

Key Points
-
•
Ongoing endothelial activation is common in children with SCD despite disease-modifying therapy with hydroxycarbamide or blood transfusion.
-
•
Persistent VWF–ADAMTS13 axis dysfunction identifies a subgroup of treated SCD children at increased risk of vaso-occlusive complications.
Visual Abstract
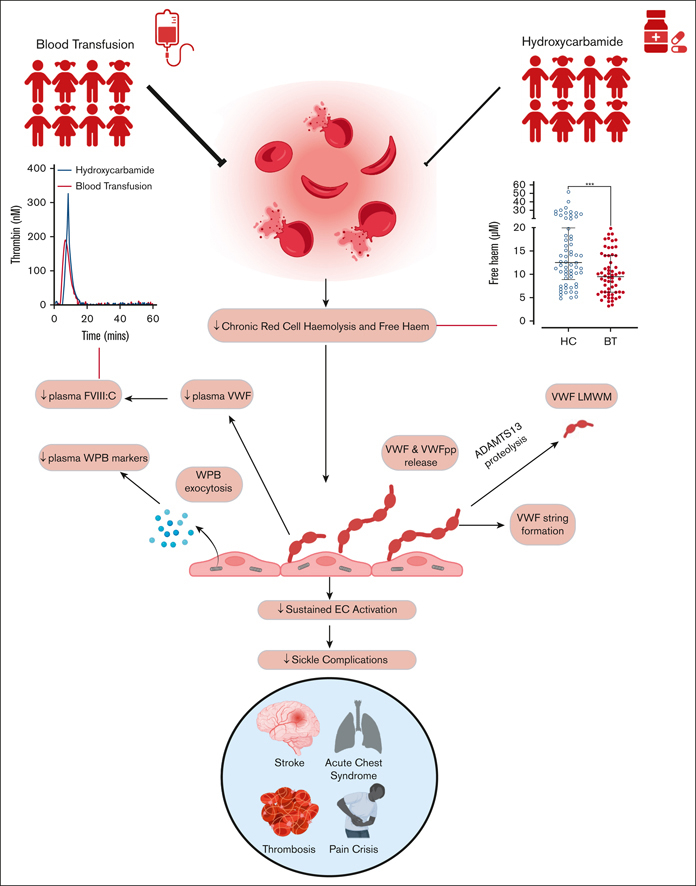
Abstract
Previous studies have reported elevated von Willebrand factor (VWF) levels in patients with sickle cell disease (SCD) and demonstrated a key role for the VWF-ADAMTS13 axis in the pathobiology of SCD vaso-occlusion. Although blood transfusion is the gold standard for stroke prevention in SCD, the biological mechanisms underpinning its improved efficacy compared with hydroxycarbamide are not fully understood. We hypothesized that the improved efficacy of blood transfusion might relate to differences in VWF–ADAMTS13 axis dysfunction. In total, 180 children with a confirmed diagnosis of SCD (hemoglobin SS) on hydroxycarbamide (n = 96) or blood transfusion (n = 84) were included. Despite disease-modifying treatment, plasma VWF and VWF propeptide were elevated in a significant proportion of children with SCD (33% and 47%, respectively). Crucially, all VWF parameters were significantly higher in the hydroxycarbamide compared with the blood transfusion cohort (P < .05). Additionally, increased levels of other Weibel-Palade body–stored proteins, including factor VIII (FVIII), angiopoietin-2, and osteoprotegerin were observed, indicated ongoing endothelial cell activation. Children treated with hydroxycarbamide also had higher FVIII activity and enhanced thrombin generation compared with those in the blood transfusion cohort (P < .001). Finally, hemolysis markers strongly correlated with VWF levels (P < .001) and were significantly reduced in the blood transfusion cohort (P < .001). Cumulatively, to our knowledge, our findings demonstrate for the first time that despite treatment, ongoing dysfunction of the VWF–ADAMTS13 axis is present in a significant subgroup of pediatric patients with SCD, especially those treated with hydroxycarbamide.
Introduction
Sickle cell disease (SCD) is characterized by chronic hemolytic anemia, inflammatory vasculopathy, and intermittent episodes of acute vaso-occlusive crises.1,2 Although the pathophysiology underlying microvascular occlusion in SCD remains incompletely understood, accumulating evidence suggests it involves interactions between sickle erythrocytes, leucocytes, platelets, and vascular endothelial cells.3, 4, 5, 6, 7 von Willebrand factor (VWF) is synthesized within endothelial cells and circulates in normal plasma as a series of heterogeneous multimers.8 After endothelial cell synthesis, VWF is either constitutively secreted into the plasma, or stored within intracellular Weibel-Palade bodies as high-molecular-weight multimers.8,9 Under normal conditions, plasma VWF multimer distribution is regulated by ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, number 13).8 VWF plays key roles in normal hemostasis, by facilitating platelet recruitment to sites of vascular injury, and by acting as a carrier molecule for procoagulant factor VIII (FVIII).8, 9, 10, 11 In addition, VWF can also bind to both neutrophils12 and macrophages,13,14 and plays an important role in linking primary hemostasis to innate immune responses.15,16
Previous studies have reported elevated plasma VWF antigen (VWF:Ag) levels in children and adults with SCD, with significantly higher VWF levels in patients with severe (hemoglobin SS [HbSS] or HbSβ0) compared with milder (HbSC or HbSβ+) genotypes.17, 18, 19 Moreover, plasma VWF:Ag levels were further elevated in patients with SCD presenting with acute vaso-occlusive crisis.20, 21, 22, 23 Pathological accumulation of ultralarge VWF multimers in the plasma of patients with SCD has also been reported.24,25 The biological mechanisms responsible for these hyperreactive ultralarge VWF multimers in patients with SCD remains poorly understood but mild decreases in plasma ADAMTS13 levels have been described.17,23,25 Importantly, a significant negative correlation was also observed between plasma ADAMTS13 levels and silent cerebral infarcts in patients with HbSS and HbSβ0.17
The combination of markedly elevated VWF:Ag levels and hyperreactive ultralarge VWF multimers raises the possibility that endothelial cells (EC)-derived VWF may play a direct role in the pathogenesis of SCD vaso-occlusion. This concept is supported by a number of independent observations. First, studies have demonstrated that a significant proportion of plasma VWF in patients with SCD circulates in an active confirmation that promotes platelet GpIbα binding.24,25 Importantly, platelets have also been implicated in facilitating adhesion of sickle erythrocytes to activated endothelial cells. Second, ultralarge VWF secreted from activated endothelial cells has been shown to directly bind and tether sickle erythrocytes in shear-based experiments.26,27 Third, recent studies have demonstrated that VWF plays a direct role in regulating hemolysis, vascular occlusion, and organ damage after tumor necrosis factor–induced vaso-occlusive crises in a murine SCD model.28 Consistently, Rossato et al also recently showed that treatment with recombinant ADAMTS13 significantly reduced acute SCD-related hemolysis and organ damage in a humanized SCD mouse model.29 Collectively, these data suggest a key role for the VWF–ADAMTS13 axis in regulating the pathobiology underlying vaso-occlusive crises in SCD.30
Although novel drugs have recently been approved for SCD, the mainstays of pediatric therapy remain hydroxycarbamide (HC) and chronic blood transfusion (BT). The efficacy of BT for stroke prevention in SCD was highlighted by the STOP study, which demonstrated a 90% reduction in risk of stroke in children at high risk.31 Consequently, consensus guidelines recommend regular transfusion to maintain HbS of <30% in children with SCD who are at high risk.32,33 Critically however, although BT is the gold standard for stroke prevention in SCD, the biological mechanisms underpinning its improved efficacy compared with HC are not fully understood. In this study, we investigated the hypothesis that the clinical efficacy of BT over HC in children with SCD might relate in part to differences in endothelial cell activation and VWF–ADAMTS13 axis dysfunction.
Materials and methods
Patients
The Sickle Vascular Ireland Consortium study was approved by the ethics committee (Medical Research; GEN/696/18) at Children’s Health Ireland at Crumlin. Children with SCD were recruited from the Comprehensive Sickle Cell Disease Centre at Children’s Health Ireland, between April 2019 and March 2020. Indications for BT were abnormal transcranial Doppler velocity of the middle cerebral artery (>200 cm/s) and/or a history of stroke or ischemic changes on magnetic resonance imaging. Informed written consent was obtained from parents/legal guardians, and written assent obtained from all children. Inclusion criteria were children aged ≥4 years with a confirmed diagnosis of SCD (HbSS), in steady state, established on disease modification therapy (either HC or BT) for at least 6 months. Exclusion criteria were children aged <4 years, non-HbSS sickle genotypes, and recent (<3 months) vaso-occlusive crisis. Samples were collected 24 hours before the next transfusion for children receiving BT, and at outpatient review for those receiving HC. Venous blood samples were collected into 3.2% sodium citrate tubes and platelet-poor plasma generated by double-spun centrifugation at 3000g for 10 minutes. EDTA and serum tubes were obtained for full blood count, Hb electrophoresis, and biochemistry profiles.
Coagulation, thrombin generation, and Weibel-Palade body protein assays
Plasma levels of VWF:Ag, VWF propeptide (VWFpp), and VWF collagen binding (VWF:CB) activity were measured by enzyme-linked immunosorbent assay, as previously described.34 Thrombin generation was performed in a Fluouroskan Ascent Fluorometer with Thrombinoscope software (Stago) using platelet poor plasma low reagent (1 pM tissue factor, 4 mM phospholipids). FVIII coagulant activity (FVIII:C) levels were measured by 1-stage clotting assay on the ACL Top 750CTS analyzer (Instrumentation Laboratory) using HemosIL SynthasIL APTT reagent. ADAMTS13 activity was quantified using a commercial FRETS-VWF73 assay (Peptides International, Inc). VWF multimer analysis was performed by electrophoresis using 1.8% agarose gels, as previously described.35 Plasma free heme concentration was measured using a colorimetric assay (Sigma Aldrich) and plasma angiopoietin-2, osteoprotegerin, platelet factor 4 (PF4), and interleukin-6 (IL-6) levels were all measured using commercial enzyme-linked immunosorbent assays according to the manufacturers’ instructions (R&D Systems, Abingdon, Oxfordshire, United Kingdom).
Red cell phosphatidylserine (PS) exposure and VWF binding
Whole blood was collected into 3.8% sodium citrate tubes from healthy volunteers and patients with SCD. For this assay, patients receiving BTs were all on a 3-weekly transfusion program, with blood samples obtained immediately before commencing their next transfusion. As is standard of care in our institution, red cell components used for pediatric top-up transfusions in SCD transfusion are <10 days old. As before, patients taking HC had blood samples collected at time of outpatient review. Platelet-rich plasma was separated from the erythrocyte-rich pellet by centrifugation at 210g for 10 minutes. The erythrocyte pellet was washed 3 times in Ringer’s buffer (125 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 2.5 mmol/L CaCl2, 5 mmol/L glucose, and 32.2 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) and then resuspended at a concentration of 5 × 107 cells per mL. To measure PS exposure on erythrocyte membranes, red blood cells (RBCs) were stained with fluorochrome-labeled annexin V–fluorescein isothiocyanate (Biolegend). To measure the VWF-binding capacity of RBCs, recombinant VWF (75 μg/mL, Vonvendi) was incubated with RBCs for 30 minutes at 37°C. Fc receptors were blocked using Fc Block (BD Biosciences), and RBCs stained with polyclonal rabbit anti-human VWF (Agilent) followed by anti-rabbit immunoglobulin G Alexa Fluor 488 (ThermoFisher Scientific) and fixable Far Red dye (ThermoFisher Scientific) for dead cells stain. To measure VWF-bound to RBC, unwashed RBCs were incubated in 1% bovine serum albumin (Sigma) for 30 minutes at 37°C, followed by staining with polyclonal rabbit anti-human VWF (Agilent) for 30 minutes and anti-rabbit immunoglobulin G Alexa Fluor 488 (ThermoFisher Scientific). Flow cytometry was performed on an Attune NxT, and results were analyzed by FlowJo version 10.8.1 Software (BD Life Sciences; see supplemental Figures 1 and 2 for gating strategies).
Data presentation and statistical analysis
All experimental data and statistical analysis were performed using the GraphPad Prism program (GraphPad Prism version 9.30; GraphPad Software, Inc, San Diego, CA) or SPSS Statistics version 24 (IBM Corp, Armonk, NY). Data were expressed as median values ± interquartile range (IQR), unless otherwise indicated. To assess statistical differences, data were analyzed using unpaired 2-tailed Student t test for normative data or Mann-Whitney U test for nonnormative data, and by analysis of variance to compare means of ≥3 groups after Kolmogorov-Smirnov or Shapiro-Wilk tests for normality. Correlation testing was performed using the Spearman rank correlation test. Mediation analyses were performed using multiple regression models. For all statistical tests, P values <.05 were considered significant.
Results
Baseline patient demographic and laboratory data
A total of 180 children with SCD (median age, 12 years [IQR, 9-16]; 46% male) were included. Of the total cohort, 84 children were on BTs and 96 children on HC therapy at enrollment (Table 1). Clinical demographics at time of study entry are presented in Table 1 including indication, age at commencement of HC treatment or BT, and duration for each therapy. All children with SCD aged >12 months were offered HC in line with consensus clinical guidelines. The majority commenced with BTs for abnormal transcranial Dopplers (47%), followed by ischemic changes (silent stroke) on magnetic resonance imaging (27%), recurrent crisis episodes despite HC (18%), and overt stroke (8%) (Table 1). The mean duration of HC and BT was 65.1 (standard deviation, 37.7) and 67.7 (standard deviation, 50.3) months, respectively. The average HC dose was 19 mg/kg (range, 10-33 mg/kg). All children receiving BTs were on simple transfusion, and none had thrombotic complications. Laboratory investigations at time of study entry are presented in Table 2. Hb, red cell count, and hematocrit levels were significantly lower in the HC cohort compared with the BT cohort (Table 2). Similarly, total white cell and neutrophil counts were also reduced in the HC cohort. In contrast, mean corpuscle volume, and HbS and HbF levels were all significantly higher in children treated with HC compared with those treated with BT (Table 2).
Table 1.
Demographic and clinical parameters of the pediatric SCD cohort
| Demographics at study entry | Total cohort (N = 180) | HC (n = 96) | BT (n = 84) |
|---|---|---|---|
| Age (y), mean (SD, range) | 12 (4, 4-19) | 12 (7, 4-19) | 12 (6, 4-19) |
| Gender, male/female | 82/98 | 45/51 | 37/47 |
| ABO blood group, O/non-O (%) | 60.5/39.5 | 60/40 | 61/39 |
| Age (y) at starting therapy, mean (SD) | 7.1 (4.7) | 6.2 (3.9) | 8.7 (5.3) |
| Duration of therapy (mo), mean (SD) | 66.0 (42.9) | 65.1 (37.7) | 67.7 (50.3) |
| HC dose (mg/kg), mean (range) | NA | 19 (10-33) | NA |
| Indication for therapy (%) | |||
| Routinely offered at age >1 y | 100 | NA | |
| Abnormal TCD | NA | 47 | |
| Ischemia on MRI (silent stroke) | NA | 27 | |
| Stroke | NA | 8 | |
| Recurrent crises despite HC | NA | 18 | |
MRI, magnetic resonance imaging; NA, not applicable; SD, standard deviation; TCD, transcranial Doppler.
Table 2.
Laboratory parameters of the pediatric SCD cohort
| Laboratory parameters at study entry, median (IQR) | Normal pediatric reference range | Total cohort (N = 180) | HC (n = 96) | BT (n = 84) | P value |
|---|---|---|---|---|---|
| Hb (g/dL) | 115-155 | 94 (84-101) | 87 (80-95) | 98 (93-106) | <.0001 |
| MCV (fL) | 77-96 | 86 (82-94) | 94 (86-99) | 85 (83-86) | <.0001 |
| RCC (×109/L) | 4-5.2 | 3.1 (2.5-3.5) | 2.6 (2.3-3) | 3.4 (3.2-3.6) | <.0001 |
| HCT (%) | 35-45 | 27 (23-29) | 24 (22-260) | 28 (27-30) | <.0001 |
| MCH (pg) | 25-33 | 30 (29-34) | 33 (31-37) | 29 (28-30) | <.0001 |
| MCHC (g/L) | 315-370 | 352 (343-362) | 361 (352-368) | 346 (340-350) | <.0001 |
| Platelets (×109/L) | 150-450 | 291 (204-389) | 313 (213-405) | 243 (184-365) | .007 |
| Leucocytes (×109/L) | 4.5-13.5 | 8.2 (6.3-9.7) | 7.6 (6-9.1) | 8.7 (6.7-11.1) | .0015 |
| Neutrophils (×109/L) | 1.5-8.0 | 3.7 (2.9-5.4) | 3.1 (2.2-4.1) | 5.1 (3.5-6.7) | <.0001 |
| HbS, % | NA | 58 (21-77) | 76 (72-90) | 20 (14-27) | <.0001 |
| HbF, % | 0-2 | 10 (3-16) | 16 (12-21) | 2.4 (1.4-4) | <.0001 |
| Reticulocyte (×109/L) | 14-99 | 184 (141-256) | 171 (141-228) | 200 (138-285) | .09 |
| Reticulocyte (%) | 0.4-1.9 | 6 (5-9) | 6 (5-9) | 6 (4-8) | .13 |
| Bilirubin (μmol/L) | 0-21 | 36 (25-55) | 33 (21-51) | 40 (27-57) | .02 |
| LDH (U/L) | 230-600 | 807 (290-1051) | 898 (689-1052) | 678 (515-968) | .0007 |
| Urea (mmol/L) | 4 (2.9-4.7) | 3 (2.4-3.6) | 5 (3.8-6) | <.0001 | |
| Creatinine (μmol/L) | 40 (31-50) | 38 (29-47) | 44 (35-51) | .01 | |
| AST (U/L) | <40 | 41 (33-54) | 43 (34-54) | 38 (32-53) | .22 |
| ALT (U/L) | <30 | 19 (14-26) | 20 (14-26) | 18 (13-24.3) | .46 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; HCT, hematocrit; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscle value; RCC, red cell count.
VWF levels in patients with SCD treated with HC compared with BT
Despite HC or BT treatment, we observed that plasma VWF:Ag levels and VWF:CB activity were significantly elevated in a significant proportion of our pediatric cohort (33% and 20%, respectively; Figure 1A-B). Marked interindividual heterogeneity in plasma VWF levels was apparent in both subgroups (total, VWF:Ag range, 0.41-2.71; HC, VWF:Ag range, 0.55-2.35 IU/mL; BT, VWF:Ag range, 0.41-2.71 IU/mL). Importantly, plasma VWF:Ag (Figure 1A) and VWF:CB activity (Figure 1B) levels were both significantly higher in the HC cohort compared with the BT cohort (total, median VWF:Ag, 1.14 IU/mL; HC, 1.28 IU/mL vs BT, 1.03 IU/mL; P = .0005; total, median VWF:CB, 1.34; HC, 1.46 IU/mL vs BT, 1.16 IU/mL; P = .002). Although high rates of self-reported compliance with HC therapy have previously been identified in our institution,36 comparing the highest and lowest quartiles of HbF in the HC cohort did not reveal significant differences in VWF parameters (supplemental Figure 3A-B). Conversely, among children receiving BT, VWF:Ag and VWF:CB levels were significantly higher in children within the highest HbS quartile compared with the lowest quartile (VWF:Ag, 1.11 vs 0.81 IU/mL; P = .03; and VWF:CB 1.50 vs 1.14 IU/mL; P = .04; supplemental Figure 3C-D).
Figure 1.

Plasma VWF levels in children with SCD treated with hydroxycarbamide compared to blood transfusion. Comparisons between children with SCD treated with HC or BT are shown for plasma levels of (A) VWF:Ag, (B) VWF:CB, and (C) VWFpp. (D) VWF:Ag levels are shown comparing group O and non-O blood groups. Dotted red lines denote the upper and lower limit of the local reference range, with green shaded area falling within normal limits. Data are presented as median and the IQR. Comparisons between groups were assessed by the Mann-Whitney U test. Correlations between plasma VWF:Ag levels and (E) Hb and (F) LDH are shown. Correlations were evaluated using the Spearman rank correlation test; ∗∗P < .01, ∗∗∗P < .001, and ∗∗∗∗P < .0001.
Plasma VWFpp levels have been used to gain insights into acute endothelial cell activation in other microangiopathic conditions.37, 38, 39 Consistent with the VWF:Ag data, plasma VWFpp levels were significantly increased in 47% of the total SCD cohort despite their disease modification therapy, and also displayed marked heterogeneity (total cohort, median VWFpp, 1.40 IU/mL [IQR, 0.94-2.0]; Figure 1C). In addition, VWFpp levels were also significantly higher in the HC cohort compared with BT cohort (median, VWFpp 1.64 vs 1.14 IU/mL; P = .002).
In spite of the increased VWF levels in our pediatric SCD cohort, an ABO effect was still apparent with significantly reduced plasma VWF:Ag and VWF:CB in blood group O compared with children with SCD in non-O blood groups (median VWF:Ag 1.02 vs 1.32 IU/mL, P < .0001; median VWF:CB 1.20 vs 1.44 IU/mL, P = .0005; Figure 1D; supplemental Figure 4A). In keeping with the hypothesis that ABO blood group influences VWF clearance in children with SCD, no effect of ABO on plasma VWFpp levels was seen (supplemental Figure 4B). Finally, we observed significant correlations between VWF parameters and established biomarkers of hemolysis severity (including hemoglobin and lactate dehydrogenase [LDH] levels; Figure 1E-F). Taken together, these findings indicate that a significant proportion of children with SCD maintained on disease-modifying therapy with either HC or BT continue to have significant increases in plasma VWF:Ag and VWFpp levels. Furthermore, these increased plasma VWF levels are significantly more common in children with SCD treated with HC compared with those treated with BT.
VWF multimers in patients with SCD treated with HC compared with BT
Next, we investigated the effects of SCD disease-modifying treatment on plasma VWF multimer distribution and ADAMTS13 activity. High-molecular-weight VWF multimers were significantly reduced (P < .0001) in children with SCD treated with HC compared with the cohort maintained on BT, or normal pooled plasma (Figure 2A-B). In addition to the loss of high-molecular-weight VWF multimers, significant increases in both intermediate- and low-molecular-weight VWF multimers were observed in the HC-treated compared with BT-treated subgroups (Figure 2C-D). Despite the differences in VWF multimer distribution, plasma ADAMTS13 activity levels were similar in the BT and HC cohorts (Figure 3A). Importantly, however, the VWF/ADAMTS13 ratios of >1.0 were observed in 74% of children in the HC compared with 60% of children in the BT cohort (total median VWF/ADAMTS13 ratio, 1.25; HC 1.41 vs BT 1.10; P = .29; Figure 3B). PF4 and IL-6 have both been proposed as inhibitors of ADAMTS13 activity.40, 41, 42 Interestingly, PF4 and IL-6 levels were both significantly elevated in the HC- vs the BT-treated SCD cohorts (total median PF4 728 ng/mL, HC 971.9 vs BT 579.7 ng/mL, P = .0015; total median IL-6, 9.3 pg/mL; HC 45.1 vs BT 9.4 pg/mL; P = .0003; Figure 3C-D). Collectively, these data demonstrate that dysfunctional VWF multimer regulation persists in a significant subgroup of children with SCD on disease-modifying treatments, and that these VWF multimer abnormalities are more marked in children with SCD managed with HC rather than with BT. In addition, the marked increase in PF4 levels suggest significant ongoing platelet activation in a significant subgroup of children with SCD on HC or BT therapy.
Figure 2.

VWF multimer distribution in children with SCD treated with hydroxycarbamide compared to blood transfusion. (A) Representative images of VWF multimer distribution in HC and BT cohorts. Percentage of (B) high-, (C) intermediate-, and (D) low-molecular-weight VWF multimers. Comparisons between groups were assessed by the Mann-Whitney U test. Dotted red lines represent the percentage of high-, intermediate-, and low-molecular-weight multimers in normal pooled plasma (NPP); ∗P < .05, ∗∗∗P < .001, and ∗∗∗∗P < .0001.
Figure 3.

ADAMTS13 activity in SCD children treated with hydroxycarbamide compared to blood transfusion. (A)ADAMTS13 activity and (B) VWF:Ag/ADAMTS13 ratio are compared between the HC and BT cohorts. Dotted green line represents the local normal VWF:Ag/ADAMTS13 ratio (1.0). Plasma levels of PF4 (C) and IL-6 (D) are compared between the HC and BT cohorts. Comparisons between groups were assessed by the Mann-Whitney U test; ns, not significant; ∗∗P < .01 and ∗∗∗P < .001.
Endothelial cell activation and Weibel-Palade body exocytosis in patients with SCD treated with HC vs BT
Previous studies have used VWFpp/VWF:Ag ratios to assess VWF clearance rates.39,43 We observed that plasma VWFpp/VWF:Ag ratios were not significantly reduced in children with SCD (Figure 4A). Furthermore, VWFpp/VWF:Ag ratios were similar in HC- and BT-treated cohorts. Together, these findings suggest that the increased plasma VWF levels in pediatric SCD are predominantly attributable to increased endothelial cell secretion. To further address this hypothesis, we investigated plasma levels of other Weibel-Palade body–stored proteins including FVIII, angiopoietin-2, and osteoprotegerin, respectively. Interestingly, significantly increased plasma FVIII:C levels were seen in 65% of our total SCD pediatric cohort (median FVIII:C = 1.48 IU/mL; range, 0.62-2.82 IU/mL) despite the use of disease modification therapy (Figure 4B). In addition, FVIII:C levels were significantly higher in children being treated with HC compared those treated with BT (median, 1.71 vs 1.21 IU/mL; P < .0001; Figure 4B). Although plasma FVIII:C levels correlated significantly with VWF:Ag levels (r = 0.80; P < .001; Figure 4C), the FVIII:C/VWF:Ag ratio was elevated and was significantly higher in the HC vs BT cohort (total median FVIII:C/VWF:Ag ratio 1.34, HC 1.38 vs BT 1.26, P = .0056; Figure 4D). Similarly, plasma levels of both angiopoietin-2 and osteoprotegerin were also both significantly more elevated in children with SCD treated with HC compared with those treated with BT (total median angiopoietin-2, 3162 pg/mL; HC 4214 vs BT 2458 pg/mL; P = .0002; total median osteoprotegerin, 1690 pg/mL; HC 1825 vs BT 1507 pg/mL; P = .0065; Figure 4E-F). Cumulatively, these data suggest that despite disease-modifying therapy, a significant proportion of pediatric patients with SCD have persistent endothelial cell activation and consequent Weibel-Palade body exocytosis. This endothelial cell activation is more effectively but not completely suppressed in children receiving BT than those receiving HC treatment.
Figure 4.

VWF-ADAMTS13 axis and Weibel Palade body exocytosis in SCD children treated with hydroxycarbamide compared to blood transfusion. Comparisons between HC and BT cohorts are shown including (A) VWFpp/VWF:Ag ratio and (B) FVIIl:C levels. (C) Correlation between FVIII:C and VWF:Ag. (D) Comparisons between HC and BT cohorts are shown for FVIII:C/VWF:Ag ratio, (E) angiopoietin-2, and (F) osteoprotegerin. Comparisons between groups were assessed by the Mann-Whitney U test. Correlations were evaluated using the Spearman rank correlation test; ns, not significant; ∗∗P < .01, ∗∗∗P < .001, and ∗∗∗∗P < .0001.
Thrombin generation and hemolysis in patients with SCD treated with HC compared with BT
Because elevated plasma FVIII:C levels constitute a dose-dependent risk factor for venous thromboembolism,44 we investigated thrombin generation in platelet-poor plasma from the pediatric SCD cohort. Overall, significantly enhanced thrombin generation was observed in the HC subgroup compared with the BT subgroup (Figure 5A). Endogenous thrombin potential (total, median, 993 nM/min; BT 980 nM/min vs HC 1041 nM/min; P = .0006; Figure 5B), peak thrombin levels (total, mean, 169 nM; BT 162.6 nM vs HC 181.5 nM; P = .008; Figure 5C), and velocity index (total, median, 52.7 nM/min; BT 44.1 nM/min vs HC 58.8 nM/min; P = .019; Figure 5D) were all significantly reduced in patient in the BT cohort compared with those in the HC cohort. In addition, plasma FVIII:C levels correlated with peak thrombin generation (P < .001; Figure 5E). Together, these findings suggest that, in all children with SCD, disease-modifying agents, in particular HC therapy, do not effectively ameliorate enhanced thrombin generation. Furthermore, the ongoing increase in thrombin generation is likely mediated, at least in part, through endothelial cell activation, resulting in elevated plasma FVIII:C levels.
Figure 5.

Thrombin generation in children with SCD treated with hydroxycarbamide compared to blood transfusion. Comparisons between the HC and BT cohorts are shown including (A) thrombin generation curves from representative individuals within each cohort; (B) the quantitative parameters of endogenous thrombin potential, (C) peak thrombin, and (D) velocity index. Comparisons between groups were assessed by the Mann-Whitney U test. (E) Correlation between plasma FVIII:C levels and peak thrombin generated. Correlations were evaluated using the Spearman rank correlation test; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, and ∗∗∗∗P < .0001.
Finally, we investigated potential mechanisms that might contribute to ongoing endothelial cell activation and Weibel-Palade body exocytosis in children with SCD, particularly in the HC-treated cohort. Importantly, previous studies have implicated free heme in modulating hemolysis-induced end-organ damage in SCD.45 Overall, hemolysis was significantly reduced in children treated with BT vs those treated with HC (total, media LDH 807 U/L; BT 666 U/L vs HC 940 U/L; P < .0001; Figure 6A). Consistently, free heme levels were also lower in the BT cohort (total, median 10.2 μM; BT 9.7 μM vs HC 11.6 μM; P = .008; Figure 6B). Although marked interindividual variability in free heme levels was seen in both the HC (range, 4.9-51.8 μM) and BT (range, 3.2-19.7 μM) cohort, plasma free heme concentrations of ≥20 μM (previously reported to increase VWF secretion from endothelial cells)46 were observed in 26% of the HC cohort but 0% of the BT children (Figure 6B). Finally, although elevated free hemoglobin has been described to inhibit ADAMTS13 activity in vitro, we observed no significant association between increased plasma free heme and reduced ADAMTS13 activity (P = .13). (supplemental Figure 5).
Figure 6.

Erythrocyte PS exposure and VWF-binding in SCD patients treated with hydroxycarbamide compared to blood transfusion. Comparisons between the HC and BT cohorts are shown for (A) LDH levels and (B) free heme levels. Comparisons between groups were assessed by the Mann-Whitney U test. (C) Washed RBCs were prepared from children with SCD on HC or BT therapy, and flow cytometry used to assess PS exposure, together with VWF-binding capacity. (D) Representative examples of annexin V binding for washed RBCs from a healthy (HbAA) control compared with children with SCD on either HC or BT treatment. (E) Data are presented as mean of the mean fluorescence intensity (MFI) of PS exposure on RBCs normalized to control for HC and BT subgroups. Statistical analyses were performed using the Mann-Whitney U test. (F) Representative examples of recombinant VWF (rVWF) binding for washed RBCs from a healthy (HbAA) control compared with children with SCD on either HC or BT treatment. (G) Data are presented as mean MFI of rVWF binding to washed RBCs normalized to control for HC and BT subgroups. Statistical analyses were performed using the Mann-Whitney U test. (H) To determine whether VWF interacts with sickle RBCs in vivo, unwashed RBCs from children on BT and HC treatment were isolated, and bound VWF assessed by flow cytometry. Statistical analyses were performed using the Mann-Whitney U test with a significant P value < .05; ∗∗∗P < .001 and ∗∗∗∗P < .0001.
Erythrocytes from children treated with HC demonstrate increased PS exposure and enhanced VWF binding
Recent studies have shown that RBCs from patients with SCD demonstrate increased PS exposure compared with RBCs from healthy individuals.47 Moreover PS-exposing RBCs can bind VWF strings, promoting RBC–endothelial cell adhesion and vaso-occlusion.27,48 Therefore, we examined PS exposure and VWF-binding potential for washed RBCs derived from our pediatric SCD cohorts on BT or HC therapy (Figure 6C). PS exposure was significantly enhanced on RBCs from children treated with HC compared with those treated with BT (mean fluorescence intensity: HC 158.5 vs BT 122.0; P < .05; Figure 6D-E). Healthy HbAA control RBCs demonstrated minimal binding to recombinant VWF (Figure 6F). In contrast, RBCs from children with SCD demonstrated interindividual variability in their VWF-binding capacity (Figure 6F). Nevertheless, significantly increased RBC binding capacity for VWF was evident for washed RBCs from children treated with HC compared with RBCs from children treated with BT (mean fluorescence intensity: HC 174.6 vs BT 138.4; P < .05; Figure 6F-G). Given these findings, we finally assessed whether endogenous VWF may be bound to circulating RBCs in children with SCD. Interestingly, significantly enhanced bound VWF was observed on unwashed RBCs from children treated with HC compared with those treated with BT (Figure 6H). Together, these findings illustrate that the persistently elevated plasma VWF levels seen in a significant subgroup of children with SCD, despite their disease-modifying therapy, can directly interact with PS-positive RBCs and thereby play roles in multiple aspects of SCD pathogenesis including hemolysis and vaso-occlusion.
Mediation analysis to unravel interfactorial relationships
To better understand the relationships between treatment (BT vs HC), hemolysis, free heme, endothelial cell activation, and increased plasma VWF levels in children with SCD, we proceeded to perform mediation analysis (Table 3). Multivariate analysis demonstrated that treatment with either HC or BT was significantly associated (P = .009) with plasma VWF levels, independent of age and blood group. HbS level was also independently associated with plasma VWF:Ag level (P = .003) but was highly correlated with treatment (r = −0.913, P < .001). Cumulatively, these data therefore suggest that the effect of HC or BT treatment on plasma VWF:Ag levels is mediated via HbS. After addition of hemolysis markers to the model, both LDH and free heme were independently associated with VWF:Ag levels (P = .027 and P = .045, respectively). This indicates that not only the rate of hemolysis but also the amount of free heme correlate with increased VWF secretion, which is in line with our in vitro findings. After addition of angiopoietin-2 (as marker for endothelial cell activation) in the model, the association between treatment, LDH, free heme, and VWF disappeared, again in keeping with our previous results that increased VWF levels are primarily attributable to endothelial cell activation. Importantly, we observed that LDH as an outcome was associated with HbS and plasma VWF:Ag levels as independent factors (P = .044 and P = .001, respectively), suggesting that there is a feedback mechanism through which VWF (and endothelial cell activation) may affect hemolysis. This concept is consistent with our flow cytometry findings (Figure 6). Lastly, free heme was associated with LDH, independent of treatment and HbS, confirming that the amount of free heme is primarily determined by the rate of hemolysis. The overall conclusions of this mediation analysis are illustrated in Figure 7.
Table 3.
Outcome of multiple regression analyses with VWF, LDH, and free heme as dependent variables
| Outcomes of mediation analysis | ||
|---|---|---|
| Outcome: VWF | B (95% CI) | P value |
| Age | 0.5 (−1.1 to 2.0) | .552 |
| Blood group (non-O) | 30.2 (17.3-43.2) | <.001 |
| Treatment (BT) | −17.0 (−29.7 to −4.3) | .009 |
| HbS is independently associated with VWF | ||
| Age | 0.1 (−1.4 to 1.7) | .854 |
| Blood group (non-O) | 29.6 (16.6-42.5) | <.001 |
| HbS | 0.3 (0.1-0.6) | .003 |
| Treatment and HbS nullify each other’s effects | ||
| Age | 0.1 (−1.4 to 1.7) | .877 |
| Blood group (non-O) | 29.5 (16.4-42.5) | <.001 |
| Treatment (BT) | 4.2 (−27.2 to −35.7) | .791 |
| HbS | 0.4 (−0.1 to 1.0) | .143 |
| LDH and free heme are independently associated with VWF | ||
| Age | 0.9 (−1.7 to 3.6) | .483 |
| Blood group (non-O) | 47.1 (27.9-66.3) | <.001 |
| Treatment (BT) | −24.2 (−43.2 to −5.3) | .013 |
| LDH (×10−2) | 3.2 (0.4-6.0) | .027 |
| Free heme | −1.0 (−2.0 to 0.0) | .045 |
| Addition of angiopoietin-2 nullifies the effect of treatment, LDH, and free heme | ||
| Age | 0.5 (−2.9 to 3.9) | .766 |
| Blood group (non-O) | 39.6 (13.9-65.3) | .003 |
| Treatment (BT) | −24.2 (−50.1 to 1.7) | .067 |
| LDH (×10−2) | 0.5 (−3.3 to 4.2) | .807 |
| Free heme | −0.9 (−2.7 to 0.9) | .311 |
| Angiopoietin-2 (×10−3) | 6.3 (0.2-12.4) | .043 |
| HbS and VWF are independently associated with LDH | ||
| Outcome: LDH | ||
| HbS | 1.8 (0.1-3.5) | .044 |
| VWF | 1.9 (0.8-3.0) | .001 |
| LDH is independently associated with free heme | ||
| Outcome: free heme | ||
| Treatment | −2.2 (−11.1 to 6.9) | .636 |
| HbS | 0.0 (−0.2 to 0.1) | .909 |
| LDH | 0.5 (0.1-1.0) | .022 |
CI, confidence interval.
Figure 7.

Representation of proposed multifactorial mechanisms through which HC or BT disease-modifying therapies affect endothelial cell activation VWF–ADAMTS13 axis dysfunction in children with SCD. Key steps highlighted in red. (1) Significantly higher free heme levels were seen in children treated with HC compared with those treated with BT. (2) Plasma concentrations of free heme present in children treated with HC were sufficient to drive Weibel-Palade body exocytosis from human umbilical vein endothelial cells in vitro. (3) LDH and free heme were both independently associated with plasma VWF levels, suggesting that rate of hemolysis and amount of free heme can affect endothelial cell activation. (4) High-molecular-weight VWF multimers were significantly reduced in children treated with HC, consistent with ongoing high-molecular-weight VWF multimers consumption. (5) Although modified by ABO blood group, increased VWF levels were predominantly attributable to increased endothelial cell secretion. (6) FVIII:C levels were significantly higher in children treated with HC, driving enhanced thrombin generation. (7) Plasma levels of VWFpp, angiopoetin-2, and osteoprotegerin were all significantly elevated in children treated with HC. Downstream consequences are currently unknown. (8) Mediation analysis highlighted feedback mechanism through which increased plasma VWF levels may directly promote hemolysis.
Discussion
Previous studies have consistently reported elevated plasma VWF levels in SCD cohorts relative to healthy controls.17,18,21,22,24,25 Moreover, recent studies have defined an important role for the VWF–ADAMTS13 axis in the pathobiology of SCD vaso-occlusion.25,28,29 Our findings demonstrate, to our knowledge, for the first time, that despite treatment with either HC or BT, ongoing dysfunction of the VWF–ADAMTS13 axis is present in a significant subgroup of pediatric patients with SCD. Overall, plasma VWF levels were significantly lower in children with SCD treated with BT compared with those on maintenance HC therapy. This difference is noteworthy given that children with SCD perceived at highest stroke risk had originally been selected for BT treatment. Thus, these children at high risk, might have been anticipated to have higher levels of endothelial cell activation at baseline. Critically however, although median VWF levels were reduced in the BT subgroup, 25% of the BT subgroup and 44% of the HC subgroup still had plasma VWF:CB activity levels of >1.5 IU/m. Importantly, significant correlations between VWF levels and biomarkers of ongoing hemolysis (including Hb and LDH levels) were also observed. Cumulatively, these findings demonstrate that a subgroup of children with SCD managed with either HC or BT continue to have sustained increases in plasma VWF antigen and VWF functional activity despite their disease-modifying therapy. Given the evidence that VWF plays an important role in vaso-occlusive crisis pathogenesis, these data are of direct translational relevance.
VWF functional activity is regulated by multimeric size, with high-molecular-weight multimers having enhanced affinity for platelets. An increase in high-molecular-weight multimers and the presence of abnormal ultralarge VWF multimers has been reported in adult patients with SCD.21,24,25 In contrast, we found that high-molecular-weight multimers were significantly reduced in children on HC treatment, although intermediate- and low-molecular-weight multimers were increased. Similar loss of high-molecular-weight multimers has been described in patients with acute thrombotic thrombocytopenic purpura and severe COVID-19 in which it was attributed to consumption of hyperadhesive high-molecular-weight multimers binding to GPIbα.49,50 Our data suggest that high-molecular-weight multimer VWF consumption may be ongoing in a subgroup of children with SCD treated with HC. Consistent with that concept, plasma PF4 levels were also significantly elevated in the HC subgroup, suggesting ongoing platelet activation in these children. Despite the differences in VWF multimers in SCD, no significant reduction in plasma ADAMTS13 levels was evident in either the HC treatment or BT treatment cohorts. Nevertheless, plasma levels of several ADAMTS13 inhibitors (including free heme, IL-6, and PF4) were all significantly elevated in the those treated with HC compared with those in the BT cohort. Thus, not only are plasma VWF levels increased in a subgroup of children with SCD despite their disease-modifying treatment but there are also abnormalities in normal VWF multimer regulation in vivo. Furthermore, there is also evidence of significant ongoing platelet activation.
Given the biological importance of VWF in vaso-occlusive crises, we investigated whether increased plasma VWF levels in children with SCD undergoing treatment were due to endothelial cell activation with enhanced VWF secretion and/or attenuated VWF clearance. Previous studies have demonstrated that plasma VWFpp constitutes a sensitive and specific marker of acute endothelial cell activation.39 To date, only 1 other study assessed VWFpp levels in a pediatric SCD cohort. van der Land et al found that VWFpp levels were increased in children with SCD at steady state, but their study excluded children with SCD on either HC or BT therapy.19 Importantly, our findings demonstrate that despite their disease-modifying therapy, plasma VWFpp levels remain significantly elevated in 32% of the BT subgroup and 60% of the HC subgroup. Consistent with the hypothesis of sustained endothelial cell activation, we observed that plasma levels of other proteins stored within Weibel-Palade bodies (including FVIII, angiopoietin-2, and osteoprotegerin) in endothelial cells were also significantly increased in children treated with HC compared with those treated with BT. Recent studies have shown that angiopoietin-2 and osteoprotegerin have distinct biological roles in regulating endothelial cell survival, permeability, and angiogenesis.51, 52, 53 Furthermore, osteoprotegerin can directly trigger upregulation of intercellular adhesion molecule-1, vascular cell adhesion protein-1, and E-selectin on endothelial cells.53 Finally, both angiopoietin-2 and osteoprotegerin have been shown to bind to the A1 domain of VWF and thereby influence platelet recruitment to VWF strings.54,55 Additional studies will be required to define the downstream consequence of persistent secretion of proinflammatory and proangiogenic Weibel-Palade body cargo proteins from activated endothelial cells in pediatric SCD. Collectively, our data demonstrate that ongoing acute endothelial cell activation and Weibel-Palade body exocytosis are common in children with SCD despite treatment with HC or BT therapies. This endotheliopathy is significantly more marked in children treated with HC and constitutes the predominant mechanism underlying persistent increases in plasma VWF-FVIII levels.
This study has a number of limitations. Inclusion of individuals on BT and HC treatment who were willing to consent to the study introduces potential for selection bias because nonadherent patients and families were less likely to participate. Another limitation is that because children were established on therapy, evaluating longitudinal impacts of therapy on VWF was not possible. However, because BT is the gold standard for stroke prevention in children with high-risk disease, randomizing children with indications for BT to HC would be unethical, hence precluding a randomized study in this setting. Conversely, compared with previous studies, our study has a number of important strengths. First, the number of patients with SCD included in our study (n = 180) is much larger than most previous studies. Second, we have only studied children with severe SCD (HbSS), whereas previous studies typically enrolled both adults and children and included a variety of SCD genotypes. Because aging in SCD is associated with significant vasculopathy, this has the potential to affect VWF–ADAMTS13 findings. Third, in addition to assessing VWF levels, multimers, and ADAMTS13 levels, we have also investigated the relative importance of (i) sustained endothelial cell activation and Weibel-Palade body secretion and (ii) alterations in VWF clearance rates in contributing to elevated VWF-FVIII levels in children with SCD. Fourth, and perhaps most importantly, our study is, to our knowledge, the first to specifically focus on examining how HC or BT therapies affect the VWF–ADAMTS13 axis dysfunction and EC activation in children with SCD.
Multifactorial mechanisms contribute to endothelial cell activation in children with SCD.3,4,7,56 Importantly, chronic hemolysis has been shown to result in the release of free heme in SCD plasma.57 Free heme has previously been proposed to promote endothelial cell activation and trigger Weibel-Palade body exocytosis.58,59 We observed reduced hemolysis and significantly lower free heme levels in children with SCD treated with BT compared with HC C. Importantly, we further showed that elevated plasma VWF levels in children with children with SCD can directly interact with PS-positive RBCs and thereby influence ongoing intravascular hemolysis, free heme levels, endothelial cell activation and ultimately risk of vaso-occlusion.
In addition to vaso-occlusive crises, SCD is also associated with significant increased risk for both arterial and venous thromboembolic events.60, 61, 62, 63, 64 Elevated plasma FVIII levels >1.5 IU/mL have been shown to constitute a dose-dependent risk factor for venous and arterial thrombosis.65 In keeping with persistent endothelial cell activation, we observed plasma FVIII:C levels >1.5 IU/mL in 74% of the HC-treated compared with only 20% of the BT-treated subgroup respectively. Thus, 46% of our total pediatric SCD cohort had significantly increased FVIII:C levels despite their disease-modifying treatment. Consistent with their elevated FVIII:C levels, we observed significantly enhanced thrombin generation (endogenous thrombin potential, peak thrombin, velocity index) in HC compared with BT patients. Based on these findings, persistent EC activation in children with SCD despite treatment has the potential to increase risk of both vaso-occlusive crises and thrombotic complications.
In conclusion, this large study of VWF biology in pediatric SCD directly compared endotheliopathy and coagulation dysfunction in children treated with HC or BT. We demonstrate that BT therapy is associated with significantly attenuated endothelial cell and coagulation activation, normal VWF multimers, and decreased thrombin generation compared with HC treatment. Importantly, however, we further show that a significant subgroup of children with SCD treated with either HC or BT continue to have ongoing endothelial cell activation despite their disease-modifying treatment. Further studies will be necessary to determine whether these persistent abnormalities in VWF levels, VWF multimer distribution, FVIII, and/or osteoprotegerin can be used as novel biomarkers to identify children with SCD who remain at increased risk of vaso-occlusive crisis who may benefit from alternative treatments.
Conflict-of-interest disclosure: J.S.O. has served on the speaker’s bureau for Baxter, Bayer, Novo Nordisk, Sobi, Boehringer Ingelheim, Leo Pharma, Takeda, and Octapharma; served on the advisory boards of Baxter, Sobi, Bayer, Octapharma CSL Behring, Daiichi Sankyo, Boehringer Ingelheim, Takeda, and Pfizer; and received research grant funding awards from 3M, Baxter, Bayer, Pfizer, Shire, Takeda, and Novo Nordisk. F.A. received research support from CSL Behring, Takeda, Octapharma, and Sobi, and received travel grants from Sobi. The remaining authors declare no competing financial interests.
Acknowledgments
This work was performed within the Irish Clinical Academic Training Programme, supported by the Wellcome Trust and the Health Research Board (grant number 203930/B/16/Z), the Health Service Executive, National Doctors Training and Planning, and the Health and Social Care, Research and Development Division, Northern Ireland. J.S.O. is supported by National Children’s Research Centre Project award H/19/1 and Science Foundation Ireland Frontiers for the Future award 20/FFP-A/8952.
Authorship
Contribution: H.F., R.I.B., R.J.S.P., C.M., and J.S.O. designed the research, performed experiments, and analyzed the data; R.G., H.C., C.S., N.N., and E.T. contributed to study design and patient enrollment; A.A., F.A., D.D., S.W., E.K., A.R., G.L., and M.B. performed experiments; and all authors contributed to literature review, final draft writing and critical revision, have participated sufficiently in this work, take public responsibility for the content, and have made substantial contributions to this research.
Footnotes
The data that support the findings of this study are available upon reasonable request from the corresponding author, James O’Donnell (jamesodonnell@rcsi.ie).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 2.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4 doi: 10.1038/nrdp.2018.10. [DOI] [PubMed] [Google Scholar]
- 3.Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9(2):101–106. doi: 10.1097/00062752-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Frenette PS. Sickle cell vasoocclusion: heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation. 2004;11(2):167–177. [PubMed] [Google Scholar]
- 5.Embury SH. The not-so-simple process of sickle cell vasoocclusion. Microcirculation. 2004;11(2):101–113. doi: 10.1080/10739680490278277. [DOI] [PubMed] [Google Scholar]
- 6.Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci U S A. 1989;86(9):3356–3360. doi: 10.1073/pnas.86.9.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–809. doi: 10.1182/blood-2015-09-618538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015;125(13):2019–2028. doi: 10.1182/blood-2014-06-528406. [DOI] [PubMed] [Google Scholar]
- 9.Leebeek FWG, Eikenboom JCJ. Von Willebrand’s disease. N Engl J Med. 2017;376(7):701–702. doi: 10.1056/NEJMc1616060. [DOI] [PubMed] [Google Scholar]
- 10.Terraube V, O'Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16(1):3–13. doi: 10.1111/j.1365-2516.2009.02005.x. [DOI] [PubMed] [Google Scholar]
- 11.Turecek PL, Johnsen JM, Pipe SW, O'Donnell JS, i Psg Biological mechanisms underlying inter-individual variation in factor VIII clearance in haemophilia. Haemophilia. 2020;26(4):575–583. doi: 10.1111/hae.14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pendu R, Terraube V, Christophe OD, et al. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108(12):3746–3752. doi: 10.1182/blood-2006-03-010322. [DOI] [PubMed] [Google Scholar]
- 13.van Schooten CJ, Shahbazi S, Groot E, et al. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 2008;112(5):1704–1712. doi: 10.1182/blood-2008-01-133181. [DOI] [PubMed] [Google Scholar]
- 14.Chion A, O'Sullivan JM, Drakeford C, et al. N-linked glycans within the A2 domain of von Willebrand factor modulate macrophage-mediated clearance. Blood. 2016;128(15):1959–1968. doi: 10.1182/blood-2016-04-709436. [DOI] [PubMed] [Google Scholar]
- 15.Drakeford C, Aguila S, Roche F, et al. von Willebrand factor links primary hemostasis to innate immunity. Nat Commun. 2022;13(1):6320. doi: 10.1038/s41467-022-33796-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petri B, Broermann A, Li H, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116(22):4712–4719. doi: 10.1182/blood-2010-03-276311. [DOI] [PubMed] [Google Scholar]
- 17.Colombatti R, De Bon E, Bertomoro A, et al. Coagulation activation in children with sickle cell disease is associated with cerebral small vessel vasculopathy. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0078801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nur E, van Beers EJ, Martina S, et al. Plasma levels of pentraxin-3, an acute phase protein, are increased during sickle cell painful crisis. Blood Cells Mol Dis. 2011;46(3):189–194. doi: 10.1016/j.bcmd.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 19.van der Land V, Peters M, Biemond BJ, Heijboer H, Harteveld CL, Fijnvandraat K. Markers of endothelial dysfunction differ between subphenotypes in children with sickle cell disease. Thromb Res. 2013;132(6):712–717. doi: 10.1016/j.thromres.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Hatzipantelis ES, Pana ZD, Gombakis N, et al. Endothelial activation and inflammation biomarkers in children and adolescents with sickle cell disease. Int J Hematol. 2013;98(2):158–163. doi: 10.1007/s12185-013-1392-y. [DOI] [PubMed] [Google Scholar]
- 21.Schnog JJ, Kremer Hovinga JA, Krieg S, et al. ADAMTS13 activity in sickle cell disease. Am J Hematol. 2006;81(7):492–498. doi: 10.1002/ajh.20653. [DOI] [PubMed] [Google Scholar]
- 22.van Beers EJ, Schaap MC, Berckmans RJ, et al. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica. 2009;94(11):1513–1519. doi: 10.3324/haematol.2009.008938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demagny J, Driss A, Stepanian A, et al. ADAMTS13 and von Willebrand factor assessment in steady state and acute vaso-occlusive crisis of sickle cell disease. Res Pract Thromb Haemost. 2021;5(1):197–203. doi: 10.1002/rth2.12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Hobbs WE, Le J, Lenting PJ, de Groot PG, Lopez JA. The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood. 2011;117(13):3680–3683. doi: 10.1182/blood-2010-08-302539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sins JWR, Schimmel M, Luken BM, et al. Dynamics of von Willebrand factor reactivity in sickle cell disease during vaso-occlusive crisis and steady state. J Thromb Haemost. 2017;15(7):1392–1402. doi: 10.1111/jth.13728. [DOI] [PubMed] [Google Scholar]
- 26.Kaul DK, Nagel RL, Chen D, Tsai HM. Sickle erythrocyte-endothelial interactions in microcirculation: the role of von Willebrand factor and implications for vasoocclusion. Blood. 1993;81(9):2429–2438. [PubMed] [Google Scholar]
- 27.Nicolay JP, Thorn V, Daniel C, et al. Cellular stress induces erythrocyte assembly on intravascular von Willebrand factor strings and promotes microangiopathy. Sci Rep. 2018;8(1) doi: 10.1038/s41598-018-28961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Shao B, Gao L, et al. Endothelial VWF is critical for the pathogenesis of vaso-occlusive episode in a mouse model of sickle cell disease. Proc Natl Acad Sci U S A. 2022;119(34) doi: 10.1073/pnas.2207592119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossato P, Federti E, Matte A, et al. Evidence of protective effects of recombinant ADAMTS13 in a humanized model of sickle cell disease. Haematologica. 2022;107(11):2650–2660. doi: 10.3324/haematol.2021.280233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellsworth P, Sparkenbaugh EM. Targeting the von Willebrand factor-ADAMTS-13 axis in sickle cell disease. J Thromb Haemost. 2023;21(1):2–6. doi: 10.1016/j.jtha.2022.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 32.Davis BA, Allard S, Qureshi A, et al. Guidelines on red cell transfusion in sickle cell disease. Part I: principles and laboratory aspects. Br J Haematol. 2017;176(2):179–191. doi: 10.1111/bjh.14346. [DOI] [PubMed] [Google Scholar]
- 33.Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4(2):327–355. doi: 10.1182/bloodadvances.2019001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larkin D, de Laat B, Jenkins PV, et al. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand multimers and ADAMTS13 inhibition. PLoS Pathog. 2009;5(3) doi: 10.1371/journal.ppat.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budde U, Schneppenheim R, Plendl H, Dent J, Ruggeri ZM, Zimmerman TS. Luminographic detection of von Willebrand factor multimers in agarose gels and on nitrocellulose membranes. Thromb Haemost. 1990;63(2):312–315. [PubMed] [Google Scholar]
- 36.Fogarty H, Gaul A, Syed S, et al. Adherence to hydroxyurea, health-related quality of life domains and attitudes towards a smartphone app among Irish adolescents and young adults with sickle cell disease. Ir J Med Sci. 2022;191(2):809–816. doi: 10.1007/s11845-021-02588-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward SE, Curley GF, Lavin M, et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): evidence of acute and sustained endothelial cell activation. Br J Haematol. 2021;192(4):714–719. doi: 10.1111/bjh.17273. [DOI] [PubMed] [Google Scholar]
- 38.O'Sullivan JM, Preston RJ, O'Regan N, O'Donnell JS. Emerging roles for hemostatic dysfunction in malaria pathogenesis. Blood. 2016;127(19):2281–2288. doi: 10.1182/blood-2015-11-636464. [DOI] [PubMed] [Google Scholar]
- 39.Haberichter SL. von Willebrand factor propeptide: biology and clinical utility. Blood. 2015;126(15):1753–1761. doi: 10.1182/blood-2015-04-512731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104(1):100–106. doi: 10.1182/blood-2004-01-0107. [DOI] [PubMed] [Google Scholar]
- 41.Nazy I, Elliott TD, Arnold DM. Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br J Haematol. 2020;190(4):594–598. doi: 10.1111/bjh.16553. [DOI] [PubMed] [Google Scholar]
- 42.Fogarty H, Ward SE, Townsend L, et al. Sustained VWF-ADAMTS-13 axis imbalance and endotheliopathy in long COVID syndrome is related to immune dysfunction. J Thromb Haemost. 2022;20(10):2429–2438. doi: 10.1111/jth.15830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Sullivan JM, Ward S, Lavin M, O'Donnell JS. von Willebrand factor clearance - biological mechanisms and clinical significance. Br J Haematol. 2018;183(2):185–195. doi: 10.1111/bjh.15565. [DOI] [PubMed] [Google Scholar]
- 44.O'Donnell J, Laffan M. Elevated plasma factor VIII levels--a novel risk factor for venous thromboembolism. Clin Lab. 2001;47(1-2):1–6. [PubMed] [Google Scholar]
- 45.Gbotosho OT, Kapetanaki MG, Kato GJ. The worst things in life are free: the role of free heme in sickle cell disease. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.561917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 47.Whelihan MF, Lim MY, Mooberry MJ, et al. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost. 2016;14(10):1941–1952. doi: 10.1111/jth.13416. [DOI] [PubMed] [Google Scholar]
- 48.Smeets MW, Bierings R, Meems H, et al. Platelet-independent adhesion of calcium-loaded erythrocytes to von Willebrand factor. PLoS One. 2017;12(3) doi: 10.1371/journal.pone.0173077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moake JL, Chow TW. Thrombotic thrombocytopenic purpura: understanding a disease no longer rare. Am J Med Sci. 1998;316(2):105–119. doi: 10.1097/00000441-199808000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Ward SE, Fogarty H, Karampini E, et al. ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J Thromb Haemost. 2021;19(8):1914–1921. doi: 10.1111/jth.15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel-Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17(1):6–18. doi: 10.1111/jth.14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Randi AM, Smith KE, Castaman G. von Willebrand factor regulation of blood vessel formation. Blood. 2018;132(2):132–140. doi: 10.1182/blood-2018-01-769018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mangan SH, Van Campenhout A, Rush C, Golledge J. Osteoprotegerin upregulates endothelial cell adhesion molecule response to tumor necrosis factor-alpha associated with induction of angiopoietin-2. Cardiovasc Res. 2007;76(3):494–505. doi: 10.1016/j.cardiores.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shahbazi S, Lenting PJ, Fribourg C, Terraube V, Denis CV, Christophe OD. Characterization of the interaction between von Willebrand factor and osteoprotegerin. J Thromb Haemost. 2007;5(9):1956–1962. doi: 10.1111/j.1538-7836.2007.02681.x. [DOI] [PubMed] [Google Scholar]
- 55.Mobayen G, Smith K, Ediriwickrema K, et al. von Willebrand factor binds to angiopoietin-2 within endothelial cells and after release from Weibel-Palade bodies. J Thromb Haemost. 2023;21(7):1802–1812. doi: 10.1016/j.jtha.2023.03.027. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Jing F, Yi W, et al. HO-1(hi) patrolling monocytes protect against vaso-occlusion in sickle cell disease. Blood. 2018;131(14):1600–1610. doi: 10.1182/blood-2017-12-819870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagener FA, Abraham NG, van Kooyk Y, de Witte T, Figdor CG. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol Sci. 2001;22(2):52–54. doi: 10.1016/s0165-6147(00)01609-6. [DOI] [PubMed] [Google Scholar]
- 58.Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123(3):377–390. doi: 10.1182/blood-2013-04-495887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camus SM, De Moraes JA, Bonnin P, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood. 2015;125(24):3805–3814. doi: 10.1182/blood-2014-07-589283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shet AS, Lizarralde-Iragorri MA, Naik RP. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica. 2020;105(10):2368–2379. doi: 10.3324/haematol.2019.239350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naik RP, Streiff MB, Haywood C, Jr., Segal JB, Lanzkron S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J Thromb Haemost. 2014;12(12):2010–2016. doi: 10.1111/jth.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 63.Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol. 2013;162(1):3–14. doi: 10.1111/bjh.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sparkenbaugh E, Pawlinski R. Prothrombotic aspects of sickle cell disease. J Thromb Haemost. 2017;15(7):1307–1316. doi: 10.1111/jth.13717. [DOI] [PubMed] [Google Scholar]
- 65.Jenkins PV, Rawley O, Smith OP, O'Donnell JS. Elevated factor VIII levels and risk of venous thrombosis. Br J Haematol. 2012;157(6):653–663. doi: 10.1111/j.1365-2141.2012.09134.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.