

Abstract
Mangrove sediment is a crucial component in the global mercury (Hg) cycling and acts as a hotspot for methylmercury (MeHg) production. Early evidence has documented the ubiquity of well-studied Hg methylators in mangrove superficial sediments; however, their diversity and metabolic adaptation in the more anoxic and highly reduced subsurface sediments are lacking. Through MeHg biogeochemical assay and metagenomic sequencing, we found that mangrove subsurface sediments (20–100 cm) showed a less hgcA gene abundance but higher diversity of Hg methylators than superficial sediments (0–20 cm). Regional-scale investigation of mangrove subsurface sediments spanning over 1500 km demonstrated a prevalence and family-level novelty of Hg-methylating microbial lineages (i.e., those affiliated to Anaerolineae, Phycisphaerae, and Desulfobacterales). We proposed the candidate phylum Zixibacteria lineage with sulfate-reducing capacity as a currently understudied Hg methylator across anoxic environments. Unlike other Hg methylators, the Zixibacteria lineage does not use the Wood–Ljungdahl pathway but has unique capabilities of performing methionine synthesis to donate methyl groups. The absence of cobalamin biosynthesis pathway suggests that this Hg-methylating lineage may depend on its syntrophic partners (i.e., Syntrophobacterales members) for energy in subsurface sediments. Our results expand the diversity of subsurface Hg methylators and uncover their unique ecophysiological adaptations in mangrove sediments.
Subject terms: Microbial ecology, Bacterial genetics
Introduction
The production of neurotoxic methylmercury (MeHg) by microorganisms is mediated by the functional proteins encoded by the two-gene cluster hgcA and hgcB [1]. The presence of the hgcAB gene pair is a reliable indicator of a microbe’s ability to methylate mercury (Hg) [2], and has previously been linked to sulfate-reducing bacteria (SRB) [1], iron-reducing bacteria (IRB) [3], methanogens [4], and other anaerobes [2]. The discovery of the hgcAB gene pair has significantly expanded the known diversity of microorganisms capable of producing MeHg [5]. For instance, recent studies investigating the subsurface biosphere [6–8] have identified Hg-methylating microbes from several phyla, including Lokiarchaeota, Aminicenantes, Kiritimatiellaeota, Spirochaetes, Marinimicrobia, and Actinobacteria, that were previously not thought to be associated with MeHg production. The subsurface biosphere is a major reservoir of phylogenetically and metabolically diverse microorganisms that have largely yet to be characterized [9]. Despite the detection of bioaccumulated MeHg in this environment [2], the microbiota responsible for mediating MeHg cycling in the subsurface biosphere remains an elusive “dark matter”.
Mangrove sediment is a crucial component in the global Hg cycling, serving as ideal niches of microbial Hg methylation [10]. Previous research has primarily focused on MeHg biogeochemistry in the superficial sediments of mangroves, revealing the presence of well-known Hg methylators [11]. Recent findings have demonstrated that the subsurface microbial communities showed apparent acclimation to Hg, and Hg methylation may be favored by more reducing conditions (i.e., in the deeper layers) [10, 12]. However, whether the more anoxic and highly reducing condition in mangrove subsurface sediments can lead to intensive MeHg production remains unknown, as does the identity of the subsurface microorganisms responsible for Hg methylation. Our prior research, indicating that the more reducing conditions in the subsurface layer favor anaerobes (i.e., SRB and methanogens) [13], raises the possibility of a more diverse Hg-methylating microbial community in the mangrove subsurface sediments.
Mangrove subsurface sediments have long been recognized as energy-limited [14–16], since more recalcitrant organic matters are trapped in deeper sediments with physical forces (i.e., hydrodynamics and gravity) [17]. However, owing to the lack of cultured relatives, much remains unknown about the metabolic feature and adaptive strategy of subsurface Hg methylators under such energy-limited conditions. In recent years, high-throughput culture-independent sequencing has provided a window into the metabolic novelty of subsurface-dwelling microorganisms [18–20]. The discovery of novel methanogen genomes from the terrestrial subsurface has uncovered their diverse ecophysiological adaptations with unexpected metabolic features (i.e., the potential for extracellular electron transfer) [21]. In energy-limited conditions, syntrophic relationships have been proposed as a common strategy for mediating biogeochemical cycles [22, 23]. A recent study has provided evidence that the SRB-methanogen syntrophy via interspecies electron transfer dramatically stimulated Hg methylation in co-cultures [24]. However, the potential for syntrophic relationships in energy-limited mangrove subsurface sediments for Hg methylation remains uncertain. With the continued advancement of high-throughput sequencing, the integration of microbial genomic information into syntrophic models will enhance our understanding of the adaptive strategies of Hg methylators in the subsurface biosphere.
In this study, we used MeHg measurements, Hg isotope assays, and metagenomic sequencing to examine the levels of MeHg, phylogenetic diversity, and metabolic adaptations of Hg methylators in 100-cm mangrove sediment columns. Our findings indicate elevated diversity of Hg methylators in subsurface layers. This prompted us to investigate the regional-scale distribution of these subsurface-dwelling Hg methylators across southeastern mangrove regions of China, spanning over 1500 km. Our most significant discovery is the identification of the candidate phylum Zixibacteria as a currently understudied Hg methylator in anoxic subsurface environments, with unexpected metabolic and lifestyle features, such as Hg methylation independent of the Wood–Ljungdahl pathway but involved in methionine synthesis, benzoyl-CoA, and syntrophic pathways. This study significantly expands the diversity of subsurface-dwelling Hg methylators and sheds light on their unique metabolic adaptations in mangrove sediments.
Materials and methods
Mangrove sediment sampling
The sampling site is located at Qi’ao Mangrove Wetland Park (22° 26′ 12.28′′ N, 113° 38′ 26.12′′ E), Guangdong Province, China. The park was characterized by a mean annual temperature of 22.4 °C and annual precipitation range of 1700–2200 mm. The tidal regime was irregular semidiurnal, with average high and low tide levels of 0.17 m and −0.14 m, respectively [25]. We collected partially air-exposed sediments from the native mangrove species (Kandelia obovata) in the park, in August 2019. Five replicate sediment cores were obtained using a 1-m long PVC sampling column after ebb. The cores were sliced into 10 depth intervals (0–5, 5–10, 10–15, 15–20, 20–30, 30–40, 40–50, 50–60, 60–80, and 80–100 cm), yielding 50 samples in total. The sliced sediments were stored in a portable cooler at 4 °C and transported back to the laboratory within 24 h for further analysis. Each sample was divided into two sub-samples for different purposes: one for biogeochemical properties analysis, stored at 4 °C, and the other for DNA extraction, stored at −80 °C.
Biogeochemical properties analyses
Sulfate anion in the sediments was analyzed using a Thermo Scientific Dionex ICS-1100 Ion Chromatography System, which consisted of a guard column (IonPac AG14A), an analytical column (IonPac AS14A), a suppressor (Dionex ERS 500), and a column heater (Dionex ICS-1100 Column Heater). CH4 measurement was carried out by placing the sediment samples in 20 mL serum bottles containing 5 mL of 1 M NaOH-buffered deionized water [26]. The porewater CH4 concentrations were then measured using an online GC–Mass (GCMS-QP2010 SE). Fe (II) concentrations were measured directly, whereas Fe (III) concentrations were calculated as the difference between total Fe and Fe (II) concentrations. Total Fe and Fe (II) were determined using iron-phenanthroline spectrophotometry, which relies on the Fe2+-dependent optical absorbance of bathophenanthrolinedisulfonic acid (BPS) at 535 nm at pH 5.4 [27].
Mercury analysis
All analyses for Hg and MeHg concentration as well as total Hg (HgT) stable isotope were performed as described in previous studies [28, 29]. The homogenized and freeze-dried sediment samples were prepared for Hg concentration and isotope analysis using a double-stage tube furnace in conjunction with 40% anti-aqua regia trapping solution (HNO3/HCl = 2/1, v/v) [30]. All digests were heated to 95 °C for 8 h. Standard reference materials (GSS-5, Hg concentration: 290 ± 30 ng/g) and method blanks were processed similarly to the samples. The former yielded Hg recoveries of 109 ± 4% and the latter showed Hg concentrations lower than the detection limit, precluding lab contamination. The HgT and MeHg contents in the digested sample were measured by a MERX automated HgT analytical system (Brooks Rand Laboratories, USA) and inductively coupled plasma mass spectrometry (ICP-MS) according to USEPA standard methods 1631E and 1630, respectively [31, 32].
For HgT stable isotope analyses, the preconcentrated solutions were diluted to 0.5 ng/mL with an acid concentration of 10–20% prior to Hg isotope analysis using a Neptune Plus MC-ICP-MS. Hg isotope ratios were reported following the convention proposed by Blum and Bergquist [33]. To ensure that pre-concentration processes did not induce fractionation [34], the National Institute of Standards and Technology (NIST) 3133 was also run before and after each sample. The mass-dependent fractionation (MDF) is expressed in δ202Hg notation in units of ‰ referenced to the NIST-3133:
| 1 |
The mass-independent fractionation (MIF) is reported in Δ notation, which describes the difference between the measured δxxxHg and the theoretically predicted δxxxHg value in units of ‰:
| 2 |
β is 0.252 for 199Hg, 0.5024 for 200Hg, and 0.752 for 201Hg. NIST-3177 secondary standard solutions, diluted to 0.5 ng/mL Hg with 10% HCl, were measured before and after each sample. The overall average and uncertainty values of NIST-3177 (δ202Hg: −0.53 ± 0.06‰; Δ199Hg: −0.15 ± 0.01‰; Δ200Hg: 0.02 ± 0.03‰; Δ201Hg: 0.00 ± 0.03‰; 2 SD, n = 8) and GSS-5 (δ202Hg: −1.76 ± 0.03‰; Δ199Hg: −1.60 ± 0.02‰; Δ200Hg: 0.03 ± 0.03‰; Δ201Hg: −0.29 ± 0.03‰; 2 SD, n = 8) agree well with previous results [28, 29]. The larger values of 2-SD between NIST-3177 and GSS-5 are used to reflect maximum analytical uncertainties.
For Hg isotope fractionation, the previous study summarized the research published up until the year 2011, with the aim of providing a basic framework for understanding MDF and MIF in the environment [35]. Nearly all kinetic reactions involving Hg produce products with lower δ202Hg (i.e., isotopically lighter) and leave a residual pool of reactants with higher δ202Hg (i.e., isotopically heavier). In contrast, microbial reactions do not produce significant MIF (i.e., Δ199Hg does not differ between products and reactants). As these reactions proceed, the ratio of Δ199Hg to δ202Hg can be diagnostic of the type of reaction. The overview of the general patterns in isotope fractionation is detailed in Supplementary Table S1.
DNA extraction
The whole community DNA was extracted from 5.0 g mangrove sediment and purified using a combined protocol of sodium dodecyl sulfate extraction method [36] and Power Soil DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, California, USA) according to the manual. The DNA purity was checked by NanoDrop ND-2000 Spectrophotometer (Thermo Fisher Scientific, MA, USA), and ratios of 260/280 and 260/230 were about 1.80 and 1.70, respectively. The DNA concentrations were quantified using a fluorescent method (Qubit 4 Fluorometer, Thermo Scientific, USA).
Metagenomic sequencing and processing
For metagenomic sequencing, 1.0 μg of DNA from each sample was prepared using the NEBNext DNA Library Prep Kit. Samples were individually sequenced with the NextSeq550 platform (Illumina, San Diego, CA, USA), resulting in 2 × 150 bp paired-end reads with an average of 12.47 Gbp per sample. Low-quality (quality score ≤ 38, base N > 10 bp, the overlap length between adapter and reads >15 bp) paired-end reads were filtered. After trimming the raw sequencing reads using BBDuk (https://jgi.doe.gov/data-and-tools/bbtools/, parameters: ktrim = r k = 28 mink = 12 hdist = 1 tbo = t tpe = t qtrim = rl trimq = 20 minlength = 70), genome assembly and binning were performed according to the MetaWRAP pipeline v1.3.0 [37]. The sequences were assembled with MEGAHIT (v1.1.3; parameters: -m 500) to generate contigs [38]. Genome binning of assembled contigs was done using MetaBAT2 (v2.12.1) [39] and MaxBin2 (v2.2.6) [40], and the resulting metagenome-assembled genomes (MAGs) were consolidated with the Bin_refinement module. The Reassemble_bins module further improved the consolidated bin sets to generate MAGs (parameters: -m 800). The quality of MAGs was evaluated with CheckM v1.0.12 [41]. Only MAGs with a completeness of over 50% and a contamination level below 10% were analyzed further. Representative MAGs were chosen using dRep (v3.4.0, parameters: dRep dereplicate -sa 0.99 -nc 0.1) [42]. The abundance of each MAG was expressed as genome copies per million reads and calculated using CoverM v0.6.1 genome mode (https://github.com/wwood/CoverM). Taxonomic assignments of the MAGs were performed using the GTDB-Tk v1.3.0 [43].
Search for hgcAB genes
In this study, we utilized the Hg-MATE-Db [44], an up-to-date database of Hg methylators from pure culture and environmental datasets, to search for hgcAB genes. The search was conducted using HMMER v3.3.2 [45], and the results were further confirmed by examining the presence of conserved motifs (N(V/I)WCA(A/G) in hgcA and (CX2CX2CX3C) in hgcB, respectively. The hgcAB sequences were clustered at 100% identity using CD-HIT to remove redundancy [46].
Relative and normalized hgcA abundance calculation
To quantify the relative abundance of hgcA gene, we calculated the average number of aligned reads that overlapped each position on the contig. The hgcA index was built using the Salmon v0.8.1 index mode [47]. Reads were mapped to the hgcA using Salmon in the quant mode. To analyze the normalized hgcA abundance, we needed to estimate total cell counts, whereas sequencing data can generate only relative abundance values (in the absence of an internal standard). We began with the general equation:
| 3 |
This approach has been discussed and tested in-depth previously [48], where DNA yield was utilized as an estimate of microbial load in a sample. DNA yield is not a perfect estimate of microbial load due to potential ploidy [49] and bias in sequencing representation depending on the DNA extraction method [50]. But in this study, all samples were extracted using the same DNA extraction kit, and DNA yield was measured using a fluorometry-based method (Qubit 4 fluorometer dsDNA HS Assay), making it the best available measurement of microbial load (Supplementary Fig. S1).
Phylogenetic analyses
Hg methylator genomes and hgcA genes recovered in this research were analyzed and compared with reference sequences from Hg-MATE-Db. MAFFT (--auto) [51], trimAl (v1.4.rev15) [52], and IQ-TREE 2 (parameters: -st AA -alrt 1000 -bb 1000) [53] were used for sequence alignment, trimming, and construction of phylogenetic trees, respectively. The phylogenetic trees for Hg methylator genomes were inferred by a concatenated set of specific single-copy marker genes in the GTDB (https://gtdb.ecogenomic.org/). The orthologs of these marker genes in the Hg methylator and the reference genomes were identified using GTDB-Tk based on HMMER. The results were edited and modified using iTOL [54] and Adobe Illustrator, respectively. The pairwise genomic ANI values were calculated by FastANI (v1.33, https://github.com/ParBLiSS/FastANI) with default parameters based on the assemblies [55]. FastANI was targeted to estimate ANI in the 70–100% identity range.
Functional characterization of genomes
Gene prediction for all MAGs was performed using Prodigal v2.6.3 (default settings) [56]. The predicted genes were characterized using McycDB [57], SCycDB [58], and FeGenie [59]. MAGs and reference genomes were also annotated using the KEGG-based annotation program METABOLIC v4.0 [60], and custom databases were searched using DIAMOND v2.0.15 [61] and HMMER. Hydrogenases were identified using METABOLIC, consisting of NiFe-, FeFe-, and Fe-hydrogenase catalytic subunits [62]. Putative hydrogenase sequences identified in the search were then uploaded to the HydDB webserver [62] to identify and remove non-hydrogenases. Anaerobic hydrocarbon degradation genes were identified using a HMMER database (hmmbuild, hmmpress, and hmmsearch modes) of reference sequences from the anaerobic hydrocarbon degradation database [63]. Prokaryotic Hg resistance genes (merAB) were identified using publicly available homologs of merAB [64] and the search custom-markers function using HMMER v3.3.2. The distribution of biogeochemically important genes identified using these methods is shown in Supplementary Data 1. To detect multi-heme cytochromes, we utilized the Python script “cytochrome_stats.py” available for download at https://github.com/bondlab/scripts [65]. This script counts the occurrence of the heme-binding motif, CXXCH, in protein sequences. Proteins with more than three CXXCH motifs were considered as multi-heme cytochromes, following the established criteria [65, 66]. The detailed information of multi-heme cytochromes is shown in Supplementary Data 2. The annotation of Wood-Ljungdahl pathway, methionine synthesis, and benzoyl-CoA reduction was based on previous studies [67–70]. The typical cobalamin-dependent methyltransferases were predicted using KofamKOALA (release 106.0, https://www.genome.jp/tools/kofamkoala/, parameters: E‐value ≤ 1e−5) [71, 72].
Protein AlphaFold2 modeling
The three-dimensional structures of the putative HgcAB and MetH protein sequences found in this study were built and refined upon the official release of the source code and monomer neural network models of AlphaFold2 (version 2.0.1) [73]. The transmembrane regions in HgcA were predicted by CCTOP [74]. Multiple structure alignment for different taxa was performed using mTM-Align [75]. The cofactor cobalamin was placed in the binding pocket by superposing the HgcA and CFeSP (PDB entry 4DJF) [76]. Consistent with the expected Cys coordination patterns from other dicluster ferredoxins, such as that from Clostridium acidurici (PDB entry 2FDN) [77], preliminary de novo models of HgcB with coevolution restraints suggest that one [4Fe-4S] cluster was bound to Cys20, Cys23, Cys26, and Cys60 and another was bound to Cys50, Cys53, Cys56, and Cys30. The preliminary models of the HgcAB complexes were generated by PyMOL 2.5.2 (www.pymol.org), and complexes were energy minimized using the Schrodinger Suite 2021-4 (Schrödinger, LLC, Portland, OR) energy minimization server. The C-terminal tail of HgcB was also introduced at this step. The alignment of HgcA AlphaFold2 models between Zixibacteria and six confirmed Hg-methylating bacterial strains was performed by PyMOL 2.5.2.
Regional-scale distribution of Hg methylators in mangrove subsurface sediments along southeastern coastal China
A total of 27 samples were collected in mangrove sediments along southeastern coastal China, with 7 samples from Hainan (5 replicates for 30–40 cm, 2 replicates for 80–100 cm), 10 samples from Fujian (5 replicates for both 30–40 cm and 80–100 cm), 10 samples from Zhejiang (5 replicates for both 30–40 cm and 80–100 cm). The sampling sites were located at Bamen Bay Mangrove National Wetland Park (19°33′25.2“N, 110°50′20.40″E) with a mean temperature of 26.5 °C in Hainan Province, Zhangjiang Estuary Mangrove National Nature Reserve (23°55′17.18″N, 117°25′11.04″E) with a mean temperature of 28.5 °C in Fujian Province, and Ximen Island (28°20′45.15′′N, 121°10′58.27′′E) with a mean temperature of 27.0 °C in Zhejiang Province, China, respectively. The methods for DNA extraction and initial metagenomic analysis to obtain MAGs were conducted as described above. Also, we performed a series of analyses, including prodigal, hgcAB genes annotation, and conserved motifs examination, on the MAGs obtained from these three sampling points to examine the presence of Hg methylators, following the methods described above.
Global distribution
The distribution of representatives of the Zixibacteria lineage across various environments was estimated following the methodology outlined in [78]. In brief, the longest 16 S rRNA gene sequence in Zixibacteria MAGs obtained from this study was utilized for a homology-based search against the 16 S rRNA Public Assembled Metagenomes database of the Integrated Microbial Genomes and Microbiomes (IMG/M v7) system [79], employing the IMG BLAST Tool. BLAST hits with sequence identity greater than 75% (phylum threshold) [80], and the associated metadata (longitude, latitude, and habitat type) were subsequently retrieved from IMG for further analysis.
HgcA-MetH protein interaction simulation and analysis
The HDOCK server (https://hdock.phys.hust.edu.cn/) was utilized to predict a binding conformation between HgcA and MetH [81]. The HDOCK algorithm incorporates both template-based and free approaches, providing cross-validation [81]. This method involves an attempt to sample all conceivable binding modes between two separate protein structures. Given the typical absence of binding site information, a global docking approach is employed to sample assumed binding modes, involving six degrees of freedom (three rotational and three translational).
Subsequently, a scoring function, comprising docking and confidence scores, was applied to rank the sampled binding modes based on the sampling process and the resulting binding conformations [82, 83]. The docking score signifies the likelihood of a binding model, with more negative scores indicating a higher probability of binding models. The confidence score was computed using the formula:
| 4 |
A confidence score exceeding 0.70 indicates a strong likelihood of binding between the two molecules. The confidence score between 0.50 and 0.70 suggests a potential binding interaction between the two molecules. A confidence score below 0.50 indicates a lower probability of binding between the two protein molecules. To generate a 2D residue-residue interaction plot depicting the entire interface of a protein-protein complex, we inputted the complex into LigPlot [84].
Co-occurrence analysis
The co-occurrence network was based on the full-length 16 S rRNA amplicon sequencing data. For full-length 16 S rRNA amplicon sequencing, both the forward and reverse primers were tailed with sample-specific PacBio barcode sequences to allow for multiplexed sequencing. The KAPA HiFi Hot Start DNA Polymerase (KAPA Biosystems) was used to perform 20 cycles of PCR amplification, with denaturing at 95 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 60 s. Post-amplification quality control was performed on a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Amplified DNA from the sediment samples was then pooled in equimolar concentration. SMRTbell libraries were prepared from the amplified DNA by blunt ligation according to the manufacturer’s instructions (Pacific Biosciences). Purified SMRTbell libraries from the mangrove sediment bacterial communities were sequenced on PacBio Sequel cells. The DADA2 method is an algorithm for the inference of the amplicon sequence variants (ASVs) present in a sample from the library of noisy reads generated by amplicon sequencing [85]. The standard processing steps in the DADA2 workflow include quality filtering, dereplication, learning the dataset-specific error model, ASV inference, chimera removal, and taxonomic assignment against a SILVA138 database [86]. After processing the full-length 16 S rRNA amplicon sequencing data, we investigated the co-occurrence network using FastSpar (v1.0) algorithm [87]. All p values were corrected using the Benjamini–Hochberg multiple correction method. Afterward, all the statistically significant correlated taxa (p < 0.001) were used to infer the network and visualized using Gephi (version 0.10.1, https://gephi.org/) [88].
Statistical analysis
Statistical analysis and plotting were performed using R software version 4.2.1 (www.r-project.org) and associated packages, including vegan, spaa, ggtrendline, gggenes, ggmsa, mapchina, ggcor, and ggplot2 [89–92]. Richness was calculated using the spa package. The analysis of the non-metric multidimensional scaling (NMDS) was performed using the vegan and ggcor packages. Correlations were evaluated by a linear fit and Spearman test using the ggtrendline and vegan packages. Alignments of the hgcAB amino acid sequences were plotted using the ggmsa package, while the genomic context was visualized with the gggenes package. The map of China was produced using the mapchina package.
Results and discussion
Microbial Hg methylation in mangrove sediments
We investigated the variations in HgT and MeHg across five mangrove sediment columns (0–100 cm) collected in Guangdong Province, China. Across the mangrove sediment columns, the average HgT concentration was 420.06 ± 95.49 ng/g, and an increase in the concentration of HgT was observed from the surface to 20–30 cm (Fig. 1A). However, the concentration of MeHg was found to peak at 0–15 cm (3.27 ng/g), followed by a gradual decrease towards the bottom of the sediment columns. The MeHg/HgT ratio, a proxy for the potential HgT available for MeHg production [93], showed a constant decrease with increasing sediment depth, with an average value of 0.67 ± 0.09% (Fig. 1A). This falls within the typical range of 0.11−7.13% observed in natural mangrove ecosystems [10]. The in-depth profiles of sulfate, CH4, and iron (referring to the iron (III)/iron (II) ratio) were found to reflect that of MeHg across the mangrove sediment columns (Supplementary Fig. S2), which are key factors involved in Hg methylation [94].
Fig. 1. The Hg profiles in mangrove sediments.
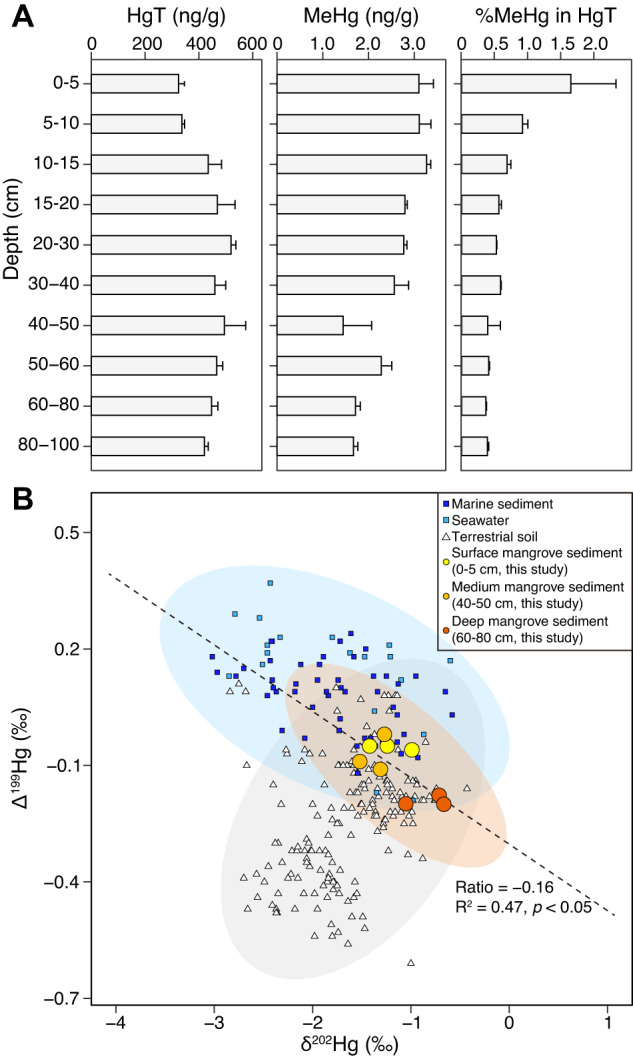
A The variations of total dissolved Hg and MeHg in the mangrove sediments column. B The relationship between δ202Hg and Δ199Hg in mangrove sediments, with reference data [96–102] from soil, marine sediments, and seawater ecosystems. The gray shaded area represents the terrestrial reservoir (soil), the light purple shaded area represents the marine reservoir (marine sediments and seawater), and the light orange shaded area represents the mangrove sediments. The trend of δ202Hg versus Δ199Hg with a near-zero ratio (Ratio = −0.16) in mangrove sediments indicates a significant contribution of microbial actions to the Hg methylation. The shadow indicates the 95% confidence interval.
Through Hg isotopic analyses, we investigated Hg sources in mangrove sediments by analyzing surface (0–5 cm), medium (40–50 cm), and deep (60–80 cm) sediment layers (three replicates each). The Δ199Hg, Δ200Hg, Δ201Hg, and δ202Hg values were determined using mass-independent fractionations (MIF) and large mass-dependent fractionations (MDF) [95]. Our data showed that the sediment across different depths exhibited similar δ202Hg ranges (−1.52 to −0.67‰, Fig. 1B and Supplementary Table S2), which were ~14 times larger than the 2 standard deviations (±0.06‰) analytical uncertainty for δ202Hg. This distinctive negative MDF of 202Hg in mangrove sediments was likely due to the mixture of δ202Hg sources in the land-ocean connection [13]. To further support this finding, we compared our results with 247 reported isotope values [96–102] of Δ199Hg and δ202Hg from terrestrial soil (177), marine sediments, and seawater (70), and found that the isotope values of mangrove sediments were in between the values from terrestrial soil and seawater (Fig. 1B). This result indicates that mangrove sediments received terrestrial- and marine-sourced Hg probably due to complex hydrodynamic (either surface runoff or tides and waves) [103].
Our dataset showed a limited MIF of 200Hg (−0.09 to 0.07‰; Supplementary Table S2), which is close to the analytical uncertainty for Δ200Hg (±0.03‰). As microbial Hg methylation does not result in odd-MIF shifts [104], the near-zero odd-MIF values detected suggest that the Hg methylation in mangrove sediments is predominantly driven by microbial reactions. To verify this hypothesis, we determined the Δ199Hg/δ202Hg ratio, which serves as a diagnostic of Hg methylation reaction types [35], with values of ~1.2–2.4 and ~0 expected for photochemical and microbial reactions, respectively (Supplementary Table S1). Our analyses revealed a strong correlation between Δ199Hg and δ202Hg in the studied samples (R2 = 0.47, p < 0.05), and the sediment Δ199Hg/δ202Hg ratio was −0.16 (Fig. 1B), indicating a substantial contribution of microbial actions to Hg methylation in mangrove sediments.
Composition and distribution of Hg methylators in mangrove subsurface sediments
Through metagenomic analysis, we identified 432 distinct hgcA sequences with conserved motifs (N(V/I)WCA(A/G)) in mangrove sediment columns and quantified their normalized abundance. We observed a notable pattern where the total normalized abundance of hgcA sequences was comparatively elevated above 20 cm and decreased below this depth (as depicted in Fig. 2A). Correlation analyses confirmed a strong association between the total normalized abundance of hgcA sequences and the concentration of MeHg in the sediments (Supplementary Fig. S3; Spearman ρ = 0.79; p < 0.0001). This finding further supports the significant role of microbial Hg methylation in mangrove sediments.
Fig. 2. The diversity and prevalence of Hg methylators in mangrove subsurface sediments.

A Normalized abundance and species richness of hgcA gene. B Occurrence frequency of representative hgcA genes from different taxonomic groups. Taxonomic classifications of the Hg methylators are labeled by different colors. C Regional-scale distribution of Hg methylators in mangrove sediments in southeastern coastal China. The samples were collected from Hainan (n = 7), Guangdong (n = 50), Fujian (n = 10), and Zhejiang (n = 10). The number of metagenomic samples is indicated by n. Taxonomic classifications of the Hg methylators are labeled by different colors, which are consistent with Fig. 2B. Among them, the gray color represents the Hg-methylating Dehalococcoidia solely present in mangrove sediment samples from Hainan. White indicates the absence of a taxonomic group at the sampling site. D Energy metabolism redox pairs of each Hg-methylating lineage.
We found that hgcA sequences in the subsurface sediments (below 20 cm) displayed a higher species richness than those in the surface (above 20 cm) (Fig. 2A, B), pointing to an elevated diversity of Hg methylators in the mangrove subsurface sediments. To verify this observation, we conducted a detailed analysis of the composition and variation of Hg methylators across the mangrove sediment columns. Our metagenomic study identified 65 medium- and high-quality MAGs with conserved hgcA (N(V/I)WCA(A/G), Supplementary Fig. S4) and hgcB (CX2CX2CX3C, Supplementary Fig. S5) motifs in the samples collected from all mangrove sediment layers. These MAGs represented a diverse range of taxa (Fig. 2B and Supplementary Data 3), including Acidobacteria (5), Anaerolineae (9), Bacteroidales (2), candidate division Zixibacteria (6; hereafter referred to as Zixibacteria throughout the manuscript), Desulfobacterales (6), GWC2-55-46 (4), Methanomicrobia (6), MBNT15 (7), Nitrospirae (8), Phycisphaerae (6), and Syntrophobacterales (6). We also observed a clear divergence in the distribution pattern of Hg methylators at the partition depth of 20 cm (Adonis test, R2 = 0.19, p < 0.001; Anosim test, R = 0.58, p < 0.001; Supplementary Fig. S6), with most of these MAGs exhibiting elevated occurrence frequencies in subsurface sediments (Fig. 2B).
To assess the regional-scale distribution of these Hg methylators, we collected and analyzed 27 additional mangrove subsurface sediment samples from three other provinces (Zhejiang, Fujian, and Hainan) along the southeastern coast of China, covering a distance of more than 1500 km (Fig. 2C). In addition to 65 MAGs recovered in Guangdong, metagenomic analyses of these 27 samples also generated 30, 15, and 38 hgcA-carrying MAGs in Zhejiang, Fujian, and Hainan, respectively. Three types of Hg methylators (Desulfobacterales, Anaerolineae, and Syntrophobacterales) were detected in all four sampling provinces (Fig. 2C). These members were the dominant Hg methylators in mangrove subsurface sediments, with the cumulative relative abundance of 26.5–91.2% (Supplementary Fig. S7). Owing to their dominance and environmental adaptability [105, 106], these Hg methylators may largely contribute to the ecological interception effect of mangrove sediments on terrestrial- or marine-sourced Hg by microbial methylation [10]. However, hgcA-carrying Zixibacteria, Dehalococcoidia, and GWC2-55-46 were only detected in mangrove subsurface sediments from Guangdong, Hainan, and Guangdong, respectively (Fig. 2C). Together with a recent discovery of Zixibacteria MAGs with hgcA in another mangrove sediment area of Guangdong [8], our findings point toward the geographic selectivity of specific Hg methylators across mangrove sediments.
Metabolic adaptation of Hg methylators in mangrove subsurface sediments
The adaptation of Hg methylators to mangrove subsurface sediments requires tolerance to intermittent low levels of O2 and energy exposure [16]. Our functional annotation revealed that the hgcA-carrying MAGs in mangrove sediments contained multiple terminal oxidases (Fig. 2D; Supplementary Data 1). Nearly all hgcA-carrying MAGs except Methanomicrobia contained the complete operon encoding the cytochrome bd complex, which has a high affinity for O2 and supports adaptation to low-oxygen environments [107]. Furthermore, most members in the GWC2-55-46 and MBNT15 groups contained genes encoding cytochrome c cbb3-type (ccoPQNO) oxidase (Supplementary Data 1), which could exhibit high affinity for O2 and enable bacteria to respire O2 under both oxic and suboxic conditions [108]. The presence of genes involved in anaerobic hydrocarbon degradation, such as those encoding putative anaerobic benzene carboxylase (abcA) and phenylphosphate carboxylase (ppcAB), provides additional evidence for the anaerobic lifestyle of Hg methylators in mangrove subsurface sediments.
Consistent with the previously documented feature of Hg methylators [109], the diverse Hg methylators prevailing in mangrove subsurface sediments can also use versatile electron acceptors (Fig. 2D). First, most MAGs belonging to Zixibacteria, Nitrospirae, Syntrophobacterales, and Desulfobacterales contained the key genes involved into the complete sulfate-reducing pathway (i.e., sat, aprAB, and dsrABD), which has been previously shown to support Hg methylation in mangrove sediments [93]. Second, consistent with the traditional view [4], the hgcA-carrying Methanomicrobia MAGs encoded functional genes responsible for CO2 reduction to CH4. Third, the GWC2-55-46, MBNT15, Nitrospirae, and Desulfobacterales MAGs harbored genes encoding nitrate/nitrite reductase to support Hg methylation potentially. Last, the Nitrospirae and Syntrophobacterales MAGs also contained porin-cytochrome modules (mtrAB), suggesting iron as an alternative terminal electron acceptor in these Hg methylators.
Phylogenetic diversity of Hg methylators in mangrove subsurface sediments
A meta-omic study has recently provided an updated catalog of Hg methylators, the Hg-MATE-Db, using both pure culture and environmental datasets [44]. This has expanded our understanding of the phylogenetic diversity of Hg methylators. Nine microbial groups were identified as having the highest diversity of Hg-methylating microorganisms across different environments [44], including paddy soils, brackish water, lake water, and reservoir and lake sediments. These groups belong to Acidobacteria, Bacteroidetes, Desulfobacterales, Chloroflexi, Euryarchaeota, Firmicutes, Nitrospirae, Spirochaetes, and PVC lineages.
To uncover the phylogenetic diversity of Hg methylators in mangrove sediments, a phylogenetic tree was constructed by integrating our detected hgcA sequences into Hg-MATE-Db (Fig. 3). Results showed that 65 hgcA-carrying MAGs in mangrove sediments were affiliated with seven microbial groups with the highest diversity (excluding Firmicutes and Spirochaetes lineages), as well as two additional groups (GWC2-55-46 and MBNT15 lineages). The number of microbial groups with the highest diversity in mangrove sediments was found to be second only to paddy soils, but higher than in brackish water, lake water, and sediments from reservoirs and lakes (Fig. 3 and Supplementary Table S3). These findings, together with more than 90% of these hgcA-carrying MAGs with a high occurrence frequency below 20 cm (Fig. 2B), point to a high phylogenetic diversity of Hg methylators in mangrove subsurface sediments.
Fig. 3. Maximum-likelihood phylogenetic tree of HgcA protein sequences.

Maximum-likelihood tree (IQ-TREE 2, LG + F + I + I + R9) was constructed using an alignment of HgcA proteins from hgcA-carrying MAGs and Hg-MATE-Db. The representative hgcA sequences recovered in this study are indicated by stars. The Hg-methylating lineages are labeled with colored circles, indicating their distribution in different environments. The scale bar represents one amino acid substitution per sequence position.
To assess the taxonomic novelty of these Hg methylators, we performed comparative genome analyses of average nucleotide identity (ANI) using our hgcA-carrying MAGs and the reference sequences in the Hg-MATE-Db (Fig. 3 and Supplementary Figs. S8–S17). Our results showed that Nitrospirae MAGs in our dataset and Hg-MATE-Db were largely syntenic and shared up to 99.4% ANI, suggesting the conservation of Hg-methylating Nitrospirae across different environments. On the other hand, our Methanomicrobia, GWC2-55-46, MBNT15, and Acidobacteria MAGs shared around 80% ANI with reference genomes, implying that these MAGs could be previously unreported species with Hg-methylating capacity. Notably, the Anaerolineae, Phycisphaerae, Desulfobacterales, and Bacteroidales MAGs shared less than 70.0% ANI against their reference genomes, indicating that they form distinct subfamilies and differ from previously known ones when following the proposed guidelines [110, 111]. These findings suggest that mangrove subsurface sediment is a promising source for identifying novel microorganisms and broadening the repertoire of Hg methylators.
Candidate phylum Zixibacteria is an understudied Hg methylator in diverse anoxic environments
Based on our phylogenetic analyses against the Hg-MATE-Db, we discovered a surprising finding: the Zixibacteria lineage, located between Bacteroidales and Phycisphaerae in the phylogenic tree (Fig. 3), has not been included in the updated catalog of Hg methylators. However, we found that the relative abundance of Zixibacteria MAGs ranked among the top 9 Hg-methylator taxa across all depths, and even ranked 5th at 30–50 cm depths (up to 9.1%, Supplementary Fig. S7) by mapping all the metagenomic reads against the genomic assemblies. Previous study has also identified subsurface-dwelling Zixibacteria lineage as a crucial hub member in mangrove sediment microbial communities, not only interacting frequently with Deltaproteobacteria but also providing metabolites for Bacteroidetes and Chloroflexi [112]. The latter three phyla have been often classified as the microbial taxa of putative Hg methylators in mangrove sediments [8]. Especially for the Syntrophobacteraceae members in Deltaproteobacteria, a recent cultivation study showed that they could enhance Hg methylation by interacting with their syntrophic partners [24]. Thus, we endeavored to decipher whether and how the “hub” Zixibacteria members methylate Hg in mangrove sediments, which is crucial for expanding the diversity of Hg methylators and understanding the microbial mechanism of Hg methylation in the subsurface biosphere.
As no cultivated representative of Zixibacteria is currently available for functional assays, we firstly investigated its potential in Hg methylation by comparing the HgcAB protein sequences from six Zixibacteria MAGs (average completeness 86.7%, average GC counts 53.2%; Supplementary data 4) against six confirmed Hg-methylating bacterial strains isolated from anaerobic environments (Supplementary Table S4) [113–117]. Our results showed that the genetic context of HgcAB sequences and the conservation of respective orthologous gene clusters in six Zixibacteria MAGs were all consistent with the confirmed Hg-methylating bacterial strains (Fig. 4A). The alignment of the HgcA protein sequences has revealed that HgcA is a cobalamin-dependent protein with a highly conserved motif in the CfsA subunit of CFeSP. Aside from HgcA, HgcB is also required for methylation activity [1], and its protein sequence alignment revealed the presence of two CXXCXXCXXXCP motifs, which are known to bind [4Fe-4S] clusters [1].
Fig. 4. Exploration of HgcAB proteins in candidate phylum Zixibacteria.

A Multiple sequence alignments of 12 HgcA and HgcB orthologs from candidate phylum Zixibacteria and six confirmed Hg-methylating bacterial strains. Red boxes indicate highly conserved regions including the putative cap helix [consensus sequence motif, N(V/I)WCA(A/G)GK] in HgcA and two strictly conserved CX2CX2CX3C motifs characteristic of [4Fe-4S] clusters in HgcB. Abbreviations for amino acid residues: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; Y, Tyr; X, any amino acid. B AlphaFold2 model of Zixibacteria HgcA protein, with an enlarged view of the functional domain bound to cobalamin. The ionic interaction between cysteine and cobalt is represented through a dotted box. C AlphaFold2 model of Zixibacteria HgcB protein in complex with two [4Fe4S] clusters.
We constructed AlphaFold2 models of the putative HgcA and HgcB proteins in Zixibacteria. Our analysis of the putative HgcA protein structure in Zixibacteria revealed a globular domain at the N-terminus and five transmembrane spanning helices at the C-terminus (Fig. 4B). By superposing the cobalamin onto the HgcA AlphaFold2 model in Zixibacteria, we found a strong H-bonding network between HgcA protein and cobalamin (Fig. 4B), which was thought to be bound with HgcA protein for catalytic methylation activity [6]. Importantly, the HgcA AlphaFold2 models in Zixibacteria and six confirmed Hg-methylating bacteria were aligned with a striking level of structural concordance (Supplementary Fig. S18). In addition to the HgcA, our analysis of the HgcB protein structure in Zixibacteria also revealed the binding of its protein with 2[4Fe-4S] clusters at the N-terminal by molecular stacking (Fig. 4C), showing a high similarity with the functionally validated Hg-methylating strain [1]. Together, our results provide genomic and structural support for the functional potential of Zixibacteria lineage in Hg methylation.
In an effort to understand the extent of Hg-methylating capacity in Zixibacteria lineage, we conducted a broader search of the NCBI SRA metagenomic datasets and obtained 135 medium- and high-quality Zixibacteria MAGs belonging to eight described orders (Fig. 5, Supplementary Data 4, 5). Among all these currently available Zixibacteria MAGs, 18 MAGs were identified to carry hgcA with conserved motifs and were distributed within six different orders, with the exception of DG-27 and MSB-5A5 orders. Our functional annotation showed that more than 70% of hgcA-carrying MAGs contained genes encoding sulfate reduction, while DG-27 and MSB-5A5 orders contained no MAGs with the potential for sulfate reduction. These results suggest that the Zixibacteria members primarily perform sulfate reduction to support Hg methylation in anoxic environments, as hgcA-carrying MAGs in Zixibacteria did not possess the capability to utilize other commonly reported electron acceptors such as iron, NO3−, NO2−, or CO2 (Supplementary Data 6).
Fig. 5. Phylogeny and environmental sources of candidate phylum Zixibacteria.

The maximum-likelihood tree was constructed using an alignment of maker genes from Zixibacteria genomes obtained in this study and from the NCBI SRA database. The genomes with hgcA sequences are highlighted in red. The diverse sources of Zixibacteria genomes are depicted in the outer circle with different colors representing the distinct environments. The capacity of sulfate reduction is indicated by brown circles, and the scale bar indicates substitutions per site. The tree was constructed using 1000 ultrafast bootstrap replicates, with black dots representing values greater than 10% at the nodes.
The environmental distribution of Zixibacteria was thoroughly investigated by screening homologous 16 S rRNA gene sequences against all available metagenomes within the IMG/M database. This search generated a total of 710 unique occurrences. The categorization of sampling locations revealed a broad environmental distribution of the Zixibacteria lineage, spanning 17 different types of environments and including natural, host-associated, and engineered systems (Supplementary Fig. S19). Of them, the hgcA-carrying Zixibacteria were mainly recovered from natural environments, including mangrove sediment, marine sediments, cold seep, hydrothermal vent, estuarine sediment, groundwater, microbial mat, and deep mine (Fig. 5). In addition to mangrove sediment (this study and Zhang et al.) [8], the Zixibacteria lineage in other environments have not been reported to carry hgcA gene, highlighting its currently understudied roles in Hg methylation across most anoxic environments. However, no clear environment-specific distribution was observed across the six different orders (Fig. 5), suggesting that hgcA-carrying Zixibacteria are well adapted to diverse anoxic environments.
Unique metabolic and lifestyle features of candidate phylum Zixibacteria in Hg methylation
During Hg methylation, the methyl group (as CH3+) in MeHg is widely thought to be transferred to HgcA by a cobalamin-dependent methyltransferase (i.e., CFeSP methyltransferase from the Wood-Ljungdahl pathway) [1, 118, 119]. However, we found all Zixibacteria MAGs lacking genes for the entire Wood-Ljungdahl pathway (Fig. 6A and Supplementary Data 7). To investigate the putative CH3+ transfer within the Zixibacteria lineage, we conducted a search for typical cobalamin-dependent methyltransferases [72, 119–127]. Our results revealed that the Hg-methylating Zixibacteria lineage only encoded the cobalamin-dependent methyltransferase involved in the methionine synthesis pathway, rather than the Wood-Ljungdahl pathway, coenzyme M pathway, or one carbon folate cycle (Supplementary Data 8). In particular, all six different orders in Zixibacteria consistently encoded the MetH (Fig. 6B), a cobalamin-dependent methyltransferase catalyzing CH3+ transfer from 5-methyl-tetrahydrofolate to homocysteine during the methionine synthesis pathway [128]. Considering that CH3+ in MeHg is traditionally obtained from 5-methyl-tetrahydrofolate by cobalamin-dependent methyltransferase [1, 118, 119], we hypothesized that the MetH in Hg-methylating Zixibacteria may have the potential to transfer CH3+ from 5-methyl-tetrahydrofolate to HgcA, forming CH3-cob(III)alamin-HgcA (Fig. 6B). To validate this hypothesis, we performed docking predictions for HgcA and MetH. For the most reliable docking model of HgcA and MetH (confidence score: 0.93), the interaction interface was defined as those amino acid residues within 5.1 Å of the surface volume encapsulating the atom positions (Fig. 6C and Supplementary Data 9). The docking score of −280.66 kcal/mol indicates a strong interaction of HgcA and MetH, which is mediated by 26 hydrogen bonds, 7 hydrophobic interactions, and 1 salt bridge (Fig. 6C and Supplementary Fig. S20). Among them, Arg in MetH exhibited the highest binding activity to HgcA, and the closest atom distance between MetH and HgcA was 1.4 Å in a hydrogen bond between Arg156 and Ser83 (Supplementary Data 9). These observations suggest that hydrogen bonding interactions associated with Arg side chains are likely important for HgcA and MetH protein binding.
Fig. 6. Detailed mechanism of Hg methylation in candidate phylum Zixibacteria.

A Metagenomics-based mechanism reconstruction of Hg-methylating Zixibacteria. Abbreviation: Fd, ferredoxin. The blue balls with “e” indicate electrons. B Potential mechanism as used for the methionine biosynthesis pathway to donate methyl groups to Hg methylation across all orders within Hg-methylating Zixibacteria. The dashed arrows signify proposed steps that require experimental validation. C Schematic representation of HgcA-MetH interaction in the 3D structure. All of the top 5 candidate models show confidence scores greater than 0.9. The visualization was accomplished using PyMOL.
Furthermore, Zixibacteria MAGs encoded the benzoyl-CoA reductase, which was reported to be structurally similar to the CODH/acetyl-CoA synthases in Wood-Ljungdahl pathway and showed a strong correlation with HgT and MeHg levels [129, 130]. The benzoyl-CoA reduction enables the Hg methylators to adapt to environments contaminated by hydrocarbons [131], which is of particular significance as hydrocarbons are frequently found in mangrove sediments, as a result of industrial discharge, shipping activities, and urban runoff [132]. These findings indicate that the Zixibacteria lineage has the potential to play a role in Hg methylation, with the added benefit of removing hydrocarbons from contaminated mangrove sediments.
Energy limitation of mangrove subsurface sediments has been widely acknowledged [14–16], but the provision of energy is essential for Hg methylation [109]. To address this issue, the researchers have proposed syntrophic microbial interaction as a major energy source for Hg methylation in energy-limited environments [24]. Through an examination of microbial syntrophic features, our study found evidence for the presence of these partnerships in mangrove subsurface sediments. The Zixibacteria MAGs, which carry the hgcA gene and have the sulfate-reducing capacity, were found to contain genes involved in interspecies electron transfer (including multi-heme cytochromes, conductive filaments and molecular hydrogen; Supplementary Data 10), the significant features for a syntrophic lifestyle [22, 133]. Also, we here recovered several Syntrophobacterales MAGs, which not only possessed genes involved in interspecies electron transfer (Supplementary Data 10), but also were dominant in mangrove subsurface sediments (up to 13.1% in the Hg-methylating genomic assemblies, Supplementary Fig. S7). In addition, our 16 S rRNA gene-based microbial network inference and metagenomic-based linear regression revealed the positive co-occurrence between the Syntrophobacterales and Zixibacteria lineages (Supplementary Figs. 21, 22), further supporting the potential for syntrophic partnerships in the mangrove subsurface sediments.
Further evidence for their potential syntrophic relationship was the absence of cobalamin biosynthetic genes in the Hg-methylating Zixibacteria. This was inconsistent with our AlphaFold2 model (Fig. 4A; indicating the participation of cobalamin in Hg methylation) and metabolic predictions (Fig. 6; with cobalamin-dependent methyltransferase involved in methionine synthase). However, the syntrophic Syntrophobacterales lineage contained all relevant genes for cobalamin biosynthesis (Supplementary Data 11), potentially leading to its interactions with Hg-methylating Zixibacteria [134]. Finally, both the co-occurrence pattern and the paired genomic analyses suggest that the Zixibacteria lineage has a unique metabolic strategy for Hg methylation in mangrove subsurface sediments, dependent on a syntrophic relationship with another organism in energy-limited conditions. Future cultivation experiments with Hg methylators isolated from mangrove sediments will help to verify this hypothesis and further clarify the nature of their syntrophic interactions.
Supplementary information
Acknowledgements
This study was supported by the National Natural Science Foundation of China (32000070, 52070196, 32370113), the Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (SML2020SP0004), the Innovation Group Project of Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (311022011), the Guangdong Natural Resources Department Contract (GDNRC[2021]62), the Guangdong Basic and Applied Basic Research Foundation (2019A1515011406), and Guangzhou Science and Technology Plan Projects (202002030454).
Author contributions
CW conceived and designed the study. SFL, NLP, RWH, ZYZ, and RHC performed the laboratory work and detailed the sampling. SFL and NLP carried out the bioinformatics and statistical analysis. SFL and CW wrote the first draft of the manuscript. SFL, CW, and ZLH discussed results and edited. All authors read and approved the final version of the manuscript.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary File 1 and 2. The raw reads of metagenomes and 16 S rRNA amplicon sequencing were submitted to the National Center for Biotechnology Information Short Reads Archive (NCBI SRA) database under the project PRJNA798446, PRJNA894374, PRJNA958041, and the National Omics Data Encyclopedia (NODE) under the project OEX012906. All Zixibacteria genomes from this study or NCBI SRA database were shared in Zenodo (10.5281/zenodo.7998010).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-023-01544-4.
References
- 1.Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Jr, Smith SD, et al. The genetic basis for bacterial mercury methylation. Science. 2013;339:1332–5. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 2.Gilmour CC, Podar M, Bullock AL, Graham AM, Brown SD, Somenahally AC, et al. Mercury methylation by novel microorganisms from new environments. Environ Sci Technol. 2013;47:11810–20. doi: 10.1021/es403075t. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Janssen SE, Schaefer JK, Yee N, Reinfelder JR. Tracing the uptake of Hg(II) in an iron-reducing bacterium using mercury stable isotopes. Environ Sci Technol Lett. 2020;7:573–8. doi: 10.1021/acs.estlett.0c00409. [DOI] [Google Scholar]
- 4.Gilmour CC, Bullock AL, McBurney A, Podar M, Elias DA. Robust mercury methylation across diverse methanogenic Archaea. mBio. 2018;9:e02403–17. doi: 10.1128/mBio.02403-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Podar M, Gilmour CC, Brandt CC, Soren A, Brown SD, Crable BR, et al. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci Adv. 2015;1:e1500675. doi: 10.1126/sciadv.1500675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin H, Ascher DB, Myung Y, Lamborg CH, Hallam SJ, Gionfriddo CM, et al. Mercury methylation by metabolically versatile and cosmopolitan marine bacteria. ISME J. 2021;15:1810–25. doi: 10.1038/s41396-020-00889-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDaniel EA, Peterson BD, Stevens SLR, Tran PQ, Anantharaman K, McMahon KD. Expanded phylogenetic diversity and metabolic flexibility of mercury-methylating microorganisms. mSystems. 2020;5:e00299–00220. doi: 10.1128/mSystems.00299-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C-J, Liu Y-R, Cha G, Liu Y, Zhou X-Q, Lu Z, et al. Potential for mercury methylation by Asgard archaea in mangrove sediments. ISME J. 2023;17:478–85. doi: 10.1038/s41396-023-01360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Probst AJ, Weinmaier T, Raymann K, Perras A, Emerson JB, Rattei T, et al. Biology of a widespread uncultivated archaeon that contributes to carbon fixation in the subsurface. Nat Commun. 2014;5:5497. doi: 10.1038/ncomms6497. [DOI] [PubMed] [Google Scholar]
- 10.Lei P, Zhong H, Duan D, Pan K. A review on mercury biogeochemistry in mangrove sediments: hotspots of methylmercury production? Sci Total Environ. 2019;680:140–50. doi: 10.1016/j.scitotenv.2019.04.451. [DOI] [PubMed] [Google Scholar]
- 11.De Lipthay JR, Rasmussen LD, Oregaard G, Simonsen K, Bahl MI, Kroer N, et al. Acclimation of subsurface microbial communities to mercury. FEMS Microbiol Ecol. 2008;65:145–55. doi: 10.1111/j.1574-6941.2008.00501.x. [DOI] [PubMed] [Google Scholar]
- 12.Frohne T, Rinklebe J, Langer U, Du Laing G, Mothes S, Wennrich R. Biogeochemical factors affecting mercury methylation rate in two contaminated floodplain soils. Biogeosciences. 2012;9:493–507. doi: 10.5194/bg-9-493-2012. [DOI] [Google Scholar]
- 13.Luo Z, Zhong Q, Han X, Hu R, Liu X, Xu W, et al. Depth-dependent variability of biological nitrogen fixation and diazotrophic communities in mangrove sediments. Microbiome. 2021;9:212. doi: 10.1186/s40168-021-01164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almahasheer H, Duarte CM, Irigoien X. Nutrient limitation in central Red Sea mangroves. Front Mar Sci. 2016;3:271. doi: 10.3389/fmars.2016.00271. [DOI] [Google Scholar]
- 15.Kuhn AL, Kominoski JS, Armitage AR, Charles SP, Pennings SC, Weaver CA, et al. Buried hurricane legacies: increased nutrient limitation and decreased root biomass in coastal wetlands. Ecosphere. 2021;12:e03674. doi: 10.1002/ecs2.3674. [DOI] [Google Scholar]
- 16.Li C-H, Zhou H-W, Wong Y-S, Tam NF-Y. Vertical distribution and anaerobic biodegradation of polycyclic aromatic hydrocarbons in mangrove sediments in Hong Kong, South China. Sci Total Environ. 2009;407:5772–9. doi: 10.1016/j.scitotenv.2009.07.034. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Shi T, Liu J, He Z, Thomas H, Dong H, et al. Eco-engineering approaches for ocean negative carbon emission. Sci Bull. 2022;67:2564–73. doi: 10.1016/j.scib.2022.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. doi: 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hug LA, Thomas BC, Sharon I, Brown CT, Sharma R, Hettich RL, et al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ Microbiol. 2016;18:159–73. doi: 10.1111/1462-2920.12930. [DOI] [PubMed] [Google Scholar]
- 20.Emerson JB, Thomas BC, Alvarez W, Banfield JF. Metagenomic analysis of a high carbon dioxide subsurface microbial community populated by chemolithoautotrophs and bacteria and archaea from candidate phyla. Environ Microbiol. 2016;18:1686–703. doi: 10.1111/1462-2920.12817. [DOI] [PubMed] [Google Scholar]
- 21.Reji L, Cardarelli EL, Boye K, Bargar JR, Francis CA. Diverse ecophysiological adaptations of subsurface Thaumarchaeota in floodplain sediments revealed through genome-resolved metagenomics. ISME J. 2022;16:1140–52. doi: 10.1038/s41396-021-01167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murali R, Yu H, Speth D, Wu F, Metcalfe KS, Cremiere A, et al. Physiological adaptation of sulfate reducing bacteria in syntrophic partnership with anaerobic methanotrophic archaea. bioRxiv. 10.1101/2022.11.23.517749 (2022).
- 23.Pernthaler A, Dekas AE, Brown CT, Goffredi SK, Embaye T, Orphan VJ. Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics. Proc Natl Acad Sci USA. 2008;105:7052–7. doi: 10.1073/pnas.0711303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu R-Q, Reinfelder JR, Hines ME, Barkay T. Syntrophic pathways for microbial mercury methylation. ISME J. 2018;12:1826–35. doi: 10.1038/s41396-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu C, Feng J, Liu K, Wang G, Zhu Y, Chen H, et al. Changes of ecosystem carbon stock following the plantation of exotic mangrove Sonneratia apetala in Qi’ao Island, China. Sci Total Environ. 2020;717:137142. doi: 10.1016/j.scitotenv.2020.137142. [DOI] [PubMed] [Google Scholar]
- 26.Mason OU, Case DH, Naehr TH, Lee RW, Thomas RB, Bailey JV, et al. Comparison of archaeal and bacterial diversity in methane seep carbonate nodules and host sediments, Eel River Basin and Hydrate Ridge, USA. Micro Ecol. 2015;70:766–84. doi: 10.1007/s00248-015-0615-6. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto Y, Chen R, Anikeeva P, Jasanoff A. Engineering intracellular biomineralization and biosensing by a magnetic protein. Nat Commun. 2015;6:8721. doi: 10.1038/ncomms9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin R, Chen D, Pan X, Deng C, Chen L, Song X, et al. Mantle Hg isotopic heterogeneity and evidence of oceanic Hg recycling into the mantle. Nat Commun. 2022;13:948. doi: 10.1038/s41467-022-28577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L, Liu C, Yin Y, Liu G, Li Y, Cai Y. Mass budget of mercury (Hg) in the seawater of Eastern China Marginal Seas: importance of the sediment–water transport processes. Environ Sci Technol. 2022;56:11418–28. doi: 10.1021/acs.est.2c03261. [DOI] [PubMed] [Google Scholar]
- 30.Zerkle AL, Yin R, Chen C, Li X, Izon GJ, Grasby SE. Anomalous fractionation of mercury isotopes in the Late Archean atmosphere. Nat Commun. 2020;11:1709. doi: 10.1038/s41467-020-15495-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anonymous, Telliard WA, Gomez-Taylor M Method 1631, Revision E: mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry. United States Environmental Protection Agency, Office of Water, 4303. EPA-821-R-02-019, (2002).
- 32.Anonymous, Telliard WA Method 1630: methyl mercury in water by distillation, aqueous ethylation, purge and trap, and cold vapor atomic fluorescence spectrometry. US Environmental Protection Agency-Office of Water. (1998).
- 33.Blum JD, Bergquist BA. Reporting of variations in the natural isotopic composition of mercury. Anal Bioanal Chem. 2007;388:353–9. doi: 10.1007/s00216-007-1236-9. [DOI] [PubMed] [Google Scholar]
- 34.Janssen SE, Lepak RF, Tate MT, Ogorek JM, DeWild JF, Babiarz CL, et al. Rapid pre-concentration of mercury in solids and water for isotopic analysis. Anal Chim Acta. 2019;1054:95–103. doi: 10.1016/j.aca.2018.12.026. [DOI] [PubMed] [Google Scholar]
- 35.Blum JD. Applications of stable mercury isotopes to biogeochemistry. Handb Environ Isot Geochem. 2012;I:229–45. [Google Scholar]
- 36.Maheshwari S, Wang J, Barbash DA. Recurrent positive selection of the Drosophila hybrid incompatibility gene Hmr. Mol Biol Evol. 2008;25:2421–30. doi: 10.1093/molbev/msn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11. doi: 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 39.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2015;32:605–7. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 41.Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2019;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capo E, Peterson BD, Kim M, Jones DS, Acinas SG, Amyot M, et al. A consensus protocol for the recovery of mercury methylation genes from metagenomes. Mol Ecol Resour. 2023;23:190–204. doi: 10.1111/1755-0998.13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He C, Keren R, Whittaker ML, Farag IF, Doudna JA, Cate JHD, et al. Genome-resolved metagenomics reveals site-specific diversity of episymbiotic CPR bacteria and DPANN archaea in groundwater ecosystems. Nat Microbiol. 2021;6:354–65. doi: 10.1038/s41564-020-00840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knudsen BE, Bergmark L, Munk P, Lukjancenko O, Priemé A, Aarestrup FM, et al. Impact of sample type and DNA isolation procedure on genomic inference of microbiome composition. mSystems. 2016;1:e00095–00016. doi: 10.1128/mSystems.00095-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. A bioinformatician’s guide to metagenomics. Microbiol Mol Biol Rev. 2008;72:557–78. doi: 10.1128/MMBR.00009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qian L, Yu X, Zhou J, Gu H, Ding J, Peng Y, et al. MCycDB: a curated database for comprehensively profiling methane cycling processes of environmental microbiomes. Mol Ecol Resour. 2022;22:1803–23. doi: 10.1111/1755-0998.13589. [DOI] [PubMed] [Google Scholar]
- 58.Yu X, Zhou J, Song W, Xu M, He Q, Peng Y, et al. SCycDB: a curated functional gene database for metagenomic profiling of sulphur cycling pathways. Mol Ecol Resour. 2021;21:924–40. doi: 10.1111/1755-0998.13306. [DOI] [Google Scholar]
- 59.Garber AI, Nealson KH, Okamoto A, McAllister SM, Chan CS, Barco RA, et al. FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front Microbiol. 2020;11:37. doi: 10.3389/fmicb.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Z, Tran PQ, Breister AM, Liu Y, Kieft K, Cowley ES, et al. Metabolic: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome. 2022;10:33. doi: 10.1186/s40168-021-01213-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18:366–8. doi: 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Søndergaard D, Pedersen CNS, Greening C. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:34212. doi: 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anonymous, Callaghan AV, Wawrik B AnHyDeg: a curated database of anaerobic hydrocarbon degradation genes. GitHub. (2016). https://github.com/AnaerobesRock/AnHyDeg.
- 64.Christakis CA, Barkay T, Boyd ES. Expanded diversity and phylogeny of mer genes broadens mercury resistance paradigms and reveals an origin for MerA among thermophilic archaea. Front Microbiol. 2021;12:682605. doi: 10.3389/fmicb.2021.682605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Badalamenti JP, Summers ZM, Chan CH, Gralnick JA, Bond DR. Isolation and genomic characterization of Desulfuromonas soudanensis WTL, a metal-and electrode-respiring bacterium from anoxic deep subsurface brine. Front Microbiol. 2016;7:913. doi: 10.3389/fmicb.2016.00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hernsdorf AW, Amano Y, Miyakawa K, Ise K, Suzuki Y, Anantharaman K, et al. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017;11:1915–29. doi: 10.1038/ismej.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adam PS, Borrel G, Gribaldo S. An archaeal origin of the Wood–Ljungdahl H4MPT branch and the emergence of bacterial methylotrophy. Nat Microbiol. 2019;4:2155–63. doi: 10.1038/s41564-019-0534-2. [DOI] [PubMed] [Google Scholar]
- 68.Adam PS, Borrel G, Gribaldo S. Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc Natl Acad Sci USA. 2018;115:E1166–E1173. doi: 10.1073/pnas.1716667115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferla MP, Patrick WM. Bacterial methionine biosynthesis. Microbiology. 2014;160:1571–84. doi: 10.1099/mic.0.077826-0. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J-W, Dong H-P, Hou L-J, Liu Y, Ou Y-F, Zheng Y-L, et al. Newly discovered Asgard archaea Hermodarchaeota potentially degrade alkanes and aromatics via alkyl/benzyl-succinate synthase and benzoyl-CoA pathway. ISME J. 2021;15:1826–43. doi: 10.1038/s41396-020-00890-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2019;36:2251–2. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matthews RG, Koutmos M, Datta S. Cobalamin-dependent and cobamide-dependent methyltransferases. Curr Opin Struct Biol. 2008;18:658–66. doi: 10.1016/j.sbi.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobson L, Reményi I, Tusnády GE. CCTOP: a consensus constrained topology prediction web server. Nucleic Acids Res. 2015;43:W408–W412. doi: 10.1093/nar/gkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong R, Pan S, Peng Z, Zhang Y, Yang J. mTM-align: a server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Res. 2018;46:W380–W386. doi: 10.1093/nar/gky430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kung Y, Ando N, Doukov TI, Blasiak LC, Bender G, Seravalli J, et al. Visualizing molecular juggling within a B12-dependent methyltransferase complex. Nature. 2012;484:265–9. doi: 10.1038/nature10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dauter Z, Wilson KS, Sieker LC, Meyer J, Moulis J-M. Atomic resolution (0.94 Å) structure of Clostridium acidurici ferredoxin. Detailed geometry of [4Fe-4S] clusters in a protein. Biochemistry. 1997;36:16065–73. [DOI] [PubMed]
- 78.Gavriilidou A, Avcı B, Galani A, Schorn MA, Ingham CJ, Ettema TJG, et al. Candidatus Nemesobacterales is a sponge-specific clade of the candidate phylum Desulfobacterota adapted to a symbiotic lifestyle. ISME J. 10.1038/s41396-023-01484-z (2023). [DOI] [PMC free article] [PubMed]
- 79.Chen I-MA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, et al. The IMG/M data management and analysis system v.7: content updates and new features. Nucleic Acids Res. 2022;51:D723–32. doi: 10.1093/nar/gkac976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 81.Yan Y, Tao H, He J, Huang S-Y. The HDOCK server for integrated protein–protein docking. Nat Protoc. 2020;15:1829–52. doi: 10.1038/s41596-020-0312-x. [DOI] [PubMed] [Google Scholar]
- 82.Yan Y, Wen Z, Wang X, Huang SY. Addressing recent docking challenges: a hybrid strategy to integrate template-based and free protein-protein docking. Proteins. 2017;85:497–512. doi: 10.1002/prot.25234. [DOI] [PubMed] [Google Scholar]
- 83.Huang SY, Zou X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins. 2008;72:557–79. doi: 10.1002/prot.21949. [DOI] [PubMed] [Google Scholar]
- 84.Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 85.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watts SC, Ritchie SC, Inouye M, Holt KE. FastSpar: rapid and scalable correlation estimation for compositional data. Bioinformatics. 2018;35:1064–6. doi: 10.1093/bioinformatics/bty734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. Proc Int AAAI Conf Web Soc Media. 2009;3:361–2. doi: 10.1609/icwsm.v3i1.13937. [DOI] [Google Scholar]
- 89.Wickham H. ggplot2. Wiley Interdiscip Rev Comput Stat. 2011;3:180–5.
- 90.Zhang J, Ma K. spaa: an R package for computing species association and niche overlap. Res Prog Biodivers Conserv China. 2014;10:165–74. [Google Scholar]
- 91.Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, et al. The vegan package. Community Ecol package. 2007;10:719. [Google Scholar]
- 92.Zhou L, Feng T, Xu S, Gao F, Lam TT, Wang Q, et al. ggmsa: a visual exploration tool for multiple sequence alignment and associated data. Brief Bioinform. 2022;23:bbac222. doi: 10.1093/bib/bbac222. [DOI] [PubMed] [Google Scholar]
- 93.Wu H, Ding Z, Liu Y, Liu J, Yan H, Pan J, et al. Methylmercury and sulfate-reducing bacteria in mangrove sediments from Jiulong River Estuary, China. J Environ Sci. 2011;23:14–21. doi: 10.1016/S1001-0742(10)60368-3. [DOI] [PubMed] [Google Scholar]
- 94.Yu R-Q, Flanders J, Mack EE, Turner R, Mirza MB, Barkay T. Contribution of coexisting sulfate and iron reducing bacteria to methylmercury production in freshwater river sediments. Environ Sci Technol. 2012;46:2684–91. doi: 10.1021/es2033718. [DOI] [PubMed] [Google Scholar]
- 95.Blum JD, Sherman LS, Johnson MW. Mercury isotopes in earth and environmental sciences. Annu Rev Earth Planet Sci. 2014;42:249–69. doi: 10.1146/annurev-earth-050212-124107. [DOI] [Google Scholar]
- 96.Demers JD, Blum JD, Zak DR. Mercury isotopes in a forested ecosystem: Implications for air-surface exchange dynamics and the global mercury cycle. Glob Biogeochem Cy. 2013;27:222–38. doi: 10.1002/gbc.20021. [DOI] [Google Scholar]
- 97.Biswas A, Blum JD, Bergquist BA, Keeler GJ, Xie Z. Natural mercury isotope variation in coal deposits and organic soils. Environ Sci Technol. 2008;42:8303–9. doi: 10.1021/es801444b. [DOI] [PubMed] [Google Scholar]
- 98.Zheng W, Obrist D, Weis D, Bergquist BA. Mercury isotope compositions across North American forests. Glob Biogeochem Cy. 2016;30:1475–92. doi: 10.1002/2015GB005323. [DOI] [Google Scholar]
- 99.Jiskra M, Wiederhold JG, Skyllberg U, Kronberg RM, Kretzschmar R. Source tracing of natural organic matter bound mercury in boreal forest runoff with mercury stable isotopes. Environ Sci Process Impacts. 2017;19:1235–48. doi: 10.1039/C7EM00245A. [DOI] [PubMed] [Google Scholar]
- 100.Štrok M, Baya PA, Hintelmann H. The mercury isotope composition of Arctic coastal seawater. CR Geosci. 2015;347:368–76. doi: 10.1016/j.crte.2015.04.001. [DOI] [Google Scholar]
- 101.Yin R, Xu L, Lehmann B, Lepak RF, Hurley JP, Mao J, et al. Anomalous mercury enrichment in Early Cambrian black shales of South China: mercury isotopes indicate a seawater source. Chem Geol. 2017;467:159–67. doi: 10.1016/j.chemgeo.2017.08.010. [DOI] [Google Scholar]
- 102.Grasby SE, Shen W, Yin R, Gleason JD, Blum JD, Lepak RF, et al. Isotopic signatures of mercury contamination in latest Permian oceans. Geology. 2017;45:55–58. doi: 10.1130/G38487.1. [DOI] [Google Scholar]
- 103.Maghsodian Z, Sanati AM, Tahmasebi S, Shahriari MH, Ramavandi B. Study of microplastics pollution in sediments and organisms in mangrove forests: a review. Environ Res. 2022;208:112725. doi: 10.1016/j.envres.2022.112725. [DOI] [PubMed] [Google Scholar]
- 104.Kritee K, Blum JD, Barkay T. Mercury stable isotope fractionation during reduction of Hg(II) by different microbial pathways. Environ Sci Technol. 2008;42:9171–7. doi: 10.1021/es801591k. [DOI] [PubMed] [Google Scholar]
- 105.Atashgahi S, Aydin R, Dimitrov MR, Sipkema D, Hamonts K, Lahti L, et al. Impact of a wastewater treatment plant on microbial community composition and function in a hyporheic zone of a eutrophic river. Sci Rep. 2015;5:17284. doi: 10.1038/srep17284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Madueño L, Starevich VA, Agnello AC, Coppotelli BM, Laprida C, Vidal NC, et al. Assessment of biological contribution to natural recovery of anthropized freshwater sediments from Argentina: autochthonous microbiome structure and functional prediction. Front Microbiol. 2021;12:601705. doi: 10.3389/fmicb.2021.601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta Bioenerg. 2011;1807:1398–413. doi: 10.1016/j.bbabio.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Visser JM, de Jong GAH, de Vries S, Robertson LA, Kuenen JG. cbb3-type cytochrome oxidase in the obligately chemolithoautotrophic Thiobacillus sp. W5. FEMS Microbiol Lett. 1997;147:127–32. doi: 10.1111/j.1574-6968.1997.tb10231.x. [DOI] [Google Scholar]
- 109.Regnell O, Watras CJ. Microbial mercury methylation in aquatic environments: a critical review of published field and laboratory studies. Environ Sci Technol. 2018;53:4–19. doi: 10.1021/acs.est.8b02709. [DOI] [PubMed] [Google Scholar]
- 110.Turner D, Kropinski AM, Adriaenssens EM. A roadmap for genome-based phage taxonomy. Viruses. 2021;13:506. doi: 10.3390/v13030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Naas AE, Solden LM, Norbeck AD, Brewer H, Hagen LH, Heggenes IM, et al. “Candidatus Paraporphyromonas polyenzymogenes” encodes multi-modular cellulases linked to the type IX secretion system. Microbiome. 2018;6:44. doi: 10.1186/s40168-018-0421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Du H, Pan J, Zou D, Huang Y, Liu Y, Li M. Microbial active functional modules derived from network analysis and metabolic interactions decipher the complex microbiome assembly in mangrove sediments. Microbiome. 2022;10:224. doi: 10.1186/s40168-022-01421-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kerin EJ, Gilmour CC, Roden E, Suzuki M, Coates J, Mason R. Mercury methylation by dissimilatory iron-reducing bacteria. Appl Environ Microbiol. 2006;72:7919–21. doi: 10.1128/AEM.01602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gilmour CC, Elias DA, Kucken AM, Brown SD, Palumbo AV, Schadt CW, et al. Sulfate-reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation. Appl Environ Microbiol. 2011;77:3938–51. doi: 10.1128/AEM.02993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brown SD, Gilmour CC, Kucken AM, Wall JD, Elias DA, Brandt CC, et al. Genome sequence of the mercury-methylating strain Desulfovibrio desulfuricans ND132. J Bacteriol. 2011;193:2078–9. doi: 10.1128/JB.00170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Graham AM, Bullock AL, Maizel AC, Elias DA, Gilmour CC. Detailed assessment of the kinetics of Hg-cell association, Hg methylation, and methylmercury degradation in several Desulfovibrio species. Appl Environ Microbiol. 2012;78:7337–46. doi: 10.1128/AEM.01792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.King JK, Kostka JE, Frischer ME, Saunders FM. Sulfate-reducing bacteria methylate mercury at variable rates in pure culture and in marine sediments. Appl Environ Microbiol. 2000;66:2430–7. doi: 10.1128/AEM.66.6.2430-2437.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Drake H, Hu S, Wood H. Purification of five components from Clostridium thermoaceticum which catalyze synthesis of acetate from pyruvate and methyltetrahydrofolate. Properties of phosphotransacetylase. J Biol Chem. 1981;256:11137–44. doi: 10.1016/S0021-9258(19)68568-6. [DOI] [PubMed] [Google Scholar]
- 119.Doukov T, Seravalli J, Stezowski JJ, Ragsdale SW. Crystal structure of a methyltetrahydrofolate-and corrinoid-dependent methyltransferase. Structure. 2000;8:817–30. doi: 10.1016/S0969-2126(00)00172-6. [DOI] [PubMed] [Google Scholar]
- 120.Burke SA, Krzycki JA. Reconstitution of monomethylamine: coenzyme M methyl transfer with a corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem. 1997;272:16570–7. doi: 10.1074/jbc.272.26.16570. [DOI] [PubMed] [Google Scholar]
- 121.Burke SA, Lo SL, Krzycki JA. Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine. J Bacteriol. 1998;180:3432–40. doi: 10.1128/JB.180.13.3432-3440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ferguson DJ, Gorlatova N, Grahame DA, Krzycki JA. Reconstitution of dimethylamine: coenzyme M methyl transfer with a discrete corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem. 2000;275:29053–60. doi: 10.1074/jbc.M910218199. [DOI] [PubMed] [Google Scholar]
- 123.Naidu D, Ragsdale SW. Characterization of a three-component vanillate O-demethylase from Moorella thermoacetica. J Bacteriol. 2001;183:3276–81. doi: 10.1128/JB.183.11.3276-3281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gottschalk G, Thauer RK. The Na+-translocating methyltransferase complex from methanogenic archaea. Biochim Biophys Acta Bioenerg. 2001;1505:28–36. doi: 10.1016/S0005-2728(00)00274-7. [DOI] [PubMed] [Google Scholar]
- 125.Tallant TC, Paul L, Krzycki JA. The MtsA subunit of the methylthiol: coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide: coenzyme M methyl transfer. J Biol Chem. 2001;276:4485–93. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 126.Sauer K, Thauer RK. Methanol: coenzyme M methyltransferase from Methanosarcina barkeri: identification of the active‐site histidine in the corrinoid‐harboring subunit MtaC by site‐directed mutagenesis. Eur J Biochem. 1998;253:698–705. doi: 10.1046/j.1432-1327.1998.2530698.x. [DOI] [PubMed] [Google Scholar]
- 127.Paul L, Ferguson DJ, Jr, Krzycki JA. The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons. J Bacteriol. 2000;182:2520–9. doi: 10.1128/JB.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Koutmos M, Datta S, Pattridge KA, Smith JL, Matthews RG. Insights into the reactivation of cobalamin-dependent methionine synthase. Proc Natl Acad Sci USA. 2009;106:18527–32. doi: 10.1073/pnas.0906132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gionfriddo CM, Tate MT, Wick RR, Schultz MB, Zemla A, Thelen MP, et al. Microbial mercury methylation in Antarctic sea ice. Nat Microbiol. 2016;1:16127. doi: 10.1038/nmicrobiol.2016.127. [DOI] [PubMed] [Google Scholar]
- 130.Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW. Life on carbon monoxide: X-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc Natl Acad Sci USA. 2001;98:11973–8. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vigneron A, Cruaud P, Aubé J, Guyoneaud R, Goñi-Urriza M. Transcriptomic evidence for versatile metabolic activities of mercury cycling microorganisms in brackish microbial mats. NPJ Biofilms Microb. 2021;7:1–11. doi: 10.1038/s41522-021-00255-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ranjan RK, Routh J, Ramanathan A, Val Klump J. Polycyclic aromatic hydrocarbon fingerprints in the Pichavaram mangrove–estuarine sediments, southeastern India. Org Geochem. 2012;53:88–94. doi: 10.1016/j.orggeochem.2012.08.007. [DOI] [Google Scholar]
- 133.Zehnle H, Laso-Pérez R, Lipp J, Riedel D, Benito Merino D, Teske A, et al. Candidatus Alkanophaga archaea from Guaymas Basin hydrothermal vent sediment oxidize petroleum alkanes. Nat Microbiol. 2023;8:1199–212. doi: 10.1038/s41564-023-01400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sultana S, Bruns S, Wilkes H, Simon M, Wienhausen G. Vitamin B12 is not shared by all marine prototrophic bacteria with their environment. ISME J. 2023;17:836–45. doi: 10.1038/s41396-023-01391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary File 1 and 2. The raw reads of metagenomes and 16 S rRNA amplicon sequencing were submitted to the National Center for Biotechnology Information Short Reads Archive (NCBI SRA) database under the project PRJNA798446, PRJNA894374, PRJNA958041, and the National Omics Data Encyclopedia (NODE) under the project OEX012906. All Zixibacteria genomes from this study or NCBI SRA database were shared in Zenodo (10.5281/zenodo.7998010).