

Abstract
R‐loops represent a major source of replication stress, but the mechanism by which these structures impede fork progression remains unclear. To address this question, we monitored fork progression, arrest, and restart in Saccharomyces cerevisiae cells lacking RNase H1 and H2, two enzymes responsible for degrading RNA:DNA hybrids. We found that while RNase H‐deficient cells could replicate their chromosomes normally under unchallenged growth conditions, their replication was impaired when exposed to hydroxyurea (HU) or methyl methanesulfonate (MMS). Treated cells exhibited increased levels of RNA:DNA hybrids at stalled forks and were unable to generate RPA‐coated single‐stranded (ssDNA), an important postreplicative intermediate in resuming replication. Similar impairments in nascent DNA resection and ssDNA formation at HU‐arrested forks were observed in human cells lacking RNase H2. However, fork resection was fully restored by addition of triptolide, an inhibitor of transcription that induces RNA polymerase degradation. Taken together, these data indicate that RNA:DNA hybrids not only act as barriers to replication forks, but also interfere with postreplicative fork repair mechanisms if not promptly degraded by RNase H.
Keywords: replication stress, R‐loops, RNase H, transcription‐replication conflicts
Subject Categories: DNA Replication, Recombination & Repair; RNA Biology
Toxic RNA:DNA hybrids not only act as roadblocks for replication fork progression, but also interfere with fork restart after HU‐/MMS‐induced stalling.
Introduction
DNA replication and transcription are essential cellular processes that operate on the same DNA template and inevitably interfere with each other. Transcription‐replication conflicts (TRCs) represent the major source of spontaneous genomic instability in all organisms, from bacteria to human (Gomez‐Gonzalez & Aguilera, 2019). In eukaryotes, TRCs occur most frequently when the transcription and replication machineries collide head‐on. Cells deploy effective strategies to avoid these conflicts (Kemiha et al, 2021; Lalonde et al, 2021). However, pathological situations that alter the coordination between replication and transcription invariably increase the frequency of TRCs. For instance, altered transcription and replication origin usage caused by deregulated oncogenic pathways induce replication‐dependent genomic instability and contributes to cancer development (Stork et al, 2016; Macheret & Halazonetis, 2018; Bowry et al, 2021).
A large body of evidence indicates that three‐stranded nucleic acid structures called R‐loops play a central role in TRCs (Brickner et al, 2022). R‐loops consist of an RNA:DNA hybrid and a displaced single‐stranded DNA (ssDNA). They form during transcription when the nascent RNA reanneals with its template DNA, leaving the non‐template DNA unpaired (Garcia‐Muse & Aguilera, 2019; Brickner et al, 2022). Two different classes of R‐loops have been described at transcription start sites (TSS) and transcription termination sites (TTS), occupying up to 5% of the human genome (Sanz Lionel et al, 2016; Castillo‐Guzman & Chédin, 2021). These structures play important physiological roles such as gene expression control, immunoglobulin class switch recombination, mitochondrial DNA replication or gene editing (Garcia‐Muse & Aguilera, 2019). However, when R‐loop homeostasis is perturbed, pathological R‐loops can alter chromatin structure and affect genome integrity (Garcia‐Muse & Aguilera, 2019).
A growing number of factors suppressing or resolving R‐loops have been identified, which includes nucleases, helicases, topoisomerases, RNA processing factors and chromatin modifiers (Brickner et al, 2022). RNase H1 and H2 are two main regulators of R‐loop homeostasis that specifically degrade the RNA moiety of RNA:DNA hybrids (Hyjek et al, 2019). RNase H1 is a conserved monomeric enzyme that is recruited to R‐loops via its interaction with RNA:DNA hybrids and with the ssDNA‐binding factor RPA on the displaced strand (Nguyen et al, 2017). RNase H2 is composed of three subunits called Rnh201RNASEH2A, Rnh202RNASEH2B and Rnh203RNASEH2C. Unlike RNase H1, RNase H2 is recruited to replication foci in vivo through the binding of its RNASEH2B subunit to PCNA (Bubeck et al, 2011). RNase H2 is also able to excise ribonucleoside monophosphates (rNMPs) incorporated into DNA, through a process called ribonucleotide excision repair (RER; Lazzaro et al, 2012; Reijns et al, 2012; Williams et al, 2016). In budding yeast, RNase H2 executes its essential functions in G2/M, whereas RNase H1 acts throughout the cell cycle (Lockhart et al, 2019). In the absence of RNase H2, rNMPs are aberrantly processed by DNA topoisomerase I (Top1), leading to deletions and double‐strand breaks (DSBs; Kim et al, 2011; Williams et al, 2013; Huang et al, 2017; Cerritelli et al, 2020).
Senataxin is another well‐characterized enzyme involved in the regulation of R‐loops and TRCs (Groh et al, 2017). Mutations in Senataxin have been implicated in neurodegenerative disorders and are associated with altered R‐loop metabolism and deregulated transcription (Skourti‐Stathaki et al, 2011). Sen1, the budding yeast ortholog of Senataxin, has been proposed to promote the dissolution of RNA:DNA hybrids (Mischo et al, 2011; Alzu et al, 2012; San Martin‐Alonso et al, 2021). Like RNase H2, Sen1 associates with the replisome (Appanah et al, 2020). Recent evidence also indicates that Sen1 removes RNAPII at sites of transcription‐transcription conflicts (Zardoni et al, 2021; Aiello et al, 2022).
A key question in the field is what distinguishes harmful R‐loops from physiological ones in the context of transcription‐replication conflicts. It is worth noting that the most deleterious TRCs result from head‐on collisions (Hamperl et al, 2017; Lalonde et al, 2021) and that complex eukaryotic genomes are functionally organized to ensure that active genes are replicated in a codirectional manner (Petryk et al, 2016; Chen et al, 2019; Promonet et al, 2020). A recent genome‐wide study of the distribution of R‐loops and replication stress marks in the human genome showed that the vast majority of R‐loops do not interfere with DNA replication under normal growth conditions (Promonet et al, 2020). Interestingly, the R‐loops present at the TTS of highly expressed head‐on genes activate the checkpoint kinase ATR, but cause DSBs and γ‐H2AX only in Top1‐deficient cells (Tuduri et al, 2009; Promonet et al, 2020). Altogether, these data indicate that replication forks pause when they encounter converging transcription, presumably because of the accumulation of positive DNA supercoiling, but do not break or collapse unless they are unable to resolve topological constraints (Promonet et al, 2020). These results also indicate that cells are very effective at stabilizing and restarting forks encountering transcription and R‐loops. This is likely due to the fact that stressed forks activate ATRMec1, which orchestrates fork repair mechanisms and promotes the displacement of RNA polymerases blocking fork progression (Im et al, 2014; Poli et al, 2016; Landsverk et al, 2020; Hurst et al, 2021).
In a head‐on orientation, the RNA:DNA hybrid component of an R‐loop is located on the opposite strand to the converging replicative helicase and does not impede replication, unless the displaced ssDNA strand has the ability to form fork‐blocking secondary structures such as G‐quartets (Kumar et al, 2021). This raises the question of how R‐loops interfere with replication fork progression. In principle, R‐loops and associated RNAPII could act as roadblocks (Chappidi et al, 2020; Zardoni et al, 2021; Aiello et al, 2022). Alternatively, RNA:DNA hybrids may interfere with fork restart mechanisms acting behind stressed forks (Barroso et al, 2019; Šviković et al, 2019). The existence of postreplicative RNA:DNA hybrids is supported by a recent electron microscopy study showing that RNA:DNA hybrids are found behind and not ahead of arrested forks at bacterial TRCs (Stoy et al, 2023).
The mechanisms by which cells process and restart arrested forks have been extensively studied in the presence of genotoxic agents such as hydroxyurea (HU) or methyl methanesulfonate (MMS). It is now well established that stressed forks are extensively remodeled through a process involving fork reversal, in which nascent DNA strands reanneal to form a four‐way structure resembling a Holliday junction (Neelsen & Lopes, 2015). Fork reversal is mediated by the RAD51 recombinase and by different DNA translocases, including SMARCAL1, HLTF and ZRANB3 (Quinet et al, 2017; Liu et al, 2023). Since the regressed arm resembles a one‐ended DSB, it is susceptible to nucleolytic degradation. To prevent excessive degradation, nascent DNA is protected by homologous recombination (HR) factors such as BRCA1 and BRCA2 (Tye et al, 2020). As with DSB repair, the controlled resection of nascent DNA at stalled forks may contribute to HR‐mediated fork restart (Pasero & Vindigni, 2017; Teixeira‐Silva et al, 2017). This resection process is initiated by MRE11 and is extended by long‐range resection nucleases (Pasero & Vindigni, 2017). Evidence from budding yeast also indicates that fork resection is important to load cohesin and to promote sister‐chromatid exchanges at stalled forks (Tittel‐Elmer et al, 2012; Delamarre et al, 2020).
Here, we have monitored the impact of RNA:DNA hybrids on fork progression, arrest and restart in budding yeast and human cells lacking RNase H activity. In yeast, we found that RNase H deficient cells were unable to generate RPA‐coated ssDNA gaps at stalled forks and efficiently resume DNA synthesis after replication stress. RPA loading was restored by the RNase H2 RED allele, which is defective for RER, but proficient in degrading the RNA in RNA:DNA hybrids. This suggests that fork processing is perturbed by the persistence of long RNA:DNA hybrids at stressed forks and not by the accumulation of rNMPs in DNA. In addition, we found that fork resection was impaired in RNase H2‐deficient human cells and was rescued by triptolide, which inhibits transcription by promoting the degradation of initiating RNAPII. Altogether, these data indicate that replication‐impeding R‐loops and RNA polymerases are removed in a coordinated manner to promote fork processing. In the absence of RNase H, we propose that RNA:DNA hybrids accumulate in front of the fork and are bypassed by the replisome, generating post‐replicative RNA:DNA hybrids that interfere with fork processing and restart.
Results
RNase H‐deficient budding yeast cells are hypersensitive to replication stress
To investigate the requirement for RNase H1 and RNase H2 in cells exposed to replication stress, we first compared the growth of wild type (WT), rnh1Δ, rnh201Δ, and rnh1Δ rnh201Δ cells in the presence of two genotoxic agents, MMS and HU. MMS blocks replication forks by alkylating DNA and HU slows down DNA synthesis by depleting dNTP pools and inducing an oxidative stress (Koc et al, 2004; Poli et al, 2012; Somyajit et al, 2017; Andrs et al, 2023). All four strains grew at the same rate in the absence of drugs. However, the growth of the rnh1Δ rnh201Δ double mutant was severely impaired by chronic exposure to low doses of MMS (Fig 1A) or HU (Fig EV1A), even though the growth of single mutants was not affected by either drug. To assess the effect of MMS and HU on S‐phase progression in the rnh1Δ rnh201Δ double mutant, we next arrested cells in G1 with α‐factor and released them into S phase in the presence or absence of a low dose of MMS or HU to induce mild replication stress without blocking cell‐cycle progression. Cells were harvested at indicated time points and DNA content was analyzed by flow cytometry. In untreated conditions, rnh1Δ rnh201Δ cells progressed through S phase with wild‐type kinetics (Figs 1B and EV1B). When wild type cells were exposed to either 0.015% MMS or 25 mM HU, bulk DNA synthesis was significantly delayed but cells managed to complete mitosis within 240 min (Figs 1B and EV1B). In contrast, rnh1Δ rnh201Δ mutants accumulated in G2/M (Figs 1B and EV1B; arrows), which may reflect the persistence of unreplicated regions. Collectively, these data suggest that RNase H1 and RNase H2 are dispensable for DNA replication under unchallenged growth conditions, but are critical to complete the cell cycle under mild replication stress, which is consistent with an earlier study (Meroni et al, 2019).
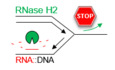
Figure 1. RNase H enzymes are required for the resection and restart of stalled replication forks in budding yeast.
- Growth of wild type and RNase H‐deficient cells on synthetic complete (SC) medium ±0.02% MMS. Spots correspond to 1:10 serial dilutions.
- Flow‐cytometry analysis of cell‐cycle progression of wild‐type and RNase H‐deficient cells exposed to MMS. Cells were synchronized in G1 with α‐factor and released into S phase in the presence of 0.015% MMS. Changes in DNA content through the cell cycle were monitored by flow cytometry at the indicated times. The arrows show the differential accumulation of cells in G2/M.
- DNA combing analysis of fork progression in the presence of MMS. Exponentially growing cells were synchronized in G1 with α‐factor and released in S phase in the presence of 0.033% MMS. Newly replicated DNA was labeled with BrdU for 45 min, then MMS was removed and fork restart was measured by DNA combing after 30 min recovery.
- Distribution of BrdU track length. Box, 25–75 percentile range. Whiskers, 10–90 percentiles range. Median is indicated in kb. ***P < 0.001, Mann–Whitney rank sum test. The DNA combing experiment was repeated twice (n = 2 biological replicates) with similar results, one representative experiment is shown. WT MMS (n = 184) and recovery (n = 99), rnh1Δ rnh201 MMS (n = 275) and recovery (n = 202).
- ChIP‐qPCR analysis of RPA enrichment around ARS306 and ARS607 in cells released for 30 min in the presence of 0.1% MMS. RPA enrichment was normalized to four unreplicated regions. Mean and SEM are indicated (n = 3 biological replicates). For statistical analysis, two‐way ANOVA was applied. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Source data are available online for this figure.
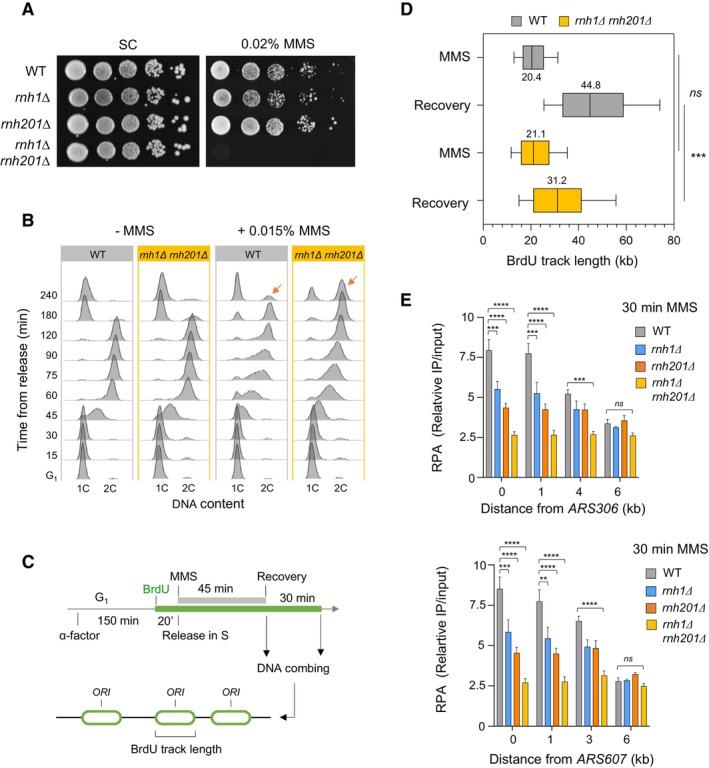
Figure EV1. RNase H enzymes are required for optimal growth and cell cycle progression in the presence of HU.
-
AGrowth of wild type and RNase H‐deficient cells on synthetic complete (SC) medium ± 50 mM HU. Spots correspond to 1:10 serial dilutions. Data for the SC + 50 mM HU used here is identical to the one in Fig 2A (WT, rnh1Δ, rnh201Δ, rnh1Δ rnh201Δ).
-
BFlow‐cytometry analysis of DNA content in wild‐type and rnh1Δ rnh201Δ cells synchronized in G1 with α‐factor and released into S phase in the presence of 25 mM HU. Arrows indicate cells accumulating in G2/M.
-
CChIP‐qPCR analysis of RPA enrichment at HU‐arrested forks in wild‐type, rnh1Δ, rnh201Δ and rnh1Δ rnh201Δ cells released synchronously into S phase for 60 min in the presence of 200 mM HU. Primer pairs correspond to regions located 1, 4, and 6 kb upstream of ARS306 and 1, 3 and 6 kb downstream of ARS607. RPA enrichment was normalized to four unreplicated regions. Mean and SEM are indicated (n = 3 biological replicates). Two‐way ANOVA was applied. *P < 0.05; **P < 0.01; ****P < 0.0001.
- D
-
E, FChIP‐qPCR analysis of Rnh1‐PK6 and Rnh201‐PK6 enrichment near ARS306 and ARS607 in wild type cells. Cells were released from G1 into S phase in medium containing 200 mM HU and were collected at the indicated timepoints. Mean and SEM are indicated (n = 3 biological replicates). Two‐way ANOVA was applied. *P < 0.05; **P < 0.01; ****P < 0.0001.
-
GChIP‐qPCR analysis of RPA enrichment at HU‐arrested forks in the indicated cells released synchronously into S phase for 60 min in the presence of 200 mM HU. Experiments were performed as indicated in panel (C) (n = 2).
-
HChIP‐qPCR analysis of relative RPA levels at ARS306 and ARS607 in cells exposed to HU. Data from Fig 2B are expressed as relative levels in mutants and wild type cells. SEM are indicated (n = 3 biological replicates).
RNase H1 and H2 are required for the recovery of stressed forks
To investigate the role of RNase H1 and H2 under replication stress conditions, wild type cells and RNase H mutants were arrested in G1 with α‐factor. Cells were then released into S phase in the presence of BrdU to label newly replicated DNA and replication fork progression was monitored by DNA combing (Fig 1C), as described previously (Tourrière et al, 2017). Cells were first exposed for 45 min to a high dose (0.033%) of MMS during BrdU labelling to induce an acute fork arrest. DNA combing analysis revealed that this treatment impeded DNA synthesis to the same extent in both wild‐type and rnh1Δ rnh201Δ strains (Fig 1D). Then, MMS was washed away and the length of replicated tracks was measured 30 min after MMS removal. BrdU tracks were significantly shorter in rnh1Δ rnh201Δ mutants after MMS removal relative to wild type cells (Fig 1D), suggesting that RNase H activity is required for the efficient restart of MMS‐arrested forks.
The formation of RPA‐coated ssDNA at stressed forks depends on RNase H
Fork restart involves extensive remodeling of replication intermediates and nascent chromatin. Central to this process is the controlled resection of nascent DNA, which depends on the MRX complex and promotes sister chromatid exchanges (Delamarre et al, 2020). To determine whether RNase H activity is required for the resection of nascent DNA in yeast cells exposed to HU and MMS, we next monitored the formation of RPA‐coated ssDNA at stressed forks. Cells were arrested in G1 with α‐factor and released synchronously into S phase for the indicated times. The presence of RPA‐coated ssDNA, which is indicative of Mre11‐dependent fork resection, was quantified by ChIP‐qPCR at forks progressing from two representative early replication origins (ARS306 and ARS607), as described previously (Delamarre et al, 2020). In HU‐ or MMS‐treated WT cells, RPA was detected up to 2–3 kb from these origins (Figs 1E, and EV1C and D), which correspond to the distance covered by forks under these replication stress conditions (Poli et al, 2012). Interestingly, levels of RPA‐coated ssDNA were strongly reduced in rnh201Δ cells and to a lesser extent in rnh1Δ cells (Figs 1E, and EV1C and D). Almost no RPA enrichment was detected at forks in the rnh1Δ rnh201Δ double mutant when cells were exposed to MMS (Figs 1E and EV1D) or HU (Fig EV1C and D). RNase H1 and RNase H2 were also detected on newly replicated DNA in HU‐treated cells (Fig EV1E and F). Together, these data indicate that RNase H1 and H2 act at stressed forks to stimulate the formation of RPA‐coated ssDNA and promote fork restart.
In addition to long RNA:DNA hybrids, RNase H1 and RNase H2 also degrade short stretches of ribonucleotides, such as RNA primers of Okazaki fragments (Hyjek et al, 2019). It has been recently reported that these RNA primers prevent the resection of nascent DNA at forks arrested at the RTS1 replication fork barrier in fission yeast and that this resection defect could be rescued in conditional mutants destabilizing the DNA polymerase α‐primase complex (Audoynaud et al, 2023). To determine whether Okazaki fragments could also prevent the formation of RPA‐coated ssDNA at HU‐arrested forks in Saccharomyces cerevisiae rnh1Δ rnh201Δ mutants, we next performed RPA ChIP‐qPCR experiments in control and rnh1Δ rnh201Δ cells bearing thermosensitive mutations of primase (pri1‐4 and pri2‐1). The destabilization of the DNA polymerase α‐primase complex reduced the amount of RPA‐coated ssDNA at HU‐arrested forks. However, it did not rescue the resection defect of rnh1Δ rnh201Δ cells (Fig EV1G). These data suggest that RNase H1 and H2 do not primarily act on Okazaki fragments to promote fork resection in HU‐arrested cells.
The RER activity of RNase H2 is dispensable for RPA loading at arrested forks
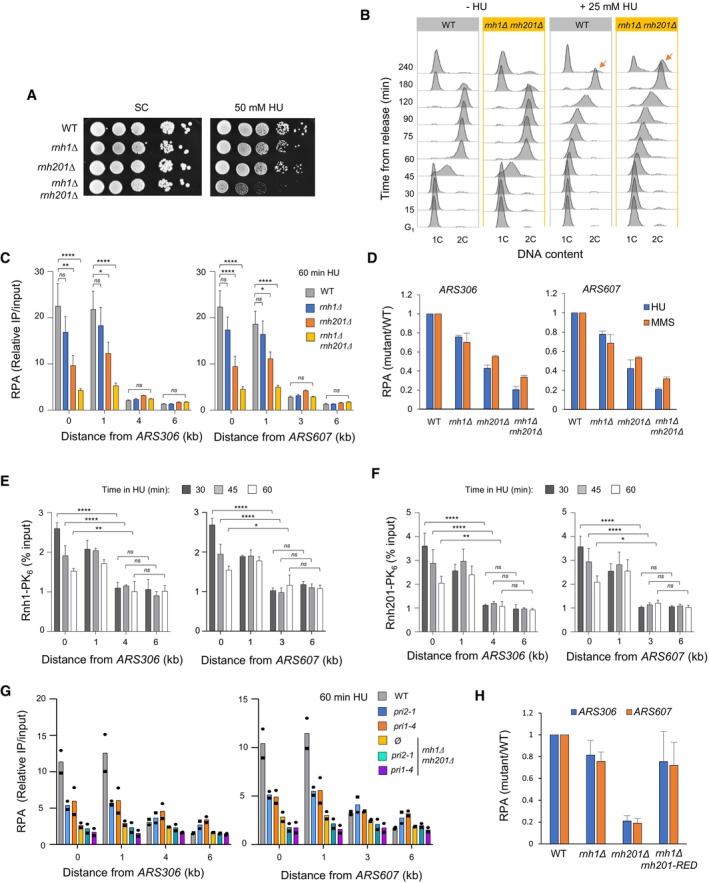
Unlike RNase H1, RNase H2 removes single ribonucleotides incorrectly incorporated into DNA during DNA synthesis through a process called RER (Williams et al, 2017; Nava et al, 2020). To determine whether RER defects could account for the lack of RPA‐coated ssDNA in HU‐arrested rnh1Δ rnh201Δ cells, we used rnh201‐RED, a separation‐of‐function allele of rnh201 that is unable to remove incorporated ribonucleotides but retains 40% of its activity against RNA:DNA hybrids (Cerritelli & Crouch, 2019). Unlike rnh1Δ rnh201Δ cells, the rnh1Δ rnh201‐RED mutant did not show increased sensitivity to HU and MMS (Fig 2A). Moreover, this mutant was proficient to form RPA‐coated ssDNA at stressed forks, to the same extent as in the rnh1Δ single mutant (Figs 2B and EV1H). The rnh1Δ rnh201‐RED mutant was also able to efficiently restart MMS‐arrested forks, unlike rnh1Δ rnh201Δ cells (Fig 2C). We therefore conclude that the fork restart defect of rnh1Δ rnh201Δ mutants stems from their inability to remove RNA:DNA hybrids rather than from the persistence of rNMPs embedded into genomic DNA.
Figure 2. The ribonucleotide excision activity of RNase H2 is dispensable for the processing of stalled forks in budding yeast.
- Growth of wild type and RNase H mutants on synthetic complete (SC) medium ± indicated drug. Spots correspond to 1:10 serial dilutions. Data for the SC and SC + 0.02%MMS used here are identical to the one in Fig 1A (WT, rnh1Δ, rnh201Δ, rnh1Δ rnh201Δ).
- ChIP‐qPCR analysis of RPA enrichment around ARS306 and ARS607 in cells released synchronously into S phase for 60 min in the presence of 200 mM HU. RPA enrichment was normalized to four unreplicated regions. Mean and SEM are indicated (n = 5 biological replicates). For statistical analysis, two‐way ANOVA was applied. **P < 0.01; ****P < 0.0001.
- DNA combing analysis of fork progression. Exponentially growing cells were synchronized in G1 with α‐factor and released into S phase in the presence of 0.1% MMS. Newly replicated DNA was labeled with BrdU for 45 min, then MMS was removed and fork restart was measured after 30 min of recovery. Graph depicts the distribution of BrdU track length. Box, 25–75 percentile range. Whiskers, 10–90 percentiles range. Median is indicated in kb. ***P < 0.001, Mann–Whitney rank‐sum test. The DNA combing experiment was repeated twice (n = 2 biological replicates) with similar results, one representative experiment is shown. WT MMS (n = 287) and recovery (n = 255), rnh1Δ rnh201 MMS (n = 212) and recovery (n = 248), rnh1Δ rnh201‐RED MMS (n = 291) and recovery (n = 147).
Source data are available online for this figure.
The replication checkpoint is fully functional in the absence of RNase H activity
The yeast Mec1ATR kinase is recruited to RPA‐coated ssDNA and activates the DNA replication checkpoint when the number of stressed forks reaches a critical threshold (Shimada et al, 2002; Bacal et al, 2018). Since RNase H activity is required to load RPA at stressed forks, we next monitored the ability of rnh1Δ rnh201Δ cells to activate the Mec1‐Rad53 pathway and repress late origins under replication stress conditions, using a genome‐wide approach. Wild type and rnh1Δ rnh201Δ cells were released synchronously into S phase for 60 min in the presence of 200 mM HU and variations in DNA copy number were determined at the genome‐wide level by deep sequencing (Fig EV2A), as reported previously (Müller et al, 2014; Fang et al, 2017). This analysis showed that late origins are efficiently repressed in rnh1Δ rnh201Δ cells, unlike in the rad53Δ mutant, used here as a control for checkpoint deficiency (Fig EV2B). Moreover, DNA combing analysis of origin usage in cells released synchronously into S phase for 90 min in the presence of HU revealed that the distance between initiation sites (inter‐origin distance) was identical in wild type and rnh1Δ rnh201Δ cells (~60 kb) in both cell types (Fig EV2C). This indicates that the lack of RNase H activity does not affect the kinetics of checkpoint activation nor the distribution of initiation events under replication stress conditions.
Figure EV2. RNase H activity is dispensable for the activation of early replication origins and the repression of late origins in the presence of HU.
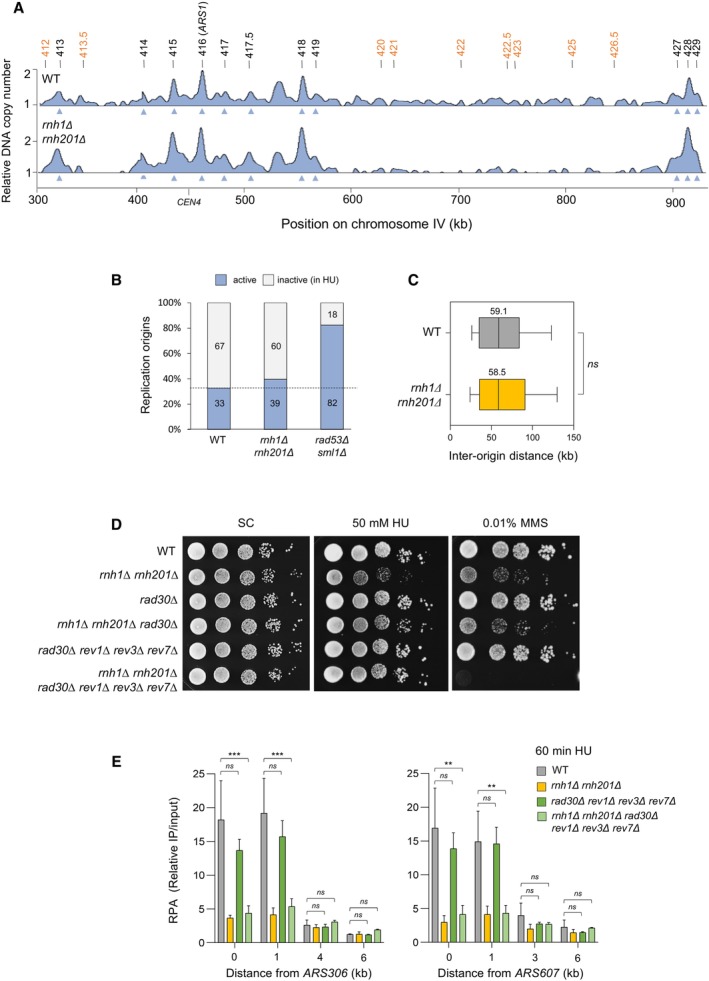
- Genome‐wide analysis of origin usage in wild‐type and rnh1Δ rnh201Δ cells released synchronously into S phase for 60 min in the presence of 200 mM HU. Relative DNA copy number was determined by deep sequencing as the ratio of normalized reads in HU and G1 cells. A representative region on chromosome IV is shown. Positions of early (black) and late (orange) origins are indicated. Arrowheads point to active origins.
- Number of active and inactive origins in the experiment shown in panel (A). The rad53Δ sml1Δ mutant is used as a positive control for the derepression of late origins in HU (Poli et al, 2012).
- Distribution of inter‐origin distances determined by DNA combing after releasing cells from G1 into S phase for 90 min in the presence of 200 mM HU. Box, 25–75 percentile range. Whiskers, 10–90 percentiles range. Median is indicated in kb. ns: not significant, Mann–Whitney rank‐sum test. The DNA combing experiment was repeated twice (n = 2 biological replicates) with similar results, one representative experiment is shown.
- Growth of the indicated strains on synthetic complete (SC) ± 50 mM HU or 0.01% MMS. Spots correspond to 1:10 serial dilutions.
- ChIP‐qPCR analysis of RPA enrichment around ARS306 and ARS607 in the indicated cells released synchronously into S phase for 60 min in the presence of 200 mM HU. RPA enrichment was normalized to four unreplicated regions. Mean and SEM are indicated (n = 3 biological replicates). For statistical analysis, two‐way ANOVA was applied. **P < 0.01; ***P < 0.001.
Fork restart defects in the absence of RNase H are not caused by translesion polymerases
It has been recently reported that the activity of translesion synthesis (TLS) DNA polymerases is toxic when rnh1Δ rnh201Δ cells are grown in the presence of HU, but not of MMS (Meroni et al, 2019). We confirmed that the slow growth of the rnh1Δ rnh201Δ mutant on HU is due at least in part to the activity of the TLS polymerase η, encoded by the RAD30 gene, as this growth defect was largely suppressed in the absence of RAD30 or all other TLS polymerases encoded by the REV1, REV3 and REV7 genes (Fig EV2D). However, the MMS sensitivity of the rnh1Δ rnh201Δ mutant was not suppressed by the absence of TLS polymerases, indicating that the suppression is specific to limiting dNTP pools (Fig EV2D). To determine whether TLS polymerases are responsible for the fork resection defect of rnh1Δ rnh201Δ cells in HU, we analyzed the formation of RPA‐coated ssDNA at forks arrested near ARS306 and ARS607 in the absence of all TLS polymerases (Fig EV2E). This analysis showed that TLS polymerases were dispensable for RPA enrichment at HU‐arrested forks and that the deletion of all TLS polymerase genes in the rnh1Δ rnh201Δ mutant did not restore stalled fork resection. We therefore conclude that the fork processing defect in the absence of RNase H1 and H2 is not caused by unscheduled TLS polymerase activity.
The overexpression of Sen1 rescues fork restart defects in rnh1Δ rnh201Δ cells
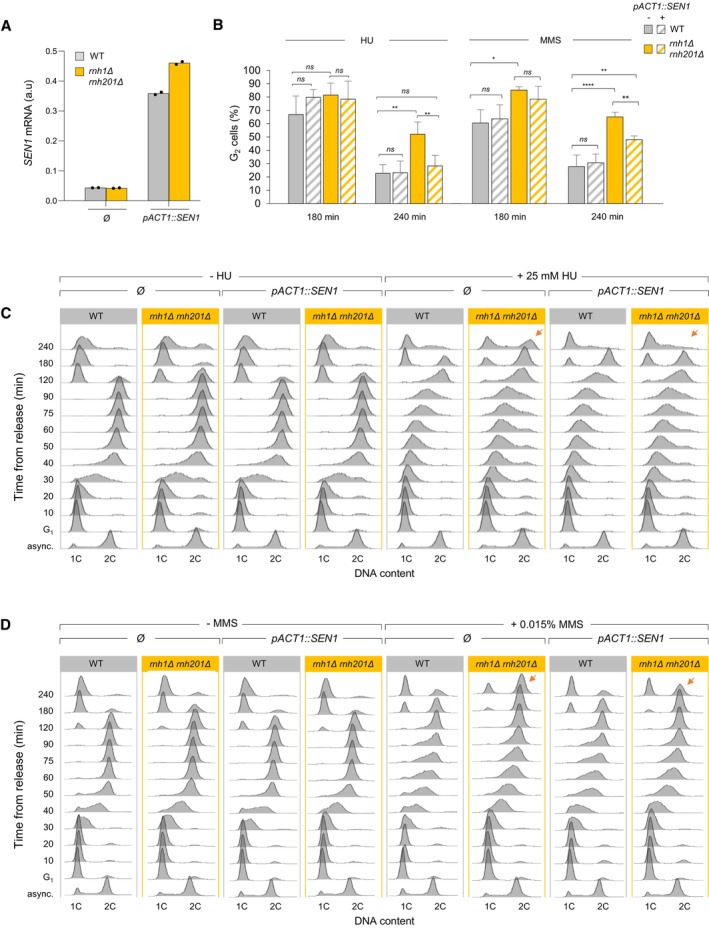
Our results suggest that the hypersensitivity of rnh1Δ rnh201Δ cells to HU and MMS is not due to ribonucleotides erroneously incorporated into DNA, unprocessed Okazaki fragments or unscheduled TLS polymerase activity. To address the possibility that it is due to cotranscriptional R‐loops, we next overexpressed Sen1, a helicase involved in the resolution of transcription‐replication conflicts (Mischo et al, 2011; Alzu et al, 2012; San Martin‐Alonso et al, 2021; Aiello et al, 2022). We placed the SEN1 gene under the control of a strong constitutive promoter (pACT1) and compared the growth of wild type and rnh1Δ rnh201Δ cells overexpressing SEN1 on HU‐ and MMS‐containing medium (Appanah et al, 2020). An 8‐ to 10‐fold increase of SEN1 expression (Fig EV3A) did not affect the growth of wild type cells in the presence of genotoxic drugs, but efficiently suppressed the growth defect of the rnh1Δ rnh201Δ mutant on both HU and MMS plates (Fig 3A). High levels of Sen1 also efficiently reduced the G2/M accumulation of RNase H‐deficient cells, as indicated by the flow cytometry analysis of DNA content in wild type and rnh1Δ rnh201Δ cells released into the cell cycle in the presence of HU or MMS (t = 240 min, Fig EV3B–D). Importantly, SEN1 overexpression also rescued fork restart defects in rnh1Δ rnh201Δ cells exposed to MMS, as measured by DNA combing experiments (Fig 3B). Altogether, our results indicate that rnh1Δ rnh201Δ cells accumulate toxic structures at HU‐ or MMS‐arrested forks that can be removed by Sen1.
Figure EV3. SEN1 overexpression alleviates the accumulation of rnh1Δ rnh201Δ cells in G2/M induced by HU and MMS exposure.
-
ASEN1 mRNA levels measured by RT–qPCR in asynchronous control cells in the indicated strains. Expression is normalized to ACT1 (n = 2 biological replicates).
-
BEffect of SEN1 overexpression on cell cycle progression in wild type and rnh1Δ rnh201Δ cells exposed to low doses of HU or MMS. Cells were arrested in G1 with α‐factor and were released into S phase in the presence of 25 mM HU or 0.015% MMS. The percentage of G2 cells at 180 and 240 min after release from G1 was calculated after flow cytometry analysis of DNA content. Mean and SEM are indicated (n = 3 biological replicates). *P < 0.05; **P < 0.01; ****P < 0.0001, two‐way ANOVA.
-
C, DRepresentative flow cytometry profiles used for calculating the percentage of G2/M cells in panel B. Arrows indicate differences due to SEN1 overexpression.
Figure 3. Increased levels of Sen1 promote fork progression in RNase H‐deficient cells exposed to MMS.
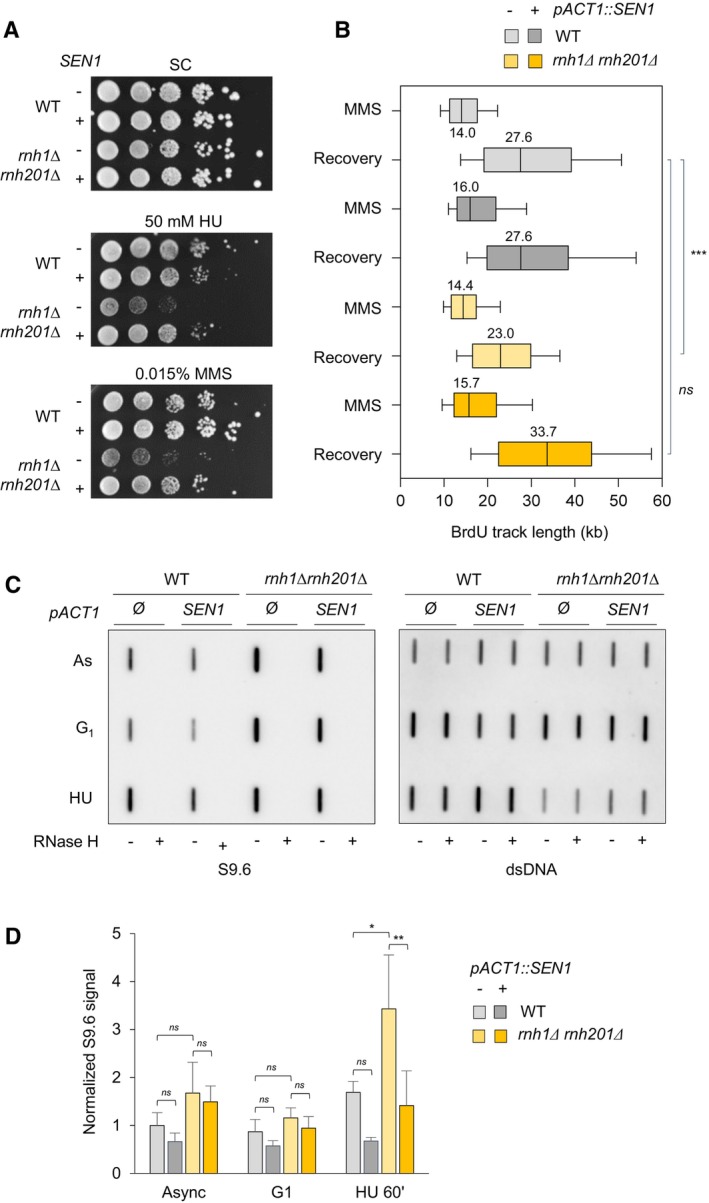
- Growth of wild type and RNase H mutants overexpressing or not SEN1 on synthetic complete (SC) medium ± indicated drug. Spots correspond to 1:10 serial dilutions.
- Exponentially growing cells were synchronized in G1 with α‐factor and released into S phase in the presence of 0.1% MMS. Newly replicated DNA was labeled with BrdU for 45 min (MMS), then MMS was removed and fork restart was measured by DNA combing after 30 min of recovery. The distribution of BrdU track length is shown. Box, 25–75 percentile range. Whiskers, 10–90 percentiles range. Median is indicated in kb. ***P < 0.001, Mann–Whitney rank‐sum test. Data from wild type and rnh1Δ rnh201Δ cells in this panel are identical to panel 2C. The DNA combing experiment was repeated twice (n = 2 biological replicates) with similar results, one representative experiment is shown. WT pACT1::SEN1 MMS (n = 201) and recovery (n = 318), rnh1Δ rnh201 pACT1::SEN1 MMS (n = 218) and recovery (n = 285).
- Slot blot analysis of RNA:DNA hybrid levels in the indicated strains. Samples from exponentially growing cells (As), α‐factor arrested cells (G1) and cells released in S phase for 60 min in the presence of 200 mM HU (HU) were treated with RNase III and RNase T1. As a control, all samples were additionally treated with RNase H. Membranes were incubated with antibodies against RNA:DNA hybrids (S9.6) or double‐stranded DNA (dsDNA).
- Intensity of the S9.6 signal, normalized to dsDNA. Mean ± SEM are indicated (n = 3 biological replicates). ns P > 0.05; *P < 0.05 and **P < 0.01, two‐way ANOVA.
Source data are available online for this figure.
RNAPII is properly evicted from chromatin in rnh1Δ rnh201Δ cells
Since RNAPII is evicted from chromatin at sites of replication‐transcription conflicts (Poli et al, 2016), we next checked whether this process is affected by the absence of RNase H and is facilitated by SEN1 overexpression. To this end, chromatin levels of Rpb1‐S2P, Rpb1‐S5P, and Rpb1‐CTD were determined by Western blotting in chromatin fractions from wild type and rnh1Δ rnh201Δ cells treated or not with HU (Fig EV4A). Rpb1 levels were normalized to the amount of Mcm2, used here as loading control for chromatin‐bound proteins and levels in SEN1‐overexpressing cells were expressed relative to control cells. This analysis revealed that levels of chromatin‐bound RNAPII in wild type and rnh1Δ rnh201Δ cells were not globally affected by SEN1 overexpression (Fig EV4B).
Figure EV4. Analysis of the levels of chromatin‐bound RNAPII around HU‐arrested forks.
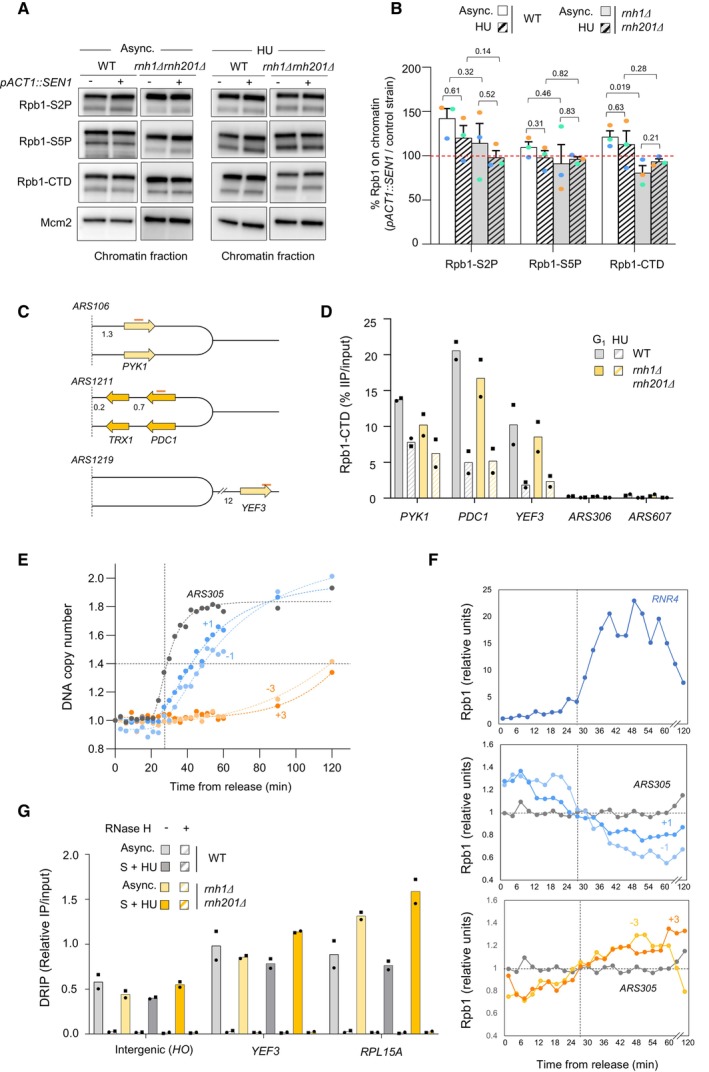
- Chromatin‐bound proteins from cells in indicated phases of the cell cycle were prepared and subjected to SDS–PAGE and immunoblotting with the indicated antibodies in a wild‐type strain that either does or does not overexpress SEN1 (Table EV1).
- Quantification of chromatin‐bound Rpb1‐S2P, Rpb1‐S5P and Rpb1‐CTD from data shown in panel F. Mcm2 is used as a loading control. Mean and SEM are indicated (n = 3 biological replicates). P‐values are indicated (paired t‐tests).
- Schematic representation of the genes exhibiting transcription‐replication conflicts.
- ChIP‐qPCR analysis comparing Rpb1‐CTD enrichment in G1‐arrested versus HU‐arrested cells (60 min, 0.2 M). Rpb1‐CTD occupancy is expressed as percentage of input DNA. Data are expressed as individual data points. Mean is indicated (n = 2 biological replicates).
- Variation of DNA copy number around the early origin ARS305 in wild type cells released synchronously into S phase after an α‐factor arrest in medium containing 200 mM HU. Samples were collected every 3 min. DNA copy number was quantified by qPCR at ARS305 and 4 other loci located at +1, +3, −1 and −3 kb from ARS305.
- ChIP‐qPCR of Rpb1‐CTD enrichment in cells collected in the experiment described in panel (E). Rpb1‐CTD occupancy is expressed as percentage of input DNA.
- DRIP‐qPCR analysis of RNA:DNA hybrid enrichment at an intergenic locus (HO) and two active genes (YEF3 and RPL15A) in wild type and rnh1Δ rnh201Δ cells ± in vitro RNase H treatment. Data are expressed as a percentage of input. Individual points are indicated (n = 2 biological replicates).
To determine whether RNase H could modulate RNAPII occupancy at specific loci upon HU exposure, wild type and rnh1Δ rnh201Δ cells were arrested in G1 and were released into S phase in the presence of HU. ChIP‐qPCR was used to quantify RNAPII levels (Rpb1‐CTD) at three representative metabolic genes replicated in a codirectional (PYK1) or in a head‐on (PDC1) manner, or were located too far from an early origin to be replicated in HU‐treated cells (YEF3; Fig EV4C). A similar reduction of chromatin‐bound Rpb1 levels was measured at all three loci in the presence of HU, regardless of the presence of RNase H1 and H2 (Fig EV4D). Very low levels of Rpb1‐CTD were also detected at the early origins ARS306 and ARS307 in these cells, before and after the HU arrest (Fig EV4D). To ensure that more subtle changes do not occur at forks during the HU treatment, we next measured RNAPII occupancy at the early origin ARS305 and at four non‐transcribed loci located at ±1 and ±3 kb from ARS305 in cells collected every 3 min after release from the G1 arrest. Variations in DNA copy number showed that ARS305 replicated 27 min after release, whereas sequences located 1 and 3 kb away replicated 20 and 80 min later, respectively (Fig EV4E). These data are consistent with the fact that replication forks do not stop in the presence of HU, but progress at a very slow rate (Poli et al, 2012). Levels of chromatin‐bound Rpb1 increased at the DNA repair gene RNR4 after 27 min, which reflects its activation in response to replication stress (Fig EV4F). In contrast, Rpb1‐CTD levels did not increase over time at ARS305 (Fig EV4F). Altogether, these data indicate that RNAPII does not accumulate in the vicinity of HU‐arrested forks and is properly evicted from highly expressed genes, independently of RNase H.
Sen1 reduces RNA:DNA Hybrid levels in HU‐treated rnh1Δ rnh201Δ cells
Next, we asked whether RNA:DNA hybrids levels increase in HU‐arrested cells in the absence of RNase H activity and whether this increase could be suppressed by SEN1 overexpression. To this end, wild type and rnh1Δ rnh201Δ cells were arrested in G1 with α‐factor and were released synchronously into S phase for 60 min in the presence of 200 mM HU, with or without SEN1 overexpression. Genomic DNA was extracted from asynchronous culture, G1 and HU‐arrested cells. Samples were treated with RNase III and RNase T1, deposited on a membrane with a slot blot apparatus and RNA:DNA hybrids were detected using the S9.6 antibody. Control samples were digested with RNase H to degrade RNA:DNA hybrids and double‐stranded DNA was used as loading control (Fig 3C). In wild type cells, we observed a two‐fold increase of RNA:DNA hybrid levels in HU‐arrested cells relative to G1 cells and this increase was even more pronounced in the rnh1Δ rnh201Δ mutant (Fig 3D). However, the overexpression of SEN1 significantly reduced RNA:DNA hybrid levels to wild type levels in HU‐treated rnh1Δ rnh201Δ cells. We therefore conclude that RNA:DNA hybrids accumulate in the yeast genome upon induction of replication stress in the absence of RNase H activity and are removed by high levels of Sen1.
RNA:DNA hybrids accumulate in front of HU‐arrested forks in RNase H‐deficient cells
Our results indicate that RNA:DNA levels increased upon HU exposure, even though HU downregulates transcription in budding yeast (Dubacq et al, 2006; Poli et al, 2016). To determine whether RNA:DNA hybrids levels could increase near HU‐arrested forks in the absence of RNase H1 and H2, we next used DRIP‐qPCR to quantify their levels at increasing distances from three early origins (ARS306, ARS607 and ARS305) and at three unreplicated loci (HO, YEF3 and RPL15A) used here as negative controls. Wild type and rnh1Δ rnh201Δ cells were arrested in G1 with α‐factor and were released synchronously into S phase for 90 min in the presence of 200 mM HU. Genomic DNA was extracted from asynchronous cultures and HU‐arrested cells. DNA was fragmented with restriction enzymes and digested or not with RNase H before immunoprecipitation with the S9.6 antibody, as described in Materials and Methods. At control loci, we detected a similar enrichment in wild type and rnh1Δ rnh201Δ cells, which was higher at expressed genes (YEF3, RPL15A) than at the intergenic HO locus and was totally sensitive to RNase H digestion (Fig EV4G). This signal increased slightly in rnh1Δ rnh201Δ mutants upon HU exposure, but to a lower extent than in slot blot experiments (Fig 3C and D). In contrast, we measured a 2‐ to 3‐fold increase in RNA:DNA hybrid levels at HU‐arrested forks in rnh1Δ rnh201Δ cells compared to wild type cells (Fig 4A). In this figure, qPCR data were expressed as the ratio of HU‐treated to asynchronous cells, but raw data are displayed in Appendix Fig S1A for ARS305. In rnh1Δ rnh201Δ mutants, the S9.6 signal was enriched from ~4 to 11 kb away from the replication origins. This higher RNA:DNA hybrids level was independent of the orientation of the underlying genes relative to fork direction (Fig 4A). Collectively, these data suggest that in rnh1Δ rnh201Δ mutants, RNA:DNA hybrid levels increase ahead of HU‐arrested forks. These structures could also be transferred behind HU‐arrested forks, but the resolution of the assay is not sufficient to demonstrate this.
Figure 4. Analysis of RNA:DNA hybrids and nascent RNA at HU‐arrested forks in budding yeast cells.
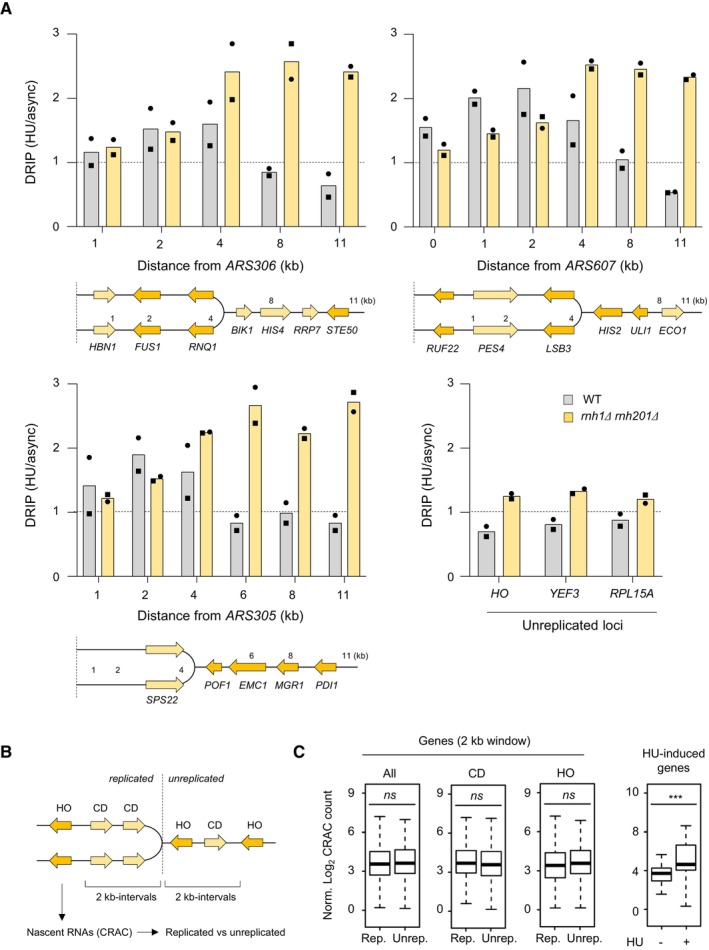
- RNA:DNA hybrids enrichment by DRIP‐qPCR around replication origins (ARS305, ARS306 and ARS607) and at RNAPII loci (HO, YEF3 and RPL15A) in wild‐type and rnh1Δ rnh201Δ cells released in S phase + HU for 90 min. Data are expressed as a fold‐enrichment in a given strain (HU/AS). Individual points are indicated (n = 2 biological replicates).
- Wild‐type cells were arrested in G1 with α‐factor and were released synchronously into S phase. The abundance of nascent RNA in the yeast genome was determined by Rpb1‐HTP CRAC as described (Aiello et al, 2022) for 2‐kb windows on both the replicated and the unreplicated side of HU‐arrested forks. Signal was measured for RNAPII transcribing in a co‐directional (CD), head‐on (HO) or both (all) configuration relative to the fork.
- Rpb1‐HTP CRAC signal computed over the whole 2‐kb interval and for coding units included in these same intervals. ns P > 0.05; ***P < 0.001, t‐test. For each boxplot, boxes represent the 25–75% quartile of the values and the central line indicates the median. Whiskers represents the minimum and maximum values on each side of the box. The number of genes analyzed per category is: codirectionnal (CD, replicated n = 153 and unreplicated n = 151), head‐on (HO, replicated n = 169 and unreplicated n = 156); HU‐induced genes (n = 42).
Source data are available online for this figure.
Pervasive transcription does not increase on nascent chromatin after HU treatment
Nascent chromatin is extensively remodeled at HU‐arrested forks to promote the resection of nascent DNA (Delamarre et al, 2020). Since open chromatin favors pervasive transcription, we next asked whether RNA synthesis increases behind HU‐arrested forks, which could interfere with the formation of RPA‐coated ssDNA. To address this question, we measured the abundance of nascent RNA in HU‐treated wild type cells using cross‐linking and analysis of cDNAs (CRAC), a method relying on UV cross‐linking of living yeast cells, purification of the elongation complexes and sequencing the nascent RNA isolated from transcription complexes to generate high‐resolution transcription maps (Granneman et al, 2009; Challal et al, 2022). DNA copy number variations were used to compare the levels of nascent RNA at genes located within 2‐kb intervals behind or ahead of HU‐arrested forks (Fig 4B). We found no difference between replicated and unreplicated regions in terms of abundance of nascent RNAs, regardless of the codirectional (CD) or head‐on (HO) orientation of transcription units (Fig 4C). The same result was obtained if the analysis was performed on all transcripts for intervals ranging from 0.1 to 2 kb (Appendix Fig S1B). In contrast, we measured an increased transcription at HU‐responsive genes, used here as positive controls, relative to untreated cells (Aiello et al, 2022). These data are consistent with ChIP‐qPCR data of RNAPII recruitment at HU‐arrested forks (Fig EV4F). Together, these results suggest that pervasive transcription is not responsible for the fork restart defects observed in the absence of RNase H.
RNase H1 and H2 are dispensable for normal fork progression in human cells
RNase H1 and RNase H2 play highly conserved functions in yeast and in human cells. To determine whether human RNases H also acts at stalled forks, we next knocked down RNase H1 and two subunits of RNase H2 (RNase H2A or RNase H2B) in HeLa cells using shRNAs or siRNAs, as indicated (Appendix Fig S2A–C) and monitored the effect of these depletions on replication forks. We first analyzed fork velocity in untreated control and RNase H‐depleted HeLa cells. Exponentially growing cells were pulse‐labeled with IdU and CldU for 15 min and DNA molecules were stretched on glass slides by DNA fiber spreading as described previously (Jackson & Pombo, 1998; Coquel et al, 2018). Then, IdU and CldU epitopes were detected using monoclonal antibodies coupled to fluorochromes and the length of at least 150 individual IdU tracks were measured on microscopy images for each condition. This analysis showed a similar rate of fork progression in control and RNase H‐deficient cells (Fig 5A), even though RNase H2‐depleted cells grew more slowly than control cells (Appendix Fig S2D).
Figure 5. RNase H2 promotes the resection of nascent DNA in HU‐treated HeLa cells.
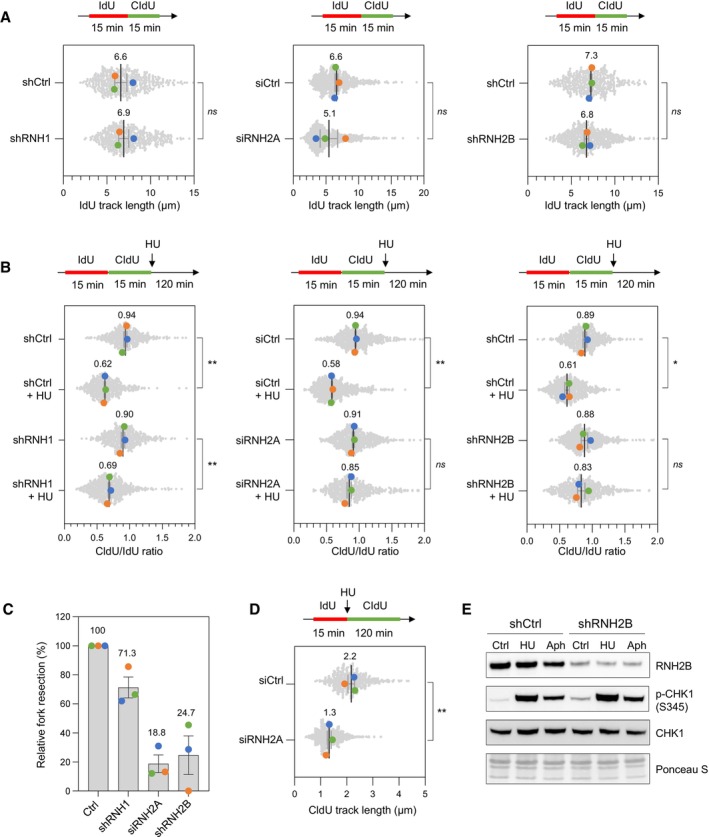
- DNA fiber analysis of replication fork speed in control HeLa cells and in cells depleted for RNase H1 (shRNH1), RNase H2A (siRNH2A) or RNase H2B (shRNH2B). Control (shCtrl), shRNH1 and shRNH2B cells were treated with doxycycline (10 μg/ml) for 72 h. Control (siCtrl) and siRNH2A cells were transfected with siRNAs for 48 h. Cells were sequentially labeled with IdU and CldU for 15 min before DNA fiber spreading. The length of IdU tracks from individual forks (~100–300 forks/condition) is shown. Mean (μm, n = 3 biological replicates) and SEM are indicated in gray. ns, non‐significant, paired t test.
- Stalled fork resection was analyzed using DNA fiber spreading. Depletion of RNase H1, RNase H2A or RNase H2B in HeLa cells was performed as indicated in panel (A). After sequential labelling of IdU and CldU for 15 min, cells were either collected immediately or treated for 2 h with 4 mM hydroxyurea (HU) before DNA fiber analysis. The lengths of the IdU and CldU tracks (~100–300 tracks/condition) were plotted as the ratio of CldU to IdU. Mean (n = 3 biological replicates) and SEM are indicated in gray. *P < 0.05; **P < 0.01; ns, non‐significant, paired t test.
- Relative extent of fork resection in control and RNase H‐deficient as determined by DNA fiber spreading (panel B). Mean ± SEM are indicated (n = 3 biological replicates).
- DNA fiber analysis fork progression in HU‐treated control and RNase H2A‐depleted cells. HeLa cells were transfected with siRNA against RNase H2A (siRNH2A) or a control sequence (siCtrl) for 48 h, and then labeled for 15 min with IdU before 4 mM HU treatment for 2 h. The distribution of CldU tracks (~100–300 tracks/condition) length is shown for three biological replicates. Mean (μm, n = 3) and SEM are indicated in gray. **P < 0.01, paired t test.
- Control (shCtrl) and RNH2B‐depleted (shRNH2B) HeLa cells were treated with doxycycline (10 μg/ml) for 72 h. Cells were then treated with or without HU (4 mM) or aphidicolin (1 μM) for 2 h. Activation of CHK1 was detected by Western blotting using anti‐phophoCHK1 (S345) antibody. Total CHK1 and ponceau staining are used as loading controls.
Source data are available online for this figure.
Human RNase H2 is required for the resection of nascent DNA at stressed forks
The fact that RNase H‐deficient yeast cells are defective for the formation of RPA‐coated ssDNA indicates that these cells are unable to resect nascent DNA. Alternatively, RNase H‐deficient cells could be proficient for resection but nascent RNA could anneal with the resulting ssDNA gaps to form stable RNA:DNA hybrids, preventing RPA binding. To discriminate between these two possibilities, we next monitored the resection of nascent DNA in human cells using DNA fiber spreading. To this end, HeLa cells were transfected for 48 h with control siRNAs (siCtrl) and siRNA directed against RNH2A (siRNH2A). Cells were labeled with two successive pulses of IdU and CldU and were grown for 120 min in the presence or the absence of HU. DNA fibers were stretched on glass slides as described above. Then, the length of IdU and CldU tracks was determined for each individual fork and expressed as the ratio of CldU to IdU (Fig 5B). In the absence of HU, this ratio was close to 1 for all cell types. In HU‐treated control cells, this ratio was significantly reduced (P < 0.001), which reflects the resection of nascent CldU tracks. Importantly, resection was reduced by 29% in RNase H1‐depleted cells relative to control cells and by 75 to 81% in cells depleted of either RNase H2A or RNase H2B (Fig 5C). In contrast, we did not detect resection defects when Senataxin (SETX) was knocked down (Appendix Fig S2E and F). These data indicate that RNase H2 is required for the resection of nascent DNA at HU‐arrested forks in human cells, as it is the case in budding yeast. To determine whether RNase H2 is required for fork progression in the presence of HU, RNase H2A‐depleted cells were labeled for 15 min with IdU and 120 min with CldU in the presence of 4 mM HU. The length of CldU tracks was 60% shorter in siRNH2A cells relative to control cells, indicating that RNase H2 promotes fork progression under replication stress conditions (Fig 5D). However, RNase H2‐depleted cells were fully proficient to activate the DNA replication checkpoint in response to HU or aphidicolin, as illustrated by the ATR‐dependent phosphorylation of the CHK1 kinase (Fig 5E).
Fork resection defects in the absence of RNase H2 are suppressed by triptolide
Our results suggest that in the absence of RNase H2, RNA:DNA hybrids do not impede the progression of replication forks in unchallenged growth conditions, but interfere with the processing of stressed forks in the presence of HU. To determine whether the formation of these toxic RNA:DNA hybrids depends on RNAPII, we monitored fork resection by DNA fiber spreading after inhibition of RNAPII activity with either DRB or triptolide. To this end, HeLa cells were transfected for 48 h with siCtrl or siRNH2A and were treated for 2 h with DRB or 1 h with triptolide before the IdU/CldU pulses and during the HU treatment (Fig 6A). Remarkably, the fork resection defect observed in siRNH2A cells was fully suppressed by triptolide, but not by DRB (Fig 6A). To explain this difference, we first quantified EU incorporation after DRB or triptolide treatment to verify that RNAPII activity was efficiently repressed under these conditions (Appendix Fig S3). We next monitored the presence of RNA:DNA hybrids in genomic DNA using the S9.6 antibody on slot blots. This analysis showed that levels of RNA:DNA hybrids were equivalent in siCtrl and siRNH2A cells and were equally reduced by the DRB and triptolide treatments (Fig 6B and C). Then, we quantified the total levels of the RPB1 subunit of RNAPII in siCtrl and siRNH2A cells treated with DRB or triptolide and found that unlike DRB, triptolide induced a drastic degradation of RNAPII (Fig 6D), which is consistent with earlier studies (Manzo et al, 2012; Tufegdžić Vidaković et al, 2020). Altogether, these data suggest that in the absence of RNase H2, RNAPII contributes to the inhibition of fork resection mediated by post‐replicative RNA:DNA hybrids.
Figure 6. Transcription inhibition with triptolide restores fork resection in RNase H2‐depleted HeLa cells.
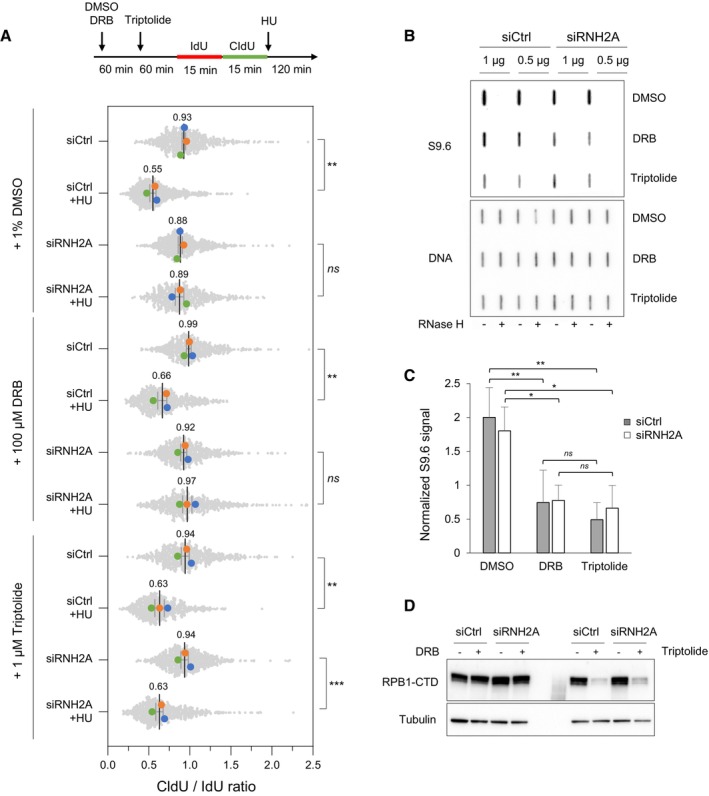
- Cells were transfected with the indicated siRNAs for 48 h and were treated with DMSO, 100 μM DRB or 1 μM triptolide for indicated time periods. Cells were labeled with two pulses of IdU and CldU for 15 min before the addition of 4 mM HU for 2 h. RNA polymerase II inhibitors were present during HU treatment. Fork resection was analyzed by DNA fiber spreading and the ratio of CldU to IdU track (~100–300 tracks/condition) length was determined in three independent experiments. Mean (n = 3) and SEM are indicated. **P < 0.01; ***P < 0.001; ns, non‐significant, paired t test.
- HeLa cells were transfected with siRNA and treated with RNAPII inhibitors as indicated in panel A, but without HU treatment. Total genomic DNA was extracted and treated as described in Materials and Methods. RNA:DNA hybrids were detected using the S9.6 antibody and RNase H‐treated samples were included as controls. Double‐stranded DNA was used as loading control. A representative image of four independent experiments is shown.
- Intensity of S9.6 signal in cells treated as indicated in panel (B), normalized to total DNA. Mean and SEM are shown (n = 4 biological replicates). ns P > 0.05; *P < 0.05 and **P < 0.01, two‐way ANOVA.
- HeLa cells were transfected with siRNA and treated with RNAPII inhibitors as indicated in panel (B). RNAPII was detected by immunoblotting using an antibody against the C‐terminal domain of the Rpb1 subunit. Tubulin was used as a loading control.
Source data are available online for this figure.
A model for the formation of toxic RNA:DNA Hybrids at paused forks
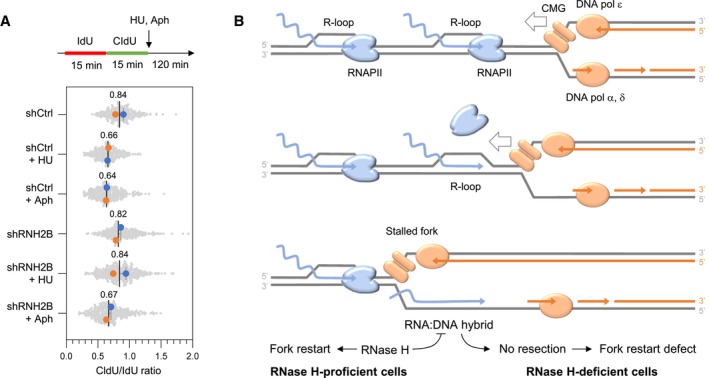
In classical model of transcription‐replication conflicts, R‐loops and RNAPII complexes interfere with fork progression by acting as replication barriers (Gomez‐Gonzalez & Aguilera, 2019; Lalonde et al, 2021). However, the fact that RNA:DNA hybrids interfere with the resection of nascent DNA indicates that toxic RNA:DNA hybrids accumulate behind the fork in the absence of RNase H activity. In a head‐on orientation, this would occur when the CMG helicase bypasses the RNA:DNA hybrid on the opposite strand, transferring it on the lagging strand.
To test this possibility experimentally, we reasoned that a drug like HU that slows but does not stop forks should promote the formation of these toxic structures whereas drugs like Aphidicolin, which completely blocks the replisome when used at high concentration, should prevent the bypass of RNA:DNA hybrids by replication forks. We first verified that 4 mM HU gradually slowed down the progression of replication forks, while 5 μM Aphidicolin completely blocked elongation after 2 h (Fig EV5A). Then, cells were labeled with two successive pulses of IdU and CldU and were exposed to either HU or Aphidicolin to measure fork resection on stretched DNA fibers (Fig 7A). In control cells, the extent of fork resection was similar in the presence of HU and Aphidicolin, which is consistent with earlier results (Coquel et al, 2018). In contrast, fork resection was only detected after Aphidicolin treatment in RNH2B‐depleted cells (Fig 7A), which supports the view that RNase H2 is dispensable for the resection of nascent DNA when the replisome is fully blocked. To confirm that this differential effect of HU and Aphidicolin on fork resection was specific to RNH2B‐deficient cells, we next monitored fork resection in the absence of SMARCAL1, a DNA translocase promoting fork reversal (Quinet et al, 2017; Liu et al, 2023) and required for the resection of nascent DNA independently of RNA:DNA hybrids. We found that SMARCAL1‐depleted cells were equally defective for fork resection in the presence of HU or aphidicolin (Fig EV5B) and that the effect of RNaseH2 and SMARCAL1 depletion on resection is epistatic (Fig EV5C). Altogether, these data argue for a model in which cotranscriptional R‐loops are converted into post‐replicative RNA:DNA hybrids when bypassed by slowed forks in a head‐on orientation. Preventing the formation of these toxic structures on the lagging strand may require a coordinated removal of RNAPII complexes and of the associated R‐loop to avoid interference with fork restart processes (Fig 7B).
Figure EV5. Effect of aphidicolin and hydroxyurea on fork velocity and on the resection of nascent DNA.
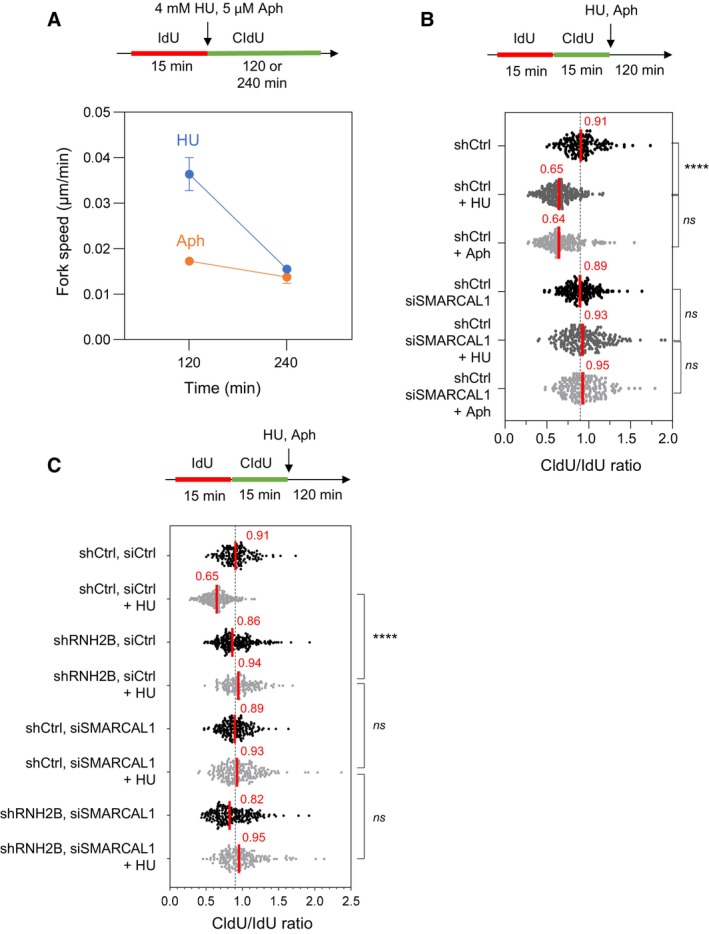
- HeLa cells were labeled for 15 min with IdU and for 120 or 240 min with CldU in the presence of 4 mM hydroxyurea (HU) or 5 μM aphidicolin (Aph). DNA fibers were stretched on glass slides and the length of CldU tracks were measured. Fork speed was calculated during the first 120 min after HU addition and between 120 and 240 min. Mean and SEM are shown for two independent experiments.
- HeLa cells were transfected with siRNA against SMARCAL1 or a control sequence (siCtrl) for 48 h. They were sequentially labeled for 15 min with IdU and for 15 min with CldU. Then, they were treated for 2 h with 4 mM hydroxyurea (HU) or 5 μM aphidicolin (Aph) before DNA fiber analysis. The lengths of the IdU and CldU tracks were plotted as the ratio of CldU to IdU. Median CldU/IdU ratios are indicated in red. ****P < 0.0001; ns, non‐significant, Mann–Whitney rank‐sum test (n = 2 biological replicates).
- DNA fiber analysis of fork resection in control HeLa cells (shCtrl) and in HeLa cells depleted for RNase H2B (shRNH2B) and transfected with siRNA against SMARCAL1 or a control sequence (siCtrl), and exposed or not to 4 mM HU. Cells were treated with doxycycline (10 μg/ml) for 24 h and were then transfected with siRNAs for 48 h in the presence of doxycycline. Cells were sequentially labeled for 15 min with IdU and for 15 min with CldU. Then, they were either collected immediately or treated for 2 h with 4 mM hydroxyurea (HU) before DNA fiber analysis. The lengths of the IdU and CldU tracks were plotted as the ratio of CldU to IdU. Median CldU/IdU ratios are indicated in red. ****P < 0.0001; ns, non‐significant, Mann–Whitney rank‐sum test (n = 2 biological replicates).
Figure 7. Cotranscriptional R‐loops are converted into post‐replicative RNA:DNA hybrids at slowed forks upon bypass by the replisome.
- HU and aphidicolin differentially affect fork resection in the absence of RNase H2. Control (shCtrl) and RNH2B‐depleted (shRNH2B) cells were treated with 10 μg/ml doxycycline for 72 h. Cells were sequentially labeled for 15 min with IdU and CldU and were exposed or not to 4 mM hydroxyurea (HU) or 5 μM aphidicolin (Aph) before DNA fiber analysis. The ratio of CldU to IdU track length is shown for two biological replicates. Mean (n = 2) and SEM are indicated in gray.
- Model: When the replisome encounters an active gene in a head‐on orientation, the replicative helicase (CMG) faces multiple RNAPII complexes at the TTS, associated or not with R‐loops. CMG is blocked by the RNAPII complex but not by the RNA:DNA hybrid, which is located on the opposite DNA strand. Mec1ATR promotes RNAPII displacement, allowing DNA synthesis on the leading strand to continue past the RNA:DNA hybrid, which is then transferred behind the fork. In the absence of RNase H2, the persistence of this structure interferes with the resection of nascent DNA and prevents fork restart. This model implies that RNAPII and the RNA:DNA hybrid must be removed in a coordinated manner to prevent the formation of toxic RNA:DNA hybrids behind the replication fork.
Source data are available online for this figure.
Discussion
R‐loops have emerged as a prominent source of endogenous replication stress in all species, from bacteria to human (Gomez‐Gonzalez & Aguilera, 2019). Because these structures are so abundant, it is unlikely that all R‐loops are equally toxic to replication forks. Therefore, an important question is what distinguishes physiological from deleterious R‐loops. A key determinant of R‐loop toxicity is their orientation relative to fork direction, with head‐on collisions being more harmful than codirectional conflicts (Hamperl et al, 2017; Lang et al, 2017; Chappidi et al, 2020; Promonet et al, 2020). However, recent in vivo and in vitro studies indicate that head‐on conflicts do not block fork progression in budding yeast (Kumar et al, 2021; Tsirkas et al, 2022). The mechanism by which head‐on R‐loops interfere with DNA replication therefore remains poorly understood.
Here, we propose that RNA:DNA hybrids do not only act as replication barriers as originally thought, but also interfere with post‐replicative processes involved in fork restart. This view arises from the analysis of DNA replication in budding yeast and human cells lacking RNase H. In both organisms, we found that RNase H activity was dispensable for normal DNA replication, but was important for the recovery of stressed forks. Indeed, the resection of nascent DNA at arrested forks was severely impaired in both yeast and human cells lacking RNase H1 and RNase H2. This activity was primarily promoted by RNase H2, presumably because it contains a PCNA‐binding domain that targets it to the replisome (Bubeck et al, 2011), unlike RNase H1. Since resection is a post‐replicative process, this implies that RNase H‐sensitive structures accumulate behind stressed forks and interfere with their processing and recovery. The existence of post‐replicative RNA:DNA hybrids is also supported by a recent electron microscopy study showing that RNA:DNA hybrids accumulate behind bacterial forks at head‐on TRCs in (Stoy et al, 2023).
RNase H2 degrade different types of substrates, including RNA:DNA hybrids, RNA primers of Okazaki fragments and ribonucleotides erroneously incorporated into DNA. In budding yeast, we found that the rnh201‐RED mutant was able to restore the formation of RPA‐coated ssDNA in rnh1Δ rnh201Δ cells, even though it is fully defective for RER. Fork restart in RNase H‐deficient cells was also rescued by the overexpression of the Sen1Senataxin helicase, which unwinds RNA:DNA hybrids but does not act on rNMPs. In contrast, nascent DNA resection at stalled forks in rnh1Δ rnh201Δ mutants could not be rescued by deletion of the TLS DNA polymerase η, whose activity is deleterious for HU‐treated rnh1Δ rnh201Δ mutants (Meroni et al, 2019). Destabilizing the pol α‐primase complex did not restore either the formation of RPA‐coated ssDNA in rnh1Δ rnh201Δ mutants, suggesting that unprocessed Okazaki fragments are not responsible for resection defects. In contrast, fork resection was restored by triptolide in RNase H2‐deficient human cells, a transcription inhibitor that does not affect the maturation of Okazaki fragments. These observations differ from a recent report showing that unprocessed RNA primers at Okazaki fragments recruit the Ku complex and prevent fork resection at the RTS1 replication barrier in fission yeast and in cells exposed to the Top1 inhibitor camptothecin (CPT; Audoynaud et al, 2023). RNase H could therefore target different structures at HU‐arrested forks and at replication barriers such as those induced by CPT and RTS1. This view is consistent with the fact that different fork restart mechanisms operate in the presence of HU and CPT and that slow forks are particularly sensitive to TRCs (Chappidi et al, 2020; Andrs et al, 2023).
Toxic RNA:DNA hybrids could result from pre‐existing R‐loops, but could also arise from de novo RNA synthesis after fork arrest. Indeed, nascent chromatin is hyperacetylated and extensively remodeled after fork arrest, which could promote pervasive transcription (Delamarre et al, 2020). This would generate stable RNA:DNA hybrids at ssDNA gaps, as proposed earlier for checkpoint and post‐replicative repair mutants (Barroso et al, 2019). However, CRAC analysis of nascent RNA at HU‐arrested forks revealed that transcription did not increase behind arrested forks relative to unreplicated regions. This observation is consistent with the fact that RNAPII levels did not increase upon HU exposure near origins and replicated genes in both control and rnh1Δ rnh201Δ mutants. Collectively, these data suggest that toxic RNA:DNA hybrids do not form de novo after HU arrest, but rather result from pre‐existing R‐loops. This view is strengthened by the observation that resection of newly synthesized DNA in human cells lacking RNase H2 was restored when the transcription inhibitor triptolide was administered prior to HU exposure.
Two main classes of cotranscriptional R‐loops have been described in human cells: class I R‐loops are short structures that form at high frequency during RNAPII pausing at promoters, whereas class II R‐loops are longer but less abundant structures that are found at TTS and throughout the gene body (Castillo‐Guzman & Chédin, 2021). Since TRCs occur preferentially at TTS in human cells (Promonet et al, 2020), it is tempting to speculate that toxic RNA:DNA hybrids that interfere with fork resection originate from class II R‐loops. Interestingly, fork resection in RNase H2‐depleted cells was not restored by DRB, another transcription inhibitor. Unlike triptolide, DRB did not induce the degradation of RNAPII in HU‐arrested cells. This suggests that RNAPII and R‐loops need to be removed in a coordinated manner in order to promote fork processing and restart.
Besides RNase H, two major players have been involved in the resolution of TRCs. ATRMec1 is activated when forks converge on the TTS of highly expressed genes in a head‐on orientation (Promonet et al, 2020) and coordinate the eviction of stalled RNAPII (Im et al, 2014; Poli et al, 2016; Landsverk et al, 2020; Hurst et al, 2021). Sen1Senataxin interacts with the replisome in budding yeast (Alzu et al, 2012; Appanah et al, 2020) and removes both RNAPII and RNA:DNA hybrids (Aiello et al, 2022). Sen1 acts during S phase and is degraded in G1 (Mischo et al, 2011; San Martin‐Alonso et al, 2021). The fact that SEN1 overexpression restored challenged fork restart in yeast rnh1Δ rnh201Δ mutants supports the view that Sen1Senataxin can functionally replace RNases H in TRC mitigation. However, Senataxin depletion in human cells did not interfere with fork resection, indicating that it is dispensable for the processing of RNA:DNA hybrids at stalled forks in RNase H‐proficient cells.
It has been reported that R‐loop levels globally increase under conditions of replication stress (Hamperl et al, 2017; Lalonde et al, 2021), but whether this increase directly relates to replication stress was unclear. Here, we have found that in RNase H‐deficient yeast cells, RNA:DNA hybrids accumulated over at least 7 kb in front of HU‐arrested forks, but not at unreplicated loci. Considering that there are approximately 200 active origins in HU‐arrested cells, these regions represent a total of nearly 3 Mb of genomic DNA, which would be sufficient to explain the global increase in RNA:DNA hybrid levels in HU‐treated rnh1Δ rnh201Δ mutants. The spatial and temporal resolution of our assay was not sufficient to determine whether some of these hybrids are also present behind HU‐arrested forks. However, the fact that these structures interfere with fork resection suggests that a subset of R‐loops is converted into post‐replicative RNA:DNA hybrids at stressed forks. These structures could either impede nucleases involved in fork resection, as reported for EXO1 by in vitro studies (Daley et al, 2020), or interfere with fork reversal, a process acting upstream of fork resection (Neelsen & Lopes, 2015). The latter view is supported by the fact that the fork reversal factor SMARCAL1 is epistatic with RNase H2 for the resection of nascent DNA, but further work is required to confirm this hypothesis.
Collectively, our data argue for a model in which the CMG helicase and the leading strand DNA polymerase bypass the RNA:DNA hybrid upon removal of RNAPII and generates a toxic structure that is degraded by RNase H to promote fork processing and restart (Fig 7B). This model is supported by the fact that resection defects are observed upon HU exposure and not in the presence of aphidicolin. Indeed, unlike HU, aphidicolin completely blocks fork progression when used at high concentration and would therefore prevent the conversion of R‐loops into toxic RNA:DNA hybrids. The view that deleterious consequences on fork structure arise from the encounter of slow‐moving forks and transcription complexes is supported by a recent study showing that HU promotes R‐loop formation at TRCs and drives fork reversal (Andrs et al, 2023). In RNase H‐deficient cells, dealing with these conflicts would be particularly challenging when slow forks encounter multiple RNAPII complexes and R‐loops in a head‐on orientation, as it is the case at TTS (Skourti‐Stathaki et al, 2011; Promonet et al, 2020). As illustrated in Fig 7B, we therefore propose that R‐loops are toxic in RNase H‐deficient cells both because they induce fork arrest along with RNAPII and because they impede restart if converted to post‐replicative RNA:DNA hybrids.
Our finding that RNA:DNA hybrids inhibit fork resection is reminiscent of their effect on DSB repair. Indeed, a large body of evidence indicates that these structures interfere with HR‐mediated DSB repair by inhibiting DNA end resection (Marnef & Legube, 2021; Brickner et al, 2022). In the absence of Senataxin, RNA:DNA hybrids persist at DSBs and favor error‐prone non‐homologous end joining over HR (Cohen et al, 2018). Whether these structures arise from pre‐existing transcription or from de novo recruitment of RNAPII is currently highly debated and many different models have been proposed to explain how RNA:DNA hybrids accumulate at DSBs (Marnef & Legube, 2021; Brickner et al, 2022). Interestingly, several lines of evidence indicate that these structures also promote HR‐mediated DSB repair (Ohle et al, 2016; Ouyang et al, 2021). It is therefore likely that RNA:DNA hybrids are needed transiently at DSBs to promote HR but then need to be removed to complete DSB repair. HR‐mediated fork repair present similarities, but also differences with DSB repair (Tye et al, 2020). Whether RNA:DNA hybrids also have both positive and negative roles in HR‐mediated fork repair by regulating nascent DNA resection is an important question that remains to be addressed.
Materials and Methods
Yeast strains, culture conditions, drop assay and flow cytometry
All strains used are listed in Table EV1. For liquid cultures, yeast extract peptone medium was supplemented with 2% glucose unless otherwise stated. MATa cells were synchronized in G1 by adding α‐factor (6 μg/ml, Biotem, No. 2968) for 170 min at 25°C unless otherwise stated. Arrest without buds was monitored by phase microscopy. G1‐arrested cells were released into S phase by filtration and resuspension in fresh medium or by the addition of 75 μg/ml Pronase (Sigma, 53702) and were treated or not with the indicated dose of HU (Sigma, H8627) or MMS (Sigma, 129925). Flow cytometry samples were prepared as previously described (Poli et al, 2016), using Sytox to label DNA. Data were acquired on a MACSQuant (Miltenyi Biotec) and analyzed with FlowJo. Drop assays were done with cells adjusted to 1 × 107 cells/ml. 10‐fold serial dilutions were spotted on YPAD or SC plates ± the indicated drug.
Protein extracts, chromatin fractionation, and Western blotting
Total protein extracts and chromatin fractionation was performed as previously described (Poli et al, 2016). Proteins were resolved by SDS–PAGE and transferred with a Trans‐Blot (Bio‐Rad). After blocking, proteins were either probed with anti‐RNAPII CTD (Abcam 8WG16, ab817), anti‐Rpb1‐S5P (Clone 3E8, Merck, 04‐1572), anti‐Rpb1‐S2P (Abcam, ab5095), anti‐Mcm2 (N‐19, Santa Cruz, sc‐9839) or anti‐tubulin (Thermo Fisher Scientific, MA1‐80017). Blots were scanned with a ChemiDoc MP (Bio‐Rad) and semi‐quantitative determination of protein level was performed using the Image J (Fiji) software using Mcm2 for normalization.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as indicated (Delamarre et al, 2020). Anti‐RPA (Agrisera, AS07214), anti‐Rpb1‐CTD (Biolegends, 8WG16) and anti‐PK (Anti‐V5 tag, AbD Serotec, MCA1360G) antibodies were used with Dynabeads Prot. A. IP/Input ratio were calculated and qPCR results were normalized on the average of four negative zones for RPA ChIP. Aspecific IP and Western blotting of the protein samples were performed for each experiment. Primers used for qPCR are listed in Table EV2.
Genome‐wide replication timing analysis
Replication timing analysis was performed as previously described (Fang et al, 2017). Genomic DNA was isolated using Qiagen genomic DNA extraction kit according to the manufacturer's instructions. DNA was fragmented using sonication (∼200‐ to 500‐base‐pair [bp] size range). Sequencing libraries were prepared using a Thru‐PLEX DNA‐seq kit (Rubicon Genomics) and sequenced on a HiSeq 4000 (Illumina). Single‐end reads of 50 bp were aligned to the S. cerevisiae genome (2011) with Bowtie, allowing only perfect matches. Relative copy number was determined as a ratio of normalized HU reads to G1 reads.
RNA extraction, RT and qPCR
Total RNA was extracted using standard hot phenol procedure as previously described (Poli et al, 2016). RT–qPCR were performed from at least two independent biological replicates, starting with 3 μg of RNA. Primers used for RT–qPCR are listed in Table EV2.
Cell culture and reagents
Human cervical adenocarcinoma HeLa cells (ATCC, CCL‐2) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C in a 5% CO2 incubator. Aphidicolin (A4487), DRB (5,6‐Dichlorobenzimidazole 1‐β‐D‐ribofuranoside, D1918), triptolide (T3652) and doxycycline hydrochloride (D3072) are from Sigma‐Aldrich.
Production of lentiviral vectors and cell transduction
HIV‐1‐derived lentiviral vectors were produced in HEK293T cells as previously described (Lin et al, 2004). Cells were seeded on poly‐D‐lysine coated plates and transfected with packaging plasmid (psPAX2, Addgene plasmid #12260): transfer vector (TRIPZ‐shCtrl; TRIPZ‐shRNH2B; TRIPZ‐shSETX, Horizon): vesicular stomatitis virus envelop plasmid (pMD2.G, Addgene plasmid #12259) at a ratio 5:3:2 by the calcium phosphate method. The culture medium was collected 48 h post‐transfection, filtrated using 0.45 μm filters and concentrated at 100 folds by ultracentrifugation at 89,000 g at 4°C for 1 h30. HeLa cells were transduced at a M.O.I. = 10 (Multiplicity of Infection) by centrifugation at 1500 g at 30°C for 1 h30 in the presence of 5 μg/ml of polybrene.
DNA combing and DNA fiber spreading
DNA combing was performed as described (Tourrière et al, 2017) using a mouse monoclonal anti‐ssDNA (Chemicon, clone 16–19) and a rat monoclonal anti‐BrdU (AbCys, clone BU1/75). Images were recorded on a Zeiss Axioimager microscope equipped with a CoolSNAP HQ CCD camera (Roper Scientific) and were processed as described (Tourrière et al, 2017).
DNA fiber spreading was performed as described previously (Jackson & Pombo, 1998; Breslin et al, 2006; Coquel et al, 2018). Briefly, subconfluent cells were sequentially labeled first with 10 μM 5‐iodo‐2′‐deoxyuridine (IdU) and then with 100 μM 5‐chloro‐2′‐deoxyuridine (CldU) for the indicated times. One thousand cells were loaded onto a glass slide (StarFrost) and lysed with spreading buffer (200 mM Tris–HCl pH 7.5, 50 mM EDTA, 0.5% SDS) by gently stirring with a pipette tip. The slides were tilted slightly and the surface tension of the drops was disrupted with a pipette tip. The drops were allowed to run down the slides slowly, then air dried, fixed in methanol/acetic acid 3:1 for 10 min, and allowed to dry. Glass slides were processed for immunostaining with mouse anti‐BrdU to detect IdU, rat anti‐BrdU to detect CldU, mouse anti‐ssDNA antibodies and corresponding secondary antibodies conjugated to various Alexa Fluor dyes. Nascent DNA fibers were visualized by using immunofluorescence microscopy (Zeiss ApoTome). The acquired DNA fiber images were analyzed by using MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices) and statistical analysis was performed with GraphPad Prism (GraphPad Software). The lengths of at least 150 tracks were measured per sample.
Detection of RNA:DNA Hybrids by slot blotting in human cells
Cells were lysed in 0.5% SDS/TE, pH 8.0 buffer containing Proteinase K overnight at 37°C. Total DNA was isolated with Phenol/Chloroform/Isoamyl alcohol extraction followed by standard ethanol precipitation. Isolated genomic DNA was then digested using a cocktail of restriction enzymes (AluI, DdeI, MboI and MseI) overnight at 37°C followed by ethanol precipitation. DNA amount was quantified using qPCR. One and two micrograms of digested DNA were treated with RNase III to remove dsRNA. One and half microgram was loaded in duplicate onto a Hybond‐N+ membrane using slot blot apparatus. Samples were deposited on two membranes, one for direct UV crosslinking at 0.12 Joules and the other for DNA denaturation. To denature DNA, membrane was incubated with denaturation buffer (0.5 M NaOH; 1.5 M NaCl) for 10 min and neutralization buffer (1 M NaCl and 0.5 M Tris, pH 7.5) for another 10 min (twice). UV crosslinked membrane was blocked with 5% skim milk in PBST (PBS; 0.1% Tween‐20) for 1 h. The RNA:DNA hybrids were detected by immunoblotting using S9.6 antibody (Abliance). Goat anti‐mouse HRP conjugate (Bio‐Rad) was used as secondary antibody (1/5,000). RNA:DNA hybrids was detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific) on Bio‐Rad ChemiDoc Touch Imaging System. dsDNA was detected using a radioactive probe against whole genomic DNA. Slot‐blots signal was quantified in ImageJ.
Detection of RNA:DNA Hybrids by slot blotting in yeast cells
Cells were spheroplasted with Zymolyase 20T (2 mg/ml, MP Biomedicals) and lysed in Buffer G2 (Qiagen) containing 5% Tween20 and 0.5% Triton 100×. Total DNA was treated with RNase T1 followed by Proteinase K treatment and recovered on a glass rod. Isolated genomic DNA was then digested using a cocktail of restriction enzymes (AluI, DdeI, MseI and HpaII; 40 U/each) overnight at 37°C. DNA amount was quantified using qPCR. One microgram of digested DNA was treated with RNase III to remove dsRNA. Half microgram was loaded in duplicate onto a Hybond‐N+ membrane using slot blot apparatus. Membranes were UV crosslinked at 0.12 Joules and blocked with 5% skim milk in PBST (PBS; 0.1% Tween‐20) for 1 h. The RNA:DNA hybrids were detected by immunoblotting using S9.6 (Abliance, 5.55 mg/ml; 1/2750) antibody or dsDNA antibody (1/10,000; ab27156 Abcam). Goat anti‐mouse HRP conjugate (Bio‐Rad) was used as secondary antibody (1/5,000). RNA:DNA hybrids and dsDNA were detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific) on Bio‐Rad ChemiDoc Touch Imaging System. Slot‐blots signal was quantified in ImageJ.
DRIP‐qPCR in yeast cells
Cultures were collected, washed with chilled water, and resuspended in 2.4 ml spheroplasting buffer (1 M Sorbitol, 2 mM Tris–HCl pH 8, 100 mM EDTA pH 8, 0.01% β‐mercaptoethanol, 2.5 mg/ml Zymolyase 20T). Spheroplasts were pelleted (5 min at 4,500 g) rinsed with water and homogeneously resuspend in 1.2 ml of Qiagen G2 buffer. Samples were treated with 4 μl of RNase T1 (Thermo Fisher Scientific) for 1 h at 37°C and 75 μl of 20 mg/ml proteinase K (Roche) for 1 h at 37°C. DNA was extracted using chloroform:isoamyl alcohol 24:1 precipitation, DNA was recovered on a glass rod and washed with 70% EtOH, resuspended gently in TE. DNA was quantified using qPCR and 12 μg of DNA was digested overnight with 40 U of AluI, DdeI, HpaII and MseI (New England BioLabs). For RNase H control, 5 μg of DNA was treated with 5 μl of RNase H (New England BioLabs) for 4 h at 37°C, Spike‐in molecules (synthetic RNA:DNA hybrid) were added to the 5 μg of DNA prior to RNase H treatment at a rate of 1 molecule/haploid genome in order to control S9.6 IP efficiency. RNA:DNA hybrids immunoprecipitation was performed with 5 μg of DNA and 10 μg of S9.6 antibody (5.55 mg/ml, Antibodies inc.) incubated overnight on a rotating wheel at 4°C. The DNA‐antibody mixture was incubated with Dynabeads protein A (Life Technologies) for 4 h at 4°C on a rotating wheel. Beads were washed five times with binding buffer and DNA was eluted in 120 μl elution buffer (50 mM Tris pH 8, 10 mM EDTA, 1% SDS) for 12 min at 65°C. Eluates were incubated for 1 h with 20 μl proteinase K (20 mg/ml) at 50°C and purified using phenol‐chloroform isoamyl alcohol 25:24:1. DNA was resuspended in 300 μl of H2O and qPCR were performed. The %IP/Input was adjusted to the %IP/Input of spike‐in molecules.
RNAPII CRAC and DNA copy number analyses
For RNAPII CRAC experiments, 2 l/condition of cells carrying an endogenously HTP‐tagged RBP1 gene were grown in exponential phase in synthetic media lacking tryptophan at 25°C. G1‐arrest was triggered at OD600 = 0.3 by three consecutive additions of 4, 8 and 4 mg of α‐factor spaced by 1 h. 20 min before release into S‐phase, cultures were supplemented with HU 200 mM. 1 h after last α‐factor addition, cells were released into S phase by depletion of α‐factor by filtration on a glass microfiber filter (pore ⌀ = 1.6 mm). Cells were rinsed while still on the filter and then resuspended in 2 l of fresh medium already supplemented with HU 200 mM and grown for 1 additional hour before UV treatment and collection. Proper cell cycle synchronization was systematically verified by FACS analysis and visualization of cell morphology at a microscope. Downstream processing was performed as previously described in (Candelli et al, 2018; Challal et al, 2018). DNA copy number analyses on the same cell culture were performed as previously described (Aiello et al, 2022). Statistical analysis was performed using t‐test and P‐values are indicated.
Author contributions
Jonathan Heuzé: Conceptualization; formal analysis; investigation; methodology; writing – review and editing. Samira Kemiha: Conceptualization; formal analysis; investigation; methodology. Antoine Barthe: Formal analysis; investigation; methodology. Alba Torán Vilarrubias: Formal analysis; investigation; methodology. Elyès Aouadi: Investigation; methodology. Umberto Aiello: Formal analysis; investigation; methodology. Domenico Libri: Supervision; funding acquisition. Yea‐Lih Lin: Conceptualization; formal analysis; investigation; methodology; writing – original draft; project administration; writing – review and editing. Armelle Lengronne: Conceptualization; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing. Jérôme Poli: Conceptualization; formal analysis; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing. Philippe Pasero: Conceptualization; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Sarah Lambert, Benjamin Pardo, Hervé Técher and Vincent Vanoosthuyse for discussions and for critical comments on the manuscript. We thank Diyavarshini Gopaul for help with the quantification of EU incorporation. We thank Benoit Palancade for help with the optimization of the DRIP assay and for sharing the DRIP spike‐in sequence in yeast. We thank Bob Crouch, Giacomo De Piccoli and Marco Muzi‐Falconi for sharing strains. This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Fondation pour la Recherche Médicale (FRM, programme Equipes 2019 to DL), Agence Nationale pour la Recherche (ANR‐16‐CE12‐0022‐01 to DL and PP; ANR‐21‐CE12‐0040‐01 to DL, ANR‐19‐CE12‐0016‐01 to PP and ANR‐22‐CE12‐0031‐01 to JP), and the Institut Universitaire de France (IUF to JP). SK thanks the French Ministère de la Recherche et de l'Enseignement Superieur (MRES) for fellowship. JH and AT were supported by ANR‐20‐CE12‐0016‐01 to AL and ANR‐19‐CE12‐0023‐03 to AL. UA was supported by the French Ministry for Education and Research, by the Fondation ARC pour la Recherche sur le Cancer, and the EUR G.E.N.E. (reference #ANR‐17‐EURE‐0013), which is part of the Université Paris Cité IdEx #ANR‐18‐IDEX‐0001 funded by the French Government through its “Investments for the Future” program.
The EMBO Journal (2023) 42: e113104
Contributor Information
Yea‐Lih Lin, Email: yea-lih.lin@igh.cnrs.fr.
Armelle Lengronne, Email: armelle.lengronne@igh.cnrs.fr.
Jérôme Poli, Email: jerome.poli@igh.cnrs.fr.
Philippe Pasero, Email: philippe.pasero@igh.cnrs.fr.
Data availability
The datasets generated in this study are available on the Gene Expression Omnibus GSE215896 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215896).
References
- Aiello U, Challal D, Wentzinger G, Lengronne A, Appanah R, Pasero P, Palancade B, Libri D (2022) Sen1 is a key regulator of transcription‐driven conflicts. Mol Cell 82: 2952–2966 [DOI] [PubMed] [Google Scholar]
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M et al (2012) Senataxin associates with replication forks to protect fork integrity across RNA‐polymerase‐II‐transcribed genes. Cell 151: 835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrs M, Stoy H, Boleslavska B, Chappidi N, Kanagaraj R, Nascakova Z, Menon S, Rao S, Oravetzova A, Dobrovolna J et al (2023) Excessive reactive oxygen species induce transcription‐dependent replication stress. Nat Commun 14: 1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appanah R, Lones EC, Aiello U, Libri D, De Piccoli G (2020) Sen1 is recruited to replication forks via Ctf4 and Mrc1 and promotes genome stability. Cell Rep 30: 2094–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoynaud C, Schirmeisen K, Ait Saada A, Gesnik A, Fernández‐Varela P, Boucherit V, Ropars V, Chaudhuri A, Fréon K, Charbonnier JB et al (2023) RNA:DNA hybrids from Okazaki fragments contribute to establish the Ku‐mediated barrier to replication‐fork degradation. Mol Cell 83: 1061–1074 [DOI] [PubMed] [Google Scholar]
- Bacal J, Moriel Carretero M, Pardo B, Barthe A, Sharma S, Chabes A, Lengronne A, Pasero P (2018) Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J 37: e99319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso S, Herrera‐Moyano E, Muñoz S, García‐Rubio M, Gómez‐González B, Aguilera A (2019) The DNA damage response acts as a safeguard against harmful DNA‐RNA hybrids of different origins. EMBO Rep 20: e47250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowry A, Kelly RDW, Petermann E (2021) Hypertranscription and replication stress in cancer. Trends Cancer 7: 863–877 [DOI] [PubMed] [Google Scholar]
- Breslin C, Clements PM, El‐Khamisy SF, Petermann E, Iles N, Caldecott KW (2006) Measurement of chromosomal DNA single‐strand breaks and replication fork progression rates. Methods Enzymol 409: 410–425 [DOI] [PubMed] [Google Scholar]
- Brickner JR, Garzon JL, Cimprich KA (2022) Walking a tightrope: the complex balancing act of R‐loops in genome stability. Mol Cell 82: 2267–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck D, Reijns MA, Graham SC, Astell KR, Jones EY, Jackson AP (2011) PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res 39: 3652–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelli T, Challal D, Briand JB, Boulay J, Porrua O, Colin J, Libri D (2018) High‐resolution transcription maps reveal the widespread impact of roadblock termination in yeast. EMBO J 37: e97490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo‐Guzman D, Chédin F (2021) Defining R‐loop classes and their contributions to genome instability. DNA Repair (Amst) 106: 103182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ (2019) RNase H2‐RED carpets the path to eukaryotic RNase H2 functions. DNA Repair (Amst) 84: 102736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Iranzo J, Sharma S, Chabes A, Crouch RJ, Tollervey D, El Hage A (2020) High density of unrepaired genomic ribonucleotides leads to Topoisomerase 1‐mediated severe growth defects in absence of ribonucleotide reductase. Nucleic Acids Res 48: 4274–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challal D, Barucco M, Kubik S, Feuerbach F, Candelli T, Geoffroy H, Benaksas C, Shore D, Libri D (2018) General regulatory factors control the fidelity of transcription by restricting non‐coding and ectopic initiation. Mol Cell 72: 955–969 [DOI] [PubMed] [Google Scholar]
- Challal D, Colin J, Villa T, Libri D (2022) A modified cross‐linking analysis of cDNAs (CRAC) protocol for detecting RNA‐protein interactions and transcription at single‐nucleotide resolution. Methods Mol Biol 2477: 35–55 [DOI] [PubMed] [Google Scholar]
- Chappidi N, Nascakova Z, Boleslavska B, Zellweger R, Isik E, Andrs M, Menon S, Dobrovolna J, Balbo Pogliano C, Matos J et al (2020) Fork cleavage‐religation cycle and active transcription mediate replication restart after fork stalling at co‐transcriptional R‐loops. Mol Cell 77: 528–541 [DOI] [PubMed] [Google Scholar]
- Chen YH, Keegan S, Kahli M, Tonzi P, Fenyo D, Huang TT, Smith DJ (2019) Transcription shapes DNA replication initiation and termination in human cells. Nat Struct Mol Biol 26: 67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Puget N, Lin Y‐L, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, Legube G (2018) Senataxin resolves RNA:DNA hybrids forming at DNA double‐strand breaks to prevent translocations. Nat Commun 9: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva M, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A et al (2018) SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61 [DOI] [PubMed] [Google Scholar]
- Daley JM, Tomimatsu N, Hooks G, Wang W, Miller AS, Xue X, Nguyen KA, Kaur H, Williamson E, Mukherjee B et al (2020) Specificity of end resection pathways for double‐strand break regions containing ribonucleotides and base lesions. Nat Commun 11: 3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre A, Barthe A, de la Roche S‐AC, Luciano P, Forey R, Padioleau I, Skrzypczak M, Ginalski K, Géli V, Pasero P et al (2020) MRX increases chromatin accessibility at stalled replication forks to promote nascent DNA resection and cohesin loading. Mol Cell 77: 395–410 [DOI] [PubMed] [Google Scholar]
- Dubacq C, Chevalier A, Courbeyrette R, Petat C, Gidrol X, Mann C (2006) Role of the iron mobilization and oxidative stress regulons in the genomic response of yeast to hydroxyurea. Mol Genet Genomics 275: 114–124 [DOI] [PubMed] [Google Scholar]
- Fang D, Lengronne A, Shi D, Forey R, Skrzypczak M, Ginalski K, Yan C, Wang X, Cao Q, Pasero P et al (2017) Dbf4 recruitment by forkhead transcription factors defines an upstream rate‐limiting step in determining origin firing timing. Genes Dev 31: 2405–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Muse T, Aguilera A (2019) R loops: from physiological to pathological roles. Cell 179: 604–618 [DOI] [PubMed] [Google Scholar]
- Gomez‐Gonzalez B, Aguilera A (2019) Transcription‐mediated replication hindrance: a major driver of genome instability. Genes Dev 33: 1008–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D (2009) Identification of protein binding sites on U3 snoRNA and pre‐rRNA by UV cross‐linking and high‐throughput analysis of cDNAs. Proc Natl Acad Sci USA 106: 9613–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Albulescu LO, Cristini A, Gromak N (2017) Senataxin: genome guardian at the interface of transcription and neurodegeneration. J Mol Biol 429: 3181–3195 [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA (2017) Transcription‐replication conflict orientation modulates R‐loop levels and activates distinct DNA damage responses. Cell 170: 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SYN, Williams JS, Arana ME, Kunkel TA, Pommier Y (2017) Topoisomerase I‐mediated cleavage at unrepaired ribonucleotides generates DNA double‐strand breaks. EMBO J 36: 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst V, Challa K, Jonas F, Forey R, Sack R, Seebacher J, Schmid CD, Barkai N, Shimada K, Gasser SM et al (2021) A regulatory phosphorylation site on Mec1 controls chromatin occupancy of RNA polymerases during replication stress. EMBO J 40: e108439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyjek M, Figiel M, Nowotny M (2019) RNases H: structure and mechanism. DNA Repair (Amst) 84: 102672 [DOI] [PubMed] [Google Scholar]
- Im JS, Keaton M, Lee KY, Kumar P, Park J, Dutta A (2014) ATR checkpoint kinase and CRL1betaTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev 28: 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemiha S, Poli J, Lin YL, Lengronne A, Pasero P (2021) Toxic R‐loops: cause or consequence of replication stress? DNA Repair (Amst) 107: 103199 [DOI] [PubMed] [Google Scholar]
- Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks‐Robertson S (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332: 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc A, Wheeler LJ, Mathews CK, Merrill GF (2004) Hydroxyurea arrests DNA replication by a mechanism that preserves Basal dNTP pools. J Biol Chem 279: 223–230 [DOI] [PubMed] [Google Scholar]
- Kumar C, Batra S, Griffith JD, Remus D (2021) The interplay of RNA:DNA hybrid structure and G‐quadruplexes determines the outcome of R‐loop‐replisome collisions. eLife 10: e72286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde M, Trauner M, Werner M, Hamperl S (2021) Consequences and resolution of transcription‐replication conflicts. Life (Basel) 11: 637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsverk HB, Sandquist LE, Bay LTE, Steurer B, Campsteijn C, Landsverk OJB, Marteijn JA, Petermann E, Trinkle‐Mulcahy L, Syljuåsen RG (2020) WDR82/PNUTS‐PP1 prevents transcription‐replication conflicts by promoting RNA polymerase II degradation on chromatin. Cell Rep 33: 108469 [DOI] [PubMed] [Google Scholar]
- Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, Merrikh H (2017) Replication‐transcription conflicts generate R‐loops that orchestrate bacterial stress survival and pathogenesis. Cell 170: 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Novarina D, Amara F, Watt Danielle L, Stone Jana E, Costanzo V, Burgers Peter M, Kunkel Thomas A, Plevani P, Muzi‐Falconi M (2012) RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell 45: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YL, Noel D, Mettling C, Reant B, Clot J, Jorgensen C, Corbeau P (2004) Feline immunodeficiency virus vectors for efficient transduction of primary human synoviocytes: application to an original model of rheumatoid arthritis. Hum Gene Ther 15: 588–596 [DOI] [PubMed] [Google Scholar]
- Liu W, Saito Y, Jackson J, Bhowmick R, Kanemaki MT, Vindigni A, Cortez D (2023) RAD51 bypasses the CMG helicase to promote replication fork reversal. Science 380: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Pires VB, Bento F, Kellner V, Luke‐Glaser S, Yakoub G, Ulrich HD, Luke B (2019) RNase H1 and H2 are differentially regulated to process RNA‐DNA hybrids. Cell Rep 29: 2890–2900 [DOI] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD (2018) Intragenic origins due to short G1 phases underlie oncogene‐induced DNA replication stress. Nature 555: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo SG, Zhou ZL, Wang YQ, Marinello J, He JX, Li YC, Ding J, Capranico G, Miao ZH (2012) Natural product triptolide mediates cancer cell death by triggering CDK7‐dependent degradation of RNA polymerase II. Cancer Res 72: 5363–5373 [DOI] [PubMed] [Google Scholar]
- Marnef A, Legube G (2021) R‐loops as Janus‐faced modulators of DNA repair. Nat Cell Biol 23: 305–313 [DOI] [PubMed] [Google Scholar]
- Meroni A, Nava GM, Bianco E, Grasso L, Galati E, Bosio MC, Delmastro D, Muzi‐Falconi M, Lazzaro F (2019) RNase H activities counteract a toxic effect of Polymerase eta in cells replicating with depleted dNTP pools. Nucleic Acids Res 47: 4612–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gomez‐Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ (2011) Yeast Sen1 helicase protects the genome from transcription‐associated instability. Mol Cell 41: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CA, Hawkins M, Retkute R, Malla S, Wilson R, Blythe MJ, Nakato R, Komata M, Shirahige K, de Moura APS et al (2014) The dynamics of genome replication using deep sequencing. Nucleic Acids Res 42: e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava GM, Grasso L, Sertic S, Pellicioli A, Muzi Falconi M, Lazzaro F (2020) One, no one, and one hundred thousand: the many forms of ribonucleotides in DNA. Int J Mol Sci 21: 1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220 [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L (2017) Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol Cell 65: 832–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohle C, Tesorero R, Schermann G, Dobrev N, Sinning I, Fischer T (2016) Transient RNA‐DNA hybrids are required for efficient double‐strand break repair. Cell 167: 1001–1013 [DOI] [PubMed] [Google Scholar]
- Ouyang J, Yadav T, Zhang JM, Yang H, Rheinbay E, Guo H, Haber DA, Lan L, Zou L (2021) RNA transcripts stimulate homologous recombination by forming DR‐loops. Nature 594: 283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Vindigni A (2017) Nucleases acting at stalled forks: how to reboot the replication program with a few shortcuts. Annu Rev Genet 51: 477–499 [DOI] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d'Aubenton‐Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C‐L, Hyrien O (2016) Replication landscape of the human genome. Nat Commun 7: 10208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbe L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P (2012) dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Gerhold CB, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner KP et al (2016) Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 30: 337–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promonet A, Padioleau I, Liu Y, Sanz L, Biernacka A, Schmitz AL, Skrzypczak M, Sarrazin A, Mettling C, Rowicka M et al (2020) Topoisomerase 1 prevents replication stress at R‐loop‐enriched transcription termination sites. Nat Commun 11: 3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Lemaçon D, Vindigni A (2017) Replication fork reversal: players and guardians. Mol Cell 68: 830–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F et al (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martin‐Alonso M, Soler‐Oliva ME, García‐Rubio M, García‐Muse T, Aguilera A (2021) Harmful R‐loops are prevented via different cell cycle‐specific mechanisms. Nat Commun 12: 4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz Lionel A, Hartono Stella R, Lim Yoong W, Steyaert S, Rajpurkar A, Ginno Paul A, Xu X, Chédin F (2016) Prevalent, dynamic, and conserved R‐loop structures associate with specific epigenomic signatures in mammals. Mol Cell 63: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Pasero P, Gasser SM (2002) ORC and the intra‐S‐phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16: 3236–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti‐Stathaki K, Proudfoot Nicholas J, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2‐dependent termination. Mol Cell 42: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Gupta R, Sedlackova H, Neelsen KJ, Ochs F, Rask MB, Choudhary C, Lukas J (2017) Redox‐sensitive alteration of replisome architecture safeguards genome integrity. Science 358: 797–802 [DOI] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, Cimprich KA (2016) Co‐transcriptional R‐loops are the main cause of estrogen‐induced DNA damage. eLife 5: e17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoy H, Zwicky K, Kuster D, Lang KS, Krietsch J, Crossley MP, Schmid JA, Cimprich KA, Merrikh H, Lopes M (2023) Direct visualization of transcription‐replication conflicts reveals post‐replicative DNA:RNA hybrids. Nat Struct Mol Biol 30: 348–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan‐Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE (2019) R‐loop formation during S phase is restricted by PrimPol‐mediated repriming. EMBO J 38: e99793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira‐Silva A, Ait Saada A, Hardy J, Iraqui I, Nocente MC, Fréon K, Lambert SAE (2017) The end‐joining factor Ku acts in the end‐resection of double strand break‐free arrested replication forks. Nat Commun 8: 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittel‐Elmer M, Lengronne A, Davidson Marta B, Bacal J, François P, Hohl M, Petrini John HJ, Pasero P, Cobb Jennifer A (2012) Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol Cell 48: 98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière H, Saksouk J, Lengronne A, Pasero P (2017) Single‐molecule analysis of DNA replication dynamics in budding yeast and human cells by DNA combing. Bio Protoc 7: e2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirkas I, Dovrat D, Thangaraj M, Brouwer I, Cohen A, Paleiov Z, Meijler MM, Lenstra T, Aharoni A (2022) Transcription‐replication coordination revealed in single live cells. Nucleic Acids Res 50: 2143–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve‐Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C et al (2009) Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11: 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufegdžić Vidaković A, Mitter R, Kelly GP, Neumann M, Harreman M, Rodríguez‐Martínez M, Herlihy A, Weems JC, Boeing S, Encheva V et al (2020) Regulation of the RNAPII pool is integral to the DNA damage response. Cell 180: 1245–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye S, Ronson GE, Morris JR (2020) A fork in the road: where homologous recombination and stalled replication fork protection part ways. Semin Cell Dev Biol 113: 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA (2013) Topoisomerase 1‐mediated removal of ribonucleotides from nascent leading‐strand DNA. Mol Cell 49: 1010–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Lujan SA, Kunkel TA (2016) Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol 17: 350–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Gehle DB, Kunkel TA (2017) The role of RNase H2 in processing ribonucleotides incorporated during DNA replication. DNA Repair 53: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zardoni L, Nardini E, Brambati A, Lucca C, Choudhary R, Loperfido F, Sabbioneda S, Liberi G (2021) Elongating RNA polymerase II and RNA:DNA hybrids hinder fork progression and gene expression at sites of head‐on replication‐transcription collisions. Nucleic Acids Res 49: 12769–12784 [DOI] [PMC free article] [PubMed] [Google Scholar]