

How pS727 regulates STAT3 activation-inactivation cycles
Keywords: inactivation, intramolecular protein–protein interaction, nuclear export, pY705 dephosphorylation, pY705, SH2 interaction
Abstract
Signal transducer and activator of transcription 3 (STAT3) is involved in many biological processes, including immunity and cancer. STAT3 becomes phosphorylated at Tyr705 and Ser727 on IL-6 stimulation. Phospho-Tyr705 (pY705) stabilizes the STAT3 dimer with reciprocal interactions between pY705 and the SH2 of the other molecule and phospho-Ser727 (pS727) accelerates pY705 dephosphorylation. We study how pS727 regulates STAT3 in both structural and biological perspectives. Using STAT3 reconstituted in HepG2-stat3-knockout cells, we show that pS727, together with a handshake N-terminal domain (NTD) interaction, causes rapid inactivation of STAT3 for pY705 dephosphorylation and a chromosome region maintenance 1 (CRM1)-independent nuclear export, which is critical for faithful STAT3 response to the cellular signals. The various N-terminal tags, GFP-related Ruby and FLAG, rendered the export CRM1-dependent and especially FLAG-tag caused nuclear accumulation of STAT3, indicating the presence of conformational changes in inactivation. Impaired reactivation of STAT3 by S727A or FLAG-tag delayed or inhibited the IL-6-induced saa1 mRNA expression, respectively. The detailed analysis of the pY705–SH2 structure identified the C-terminal tail (CTT) from L706 to P715 as a key regulator of the CTT–CTT intermolecular and the CTT–SH2 intramolecular interactions that support pY705–SH2 association. The functional studies using multiple STAT3 mutants indicated that the degree of the two interactions determines the stability of pY705–SH2 interaction. Importantly, Pro715 was critical for the pS727's destabilizing activity and the known phosphorylation and acetylation at the CTT structurally inhibited the pY705–SH2 interaction. Thus, pS727 triggers pY705–SH2 dissociation by weakening the supportive interactions likely through CTT modulation, inducing rapid cycles of STAT3 activation–inactivation for proper function of STAT3.
Introduction
The signal transducer and activator of transcription (STAT) family members are key mediators of signals generated by cytokine receptor-associated tyrosine kinases that lead to gene-expression programs (1). Seven STAT proteins, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6, are known (2). All of them are activated by phosphorylation at a specific tyrosine residue critical for dimerization (2). Among them, STAT3 is activated by the IL-6 family cytokines and many growth factors (2, 3), and is involved in a variety of immune responses, including Th17 development, immunoglobulin production in B cells, and in other cellular responses of normal and tumor cells by regulating cell proliferation, survival, cell differentiation and even cellular senescence (3–6). Upon stimulation, STAT3 is phosphorylated at Tyr705 (pY705) by receptor-associated Jak kinases, Src family kinases or receptor-type tyrosine kinases (1, 3), and at Ser727 by multiple Ser/Thr kinases (7–9).
Tyrosine-phosphorylated STAT3 forms a stable parallel dimer with reciprocal interactions between the pY705-containing region and the SH2 domain of the other STAT3 molecule, which is required for stimulation-induced gene activation (2, 10). Phospho-Ser727 (pS727) has been shown to enhance STAT3's transcriptional activity (7, 11), and later pS727 was shown to shorten the duration of STAT3's transcriptional activity for the socs3 gene by enhancing pY705 dephosphorylation mainly through TC45 in the nucleus (12). Interestingly, apart from its transcriptional activity in the nucleus, STAT3 has been shown to have distinct activity in the mitochondria (13, 14). Mitochondrial STAT3 needs pS727 but not pY705 nor DNA-binding activity for its role in enhancing electron respiratory chain activity and Ras-dependent tumorigenesis (13, 14). Therefore, it is important to elucidate both the mechanisms by which once-activated STAT3 is inactivated in a pS727-dependent manner and the role of pS727 in the regulation of STAT3 activities for gene activation.
Various aspects of activation–inactivation cycles of STAT3 have been investigated after studies on STAT1 (15–23). Most STAT3, reportedly, forms a homo-dimer in the absence of pY705 (24), and is distributed in both the cytoplasm and nucleus with continuous shuttling between the two compartments independently of pY705 (25–27). Later, it was reported that the N-terminal domain (NTD) is required to form dimers of unstimulated STAT3 (23). Activated STAT3 in the form of a stable parallel dimer enters the nucleus by the importin-α/β pathway (26, 28), accumulates in the nucleus for some time to activate its target genes likely by reducing the export (23) and eventually becomes inactivated to undergo pY705 dephosphorylation and nuclear export (2). The once-inactivated and exported STAT3 is reactivated by the remaining signals for activation (29). However, it remains unclear how once-activated nuclear STAT3 is inactivated and proceed to pY705 dephosphorylation and nuclear export and which processes of STAT3 inactivation are regulated by pS727. After the report on the structure of unphosphorylated STAT1 showing a tetramer that is composed of two antiparallel dimers (30), conformational changes of STAT1 during inactivation have been studied (31, 32). Mertens et al. presented a model for the STAT1 inactivation process showing that once-activated STAT1 undergoes spatial reorientation of the monomers using the NTD, called the antiparallel dimer, for tyrosine dephosphorylation (32). By contrast, little is known about triggering events and possible conformational changes of STAT3 during its inactivation process. Ren et al. reported that recombinant STAT3 core bodies devoid of both the NTD and the C-terminal domain do not make homodimers and argued that antiparallel dimer formation is unlikely for STAT3 during inactivation (33). Although the dephosphorylation of STAT3 pY705 by the nuclear phosphatase TC45 is associated with the nuclear export of once-activated STAT3, like the case of STAT1 (15, 16, 23, 34–36), it is not known whether pY705 dephosphorylation in the nucleus is prerequisite for inactivation leading to the post-activation nuclear export of STAT3. Another obscure inactivation process is the selection of export systems, the chromosome region maintenance 1 (CRM1)-dependent or the CRM1-independent system, by unstimulated STAT3 and post-activated STAT3. The STAT3s variously tagged with GFP or related proteins or short peptides at the C- or N-terminus mostly showed CRM1-dependent post-activation nuclear export (27, 29, 37–39), while the nucleocytoplasmic shuttling of GFP-tagged STAT3 as well as untagged STAT3 in unstimulated cells seemed CRM1-independent (27, 38). This difference suggests that STAT3 may select the export systems, depending on the status of stimulation and/or the conformations they would take under the conditions. In addition, we need to be careful in interpreting the results obtained with the use of GFP-tagged STAT3s, because it was reported by Meyer et al. that a GFP-tag at the C-terminus of STAT1 caused a prolonged delay in the nuclear export of once-activated STAT1 compared with that of untagged STAT1 (40).
Here we have investigated how pS727 regulates the STAT3 inactivation and its function after stimulation with IL-6 in both biological and structural perspectives. In contrast to the currently prevailing models of the activation–inactivation cycle of STAT3 (22, 23, 37), we show that a pS727-dependent event(s) in cooperation with an NTD–NTD dimer causes a rapid inactivation of STAT3 leading to pY705 dephosphorylation and a CRM1-independent nuclear export. Inhibition of the cycle of pS727-dependent STAT3 activation–inactivation results in delay or inhibition of IL-6-induced gene activation in response to the cellular signals. Moreover, we provide the structural basis for the pY705–SH2 interaction that is supported by the two intermolecular and intramolecular interactions commonly involving the C-terminal tail (CTT) from L706 to P715 and reveal that pS727 regulates the dissociation of pY705–SH2 by modulating the CTT in a Pro715-dependent manner.
Methods
Cell culture and establishment of gene knockout of HepG2 and 293T-G133 cell lines
The HepG2 human hepatocellular carcinoma cell line and its derivative cell lines were grown in Dulbecco's Modified Eagle's Media (DMEM) with 10% fetal calf serum (FCS) (Gibco, Grand Island, NY, USA) at 37°C. HEK293T cells and HEK293T-G133 cells (11) were grown in DMEM with 5% FCS. HepG2-stat3-knockout (HepG2-SKO) and HepG2-stat3, tc45 double-knockout (HepG2-DKO) cells were established using the CRISPR/Cas9 system (41). The pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Dr Feng Zhang (Addgene plasmid #42230). Guide RNA sequences for the stat3 and the tc45 genes were 5′-agattgcccggattgtggc-3′ and 5′-catgcccaccaccatcgag-3′, respectively. HepG2 cells were transiently transfected with pX330-U6-guideRNA-stat3-Cas9 and pEF-puro and 36 h later, transfected cells were selected with puromycin for 2 days. The surviving cells were expanded and tested for the presence of STAT3 protein with an immunoblot analysis. The loss-of-function alleles of the stat3 gene of the two clones were verified by DNA sequencing. The tc45-knockout was generated by transfecting pX330-U6-guideRNA-tc45-Cas9 and pEF-Puro to one of the HepG2-SKO clones. The knockout alleles of the HepG2-stat3KO-tc45KO (DKO) clones were verified by DNA sequencing. Lentiviral preparations expressing various STAT3 mutants were done as described previously (12) and used to infect HepG2-SKO and HepG2-DKO cells. Cell lines expressing STAT3s at levels 2- to 3-fold higher than that of endogenous STAT3 in unstimulated HepG2 cells were chosen. HEK293T-G133-stat3-knockout (293T-G133SKO) cells were established using 293T-G133 cells as above and used to make various 293T-G133-SKO cells expressing STAT3 wild type (WT) or STAT3 S727A.
Antibodies and reagents
Antibodies against STAT3 and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to phospho-STAT3 Y705 and to phospho-STAT3 S727 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-STAT3 monoclonal antibody (MAB1799) for immunohistochemistry was from Bio-Techne, R&D Systems (Saint Louis, MO, USA). Goat anti-mouse IgG antibody conjugated with horseradish peroxidase and goat anti-rabbit IgG antibody conjugated with HRP were from Invitrogen (Frederick, MD, USA). Anti-mouse antibody conjugated with Alexa Fluor594 was from Life Technologies (Carlsbad, CA, USA). Staurosporine was from Calbiochem (EMD Millipore Corp., Billerica, MA, USA). Leptomycin B (LMB) was purchased from Focus Biomolecules (Plymouth Meeting, PA, USA). Human recombinant IL-6 was a gift from the Ajinomoto Research Institute (Tokyo, Japan). All other reagents were purchased from Nacalai Tesque Inc. (Kyoto, Japan).
Plasmid and lentiviral vector preparation
WT and mutant STAT3s, including STAT3 S727A, STAT3 S727D, STAT3 K709E, STAT3 T714A, STAT3 M660T, STAT3 Y657C, STAT3 K658Y, STAT3 Y640F, STAT3 P715A, STAT3 K709E/P715A, STAT3 T714A/P715A, STAT3 K709E/S727A, STAT3 T714A/S727A, STAT3 Y640F/K709E, STAT3 K658Y/K709E, STAT3 Y640F/T714A and STAT3 K658Y/T714A, were made using the quick-change mutation system (Stratagene, La Jolla, CA, USA). The NTD deletion was made with PCR, and FLAG-tag was added at the N-terminus to stabilize the NTD-deleted STAT3 protein. The DNA sequences of each mutant were verified by DNA sequencing. Each Stat3 cDNA was transferred into a lentiviral vector CSII-EF-MCS-IRES-Venus (a gift from H. Miyoshi, RIKEN, Tsukuba, Japan). Untagged STAT3 and its mutants were used throughout this study except for some experiments where FLAG-tag or GFP-related Ruby tag were added at the N-terminus of STAT3 WT. pEGFP-N1 was purchased from Clontech (Takara Bio, Kusatsu, Japan). mRuby2-C1 was a gift from Michael Davidson (Addgene plasmids #54768). The linker region, N-linker, between the tag and STAT3 at the N-terminus was ELLQPGGGGSAPEF. pEGFP-N-linker was used to make pEGFP-HIV-REV NES, which contained a sequence encoding LQLPPLERLTLD, a NES of HIV-Rev (42).
Western blotting
Cells were plated at 1.5 × 106 cells per 6-cm dish, then stimulated on the next day with 20 ng ml−1 IL-6 for the indicated times. Some cells were stimulated with IL-6 for 15 min, followed by 0.5 μM staurosporine treatment for the indicated times. Whole cell lysates were prepared and subjected to immunoblot analysis as described previously (12). Anti-STAT3, anti-phospho-STAT3 Y705, anti-phospho-STAT3 S727 and anti-β-actin antibodies were used for blotting. Signals were detected with Chemi-Lumi One (Nacalai Tesque).
Immunohistochemistry
Cells were plated on glass coverslips in 6-well plates at 3.0 × 105 cells per well, and then stimulated on the next day with 20 ng ml−1 IL-6 for the indicated times, or with IL-6 for 15 min followed by 0.5 μM staurosporine treatment for the indicated times. Cells were washed twice with cold phophate-buffered saline (PBS) and fixed with 3.7% formalin at 4°C for 1 h. The cells were then incubated with 0.5% TritonX-100 for 5 min to permeabilize the cell membrane, and blocked with 5% milk in cold PBS (w/v) overnight at 4°C. Monoclonal anti-STAT3 antibody (diluted to 1.5 μg ml−1 in 5% milk/PBS) was added to the coverslips and incubated overnight at 4°C. The coverslips were then washed three times with PBS, and an Alexa Fluor594-conjugated secondary antibody diluted 1:500 in 5% milk/PBS was added to the coverslips at room temperature for 1 h in the dark. The coverslips were washed three times with PBS, then incubated with DAPI at room temperature for 5 min to stain DNA. Images were collected with a Zeiss LSM 700 confocal microscope through a ×63 lens.
Cellular distribution of STAT3 and data analysis
The distribution of STAT3 in the nucleus relative to that in the whole cell area was measured from images of STAT3-expressing cells using ImageJ (43), and >25 cells were analyzed for each condition. Experiments were repeated more than three times. The average ratio of the nuclear STAT3 signal to the total STAT3 signal (depicted as %) is shown together with a representative picture for each condition. The quantified data sets of the nuclear distribution as ratios under each condition, obtained as described above, were analyzed by EZR (44) with one-way ANOVA to compare each data set to others. Pairwise comparisons using t-tests with pooled standard deviation were used. P-values <0.001 and 0.01 are shown as *** and **, respectively, in the figures.
Time-lapse live-cell imaging analysis
For time-lapse imaging of fluorescent protein in living cells, we used an Olympus FV10i confocal microscope (Olympus, Tokyo, Japan) with a cell-culture unit, which enabled maintaining the stage at 37°C with 5% CO2. Two channels (DsRed and Transmitted) of images were captured every 5 or 10 min for up to indicated times. Images of representative cells at indicated time points were shown.
Quantitative real-time PCR
Total RNA was isolated with Sepasol RNA I super (Nacalai Tesque). The reverse transcriptase reaction was done using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). RT–PCR was performed with THUNDERBIRD SYBR qPCR Mix (Toyobo) and the ABI 7500 Fast real-time PCR system (Applied Biosystems). The data were analyzed using the 7500-software (Ver.2.03, Applied Biosystems), and the quantities of expression relative to GAPDH mRNA were determined from two independent experiments. Primers used for PCR are as follows: hGAPDH (5′-GCACCGTCAAGGCTGAGAAC-3′ and 5′-TGGTGAAGACGCCAGTGGA-3′), hSOCS3 (5′-GGA ATGTAGCAGCGATGGAA-3′ and 5′-GCCCCTGTCCAGC CCAATAC-3′) and hSAA1 (5′-TTCACTCTGCTCTCAGGAGATCTG-3′ and 5′-GTGTACCCTCTCCCCGCTTT-3′).
Structural analysis
The PDB file for the STAT3β dimer on DNA, 1BG1, was used to analyze the intermolecular and intramolecular interactions by PIC, the protein interactions calculator (45). The interface of STAT3 dimerization was visualized with the PyMOL (molecular graphics system version 2.0.7, DeLano Scientific). MutaBind (46) was used to generate modified PDB files according to given mutations. The names of atoms in the amino acid residues are shown according to the guidelines of the International Union of Pure and Applied Chemistry (IUPAC).
Results
pS727 accelerates inactivation of activated STAT3 leading to both pY705 dephosphorylation and post-activation nuclear export
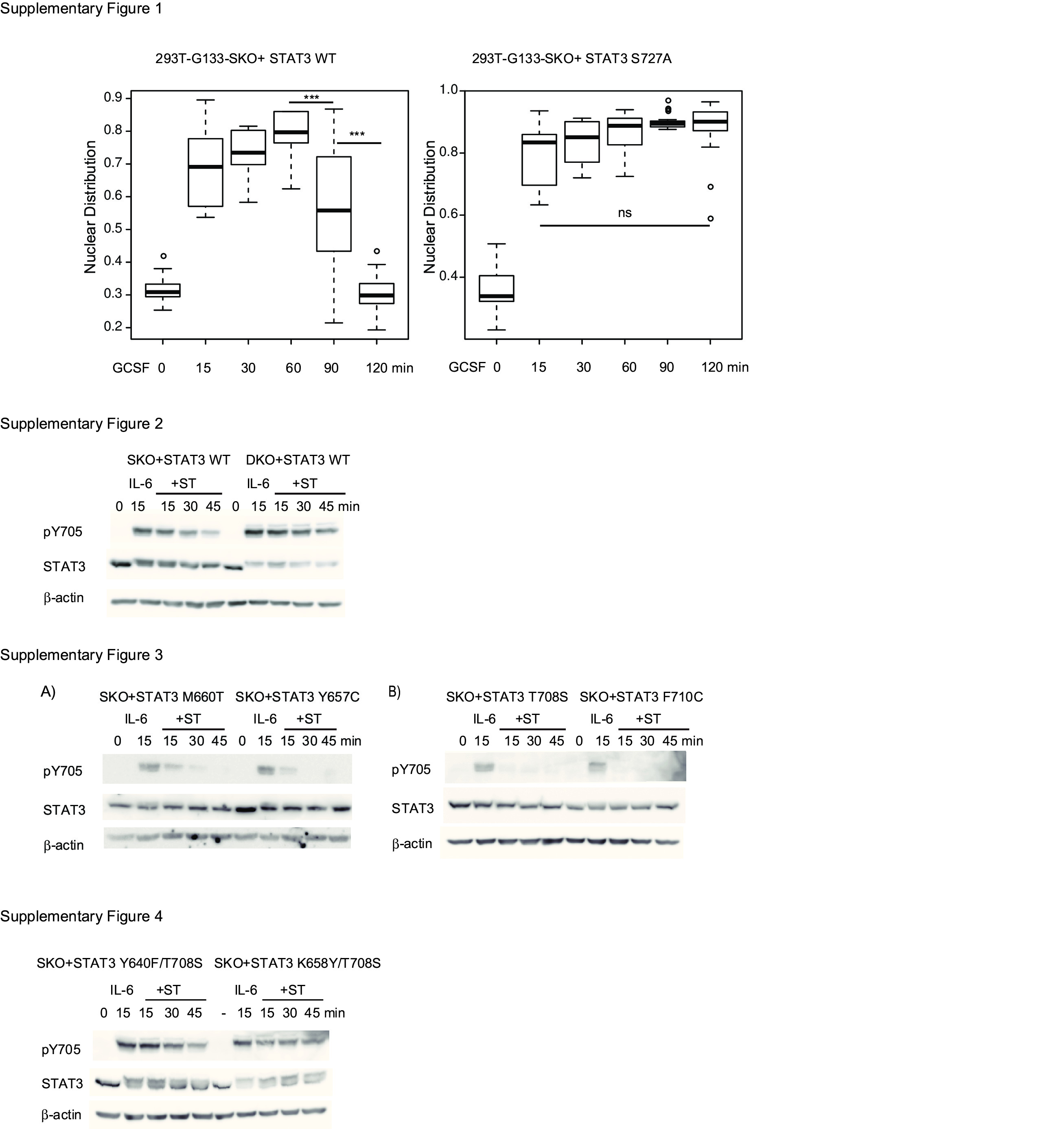
Although pS727 of STAT3 has been reported to enhance STAT3's transcription activity (7, 11) and cause rapid dephosphorylation of pY705 mainly through TC45 in the nucleus (12), how pS727 exerts such functions remains unknown. To address this problem, we thought it important to understand how once-activated nuclear STAT3 with pY705 and pS727 is inactivated for pY705 dephosphorylation and nuclear export. We first established HepG2-stat3-knockout (SKO) cell lines and reconstituted HepG2-SKO cells with untagged WT STAT3, STAT3 S727A or STAT3 S727D, whose expression levels were 2- to 3-fold higher than that of endogenous STAT3 in the unstimulated HepG2 cells; these were still within physiological variation observed in IL-6-stimulated HepG2 cells (47) and in various cells stimulated with IL-6 in mice (48) and were chosen to reduce the experimental errors due to the non-specific signals detected by a monoclonal anti-STAT3 antibody in the immunohistochemistry (Fig. 1B, C and E). The cells were stimulated with IL-6 for various times up to 120 or 180 min. Immunoblot analysis showed that the exogenously expressed STAT3 WT was fully phosphorylated at Y705 as well as at S727 in 15 min of IL-6 stimulation. The WT STAT3 started to lose the pY705 at around 60 min with the lowest level between 90 and 120 min, but sustained pS727 for a little longer time with a slight decrease starting from 60 min compared with pY705 changes (Fig. 1A, see also Fig. 3A), while STAT3 S727A showed sustained levels of pY705 up to 120 min (Fig. 1A), consistent with the previous report by Wakahara et al. (12). Compared with STAT3 WT, STAT3 S727D showed a little delay in the pY705 dephosphorylation with a low level remaining at 120 min, indicating that the S727D mutation only partially mimics the activity of phospho-Ser727. It is noted that stimulated STAT3 shows a little retardation in the mobility in sodium dedecyl sulfate- polyacrylamide gel electrophoresis that is caused by unknown modifications depending on the presence of pS727, since STAT3 S727A does not show such retardation after stimulation (Fig. 1A, 11, 12). The cellular distribution of STAT3 was then examined by immunohistochemistry, and the results were quantified as described in the Methods. Representative images and the average percentages of the nuclear distribution under each condition are shown in Fig. 1(B), and the data were statistically analyzed (shown as box plots in Fig. 1C). Approximately 30–40% of the STAT3 showed a nuclear distribution in unstimulated cells. After stimulation, most of the activated STAT3, STAT3 S727A and STAT3 S727D were detected in the nucleus within 15 min. STAT3 remained mostly localized to the nucleus for the first 45 min, and the nuclear STAT3 started to decrease at 60 min and showed an obvious cytoplasmic localization at 90 and 120 min. In contrast, STAT3 S727A mostly remained in the nucleus even at 120 min of stimulation and partially showed cytoplasmic distribution at 180 min. STAT3 S727D showed significant post-activation nuclear export after 90 min, consistent with the partial mimicking activity of S727D. The changes over time in the levels of pY705 and the cellular distribution of endogenous STAT3 in HepG2 cells in response to IL-6 were also examined. Endogenous STAT3 in HepG2 showed essentially the same kinetic changes in pY705 levels (Fig. 1D) and cellular distribution of STAT3 (Fig. 1E) as those of the untagged STAT3 in the SKO cells except that the percentages of the nuclear distribution of IL-6-stimulated endogenous STAT3 were lower at any time point compared with those of untagged STAT3 in SKO cells (Fig. 1B and C). This underestimation likely resulted from non-specific signals in the cytoplasm of SKO cells detected by the antibody (data not shown). Importantly, the rapid pS727-dependent inactivation of STAT3 was not unique to HepG2 cells but was observed in other lineages of cells, since the same results were observed in the 293T cell-based system: the stat3 genes were knocked-out and the resulting stat3-ko cells were reconstituted with STAT3 WT or STAT3 S727A (Supplementary Figure 1). Together, these results indicated that pS727 accelerated the inactivation of activated STAT3 to the completion of the first activation–inactivation cycle within 60–90 min as a whole in response to IL-6.
Fig. 1.

pS727-dependent STAT3 inactivation for both pY705 dephosphorylation and post-activation nuclear export. (A) Changes in the pY705 level over time. HepG2-SKO cells expressing WT STAT3 (SKO+STAT3 WT), STAT3 S727A (SKO+STAT3 S727A) or STAT3 S727D (SKO+STAT3 S727D) were stimulated with 20 ng ml−1 IL-6 for the indicated times, and whole cell extracts (WCEs) were subjected to immunoblot analysis with antibodies to phospho-Tyr705, phospho-Ser727, STAT3 and β-actin. Experiments were repeated three times. (B) Cellular distribution of STAT3. Cells were subjected to immunohistochemistry using a monoclonal antibody to STAT3 (red) and to DNA staining with DAPI (blue) and observed by confocal microscopy. One representative picture of each condition from >25 pictures is shown. The average percentage of nuclear STAT3 for each condition is shown. The scale bar in the first picture shows 10 μm. (C) STAT3 nuclear distribution. The data of the nuclear distribution of STAT3 in the indicated condition (shown as a box plot) were analyzed with one-way ANOVA. Pairwise comparisons using t-tests with pooled standard deviation were used. P-value <0.001 or <0.01 is depicted as *** or **, respectively, with a line showing the compared data sets. P-value >0.05 is shown as ns. The experiments in (B) and (C) were performed at least three times with similar results. (D) Changes in the pY705 level of endogenous STAT3 over time. HepG2 cells were stimulated with IL-6 for the indicated times, and WCEs were subjected to immunoblot analysis as in (A). (E) Nuclear distribution of endogenous STAT3 in HepG2 cells stimulated with IL-6 for the indicated times. The experiments were repeated twice with similar results.
Fig. 3.

pS727-dependent rapid cycle of activation/inactivation through the CRM1-independent system is required for proper gene expression. (A) Live-cell images were used to evaluate the effectiveness of 10 ng ml−1 LMB in inhibiting the CRM1-dependent nuclear export of EGFP-Rev NES in HepG2 cells. Live-cell images of five cells were taken every minute for 90 min using an Olympus FV10. Pictures of representative cells at the indicated time points are shown. (B) Cellular distribution of STAT3. HepG2-SKO cells expressing STAT3 untreated or pre-treated with LMB for 15 min were stimulated with IL-6 for the indicated times. The nuclear distribution of STAT3 was evaluated as in Fig. 1(B) and the data of each condition were analyzed as in Fig. 1(C). The experiments were repeated three times with similar results. (C) Live-cell images of Ruby-tagged STAT3. HepG2-SKO cells expressing STAT3 tagged with N-terminal Ruby (Ruby-STAT3) untreated or pre-treated with LMB for 15 min were stimulated with IL-6 for 300 min (left panel). Live-cell images of 10 fields were taken every 10 min up to 300 min. Pictures of representative cells at indicated time points are shown. The scale bar in the first picture shows 10 μm. The nuclear distribution of Ruby-STAT3 in the two cells (C1 and C2) are shown below the pictures. Changes in the pY705 level of Ruby-STAT3 are shown in the right panel. (D) Cellular distribution of FLAG-STAT3 (left panel). HepG2-SKO cells expressing FLAG-STAT3 untreated or pre-treated with LMB for 15 min were stimulated with IL-6 for the indicated times. The nuclear distribution of FLAG-STAT3 was evaluated and analyzed as in (B). The experiments were repeated twice with similar results. The P-value < 0.001, < 0.01 or < 0.05 is shown ***, ** or * with a line. Changes in the pY705 and pS727 levels of FLAG-STAT3 are shown in the right panel. (E) IL-6-induced mRNA expression of the two STAT3-target genes by untagged STAT3 WT, STAT3 S727A or FLAG-tagged STAT3 WT. HepG2-SKO cells expressing STAT3, STAT3 S727A or FLAG-STAT3 were stimulated with IL-6 for 0, 45, 180 or 360 min and the total RNAs from each condition were used to measure the amount of socs3 and saa1 mRNAs relative to that of gapdh mRNA. Data are expressed as relative levels to those of each mRNA in the unstimulated HepG2-SKO cells expressing STAT3 WT. The average numbers from three independent experiments with standard deviation of around 10% of the values are shown. The STAT3 levels of the three cell lines were shown in the left panel.
pS727-dependent event(s) cooperates with the NTD dimerization in the initiation of STAT3 inactivation prior to pY705 dephosphorylation
To test whether pY705 dephosphorylation by TC45 in the nucleus is required for pS727-dependent STAT3 inactivation, we established a HepG2-stat3-tc45-double-knockout (DKO) cell line and reconstituted the line with WT STAT3. We first compared the kinetic changes of pY705 and pS727 between the two cell lines, SKO+STAT3 WT and DKO+STAT3 WT. STAT3 WT in DKO cells showed slower decrease in the levels of pY705 and pS727 after 90 and 120 min of IL-6 stimulation compared with STAT3 WT in SKO cells (Fig. 2A). The appearance of cytoplasmic STAT3 WT at 60, 90 and 120 min in DKO cells (Fig. 2B and C) was coincident with the slow decline in pY705 levels of STAT3 WT in DKO cells (Fig. 2A). These results indicated that although TC45 contributes to the rapid STAT3 inactivation, the post-activation nuclear export occurred without pY705 dephosphorylation by TC45 in the nucleus and that the pY705 dephosphorylation could be done by cytoplasmic phosphatases other than TC45 after the nuclear export in DKO cells. Taken together, these results supported the notion that a pS727-dependent event(s) in the activated STAT3 dimer induces a change toward inactivation steps needed for both pY705 dephosphorylation and post-activation nuclear export.
Fig 2.

pS727-dependent rapid STAT3 inactivation does not require pY705 dephosphorylation by TC45 but requires the NTD–NTD interaction involving L78. (A) Changes in the pY705 level over time. HepG2-SKO cells expressing WT STAT3 (SKO+STAT3) and HepG2-stat3-tc45-double knockout cells (HepG2-DKO) cells expressing WT STAT3 (DKO+STAT3 WT) were stimulated with 20 ng ml−1 IL-6 for the indicated times, and whole cell extracts (WCEs) were subjected to immunoblot analysis as in Fig. 1(A). The experiments were repeated three times. (B) Cellular distribution of STAT3. HeoG2-DKO cells expressing WT STAT3 were stimulated with IL-6 for the indicated times. Cells were subjected to immunohistochemistry as in Fig. 1(B). The representative picture and average percentage of nuclear STAT3 for each condition are shown. The experiments (B and C) were repeated twice with similar results. (C) STAT3 nuclear distribution. The data of the nuclear distribution of STAT3 in the indicated condition (shown as box plot) were analyzed with one-way ANOVA. The P-value <0.001 (***) is shown with a line. (D) Changes in the pY705 level and pS727 of STAT3 WT (SKO+STAT3 WT), pY705 levels of STAT3 S727A (SKO+STAT3 S727A), FLAG-tagged NTD-deleted STAT3 (STAT3 dNTD) and STAT3 L78R mutant (STAT3 L78R) in HepG2-SKO cells. HepG2-SKO cells expressing STAT3 WT, STAT3 S727A, STAT3 dNTD or STAT3 L78R were stimulated with IL-6 at 20 ng ml−1 for 15 min, followed by treatment with ST at 500 nM for the indicated times. WCEs were subjected to immunoblot analysis as in Fig. 1(A). The experiments were repeated three times. Note the pS727 levels and the mobility changes of the STAT3.
Since we previously observed that an N-terminally truncated STAT3 mutant, STAT3-162-770, showed sustained pY705 regardless of the presence of pS727 (12), we tested the role of the NTD of STAT3 in STAT3 inactivation. We added a FLAG-tag at the N-terminus of STAT3 mutant with an NTD deletion to suppress its degradation (data not shown). The STAT3 L78R mutant was also tested because the L78R mutation, found in the patients with inflammatory hepatocellular adenoma (49), is known to disrupt the major intermolecular NTD–NTD interaction (23). To stop IL-6 signals, we used staurosporine (abbreviated as ST hereafter), an inhibitor of various kinases (50) including JAK and other protein tyrosine and Ser/Thr kinases; this drug has been used to examine the inactivation process of STAT1 and STAT3 after ST-induced inhibition of cytokine actions (15, 51). Treating cells with ST after 15 min of IL-6 stimulation slightly accelerated dephosphorylation of pY705 in STAT3 WT without altering the course of pS727 changes (Fig. 2D). STAT3 S727A did not show pY705 dephosphorylation after 45 min of ST treatment (12, Fig. 2D). The ST treatment even for 60 min caused only a little decrease in the pY705 in both the NTD-deletion mutant and STAT3 L78R (Fig. 2D) despite the presence of pS727 (data not shown but see the mobility shift of these STAT3), indicating that the handshake type NTD–NTD interaction (52) using the interface containing Leu78 was required for the rapid STAT3 inactivation. Taken together, these results suggested that the pS727-dependent event and the handshake type of NTD–NTD interaction might cooperatively initiate the STAT3 inactivation likely through dissociation of pY705–SH2 interaction, leading to the next step for inactivation.
Post-activation nuclear export of STAT3 is mainly CRM1-independent, while N-terminal tags, GFP-related Ruby and FLAG, render the export CRM1-dependent likely by inhibiting a conformational change
Upon stimulation with IL-6 or gp130-signals, STAT3 unexpectedly showed very rapid inactivation in HepG2 and 293T cells, in contrast to the reports showing the slow inactivation of STAT3 as observed with GFP-tagged STAT3 (27). Activated nuclear STAT3, tested with variously tagged STAT3, has been reported to undergo nuclear export through CRM1 (27, 29, 37–39), while unstimulated STAT3 shuttles between the cytoplasm and nucleus independently of tyrosine phosphorylation (25, 26) likely in a CRM1-independent manner (27). To understand how STAT3 completes the first cycle of activation–inactivation in a pS727-dependent manner in a short time, we evaluated the use of CRM1 by activated untagged STAT3. Since treatment of cells with LMB, a CRM1 inhibitor, for longer than 4–7 h was shown to enhance nuclear distribution of unstimulated STAT3, we first tested how fast LMB inhibited the CRM1-dependent nuclear export of EGFP-Rev-NES in HepG2 cells. As shown in Fig. 3(A), LMB started to inhibit the cytoplasmic distribution of EGFP-Rev-NES in several minutes and the effect reached a plateau within 15 min. We then added LMB to cells 15 min before IL-6 stimulation. LBM treatment, however, showed no significant inhibition of the post-activation nuclear export of untagged STAT3 at any time points of IL-6 stimulation until post-activation nuclear export of the first cycle completed by 90 min (Fig. 3B), indicating that the post-activation nuclear export of STAT3 was mainly CRM1-independent. This result was quite a contrast to the reported slow and CRM1-dependent post-activation nuclear export of variously tagged STAT3 (27, 29, 37, 38). To obtain insight into the inactivation mechanism of STAT3, we tested two different tags, a GFP-related Ruby and a short peptide FLAG, fused at the N-terminus of STAT3 to see if they alter the inactivation processes (Fig. 3C and D). A live-cell imaging analysis was used to chase the movements of Ruby-STAT3 fusion proteins in single cells (Fig. 3C). The Ruby-STAT3 showed activation–inactivation cycles with a duration of roughly 120–150 min, depending on cells (see the calculated % nuclear distribution of Ruby-STAT3 at the indicated times from two cells, C1 and C2, shown below the picture of Fig. 3(C)), where the post-activation nuclear export of Ruby-STAT3 was inhibited by LMB, indicating that the post-activation nuclear export of Ruby-STAT3 was dependent on a CRM1. Ruby-STAT3 showed pY705 dephosphorylation indistinguishably from that of STAT3 in Fig. 1(A) (Fig. 3C). FLAG-STAT3 showed a very rapid dephosphorylation of pY705 and pS727 (Fig. 3D, right panel) compared with those of STAT3 WT in Fig. 1(A). FLAG-STAT3, however, showed a peculiar pattern of nuclear accumulation of STAT3 after stimulation: only a small fraction, about 15–20%, of nuclear FLAG-STAT3 underwent nuclear export at 90 min in a CRM1-dependent manner (Fig. 3D). These results collectively suggested that the two N-terminal tags, FLAG and Ruby, might block the transition from an inactivated form for CRM1-dependent nuclear export and pY705 dephosphorylation to a form for CRM1-independent nuclear export. Considering the report showing that unphosphorylated STAT3 core proteins bound on DNA form a dimer with the reciprocal interaction between unphosphorylated Y705 and SH2 (53), once-activated FLAG-STAT3, but not of Ruby-STAT3, might easily form a parallel dimer for nuclear retention after loss of phosphorylation at both Y705 and S727.
The pS727-dependent rapid cycles of STAT3 activation–inactivation are critical for the IL-6-induced gene activation
As shown in Fig. 3(C), STAT3 could be activated repeatedly in response to the remaining signals in the cells and the rapid inactivation should be critical for the nature of the JAK-STAT signal transducing systems. To examine the role of repeated cycles of STAT3 activation–inactivation in IL-6-induced gene expression programs, we used STAT3 WT and two STAT3 mutants, STAT3 S727A and FLAG-STAT3, and compared the IL-6-induced mRNA expression of two STAT3 target genes (54, 55), the socs3 and saa1 (serum amyloid A1). The two genes were activated in response to IL-6 in a STAT3-dependent manner with different kinetic induction without de novo protein synthesis (data not shown). In response to IL-6, the socs3 gene was rapidly activated by all of the three types of STAT3 with a little different kinetic change; STAT3 WT induced the largest mRNA expression at 45 min, followed by a decline at 3 and 6 h, and FLAG-STAT3 also rapidly induced socs3 mRNA at 45 min, followed by a sharp decline at 3 and 6 h. STAT3 S727A caused a little low but sustained level of mRNA from 45 min to 6 h, as reported by Wakahara et al. (12). The socs3 mRNA levels seemed to reflect the amount of activated STAT3 in the nucleus. On the other hand, STAT3 WT started to activate the saa1 gene from 45 min with its level reaching a plateau between 45 min and 3 h, while STAT3 S727A did not induce saa1 mRNA at 45 min but induced it well after 3 h and increased it at 6 h to the level comparable to that of STAT3 WT at 3 h, indicating a delay in the STAT3-induced saa1 activation by STAT3 S727A. The saa1 gene was poorly activated at all time points from 45 min to 6 h by FLAG-STAT3 (Fig. 3E). These results may suggest that both repeated activation and accumulation of activated STAT3 for enough time might be required for activation of the saa1 gene. It is likely that the repeated cycle of STAT3 activation has a role in continuous socs3 mRNA expression at 3 and 6 h. Thus, pS727-dependent repeated cycles of STAT3 activation–inactivation allow STAT3 to faithfully respond to the cellular signals.
The two intramolecular and intermolecular interactions involving the CTT from L706 to P715 support the stable pY705–SH2 interaction
The above results showed that pS727 triggers the initiation of STAT3 inactivation prior to pY705 dephosphorylation (Fig. 2). Then we searched for the target of the pS727-dependent event that likely regulates dissociation of the pY705–SH2 interaction by analyzing the structure of the pY705–SH2 interaction. We analyzed the structure of an N-terminally truncated STAT3β dimer on DNA (10) by using online software PIC (45) and graphical software PyMol. As depicted in Fig. 4(A), the residues 689 to 701, which link the SH2 domain with the phosphotyrosine-containing region (residues 702 to 715), are poorly ordered. A long region containing 55 residues from Thr716 to Met770 specific to STAT3α or a short seven residues from Phe716 to Lys722 specific to STAT3β follows the pY705-containing peptide. We named the region from Leu706 to Pro715 the CTT because this region is involved in both the intermolecular CTT–CTT and the intramolecular CTT-own SH2 protein–protein interactions and possibly functions as a regulator, as shown in Table 1. The structural basis of the pY705–SH2 interaction in the STAT3 dimer is depicted in Fig. 4(B). As shown in Table 1 and Fig. 4(B), the intermolecular interaction takes place between the proximal part of CTTs from Thr708 to Cys712 in the two molecules, and the intramolecular interaction is made between the distal part of CTT from Lys709 to Pro715 and its own SH2 domain. The CTT–SH2 interaction is composed of multiple bindings (Table 1; Fig. 4B): a hydrogen bond between the atom N in the side chain of K709 and the atom O of the main chain of R688 (α in Fig. 4B), four hydrophobic interactions between the two residues, V713 and P715, in the CTT and the three residues, Y657, M660 and L666, in the BG loop from AA655-667 (10) (β in Fig. 4B) of the SH2, a hydrogen bond between the atom O of the side chain of T714 and the OH of the side chain of Y657 (γ in Fig. 4B) and a hydrogen bond between the atom S of the side chain of Cys712 and the OH of the side chain of Y640 (δ in Fig. 4B). Thus, the two layers of interactions involving the CTT, the intermolecular CTT–CTT interaction (② in Fig. 4B) and the intramolecular CTT–SH2 interaction (③ in Fig. 4B), likely give a basis for the final pY705–SH2 interaction (① in Fig. 4B). Importantly, multiple STAT3-inactivating mutants cluster in the CTT and the BG loop region of the SH2 domain responsible for the CTT–SH2 interaction and the CTT–CTT interaction, including K709E, T708S, F710C, I711T, C712T, V713L/M, T714A, M660T and Y657F/C found in the patients with hyper IgE syndrome due to insufficient STAT3 activity (56, 57). Some active STAT3 mutants found in tumors or cancers (58), including K658Y, D661V and Y640F (49, 59), cluster also in the BG loop or a region of the SH2 responsible for the CTT–SH2 association. These findings suggest the possibility that the CTT may be a key regulatory region which receives signal-dependent information, such as phosphorylation and acetylation, for dissociation of the pY705–SH2 interaction.
Fig. 4.

The CTT from L706 to P715 plays a role for the pY705–SH2 interaction by being involved in the intramolecular CTT–SH2 and intermolecular CTT–CTT interactions. (A) Schematic diagram of the C-terminal regions of STAT3. The SH2 domain is connected to the Y705-containing peptide by the disordered region (689–701), followed by the CTT from L706 to P715 and the C-terminal transcription-activating domain with S727 in STAT3α or the short peptide of seven residues in STAT3β. (B) Structure of the pY705–SH2 interaction. A part of the SH2 domain of molecule A in green ribbon structure, the pY705–CTT of molecule A as green sticks and the pY705–CTT of molecule B as orange sticks are shown using PyMol. The Y657, M660 and L666 of molecule A are shown as yellow sticks. The CTT–CTT intermolecular interaction (②) and the CTT–SH2 intramolecular interaction (interaction between the distal part of the CTT from K709 to P715 and the BG loop of its own SH2, ③) and the pY705–SH2 interaction (①) are shown. The CTT–SH2 interaction is composed of four major interactions, shown as α, β, γ and δ. Hydrogen bonds and polar interactions are shown as dashed lines and hydrophobic interactions are shown as arcs. Note the differences in the orientations of the multiple interactions for the CTT–SH2 interaction (α, β and γ) and that of the C712–Y640 interaction (δ).
Table 1.
Detailed molecular interactions involved in the pY705-SH2 interaction
| Intermolecular interaction | |||
|---|---|---|---|
| Molecule B | Molecule A | Type of interaction | |
| PTM–SH2 | pY705 | K591 | Polar interaction |
| pY705 | R609 | Polar interaction | |
| pY705 | S611 | Polar interaction | |
| pY705 | S613 | Polar interaction | |
| L706 | W623 | Hydrophobic interaction | |
| L706 | V637 | Hydrophobic interaction | |
| K707 | E638 | Ionic interaction | |
| F710 | Y640 | Hydrophobic interaction | |
| F710 | Y640 | Aromatic–aromatic interaction | |
| F710 | M648 | Hydrophobic interaction | |
| CTT–CTT | T708 OG1 | C712 SG | Hydrogen bond (side chain–side chain) |
| K709 N | C712 SG | Hydrogen bond (side chain–main chain) | |
| K709 O | C712 SG | Hydrogen bond (side chain–main chain) | |
| F710 | C712 | Aromatic–sulfur interaction | |
| F710 N | C712 SG | Hydrogen bond (main chain–side chain) | |
| I711 N | I711 O | Hydrogen bond (main chain–main chain) | |
| I711 O | I711 N | Hydrogen bond (main chain–main chain) | |
| Intramolecular interaction | |||
| Molecule A | Molecule A | Type of interaction | |
| CTT-own SH2 | K709 NZ | R688 O | Hydrogen bond (side chain–main chain) |
| I711 | M648 | Hydrophobic interaction | |
| C712 SG | Y640 OH | Hydrogen bond (side chain–side chain) | |
| V713 | Y657 | Hydrophobic interaction | |
| V713 | M660 | Hydrophobic interaction | |
| V713 | L666 | Hydrophobic interaction | |
| T714 OG1 | Y657 OH | Hydrogen bond (side chain–side chain) | |
| P715 | M660 | Hydrophobic interaction | |
STAT3-inactivating mutants causing weaker intramolecular CTT–SH2 interaction or weaker intermolecular CTT–CTT interaction result in rapid inactivation even in TC45-deficient cells, independently of pS727
To examine the role of the two interactions in supporting the pY705–SH2 interaction, we first tested the two dominant-negative mutants, STAT3 K709E and STAT3 T714A (56, 57). MutaBind (46) was used to obtain a recalculated PDB file according to a given point mutation in one chain of the protein dimer. This new PDB file was then subjected to analysis with PyMol and PIC. Structural analysis revealed that the K709E mutation would remove the hydrogen bond between the atom N of the side chain of K709 and the main chain O of R688, while the T714A mutation would remove the hydrogen bond between the atom O of the side chain of T714 and the OH of the side chain of Y657, indicating that these two mutations are likely to weaken the intramolecular association of the CTT with the SH2 (the effects of mutations were summarized in Table 2). We then examined the phenotypes of these mutants by examining the degree of pY705 dephosphorylation and the cellular distribution of STAT3. Both STAT3 K709E and STAT3 T714A showed very rapid loss of pY705 in both TC45-positive SKO and TC45-deficient DKO cells immediately after inhibition with ST following IL-6 stimulation (Fig. 5A), compared with those of WT STAT3 in SKO (Fig. 3D and Supplementary Figure 2) and in DKO (Supplementary Figure 2). The pY705 levels relative to the STAT3 protein in both mutant STAT3s at 15 min of IL-6 stimulation were lower in TC45-positive cells than those in TC45-deficient cells, indicating that these mutant STAT3s underwent a rapid inactivation in the nucleus even during the first 15 min of IL-6 stimulation. The rapid loss of pY705 in both mutants in DKO cells immediately after ST treatment suggested that these two mutants, K709E and T714A, were quickly inactivated and exported to the cytoplasm immediately after ST treatment. Consistent with the rapid pY705 dephosphorylation in both mutant STAT3s under TC45 deficiency (Fig. 5A), these mutant STAT3s underwent rapid nuclear export within 10 min of ST treatment after 15 min of IL-6 stimulation in HepG2-DKO cells (Fig. 5B and C). These results indicated that these mutants could be activated well and then rapidly inactivated presumably through a reduced intramolecular association between the distal part of CTT and the SH2 domain. Importantly, the accelerated pY705 dephosphorylation of STAT3 K709E and STAT3 T714A occurred in the absence of pS727 (Fig. 5D), indicating that these STAT3 mutants with the weakened CTT–SH2 interaction underwent rapid inactivation independently of pS727. Other inactive mutants, including T708S, F710C, Y657C and M660T, were also found to be rapidly inactivated like K709E and T714A mutants (Supplementary Figure 3A and B). These mutants also showed rapid pY705 dephosphorylation in HepG2-DKO (data not shown). The effects of those mutations are summarized in the Table 2. Among them, T708S and F710C inhibit the CTT–CTT interaction. These results supported the notion that any mutants leading to weaker interaction at either CTT–SH2 or CTT–CTT cause rapid inactivation of STAT3.
Table 2.
Changes of interactions by mutations in the interaction interface and CTT modulation
| Loss of interaction by mutations in the interaction interface | Effect | |
|---|---|---|
| T708S | T708 OG1–C712 SG interaction | Inactivationa |
| K709E | K709–R688 interaction | Inactivationa |
| T714A | T714–Y657 interaction | Inactivationa |
| Y657C | T714–Y657 interaction | Inactivationa |
| F710C | F710–C712 interaction | Inactivationa |
| M660T | M660–V713, M660–P715 | Inactivationa |
| Y640F | C712–Y640 interaction | Activationa |
| Creation of hydrophobic interactions by mutation | ||
| K658Y | Y658–L666 | Activationa |
| Y658–P669 | ||
| Y658–I711 | ||
| Y658–V713 | ||
| Loss of interaction by CTT modification | ||
| K707 acetylation | K707–E638 interaction | Inactivationb |
| K709 acetylation | K709–R688 interaction | Inactivationb |
| T708 phosphorylation | T708–C712 interaction | Inactivationb |
| T714 phosphorylation | T714–Y657 interaction | Inactivationb |
aExperimentally verified.
bNot proved yet, but likely.
Fig. 5.

The inactive CTT mutants, STAT3 K709E and STAT3 T714A, show rapid inactivation independently of pS727. (A) Changes in the pY705 levels of STAT3 K709E and STAT3 T714A in HepG2-SKO cells and HepG2-DKO cells. HepG2-SKO cells and HepG2-DKO cells expressing STAT3 K709E, or STAT3 T714A were stimulated with IL-6 at 20 ng ml−1 for 15 min, followed by treatment with ST at 500 nM for the indicated times. Whole cell extracts (WCEs) were subjected to immunoblot analysis as in Fig. 1(A). The experiments were repeated three times with similar results. (B and C) Cellular distribution of the two STAT3-inactivating mutants. HepG2-DKO cells expressing STAT3 K709E or T714A were stimulated with IL-6 for the indicated times or stimulated with IL-6 for 15 min and treated with ST for 10 or 20 min. Cells were subjected to immunohistochemistry and observed by confocal microscopy as in Fig. 1(B). The data of the nuclear distribution of STAT3 in the indicated condition (shown as a box plot) were analyzed with one-way ANOVA. STAT3 K709E in DKO cells (B) and STAT3 T714A in DKO cells (C). The experiments were repeated twice with similar results. (D) Changes in the pY705 levels in STAT3 K709E/S727A and STAT3 T714A/S727A. Cells were stimulated with IL-6 for 0 or 15 min, or with IL-6 for 15 min, followed by treatment with ST for the indicated times. WCEs were immunoblotted as in Fig. 1(A). The experiments were repeated three times with similar results.
STAT3-activating mutants causing stronger CTT–SH2 interaction showed sustained STAT3 activation despite pS727, while pS727 might work through P715-dependent modification of the CTT to dissociate the pY705–SH2 interaction
To further examine the regulatory role of the CTT–SH2 interaction in the activation of STAT3, we chose two activating mutants, Y640F and K658Y (49). The structural analysis of these mutations using MutaBind and PIC revealed that the K658Y mutation created new hydrophobic interactions between Y658 and each of the residues I711, V713, L666 and P669, favoring stronger association between the CTT and the SH2 (Table 2). The analysis also showed that the Y640F mutation disrupted the hydrogen bond between the side chains of C712 and Y640, while the mutation preserved the hydrophobic interaction and the aromatic–aromatic interaction between F710 and the mutated F640 (Table 2). The structural analysis, together with the positions of the C712–Y640 interaction (δ in Fig. 4B), suggested that the loss of a hydrogen bond between C712 and Y640 would enhance the association of the distal part of the CTT with SH2, since the hydrogen bond positions in the direction opposite from that of the CTT–SH2 interaction made by the other three interactions described above (α, β and γ in Fig. 4B). The loss of a hydrogen bond between C712 and Y640 would likely make the pY705-containing peptide closer to the SH2 domain, thereby enhancing the pY705–SH2 interaction. These predicted structural changes in both mutants are consistent with the actual results showing that these STAT3 mutants sustained pY705 even after ST treatment for 60 min (Fig. 6A). The stabilizing effect of these mutants was confirmed by testing STAT3 double mutants, STAT3 Y640F/K709E, STAT3 K658Y/K709E, STAT3 Y640F/T714A and STAT3 K658Y/T714A, showing that both Y640F and K658Y differentially stabilized the inactive K709E or T714A mutants (Fig. 6B and C). Both active mutants stabilized the T708S mutant (Supplementary Figure 4). Taken together, these results suggested that the overall degree of the intramolecular and intermolecular associations involving the CTT determines the stability of the STAT3 pY705–SH2 interaction, and importantly the enhanced association of the CTT–SH2 by the active mutants negates the destabilizing effect of pS727. The STAT3 mutants containing Y640F or K658Y showed pS727 in unstimulated cells (data not shown, see also a mobility shift of STAT3 in Fig. 6A–C).
Fig. 6.

The active mutants, STAT3 Y640F and STAT3 K658Y, stabilize pY705–SH2 by enhancing the CTT–SH2 interaction, while the P715A mutant functionally inhibits the pS727's destabilizing activity. Changes in the pY705 levels in STAT3 Y640F and STAT3 K658 (A), Y640F/K709E and K658Y/K709E (B), Y640F/T714A and K658Y/T714A (C), P715A (D), P715A/K709E and P715A/T714A (E). Cells were stimulated with IL-6 for 0 or 15 min, or with IL-6 for 15 min, followed by treatment with ST for the indicated times. Whole cell extracts were immunoblotted as in Fig. 1(A). The experiments were repeated three times with similar results.
On the basis of the above results, we hypothesized that the physiological pS727-dependent inactivation of STAT3 might be triggered by pS727-dependent modulation of the CTT that weakens either the CTT–SH2 interaction or the CTT–CTT interaction. One possible modulation is phosphorylation of the CTT at T708 or T714 (58, 59) and another is acetylation of the two lysine residues, K707 and K709, in the CTT by p300/CBP (60). The phosphorylation at either T708 or T714 is expected to reduce the level of dimerization, on the basis of the dimer interface depicted in Fig. 4(B) and shown in Table 1. In particular, the phosphorylation of T714 would expand the distance between T714 and Y657, thereby weakening the association of the distal part of CTT with the SH2. Importantly, STAT3 T714 was previously shown to be phosphorylated dependently on pSer727 through GSK-3 α/β in human endothelial cells stimulated with a combination of epidermal growth factor and protease-activated receptor-1 (61). In that study, IL-6 was shown to phosphorylate T714 in the endothelial cells, although the level of pT714 was low compared with those of pY705 and pS727 (61). We probed the possible role of pT714 by using the P715A mutation, which likely inhibits phosphorylation at T714 by any proline-directed kinases but shows no change in protein–protein interactions in the pY705–SH2 according to our analysis using MutaBind and PIC programs (data not shown). The P715A mutation inhibited the ST-induced inactivation of STAT3 with keeping pS727 (see a mobility shift of STAT3 in Fig. 6D), but the same mutation did not affect the inactivation of STAT3 K709E nor that of STAT3 T714A (Fig. 6E), which was quite a contrast to the stabilizing effects of Y640F and K658Y (Fig. 6B and C), suggesting that Pro715 may functionally mediate pS727's effect without enhancing the CTT–SH2 interaction. Although phospho-T714 is a possible and likely candidate for the pS727-dependent event mediating the destabilizing effect, it is still possible that an as-yet unidentified Pro715-dependent modification of the CTT might be involved in the pS727-dependent inactivation of WT STAT3. The acetylation on K709 would weaken the CTT–SH2 interaction by disrupting the K709–R688 interaction and that on K707 would inhibit the PTM–SH2. Our results collectively indicated that pS727 enhances dissociation of the pY705–SH2 interaction by weakening the CTT–SH2 association and/or the CTT–CTT association likely through Pro715-dependent CTT modulation.
Discussion
Cells are often stimulated by extracellular signals to survive, function, proliferate or differentiate. STAT3 is one of the transcription factors that mediate cellular signals generated by extracellular cytokines. The current study reconfirmed that the rapid cycling of STAT3 between the cytoplasm and nucleus both in resting cells and in stimulated cells is an important feature as a signal-transducing transcription factor. In contrast to the shuttling mechanism of unstimulated STAT3, once-activated STAT3 undergoes inactivation processes to pY705 dephosphorylation and to post-activation nuclear export in a pS727-dependent manner. The rapid cycle makes it possible for STAT3 to faithfully respond to the information on the intensity and duration of signals both for pY705 and pS727 to cause specific outcomes. A study by Braun et al. showed that the differences of the duration of receptor activation and STAT3 activation in dendritic cells in response to IL-6 and IL-10 determine the different outcomes of pro-inflammatory or anti-inflammatory responses, respectively (62). The rapid cycling of STAT3 activation–inactivation characterized here should be important for determining the STAT3 function in response to cellular signals generated by IL-6 or IL-10.
We studied how pS727 causes rapid cycles of STAT3 activation–inactivation through CRM1-independent export and revealed the structural basis of pS727-dependent dissociation of the pY705–SH2 interaction. A multistep model of STAT3 inactivation together with the role of pS727 is depicted in Fig. 7. The STAT3 inactivation process involves the following steps: (i) the pS727-dependent event(s) and the handshake type NTD–NTD interaction trigger the STAT3 inactivation, changing from the activated form (A) to the inactive form of STAT3 (B) with pY705 being dissociated from the SH2 of its partner, (ii) an unknown process changes the form (B) to another inactivated form of STAT3 (C), which is used for pY705 dephosphorylation by TC45 and CRM1-dependent nuclear export, (iii) a further conformational change brings the (C) form to the final inactivated form (C′). The transition from (C) to (C′) is efficiently blocked by any N-terminal tags, GFP-related Ruby or a small peptide FLAG-tag. The inactivated form (C′) uses a CRM1-independent system for its nuclear export and seems the major form for the post-activation nuclear export. Activated nuclear STAT3 should go through this series of conformational changes in 60–90 min, possibly at much earlier times judged by the induction of saa1 mRNA (Fig. 3E). It is possible that the inactivation process may be regulated by unidentified regulators. A possible regulator is an ST-sensitive kinase(s) or its downstream kinase(s), since a certain kinase, directly or indirectly inhibited by ST, seems to stabilize the activated STAT3 in the nucleus, considering that once-activated nuclear STAT3 mutants, STAT3 K709E and STAT3 T714A, were rapidly inactivated by ST for nuclear export and pY705 dephosphorylation in the cytoplasm in DKO cells (Fig. 5A–C). Acetylation of STAT3 at K49 and K87 has been shown to accelerate nuclear export of STAT3 and activate an acute-phase response gene hAGT, as reported by Ray et al. (63). It is possible that the acetylation at the N-terminus of STAT3 may be involved in the repeated activation of STAT3 for proper gene activation.
Fig. 7.

Model for the pS727- and NTD-dependent inactivation of STAT3, involving multiple steps leading to pY705 dephosphorylation and a selection of nuclear export system. The activated form of STAT3 (A) shows a parallel dimer stabilized by the reciprocal pY705–SH2 interactions with their NTDs apart. The activated STAT3 undergoes inactivation in a pS727-dependent manner with the assistance of the handshake type NTD–NTD dimerization to become the intermediate inactivated form (B). The dissociation of the pY705–SH2 and the NTD–NTD interaction cause a further change to become inactive conformation (C) that allows for TC45-dependent pY705 dephosphorylation and CRM1-dependent nuclear export, followed by a transition to another inactive conformation (C′) that allows for CRM1-independent nuclear export. The transition from (C) to (C′) can be inhibited by any N-terminal tags. pS727 enhances the dissociation of the pY705–SH2 interaction by weakening the intramolecular CTT–SH2 and/or intermolecular CTT–CTT interaction likely through modulating the CTT in a P715-dependent manner. pS727+α, pS727-dpendent event(s); TAD, transactivation domain; NTD, N-terminal domain. Core body is composed of the coiled-coil domain, DNA-binding domain, linker domain and SH2 domain.
The sequential changes in the STAT3 conformations, from (C) to (C′), may explain how pY705 dephosphorylation precedes the major type of CRM1-independent nuclear export (Figs 1–3). Considering the likely position of the handshake NTD dimer relative to the core bodies of STAT3 that has been simulated from that of unstimulated STAT1 (64), it is likely that the NTD–NTD dimer may stabilize the STAT3 dimer with the dissociated C-termini (B in the model) for further inactivation processes. Although it is argued that STAT3 does not make anti-parallel dimers during inactivation (33), the consequences of the Ruby or FLAG-tagging to STAT3, including alterations in the selection of nuclear export mechanisms and a delay in the nuclear export, strongly suggested the occurrence of complex conformational changes of STAT3 dimers during inactivation. The entities of each STAT3 form (B), (C) and (C′), the nuclear export systems and the underlying mechanisms for the sequential changes should be clarified to prove this model.
The importance of repeated activation of STAT3 in gene activation has been reported by Herrmann et al. using a system of STAT3-CFP-YFP with inhibition of its nuclear export using a CRM1 inhibitor (29). Our study confirmed the role of repeated activation of STAT3 in activating STAT3 target genes, both the socs3 and saa1 genes. This result highlighted the importance of pS727 in determining the STAT3 activities in response to the cellular signals generated by extracellular signals.
Our structural and functional analyses, based on the experiments using inactive mutants, active mutants and double mutants with a combination of one active mutant and one inactive mutant, supported the notion that the degree of the CTT–CTT intermolecular interaction and the CTT–SH2 intramolecular interaction determines the pY705–SH2 interaction for STAT3 stability. Importantly, the pS727-dependent event is likely to use this regulatory system by modulating the CTT to accelerate the pY705–SH2 dissociation. On the pS727-dependent CTT modulation, we proposed that P715 may play an important role to mediate pS727's action since P715A mutation inhibited the pS727's destabilizing activity without causing structural changes judged by both structural analysis of STAT3 P715A, and the pY705 dephosphorylation assay using STAT3 K709E/P715A and STAT3 T714A/P715A mutants (Fig. 6E). Although the likely modification mediating pS727's effect is phosphorylation at T714, it is possible that an unidentified Pro715-dependent modification in addition to or independently of T714 phosphorylation may mediate the pS727's effect. Further study will be needed to identify such modification. In our preliminary study, we examined the phosphorylation sites in the STAT3 K709E/Y640F mutant without or with IL-6 stimulation for 15 min and observed multiple phosphorylations in the C-terminal region of the mutant, including those of T708, T714 and S727 in unstimulated cells and those of T721, S727 and Y705 in stimulated cells (H. Zhao, T. Shiromizu, T. Tomonaga and K. Nakajima, unpublished data). This observation indicated that activated STAT3 K709E/Y640F with enhanced pY705–SH2 interaction (Fig. 6B) actually removed phosphorylations at T708 and T714, supporting the notion that phosphorylation at T708 or T714 of the CTT would cause dissociation of pY705–SH2 interaction (Table 2).
The role of a stabilizing intramolecular interaction between the C-terminal region and the SH2 region has been reported with STAT5 (65). That study shows that STAT5 F706 intramolecularly interacts with an SH2 region through multiple hydrophobic interactions, and the intramolecular interaction is required for the dimerization of STAT5 (65). It is not known, however, whether the intramolecular interaction between the distal part of the C-terminal segment and its own SH2 is a target of regulation during the process of physiological inactivation, like the case of STAT3. Although STAT1 has a structure similar to the BG loop of STAT3, the protein interaction interface of its dimer differs slightly from that of STAT3, in that STAT1 does not have an apparent intramolecular association (unpublished data obtained through the analysis of 1BF5 (66) by PIC), suggesting that the pS727-dependent regulation of STAT inactivation may be unique to STAT3. This possibility is currently under investigation.
Both pS727 and the acetylation of lysine residues including K707 and K709 have been shown to enhance STAT3's recruitment to and STAT3's activity in the mitochondria (67, 68). Our results showing that once-activated STAT3 with pY705, pS727 and modified CTT is rapidly inactivated for pY705 dephosphorylation and nuclear export is consistent with the above STAT3's recruitment to the mitochondria and its activity in the mitochondria. Considering the mechanisms of action of pS727 and the CTT modulation, the conformations of STAT3 with loosened C-termini might be suitable for its translocation to the mitochondria and its function there. However, when cells have enough activity to reactivate STAT3, activated STAT3 will work in the nucleus for gene activation.
Funding
This work was supported by JSPS KAKENHI grant number JP17K08639 (K.N.).
Supplementary Material
{kind=link}
Acknowledgements
We are grateful to Akira Inoue for discussion and Junko Kanbara for technical assistance. We also thank Mamoru Suzuki for the use of PDB files and PyMol, and Fuminori Tokunaga and the members of his laboratory for discussions. The sequence analysis, western blot analysis and immunohistochemistry analysis were performed in the Research support platform of Osaka City University Graduate School of Medicine.
Conflicts of interest statement: the authors declared no conflicts of interest.
References
- 1. Darnell, J. E., Jr, Kerr, I. M. and Stark, G. R. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415. [DOI] [PubMed] [Google Scholar]
- 2. Darnell, J. E., Jr. 1997. STATs and gene regulation. Science 277:1630. [DOI] [PubMed] [Google Scholar]
- 3. Hirano, T., Nakajima, K. and Hibi, M. 1997. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 8:241. [DOI] [PubMed] [Google Scholar]
- 4. Yu, H., Pardoll, D. and Jove, R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakajima, K., Yamanaka, Y., Nakae, K.et al. 1996. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 15:3651. [PMC free article] [PubMed] [Google Scholar]
- 6. Kojima, H., Kunimoto, H., Inoue, T. and Nakajima, K. 2012. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 11:730. [DOI] [PubMed] [Google Scholar]
- 7. Wen, Z., Zhong, Z. and Darnell, J. E., Jr. 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241. [DOI] [PubMed] [Google Scholar]
- 8. Kojima, H., Sasaki, T., Ishitani, T.et al. 2005. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFbeta-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc. Natl Acad. Sci. USA 102:4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Decker, T. and Kovarik, P. 2000. Serine phosphorylation of STATs. Oncogene 19:2628. [DOI] [PubMed] [Google Scholar]
- 10. Becker, S., Groner, B. and Müller, C. W. 1998. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 394:145. [DOI] [PubMed] [Google Scholar]
- 11. Abe, K., Hirai, M., Mizuno, K.et al. 2001. The YXXQ motif in gp 130 is crucial for STAT3 phosphorylation at Ser727 through an H7-sensitive kinase pathway. Oncogene 20:3464. [DOI] [PubMed] [Google Scholar]
- 12. Wakahara, R., Kunimoto, H., Tanino, K.et al. 2012. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells 17:132. [DOI] [PubMed] [Google Scholar]
- 13. Gough, D. J., Corlett, A., Schlessinger, K., Wegrzyn, J., Larner, A. C. and Levy, D. E. 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wegrzyn, J., Potla, R., Chwae, Y. J.et al. 2009. Function of mitochondrial Stat3 in cellular respiration. Science 323:793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haspel, R. L., Salditt-Georgieff, M. and Darnell, J. E., Jr. 1996. The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO J. 15:6262. [PMC free article] [PubMed] [Google Scholar]
- 16. Haspel, R. L. and Darnell, J. E., Jr. 1999. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc. Natl Acad. Sci. USA 96:10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Köster, M. and Hauser, H. 1999. Dynamic redistribution of STAT1 protein in IFN signaling visualized by GFP fusion proteins. Eur. J. Biochem. 260:137. [DOI] [PubMed] [Google Scholar]
- 18. Begitt, A., Meyer, T., van Rossum, M. and Vinkemeier, U. 2000. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl Acad. Sci. USA 97:10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McBride, K. M., McDonald, C. and Reich, N. C. 2000. Nuclear export signal located within the DNA-binding domain of the STAT1transcription factor. EMBO J. 19:6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meyer, T., Hendry, L., Begitt, A., John, S. and Vinkemeier, U. 2004. A single residue modulates tyrosine dephosphorylation, oligomerization, and nuclear accumulation of stat transcription factors. J. Biol. Chem. 279:18998. [DOI] [PubMed] [Google Scholar]
- 21. Reich, N. C. and Liu, L. 2006. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6:602. [DOI] [PubMed] [Google Scholar]
- 22. Reich, N. C. 2013. STATs get their move on. JAKSTAT 2:e27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Domoszlai, T., Martincuks, A., Fahrenkamp, D., Schmitz-Van de Leur, H., Küster, A. and Müller-Newen, G. 2014. Consequences of the disease-related L78R mutation for dimerization and activity of STAT3. J. Cell Sci. 127:1899. [DOI] [PubMed] [Google Scholar]
- 24. Braunstein, J., Brutsaert, S., Olson, R. and Schindler, C. 2003. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 278:34133. [DOI] [PubMed] [Google Scholar]
- 25. Pranada, A. L., Metz, S., Herrmann, A., Heinrich, P. C. and Müller-Newen, G. 2004. Real time analysis of STAT3 nucleocytoplasmic shuttling. J. Biol. Chem. 279:15114. [DOI] [PubMed] [Google Scholar]
- 26. Liu, L., McBride, K. M. and Reich, N. C. 2005. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl Acad. Sci. USA 102:8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhattacharya, S. and Schindler, C. 2003. Regulation of Stat3 nuclear export. J. Clin. Invest. 111:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ushijima, R., Sakaguchi, N., Kano, A.et al. 2005. Extracellular signal-dependent nuclear import of STAT3 is mediated by various importin alphas. Biochem. Biophys. Res. Commun. 330:880. [DOI] [PubMed] [Google Scholar]
- 29. Herrmann, A., Vogt, M., Mönnigmann, M.et al. 2007. Nucleocytoplasmic shuttling of persistently activated STAT3. J. Cell Sci. 120:3249. [DOI] [PubMed] [Google Scholar]
- 30. Mao, X., Ren, Z., Parker, G. N.et al. 2005. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell 17:761. [DOI] [PubMed] [Google Scholar]
- 31. Zhong, M., Henriksen, M. A., Takeuchi, K.et al. 2005. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc. Natl Acad. Sci. USA 102:3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mertens, C., Zhong, M., Krishnaraj, R., Zou, W., Chen, X. and Darnell, J. E., Jr. 2006. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 20:3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren, Z., Mao, X., Mertens, C.et al. 2008. Crystal structure of unphosphorylated STAT3 core fragment. Biochem. Biophys. Res. Commun. 374:1. [DOI] [PubMed] [Google Scholar]
- 34. ten Hoeve, J., de Jesus Ibarra-Sanchez, M., Fu, Y.et al. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 22:5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer, T., Marg, A., Lemke, P., Wiesner, B. and Vinkemeier, U. 2003. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev. 17:1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamamoto, T., Sekine, Y., Kashima, K.et al. 2002. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem. Biophys. Res. Commun. 297:811. [DOI] [PubMed] [Google Scholar]
- 37. Martincuks, A., Fahrenkamp, D., Haan, S., Herrmann, A., Küster, A. and Müller-Newen, G. 2016. Dissecting functions of the N-terminal domain and GAS-site recognition in STAT3 nuclear trafficking. Cell Signal. 28:810. [DOI] [PubMed] [Google Scholar]
- 38. Muromoto, R., Sekine, Y., Imoto, S.et al. 2008. BART is essential for nuclear retention of STAT3. Int. Immunol. 20:395. [DOI] [PubMed] [Google Scholar]
- 39. Sato, N., Tsuruma, R., Imoto, S.et al. 2005. Nuclear retention of STAT3 through the coiled-coil domain regulates its activity. Biochem. Biophys. Res. Commun. 336:617. [DOI] [PubMed] [Google Scholar]
- 40. Meyer, T., Begitt, A. and Vinkemeier, U. 2007. Green fluorescent protein-tagging reduces the nucleocytoplasmic shuttling specifically of unphosphorylated STAT1. FEBS J. 274:815. [DOI] [PubMed] [Google Scholar]
- 41. Shalem, O., Sanjana, N. E., Hartenian, E.et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wen, W., Meinkoth, J. L., Tsien, R. Y. and Taylor, S. S. 1995. Identification of a signal for rapid export of proteins from the nucleus. Cell 82:463. [DOI] [PubMed] [Google Scholar]
- 43. Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanda, Y. 2013. Investigation of the freely available easy-to-use software ‘EZR' for medical statistics. Bone Marrow Transplant. 48:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tina, K. G., Bhadra, R. and Srinivasan, N. 2007. PIC: protein interactions calculator. Nucleic Acids Res. 35:W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li, M., Simonetti, F. L., Goncearenco, A. and Panchenko, A. R. 2016. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 44:W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ichiba, M., Nakajima, K., Yamanaka, Y., Kiuchi, N. and Hirano, T. 1998. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J. Biol. Chem. 273:6132. [DOI] [PubMed] [Google Scholar]
- 48. Narimatsu, M., Maeda, H., Itoh, S.et al. 2001. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol. Cell. Biol. 21:6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pilati, C., Amessou, M., Bihl, M. P.et al. 2011. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 208:1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karaman, M. W., Herrgard, S., Treiber, D. K.et al. 2008. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26:127. [DOI] [PubMed] [Google Scholar]
- 51. Schindler, C., Shuai, K., Prezioso, V. R. and Darnell, J. E., Jr. 1992. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257:809. [DOI] [PubMed] [Google Scholar]
- 52. Hu, T., Yeh, J. E., Pinello, L.et al. 2015. Impact of the N-terminal domain of STAT3 in STAT3-dependent transcriptional activity. Mol. Cell. Biol. 35:3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nkansah, E., Shah, R., Collie, G. W.et al. 2013. Observation of unphosphorylated STAT3 core protein binding to target dsDNA by PEMSA and X-ray crystallography. FEBS Lett. 587:833. [DOI] [PubMed] [Google Scholar]
- 54. Auernhammer, C. J., Bousquet, C. and Melmed, S. 1999. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc. Natl Acad. Sci. USA 96:6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hagihara, K., Nishikawa, T., Isobe, T., Song, J., Sugamata, Y. and Yoshizaki, K. 2004. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem. Biophys. Res. Commun. 314:363. [DOI] [PubMed] [Google Scholar]
- 56. Woellner, C., Gertz, E. M., Schäffer, A. A.et al. 2010. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J. Allergy Clin. Immunol. 125:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chandesris, M. O., Melki, I., Natividad, A.et al. 2012. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 91:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cerami, E., Gao, J., Dogrusoz, U.et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koskela, H. L., Eldfors, S., Ellonen, P.et al. 2012. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 366:1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nie, Y., Erion, D. M., Yuan, Z.et al. 2009. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 11:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waitkus, M. S., Chandrasekharan, U. M., Willard, B.et al. 2014. Signal integration and gene induction by a functionally distinct STAT3 phosphoform. Mol. Cell. Biol. 34:1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Braun, D. A., Fribourg, M. and Sealfon, S. C. 2013. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J. Biol. Chem. 288:2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ray, S., Boldogh, I. and Brasier, A. R. 2005. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 129:1616. [DOI] [PubMed] [Google Scholar]
- 64. Sgrignani, J., Olsson, S., Ekonomiuk, D.et al. 2015. Molecular determinants for unphosphorylated STAT3 dimerization determined by integrative modeling. Biochemistry 54:5489. [DOI] [PubMed] [Google Scholar]
- 65. Fahrenkamp, D., Li, J., Ernst, S.et al. 2016. Intramolecular hydrophobic interactions are critical mediators of STAT5 dimerization. Sci. Rep. 6:35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen, X., Vinkemeier, U., Zhao, Y., Jeruzalmi, D., Darnell, J. E., Jr and Kuriyan, J. 1998. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93:827. [DOI] [PubMed] [Google Scholar]
- 67. Tammineni, P., Anugula, C., Mohammed, F., Anjaneyulu, M., Larner, A. C. and Sepuri, N. B. 2013. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 288:4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu, Y. S., Liang, J. J., Wang, Y.et al. 2016. STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci. Rep. 6:39517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.