


Abstract
Salmonella enterica serovar Choleraesuis (S.Choleraesuis), a highly invasive serovar among non-typhoidal Salmonella, usually causes sepsis or extra-intestinal focal infections in humans. S.Choleraesuis infections have now become particularly difficult to treat because of the emergence of resistance to multiple antimicrobial agents. The 4.7 Mb genome sequence of a multidrug-resistant S.Choleraesuis strain SC-B67 was determined. Genome wide comparison of three sequenced Salmonella genomes revealed that more deletion events occurred in S.Choleraesuis SC-B67 and S.Typhi CT18 relative to S.Typhimurium LT2. S.Choleraesuis has 151 pseudogenes, which, among the three Salmonella genomes, include the highest percentage of pseudogenes arising from the genes involved in bacterial chemotaxis signal-transduction pathways. Mutations in these genes may increase smooth swimming of the bacteria, potentially allowing more effective interactions with and invasion of host cells to occur. A key regulatory gene of TetR/AcrR family, acrR, was inactivated through the introduction of an internal stop codon resulting in overexpression of AcrAB that appears to be associated with ciprofloxacin resistance. While lateral gene transfer providing basic functions to allow niche expansion in the host and environment is maintained during the evolution of different serovars of Salmonella, genes providing little overall selective benefit may be lost rapidly. Our findings suggest that the formation of pseudogenes may provide a simple evolutionary pathway that complements gene acquisition to enhance virulence and antimicrobial resistance in S.Choleraesuis.
INTRODUCTION
Salmonella are important pathogens in humans and animals. Taxonomically, there are two species in the genus Salmonella: S.enterica (six subspecies) and S.bongori (one subspecies). Members of the seven subspecies can be serotyped into one of more than 2500 different serovars according to somatic (O) and flagellar (H) antigens. Some serovars such as S.Typhi and S.Paratyphi are highly adapted to humans without other known natural hosts, while others such as S.Typhimurium have a broad host range and infect a wide variety of animal hosts. A few serovars such as S.Choleraesuis have a narrow host range and occasionally infect humans. Nevertheless, when S.Choleraesuis infects humans, it usually causes invasive infections (1,2). It is one of the most common etiologic organisms for infective aneurysms, a devastating endovascular infection in humans (1,3). S.Choleraesuis also can cause swine paratyphoid, with clinical manifestations of enterocolitis and septicemia (4). Furthermore, it is a common bacterial isolate from swine with porcine reproductive and respiratory syndrome (4–6), which is now found worldwide and leads to considerable economic loss in the swine industry (5).
Antimicrobial therapy is essential in the treatment of S.Choleraesuis infections in humans. The emergence of resistance to ampicillin, chloramphenicol, trimethoprim-sulfamethoxazole, and notably, fluoroquinolone antibiotics in S.Choleraesuis has aroused concern on the use of these agents for the treatment of systemic infections caused by this organism (7). A multidrug-resistant strain of S.Choleraesuis was recently isolated from a patient with sepsis (8). This isolate, SC-B67, was resistant to all antimicrobial agents commonly used to treat patients with invasive salmonellosis, including ceftriaxone [minimal inhibitory concentration (MIC), 16 μg/ml] and ciprofloxacin (MIC, 32 μg/ml) (8). To investigate the virulence factors, host-specificity and mechanism of antimicrobial resistance of S.Choleraesuis, we sequenced the whole genome of S.Choleraesuis SC-B67. This work enabled us to perform comparative analyses with two other sequenced serovars, S.Typhi and S.Typhimurium (9–11). The three Salmonella serovars compared here are associated with three distinct human disease syndromes: typhoid fever caused by S.Typhi, self-limited enterocolitis by S.Typhimurium, and septicemic diseases with little involvement of the gastrointestinal tract by S.Choleraesuis. In this study, we discovered significant differences in genomic composition and organization of these serovars, and we identified several mechanisms underlying the unique pathogenicity and antimicrobial resistance mechanism of S.Choleraesuis.
MATERIALS AND METHODS
Genome sequencing and analysis
S.Choleraesuis strain SC-B67 was isolated from a patient with sepsis in 2002. Whole-genome libraries in pUC18 were prepared from genomic DNA as described earlier (12). Random clones were sequenced by using dye-terminator chemistry and data collected on MegaBASE 1000 automated sequencers. The total number of reads (>200 bp) was 66 494, with an average read length of 540 bp. The Phred/Phrap/Consed software package was used for quality assessment and sequence assembly (13–15). Gaps were filled by sequencing opposite ends of linking clones and the use of several PCR-based techniques and primer walking methods. The final assembly was verified by comparison with restriction-enzyme digest patterns of I-CeuI, AvrII and XbaI. Furthermore, we performed PCR on genomic DNA to amplify regions covering the pseudogenes. All the sequence alterations were confirmed by re-sequencing the PCR products.
The potential CDS were established with GLIMMER 2.1 (16) and the annotated open reading frames (ORFs) of three known Salmonella genomes (9–11); both predicted CDS and putative intergenic sequences were subjected to further manual inspections. ORFs that contain insertion, deletion or mutation to a stop codon compared with those known Salmonella genes were selected as pseudogenes. To the 4445 determined ORFs, exhaustive BLAST searches with an incremental stringency against NCBI non-redundant protein database were performed to determine their homology (17). To further investigate the function of each protein we used the InterProScan to search against the InterPro protein family database, which included PROSITE, PRINTS, Pfam, ProDom, SMART, TIGRFAMs, PIR SuperFamily and SUPERFAMILY (18). The up-to-date KEGG pathway database (ftp://ftp.genome.ad.jp/pub/kegg/genomes) was used for pathway analysis. Genes from 127 bacteria were gathered and classified into 127 pathways, to which we searched the pseudogenes in the BLAST suite, and the best hit was selected. The BLAST search criteria were as follows: (1) ∼30% of the subject sequence was aligned and (2) E-value = 10−10 or less. Putative aliens were detected by finding anomalous gene regions in genomic characterizations (19), and the pathogenic islands were further determined according to their function annotation and comparison with other Salmonella genomes (9,10). Unique regions of a genome were identified as sequences that have no counterpart in another compared genome with MegaBlast search. Genome used for comparison are from GenBank and their accession numbers are: NC_003143, NC_004061, NC_002528, NC_004741, NC_004631, NC_004545, NC_000913, NC_004431, NC_004088, NC_003198, NC_002695, NC_002655, NC_004344, NC_004337 and NC_003197.
Electron microscopy
Scanning electron microscopy was performed to examine the flagella of S.Choleraesuis SC-B67 and S.Typhimurium LT2. In preparation, bacteria were collected from the culture plate and suspended directly to 3% glutaraldehyde–2% paraformaldehyde mixture in 0.1 mol/l cacodylate buffer (pH = 7.4) for fixation at room temperature for 1 h. After adhering to poly-l-lysine coated glass coverslips, they were then post-fixed in 1% osmium tetroxide in the same buffer at 4°C for 1 h. After dehydration with graded ethanol, the samples were critical point dried, mounted onto the specimen stub, coated with Pt/Pd alloy, and examined with Hitachi S-5000 electron microscope at 3 kV.
Inhibition assay and western blot analysis
MICs were determined by the standard agar dilution method on the Mueller-Hinton medium. The MIC was defined as the lowest concentration of the drug that completely inhibited visible growth after inoculation for 18 h at 37°C. MICs of ciprofloxacin were also determined in the presence of the efflux pump inhibitor, Phe-Arg-β-naphthylamide (Sigma), at the concentration of 80 μg/ml (22). The efflux pump protein, AcrA, expression in Salmonella strains was analyzed by western blot hybridization. Fluoroquinolone-susceptible S.Typhimurium BN18 and in vitro-selected resistant mutants BN18/21, BN18/41 and BN18/71 were used as controls in these experiments (22). For comparison, a fluoroquinolone-susceptible S.Choleraesuis isolate SC-B42, was also examined by these methods.
RESULTS AND DISCUSSION
General features of the genome
The principal features of S.Choleraesuis SC-B67 genome is composed of a 4 755 700 bp chromosome, a 138 742 bp large plasmid, designated pSC138, and a 49 558 bp virulence plasmid, designated pSCV50 (Table 1 and Figure 1A–C). The replication origin and terminus, predicted by comparison with Escherichia coli K12 (23) and confirmed by GC-skew, are near 1.647 and 4.010 Mb, respectively.
Table 1.
Features of the Salmonella genomes
| Serovar | S.Choleraesuis SC-B67 | S.Typhimurium LT2a | S.Typhi CT18b | ||
|---|---|---|---|---|---|
| Chromosome (size, bp) | 4 755 700 | 4 857 432 | 4 809 037 | ||
| G+C content (%) | 52.11 | 53 | 52.09 | ||
| Coding sequences (excluding pseudogenes) | 4445 | 4450 | 4359 | ||
| Pseudogenes | 151 | 39 | 204 | ||
| Ribosomal RNAs | 6 × (16S-23S-5S), 1 × (16S-23S-5S-5S) | 6 × (16S-23S-5S), 1 × (16S-23S-5S-5S) | 6 × (16S-23S-5S), 1 × (16S-23S-5S-5S) | ||
| tRNAs | 85 | 85 | 78 | ||
| Average gene length (bp) | 898 | 946 | 958 | ||
| Plasmid | pSC138 | pSCV50 | pSLT | pHCM1 | pHCM2 |
| Size (bp) | 138 742 | 49 558 | 93 939 | 218 150 | 106 516 |
| G+C content (%) | 51.30 | 52.10 | 53 | 47.58 | 50.60 |
| Coding sequences (excluding pseudogenes) | 178 | 51 | 108 | 249 | 131 |
| Pseudogenes | 2 | 0 | 6 | 8 | 0 |
Figure 1.

Circular representation of the S.Choleraesuis genome. (A) The chromosome. The outer scale is marked in megabases. Circles range from 1 (outer circle) to 9 (inner circle). Circles 1 and 2, genes on forward and reverse strand; circle 3, transposons, insertion sequences and prophages; circle 4, unique regions (>100 bp in length) in S.Choleraesuis genome, relative to S.Typhi CT18 and S.Typhimurium LT2; circle 5, pseudogenes; circle 6, G+C content, values >52.1% (average) are in red and smaller in blue; circle 7, GC skew (G−C/G+C), values > 0 are in gold and smaller in purple; circle 8, tRNA genes; and circle 9, Salmonella pathogenicity islands 1–6 and 9. (B) Circular representation of pSC138. For the circular diagrams, the outer scale is marked in base pairs. Circles are numbered to the same scheme as in (A). Circles 1 and 2, all genes; circle 3, transposons and insertion sequences (blue) and bacteriophages (red); circle 4, resistance genes on forward (outward orange marks) and reverse (inward orange marks) strand; circle 5, repeat sequences; circle 6, G+C content; and circle 7, GC skew. (C) Circular representation of pSCV50. Circles 1 and 2, all genes; circle 3, G+C content; and circle 4, GC skew. All genes displayed in circles 1 and 2 are color-coded by function: translation/ribosome structure/biogenesis, pink; transcription, olive drab; DNA replication/recombination/repair, forest green; cell division/chromosome partitioning, light blue; posttranslational modification/protein turnover/chaperones, purple; cell envelop biogenesis, red; cell motility/secretion, plum; inorganic ion transport/metabolism, dark sea green; signal transduction mechanisms, medium purple; energy production/conversion, dark olive green; carbohydrate transport/metabolisms, gold; amino acid transport/metabolism, yellow; nucleotide transport/metabolism, orange; coenzyme transport/metabolism, tan; lipid transport/metabolism, salmon; secondary metabolites biosynthesis/transport/catabolism, light green; defense mechanism, black; general function prediction only, dark blue; function unknown, gray.
Genome comparison
The overall comparison of S.Choleraesuis SC-B67 genome to two other sequenced Salmonella genomes is shown in Table 1 (9,10). The S.Choleraesuis SC-B67 chromosome is 101 732 bp shorter than S.Typhimurium LT2, and 53 337 bp shorter than S.Typhi CT18; however, S.Typhi CT18 genome contains a unique 134 kb DNA segment coding for Vi capsule (9,10). The overall similarity of SC-B67 genome to those of LT2 and CT18 (see Supplementary Figure S1) is demonstrated by CROSS_MATCH comparison of the genomes (P. Green, unpublished data) with NUCmer (24). They are generally collinear and share >98% homology at the DNA level; however, such co-linearity between SC-B67 and CT18 is in part broken by two major inverted segments. Rearrangements between the rrn operons are common in S.Typhi (25), but are not found in S.Typhimurium LT2 and S.Choleraesuis SC-B67. A comparison of the insertions and deletions (indels) among the three sequenced serovars indicates that such events generally involve a small number of genes (Figure 2). The three Salmonella genomes are similar in size, indicating that the acquisition of genes has been counterbalanced by deletions; however, more deletion events occurred in S.Choleraesuis and S.Typhi (Figure 2). There are three major differences between S.Typhi and the two non-typhoidal Salmonella. S.Typhi contains Vi capsule, many more deletions, and a different profile of adherence- and motility-related genes, for which it lacks sti, stf, lpf, pef and fljA, but harbors genes like sta, stg, sef and pil. Vi is important in the pathogenesis of typhoid fever because of its role in antiphagocytosis and complement evasion. It is, however, difficult to address the relatedness between the different fimbrial patterns and pathogenicity, due to the redundancy of fimbrial operons in Salmonella.
Figure 2.

Distribution of insertions and deletions (indels) among the three sequenced Salmonella genomes. The graph shows the number of the indel events plotted against the size of the inserted or deleted element (shown as number of genes), clearly indicating that most of the events involve a small number of genes. Values above the line represent genes present in one genome relative to the other; values below the line represent genes absent in one genome relative to the other.
Pseudogenes
A remarkable difference among the three serovars is the possession of 151 pseudogenes in S.Choleraesuis SC-B67 in contrast to the numbers that have been observed, 39 in S.Typhimurium LT2 and 204 in S.Typhi CT18 (9,10). It appears that S.Choleraesuis has joined those narrow host range serovars of Salmonella, such as S.Typhi and S.Paratyphi A, that have undergone genome degradation by point mutations (10,26). Most of the 151 pseudogenes in S.Choleraesuis are intact in S.Typhimurium and S.Typhi: only two shared by S.Typhimurium and 17 by S.Typhi. These pseudogenes have been inactivated through the introduction of a stop codon (68 out of 151) or a frameshift mutation (83 out of 151). The latter case involved 1- or 2-bp shift (insertion or deletion) in 76 pseudogenes, truncation in six, and transposon insertion in one. A significant number of the pseudogenes in S.Choleraesuis (64 out of 151) and S.Typhi (75 out of 204) were predicted to be metabolism-related, according to the clusters of orthologous groups of proteins (COGs) database (Table 2). The presence of such pseudogenes may be the reason that S.Choleraesuis evolved from the rest of the non-typhoidal Salmonella to become a narrow host range pathogen. Results of the cross-genome comparison of the pseudogenes of the three Salmonella genomes can be found in the website http://saldb.cgu.edu.tw.
Table 2.
Functional classes of the corresponding genes of pseudogenes according to the clusters of orthologous groups of proteins (COGs) database in the three sequenced Salmonella genomes
| S.Choleraesuis SC-B67 | S.Typhimurium LT2 | S.Typhi CT18 | |
|---|---|---|---|
| Information storage and processing | |||
| Translation, ribosomal structure and biogenesis | 1 | 1 | 1 |
| Transcription | 10 | 1 | 13 |
| Replication, recombination and repair | 7 | 6 | 29 |
| Cellular processes and signaling | |||
| Cell cycle control, cell division, chromosome partitioning | 1 | 0 | 0 |
| Defense mechanisms | 1 | 0 | 4 |
| Signal transduction mechanisms | 6 | 1 | 8 |
| Cell wall/membrane/envelope biogenesis | 11 | 1 | 13 |
| Cell motility | 5 | 0 | 9 |
| Intracellular trafficking, secretion and vesicular transport | 0 | 0 | 1 |
| Posttranslational modification, protein turnover, chaperones | 10 | 0 | 3 |
| Metabolism | |||
| Energy production and conversion | 15 | 1 | 11 |
| Carbohydrate transport and metabolism | 18 | 7 | 27 |
| Amino acid transport and metabolism | 13 | 3 | 13 |
| Nucleotide transport and metabolism | 2 | 0 | 6 |
| Coenzyme transport and metabolism | 5 | 0 | 7 |
| Lipid transport and metabolism | 3 | 0 | 4 |
| Inorganic ion transport and metabolism | 6 | 0 | 6 |
| Secondary metabolites biosynthesis, transport and catabolism | 2 | 0 | 1 |
| Poorly characterized | |||
| General function prediction only | 23 | 2 | 16 |
| Function unknown | 9 | 2 | 22 |
| Unknown | |||
| Unknown | 3 | 14 | 12 |
Notably, in S.Choleraesuis we found five pseudogenes derived from genes of fimbriae synthesis or from flagellar or chaperone-usher operons. Two other pseudogenes were derived from genes that are likely to contribute to enteropathology of S.Typhimurium: icsA encoding a Peyer's patch colonization and shedding factor and STM1669 encoding a protein homologous to invasin C of Yersinia. The mutation in these genes may have implications for bacterial adhesion to intestinal epithelial cells and may explain why S.Choleraesuis produces diarrhea more rarely than S.Typhimurium. Similar mutations in fimbriae clusters were also found in S.Typhi and S.Paratyphi A (10,26).
Furthermore, several pseudogenes (13 out of 151) arose from genes that are involved in the regulation of the virulence or metabolism-related genes. We searched the 151 pseudogenes in the KEGG database. Using the search criteria described earlier, we were able to identify appropriate pathways for 6 of the 13 regulatory genes (Table 3). S.Choleraesuis is a motile organism. We observed that it possesses a number of long, peritrichous flagella using electron microscopy (data not shown). Such flagella were also present in S.Typhimurium LT2. Motility is believed to increase the probability that bacteria reach suitable sites for invasion; chemotaxis, on the other hand, allows bacteria to sense the environmental cues, thereby modulating bacterial movement. S.Choleraesuis apparently has several pseudogenes derived from genes involved in the bacterial chemotaxis signal-transduction pathways (Table 4). In an in vitro assay, S.Choleraesuis SC-B67 showed a relatively decreased chemotactic activity, compared with S.Typhimurium LT2. In the latter case, the 24-h swarm rate increased significantly in the presence of glucose 10 mM with or without the addition of PQQ (Figure 3). Previous studies have indicated that mutations, which disrupt the normal functioning of chemotaxis, enhance the invasive phenotype of S.Typhimurium (27,28). Such invasive phenotype was also observed in vivo in a murine infection model (28). Subsequent work revealed that defects in modulatory genes of the chemotaxis response (cheA, cheR, cheW and cheY) are responsible for the hyperinvasiveness of the mutants (28). These data are most consistent with the idea that motility affects the ability of Salmonella to enter host cells. As described previously, during smooth swimming the flagella operate as a single concerted bundle to propel the bacterium forward (28,29). The mutations in chemotaxis receptors may increase smooth swimming, thus allowing more effective bacterium–epithelial cell interactions and leading to cellular entry to occur at a higher frequency. As suggested for S.Paratyphi A (26), our data also suggest that loss of the genes involved in chemotaxis pathways may play a role in hyperinvasiveness of S.Choleraesuis.
Table 3.
Pseudogenes with predicted regulatory functions and pathways involved
| Pseudogene | Classification | Pathway involved | Source | EC number or protein description |
|---|---|---|---|---|
| Pseudogene 20 | Putative transcriptional regulator, LysR family | Pyrimidine metabolism | Borelia burgdorferi | 3.5.4.5 |
| Pseudogene 42 | Sensory histidine kinase of a two-component regulatory system (NarP) | Two-component system | E.coli | 2.7.3.− |
| Pseudogene 61 | Putative transcriptional regulator, LysR family | Purine metabolism | Borelia burgdorferi | 2.7.7.7 |
| Pseudogene 66 | Membrane protein, regulator of uhpT expression | Two-component system | E.coli | Regulatory protein UhpC |
| Pseudogene 68 | Periplasmic sensor of a multi-component regulatory system (TorS) | ABC transporters, prokaryotic | E.coli | D-ribose transport system substrate-binding protein RbsB |
| Pseudogene 134 | Transcriptional regulator, LysR family | ABC transporters, prokaryotic | Borelia burgdorferi | Oligopeptide transport system substrate-binding protein OppA-1 |
Table 4.
Pseudogenes with their predicted functions involved in the bacterial chemotaxis pathways
| Pseudogene | Corresponding gene in the chemotaxis pathways | Source | EC number or protein description |
|---|---|---|---|
| Pseudogene 48 | cheY | Clostridium acetobutylicum | Chemotaxis response regulator |
| Pseudogene 68 | rbsB | E.coli | d-ribose transport system substrate-binding protein |
| Pseudogene 112 | cheA | Borrelia burgdorferi | 2.7.3.− |
| Pseudogene 122 | tar | Thermoanaerobacter tengcongensis | Methyl-accepting chemotaxis protein |
| Pseudogene 123 | cheR | Borrelia burgdorferi | 2.1.1.80 |
| Pseudogene 129 | motB | Clostridium acetobutylicum | Flagellar motor protein |
Figure 3.
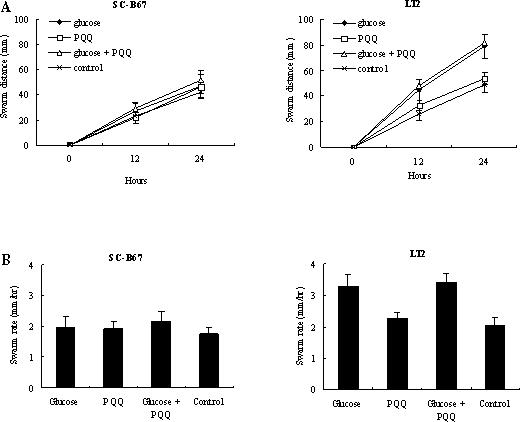
Effect of chemotactic attractants on the swarming of S.Choleraesuis SC-B67 and S.Typhimurium LT2 by using tryptone swarm tubes. (A) Swarm distance for S.Choleraesuis SC-B67 and S.Typhimurium LT2 over the time. Control indicates no addition of chemotactic attractants at the bottom of the tube. Each point represents mean ± SD. (B) The swarm rate in the presence of chemotactic attractants of each strain. S.Typhimurium LT2 swarm rate was significantly higher (*P < 0.05 by Student's t-test) in the presence of glucose (with or without PQQ). This situation was not observed in S.Choleraesuis SC-B67.
von Willebrand factor
The genome of S.Choleraesuis encodes a protein bearing homology to von Willebrand factor type A domain of humans (SC2319, vwa). The von Willebrand factor is a multimeric glycoprotein that is synthesized in megakaryocytes and endothelial cells (30). During normal hemostasis, it adheres to the subendothelial matrix following vascular damage. This vwa gene was also found in S.Typhimurium, but not in S.Typhi. Because a gene (SC1603) encoding microbial collagenase has been identified, it is reasonable to propose that collagenase-mediated vascular injury during infection and subsequent combined effect of the Vwa could lead to the development of septic vascular thrombi. This may explain the observed severe vascular sequelae of non-typhoidal Salmonella infections, namely infective aneurysm.
Pathogenicity islands
Genomic islands are regions of DNA on the chromosome of bacteria acquired via horizontal gene transfer. They are named pathogenicity, metabolic or resistance islands depending on functions of the genes on the islands. Salmonella have acquired a large number of virulence genes required during various stages of pathogenesis. In S.Choleraesuis, we have identified the five Salmonella pathogenicity islands (SPIs) shared by S.Typhimurium and S.Typhi. The average G+C content of the five SPIs in S.Choleraesuis genome is 46.62% (range, 43.55–50.33%). Of the five, we found variations in SPI-1, -3 and -5 (9,10,31). SPI-3 of S.Typhimurium harbors 10 genes, including the mgtCB operon that is required for survival in macrophages and growth in low-Mg2+ environment. Two genes, sugR and rhuM, were not found in SPI-3 of S.Choleraesuis. In addition, the high level of variability of sequence adjacent to selC among different Salmonella serovars implied that SPI-3 may be still evolving through gene acquisitions (31). SPI-1 and SPI-5 encode the most important invasion and enteropathology phenotypes of Salmonella. A 321 bp orf (SC1043) was found between pipB and pipC of SPI-5 in S.Choleraesuis, instead of the 431 bp orf (STM1089) in S.Typhimurium. Two genes (STM1092 and STM1093) between sopB and pipD of SPI-5 were absent in S.Choleraesuis. Within SPI-1 of S.Choleraesuis, while avrA and three other genes of unknown function (STM2901-STM2903) were not identified, three other unique genes (SC2841-SC2843) were present between pphB and STM2908. Two genes within SPI-1 of S.Typhimurium encoding a putative permease and a LysR transcriptional regulator are pseudogenes in S.Choleraesuis. The variation in SPI-1 and -5 may be another reason to explain why S.Choleraesuis produces fewer diarrheas when infecting humans. In addition to the SPIs 1-5, we also found SPI-6 and -9 (Figure 1A) that were previously identified in S.Typhi in the genome of S.Choleraesuis (10). We further discovered two SPIs (designated SPI-11 and -12) that had not been described previously and two metabolic islands in S.Choleraesuis (Table 5). These DNA segments have abnormal G+C contents that deviate from the genome average and are in proximity to tRNA genes or mobile elements. All the major SPIs, including SPIs 1-5 and the newly described SPIs 11-12 are highly conserved among the three Salmonella genomes (9–11,31).
Table 5.
Newly described pathogenicity and metabolic islands of Salmonella
| Islands | Sequence signature ahead of the island | Size/G+C content (%) | Number of genes | Important genes |
|---|---|---|---|---|
| Salmonella pathogenicity island-11 (SC1242-SC1258) | Gifsy-1 prophage | 14 kb/41.32 | 17 genes, 1 pseudogene | sopB, mppA, icdA, envF, msgA, envE, pagD, pagC |
| Salmonella pathogenicity island-12 (SC2251-SC2255) | tRNAPro | 6.3 kb/49.92 | 5 genes, 1 pseudogene | msgA, narP |
| Metabolic island (SC594-SC606) | tRNAArg | 13 kb/46.41 | 13 genes, 2 pseudogenes | gtrA, gtrB, cusS, ykg operon |
| Metabolic island (SC4347-SC4409) | tRNALeu | 68 kb/47.55 | 62 genes, 5 pseudogenes | yji operon, yjj operon, uxuR, bglJ |
Plasmids and antimicrobial resistance
S.Choleraesuis SC-B67 has two plasmids, pSC138 and pSCV50 (Figure 1B and C). The sequence of pSCV50 is almost identical to that of pKDSC50 (32). Compared with the virulence plasmid (pSLT) of S.Typhimurium LT2 (9,33), pSCV50 contains two major deletions in pef-RepFIIA and traT-samA. The plasmid pSCV50 is the smallest, among the seven known Salmonella virulence plasmids (32,33). The result of such reductive evolution is that pSCV50 becomes non-mobilizable but more stable (34). The plasmid pSC138 is composed of two distinguishable regions similar in size (∼68 kb and 70 kb). The 68 kb segment is a mosaic of multiple antimicrobial resistance genes, insertion sequences and transposons, suggesting that the resistance genes were acquired through multiple lateral gene transfer events under selective pressure. The other segment contains partition, replication and partial transfer regions of IncI1 plasmid ColIb-P9 (35,36). Many clinically relevant resistance genes were identified on this segment, including dhfr (trimethoprim), sulI and sulII (sulfonamide), catI and cmlA (chloramphenicol), bla (TEM-1; ampicillin), strA and aadA2 (streptomycin), tetRA (tetracycline), aph (kanamycin), sat (streptothricin), mef (macrolide), mer (mercury), ebr (ethidium bromide) and qac (quaternary ammonium compound) (see Supplementary Figure S2). A β-lactamase inhibitor-resistant β-lactamase gene, bla TEM-67, was found on this plasmid (see Supplementary Figure S2). This gene, originally identified on the plasmid pANG-1 of Proteus mirabilis, codes for an enzyme similar to TEM-2 type β-lactamase that confers the organism resistance to ampicillin as well as β-lactamase inhibitors, such as clavulanic acid (37). On the other hand, the ampC (blaCMY-2) that mediates resistance to extended-spectrum cephalosporins was found located on a transposon-like element inserted in finQ of the ColIb-P9 plasmid (8). There was no ampR, previously described in resistant Salmonella that produced AmpC β-lactamase, in the upstream region of the element (8,38). The ampR is a regulatory gene of LysR family to ampC (38). The loss of ampR, which results in high-level AmpC expression, may be associated with cephalosporin resistance phenotype of the organism (8). The pSC138 is non-conjugative, but contains oriT sequence and a defective transfer system; therefore, it is probably mobilizable (39). All of the antimicrobial resistance genes may be disseminated to other organisms through the spread of this potentially transmissible plasmid.
On the other hand, we found that ciprofloxacin resistance of S.Choleraesuis SC-B67 is conferred through mutations of gyrA and parC in the chromosome. We further verified this finding by checking many more resistant strains of S.Choleraesuis using PCR and sequencing (7), and found only when both parC and gyrA mutations are present is ciprofloxacin resistance reliably detected. Furthermore, recent reports have indicated that the AcrAB-TolC efflux system and its regulatory gene, acrR, may participate in the resistance to fluoroquinolones in Salmonella (22). It was noticed that when the acrB gene is inactivated, the resistance level to fluoroquinolones significantly reduces even when multiple target gene mutations are present (22). In S.Choleraesuis SC-B67, a pseudogene was formed through the introduction of an internal stop codon in acrR, which acts as a transcriptional suppressor to downregulate the expression of acrAB. An efflux pump inhibitor, Phe-Arg-β-naphthylamide (22), at the concentration of 80 μg/ml reduced the ciprofloxacin MIC from 32 to 8 μg/ml, suggesting the involvement of efflux pumping mechanism in fluoroquinolone resistance (Figure 4). Moreover, western blotting analysis with anti-AcrA antibody showed that SC-B67 exhibited a detectable increased level of AcrA expression, compared to a ciprofloxacin-susceptible S.Choleraesuis isolate that contains an intact acrR gene (Figure 4). The data suggested that fluoroquinolone resistance in S.Choleraesuis should be due to a combined effect of multiple target gene mutations and overexpression of AcrAB.
Figure 4.
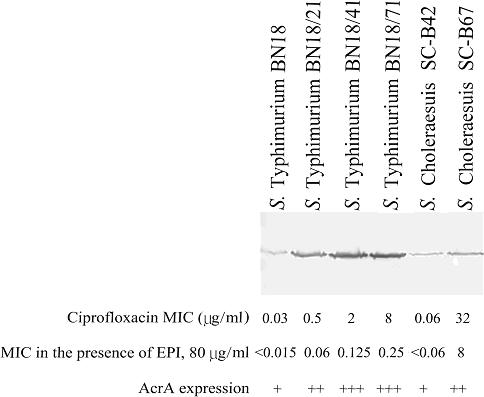
MICs of Salmonella strains with and without the efflux pump inhibitor (EPI), Phe-Arg-β-naphthylamide and western blotting analysis of AcrA expression by these strains. S.Typhimurium strains BN18, BN18/21, BN18/41, BN18/71 [see (22)], and the ciprofloxacin-susceptible S.Choleraesuis strain SC-B42 were used as controls.
In conclusion, genome comparisons of the closely related Salmonella emphasize the insights that can only be gleaned from sequencing multiple genomes of a single species. Lateral gene transfer providing basic functions to allow niche expansion in the host and environment is maintained in the evolution of different serovars of Salmonella. On the other hand, genes providing little overall selective benefit may get lost rapidly. Our study reveals that S.Choleraesuis has evolved through gene deletion and sequence alternations to become a very efficient and successful pathogen among non-typhoidal Salmonella, particularly with regard to its pathogenicity and antimicrobial resistance.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR online.
Supplementary Material
Acknowledgments
This work was supported by a grant (SMRPD32010) from the Chang Gung Memorial Hospital and Chang Gung University, Taoyuan, Taiwan. We are appreciative of the technical assistance by Steve Hsu and Kevin Wang, Tri-I Biotech, Inc., Taiwan and Axel Cloeckaert, Institut National de la Recherche Agronomique, France. Funding to pay the Open Access publication charges for this article was provided by Chang Gung Memorial Hospital.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cohen J.I., Bartlett J.A., Corey R. Extra-intestinal manifestations of Salmonella infections. Medicine. 1987;66:349–388. doi: 10.1097/00005792-198709000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Chiu C.H., Lin T.Y., Ou J.T. Predictors for extraintestinal infections of non-typhoidal Salmonella in patients without AIDS. Int. J. Clin. Pract. 1999;53:161–164. [PubMed] [Google Scholar]
- 3.Soravia-Dunand V.A., Loo V.G., Salit I.E. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin. Infect. Dis. 1999;29:862–868. doi: 10.1086/520450. [DOI] [PubMed] [Google Scholar]
- 4.Straw B.E., D'Allaire S., Mengeling W.L., Taylor D.J. Diseases of Swine, 8th edn. Ames, IA, USA: Balckwell Publishing; 1999. [Google Scholar]
- 5.Salmon S.A., Watts J.L., Case C.A., Hoffman L.J., Wegener H.C., Yancey R.J. Comparison of MICs of ceftiofur and other antimicrobial agents against bacterial pathogens of swine from the United States, Canada, and Denmark. J. Clin. Microbiol. 1995;33:2435–2444. doi: 10.1128/jcm.33.9.2435-2444.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plagemann P.G.W. Porcine reproductive and respiratory syndrome virus: origin hypothesis. Emerg. Infect. Dis. 2003;9:903–908. doi: 10.3201/eid0908.030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu C.H., Wu T.L., Su L.H., Chu C., Chia J.H., Kuo A.J., Chien M.S., Lin T.Y. Emergence in Taiwan of fluoroquinolone resistance in Salmonella enterica serovar Choleraesuis. N. Engl. J. Med. 2002;346:413–419. doi: 10.1056/NEJMoa012261. [DOI] [PubMed] [Google Scholar]
- 8.Chiu C.H., Su L.H., Chu C., Chia J.H., Wu T.L., Lin T.Y., Lee Y.S., Ou J.T. Isolation of Salmonella enterica serotype Choleraesuis resistant to ceftriaxone and ciprofloxacin. Lancet. 2004;363:1285–1286. doi: 10.1016/S0140-6736(04)16003-0. [DOI] [PubMed] [Google Scholar]
- 9.McClelland M., Sanderson K.E., Spieth J., Clifton S.W., Latreille P., Courtney L., Porwollik S., Ali J., Dante M., Du F., et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 10.Parkhill J., Dougan G., James K.D., Thomson N.R., Pickard D., Wain J., Churcher C., Mungall K.L., Bentley S.D., Holden M.T.G., et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 11.Deng W., Liou S.R., Plunkett G., III, Mayhew G.F., Rose D.J., Burland V., Kodoyianni V., Schwartz D.C., Blattner F.R. Comparative genomics of Salmonella enterica serovar Typhi strains Ty2 and CT18. J. Bacteriol. 2003;185:2330–2337. doi: 10.1128/JB.185.7.2330-2337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao Q., Tian Y., Li W., Xu Z., Xuan Z., Hu S., Dong W., Yang J., Chen Y., Xue Y., et al. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewing B., Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 14.Ewing B., Hillier L., Wendl M.C., Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 15.Gordon D., Abajian C., Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 16.Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Barrell D., Bateman A., Binns D., Biswas M., Bradley P., Bork P., et al. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 2003;31:315–318. doi: 10.1093/nar/gkg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlin S. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 2001;9:335–343. doi: 10.1016/s0966-842x(01)02079-0. [DOI] [PubMed] [Google Scholar]
- 20.Adler J. A method for measuring chemotaxis and use of the method to determine optimum conditions for chemotaxis by Escherichia coli. J. Gen. Microbiol. 1973;74:77–91. doi: 10.1099/00221287-74-1-77. [DOI] [PubMed] [Google Scholar]
- 21.De Jonge R., De Mattos M.J.T., Stock J.B., Neijssel O.M. Pyrroloquinoline quinone, a chemotactic attractant for Escherichia coli. J. Bacteriol. 1996;178:1224–1226. doi: 10.1128/jb.178.4.1224-1226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baucheron S., Imberechts H., Chaslus-Dancla E., Cloeckaert A. The AcrB multidrug transporter plays a major role in high-level fluoroquinolone resistance in Salmonella enterica serovar Typhimurium phage type DT204. Microb. Drug Resist. 2002;8:281–290. doi: 10.1089/10766290260469543. [DOI] [PubMed] [Google Scholar]
- 23.Blattner F.R., Plunkett G., III, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 24.Delcher A.L., Phillippy A., Carlton J., Salzberg S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S.L., Sanderson K. Highly plastic chromosomal organization in Salmonella typhi. Proc. Natl Acad. Sci. USA. 1996;93:10303–10308. doi: 10.1073/pnas.93.19.10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClelland M., Sanderson K.E., Clifton S.W., Latreille P., Porwollik S., Sabo A., Meyer R., Bieri T., Ozersky P., McLellan M., et al. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nature Genet. 2004;36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- 27.Khoramian F.T., Harayama K., Kutsukake K., Pechere J.C. Effect of motility and chemotaxis on the invasion of Salmonella typhimurium into HeLa cells. Microb. Pathog. 1990;9:47–53. doi: 10.1016/0882-4010(90)90039-s. [DOI] [PubMed] [Google Scholar]
- 28.Jones B.D., Lee C.A., Falkow S. Invasion by Salmonella typhimurium is affected by the detection of flagellar rotation. Infect. Immun. 1992;60:2475–2480. doi: 10.1128/iai.60.6.2475-2480.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunn J.S., Alpuche-Aranda C.M., Loomis W.P., Belden W.J., Miller S.I. Invasion by Salmonella typhimurium is affected by the detection of flagellar rotation. J. Bacteriol. 1995;177:5040–5047. doi: 10.1128/jb.177.17.5040-5047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuckwell D. Evolution of von Willebrand factor A (VWA) domains. Biochem. Soc. Trans. 1999;27:835–840. doi: 10.1042/bst0270835. [DOI] [PubMed] [Google Scholar]
- 31.Amavisit P., Lightfoot D., Browning G.F., Markham P.F. Variation between pathogenic serovars within Salmonella pathogenicity islands. J. Bacteriol. 2003;185:3624–3635. doi: 10.1128/JB.185.12.3624-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haneda T., Okada N., Nakazawa N., Kawakami T., Danbara H. Complete DNA sequence and comparative analysis of the 50-kilobase virulence plasmid of Salmonella enterica serovar Choleraesuis. Infect. Immun. 2001;69:2612–2620. doi: 10.1128/IAI.69.4.2612-2620.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu C., Hong S.F., Tsai C., Lin W.S., Liu T.P., Ou J.T. Comparative physical and genetic maps of the virulence plasmids of Salmonella enterica serovars Typhimurium, Enteritidis, Choleraesuis, and Dublin. Infect. Immun. 1999;67:2611–2614. doi: 10.1128/iai.67.5.2611-2614.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ou J.T., Lin M.Y., Chao H.L. Presence of F-like OriT base-pair sequence on the virulence plasmids of Salmonella serovars Gallinarum, Enteritidis, and Typhimurium, but absent in those of Choleraesuis and Dublin. Microb. Pathog. 1994;17:13–21. doi: 10.1006/mpat.1994.1048. [DOI] [PubMed] [Google Scholar]
- 35.Rees C.E.D., Bradley D.E., Wilkins B.M. Organization and regulation of the conjugation genes of IncI1 plasmid ColIb-P9. Plasmid. 1987;18:223–236. doi: 10.1016/0147-619x(87)90065-5. [DOI] [PubMed] [Google Scholar]
- 36.Hama C., Takizawa T., Moriwaki H., Urasaki Y., Mizobuchi K. Organization of the replication control region of plasmid ColIb-P9. J. Bacteriol. 1990;172:1983–1991. doi: 10.1128/jb.172.4.1983-1991.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naas T., Zerbib M., Girlich D., Nordmann P. Integration of a transposon Tn1-encoded inhibitor-resistant β-lactamase gene, blaTEM-67 from Proteus mirabilis, into the Escherichia coli chromosome. Antimicrob. Agents Chemother. 2003;47:19–26. doi: 10.1128/AAC.47.1.19-26.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnaud G., Arlet G., Verdet C., Gaillot O., Lagrange P.H., Philippon A. Salmonella enteritidis: AmpC plasmid-mediated inducible β–lactamase (DHA-1) with an ampR gene from Morganella morganii. Antimicrob. Agents Chemother. 1998;42:2352–2358. doi: 10.1128/aac.42.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu C., Chiu C.H., Chu C.H., Ou J.T. Nucleotide and amino acid sequences of oriT-traM-traJ-traY-traA-traL regions and mobilization of virulence plasmids of Salmonella enterica serovars Enteritidis, Gallinarum-Pullorum, and Typhimurium. J. Bacteriol. 2002;184:2857–2862. doi: 10.1128/JB.184.11.2857-2862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.