

Significance
The ERK (extracellular signal-regulated kinase) and p38 subfamilies of mitogen-activated protein kinases (MAPKs) are closely related yet typically have opposing cellular roles. Their divergent functions are attributable to interactions with unique regulatory and substrate proteins. MAPK interactions with binding partners often occur through a conserved groove within the kinase catalytic domain, which engages short linear sequence motifs within the target protein. Here, we identified hundreds of short peptide sequences from human proteins that dock onto the MAPKs ERK2 and p38α. Sequences selective for each MAPK conformed to distinct motifs, providing a mechanism for enforcing signaling specificity. Identification of these MAPK targeting sequences facilitated the identification of interacting proteins and substrates, thus enhancing our understanding of MAPK signaling networks.
Keywords: short linear motif, MAP kinase, signal transduction, substrate specificity, kinase docking interactions
Abstract
Mitogen-activated protein kinase (MAPK) cascades are essential for eukaryotic cells to integrate and respond to diverse stimuli. Maintaining specificity in signaling through MAPK networks is key to coupling distinct inputs to appropriate cellular responses. Docking sites—short linear motifs found in MAPK substrates, regulators, and scaffolds—can promote signaling specificity through selective interactions, but how they do so remains unresolved. Here, we screened a proteomic library for sequences interacting with the MAPKs extracellular signal-regulated kinase 2 (ERK2) and p38α, identifying selective and promiscuous docking motifs. Sequences specific for p38α had high net charge and lysine content, and selective binding depended on a pair of acidic residues unique to the p38α docking interface. Finally, we validated a set of full-length proteins harboring docking sites selected in our screens to be authentic MAPK interactors and substrates. This study identifies features that help define MAPK signaling networks and explains how specific docking motifs promote signaling integrity.
Mitogen-activated protein kinases (MAPKs) are Ser/Thr kinases at the bottom of three-tiered kinase cascades that are essential for transmitting extracellular signals to effect changes in cell physiology and function (1). Mammals have four canonical MAPK subfamilies, the p38, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) 1/2, and ERK5 MAPKs. The various MAPK cascades respond to different stimuli and control distinct cellular processes (2–5). For example, activation of the ERK pathway by growth factors promotes cell proliferation (2), while the stress-activated p38 MAPKs are key to stress-induced cell cycle arrest (5) and pro-inflammatory signaling in post-mitotic cells (3). Distinct biological roles played by each MAPK are in part mediated by connections with unique sets of interacting proteins including substrates, scaffolds, and regulators. While numerous substrates and binding partners have been identified for each MAPK, their broad physiological and pathological functions suggest that key components of their signaling networks remain to be discovered. A more complete understanding of how MAPKs make selective connections with substrates and other interacting proteins would facilitate the discovery of these components.
Numerous mechanisms can underlie signaling specificity by protein kinases, including subcellular compartmentalization, scaffolding, signal dynamics, and direct kinase-substrate interactions (6). One mechanism common to protein kinases involves the recognition of sequence motifs surrounding sites of phosphorylation by the catalytic cleft (7). However, since all MAPKs target a common Ser/Thr-Pro consensus sequence, catalytic site specificity is insufficient to mediate selective phosphorylation by a particular member of the family. For MAPKs, docking interactions at regions outside of the catalytic cleft reportedly control specificity and impact their biological function (8). In particular, MAPKs harbor a docking groove within the catalytic domain called the D-recruitment site (DRS), an extended shallow surface comprising several hydrophobic pockets and a negatively charged region termed the common docking (CD) site. The DRS binds to short linear motifs (SLiMs) known as D-sites, also called δ-domains or docking site for ERK and JNK, LXL (DEJL) motifs, found in MAPK substrates and other interactors (9, 10). Like other SLiMs, D-sites are generally found in intrinsically disordered regions (IDRs) of proteins and bind to MAPKs with moderate to weak affinity. While such binding interactions are optimal for mediating substrate recruitment (11), their transient nature makes them difficult to discover and as a result only a small number of D-sites have been studied.
D-sites have been subcategorized into “forward” or “reverse” sequences as defined by their binding orientation in the DRS (12). Forward D-sites, which are the most common, conform to a consensus sequence consisting of a cluster of basic residues, connected to a hydrophobic ϕ-x-ϕ motif by a variable length linker sequence (9, 12–14). Within this general sequence context, several motifs with distinct linker amino acid composition and length reportedly confer selective binding to particular MAPK subfamilies (15–19). Structural studies of MAPKs in complex with D-site peptides (D-peptides) have revealed distinct binding modes associated with reported motifs, in some cases allowing rationalization of MAPK selectivity (12, 20–28). In particular, unique features of the JNK DRS facilitate substantial discrimination between cognate and non-cognate motifs (12, 16, 19). While reverse D-site peptides from the homologous kinases RSK1 and MK2 bind with a high degree of selectivity (>10-fold) to ERK and p38 MAPKs respectively, the extent to which forward D-sites distinguish between members of these two MAPK subfamilies is less clear (12, 29). In particular, specific features within D-sites conferring selective binding to their cognate MAPKs, and how their respective docking grooves encode specificity, are not known.
Here, we define ERK2 and p38α D-site interactomes by screening a library of candidate docking sequences from the human proteome. These screens revealed distinct MAPK-selective D-site sequences and associated motifs. We show that motifs with Lys residues clustered in the D-site variable linker region conferred selectivity for p38α over ERK2. Furthermore, through exchange mutagenesis, we identified a region of the DRS largely responsible for differential binding to the two MAPKs. In addition, our screen uncovered docking sequences within several established MAPK substrates and led us to identify a set of ERK2 and p38α interactors and substrates. Overall, these studies reveal connections in MAPK signaling networks and demonstrate how these connections occur selectively.
Results
Establishment of a Yeast Two-Hybrid (Y2H) Screening Platform to Identify MAPK D-Sites.
We established a Y2H assay to probe the interaction between MAPKs and putative docking sequences using a fragment of the MAPK substrate ELK1 (residues 306 to 427) containing an essential D-site motif (30) (Fig. 1A and SI Appendix, Fig. S1). A Y2H reporter strain was co-transformed with a bait plasmid expressing the MAPK fused to the Gal4 DNA binding domain (Gal4DBD-ERK2 or Gal4DBD-p38α) and a prey plasmid expressing the ELK1 fragment fused to the Gal4 activation domain (Gal4AD-ELK1) (Fig. 1B), and assayed for growth under selective (His−) and non-selective (His+) conditions (Fig. 1C). We found that ELK1 could interact physically with ERK2, but not p38α (Fig. 1 C and D). Mutation of the D-site (L319A/E320A/L321A, ELK1ΔD) disrupted binding, confirming the interaction to be D-site dependent. We next examined whether other cognate or non-cognate docking sequences could support this interaction. As anticipated based on reported peptide binding affinities (12, 13, 19), replacing the native ELK1 D-site with that of MAP2K2 (ELK1MAP2K2-D) allowed it to interact with both ERK2 and p38α. Conversely, substitution with a JNK-selective sequence (ELK1NFAT4-D) abolished binding to either kinase (Fig. 1 C and D). Taken together, these results suggest that Y2H is a suitable strategy to discover D-site sequences that bind competitively to MAPKs.
Fig. 1.

Y2H system to detect MAPK binding to docking sites. (A) Diagram of ELK1 transcriptional activation domain construct showing the D-site (purple) with flanking linker sequence (cyan) and MAPK Ser/Thr-Pro phosphorylation sites (lollipops). (B) Docking-dependent Y2H scheme. (C) Growth under selective (−His + 50 μM 3-AT) and non-selective (+His) conditions of serially diluted cultures of strains co-expressing the indicated MAPK and ELK1 variant. (D) D-site sequences used in ELK1 constructs with their reported MAPK binding selectivity (12).
Y2H Screen to Define ERK2 and p38α D-Site Interactomes.
To identify additional sequence features that mediate selective docking to ERK2 or p38α, we prepared a custom library of ELK1 D-site exchange mutants in the Gal4AD-ELK1 prey vector background. The library incorporated a set of 11,756 SLiM sequences from the human proteome conforming to the general forward D-site sequence motif [R/K]-x0-2-[R/K]-x3-5-[ILV]-x-[FILMV] (Dataset S1), which we have previously used in a different screening platform (16). Yeast bearing Gal4DBD-MAPK bait plasmids were transformed with the prey plasmid library, and transformants were pooled and grown under selective and non-selective conditions in liquid culture. Cultures were periodically sampled, and the D-site coding sequence was PCR-amplified from extracted plasmid DNA and analyzed by next-generation sequencing (NGS) (Fig. 2A). As expected, we saw significant enrichment of specific D-site sequences by ERK2 and p38α only under selective conditions (Fig. 2 B and C and SI Appendix, Fig. S2 A and B). We assigned an enrichment score (ES) for each sequence by fitting the change in relative abundance over the first ~8 population doublings to an exponential function. ES values for the full set of library sequences correlated significantly across three replicate screens for both ERK2 [range of Pearson’s correlation coefficient (R) = 0.60 to 0.74] and p38α (R = 0.82 to 0.85) and (SI Appendix, Fig. S2 C and D). Sequences with average ES values ≥2 SDs from the mean of all sequences (Z-score ≥ 2) were defined as hits. ERK2 and p38α hits (Datasets S2–S5) included most previously known interacting sequences (18, 20, 22, 31–34) (Fig. 2 D and E and SI Appendix, Fig. S2B), though several established D-sites with low affinity for p38α escaped detection.
Fig. 2.

Pooled Y2H screening. (A) Library construction and Y2H screening workflow. (B and C) Change in frequency of selected library sequences in the ERK2 and p38α screens plotted as a function of yeast growth under non-selective (Left) and selective (Right) conditions. The most enriched sequences, several known interactors, and the parental ELK1 sequence are shown. Error bars indicate SD from three separate screens. (D and E) Waterfall plots show average ES from three independent screens in descending order for the 1,000 most enriched sequences under selective conditions. Interactors shown in (B) and (C) are highlighted. The dashed gray line indicates a Z-score of 2.
Contrary to the generally good correlation between replicates for each MAPK, average ES values from the ERK2 and p38α screens showed relatively poor correlation (R = 0.57), with many sequences enriched only in the presence of a single MAPK. Overall, screen hits included 98 sequences that were selected only by ERK2, 238 sequences selected only by p38α and 105 common hit sequences selected by both kinases (Fig. 3A and Datasets S4 and S5). To validate the results from Y2H screens, we determined ERK2 and p38α binding affinities for a set of synthetic D-peptides corresponding to the top-ranking screening hits. Peptide binding was assayed by competitive inhibition of MAPK kinase activity using a D-site-dependent fluorescent peptide substrate (35). D-peptide IC50 values ranged from 50 nM to 100 μM for at least one MAPK (Fig. 3B, SI Appendix, Fig. S3, and Dataset S6). We observed a trend for peptides to display higher affinity for p38α, including a number of common hits and a single hit sequence unique to the ERK2 screen (Fig. 3 B and C and Dataset S6). These observations suggest that the ERK2 and p38α screens may have different affinity thresholds for significant enrichment in the context of the ELK1 sequence. It is also possible that p38α has a propensity to bind more tightly to D-site peptides than does ERK2 in the context of sequences present in our library. Overall, these results confirm the selection of true MAPK biding sequences by Y2H screens and delimits ERK2 and p38α D-site interactomes within the context of our library.
Fig. 3.

Comparison of ERK2 and p38α screens plus hit validation. (A) Mean ES plotted for each sequence from the ERK2 and p38α Y2H screens. Dotted lines indicate Z ≥ 2. The number of sequences in each quadrant is indicated. (B) The plot shows mean IC50 values (three independent experiments) for inhibition of ERK2 and p38α activity by synthetic D-peptides. Blue triangles, ERK2-specific hits. Salmon circles, p38α-specific hits. Purple diamonds, common hits. (C) The bar graph shows selectivity ratios for D-peptides (indicated by corresponding gene name) calculated from IC50 values in (B). Peptide sequences and source data are in Dataset S6. Bars show mean ± SD from three independent experiments. (D) The heat map shows the net charge (at pH 7.0) and number of Arg, Lys, and Pro residues for the peptides shown in (C).
Identifying Determinants of MAPK Selectivity.
We next analyzed our dataset to identify sequence features preferably recognized by each MAPK. Considered in aggregate, ERK2 and p38α significantly selected sequences with Leu residues at positions 10 and 12, corresponding to the two residues that we required to be hydrophobic in the library design. In contrast, sequences selected by the two MAPKs differed substantially in residues over-represented at positions 7 to 9 and 13 (Fig. 4 A and B). ERK2 was significantly selective for Pro at each of these positions (Fig. 4A), while p38α was significantly selective for basic residues and against acidic residues (Fig. 4B). These differences in amino acid composition correlated with a tendency for p38α hits to carry a higher net positive charge than those of ERK2 (Fig. 4C). Furthermore, in our validation set of synthetic D-peptides, highly charged and basic residue-rich D-peptides tend to have a higher affinity for p38α over ERK2, while proline-rich D-peptides tend to prefer ERK2 (Fig. 3C).
Fig. 4.

Sequence motifs selected by ERK2 and p38α. (A and B) Probability logos (pLogos) (36) generated from multiple sequence alignments of all ERK2 or all p38α hits using the full starting library as background. (C) Heat map showing the percentage of total hits for each MAPK with a given calculated net charge at pH 7.0. (D–F) pLogos are shown corresponding to unique ERK2 hits, common hits, and unique p38α unique. (G) Distribution of motif classes for common and unique ERK2 and p38α hits.
Previous studies have defined multiple D-site motifs that bind non-selectively to both ERK and p38 (12, 15). When we subdivided our hits into those unique to a single MAPK or common to both, it became evident that sequences in each category conform to distinct motifs (Fig. 4 D–G). For example, 30% of hits unique to ERK2 had a Pro residue at position 7 (P7), most of which had an additional Pro residue at position 9 and lacked basic residues at position 11 (Fig. 4D and SI Appendix, Fig. S4). The full P-x-P-L-x-ϕ motif has been previously defined as the “Far1” motif (18, 21). ERK2 unique hits lacking Pro at position 7 (14%) generally conformed to the previously defined “MEF2A” motif class characterized by aliphatic residues at position 8 (Fig. 4G and SI Appendix, Fig. S4) (18). The MEF2A motif was also found in a substantial proportion (24%) of p38α unique hits (Fig. 4G and SI Appendix, Fig. S5) and was the dominant signature in hits common to both MAPKs (Fig. 4 E and G). However, unlike its prior definition (P/ϕ-x-L-x-L), we found MEF2A class sequences to be significantly enriched for basic residues at positions 9 and 11 (Fig. 4E and SI Appendix, Figs. S4 and S5).
In contrast to ERK2 hits, many of those unique to p38α conformed to a motif we term “Lys cluster” (K2-3-L-x-ϕ), which included basic residues clustered at positions 7 to 9. (Fig. 4F and SI Appendix, Fig. S5C). Notably, of the nine sequences in our library having three Lys residues at positions (7 to 9), seven scored as p38α hits, and the relative affinity of synthetic D-peptides for p38α over ERK2 correlated well with the number of Lys residues (Fig. 3C). Finally, a number of p38α (2%) and ERK2 (6%) hits included an L/I/V-x-x-R-R sequence motif near the N-terminus of the D-site, a motif previously noted in the D-site from the phosphatase PTPN7 (HePTP) with few other known examples (Fig. 4C) (18, 22). Overall, through these analyses, we identified hits containing previously known MAPK docking motifs and an unknown Lys motif signature promoting selective binding to p38α over ERK2.
Among the defined motifs, the Far1 and Lys cluster motifs exhibited the largest differences in representation between ERK and p38 hits (Fig. 4G). To assess the extent to which these sequence motifs interact selectively with ERK2 and p38α, we generated two D-peptides (ERK2-D-pep and p38α-D-pep) incorporating residues specifically selected by the corresponding MAPKs in the context of the Far1 and Lys cluster motifs, respectively. We determined their relative affinity for binding to ERK2 and p38α by competitive kinase assay as described above (Fig. 5A and Dataset S6). Both peptides bound their corresponding MAPKs with relatively high affinity (IC50 values ~2 μM), likely due to inclusion of favorable residues throughout the peptide sequence. Consistent with their anticipated selectivity, p38-D-pep bound p38α with 10-fold higher affinity than ERK2, while ERK-D-Pep showed a more modest 2.4-fold preference for ERK2. We then evaluated a pair of consensus peptides designed in the context of the common MEF2A motif (ERK2-MEF-pep and p38α-MEF-pep) with differing net charges (+6 and +8, respectively, at neutral pH). We found that ERK2-MEF-pep bound ERK2 and p38α with similar affinities, while p38-MEF-pep favored p38α by ninefold (Fig. 5A). These results confirm that enrichment for basic residues and high net charge promote p38α selectivity.
Fig. 5.
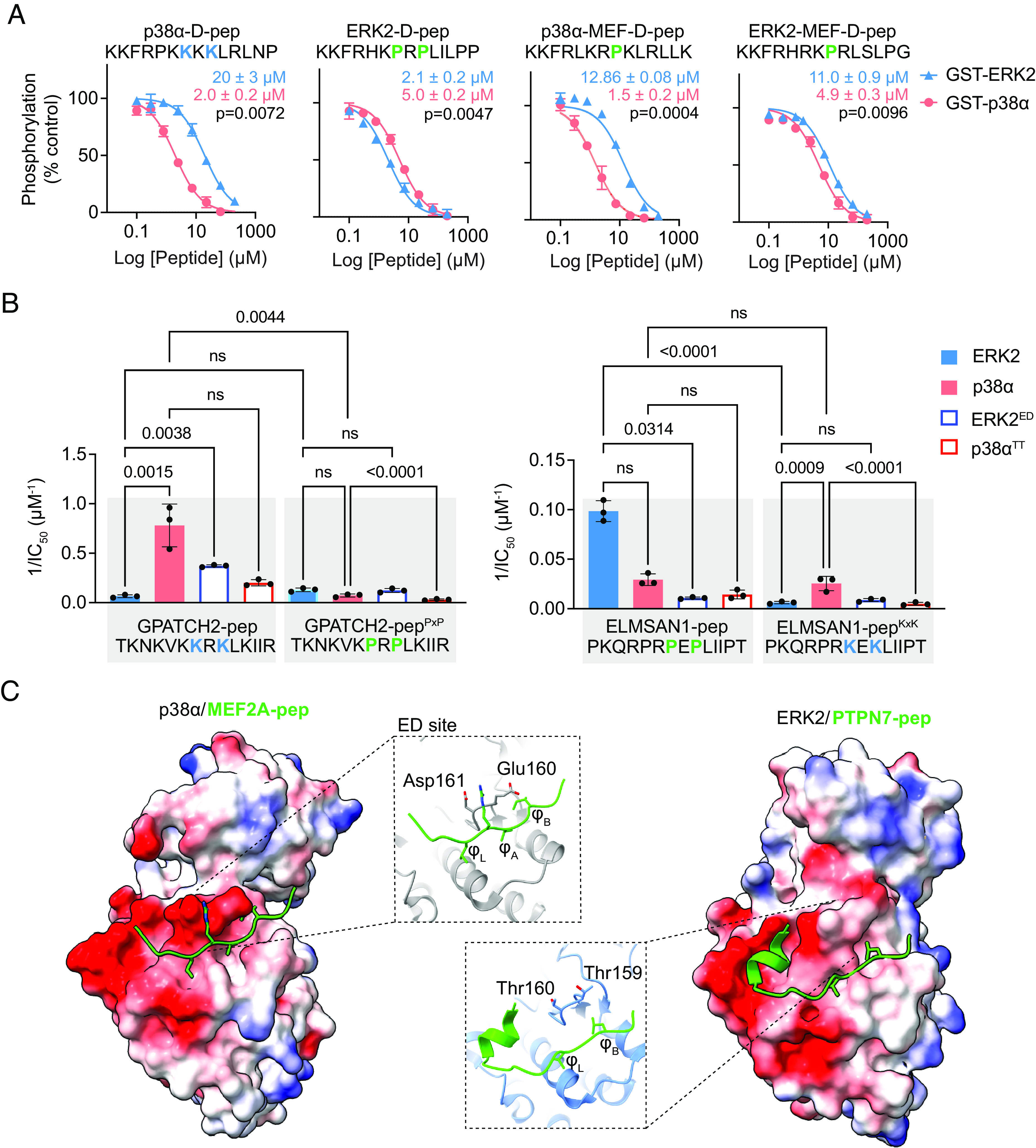
Validation of MAPK-selective D-site motifs. (A) Dose–response curves for inhibition of ERK2 (blue triangles) or p38α (salmon circles) by the indicated D-peptides. Error bars show SD from three independent experiments. Calculated IC50 values show mean ± SD. Two-tailed paired t test analysis comparing IC50 values was used to calculate P-values. (B) Inverse IC50 values for peptide inhibition of the indicated WT or mutant MAPK (full dose–response curves are in SI Appendix, Fig. S6C). Error bars represent SD from three independent experiments. The P-values shown were calculated by ordinary one-way ANOVA with Tukey’s multiple comparison test. Non-significant (ns) indicates P >0.05. (C) Structures of the p38α-MEF2A [Left, PDB: 1LEW (20)] and ERK2-PTPN7 [Right, PDB: 2GPH (22)] D-peptide complexes, showing electrostatic potentials generated in ChimeraX (37). The residues in the β7-β8 turn are labeled in the inset and shown in stick representation.
To systematically test the capacity of Lys- and Pro-rich motifs to specifically bind p38α and ERK2 respectively, we examined the impact of eliminating or exchanging these features in the context of p38 selective (GPATCH2-pep, with a native KxK motif) and ERK selective (ELMSAN1-pep, with a native PxP motif) D-peptides. As anticipated, substituting the key residues with Ala in these peptides led to a significant reduction in binding affinity to both MAPKs (SI Appendix, Fig. S6 A and B and Dataset S6). Moreover, replacement of Lys with Pro residues in the GPATCH2-pep (GPATCH2-pepPxP) caused a significant >10-fold reduction in affinity for p38α, though it did not significantly increase its affinity for ERK2 (Fig. 5B and Dataset S6). The converse substitutions in ELMSAN1-pep (ELMSAN1-pepKxK) provided >100-fold reduced affinity toward ERK2 without significant change in affinity for p38α. These results suggest that the selectivity of Pro-rich sequences for ERK2, and of Lys-rich sequences for p38α, is at least partly due to negative selection by the non-cognate kinase.
To rationalize the selectivity of p38α for Lys-rich sequences, we considered features of the DRS previously implicated in binding specificity. A negative electrostatic potential in the p38α groove spans a large surface including the ED site, localized to the MAPK β7-β8 turn (E160 and D161), and the CD region (centered around loop L16) (Fig. 5C). In contrast, the ERK2 groove has a pair of neutral residues (Thr 159-Thr160) in the β7-β8 turn and has one fewer negatively charged residue in the CD region. Tanoue et al. reported roles for both the ED and CD sites in mediating MAPK specificity in binding to reverse D-sites, with the ED site appearing to have a predominant role (34). In particular, they reported that a p38α E160T/D161T exchange mutation (p38αTT) disrupted binding to the p38-selective substrates MK2 and MK3, both with reverse D-site sequences, while the converse ERK2 ED site mutation (T159E/T160D, ERK2ED) conferred binding to those substrates (34). We thus examined whether the ED site could likewise mediate enhanced selectivity of p38α for forward D-peptides with basic residues clustered in positions 7 to 9. We generated ERK2ED and p38αTT DRS exchange mutants and examined their binding to the set of GPATCH2 and ELMSAN1 D-peptides (Fig. 5B, SI Appendix, Fig. S6C, and Dataset S6). Consistent with a role for the ED site in recognition of the KxK motif, we found that the p38αTT mutation led to a significantly large decrease in binding affinity for ELMSAN1-pepKxK, though a twofold decreased affinity for GPATCH2-pep was not statistically significant. Likewise, the ERK2ED mutant bound GPATCH2-pep about sixfold more tightly than did WT ERK2. ERK2ED mutation, however, did not promote a significant increase in binding to ELMSAN1-pepKxK. Furthermore, we found that while ERK2ED mutation caused a significant drop in affinity for ELMSAN1-pep, it had no effect on binding to GPATCH2-pepPxP. Finally, p38αTT mutation did not promote binding to either PxP-containing peptide (ELMSAN1-pep or GPATCH-pepPxP). Overall, these results suggest that as previously observed for reverse D-sites, residues located in the β7-β8 turn are important for p38α recognition of basic cluster sequences, but do not promote binding to ERK2-selective interactors.
Identification and Validation of MAPK Interactors and Substrates.
We next examined whether D-site hits from the screens could function to recruit MAPKs in the context of their corresponding full-length proteins. We first tested a set of seven common hit proteins to investigate whether they could interact with MAPKs in co-immunoprecipitation (IP) experiments. When co-expressed in HEK293T cells, we found that all but one of the tested proteins reproducibly co-immunoprecipitated with p38α, but that only a subset (GPATCH2, ISG20, ARHGEF2, and MLX-interacting protein-like [MLXIPL]) co-precipitated ERK2 above background levels (Fig. 6 A and B). For a subset of these proteins, we next examined whether the D-site sequence was required for optimal MAPK binding. We found that mutation of the MLXIPL D-site or full deletion of the ISG20 D-site decreased binding to both MAPKs to varying degrees. However, deletion of the ARHGEF2 D-site only modestly decreased ERK2 binding and actually increased its interaction with p38α (Fig. 6 C and D). These observations suggest that at least some of our hit D-sites promote interactions with ERK and p38 in the context of their respective full-length proteins but that other sites of interaction must contribute to MAPK binding. We do note that structural perturbation of ISG20 due to deletion of its D-site could underlie reduced binding to both MAPKs.
Fig. 6.

Identification of MAPK interacting proteins. (A) Immunoblots showing co-IP or whole cell lysate (WCL) samples from cells ectopically expressed ERK2 or with the indicated FLAG epitope-tagged proteins. A representative of three independent replicates is shown. (B) Co-IP of ERK2 with WT and D-site mutated or deleted (ΔD) versions of the indicated proteins. A representative immunoblot is shown at Left, and quantified data for all three independent replicates are graphed at Right. (C) The immunoblot shows co-IP of p38α with the indicated proteins, performed as in (A). (D) Co-IP of WT and ΔD mutant proteins with p38α, with quantified data from three independent experiments shown at Right.
Because D-sites often mediate substrate recruitment, we next examined whether some of these proteins could be phosphorylated by MAPKs in a manner dependent on the docking sites in radiolabel kinase assays. Two of the proteins, TACC1 and the established ERK substrate ARHGEF2 (38–40), were phosphorylated by both ERK2 and p38α. However, only phosphorylation of TACC1 was consistently decreased upon deletion of its D-site (Fig. 7 A and B). Despite interacting with p38α in cells (Fig. 6B), L3MBTL3 was only phosphorylated by ERK2, and it required the D-site for maximal phosphorylation. These data indicate that some of the D-sites identified in our screens can promote substrate recruitment in an in vitro setting.
Fig. 7.

Identification of MAPK substrates. (A and B) Radiolabel kinase assays showing phosphorylation of WT or D-site deleted (ΔD) versions of the indicated recombinant protein by either ERK2 or p38α. The graph at the bottom shows quantified levels of phosphorylation relative to the WT substrate, from 3 to 5 independent experiments. Error bars, SD. (C) TACC1 phosphorylation in HEK293T cells. Immunoblots of Phos-tag or standard SDS-PAGE show lysates of transiently transfected serum-starved cells pre-treated with 500 nM trametinib followed by 25 ng/mL EGF for 5 min as indicated. Representative of three independent experiments. (D) Quantified MS maximum peak intensity for the tryptic phosphopeptide containing phospho-Ser231 in WT or ΔD forms of TACC1 expressed in HEK293T cells and treated as in (C). Signals are shown relative to the serum-starved WT condition. Data from two independent replicates are shown.
We next examined whether either an interacting protein (MLXIPL) or an in vitro substrate (TACC1) could be phosphorylated by MAPKs in cells. HEK293T cells expressing candidate substrates were treated with EGF (with and without the MEK1/2 inhibitor trametinib) to stimulate ERK, or anisomycin (with or without the p38α/β inhibitor SB203580) to stimulate p38. As evidenced by inhibitor-sensitive mobility shift on Phos-tag or standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), TACC1 and MLXIPL were both phosphorylated in an ERK-dependent manner, and anisomycin-induced MLXIPL phosphorylation was partly dependent on p38α/β (Fig. 7C and SI Appendix, Fig. S7). Anisomycin did not induce phosphorylation of TACC1, suggesting that it may be selectively phosphorylated by ERK and not p38 MAPKs (SI Appendix, Fig. S7). Mutation of the MLXIPL D-site did not impact its mobility shift in response to either EGF or anisomycin. Furthermore, MS analysis of MLXIPL isolated from HEK293T cells revealed two EGF-induced, trametinib-sensitive Ser-Pro phosphorylation sites, neither of which were affected by D-site mutation (SI Appendix, Fig. S7). Because phosphorylation occurs independently of the docking interaction, it is therefore not clear whether this is directly mediated by MAPKs or by other kinases acting downstream. In contrast, MS analysis of TACC1 isolated from HEK293T cells revealed a single Ser-Pro phosphorylation site (Ser231) that was stimulated by EGF and blocked by trametinib treatment (Fig. 7D). Furthermore, deletion of the TACC1 D-site caused a substantial decrease in Ser231 phosphorylation in cells. Consistent with D-sites generally directing MAPKs to phosphorylate proximal sites (41), Ser231 is positioned ~40 residues downstream of the putative docking sequence. Taken together, these data suggest TACC1 to be a direct substrate of ERK, thus confirming the ability of our screening platform to identify authentic cellular MAPK substrates.
Discussion
We have reported a pooled Y2H screen to identify short proteomic sequences interacting with the MAPK DRS. Through this approach, we uncovered a large number of D-sites interacting with the MAPKs ERK2 and p38α, and we have shown that some of these sequences function, in the context of their respective full-length proteins, as binding partners and as substrates of MAPKs in cells.
Prior work has suggested that most forward D-sites bind promiscuously to both ERK2 and p38α. Indeed, we identified the so-called MEF2A motif (P/ϕ-x-L-x-ϕ) in hits selected by both of the MAPKs (18). However, we found that residues in the linker region (positions 7 to 9 in our library) of the D-site sequence can promote MAPK-selective binding. For example, ERK2 preferentially bound sequences having Pro residues located two positions upstream of the core hydrophobic motif (P-x-x-L-x-L), a feature present in several established ERK interactors including its activating kinase MAP2K1 (MEK1). In addition, we found that sequences with basic residues clustered in the linker region were highly selective for p38α. We have thus identified a forward D-site motif that can strongly distinguish between these two MAPKs.
We found that D-sites with higher net charge generally bound more selectively to p38α over ERK2. This phenomenon appeared to be driven by the presence of clustered Lys, as opposed to Arg, residues. Lys residues tend to increase conformational heterogeneity of IDRs (42) and are found more frequently than Arg both in IDRs (as annotated in the DisProt database) and in variable regions of reported linear motifs (43, 44). We speculate that the Lys cluster motif binds to p38α by forming a “fuzzy” complex, which is partly stabilized by long-range electrostatic interactions with the complementary ED site and possibly the CD region of the DRS groove. By definition, a fuzzy complex occurs when one binding partner in a protein-protein complex remains dynamic, where conformational heterogeneity drives interaction specificity (45, 46). In support of this, structural studies have revealed that D-peptides adopt a variety of binding modes when interacting with MAPKs (12, 18, 20, 22, 23, 26, 27), correlating well with proposed models of fuzzy complexes (47). While forward D-sites are anchored through interactions with their C-terminal hydrophobic ϕ-x-ϕ residues, the N-terminal region can either be conformationally stable or considerably dynamic. For example, no electron density was observed for the N-terminal basic regions of the MAP2K3 and MAP2K6 D-peptides that were co-crystalized with p38α, while NMR studies of these same complexes suggest that this region makes a significant contribution to docking interactions in solution (12, 20, 33, 48, 49). In addition, an NMR study of ERK2 bound to the substrate ETS1 suggested fuzzy interactions proximal to the D-site hydrophobic anchor (50, 51). Collectively, these studies suggest that despite making important contributions to binding affinity, clusters of basic residues in p38-selective D-sites do not need to adopt a single stable conformation when bound to the MAPK to mediate recruitment.
The importance of the ED site in mediating ERK versus p38 D-site specificity has been highlighted in previous studies, where mutations at this site can invert selectivity for binding to substrates harboring reverse D-sites (RSK1 and MK2/3) that have more stable secondary structures (12, 34). In addition, NMR and hydrogen-deuterium exchange studies of p38α/D-peptide complexes suggest that interactions involving the ED site can be more significant than those involving the CD region for peptides with basic residues clustered N-terminal to the canonical hydrophobic motif (33, 48, 49, 52). Our study provides evidence that the ED site can also mediate forward D-site selectivity, which likely contributes to functional differences between ERK and p38. In contrast, the ED site is conserved among all four p38 isoforms. We note that p38γ and p38δ, which have functions distinct from p38α (3), have an additional negatively charged residue in the αD-αE loop (Glu123 in human p38γ, equivalent to Gln120 of p38α) located proximal to the ED site. Thus, differences in the DRS could contribute to the distinct substrate repertoires of the various p38 isoforms. Future studies will be required to determine whether p38γ or p38δ target unique D-site motifs.
Our studies identified D-sites corresponding to many known MAPK binding partners and substrates (12, 53–55) but also identified a large number of other docking sequences (Datasets S4 and S5), indicating that multiple complementary approaches will be important to completely define MAPK interactomes. For example, prior research using a structure-guided in silico screen based on the p38-MEF2A peptide complex (18) predicted only 20% of D-sites selected by p38α in this study (Dataset S5). Analysis of hits selected in our Y2H screens produced a refined definition of the MEF2A motif that includes basic residues alternating with the core hydrophobic (ϕ-x-ϕ) sequence motif. This and other refined motifs defined in this work should facilitate more accurate computational searches to identify additional interactors that were excluded from our library. Furthermore, unbiased experimental screens such as those described here are not restricted to previously defined sequence motifs, and indeed a substantial number of our hits did not have clearly evident motifs beyond those comprising the generic D-site sequence. Our current work also identified 88% of the 71 p38α hit sequences reported in a recent study from our laboratory, in which we screened the same SLiM library for sequences inhibiting flux through the mammalian p38 pathway reconstituted in yeast (16). Interestingly, p38α hits identified in our prior study conformed to the MEF2A class motif, while in this study, a substantial number of hits did not. While the Y2H screens identified most previously known D-sites (Datasets S4 and S5), they failed to detect some functional low-affinity interactions, including D-sites from the p38α substrates MEF2A and MEF2C, that scored as hits in our previous screen. This suggests that D-site affinity is likely dependent on the surrounding sequence context, which differed between the two screens. Though residues surrounding the core library sequence were not predicted to directly interact with the MAPK DRS, it has been suggested that sequence context can contribute substantial binding energy to SLiM-mediated interactions (56, 57). Sequence context appears particularly important when non-interacting residues reduce the entropic penalty associated with the disorder-to-order transition occurring when a SLiM binds to a globular domain (58). Finally, a recent study reported screening a random combinatorial library of MAP2K1 D-site variants for function in promoting phosphorylation of ERK2 (59). In addition to presenting the sequence in the context of a specific ERK activator, this function-based screening approach could select for features other than simply binding to the docking groove, such as the capacity to allosterically impact ERK2 activation loop conformation or dynamics (22, 48, 52).
In addition to nominating candidate MAPK interactors, our screens have also identified cryptic docking sites present in known interaction partners, including 16 proteins annotated in a literature-curated compendium of ERK targets (55) and 18 reported p38α substrates and other binding proteins (54) (Datasets S4 and S5). We verified that several full-length proteins harboring hit sequences interact with ERK2 or p38α in cells, and we validated TACC1 as a MAPK substrate. TACC1 is a member of the transformation-associated coiled coil family of proteins that localize to centrosomes during mitosis, where they function to recruit chTOG/MSPS family proteins to influence microtubule dynamics (60). Our finding that TACC1 is a docking-dependent substrate may explain observations that overexpression causes cytoplasmic sequestration of ERK2 and inhibition of downstream signaling (61). Phosphorylation by ERK could also impact the function of TACC1 in promoting cell division and transformation (62–64). Additional studies will be needed to determine how MAPK phosphorylation of TACC1 impacts its function.
Overall, studies presented here expand the D-site mediated interactomes of ERK2 and p38α and provide insight into how SLiMs mediate selectivity in MAPK signaling networks. Understanding the full repertoire of D-site-dependent interactions is important in light of recent efforts to identify small molecules targeting MAPK docking grooves, which may serve as substrate-selective inhibitors (65–69). In addition, recurrent cancer-associated mutations in the MAPK1 gene encoding ERK2 map to the CD region and are likely to selectively disrupt phosphorylation of a subset of its substrates (70). Our approach should also be generally applicable to provide insight into understudied MAPKs that do not participate in canonical cascades (71), for which D-site interactions are poorly characterized. By providing a set of candidate substrates and interaction partners, this work will serve as a resource for future exploration of the regulation and function of these important kinases.
Materials and Methods
Plasmids.
The Y2H prey vector in pGAD-GH included the ELK1 transcriptional activation domain (residues 306 to 427) with a C-terminal V5 tag and was modified to facilitate D-site sequence substitutions and to match the linker region in the oligonucleotide library. The full ORF sequence is provided in supplementary SI Appendix, Fig. S1. Y2H bait vectors inserted rat ERK2 or p38α coding sequences into pGBT9. Plasmids for protein expression are described in SI Appendix. A complete list of plasmids and gene-specific primers is in SI Appendix, Tables S1 and S2.
D-Site Library Construction.
The human proteomic D-site library oligonucleotide pool (16) was amplified by PCR, restriction enzyme digested and ligated into pGAD-GH-ELK1 to replace the ELK1 D-site. MAX Efficiency DH10B electrocompetent Escherichia coli (ThermoFisher) transformed with the ligation product were grown on solid media overnight at 37 °C, and plasmid DNA was isolated from pooled colonies. Clone representation was established by NGS (Dataset S1).
Y2H Analysis.
Y2H growth assays of individual ELK1 variants are described in SI Appendix. For library screens, yeast harboring bait plasmids were transformed with the ELK1 D-site library plasmid pool and selected on agar plates containing SD-Leu-Trp media. Transformants were suspended in SD-Leu-Trp liquid media and grown at 30 °C. When the culture reached an OD600 of 1.0, it was used to inoculate 50 mL of selective media (SD-Leu-Trp-His + 50 μM 3-AT) and 50 mL of non-selective media (SD-Leu-Trp) at an OD600 of 0.1. Yeast cultures were subjected to two growth and dilution cycles, with 25 mL aliquots reserved for DNA extraction each time. Extracted DNA was PCR-amplified with barcoded primers and sequenced on an Illumina HiSeq 4000 instrument with 100-bp paired-end reads. NGS data analysis is described in SI Appendix. NGS results are summarized in Datasets S2 (for ERK2 screens) and S3 (for p38α screens).
Protein Expression and Purification.
Active forms of ERK2 and p38α were produced by co-expression in bacteria with constitutively active alleles of their corresponding MKK. N-terminally His6-tagged kinase substrates were produced in bacteria and isolated by one-step affinity purification. Details for protein expression and purification are provided in SI Appendix.
Peptide Kinase Assays.
Peptides encoding hit sequences were synthesized commercially (GenScript) with an added Tyr-Ala sequence at the N-terminus to facilitate quantification and used without further purification. The concentrations of peptide DMSO stocks were determined from A280 values of aqueous dilutions calculated from the extinction coefficients of tyrosine (1,280 M−1 cm−1) and tryptophan (5,600 M−1 cm−1) residues.
Competitive peptide kinase assays were performed with a MAPK D-site-dependent reporter peptide (AssayQuant Technologies Inc, #AQT0376). Reporter peptide (5 μM) was mixed with various concentrations of unlabeled hit peptides in reaction buffer [54 mM HEPES (pH 7.4), 1.2 mM DTT, 0.01% Brij-35, 1% glycerol, 0.2 mg/mL BSA, and 10 mM MgCl2]. Reactions were initiated by the addition of MAPK (2 nM GST-ERK2, 4 nM GST-p38α, 20 nM GST-ERK2T160E/T161D, or 4 nM p38αE161T/D162T) and 10 μM ATP. Fluorescence (λex 360 nm, λem 485 nm) was measured in 90-s intervals over 21 min in a Spectra M5 plate reader, and rates were calculated from the linear portion of the reaction progress curve. Data were normalized to maximum (samples lacking competitor peptide) and minimum (lowest fluorescence value per experiment or control reactions lacking kinase) values. IC50 values for competitor peptides were calculated by fitting normalized rates to a sigmoidal dose–response curve using GraphPad Prism (Version 9.3.0). Dataset S6 provides IC50 values with 95% CIs for all peptides.
Radiolabel Kinase Assays.
Radiolabel kinase assays included 0.8 μM substrate and active MAPK (1.2 nM p38α, Reaction Biology #0443-0000-3, or 20 to 100 nM ERK2) in 20 mM HEPES (pH 7.4), 10 mM MgCl2, 1 mM DTT, and 10 μM [γ-32P] ATP (0.04 μCi/μL). After incubating at 30 °C for 20 min, reactions were quenched by adding 4x SDS-PAGE loading buffer, boiled for 5 min, and subjected to SDS-PAGE. Coomassie-stained gels were dried and exposed to a phosphor screen. Radiolabel incorporation was visualized on a Molecular Imager FX phosphorimager (Bio-Rad) and quantified using QuantityOne Software (Bio-Rad version 11.0.5).
Pulldown Assays.
HEK293T cells were co-transfected with MAPK and FLAG-tagged substrate plasmids using polyethyleneimine. After 48 h, cells were washed and suspended in ice-cold PBS, pelleted, and suspended in 500 μL lysis buffer. Buffer for ERK2 co-IP with WT proteins was 10 mM PIPES (pH 6.8), 50 mM NaF, 40 mM Na4P2O7, 50 mM NaCl, 150 mM sucrose, 0.1% Triton X-100, 2 mM Na3VO4, and cOmplete™ protease inhibitor cocktail (CPIC). For all other IPs, lysis buffer was 50 mM HEPES (pH 7.4), 150 mM NaCl, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM DTT 1 mM EDTA, and CPIC. After 10 min, lysates were centrifuged (21,500 × g, 10 min). Supernatant was incubated with 30 μL anti-FLAG M2 affinity gel (Sigma, A2220) for 2 to 16 h at 4 °C. Beads were pelleted, washed once with lysis buffer and twice in wash buffer [50 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM β-glycerophosphate, 0.1 mM Na3VO4, 0.01% Igepal CA-630], resuspended in 2X SDS-PAGE loading buffer, and boiled for 5 min. Samples were subjected to SDS-PAGE and immunoblotting as described in SI Appendix.
Protein Phosphorylation in Cells.
HEK293T cells were transiently transfected with pCDNA3-FLAG-MLXIPL or pV1900-TACC1 as described above. For ERK activation, cells were starved in 0.1% FBS for 24 h prior to addition of EGF (R&D systems #236-EG, diluted in 1 mg/mL BSA/PBS) to 25 ng/μL or carrier alone for 5 min at 37 °C. To induce p38 activation, cells were stimulated with 500 nM anisomycin or 0.001% DMSO vehicle for 1 h at 37 °C. Where indicated cells were pre-treated with with 500 nM trametinib (SelleckChem #S2673) or 50 μM SB203580 (Sigma #559389) for 1 h at 37 °C prior to stimulation. Cells were placed on ice, washed once with ice-cold PBS, scraped into 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM NaF, 0.1% Triton X-100, 1 mM β-glycerophosphate, 2.5 mM Na4P2O7, 1 mM Na3VO4 with cOmplete™ protease inhibitor cocktail and 10 μg/mL pepstatin A. After 10 min, incubation lysates were clarified by centrifugation (10 min, 21,500 × g). Equal amounts of total protein as determined by BCA assay were fractionated with 5% SDS-polyacrylamide gels containing 25 nM Phos-tag reagent (Nard Institute AAL-107) and 50 μM MnCl2 or by standard SDS-PAGE, followed by immunoblotting (SI Appendix).
For MS analysis, HEK293T cells were transfected, treated, and lysed as described above, and proteins were isolated by affinity chromatography with anti-FLAG M2 gel. Samples were fractionated by SDS-PAGE, and gels were stained briefly with Coomassie and destained. TACC1 or MLXIPL bands were processed and subjected to MS as described in SI Appendix, and phosphopeptides identified are provided in Dataset S7.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Acknowledgments
We thank Titus Boggon and Lise Thomas for comments on the manuscript and members of the Turk lab for feedback on this research. We thank John Blenis and Chris Burd for providing plasmids and the Yeast Resource Center at the University of Washington for Y2H strains. We thank Ross Tomaino at the Taplin Mass Spectrometry facility for mass spectrometry analysis and help with data processing. This work was supported by NIH R01 GM135331 to B.E.T., NSF Graduate Research Fellowship 1752134 to J.T.R., and support from the China Scholarship Council to G.S.
Author contributions
J.T.R., G.S., and B.E.T. designed research; J.T.R., H.J.L., and P.L.P. performed research; J.T.R., H.J.L., and G.S. contributed new reagents/analytic tools; J.T.R. and B.E.T. analyzed data; and J.T.R. and B.E.T. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Mass spectrometry data have been deposited in ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier (72). All other data are included in the manuscript and/or supporting information.
Supporting Information
References
- 1.Raman M., Chen W., Cobb M. H., Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Lavoie H., Gagnon J., Therrien M., ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Canovas B., Nebreda A. R., Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 22, 346–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeke A., Misheva M., Remenyi A., Bogoyevitch M. A., JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 80, 793–835 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faust D., et al. , Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 10, 6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller C. J., Turk B. E., Homing in: Mechanisms of substrate targeting by protein kinases. Trends Biochem. Sci. 43, 380–394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson J. L., et al. , An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanoue T., Nishida E., Molecular recognitions in the MAP kinase cascades. Cell. Signal. 15, 455–462 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Tanoue T., Adachi M., Moriguchi T., Nishida E., A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Peti W., Page R., Molecular basis of MAP kinase regulation. Protein Sci. 22, 1698–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyla M., Gonzalez Foutel N. S., Otzen D. E., Kjaergaard M., The optimal docking strength for reversibly tethered kinases. Proc. Natl. Acad. Sci. U.S.A. 119, e2203098119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garai A., et al. , Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 5, ra74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardwell A. J., Abdollahi M., Bardwell L., Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem. J. 370, 1077–1085 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho D. T., Bardwell A. J., Grewal S., Iverson C., Bardwell L., Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J. Biol. Chem. 281, 13169–13179 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bardwell A. J., Frankson E., Bardwell L., Selectivity of docking sites in MAPK kinases. J. Biol. Chem. 284, 13165–13173 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi G., et al. , Proteome-wide screening for mitogen-activated protein kinase docking motifs and interactors. Sci. Signal. 16, eabm5518 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whisenant T. C., et al. , Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 6, e1000908 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeke A., et al. , Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol. Syst. Biol. 11, 837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bardwell A. J., Bardwell L., Two hydrophobic residues can determine the specificity of mitogen-activated protein kinase docking interactions. J. Biol. Chem. 290, 26661–26674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang C. I., Xu B. E., Akella R., Cobb M. H., Goldsmith E. J., Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Remenyi A., Good M. C., Bhattacharyya R. P., Lim W. A., The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol. Cell 20, 951–962 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Zhou T., Sun L., Humphreys J., Goldsmith E. J., Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure 14, 1011–1019 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Liu S. J., Sun J. P., Zhou B., Zhang Z. Y., Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc. Natl. Acad. Sci. U.S.A. 103, 5326–5331 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akella R., Min X., Wu Q., Gardner K. H., Goldsmith E. J., The third conformation of p38α MAP kinase observed in phosphorylated p38α and in solution. Structure 18, 1571–1578 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Ma W., et al. , Phosphorylation of DCC by ERK2 is facilitated by direct docking of the receptor P1 domain to the kinase. Structure 18, 1502–1511 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y. Y., Wu J. W., Wang Z. X., A distinct interaction mode revealed by the crystal structure of the kinase p38α with the MAPK binding domain of the phosphatase MKP5. Sci. Signal. 4, ra88 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Gogl G., Toro I., Remenyi A., Protein-peptide complex crystallization: A case study on the ERK2 mitogen-activated protein kinase. Acta Crystallogr. D Biol. Crystallogr. 69, 486–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kragelj J., et al. , Structure and dynamics of the MKK7-JNK signaling complex. Proc. Natl. Acad. Sci. U.S.A. 112, 3409–3414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng L., et al. , Multiplexing the quantitation of MAP kinase activities using differential sensing. J. Am. Chem. Soc. 144, 4017–4025 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Yang S. H., Yates P. R., Whitmarsh A. J., Davis R. J., Sharrocks A. D., The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell. Biol. 18, 710–720 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pulido R., Zuniga A., Ullrich A., PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 17, 7337–7350 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardwell A. J., Flatauer L. J., Matsukuma K., Thorner J., Bardwell L., A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 276, 10374–10386 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar G. S., Page R., Peti W., The interaction of p38 with its upstream kinase MKK6. Protein Sci. 30, 908–913 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanoue T., Maeda R., Adachi M., Nishida E., Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 20, 466–479 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson L. B., Yaffe M. B., Imperiali B., Selective mitogen activated protein kinase activity sensors through the application of directionally programmable D domain motifs. Biochemistry 53, 5771–5778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Shea J. P., et al. , pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 10, 1211–1212 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Goddard T. D., et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waheed F., et al. , Central role of the exchange factor GEF-H1 in TNF-alpha-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol. Biol. Cell 24, 1068–1082 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Thun A., et al. , Extracellular signal-regulated kinase regulates RhoA activation and tumor cell plasticity by inhibiting guanine exchange factor H1 activity. Mol. Cell. Biol. 33, 4526–4537 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujishiro S. H., et al. , ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 368, 162–167 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Fantz D. A., Jacobs D., Glossip D., Kornfeld K., Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276, 27256–27265 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Zeng X., Ruff K. M., Pappu R. V., Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 119, e2200559119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uversky V. N., The intrinsic disorder alphabet. III. Dual personality of serine. Intrinsically Disord Proteins 3, e1027032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuxreiter M., Tompa P., Simon I., Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Sharma R., Raduly Z., Miskei M., Fuxreiter M., Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 589, 2533–2542 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Tompa P., Fuxreiter M., Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Hadzi S., Loris R., Lah J., The sequence-ensemble relationship in fuzzy protein complexes. Proc. Natl. Acad. Sci. U.S.A. 118, e2020562118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar G. S., et al. , Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl. Acad. Sci. U.S.A. 115, 4655–4660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francis D. M., et al. , Structural basis of p38α regulation by hematopoietic tyrosine phosphatase. Nat. Chem. Biol. 7, 916–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piserchio A., et al. , Local destabilization, rigid body, and fuzzy docking facilitate the phosphorylation of the transcription factor Ets-1 by the mitogen-activated protein kinase ERK2. Proc. Natl. Acad. Sci. U.S.A. 114, E6287–E6296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piserchio A., et al. , Solution NMR insights into docking interactions involving inactive ERK2. Biochemistry 50, 3660–3672 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee T., et al. , Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Hornbeck P. V., et al. , PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stark C., et al. , BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unal E. B., Uhlitz F., Bluthgen N., A compendium of ERK targets. FEBS Lett. 591, 2607–2615 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Stein A., Aloy P., Contextual specificity in peptide-mediated protein interactions. PLoS One 3, e2524 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivarsson Y., Jemth P., Affinity and specificity of motif-based protein-protein interactions. Curr. Opin. Struct. Biol. 54, 26–33 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Flock T., Weatheritt R. J., Latysheva N. S., Babu M. M., Controlling entropy to tune the functions of intrinsically disordered regions. Curr. Opin. Struct. Biol. 26, 62–72 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Scheele R. A., et al. , Droplet-based screening of phosphate transfer catalysis reveals how epistasis shapes MAP kinase interactions with substrates. Nat. Commun. 13, 844 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peset I., Vernos I., The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 18, 379–388 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Lauffart B., Sondarva G. V., Gangisetty O., Cincotta M., Still I. H., Interaction of TACC proteins with the FHL family: Implications for ERK signaling. J. Cell Commun. Signal. 1, 5–15 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gergely F., et al. , The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc. Natl. Acad. Sci. U.S.A. 97, 14352–14357 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Still I. H., Hamilton M., Vince P., Wolfman A., Cowell J. K., Cloning of TACC1, an embryonically expressed, potentially transforming coiled coil containing gene, from the 8p11 breast cancer amplicon. Oncogene 18, 4032–4038 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Cully M., et al. , Transforming acidic coiled coil 1 promotes transformation and mammary tumorigenesis. Cancer Res. 65, 10363–10370 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Miller C. J., Muftuoglu Y., Turk B. E., A high throughput assay to identify substrate-selective inhibitors of the ERK protein kinases. Biochem. Pharmacol. 142, 39–45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boston S. R., et al. , Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9. BMC Cancer 11, 7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sammons R. M., et al. , A novel class of common docking domain inhibitors that prevent ERK2 activation and substrate phosphorylation. ACS Chem. Biol. 14, 1183–1194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alexa A., et al. , Peptide based inhibitors of protein binding to the mitogen-activated protein kinase docking groove. Front. Mol. Biosci. 8, 690429 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samadani R., et al. , Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRaf. Biochem. J. 467, 425–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor C. A., et al. , Functional divergence caused by mutations in an energetic hotspot in ERK2. Proc. Natl. Acad. Sci. U.S.A. 116, 15514–15523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coulombe P., Meloche S., Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta 1773, 1376–1387 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Turk B. E., Linear motif specificity in signaling through p38α and ERK2 mitogen-activated protein kinases. PRIDE. https://www.ebi.ac.uk/pride/archive/projects/PXD046248. Deposited 19 October 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Data Availability Statement
Mass spectrometry data have been deposited in ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier (72). All other data are included in the manuscript and/or supporting information.