

Abstract
Purpose:
Vaccination with dendritic cell (DC)/multiple myeloma (MM) fusions has been shown to induce the expansion of circulating multiple myeloma–reactive lymphocytes and consolidation of clinical response following autologous hematopoietic cell transplant (auto-HCT).
Patients and Methods:
In this randomized phase II trial (NCT02728102), we assessed the effect of DC/MM fusion vaccination, GM-CSF, and lenalidomide maintenance as compared with control arms of GM-CSF and lenalidomide or lenalidomide maintenance alone on clinical response rates and induction of multiple myeloma–specific immunity at 1-year posttransplant.
Results:
The study enrolled 203 patients, with 140 randomized posttransplantation. Vaccine production was successful in 63 of 68 patients. At 1 year, rates of CR were 52.9% (vaccine) and 50% (control; P = 0.37, 80% CI 44.5%, 61.3%, and 41.6%, 58.4%, respectively), and rates of VGPR or better were 85.3% (vaccine) and 77.8% (control; P = 0.2). Conversion to CR at 1 year was 34.8% (vaccine) and 27.3% (control; P = 0.4). Vaccination induced a statistically significant expansion of multiple myeloma–reactive T cells at 1 year compared with before vaccination (P = 0.024) and in contrast to the nonvaccine arm (P = 0.026). Single-cell transcriptomics revealed clonotypic expansion of activated CD8 cells and shared dominant clonotypes between patients at 1-year posttransplant.
Conclusions:
DC/MM fusion vaccination with lenalidomide did not result in a statistically significant increase in CR rates at 1 year posttransplant but was associated with a significant increase in circulating multiple myeloma–reactive lymphocytes indicative of tumor-specific immunity. Site-specific production of a personalized cell therapy with centralized product characterization was effectively accomplished in the context of a multicenter cooperative group study.
Translational Relevance.
In contrast to lenalidomide maintenance therapy alone, vaccination with patient-specific dendritic cell/tumor hybridomas following autologous transplant for multiple myeloma was uniquely associated with the statistically significant and durable expansion of myeloma specific T cells at 1 year posttransplant. Vaccination induced the clonal expansion of activated T cells against shared epitopes as determined by single-cell transcriptomics. Differences in early clinical response between the vaccine and control arms did not meet statistical significance and may require longer follow-up. Vaccine-mediated expansion of tumor-specific T cells provides a platform for myeloma-directed immunity that may be augmented through enhancement of T-cell functional activity and targeting of the immune microenvironment. The study is a first-of-its-kind, academically led, collaborative multicenter randomized trial of personalized cell therapy with site-specific production and centralized assessment of vaccine characterization and immune response.
Introduction
The advent of combination biologic therapy for multiple myeloma (MM) has significantly improved patient outcomes. Induction therapy followed by consolidation with autologous hematopoietic stem cell transplantation (auto-HCT) results in effective cytoreduction of disease and prolongs progression-free survival (1). The addition of posttransplant lenalidomide maintenance therapy further extends the duration of remission and may augment clearance of residual malignant cells (2). However, the vast majority of patients ultimately experience disease progression due to the emergence of resistant disease. The development of strategies to enhance the durability of posttransplant response remains a major unmet need in the field. Immune dysregulation and loss of antitumor immunity contribute to the disease evolution of multiple myeloma (3–7). Chimeric antigen receptor (CAR) T-cell therapy has demonstrated dramatic responses in patients with relapsed/refractory disease, illustrating the potential unique efficacy of immune effector cells in targeting multiple myeloma that is resistant to standard biologic agents (8, 9).
The development of cancer vaccines is being investigated in multiple myeloma with the goal of stimulating tumor-specific immunity within the native T-cell repertoire and restoring immune surveillance (10–12). Critical elements of vaccine design include the presence of costimulatory signaling necessary for T-cell activation and the identification of appropriate targets that demonstrate immunogenicity and tumor selectivity. Dendritic cells (DC) are the most potent antigen-presenting cells that prominently express costimulatory signaling necessary for optimal T-cell activation, cross-presentation of antigen to reactive CD8 T cells, and the development of primary immunity (13, 14). Neoantigens arising from tumor-associated mutations are a powerful substrate for vaccination, showing heightened responses with high-affinity T cells (15). Lymphopoietic reconstitution following auto-HCT is characterized by transient disruption of immune-tolerance mechanisms, thus providing a powerful platform for therapeutic vaccination (16–18). Potential synergy between lenalidomide and vaccine therapy has been demonstrated with the pneumococcal vaccine and in preclinical studies with a personalized cancer vaccine (19, 20).
We have developed a personalized cancer vaccine in which patient-derived tumor cells are fused with autologous monocyte-derived DCs to present a broad array of tumor antigens, including potentially shared epitopes and neoepitopes, in the context of DC-mediated costimulation to restore the immune repertoire against multiple myeloma and improve clinical outcomes (21). Importantly, this vaccine design offers a platform to direct an immune response in vivo against the most immunogenic targets of an individual's tumor. In a phase II study, vaccination with autologous DCs fused with primary multiple myeloma tumor cells (DC/MM fusions) in conjunction with GM-CSF after auto-HCT induced the expansion of circulating multiple myeloma–reactive lymphocytes and was associated with conversion to complete response (CR) post-auto-HCT in the absence of maintenance therapy (21). The promise of this vaccine approach is further supported by a study for acute myeloid leukemia (AML) in which vaccination of DC/AML fusions for older patients in chemotherapy-induced remission resulted in the durable expansion of tumor-reactive lymphocytes in the bone marrow and peripheral blood, with 71% of patients in ongoing remission at 5 years of follow-up (22).
This multicenter randomized phase II study examined the efficacy of the DC/MM fusion vaccine in conjunction with lenalidomide maintenance therapy after auto-HCT, compared with maintenance alone. The primary immunologic endpoint was to examine the impact of vaccination on the durable expansion of multiple myeloma–reactive T cells, as compared with that observed with lymphopoietic reconstitution in the context of lenalidomide maintenance alone. The primary clinical endpoint was to assess the capacity of vaccination to enhance clinical response at 1-year posttransplant with respect to the percentage of patients achieving CR and those converting to CR during the period of posttransplant therapy. The study offered a first-of-its-kind academic collaborative effort of personalized cell therapy using an open-source format, site-specific production, and centralized product characterization/release criteria verification in a large multicenter setting.
Patients and Methods
Study design
The Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 1401 was a randomized phase II study at 18 transplantation centers in the United States. Participating centers demonstrated proficiency in vaccine production at their respective sites after receiving in-person training. This study was registered with ClinicalTrials.gov (NCT02728102). Patients with newly diagnosed multiple myeloma, age ≤70 years, candidates for auto-HCT, and having received not more than one cycle of induction therapy were potentially eligible for enrollment. Bone marrow plasma cell infiltration ≥20% was required to ensure sufficient tumor cells for vaccine generation. Patients received standard-of-care induction therapy and auto-HCT conditioned with melphalan 200 mg/m2. The choice of pretransplant induction therapy was not prescribed by the study and commonly consisted of combination biologic therapy with a bortezomib, lenalidomide, and dexamethasone or an equivalent regimen. At approximately 2 months after auto-HCT, patients without significant ongoing transplant-related toxicity were randomly assigned in a 2:1:1 manner to vaccination with DC/MM fusions with GM-CSF and lenalidomide, lenalidomide and GM-CSF, or lenalidomide alone, stratified by disease response posttransplant (CR vs. not CR). Lenalidomide was required for 2 years following transplant and then continued as per standard-of-care maintenance. Coprimary endpoints were comparison of CR conversion across treatment arms and treatment-induced expansion of multiple myeloma–specific T cells. Multiple myeloma with high-risk features was defined by the presence of elevated LDH, β2 microglobulin > 5.5 mg/L, and/or high-risk cytogenetics, including 17p, t(4;14), t(14,20), t(14,16) del 13q, and 1p/1q abnormalities.
The studies were conducted in accordance with:
○ Declaration of Helsinki
○ Belmont Report
○ U.S. Common Rule
Written consent was obtained from each subject or each subject's guardian and human investigations were performed after approval by an Institutional Review Board and in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services, where appropriate.
Procedures
After initial enrollment, patients underwent tumor collection from 30 mL of bone marrow aspirate, which was cryopreserved for future vaccine production and immune response assessments. A minimum of 10 million CD38-expressing plasma cells, quantified by immunocytochemistry, was required for vaccine production. Approximately 2 months after auto-HCT, patients randomized to the DC/MM vaccine arm underwent leukapheresis for isolation of adherent mononuclear cells for vaccine generation. All patients started lenalidomide maintenance approximately 3 months post–auto-HCT at a dose of 10 mg a day for 28 days of a 28-day cycle. On day 1 of cycles 2, 3, and 4 of lenalidomide maintenance, patients assigned to the vaccine arm underwent subcutaneous administration of the DC/MM fusion vaccine along with 100 μg GM-CSF administered subcutaneously adjacent to the vaccine site on days 1 to 4. Patients randomized to the nonvaccine control arms received either lenalidomide alone or lenalidomide with GM-CSF at the same dose and schedule as the vaccine arm.
Vaccine generation
Vaccines were produced at each participating site using a shared manufacturing protocol. Central characterization of the DC, tumor, and vaccine components was conducted at Beth Israel Deaconess Medical Center (Boston, MA). Vaccine uniformity was enhanced through the use of common lots for all reagents. Microbiologic assessments for sterility, mycoplasma, and endotoxin were performed centrally. Adherent mononuclear cells were cultured with rhGM-CSF and IL4 for 5 to 7 days and matured with TNFα. DC and tumor cells were transiently resuspended in polyethylene glycol to generate fusion cells (6), which were quantified based on the dual expression of cell-specific markers. Release criteria included: (i) ≥20% CD38 or CD138 expression by the tumor cell prep; (ii) ≥10 million plasma cell yield from the tumor cell prep; (iii) ≥ 50% of DC prep expression of CD86; (iv) >50% viability of DC prep; (v) >15% fusion efficiency; (vi) > 50% fusion viability; and (vii) negative sterility, mycoplasma, and endotoxin assays.
Assessment of clinical and immunologic response
Multiple myeloma response and disease progression were determined using the International Myeloma Working Group (IMWG) response criteria. Minimal residual disease (MRD) analysis was performed centrally by multichannel flow cytometry (Roswell Park Core Facility). The flow cytometer employed in the data acquisition was a BD FACS Canto, 10-Color Configuration (BD Biosciences) using 8 color flow cytometry (tube 1: CD38/CD56/CD45/CD19/CD117/CD81/ CD138/CD27 and tube 2: CD38/CD56/CD45/CD19/cKappa/cLambda/CD138/ CD27). Bone marrow aspirate samples (3 mL) were collected at 1-year posttransplant from patients without overt progression. MRD analysis was reported on 112 patients excluding patients with insufficient material for the assay. MRD was determined by multi-parameter flow cytometry identifying malignant plasma cells defined as CD45 (-/dim), CD38+, CD138+, CD19−, CD56+, and kappa or lambda-restricted at a level of detection of 10−5. Adverse events were assessed using NCI CTCAE version 4.0 at prior to initiation of lenalidomide maintenance and during maintenance therapy monthly for the first 4 cycles and then at cycles 6, 9, 15, 21, and 24.
Peripheral blood samples were collected at serial time points, including at enrollment, prior to initiation of posttransplant lenalidomide maintenance, following cycle 1 of lenalidomide and prior to vaccination, following 2 cycles of lenalidomide and vaccination, and following 3 vaccinations and 8 cycles of lenalidomide maintenance (approximately 1-year posttransplant). Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation (Histopaque-1077 Hybry-Max, Sigma-Aldrich), cryopreserved, and subsequently thawed concurrently for immune analysis. Multiple myeloma–reactive T cells were quantified by determining the percentage of IFNγ-expressing CD4 and CD8 T cells following exposure to autologous tumor lysate ex vivo. Briefly, 1 × 106 PBMCs were plated in 1 mL of RPMI1640 medium supplemented with 2 mmol/L Glutamine (Gibco) 10% fetal bovine serum (R&D Systems), 50 U/mL penicillin, 50 mg/mL streptomycin (Gibco) and pulsed with 20 μL of tumor lysate obtained by repeated freeze/thaw cycles and sonication of 1 × 106 BM/tumor cells collected at time of patient enrollment into the study. T-cell expression of IFNγ was measured after 5 days of culture. For intracellular IFNγ staining, cells were incubated for 6 hours with 1 μg/mL GolgiStop (BD Biosciences) to inhibit IFNγ secretion. Cells were then stained with APC-Cy7–conjugated anti-CD4 (Molecular Probes) and PE-conjugated anti-CD8 (BD Pharmingen) antibodies. PBMCs were then permeabilized with Cytofix/Cytoperm plus (BD Bioscience) before staining with PB-conjugated anti-IFNγ antibody (Invitrogen). The percentage of CD4 and CD8 T cells expressing IFNγ was determined by flow cytometry with a Gallios instrument (Beckman Coulter). Flow cytometric analysis was performed using isotype control antibody to set the quadrants for quantification of IFN© expression.
Statistical analysis
The primary analysis compared the proportion of patients in CR/stringent CR (sCR) at 1-year posttransplant between the vaccine and nonvaccine groups using an intent-to-treat (ITT) analysis. A sample size of 66 patients in each cohort (132 total) targeted 85% power to detect an improvement in the CR/sCR rate at 1 year from 40% to 60%, using a two-sample Z test comparing proportions with a one-sided type I error of 10%. Because randomization occurred after tumor cell collection and transplant, target enrollment was 188, assuming 30% dropout between tumor cell collection and randomization. The difference in response rates was also estimated with 80% confidence intervals, consistent with the one-sided type I error of 10%. A secondary analysis stratified by disease response prior to randomization used a Cochran–Mantel–Haenszel test and stratified odds ratio. A secondary pairwise analysis compared the vaccine arm, lenalidomide/GM-CSF arm, and the lenalidomide alone arm. Proportions of patients who converted to CR and proportions with MRD present were compared using the χ2 test.
PFS (progression-free survival) and OS (overall survival) were secondary endpoints of the study. Death or disease progression was considered an event for PFS. The time to event was calculated as the time from randomization to disease progression, death, or initiation of nonprotocol anti-myeloma therapy, whichever occurred first. The Kaplan–Meier estimators were constructed for each treatment arm. PFS/OS was compared between the vaccine and the combined non-vaccine arms using the log-rank test. Patients experiencing grade 3–5 toxicities were compared between vaccine and non-vaccine group using χ2 test. Incidence of grade 2–3 infections were compared between vaccine and non-vaccine group using Gray test by cumulative incidence function, treating death as a competing risk. The proportion of patients with MRD present (MRD+) was described using frequencies at pre-randomization and 9th cycle post-randomization and compared between the vaccine arm with the nonvaccine arms combined using the χ2 test. For immune response assessments, nonparametric tests were used because of data nonnormality. A Kruskal–Wallis test was used to compare 3 groups, and Mann–Whitney test was used to compare two groups. For the within-group comparison over time, Wilcoxon signed-rank test was used. All secondary analyses, exploratory analyses, and immunologic analyses used a two-sided significance level of 5%. Analyses of clinical endpoints and primary immunologic endpoint were performed using SAS version 9.4 and R version 3.6. Analyses of secondary immunologic endpoints were calculated using Prism 8 software (GraphPad).
Single-cell immunoprofiling
To characterize the cellular landscape before and after DC/MM fusion vaccination, we performed single-cell immunoprofiling (5′ mRNA expression + V(D)J sequencing) using the 10× Genomics Chromium platform on samples collected at enrollment, 90 days posttransplant, following 2 vaccinations, and at 1-year posttransplant following 3 vaccinations. Briefly, cryopreserved PBMCs were thawed and processed using the Chromium Next GEM Single Cell 5′ Kit v2 (10X Genomics) and loaded on a Chromium Next GEM Chip K for gene expression library preparation. Full-length paired α/β TCR libraries were obtained using the Chromium Single Cell V(D)J Enrichment, Human T Cell kit. Libraries were sequenced on an Illumina sequencer.
Single-cell RNA sequencing immunoprofiling data analysis
Gene expression
Cell Ranger v.4.0.0 was used for cell assignment and gene expression quantification. Gene expression libraries were loaded, and droplets were subsetted using mitochondrial content (<20%), number of genes detected in each cell (250 < n < 5000), and total number of unique molecules detected within a cell (250 < n < 20,000). Samples derived from 13 vaccinated patients across 4 timepoints were integrated and utilized for cluster detection using Seurat v.3.2.3 (Fig. 4A; ref. 23). Variable genes were defined with the FindVariableFeatures function from Seurat, while dimensionality reduction was performed on the first 30 PCA-retrieved principal components (PC) and the top 2,000 variable genes. Cluster detection, two-dimensional representations, and batch-effect correction were performed with Harmony (23). We applied Seurat's FindClusters and FindMarkers functions on the harmonized embeddings to define the distinct cell populations and the differentially expressed markers of each cluster, respectively. Cell type characterization was performed using SingleR (24), supported by domain expert manual annotation based on canonical markers for each lineage. Comparisons in cluster relative abundances were detected using a Dirichlet multinomial regression model, taking into account the distinct time points.
Figure 4.

Lymphocyte profiles at single-cell resolution following DC/MM fusion vaccination after autoHCT. A, Bar plots portraying the number of cells captured with scRNA-seq per patient (n = 13) and time point (n = 4). B and C, Two-dimensional uniform manifold approximation and projection (UMAP) of all cells passing QC (B; n = 309,423), attributed to 47 cellular populations, and the T-cell compartment (C; n = 146,373), composed of 14 cellular populations and 9 major compartments (pooled data from multiple time points, n = 13 vaccinated patients). D, Dot plot capturing the average expression of marker genes and the percentage of cells expressing them across the distinct cell populations. Low to high average expression is presented in a gray-to-red color gradient. The size of the dot indicates the percentage of cells in each cell population expressing the specific marker genes. E, Two-dimensional uniform manifold approximation and projection (UMAP) plots of all cells in pre-/post-vaccination time points. The distinct cell populations are depicted with distinct colors, corresponding to 12 major lineages. F, Boxplots portraying cell proportion differences in the major cell types among the pre-/postvaccination time points. Each circle represents the cell-type–specific relative abundance per sample. Significantly different proportions of postvaccination time points compared with 90 days posttransplant are denoted with * (*, P < 0.05; **, P < 0.01; Dirichlet multinomial regression). A statistically significant increase in the cell proportions of CD4 and CD8 T cells in the postvaccination time points as compared with 90 days posttransplant is observed, followed by a decrease in NK cells (CD4 T cells; CD8 T cells; NK cells, 90 days posttransplant vs. after 2 vaccinations: P: 9.35 × 10−6; 6.5 × 10−3; 9.6 × 10−3, 90 days posttransplant vs. 1 year posttransplant: P: 8.7 × 10−3, Dirichlet multinomial regression).
T-cell receptor analysis
Cell Ranger v.4.0.0 V(D)J Annotation pipeline (25) was utilized to retrieve clonotype information. Cell barcodes with TCRα and/or TCRβ chains were retained, and each clonotype was characterized by the combination of the amino acid sequences of the 2 chains. The Shannon–Wiener Diversity Index was used to calculate diversity scores with the vegan package in R (26, 27). Comparisons in the abundances of TCRs among the different time points were detected using a Dirichlet multinomial regression model, taking into account the distinct time points and the distinct TCR frequency thresholds, i.e., ∼Timepoint*TCR_frequency. GLIPH2 (28) was used to detect antigen-specific T-cell groups based on CDR3b sequences. A reference set of 840,162 distinct CDR3b sequences from CD4 and CD8-naïve T cells was utilized to extract clusters of β TCR sequences that share global or local motifs. High-confident TCR clonotype clusters were prioritized compared with the reference dataset (n = 120, P < 0.0001, Fisher exact test) and further refined to select groups with at least 3 CDR3b members in >2 patients. Sequence logos were generated using the ggseqlogo package in R (29).
Data availability
The data generated in this study are available upon request from the corresponding author.
Results
Patient characteristics
The study accrued patients from July 2016 to October 2018, with 203 patients enrolled from 18 participating centers. The study schema is depicted in Fig. 1A. Sixty-three patients dropped out of the study from enrollment to randomization, concordant with the expected rate of 30% pre-specified in the protocol (Fig. 1B). Table 1 displays the demographic and baseline characteristics for 140 patients randomized by treatment group and demonstrates balanced distribution of disease prognostic factors between the assigned treatment cohorts (Supplementary table S1). High-risk features observed in the randomized patients included t(4,14), t(14,16), t(14,20), and deletion of 17p (totaling 8.8%); deletion 13/13q (8.8%);and abnormalities in 1p/1q (25.3%). Sixty-eight patients were randomized to DC/MM fusion vaccine/GM-CSF and lenalidomide maintenance, 37 to lenalidomide maintenance with GM-CSF, and 35 to the lenalidomide alone (Fig. 2). The percentage of patients randomized on stratum 1 (sCR/CR) and stratum 2 (VGPR/PR/stable disease) was 36.4% and 63.6%, respectively, and was balanced between the treatment arms. Median time from enrollment to transplant, transplant to randomization, and randomization to initiation of maintenance therapy was 5 months, 68 days, and 28 days, respectively. The median follow-up time was 26.7 months postrandomization.
Figure 1.

A, Study schema. B, CONSORT diagram.
Table 1.
Randomized patient characteristics by treatment arm allocation.
| Treatment arm | ||||
|---|---|---|---|---|
| Vaccine/GM-CSF/Lenalidomide (N = 68) N (%) | Lenalidomide/GM-CSF (N = 37) N (%) | Lenalidomide alone (N = 35) N (%) | Total (N = 140) N (%) | |
| Gender | ||||
| Female | 27 (39.7%) | 18 (48.6%) | 18 (51.4%) | 63 (45.0%) |
| Male | 41 (60.3%) | 19 (51.4%) | 17 (48.6%) | 77 (55.0%) |
| Age, y | ||||
| Median (range) | 60.2 (41.6–70.2) | 64.0 (44.2–70.2) | 59.2 (35.2–70.9) | 61.6 (35.2–70.9) |
| Black or African American | 8 (11.8%) | 2 (5.4%) | 5 (14.3%) | 27 (13.3%) |
| Karnofsky performance score | ||||
| 100 | 16 (23.5%) | 7 (18.9%) | 9 (25.7%) | 32 (22.9%) |
| 90 | 29 (42.6%) | 10 (27.0%) | 14 (40.0%) | 53 (37.9%) |
| 80 | 17 (25.0%) | 15 (40.5%) | 9 (25.7%) | 41 (29.3%) |
| 70 | 6 (8.8%) | 5 (13.5%) | 3 (8.6%) | 14 (10.0%) |
| Plasma cells percentage in BM aspirate differential | ||||
| Mean (SD) | 49.1 (23.5) | 48.9 (21.8) | 48.4 (20.2) | 48.9 (22.1) |
| Median (range) | 45 (9–100) | 40 (20–93) | 50 (20–80) | 45 (9–100) |
| Received initial systemic therapy prior to enrollment | ||||
| Yes | 7 (10.3%) | 4 (10.8%) | 3 (8.6%) | 14 (10.0%) |
| No | 61 (89.7%) | 33 (89.2%) | 32 (91.4%) | 126 (90.0%) |
| Randomization strata (1) | ||||
| sCR/CR | 24 (35.3%) | 14 (37.8%) | 13 (37.1%) | 51 (36.4%) |
| VGPR/PR/stable disease | 44 (64.7%) | 23 (62.2%) | 22 (62.9%) | 89 (63.6%) |
| Disease status at randomization (1) | ||||
| Stringent complete response | 11 (16.2%) | 6 (16.2%) | 4 (11.4%) | 21 (15.0%) |
| Complete response | 11 (16.2%) | 9 (24.3%) | 9 (25.7%) | 29 (20.7%) |
| Very good partial response | 37 (54.4%) | 15 (40.5%) | 17 (48.6%) | 69 (49.3%) |
| Partial response | 9 (13.2%) | 7 (18.9%) | 5 (14.3%) | 21 (15.0%) |
| Stable response | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Received initial systemic anti-myeloma therapy prior to randomization | ||||
| Yes | 65 (95.6%) | 35 (94.6%) | 32 (91.4%) | 132 (94.3%) |
| # Lines of systemic therapy received | ||||
| 1 | 49 (75.4%) | 28 (80.0%) | 21 (65.6%) | 98 (74.2%) |
| 2 | 11 (16.9%) | 5 (14.3%) | 7 (21.9%) | 23 (17.4%) |
| 3 | 5 (7.7%) | 1 (2.9%) | 3 (9.4%) | 9 (6.8%) |
| 4 | 0 (0.0%) | 1 (2.9%) | 1 (3.1%) | 2 (1.5%) |
| No | 3 (4.4%) | 2 (5.4%) | 3 (8.6%) | 8 (5.7%) |
| Unknown/missing | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Risk stratification | ||||
| Standard | 23 (33.8%) | 9 (24.3%) | 15 (42.9%) | 47 (33.6%) |
| High | 44 (64.7%) | 27 (73.0%) | 20 (57.1%) | 91 (65.0%) |
| Time from diagnosis date to enrollment, months | ||||
| Mean (SD) | 9.9 (6.9) | 11.2 (12.1) | 9.0 (1.7) | 10.0 (7.9) |
| Median (range) | 8 (6–53) | 8 (3–80) | 9 (7–13) | 8 (3–80) |
| Time from enrollment to transplant, months | ||||
| Mean (SD) | 5.5 (1.9) | 5.6 (1.8) | 5.6 (1.3) | 5.6 (1.7) |
| Median (range) | 5 (3–11) | 5 (4–11) | 5 (4–9) | 5 (3–11) |
| Time from transplant to randomization, days | ||||
| Mean (SD) | 66.3 (7.7) | 67.1 (9.4) | 67.7 (8.5) | 66.9 (8.3) |
| Median (range) | 68 (49–92) | 69 (50–79) | 67 (53–87) | 69 (49–92) |
| Time from randomization to maintenance initiation, days | ||||
| Mean (SD) | 30.1 (8.2) | 27.6 (9.7) | 27.8 (9.7) | 28.9 (9.0) |
| Median (range) | 30 (12–47) | 27 (12–47) | 27 (9–49) | 28 (9–49) |
Figure 2.
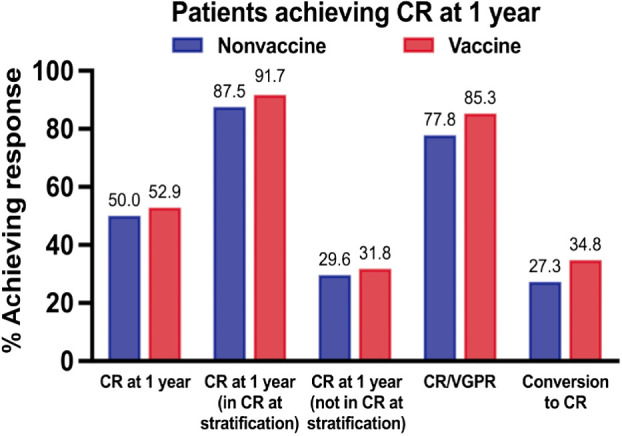
CR rates at 1 year. Percentage of patients in the vaccine and nonvaccine arms achieving CR at 1 year based on intention-to-treat. Percentage of patients achieving CR/VGPR and conversion to CR at 1 year for those patients not achieving CR at time of posttransplant randomization is also presented.
Vaccine generation
A collaborative process was established for the standardization of vaccine manufacturing, including tumor cell harvest and cryopreservation, DC generation from leukapheresis collection, creation and quantification of the DC/tumor fusion vaccine, and the process of characterizing each of the cellular products. Site validation required successful production of DC/tumor fusions in two mock runs meeting requisite fusion efficiency and sterility parameters, as outlined in the study release criteria.
Of the 140 patients undergoing randomization, the median percentage of plasma cells in bone marrow aspirate differential at time of enrollment was 45%. The mean CD86 expression and viability of the DC preparations were 80.6% and 79.3%, respectively. The mean fusion efficiency of the DC/MM product, as determined by coexpression of standard DC (CD86) and multiple myeloma (CD38) markers, was 47.9%. Mean fusion cell viability was 78.6%.
Vaccines were successfully generated for 63 of 68 patients (93%) assigned to the vaccine arm of the study. Reasons for production failure (n = 4) included insufficiently viable final cell product (n = 3) and cell culture contamination (n = 1); these four patients were treated per the lenalidomide arm. One patient never initiated maintenance therapy and later died of disease progression. Sixty-three patients received vaccines, including 57 receiving three doses, 5 receiving two doses, and 1 receiving one dose.
Clinical response
Differences in rates of clinical response between the vaccine and control arms did not reach statistical significance. Thirty-six of the 68 (52.9%) evaluable patients on the vaccine arm [80% confidence interval (CI), 44.5–61.3] and 34 of the 68 (50.0%) of the nonvaccine arms (80% CI, 41.6–58.4) achieved best response of CR/sCR at 1-year posttransplant (difference in response rates 2.9%; 80% CI, −8.8%–14.6%; P = 0.3; Fig. 2). Of the patients not achieving CR at time of randomization posttransplant, conversion to CR at 1-year was 34.8% for the vaccine arm and 27.3% for the nonvaccine arm (P = 0.4). Best response of VGPR or better at 1 year was achieved by 85.3% and 77.8% of patients on the vaccine and nonvaccine arms, respectively (P = 0.2). A secondary pairwise analysis of CR rates comparing the vaccine arm with each of the nonvaccine arms did not show a significant difference in CR rates. Assessment of MRD in the bone marrow was performed at 1-year posttransplant in patients without disease progression and with an adequate sample for analysis. In the vaccine arm, 38 of 53 (71.7%) achieved MRD-negative disease at 1-year posttransplant compared with 41 of 59 (69.5%) in the combined nonvaccine groups (P = 0.80). At 2 years, PFS in the vaccine and nonvaccine arms was 79.3% (95% CI, 67.6–87.2) and 88.2% (95% CI, 77.8–93.9), respectively (P = 0.168). OS was 97% and 98% at 2 years, respectively (Supplementary Fig. S1A and S1B).
Adverse events
The percentage of patients experiencing post-transplant grade 3–4 toxicities were 77.9%, 62.2%, and 74.3% for the vaccine, lenalidomide/GM-CSF, and lenalidomide alone arms, respectively (P = NS; Table 2). The percentage of patients experiencing grade 3–4 toxicities was 78% and 68% in the vaccine and nonvaccine cohorts (P = 0.19). There were no grade 5 toxicities in any of the cohorts. The overall grade 2–3 infection rate was 22.9% (23.5% on the vaccine arm, 13.5% on the lenalidomide/GM-CSF arm, and 31.4% on the lenalidomide alone arm, P = NS). The percentage of patients experiencing grade 2–3 infection was 26.6% and 23.2% for the vaccine and nonvaccine cohorts, respectively (P = 0.84).
Table 2.
Adverse events by treatment arm.
| Grade 3–5 toxicities | ||||||
|---|---|---|---|---|---|---|
| Vaccine (N = 68) | Lenalidomide/GM-CSF (N = 37) | Lenalidomide alone (N = 35) | ||||
| System organ class | # Events | # Participants | # Events | # Participants | # Events | # Participants |
| Auditory disorders | 1 | 1 (1.5%) | 0 | 0 (0.0%) | 0 | 0 (0.0%) |
| Blood and lymphatic disorders | 81 | 36 (52.9%) | 40 | 17 (45.9%) | 38 | 18 (51.4%) |
| Cardiovascular disorders | 4 | 3 (4.4%) | 3 | 3 (8.1%) | 2 | 2 (5.7%) |
| Endocrine disorders | 0 | 0 (0.0%) | 0 | 0 (0.0%) | 0 | 0 (0.0%) |
| GI disorders | 15 | 10 (14.7%) | 4 | 4 (10.8%) | 2 | 2 (5.7%) |
| General disorders | 5 | 5 (7.4%) | 2 | 2 (5.4%) | 2 | 2 (5.7%) |
| Hemorrhagic disorders | 0 | 0 (0.0%) | 0 | 0 (0.0%) | 0 | 0 (0.0%) |
| Hepatobiliary/pancreas disorders | 4 | 4 (5.9%) | 1 | 1 (2.7%) | 1 | 1 (2.8%) |
| Immune system disorders | 2 | 2 (2.9%) | 0 | 0 (0.0%) | 0 | 0 (0.0%) |
| Investigations | 2 | 2 (2.9%) | 0 | 0 (0.0%) | 1 | 1 (2.8%) |
| Metabolism and nutrition disorders | 7 | 7 (10.3%) | 1 | 1 (2.7%) | 3 | 2 (5.7%) |
| Musculoskeletal and connective tissue disorders | 1 | 1 (1.5%) | 1 | 1 (2.7%) | 2 | 2 (5.7%) |
| Nervous system disorders | 14 | 11 (16.2%) | 5 | 4 (10.8%) | 1 | 1 (2.8%) |
| Ocular/visual disorders | 0 | 0 (0.0%) | 0 | 0 (0.0%) | 0 | 0 (0.0%) |
| Renal disorders | 1 | 1 (1.5%) | 0 | 0 (0.0%) | 2 | 1 (2.8%) |
| Respiratory, thoracic, and mediastinal disorders | 5 | 5 (7.4%) | 1 | 1 (2.7%) | 2 | 2 (5.7%) |
| Skin and subcutaneous tissue disorders | 4 | 4 (5.9%) | 5 | 3 (8.1%) | 7 | 5 (14.3%) |
| Vascular disorders | 6 | 6 (8.8%) | 11 | 6 (16.2%) | 3 | 2 (5.7%) |
| Abnormal liver symptoms | 0 | 0 (0.0%) | 1 | 1 (2.7%) | 0 | 0 (0.0%) |
| Total | 152 | 53 (77.9%) | 75 | 23 (62.2%) | 66 | 26 (74.3%) |
Immune response assessments
The primary immunologic endpoint of the study was the impact of posttransplant vaccination as compared to maintenance alone on the percentage of circulating multiple myeloma–reactive T cells as determined by the percentage of CD8 T cells expressing IFNγ upon ex vivo exposure to autologous PBMCs pulsed with tumor lysate as a measure of antigen-specific activation (Fig. 3A). Levels were quantified for an initial cohort of 45 patients (those first completing the 1-year follow-up period and availability of adequate sample material), including 24 from the vaccine arm and 21 from the nonvaccine arms (12 patients with lenalidomide alone and 9 with lenalidomide + GM-CSF).
Figure 3.

DC/MM fusion vaccination after auto-HCT induces the expansion of multiple myeloma–reactive T cells. PBMCs obtained at enrollment (Pre) and before lenalidomide maintenance cycles 1 (PC1), 2 (PC2, vaccine initiation time point for the vaccine arm), 3 (PC3), and 9 (PC9) exposed to autologous tumor lysate were assessed by multiparameter flow cytometry for percentage of multiple myeloma–specific T cells expressing intracellular IFNγ. A, Peak change of multiple myeloma–reactive CD8 T cells for lenalidomide control (n = 12), lenalidomide + GM-CSF control (n = 9), and vaccinated patients (lenalidomide + GM-CSF + vaccine, n = 24). B, Quantification of the multiple myeloma–reactive CD8 T cells for lenalidomide control, lenalidomide + GM-CSF control, and vaccinated patients over time. C, Quantification of the multiple myeloma–reactive CD8 T cells comparing nonvaccine controls (n = 21) with vaccinated patients (lenalidomide + GM-CSF + vaccine, n = 24) over time. D, Quantification of the multiple myeloma–reactive CD4 T cells for lenalidomide control, lenalidomide + GM-CSF control, vaccinated patients. Outliers greater than Q3+1.5IQR or less than Q1–1.5IQR are omitted from the figure. Nonsignificant P value (P > 0.05) are not shown in B and D.
Immunologic response was calculated by determining the peak change from baseline values, as well as quantification of MM-reactive T cells at 1-year posttransplant in each of the treatment cohorts. The peak change in tumor-reactive CD8 T cells from baseline for the vaccine, lenalidomide alone, and lenalidomide + GM-CSF arms were 5.29%, 0.21%, and 2.25%, respectively (Kruskal–Wallis test P = 0.157, Fig. 3A). The peak change of circulating CD8 T cells for vaccine and combined non-vaccine cohort was 5.29% and 0.68%, respectively (Mann–Whitney test P = 0.065). At 1-year posttransplant, the median percentage of circulating multiple myeloma–reactive CD8 T cells was 5.80% for patients on the vaccine arm, 0.58% on the lenalidomide + GM-CSF arm, and 1.03% on the lenalidomide alone arm (Fig. 3B). Within the vaccine arm, there were statistically significant increases in multiple myeloma–reactive CD8 T cells following 3 vaccinations at 1-year posttransplant as compared to 2 time points prior to vaccination, prior to the initiation of lenalidomide maintenance and following one cycle of lenalidomide maintenance (Wilcoxon signed-rank test P = 0.040, 0.024 respectively; Fig. 3B). In contrast, within each of the control cohorts (lenalidomide + GM-CSF for 3 cycles and lenalidomide maintenance alone), there was no statistically significant increase in multiple myeloma–reactive CD8 T cells over time (Fig. 3B and C). At 1-year posttransplant, patients on the vaccine arm showed a statistically significant increase in the percentage of circulating multiple myeloma–reactive CD8 T cells as compared with the control arms (Mann–Whitney test P = 0.026 for comparison of vaccine arm to combined nonvaccine arms). A statistically significant rise in multiple myeloma–reactive CD4 T cells was observed transiently after the second vaccination (1.39% at maintenance cycle 1 to 2.00% at cycle 3 with Wilcoxon signed-rank test P = 0.029) but did not persist at 1-year posttransplant (Fig. 3D).
Further characterization of the T-cell repertoire was performed by single-cell transcriptomic analysis on a subset of 13 vaccinated patients with adequate samples for analysis. Assessment of serial peripheral blood samples from this initial cohort pre-/postvaccination differentiated key immune subsets (Fig. 4A and B) and provided a detailed picture of the T-cell landscape, including subsets of regulatory, effector, and memory compartments (Fig. 4C–E). Of note, a progressive rise in the relative abundance of CD4 T cells was observed after 2 and 3 vaccinations, as compared with the prevaccination period (prevaccination/90 days posttransplant vs. after 2 vaccinations: P < 0.001, prevaccination/90 days posttransplant versus 1-year posttransplant: P = 0.0087, Dirichlet multinomial regression, Fig. 4F). Similarly, a significant increase in CD8 T-cell abundance was observed after 2 vaccinations compared to prevaccination (prevaccination/90 days posttransplant vs. after 2 vaccinations: P = 0.006, Dirichlet multinomial regression, Fig. 4F).
Immune cell repertoire profiling by sequencing of full-length paired α/β T-cell receptors at the single-cell level demonstrated a progressive increase in the number of clonotypes (TCR clonotype frequency ≥ 1) within the activated CD4 and NK T cellular compartments and higher clonotypic expansion of the CD8 effector memory T cells (TCR clonotype frequency > 20) following 2 vaccinations and 3 vaccinations (approximately 1-year posttransplant) as compared with prevaccination levels (enrollment vs. after 2 vaccinations: P < 0.001, enrollment vs. 1-year posttransplant, after 3 vaccinations: P < 0.001, prevaccination/90 days posttransplant vs. after 2 vaccinations: P = 0.049, Dirichlet multinomial regression, Fig. 5A and B). CD8 T cells showed greater expansion than all other T-cell populations at each time point (enrollment: P = 0.0001, 90 days posttransplant: P < 0.001, after 2 vaccinations: P < 0.001, 1-year posttransplant, after 3 vaccinations: P < 0.001, Pearson χ2 test). These observations aligned with a recovery of T-cell clonal diversity following vaccination that had been decreased in the immediate posttransplant period (Fig. 5C). Consistent with these findings, a higher proportion of shared TCR β clonotype groups across the 13 patients was observed after vaccination compared with the early posttransplant period, predominantly at 1-year posttransplant after 3 vaccinations (Fig. 5D, Materials and Methods). Eighty-two clonotype clusters were observed across the patient cohort, of which 4 (69 β TCRs) were detected in more than 6 patients after 3 vaccinations (Fig. 5D).
Figure 5.

T-cell receptor clonality and α/β TCR clonotype sharing following DC/MM fusion vaccination after auto-HCT. A, T-cell receptor clonality at single-cell resolution at the indicated time points is portrayed with distinct colors in a two-dimensional uniform manifold approximation and projection (UMAP) of all T cells. T cells with TCR frequencies between 1 and 4, 5 and 20, and >20 are marked with blue (≥1), red (>4), and yellow (>20), respectively. B, Boxplots portraying the proportions of paired α/β TCRs at different clonotype-frequency thresholds (Unexpanded: α/β TCR frequency = 1, Expanded: α/β TCR frequency: 1≤ clones ≤ 4, 4 < clones ≤ 20, and 20 < clones) in the CD8 activated and T effector memory cells per distinct patient and time point. Each circle represents the TCR frequency normalized by the number of T cells per sample. Significantly different proportions of postvaccination time points compared with prevaccination (enrollment) are denoted with * (*, P < 0.05; **, P < 0.01; Dirichlet multinomial regression). A statistically significant increase in the highly expanded clonotypes (TCR-clonotype freq > 20) of CD8 effector memory T cells postvaccination as compared with prevaccination levels is observed (enrollment vs. after 2 vaccinations: P: 1.38 × 10−31, enrollment vs. 1 year posttransplant, after 3 vaccinations: P: 2.7 × 10−7, 90 days posttransplant vs. after 2 vaccinations: P: 0.049, Dirichlet multinomial regression). C, T-cell receptor Shannon diversity index per timepoint for the 13 profiled patients undergoing vaccination. (t1-t2P = 0.00049, t1-t3P = 0.36, t1-t4P = 0.64, t2-t3P = 0.016, t2-t4P = 0.092, t3-t4P = 0.3, two-tailed Wilcoxon signed-rank test adjusted P values using Benjamini–Hochberg false discovery rate). A recovery of T-cell clonal diversity following vaccination that was decreased in the immediate posttransplant period is portrayed. D, TCR clonotype-sharing among the 13 patients analyzed is portrayed at the indicated time points and with the associated quality of response noted. The number (%) of shared β TCR sequence clusters distributed across the patient cohort is depicted in a clear (low) to a red (high) color gradient. The actual number (%) of the shared clonotype clusters among the patients (>20%) is depicted in the relevant pairwise comparisons among patients. The sequence logos (right) represent 69 consensus β TCR sequences with high similarity in more than 6 patients, corresponding to 4 TCR clonotype clusters, 1 year posttransplant and after 3 vaccinations.
Discussion
There has been renewed interest in the potential for cancer vaccines to induce tumor-specific immunity within the native T-cell repertoire through the effective physiologic presentation of cancer antigens. The DC/tumor fusion vaccine has the potential advantages of inducing broad immunity against multiple antigens, including shared epitopes and neoepitopes that captures tumor heterogeneity (21, 22). Administration of cancer vaccines during lymphopoietic reconstitution posttransplant associates with enhancement of multiple myeloma–specific immunity, targeting of residual disease, and prolongation of response (30).
This multicenter randomized trial examined the clinical and immunologic impact of a personalized DC-based cancer vaccine administered with GM-CSF posttransplant in conjunction with lenalidomide maintenance, as compared to lenalidomide maintenance alone or with GM-CSF. Analysis of an initial cohort of 24 patients from the vaccine arm showed a statistically significant progressive and durable expansion of multiple myeloma–specific CD8 T cells following vaccination persisting at 1-year posttransplant. In contrast, a statistically significant expansion of multiple myeloma–specific CD8 T cells was absent in 21 patients following lenalidomide and GM-CSF or lenalidomide maintenance alone. At 1-year posttransplant, the vaccine arm showed a statistically higher median percentage of multiple myeloma–specific CD8 T cells of 5.80%, as compared with 0.70% for the combined nonvaccine group. Of note, the use of lenalidomide maintenance alone in the context of posttransplant lymphopoietic reconstitution did not result in the expansion of tumor-reactive clones in the peripheral circulation.
Consistent with these observations, single-cell transcriptomics from an initial subset of vaccinated patients revealed the expansion of CD4 and CD8 T cells, the recovery of clonal diversity, and the emergence of dominant TCRs within the activated CD8 T-cell compartment (clonotype frequency > 20) corresponding to clonal expansion after vaccination. At 1 year, TCR signature clustering was observed within the CD8 effector memory and activated T-cell compartments in vaccinated patients. Concordance of dominant TCR signatures was observed across the vaccinated patient population and is indicative of shared immunogenic targets. These findings are consistent the emergence of dominant T-cell clones that may correlate with the presence of tumor-specific T cells and provide a unique platform to interrogate potential targets mediating long-term response. While transcriptomic analysis was not performed on the samples from the maintenance alone arm, the findings from patients undergoing vaccination indicative of clonal expansion of CD8 compartment are consistent with the expansion of multiple myeloma–reactive T cells observed in the vaccine but not maintenance alone arm.
Although vaccine-mediated expansion of multiple myeloma–reactive T cells was observed at 1 year, this did not coincide with a statistically significant increase in the percentage of patients achieving CR at 1 year. The percentage of patients achieving CR + VGPR in the vaccine and nonvaccine arms was 85% and 77%, respectively, not reaching statistical significance. The percentage of patients who converted from partial response posttransplant to CR at 1 year was 34.8% and 27.3%, respectively, which also did not reach statistical significance. Vaccination in conjunction with lenalidomide maintenance was well-tolerated without manifestations of autoimmunity. The 2-year PFS did not demonstrate a statistically significant difference between the vaccine and nonvaccine arms. The clinical findings are limited by sample size in the study arms and the heterogeneity of the patient populations. Of note, 65% of the patient population met criteria for high-risk disease by cytogenetic classification that may have impacted response to immunotherapy. Importantly, the full clinical impact of vaccination and comparative changes in antitumor immunity will likely require longer follow-up.
Previous studies of cancer vaccines have shown an association between vaccine-mediated immune response and long-term impact on disease-free survival (31, 32). In the current trial, the durable expansion multiple myeloma–reactive T cells was not associated with a statistically significant improvement of CR rate at 1 year or PFS. Similarly, a recently reported phase II randomized trial of an idiotype based vaccine post-transplant for multiple myeloma, as compared with a KLH control, demonstrated upregulation of genes associated with memory and cytotoxicity and downregulation of regulatory T cell function in the vaccine arm without a statistically significant impact on the early response rate (33).
CAR T-cell therapy has shown dramatic efficacy in patients with advanced multiple myeloma associated with high rates of response, clearance of minimal residual disease, and prolonged remissions (8, 9). However, the supraphysiologic mechanism of T-cell activation may be associated with exhaustion, loss of persistence, and subsequent tumor escape (34). In contrast, cancer vaccines employ physiologic stimulation of the native T-cell repertoire to establish long-term memory but are dependent on the functional competence of the effector cell compartment and are countered by the surrounding immunosuppressive tumor microenvironment. Therapeutic efficacy of vaccination may be mitigated by the upregulation of negative costimulatory receptors on T-cell populations indicative of an exhausted phenotype or the development of terminal differentiation leading to senescence and apoptosis (11, 12). In addition, elements within the immunologic milieu, including accessory cells (myeloid-derived suppressor cells and regulatory T cells), immunosuppressive factors (indoleamine 2,3-dioxygenase, IL10), and metabolic regulation (adenosine) may neutralize tumor-specific populations.
While expansion of the MM-reactive T cells may be sufficient to buffer disease progression in settings of greater immune competence, vaccine-mediated modulation of the T-cell repertoire may require combinatorial strategies that target critical aspects of the immunosuppressive tumor microenvironment. For example, vaccine efficacy is being examined in the context of checkpoint inhibition and immunoregulatory agents (35, 36). In addition, engineered T-cell platforms such as CAR T cells are subject to resistant mechanisms but may synergize with vaccination by providing a pool of functionally competent T cells receptive to vaccine-induced physiologic stimulus for T-cell expansion and activation (37–40). Similarly, T-cell engagers (TCE) have demonstrated significant potency in multiple myeloma by creating a synapse between activated T cells and multiple myeloma cells (41). This effect may be further augmented and potentially prolonged through the introduction of vaccine-educated T cells as a substrate for TCE-mediated tumor localization and TCR-mediated lysis. As such, we have demonstrated that administration of the DC/MM hybridomas posttransplant induces the durable expansion of multiple myeloma–reactive T cells against shared epitopes and provides a critical step in the development of combination immunotherapy reliant on multiple myeloma–specific effector cells.
An important objective of this study was to determine the practicality of performing a personalized cellular immunotherapy trial as a multi-institutional academic endeavor sharing unique national expertise in cell production, vaccine characterization, and platforms for immune response analysis. Site-specific vaccine generation mirrors the process by which hematopoietic stem cell transplantation is accomplished. A critical factor is the development of clear criteria for site training and validation, shared standards of practice and reagents for vaccine production, and centralized verification of vaccine characterization. For the current study, vaccine production was successfully completed for 63 of 68 participants across 18 different centers. The trial sets an important precedent for cell-based therapeutics in an academic framework that will facilitate combined or sequential interventions for immune-based therapy for cancer.
Supplementary Material
Study representation of underserved communities
Supplemental Figure 1: (A) Progression Free Survival Kaplan Meier Curve. (B) Overall Survival Kaplan Meier Curve.
Acknowledgments
Support for this study was provided by grants #U10HL069294 and #U24HL138660 to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute and the National Cancer Institute along with contributions by Celgene Corporation and the Multiple Myeloma Research Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the abovementioned parties.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
N. Shah reports grants from Celgene/BMS, Janssen, Bluebird Bio, Sutro Biopharma, Teneobio, Poseida, Nektar, and Precision Biosciences, and other support from GSK, Amgen, Indapta Therapeutics, Sanofi, CareDx, Kite, Karyopharm, Oncopeptides, CSL Behring, Allogene, and AstraZeneca outside the submitted work. K. Anderson reports personal fees from Pfizer, AstraZeneca, and Janssen and other support from C4 Therapeutics, Dynamic Cell Therapies, Window Therapeutics, Starton Therapeutics, NextRNA, and Oncopep outside the submitted work. B. Dhakal reports grants and personal fees from Sanofi, Janssen, GSK, and BMS; personal fees from Genentech, Pfizer, Arcellx, and Karyopharm; and grants from Carsgen outside the submitted work. L. Holmberg reports grants from Seattle Genetics, Sanofi, Millennium-Takeda, Bristol-Myers Squibb, Merck, and Iteos outside the submitted work as well as royalties from UpToDate. H.M. Lazarus reports personal fees from Partner Therapeutics and Bristol-Myers Squibb outside the submitted work. E. Malek served on advisory boards for Takeda, Celgene, Janssen, Sanofi, Karyopharm, and Adaptive Biotechnology; is a speaker for Amgen, Takeda, Bristol-Myers Squibb, Karyopharm, and Adaptive Biotechnology; and received research funding from Medpacto Pharmaceuticals. P. McCarthy reports other support from Bristol-Myers Squibb, Janssen, Oncopeptides, Karyopharm, Starton, Partner Therapeutics, Takeda, BeiGene, HSC Acquisition, and GSK outside the submitted work. D. McKenna reports grants from NIH during the conduct of the study. A. Nooka reports personal fees from Adaptive Biotechnologies, Amgen, Beyond Spring, Bristol-Myers Squibb, Cellectar Biosciences, GlaxoSmithKline, Janssen, Karyopharm, Oncopeptides, ONK Therapeutics, Pfizer, Sanofi, Secura Bio, and Takeda outside the submitted work. N. Munshi reports personal fees from AbbVie, Adaptive, Amgen, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Janssen, Legend, Novartis, Pfizer, Takeda, and Dana-Farber Cancer Institute and personal fees and other support from OncoPep outside the submitted work. L. O'Donnell reports grants from NIH/NCI/BMTCTN during the conduct of the study. J. Rosenblatt reports grants from MMRF and Celgene during the conduct of the study as well as grants and other support from BMS; grants from Sanofi and Karyopharm; other support from Attivare, Parexel, Clario/Bioclinica, Amgen, Merck, Kite Pharma, Partner Tx, Imaging Endpoint, Dava Oncology, and Wolters Kluwer Health, Inc.; and grants and nonfinancial support from Celgene outside the submitted work. In addition, J. Rosenblatt has a patent for PCT/US2021059199 pending. R. Soiffer reports personal fees from BMS, Vor, Gilead, Astellas, Smart Immune, Cugene, Jasper, BlueSphere Bio, and Daiichi Sankyo outside the submitted work. D. Stroopinsky reports other support from Takeda Pharmaceuticals during the conduct of the study. I.S. Vlachos reports grants from NCI, NHLBI, Singular Genomics, Harvard Stem Cell Institute, and Massachusetts Life Sciences Center and personal fees from Mosaic, Guidepoint Global, and Next RNA and outside the submitted work. E.K. Waller reports grants from BMTCTN during the conduct of the study. M.C. Pasquini reports grants from Novartis, Kite Pharma, and Janssen and grants and personal fees from Bristol-Myers Squibb outside the submitted work. D. Avigan reports grants from MMRF and CTN (NIHLBI) during the conduct of the study as well as grants and other support from Celgene and Kite Pharma; grants from Pharmacyclics; and other support from Juno, Partners TX, Karyopharm, BMS, Aviv MedTech Ltd., Takeda, Legend Bio Tech, Chugai, Caribou Biosciences, Janssen, Parexel, Sanofi, and Kowa outside the submitted work. In addition, D. Avigan has as a patent for Personalized Cancer Vaccine and CAR T cells (PCT/US2021/059199) pending. No disclosures were reported by the other authors.
Authors' Contributions
D.J. Chung: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. N. Shah: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. J. Wu: Data curation, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. B. Logan: Investigation, methodology, writing–review and editing. L. Bisharat: Investigation, methodology, writing–review and editing. N. Callander: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. G. Cheloni: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. K. Anderson: Investigation, methodology, writing–review and editing. T. Chodon: Investigation, methodology, writing–review and editing. B. Dhakal: Investigation, methodology, writing–review and editing. S. Devine: Investigation, methodology, writing–review and editing. P. Somaiya Dutt: Investigation, methodology, writing–review and editing. Y. Efebera: Investigation, methodology, writing–review and editing. N. Geller: Investigation, methodology, writing–review and editing. H. Ghiasuddin: Investigation, methodology, writing–review and editing. P. Hematti: Investigation, methodology, writing–review and editing. L. Holmberg: Investigation, methodology, writing–review and editing. A. Howard: Investigation, methodology, writing–review and editing. B. Johnson: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. D. Karagkouni: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. H.M. Lazarus: Investigation, methodology, writing–review and editing. E. Malek: Investigation, methodology, writing–review and editing. P. McCarthy: Investigation, methodology, writing–review and editing. D. McKenna: Investigation, methodology, writing–review and editing. A. Mendizabal: Investigation, methodology, writing–review and editing. A. Nooka: Investigation, methodology, writing–review and editing. N. Munshi: Investigation, methodology, writing–review and editing. L. O'Donnell: Investigation, methodology, writing–review and editing. A.P. Rapoport: Investigation, methodology, writing–review and editing. J. Reese: Investigation, methodology, writing–review and editing. J. Rosenblatt: Investigation, methodology, writing–review and editing. R. Soiffer: Investigation, methodology, writing–review and editing. D. Stroopinsky: Investigation, methodology, writing–review and editing. L. Uhl: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. I.S. Vlachos: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. E.K. Waller: Investigation, methodology, writing–review and editing. J.W. Young: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. M.C. Pasquini: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. D. Avigan: Conceptualization, data curation, formal analysis, investigation, methodology, writing–original draft, writing–review and editing.
References
- 1. Richardson PG, Jacobus SJ, Weller EA, Hassoun H, Lonial S, Raje NS, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. N Engl J Med 2022;387:132–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med 2012;366:1770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer 2020;1:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakamura K, Smyth MJ, Martinet L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020;136:2731–40. [DOI] [PubMed] [Google Scholar]
- 5. Alrasheed N, Lee L, Ghorani E, Henry JY, Conde L, Chin M, et al. Marrow-infiltrating regulatory T cells correlate with the presence of dysfunctional CD4+PD-1+ cells and inferior survival in patients with newly diagnosed multiple myeloma. Clin Cancer Res 2020;26:3443–54. [DOI] [PubMed] [Google Scholar]
- 6. Cohen AD, Raje N, Fowler JA, Mezzi K, Scott EC, Dhodapkar MV. How to train your T cells: overcoming immune dysfunction in multiple myeloma. Clin Cancer Res 2020;26:1541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dhodapkar MV, Sexton R, Das R, Dhodapkar KM, Zhang L, Sundaram R, et al. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood 2015;126:2475–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021;384:705–16. [DOI] [PubMed] [Google Scholar]
- 9. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 2021;398:314–24. [DOI] [PubMed] [Google Scholar]
- 10. Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med 2005;11:1230–7. [DOI] [PubMed] [Google Scholar]
- 11. Avigan D, Rosenblatt J. Vaccine therapy in hematologic malignancies. Blood 2018;131:2640–50. [DOI] [PubMed] [Google Scholar]
- 12. Liegel J, Weinstock M, Rosenblatt J, Avigan D. Vaccination as immunotherapy in hematologic malignancies. J Clin Oncol 2021;39:433–43. [DOI] [PubMed] [Google Scholar]
- 13. Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 2019;10:5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santos PM, Butterfield LH. Dendritic cell-based cancer vaccines. J Immunol 2018;200:443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med 2021;27:515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gress RE, Emerson SG, Drobyski WR. Immune reconstitution: how it should work, what's broken, and why it matters. Biol Blood Marrow Transplant 2010;16:S133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang H-B, Cai L, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 2011;117:788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chung DJ, Pronschinske KB, Shyer JA, Sharma S, Leung S, Curran SA, et al. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res 2016;4:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Noonan K, Rudraraju L, Ferguson A, Emerling A, Pasetti MF, Huff CA, et al. Lenalidomide-induced immunomodulation in multiple myeloma: impact on vaccines and antitumor responses. Clin Cancer Res 2012;18:1426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luptakova K, Rosenblatt J, Glotzbecker B, Mills H, Stroopinsky D, Kufe T, et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol Immunother 2013;62:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res 2013;19:3640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci Transl Med 2016;8:368ra171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hurlbert SH. The nonconcept of species diversity: a critique and alternative parameters. Ecology 1971;52:577–86. [DOI] [PubMed] [Google Scholar]
- 27. Jost L. Partitioning diversity into independent alpha and beta components. Ecology 2007;88:2427–39. [DOI] [PubMed] [Google Scholar]
- 28. Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM. Analyzing the mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol 2020;38:1194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 2017;33:3645–7. [DOI] [PubMed] [Google Scholar]
- 30. Lacy MQ, Mandrekar S, Dispenzieri A, Hayman S, Kumar S, Buadi F, et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am J Hematol 2009;84:799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv 2018;2:224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia 2017;31:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qazilbash MH, Saini NY, Cha S-C, Wang Z, Stadtmauer EA, Baladandayuthapani V, et al. A randomized phase 2 trial of idiotype vaccination and adoptive autologous T-cell transfer in patients with multiple myeloma. Blood 2022;139:1289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Milone MC, Xu J, Chen S-J, Collins MA, Zhou J, Powell DJ, et al. Engineering enhanced CAR T-cells for improved cancer therapy. Nat Cancer 2021;2:780–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stroopinsky D, Liegel J, Bhasin M, Cheloni G, Thomas B, Bhasin S, et al. Leukemia vaccine overcomes limitations of checkpoint blockade by evoking clonal T cell responses in a murine acute myeloid leukemia model. Haematologica 2021;106:1330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nahas MR, Stroopinsky D, Rosenblatt J, Cole L, Pyzer AR, Anastasiadou E, et al. Hypomethylating agent alters the immune microenvironment in acute myeloid leukaemia (AML) and enhances the immunogenicity of a dendritic cell/AML vaccine. Br J Haematol 2019;185:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemoine J, Ruella M, Houot R. Born to survive: how cancer cells resist CAR T cell therapy. J Hematol Oncol 2021;14:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van de Donk NWCJ, Themeli M, Usmani SZ. Determinants of response and mechanisms of resistance of CAR T-cell therapy in multiple myeloma. Blood Cancer Discov 2021;2:302–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood 2018;132:1134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020;367:446–53. [DOI] [PubMed] [Google Scholar]
- 41. Ravi G, Costa LJ. Bispecific T-cell engagers for treatment of multiple myeloma. Am J Hematol 2023;98:S13–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study representation of underserved communities
Supplemental Figure 1: (A) Progression Free Survival Kaplan Meier Curve. (B) Overall Survival Kaplan Meier Curve.
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.