = 1e-5).
Construction of the gene-based pangenome of Trapa
To identify the core/dispensable/private gene sets, we clustered gene families using orthofinder v.2.2.7 [69]. Firstly, the genes containing coding sequence (CDS) with 100% similarity to other genes were removed using the cd-hit-est implemented in cd-hit v.4.8.1 [70] toolkit for each accession with parameters ‘–c 1 –aS 1’. Protein sequences of the remaining genes were then subjected to homologous searching by dimand balstp v.2.0.11.149 [71], with an E-value cutoff of 1e−5. Based on the results of this latter search, orthofinder was used for gene family clustering, with parameters ‘percentMatchCutoff = 50’ and ‘-I 1.5’. Gene clusters shared among all six accessions were defined as core gene clusters, while the gene clusters missed in more than one accessions were defined as dispensable gene clusters, and those only existed in single accession were defined as private gene clusters.
kaks_calculator v.2.0 [72] and the parallel tool paraat v.2.0 [73] were used to calculate Ka/Ks ratio to estimate the selection pressure acting on core/dispensable/private gene clusters. In addition, we performed GO enrichment analysis for each cluster at GOC website (http://geneontology.org/) under default parameters.
Construction of the graph-based pangenome of Trapa
To construct a graph-based genome for the genus Trapa, we selected the TnA genome of diploid T. natans as the ‘backbone’, as it showed the highest contiguity and completeness among the four (sub)genomes. In a next step, the PacBio long reads of the four sub(genomes) were mapped onto the TnA genome using minimap2 v.2.24 [74]. PAVs were called using cutesv v.1.0.13 [75] with the suggested parameters. All PAVs across the four (sub)genomes were merged according to instructions provided on GitHub (https://github.com/vgteam/giraffe-sv-paper/blob/master/scripts/sv). Finally, the final graph-based genome of Trapa was constructed using the variation graph (vg v.1.38.0) toolkit pipeline [39].
Mapping accuracy of graph-based genome was assessed following the pipeline of human research (https://github.com/vgteam/giraffe-svpaper/blob/master/scripts/read_simulation/). Firstly, one million read pairs were simulated and mapped to the graph and linear genomes by vg giraffe [39] and bwa-mem [48], respectively. Subsequently, we calculated the cumulative true-positive rate (TPR) and false-positive rate (FPR) at different mapping quality thresholds to assess mapping sensitivity and specificity. The results were finally visualized as the receiver operating characteristic (ROC) curve.
Assessment of genomic variation between diploids and subgenomes
The subgenomes of allotetraploid T. natans, respectively, were aligned to their progenitor diploid genomes using the nucmer program implemented in the mummer toolkit v.4.0.0 [76] with parameters ‘-maxmatch -c 100 -l 50’. The alignment results were filtered using the delta-filter program of mummer with only one-to-one alignment blocks retained using parameters ‘-1 -l 1000’. In addition, SNPs and InDels (<50 bp) were identified using show-snps (with potion ‘-clrt’) from mummer v.4.0.0 [76], and the cutesv pipeline (see above) was used to identify large insertions/deletions ( 50 bp).
50 bp).
Gene expression analysis and identification of homeolog expression bias (HEB)
Transcriptome sequencing encompassed the use of three biological replicates of the flower buds (FB), fertilized flowers (FF), juvenile fruits (JF), and leaves (L) of diploid T. natans, allotetraploid T. natans, and T. incisa. Clean reads of RNA-seq were aligned to the allotetraploid T. natans genome, using star v.2.7.9a [77]. Only uniquely mapped reads were retained and used for counting reads of the annotated genes with featurecounts v.2.0.3 [78].
HEB was identified across all 1:1 homeologous gene pairs using deseq2 v.4.2 [79]. Comparisons were made between the two homeologous gene pairs for each tissue. We performed library size normalization for gene counts with the ‘Relative Log Expression’ normalization (RLE) method, as implemented in the deseq2 package. Log 2 fold change of a given gene was calculated using the Built-in Wald test. The genes with false discovery rate (FDR) corrected P-value <0.05 were considered significantly biased. DEGs between diploid T. natans and T. incisa were identified using deseq2 v.4.2.
Identify HE regions between two subgenomes
Segmental HE regions between the two subgenomes of allotetraploid T. natans were identified according to the method of ‘assessment of read depth’ as successfully applied in the ‘Brassica napus genome project’ [31]. Briefly, Illumina paired-end reads of allotetraploid T. natans were mapped to diploid T. natans and T. incisa concatenated together. The average sequencing depth was calculated based on 10 kb non-overlapping window using bedtools v.2.30.0 [80]. Windows with read depth 1.5 times being greater than the average mapping depth of the whole genome were considered as candidate duplications generated by HEs. The windows with low or no coverage were regarded as deletions. Moreover, if adjacent windows had a depth above the set threshold and were within 50 kb (five windows), they were linked together. Only regions that extended beyond 80 kb were considered as probable HEs (see also [31]).
Supplementary Material
Acknowledgements
We thank Lin Cheng (Agricultural Genomics Institute at Shenzhen) and Shuo Cao (Agricultural Genomics Institute at Shenzhen) for their assistance with data analyses; Emmanuel Nyongesa Waswa (Wuhan Botanical Garden) for his assistance with polishing the manuscript; and two anonymous referees for valuable advice and comments that have substantially improved the manuscript. This work was supported by the collaborative program of Chinese Academy of Agricultural Sciences (CAAS)-Jinhua Academy of Agricultural Sciences, funded by Jinhua City of Zhejiang Province, and the Research Grant from Wuhan Botanic Garden (E1559901)
Contributor Information
Xinyi Zhang, Systematic and Evolutionary Botany and Biodiversity Laboratory, College of Life Sciences, Zhejiang University, Hangzhou, 310058, Zhejiang, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, 430074, Hubei, China.
Yang Chen, Systematic and Evolutionary Botany and Biodiversity Laboratory, College of Life Sciences, Zhejiang University, Hangzhou, 310058, Zhejiang, China; CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, 430074, Hubei, China.
Lingyun Wang, Provincial Key Laboratory of Characteristic Aquatic Vegetable Breeding and Cultivation, Jinhua Academy of Agricultural Sciences (Zhejiang Institute of Agricultural Machinery), Jinhua, 321000, Zhejiang, China.
Ye Yuan, Jiaxing Academy of Agricultural Sciences, Jiaxing, 314016, Zhejiang, China.
Mingya Fang, Provincial Key Laboratory of Characteristic Aquatic Vegetable Breeding and Cultivation, Jinhua Academy of Agricultural Sciences (Zhejiang Institute of Agricultural Machinery), Jinhua, 321000, Zhejiang, China.
Lin Shi, Provincial Key Laboratory of Characteristic Aquatic Vegetable Breeding and Cultivation, Jinhua Academy of Agricultural Sciences (Zhejiang Institute of Agricultural Machinery), Jinhua, 321000, Zhejiang, China.
Ruisen Lu, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing 210014, Jiangsu, China.
Hans Peter Comes, Department of Environment & Biodiversity, Salzburg University, Salzburg, 5020, Austria.
Yazhen Ma, CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, 430074, Hubei, China.
Yuanyuan Chen, CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, 430074, Hubei, China.
Guizhou Huang, State Key Laboratory of Tropical Crop Breeding, Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture; Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518124, Guangdong, China.
Yongfeng Zhou, State Key Laboratory of Tropical Crop Breeding, Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture; Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518124, Guangdong, China.
Zhaisheng Zheng, Provincial Key Laboratory of Characteristic Aquatic Vegetable Breeding and Cultivation, Jinhua Academy of Agricultural Sciences (Zhejiang Institute of Agricultural Machinery), Jinhua, 321000, Zhejiang, China.
Yingxiong Qiu, CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, 430074, Hubei, China.
Author contributions
Y.Q. and Z.Z. conceived the project. X.Z., Y.C., and Y.Q. performed experiments and coordinated research activities. X.Z., Y.C., L.W., Y.Y., L.S., and M.F. collected the samples. X.Z. analysed data. X.Z. and Y.Q. drafted the manuscript. Y.Q., H.P.C., Y.Z., and Y.M. revised and finalized the manuscript. All authors read and approved the manuscript. X.Z. and Y.C. contributed equally to this work.
Data availability
The whole genome sequencing data for diploid T. natans have been deposited under NCBI BioProject PRJNA932942 and GSA BioProject PRJCA016421. The whole genome sequencing data for diploid T. incisa have been deposited under NCBI BioProject PRJNA933001 and GSA BioProject PRJCA016421. The transcriptome sequencing data of diploid T. incisa and allotetraploid T. natans have been deposited under NCBI BioProject PRJNA941110 and PRJNA731291, respectively. The re-sequencing (57 accessions) and transcriptome sequencing data of diploid Trapa natans for genome annotation were obtained from the NCBI BioProject PRJNA725399 of Lu et al. [10].
Conflict of interests
All authors confirm that they have no conflict of interest.
References
- 1. Jain S, Dutta GS, eds. Neglected and Underutilized Crops - Towards Nutritional Security and Sustainability, Biotechnology of Neglected and Underutilized Crops. Dordrecht: Springer; 2012: p. v. [Google Scholar]
- 2. Chang Y, Liu H, Liu M. et al. The draft genomes of five agriculturally important African orphan crops. GigaScience. 2019;8:giy152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li X, Yadav R, Siddique KHM. Neglected and underutilized crop species: the key to improving dietary diversity and fighting hunger and malnutrition in Asia and the Pacific. Front Nutr. 2020;7:593711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ye C, Fan L. Orphan crops and their wild relatives in the genomic era. Mol Plant. 2021;14:27–39 [DOI] [PubMed] [Google Scholar]
- 5. Dawson IK, Powell W, Hendre P. et al. The role of genetics in mainstreaming the production of new and orphan crops to diversify food systems and support human nutrition. New Phytol. 2019;224:37–54 [DOI] [PubMed] [Google Scholar]
- 6. Takano A, Kadono Y. Allozyme variations and classification of Trapa (Trapaceae) in Japan. Aquat Bot. 2005;83:108–18 [Google Scholar]
- 7. Ding B, Jin X. Taxonomic notes on genus Trapa L. (Trapaceae) in China. Guihaia. 2015;40:1–15 [Google Scholar]
- 8. Hoque A, Davey MR, Arima S. Water chestnut: potential of biotechnology for crop improvement. J New Seeds. 2009;10:180–95 [Google Scholar]
- 9. Guo Y, Wu R, Sun G. et al. Neolithic cultivation of water chestnuts (Trapa L.) at Tianluoshan (7000-6300 cal BP), Zhejiang Province, China. Sci Rep. 2017;7:16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu R, Chen Y, Zhang X. et al. Genome sequencing and transcriptome analyses provide insights into the origin and domestication of water caltrop (Trapa spp., Lythraceae). Plant Biotechnol J. 2022;20:761–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qu M, Fan X, Hao C. et al. Chromosome-level assemblies of cultivated water chestnut Trapa bicornis and its wild relative Trapa incisa. Scientific Data. 2023;10:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaut BS, Seymour DK, Liu Q. et al. Demography and its effects on genomic variation in crop domestication. Nature Plants. 2018;4:512–20 [DOI] [PubMed] [Google Scholar]
- 13. Hämälä T, Wafula EK, Guiltinan MJ. et al. Genomic structural variants constrain and facilitate adaptation in natural populations of Theobroma cacao, the chocolate tree. Proc Natl Acad Sci. 2021;118:e2102914118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kou Y, Liao Y, Toivainen T. et al. Evolutionary genomics of structural variation in asian rice (Oryza sativa) domestication. Mol Biol Evol. 2020;37:3507–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Della Coletta R, Qiu Y, Ou S. et al. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021;22:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Danilevicz MF, Tay Fernandez CG, Marsh JI. et al. Plant pangenomics: approaches, applications and advancements. Curr Opin Plant Biol. 2020;54:18–25 [DOI] [PubMed] [Google Scholar]
- 17. Torkamaneh D, Lemay M-A, Belzile F. The pan-genome of the cultivated soybean (PanSoy) reveals an extraordinarily conserved gene content. Plant Biotechnol J. 2021;19:1852–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y, Du H, Li P. et al. Pan-genome of wild and cultivated soybeans. Cell. 2020;182:162–176.e13 [DOI] [PubMed] [Google Scholar]
- 19. Qin P, Lu H, Du H. et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell. 2021;184:3542–3558.e16 [DOI] [PubMed] [Google Scholar]
- 20. Zhao Q, Feng Q, Lu H. et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat Genet. 2018;50:278–84 [DOI] [PubMed] [Google Scholar]
- 21. Gui S, Wei W, Jiang C. et al. A pan-zea genome map for enhancing maize improvement. Genome Biol. 2022;23:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li L, Zhang Z, Wang Z. et al. Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Mol Plant. 2022;15:488–503 [DOI] [PubMed] [Google Scholar]
- 23. Tao Y, Luo H, Xu J. et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nature Plants. 2021;7:766–73 [DOI] [PubMed] [Google Scholar]
- 24. Yu J, Golicz AA, Lu K. et al. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol J. 2019;17:881–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao J, Bayer PE, Ruperao P. et al. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol J. 2020;18:1946–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Catlin NS, Josephs EB. The important contribution of transposable elements to phenotypic variation and evolution. Curr Opin Plant Biol. 2022;65:102140 [DOI] [PubMed] [Google Scholar]
- 27. Ou S, Collins T, Qiu Y. et al. Differences in activity and stability drive transposable element variation in tropical and temperate maize. bioRxiv. 2022; 2022.10.09.511471, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mérot C, Oomen RA, Tigano A. et al. A roadmap for understanding the evolutionary significance of structural genomic variation. Trends Ecol Evol. 2020;35:561–72 [DOI] [PubMed] [Google Scholar]
- 29. Graham SA, Hall J, Sytsma K. et al. Phylogenetic analysis of the Lythraceae based on four gene regions and morphology. Int J Plant Sci. 166:995–1017 [Google Scholar]
- 30. Berger BA, Kriebel R, Spalink D. et al. Divergence times, historical biogeography, and shifts in speciation rates of Myrtales. Mol Phylogenet Evol. 2016;95:116–36 [DOI] [PubMed] [Google Scholar]
- 31. Chalhoub B, Denoeud F, Liu S. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–3 [DOI] [PubMed] [Google Scholar]
- 32. Alfasane MA, Khondker M, Rahman MM. Biochemical composition of the fruits of water chestnut (Trapa bispinosa Roxb.). Dhaka Univ J Biol Sci. 2011;20:95–8 [Google Scholar]
- 33. Subbahmanyan V, Rama RG, Kuppuswamy S. et al. Nutritive value of water chestnut (Singhara). Bull Cent Food Tech Res Inst. 1954;3:134–5 [Google Scholar]
- 34. Hurgobin B, Golicz AA, Bayer PE. et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018;16:1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bayer PE, Scheben A, Golicz AA.et al. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol J. 2021,19:2488–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li A, Wang J, Sun K. et al. Two reference-quality sea snake genomes reveal their divergent evolution of adaptive traits and venom systems. Mol Biol Evol. 2021;38:4867–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou Y, Minio A, Massonnet M. et al. The population genetics of structural variants in grapevine domestication. Nature Plants. 2019;5:965–79 [DOI] [PubMed] [Google Scholar]
- 38. Ding B, Jin X.. Taxonomic notes on genus Trapa L. (Trapaceae) in China. Guihaia 2019;40:1–15. [Google Scholar]
- 39. Sirén J, Monlong J, Chang X. et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science. 2021;374:abg8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crysnanto D, Pausch H. Bovine breed-specific augmented reference graphs facilitate accurate sequence read mapping and unbiased variant discovery. Genome Biol. 2020;21:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou Y, Zhang Z, Bao Z. et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature. 2022;606:527–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zumajo-Cardona C, Aguirre M, Castillo-Bravo R. et al. Maternal control of triploid seed development by the TRANSPARENT TESTA 8 (TT8) transcription factor in Arabidopsis thaliana. Sci Rep. 2023;13:1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bird KA, VanBuren R, Puzey JR. et al. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 2018;220:87–93 [DOI] [PubMed] [Google Scholar]
- 44. Sun Y, Liu Y, Shi J. et al. Biased mutations and gene losses underlying diploidization of the tetraploid broomcorn millet genome. Plant J. 2023;113:787–801 [DOI] [PubMed] [Google Scholar]
- 45. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng H, Concepcion GT, Feng X. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18:170–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vasimuddin M, Misra S, Li H. et al. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. IEEE International Parallel and Distributed Processing Symposium. 2019;314–24 [Google Scholar]
- 49. Zhang X, Zhang S, Zhao Q. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants. 2019;5:833–845 [DOI] [PubMed] [Google Scholar]
- 50. Durand NC, Robinson JT, Shamim MS. et al. Juicebox provides a visualization system for hi-C contact maps with unlimited zoom. Cell Systems. 2016;3:99–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Simão FA, Waterhouse RM, Ioannidis P. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2 [DOI] [PubMed] [Google Scholar]
- 52. Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7 [DOI] [PubMed] [Google Scholar]
- 53. Koren S, Walenz BP, Berlin K. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ou S, Su W, Liao Y. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yan H, Bombarely A, Li S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics. 2020;36:4269–75 [DOI] [PubMed] [Google Scholar]
- 56. Stanke M, Keller O, Gunduz I. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–9 [DOI] [PubMed] [Google Scholar]
- 58. Burge C, Karlin S. Prediction of complete gene structures in human genomic. J Mol Biol. 1997;268:78–94 [DOI] [PubMed] [Google Scholar]
- 59. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2007;18:4.3.1–4.3.28. [DOI] [PubMed] [Google Scholar]
- 61. Healey AL, Shepherd M, King GJ. et al. Pests, diseases, and aridity have shaped the genome of Corymbia citriodora. Commun Biol. 2021;4:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Myburg AA, Grattapaglia D, Tuskan GA. et al. The genome of Eucalyptus grandis. Nature. 2014;510:356–62 [DOI] [PubMed] [Google Scholar]
- 63. Yuan Z, Fang Y, Zhang T. et al. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol J. 2018;16:1363–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gertz EM, Yu Y-K, Agarwala R. et al. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–75 [DOI] [PubMed] [Google Scholar]
- 66. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Trapnell C, Roberts A, Goff L. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haas BJ, Salzberg SL, Zhu W. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fu L, Niu B, Zhu Z. et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60 [DOI] [PubMed] [Google Scholar]
- 72. Wang D, Zhang Y, Zhang Z. et al. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom, Proteom Bioinform. 2010;8:77–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang Z, Xiao J, Wu J. et al. ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem Biophys Res Commun. 2012;419:779–81 [DOI] [PubMed] [Google Scholar]
- 74. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jiang T, Yongzhuang L, Jiang Y. et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020;21:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Marçais G, Delcher AL, Phillippy AM. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018;14:e1005944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dobin A, Davis CA, Schlesinger F. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30 [DOI] [PubMed] [Google Scholar]
- 79. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome sequencing data for diploid T. natans have been deposited under NCBI BioProject PRJNA932942 and GSA BioProject PRJCA016421. The whole genome sequencing data for diploid T. incisa have been deposited under NCBI BioProject PRJNA933001 and GSA BioProject PRJCA016421. The transcriptome sequencing data of diploid T. incisa and allotetraploid T. natans have been deposited under NCBI BioProject PRJNA941110 and PRJNA731291, respectively. The re-sequencing (57 accessions) and transcriptome sequencing data of diploid Trapa natans for genome annotation were obtained from the NCBI BioProject PRJNA725399 of Lu et al. [10].