

Significance
We wanted to determine the dose-dependent effects of TGF-β signals on memory CD8 T cell reactivation given the relevance in the context of repeated infections with pathogens as well as tumor responses. To our surprise, and contrary to the prevailing notion that TGF-β is a master suppressor of CD8 T cell effector functions, we found that TGF-β modulates several aspects of memory CD8 T cell function in a dose-dependent manner and based on the strength of the reactivation signal. We discuss the implications of our findings in the context of recurrent infections, autoimmunity, and cancer and highlight the clinical relevance: Therapeutic blocking of TGF-β signals for memory T cells is likely to have unwanted side effects such as altered trafficking.
Keywords: CD8 T cell, TGF beta, memory T cell, trafficking, effector
Abstract
Transforming growth factor β (TGF-β) directly acts on naive, effector, and memory T cells to control cell fate decisions, which was shown using genetic abrogation of TGF-β signaling. TGF-β availability is altered by infections and cancer; however, the dose-dependent effects of TGF-β on memory CD8 T cell (Tmem) reactivation are still poorly defined. We examined how activation and TGF-β signals interact to shape the functional outcome of Tmem reactivation. We found that TGF-β could suppress cytotoxicity in a manner that was inversely proportional to the strength of the activating TCR or proinflammatory signals. In contrast, even high doses of TGF-β had a comparatively modest effect on IFN-γ expression in the context of weak and strong reactivation signals. Since CD8 Tmem may not always receive TGF-β signals concurrently with reactivation, we also explored whether the temporal order of reactivation versus TGF-β signals is of importance. We found that exposure to TGF-β before or after an activation event were both sufficient to reduce cytotoxic effector function. Concurrent ATAC-seq and RNA-seq analysis revealed that TGF-β altered ~10% of the regulatory elements induced by reactivation and also elicited transcriptional changes indicative of broadly modulated functional properties. We confirmed some changes on the protein level and found that TGF-β-induced expression of CCR8 was inversely proportional to the strength of the reactivating TCR signal. Together, our data suggest that TGF-β is not simply suppressing CD8 Tmem but modifies functional and chemotactic properties in context of their reactivation signals and in a dose-dependent manner.
The pleiotropic functions of TGF-β have been described in a wealth of literature and include roles in angiogenesis, wound healing, cancer, and regulating immune responses (1, 2). TGF-β1 [often referred to as TGF-β since it is the most prevalent and studied isoform (3)] is typically considered to be a powerful suppressor of the immune response (3). Most immune cells express the TGFβ type I and type II serine/threonine kinase receptors (also referred to as TβRI and TβR2 or TGF-βRI and TGF-βRII) and are thus able to respond to TGF-β signals (2). TGF-β affects T cells at all stages of development, starting in the thymus during T cell development, T cell homeostasis in the periphery, as well as T cell differentiation following activation (4).
Two mouse model approaches have been widely used to define the consequences of TGF-β signaling on T cell fate and differentiation: First, transgenic mice expressing a dominant negative form of the TGF-β receptor II (dnTGβRII) under the control of the CD4 promoter that lacks the CD8 silencer (5) or a CD2 promoter (6), thus allowing for expression in CD4 and CD8 T cells in these mouse lines. A follow-up study with the CD4-dnTGFβRII mice revealed that the dominant negative receptor still had some signaling capacity (possibly independent of bona fide TGF-β receptor activation) (7), which somewhat complicates the interpretation of studies that used these mice. Second, mice bearing TGF-βr2 alleles with flanking loxP sites (floxed TGF-βRII) allow for conditional deletion in T cells by crossing to mice expressing Cre recombinase under control of the Cd4 promoter that is active in thymocytes (CD4-cre) (8, 9) or expressing Cre under control of the distal Lck promoter active in mature, naive T cells (dLck-cre) (10). Of note, these distinct approaches to abrogating TGF-β signaling in T cells also had distinct disease phenotypes, which ultimately helped separate the roles of TGF-β signals during thymic selection and maintenance of tolerance in the periphery (8–10). To study the consequences of TGF-β signals during the effector stage flox TGF-βRII mice were crossed to mice expressing Cre under control of the Granzyme B locus (granzyme B-cre), which revealed a role for TGF-β in controlling the number of short-lived effector cells (7). To study the effect on Tmem, flox TGF-βRII mice were crossed to mice expressing Cre fused to the ligand binding domain of the estrogen receptor (ER-cre) (11). Tamoxifen-induced Cre-mediated deletion of TGF-βRII during the CD8 Tmem stage revealed that TGF-β signals are required for the maintenance of the CD8 Tmem transcriptional program and function (11).
An inherent limitation of these powerful genetic approaches is that deletion or expression of a dominant negative form of a receptor precludes studying dose-dependent effects of the ligand. For cytokines and T cells, it is noteworthy that the effect of a signal on T cell fate decisions does not necessarily follow a titration curve, but can result in a quantal—all or none—outcome (12). In context of TGF-β, the potential dose-dependent effects on T cells at various stages of differentiation are still poorly defined. This is at least in part due to the challenge of measuring biologically active TGF-β (13). TGF-β is abundant in blood and tissues, but most of the TGF-β in blood and tissues is present as a complex with latency-associated peptide (LAP) and latent TGF-β-binding proteins (LTBPs), respectively. Once activated by integrins or other signals, the receptor-binding site of TGF-β is exposed and TGF-β becomes active. Measuring the availability of the latent and active form of TGF-β is possible using ELISAs and reporter cells (13–15) but often varies based on the reagents and protocols used (13). Thus, the concentration range of biologically active TGF-β in health versus disease is still poorly defined.
We were specifically interested in potential concentration-dependent effects of TGF-β on CD8 Tmem in the context of reactivation. CD8 Tmem reactivation is typically considered in the context of repeated infections with pathogens but is also highly relevant in the context of tumor responses: Vaccines that elicit immune responses against tumor antigens have had promising results and generate memory T cells (16, 17), and tumor-specific memory CD8 T cells responding to PD-1/PD-L1 blockade reside in the tumor-draining lymph node (18). We thus wanted to examine whether the effect of TGF-β on memory CD8 T cell reactivation is 1) potentially distinct from its role during T cell priming and 2) TGF-β dose- and 3) activation-signal dependent. Since TGF-β has been reported to inhibit IFN-γ production by cytokine-activated memory CD8 T cells (19, 20), we wanted to define whether the type of activating signal (T cell receptor- vs. cytokine-mediated) yields distinct responses to TGF-β signals.
Since the mouse model has been so widely used to define the effects of TGF-β signaling, we also used a mouse model system to generate a CD8 Tmem population with a well-defined T cell receptor specific for an epitope of chicken ovalbumin (OT-I T cells). We utilized OT-I Tmem to define how low to high concentrations of TGF-β signals affect the functional properties of CD8 Tmem across a range of reactivation signals (weak to strong TCR activating and cytokine-driven activation). We found that TGF-β was not broadly immunosuppressive, but rather altered functional and chemotactic properties in a dose- and reactivation context-dependent manner. TGF-β could suppress cytotoxicity in a manner that was inversely proportional to the strength of the activating TCR or proinflammatory signal. In contrast, TGF-β had a rather modest effect on IFN-γ expression. Importantly, TGF-β was not merely suppressing aspects of effector function, it directly increased expression of some chemokine receptors, including CCR8. TGF-β induced the expression of CCR8 in CD8 Tmem regardless if reactivation occurred via TCR or cytokines. Interestingly, induction of expression was inversely proportional to the suppression in cytotoxicity and most effective in CD8 Tmem reactivated by a weak TCR signal. We discuss the implication of our findings in context of CD8 Tmem reactivation in response to infections and in tumors.
Results
TGF-β Strongly Inhibits Cytotoxic Function but Not IFN-γ Production by CD8 Tmem in a Dose-Dependent manner.
To generate a population of CD8 Tmem with known Ag- specificity, we transferred congenically marked OT-I T cells, which recognize the SIINFEKL (N4) epitope of chicken ovalbumin (OVA) bound to the MHC class I molecule H-2Kb, into C57BL/6J mice. We then infected these mice with OVA-expressing vesicular stomatitis virus (VSV-OVA) and waited at least 60 d before isolating cells from these OT-I memory mice. As a first step, we wanted to define the effect of a high dose (100 ng/mL) of TGF-β in context of a very strong reactivating signal: We isolated T cells from spleen and lymph nodes (LN) of OT-I memory mice followed by ex vivo stimulation with plate-bound anti-CD3/28 antibodies (CD3/28) for 24 h with or without TGF-β (Fig. 1A). Using flow cytometry, we found that TGF-β was sufficient to reduce IFN-γ expression, but greatly diminished GzmB expression in reactivated OT-I memory T cells (Fig. 1 B and C).
Fig. 1.

TGF-β preferentially inhibits cytotoxicity of memory CD8+ T cells in a dose-dependent manner. (A) Schematic of naive OT-I CD8+ T cell adoptive transfer, memory OT-I T cell generation with VSV-OVA, T cell isolation with magnet-activated cell sorting (MACS) from Ag-experienced OT-I memory mice, and subsequent ex vivo stimulation and analysis. Stimulation was 24 h with plate-bound anti-CD3 and anti-CD28 (CD3/28) in the presence or absence of TGF-β at 100 ng/mL. (B) Representative expression and gating of IFN-γ and GzmB in OT-I Tmem after stimulation. (C) IFN-γ and GzmB frequencies. Each point represents an individual animal, with connecting lines across points from the same animal (n = 14 animals). Statistical significances were calculated using paired t tests. (D) Frequencies of IFN-γ and GzmB in OT-I Tmem post 24 h stimulation with CD3/28 in the presence of titrated TGF-β (n = 7). TGF-β was titrated in twofold dilutions starting with 20 ng/mL and ending with 0.04 ng/mL, in fivefold dilutions starting with 20 ng/mL and ending at 0.032 ng/mL, and in fivefold dilutions starting with 1 ng/mL and ending at 0.0016 ng/mL. Each point represents an individual animal with connecting lines across points from the same animal. Data shown are from 6 to 14 independent experiments.
Next, we titrated the concentration of TGF-β to assess its dose-dependent effects. As a reference value, the total TGF-β1 in mouse spleen has been reported to be ~1,000 ng/g spleen (21) (SI Appendix, Fig. S1A), but active TGF-β is often only a fraction of total TGF-β (13). We observed again that IFN-γ was fairly resistant to TGF-β as only concentrations above 1 ng/mL appeared to have at least a modest effect (Fig. 1D). In contrast, cytotoxicity was much more susceptible to TGF-β-mediated suppression as a dose of 1.3 ng/mL was sufficient to decrease the frequency of granzyme B expressing OT-I T cells twofold, indicated as the “1/2 Max” value. We also examined activation-associated protein biomarkers in OT-I Tmem and found that the frequency of cells expressing Programmed cell death protein 1 (PD-1) and median fluorescence intensity (MedFI) of the transcription factor T cell factor 1 (TCF1) were very modestly but significantly increased by TGF-β. In contrast, the frequency of Ki67-expressing Tmem and the MedFI of TOX did not significantly change (SI Appendix, Fig. S1B).
Finally, we also assessed the effect on reactivation of endogenous CD8 Tmem. We found that in the presence of TGF-β, reactivated endogenous Tmem had modestly reduced IFN-γ, but starkly decreased GzmB frequencies (SI Appendix, Fig. S1C) thus mirroring our OT-I T cell data. Similarly, we found that the frequency of PD-1+ CD8 Tmem slightly increased, while the frequency of Ki67, MedFI Tox, and MedFI TCF1 did not change (SI Appendix, Fig. S1D). We observed similar effects of TGF-β when we recapitulated our ex vivo experimental approach with human CD8 Tmem (SI Appendix, Fig. S2A). Of note, human PBMCs contain effector memory CD8 T cells which express granzyme B prior to reactivation. TGF-β did not appear to affect this steady-state expression pattern (SI Appendix, Fig. S2B).
TGF-β Is not Sufficient to Fully Suppress Cytotoxicity when CD8+ Tmem Are Reactivated by Strong TCR Signals or Cytokines.
Cross-linking of the TCR by a monoclonal antibody delivers a very strong reactivation signal. To assess the effects of TGF-β in cells reactivated via their TCR triggered through peptide/MHC complexes, we compared OT-I Tmem reactivated by SIINFEKL (N4) and SIIQFEKL (Q4) peptides. N4 (SIINFEKL) bound to H-2Kb is a strong agonist for OT-I T cells, while the variant Q4 (SIIQFEKL) binds equally well to H-2Kb but is only a weak agonist for OT-I T cells (22). As an alternative reactivation signal, we also stimulated OT-I Tmem with a combination of IL-12, IL-15, and IL-18 (IL-12/15/18; Cyt) to induce reactivation in a TCR agonist-independent manner. We found that IFN-γ expression was again only modestly affected in all experimental conditions (Fig. 2A), while TGF-β essentially ablated cytotoxic function in N4- and Q4-reactivated OT-I Tmem (Fig. 2A and SI Appendix, Fig. S1 E and F). Cytokine-mediated reactivation yielded outcomes comparable to TCR cross-linking with and without cytokine treatment (SI Appendix, Fig. S1 E and F). Finally, we titrated TGF-β in context of reactivation with N4, Q4, and Cyt stimulation (Fig. 2 B and C). When we reactivated OT-I Tmem with either N4 or Q4 peptide, we found that a much lower concentration of TGF-β was sufficient to reduce granzyme B expression (0.16 and 0.09 ng/mL of TGF-β reduce the frequency of gzmB+ OT-I Tmem 2× fold for N4 and Q4, respectively, compared to 0.99 ng/mL after cytokine reactivation), but the impact on IFN-γ expression was again much more limited across all restimulation conditions.
Fig. 2.
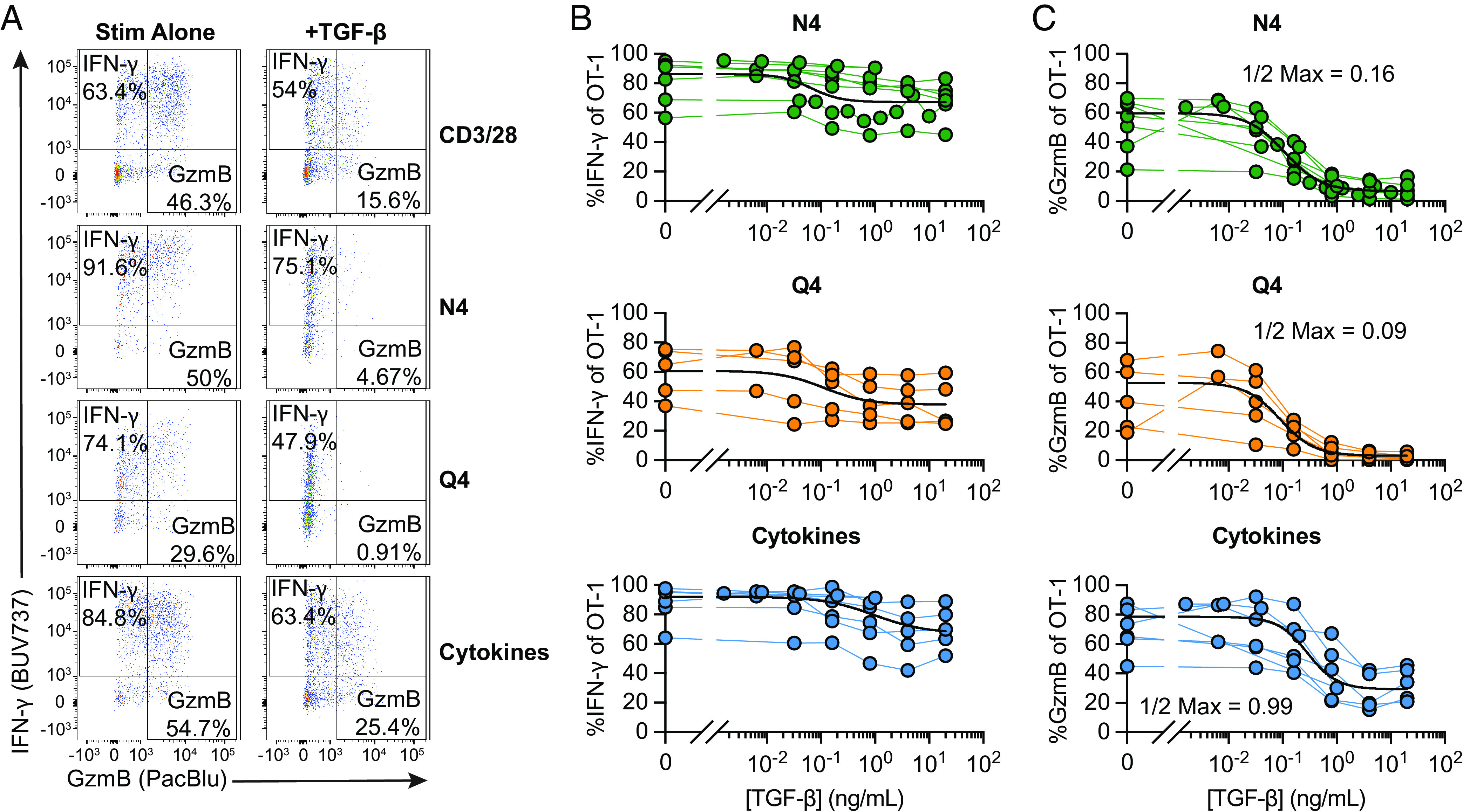
Strongly activated memory CD8+ T cells are less susceptible to TGF-β-mediated suppression. Stimulations were 24 h with CD3/28, 100 nM SIINFEKL (N4), 100 nM SIIQFEKL (Q4), rIL-12, rIL-15, and rIL-18 in combination, with rIL12 and rIL-15 at 100 ng/mL and rIL-18 at 0.5 ng/mL (“Cyt”), and TGF-β at 20 ng/mL. (A) Representative gating of IFN-γ and GzmB staining in OT-I Tmem at 24 h post stimulation with CD3/28, N4, Q4, or Cyt in the presence or absence of TGF-β. (B) Frequencies of IFN-γ and (C) GzmB of OT-I T cells 24 h post indicated stimulation condition in the presence of titrated TGF-β (N4 conditions are depicted from n = 7 animals, Q4 from n = 5, and Cytokines from n = 6). Each point represents an individual animal with connecting lines across points from the same animal. TGF-β was titrated in twofold dilutions starting with 20 ng/mL and ending with 0.04 ng/mL, in fivefold dilutions starting with 20 ng/mL and ending at 0.032 ng/mL, and in fivefold dilutions starting with 1 ng/mL and ending at 0.0016 ng/mL (for N4 and Cyt). Calculated ½ Max inhibitory capacity values indicated. Data shown are from 4 to 6 independent experiments.
Finally, we also titrated the proinflammatory cytokines to determine the relationship between TGF-β and strength of the reactivating proinflammatory signals. We used IL-12/15/18 to elicit strong IFN-γ production and found that TGF-β could reduce IFN-γ twofold when less than 10 ng/mL of each cytokine were available (SI Appendix, Fig. S3). To elicit strong granzyme B expression upon reactivation, we exposed OT-I Tmem to IL-12/IL-15—granzyme B expression was again much more susceptible to inhibition by TGF-β and essentially completely inhibited unless more than 25 ng/mL of each cytokine was present (SI Appendix, Fig. S3, Right).
Together, these data indicate that the inhibitory effect of TGF-β on reactivation-induced cytotoxicity can be tuned by the concentration of TGF-β as well as the strength of the activating signal, while IFN-γ production is comparatively resistant to TGF-β-mediated suppression.
TGF-β Can Still Affect Function if Reactivation Signals Temporally Precede the TGF-β Signal.
In these previous experiments, we provided reactivation and TGF-β signals at the same time, but we considered that CD8 Tmem may receive activating signals before or after a TGF-β signal (for example, reactivation in a lymph node followed by a high dose TGF-β exposure in the tissue). To test whether TGF-β could inhibit the cytotoxicity of already reactivated OT-I Tmem, we modified the ex vivo stimulation conditions to include two additional experimental conditions: first, reactivate the OT-I Tmem, followed by adding TGF-β either 6 h or 12 h after the reactivation stimulation (TGF-β 6 h, 12 h; Fig. 3A). We found that both the 6-h and 12-h delay between reactivation signal and TGF-β exposure inhibited IFN-γ to the same extent as the positive control (TGF at 0 h) in N4- and Q4-activated memory CD8+ T cells (Fig. 3B). IFN-γ was also attenuated by TGF-β when added 12 h after anti-CD3/CD28-mediated reactivation, while a 12-h delay had essentially no effect on IFN-γ production in the cytokine-mediated reactivation condition (Fig. 3B). For GzmB, we found that the frequency of gzmB+ OT-I Tmem increased the longer the delay between reactivation and TGF-β addition for N4 and Q4-mediated reactivation. For the CD3/CD28 and cytokine stimulation conditions, the 0-h control and 6-h delay groups were similar (Fig. 3C), while OT-I Tmem in the 12-h delay condition had more gzmB+ cells than the 0-h control group, but less than the no TGF-β control (Fig. 3C). To determine whether IFN-γ expression at the 24-h analysis time point reflects an overall decrease in IFN-γ production or altered IFN-γ production kinetics, we measured the concentrations of IFN-γ in the culture supernatant. We observed similar trends of reduction in these experimental groups, but these were not statistically significant (SI Appendix, Fig. S4A). Together, these data indicate that TGF-β can effectively limit cytotoxic function after CD8 Tmem have already been reactivated, particularly in context of reactivation with a low-affinity ligand, while only modestly affecting IFN-γ production.
Fig. 3.
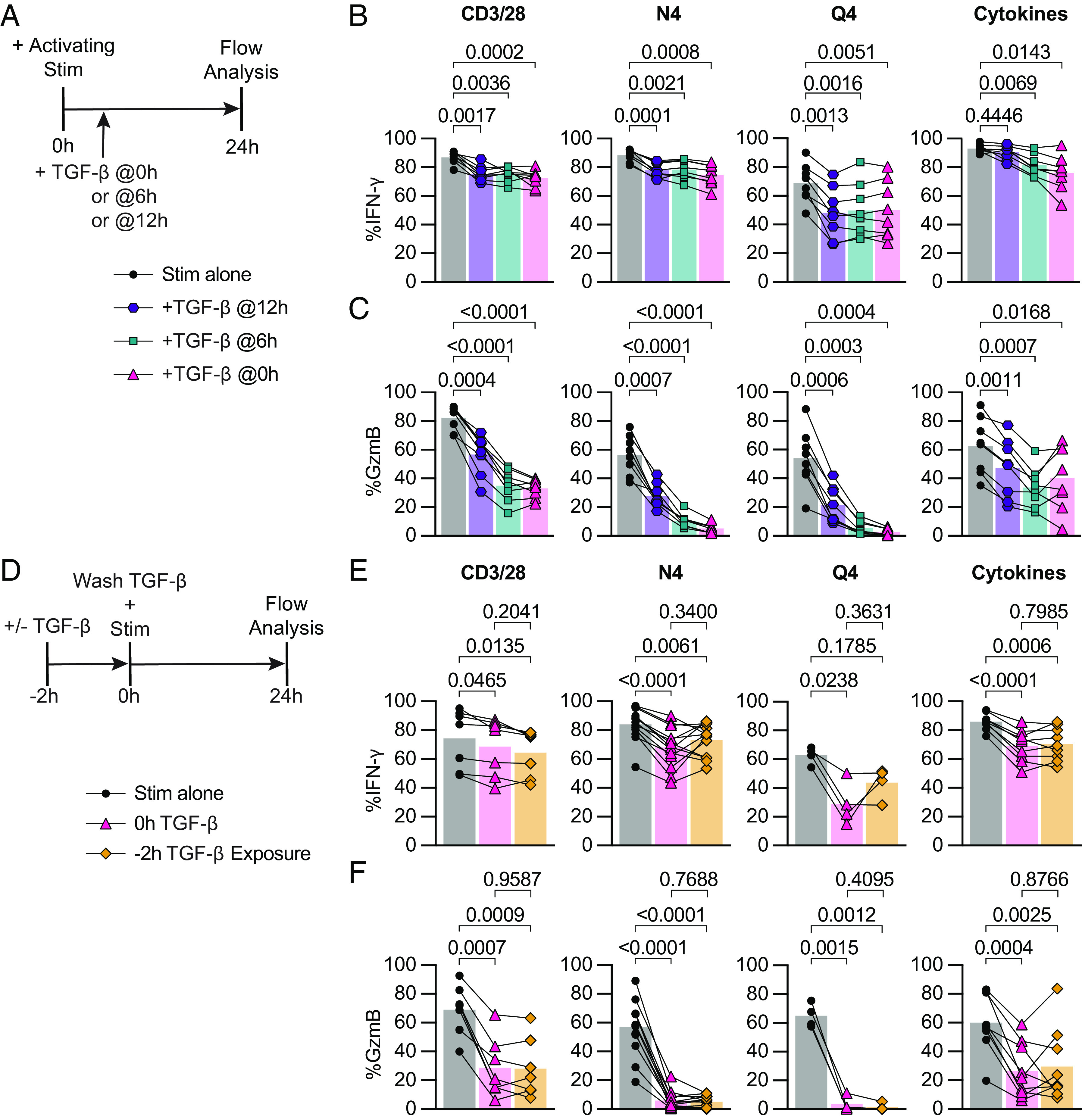
TGF-β inhibits cytotoxicity from recently reactivated memory CD8+ T cells, and short-term exposure to TGF-β inhibits cytotoxicity of subsequently activated memory CD8+ T cells. (A) Schematic of ex vivo stimulation; cells were stimulated for 24 h with CD3/28, N4, Q4, Cyt, and TGF-β at 100 ng/mL. TGF-β was added 0 h, 6 h, or 12 h poststart of activating stimulation. (B) Frequencies of IFN-γ and (C) GzmB in OT-I Tmem compared across stimulation conditions with TGF-β addition at indicated time points (n = 8 animals). (D) Schematic of ex vivo stimulation of isolated T cells from OT-I memory mice. Cells were treated with 100 ng/mL TGF-β or media alone for 2 h, the TGF-β was then washed out (down to 0.001 ng/mL), immediately followed by 24 h of activating stimulation. Stimulations were CD3/28, N4, Q4, Cyt, and TGF-β at 100 ng/mL. (E) Frequencies of IFN-γ and (F) GzmB in OT-I T cells compared across stimulation conditions. CD3/28 data depicted are from n = 7 animals, N4 from n = 13, Q4 from n = 4, and Cyt from n = 9. All indicated statistical significances were calculated using one-way ANOVA with Tukey’s multiple comparison test. Data shown are from 3 to 10 independent experiments.
Short-Term Exposure to TGF-β Is Sufficient to Inhibit Cytotoxicity of Subsequently Activated Memory CD8+ T Cells.
Next, we reversed the order of signals and asked whether a brief exposure to TGF-β prior to reactivation could also inhibit the subsequent CD8 Tmem effector response (for example, exposure to TGF-β in the tissue prior to tissue egress into the draining LN). We pre-exposed OT-I Tmem to TGF-β for 2 h, washed out the TGF-β, and then stimulated these cells for 24 h (Fig. 3D). Interestingly, we found that IFN-γ was inhibited to a similar extent in the 2-h pre-exposure condition as in the 0 h (TGF-β added with stimulation for 24 h) positive control TGF-β condition following cytokine stimulation (Fig. 3E). Similarly, in the CD3/28 stimulation condition, an average of 74.3% of OT-I Tmem expressed IFN-γ, which decreased to 68.6% and 64.5% in the 0 h TGF-β and 2 h exposure conditions, respectively. In the N4 and Q4 conditions, the pre-exposure had a different effect: IFN-γ expression increased from 67.9 to 73.1% (N4) and 29.0 to 43.8% (Q4) in the 0 h TGF-β vs. 2 h pre-exposure conditions (Fig. 3E). Surprisingly, we found that the 2 h pre- exposure to TGF-β was sufficient to inhibit GzmB expression to the same drastic extent as prolonged TGF-β exposure. In the CD3/28 stimulation, GzmB expression, on average, decreased twofold from 69.1 to 28.7%, and 28.0% in the 0 h TGF-β and 2 h exposure conditions, respectively. The frequency of granzyme B+ OT-I Tmem reactivated by N4 decreased from 57.0 to 6% with 0h TGF-β or 5.2% with 2 h exposure. OT-I Tmem cells reactivated by Q4 had an even greater 20-fold reduction in GzmB expression, from 65.0 to 3.5% and 1.5% in the 0 h TGF-β and 2 h exposure conditions, respectively. In line with previous data, the cytokine-activated OT-I Tmem exhibited suppression similar to CD8 Tmem reactivated by TCR cross-linking, with GzmB expression decreasing from 60.1% to only 26.5% and 29.6% in the 0 h TGF-β and 2 h exposure conditions. (Fig. 3F).
We next examined whether the TGF-β exposed Tmem could regain full cytotoxic function after a short rest period. To test this, after the 2-h exposure to TGF-β, we rested the T cells in fresh media for 4 h before addition of activating stimulation for 24 h. We found that even after resting for 4 h post-TGF-β exposure, GzmB expression could not be rescued, maintaining the twofold reduction in the CD3/28 condition, the 10-fold reduction in the N4 condition, and 20-fold reduction the Q4 condition (SI Appendix, Fig. S4B). In contrast, IFN-γ expression was similarly reduced when compared to the 0 h and 2 h exposure conditions (SI Appendix, Fig. S4C).
Overall, these data indicate that a short exposure to TGF-β is sufficient to control CD8 Tmem cytotoxic effector function for at least 24 h. To elucidate how this may occur, we next examined how TGF-β alters chromatin accessibility and the transcriptome of CD8 Tmem.
Brief Exposure to TGF-β Is Sufficient to Epigenetically and Transcriptionally Alter Memory CD8+ T Cells.
We concomitantly interrogated the epigenetic and transcriptional effects of TGF-β on reactivated memory CD8+ T cells. We set up a short TGF-β exposure condition (±2 h of TGF-β in the absence of stimulation) and a 24 h ex vivo restimulation condition (N4 ± TGF-β). OT-I Tmem from the same experiment were analyzed in parallel using ATAC- and RNA-sequencing (Fig. 4A) akin to recently published NK cell study examining cytokine signaling networks (23). Of note, we also assessed granzyme B and IFN-γ protein expression in parallel, thus allowing us to link these protein, transcript, and epigenetic datasets. We first used chromVar analysis of the ATAC-seq data to globally assess changes in SMAD transcription factor activity and confirm that we can detect TGF-β treatment-mediated changes. This revealed that a 2-h exposure to TGF-β was sufficient to detect increases in chromatin accessibility at regions containing motifs bound by the SMAD family, which are the downstream transcriptional factors of the TGF-βR complex (24), compared to media alone (Fig. 4B). The effect of TGF-β on SMAD TF motif-associated chromatin accessibility was more pronounced in the 24 h stimulation condition (Fig. 4B). We next assessed changes in differentially accessible (individual) peaks using a pairwise comparison approach (Fig. 4C). Of note, reactivation itself altered nearly 26,000 regulatory elements, which is about 25% of the regulatory elements in our global peak set of 99,317 peaks. We detected 2,286 differentially accessible peaks following reactivation with TGF-β and 369 differentially accessible peaks in the reactivation without TGF-β condition (Fig. 4C). Thus, TGF-β affects about 10% of the regulatory elements that are altered during reactivation. Similarly to the ATAC-seq data, we also detected some transcriptional changes in our ±2 h TGF-β group (28 down and 46 up) and a more substantial change (134 down 378 up) in the transcriptome after 24 h of N4 stimulation ± TGF-β (Fig. 4 D and E). Consistent with our flow cytometry findings, we found that GzmB was significantly decreased while IFN-γ was only minimally affected in the N4 + TGF-β condition at 24 h (Fig. 4F). Interestingly, we also found significant decreases in GzmC and Prf1 (Fig. 4F). The ATAC-seq data indicate that there are no significant changes in accessibility for at the Ifng, Gzmb, Gzmc, or Prf1 loci, all of which have decreased transcriptional abundance in the N4 + TGF-β group (Fig. 4G). As in our previous experiments, GzmB protein expression was decreased in these experiments as well (SI Appendix, Fig. S5A). Together, these data indicate that TGF-β can alter over 2,500 regulatory elements during reactivation, but not all transcriptional changes are necessarily caused by epigenetic changes.
Fig. 4.
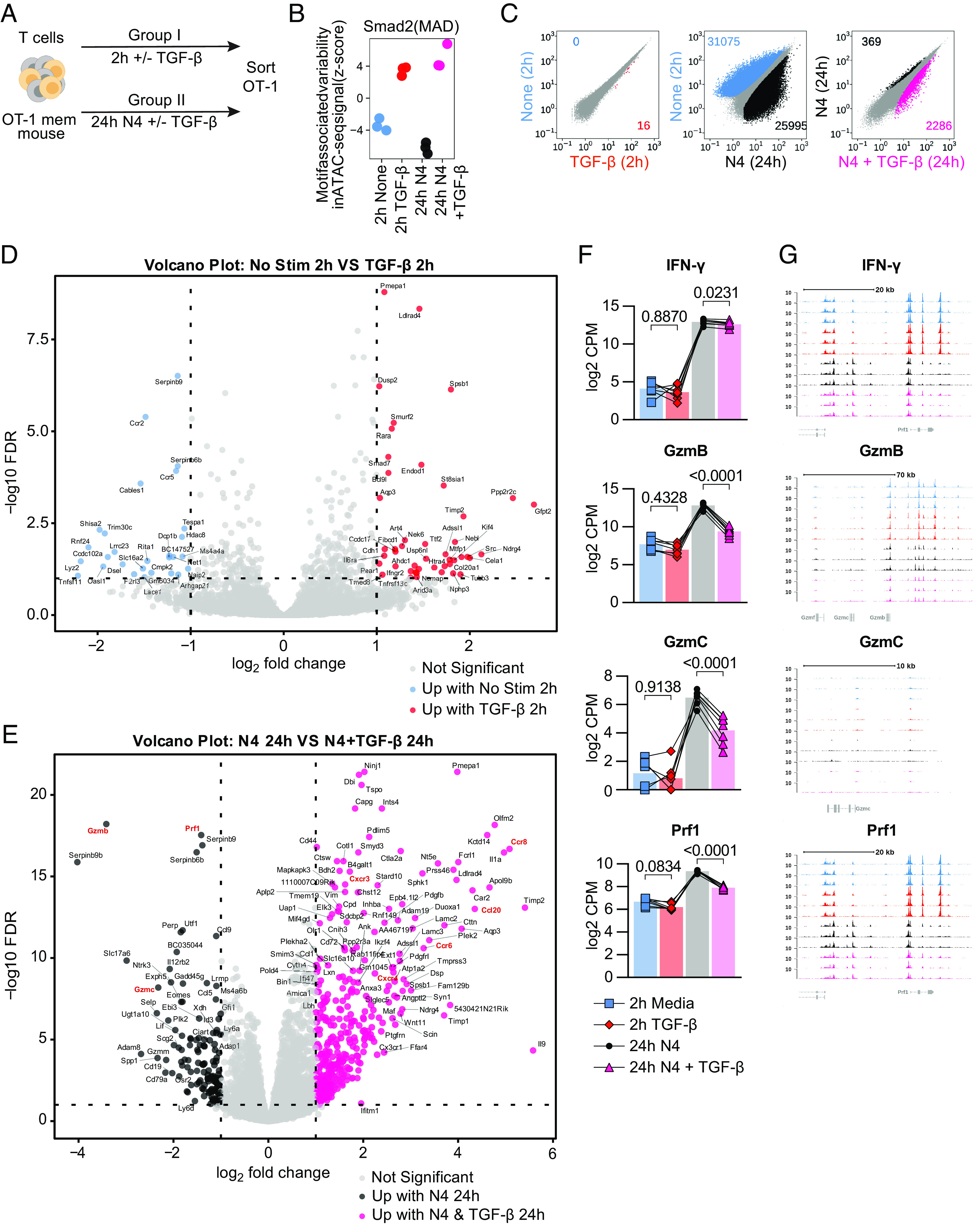
TGF-β epigenetically and transcriptionally alters memory CD8 T cell function. (A) Schematic for T cell isolation with MACS from Ag-experienced OT-I memory mice, subsequent ex vivo stimulation, and sorting of Live, CD8+, CD45.1+ OT-I Tmem cells. Sorted OT-I Tmem were processed immediately for ATAC- and RNA- seq library preparation and sequencing. Stimulations were 2 h with media alone or TGF-β at 100 ng/mL and 24 h 100 nM N4 with or without TGF-β at 100 ng/mL. (B) chromVAR analysis of ATAC-seq signal (z-score) with SMAD transcription factor motifs. The colors are indicative of the experimental condition and also used in C. (C) Scatterplots comparing differentially accessible chromatin regions for pairs of stimulation conditions (from left to right): 2 h untreated vs. 2 h + TGF-β; 2 h untreated vs. 24 h reactivated; 24 h reactivated vs. 24 h reactivated + TGF-β. Plots compare the normalized signal in counts/per peak at all regions across the genome to identify individual peaks that meet the criteria for differential accessibility based (at least 1.5-fold estimated change and adjusted P value of less than 0.01). The number of differentially accessible peaks is shown for each condition in each plot. (D) Volcano plot depicting differentially expressed genes between 2-h media and (E) Twenty-four-hour 100 ng/mL N4 stimulation conditions: with TGF-β or without TGF-β. DE gene cutoff values were adj P value 0.1, Log FC > 1 and < −1. (F) Selected DE genes from RNAseq and indicated statistical significance. (G) Chromatin accessibility of selected genes from ATAC-seq. All data depicted are from n = 7 animals and 2 independent experiments.
TGF-β Alters the Chemotactic Properties of Memory CD8 T Cells.
Several chemokines and chemokine receptors were also altered by TGF-β, including increased transcript expression of CCR8, CXCR3, CCR6, CXCR4, and CCL20. Of note, we detected a change in chromatin accessibility for CCR8, CXCR3, CCR6, CXCR4, and CCL20 indicating that the TGF-β-induced differences in chemokine transcripts may be due to the increased access of their loci (Fig. 5 A and B). Next, we examined whether these alterations also resulted in changed protein expression in a set of follow-up experiments. We performed ex vivo stimulations on OT-I Tmem as described in Figs. 3A and 4A. We found a modest increase of CXCR3 when OT-I Tmem were reactivated via their TCR and in the presence of TGF-β (SI Appendix, Fig. S6A). In contrast, the changes for CCR8 were much more pronounced: We found that CCR8 had distinct low and high expression patterns dependent on the stimulation condition, and we gated these populations accordingly (SI Appendix, Fig. S9B). In context of TCR-mediated reactivation, TGF-β greatly increased the frequency of CCR8hi expressing OT-I Tmem but was most pronounced in the N4 and Q4-reactivated groups (Fig. 5C). Of note, this occurred even when TGF-β was added 6 or 12 h after the reactivation stimulus (Fig. 5D). In contrast, TGF-β only elicited a substantial CCR8hi expressing OT-I Tmem population when given concurrently with the cytokines (Fig. 5D). Similarly, pre-exposure of OT-I Tmem to TGF-β for 2 h followed by washing out the TGF-β and reactivation with N4 or Q4 was sufficient to induce CCR8 expression that was nearly indistinguishable from the positive control groups (SI Appendix, Fig. S6 B and C). We also measured CCR8 expression in context of a TGF-β titration and found that the weaker the TCR activating signal, the higher CCR8hi expression frequency among OT-I Tmem (SI Appendix, Fig. S6D). Thus, the CCR8 expression pattern is a negative mirror of the granzyme B expression data.
Fig. 5.
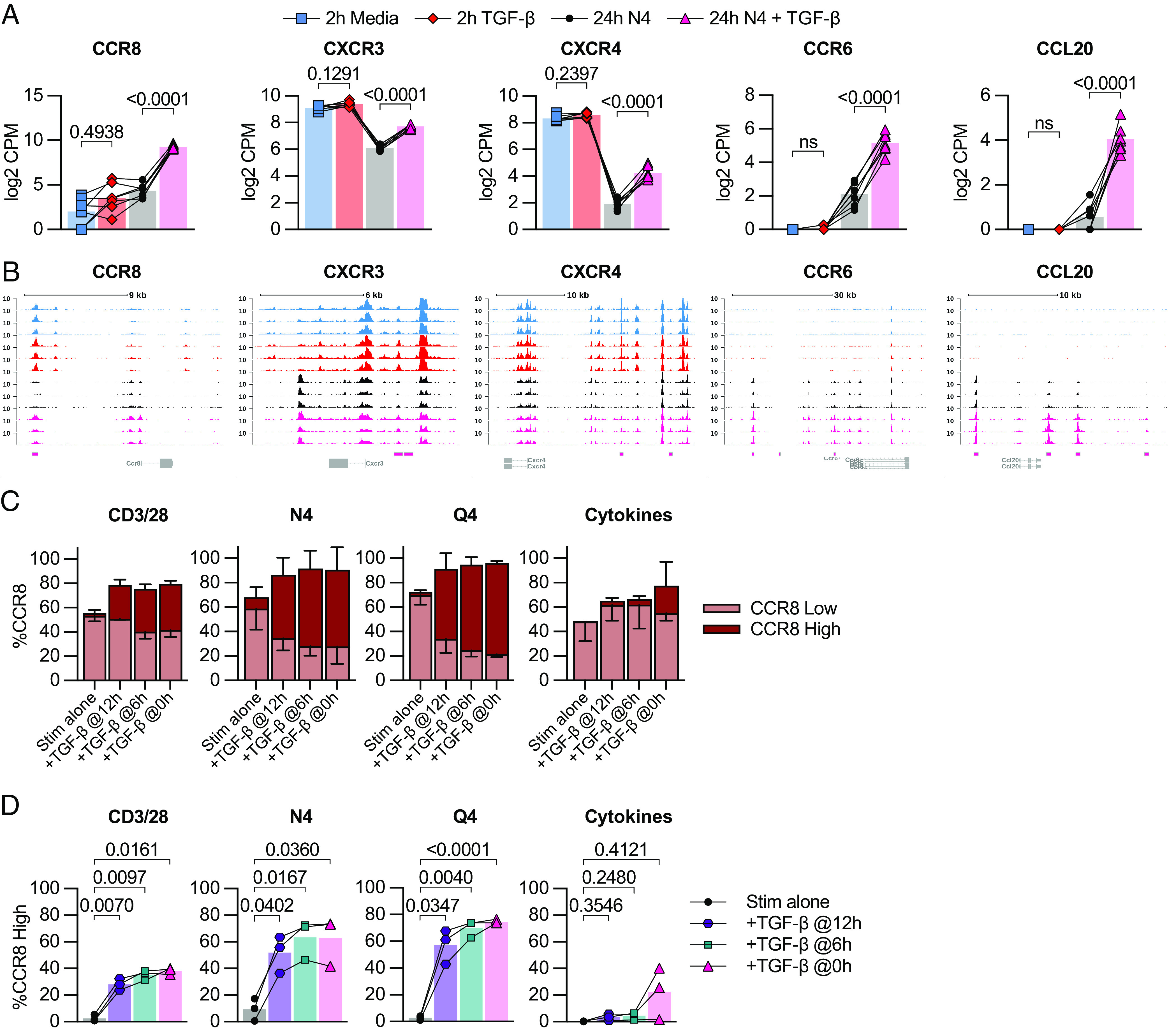
TGF-β epigenetically and transcriptionally alters memory CD8 T cell chemotaxis. (A) Selected DE genes from RNA-seq and indicated statistical significance. (B) Chromatin accessibility of selected genes from ATAC-seq. (C) Frequency of low and high CCR8 expression by flow cytometry in OT-I Tmem across 24-h stimulation conditions with TGF-β addition at indicated time points. (D) Frequency of CCR8-high expression plotted individually and indicated statistical significance. Stimulations in C and D are from the same experiments shown in Fig. 3A. Indicated statistical significances were calculated using one-way ANOVA with Tukey’s multiple comparison test. In A and B, RNA- and ATAC-seq data depicted are from n = 7 animals. In C and D, data depicted are from n = 3. Data shown are from 2 independent experiments.
Overall, these data highlight that TGF-β can modify the chemotactic properties of reactivated CD8 Tmem in a dose-dependent and reactivation signal-dependent manner.
Discussion
Genetic ablation approaches of TGF-β receptor signaling have provided a set of important tools to demonstrate that TGF-β signals directly act on T cells during priming and control survival, differentiation, effector function, and formation of tissue-resident T cells (5, 6, 8–11, 25). A study by Ma and Zhang demonstrated that TGF-β signals are necessary for the proper maintenance of functional memory T cells (11), which has recently also been extended to chronic infections (26, 27). Importantly, dose-dependent effects of a ligand cannot be assessed with these genetic ablation models. TGF-β availability changes during inflammatory processes (28), but how these changes impact memory T cell function is poorly understood. We thus wanted to assess how low to high concentrations of TGF-β affect CD8 Tmem function in context of different reactivation signals. Reactivation of memory CD8 T cells is a critical component of providing protection against infections (29, 30), PD-1/PD-L1 induced antitumor responses (18) as well as vaccines targeting cancer (16). TGF-β has been reported to inhibit Ca2+ influx (31) thus indicating that the reactivation signal itself may affect the consequences of TGF-β signaling. The quality of the TCR signal controls the downstream transcriptional changes (32), and we considered that CD8 Tmem can be reactivated by a range of different TCR- as well as cytokine-mediated signals. To simultaneously manipulate reactivation and TGF-β signals, we needed to generate CD8 Tmem with intact TGF-β signaling and then control TGF-β and reactivation signals in an ex vivo setup.
When we reactivated OT-I Tmem by TCR cross-linking by plate-bound antibodies, TGF-β very effectively inhibited granzyme B expression (1.3 ng/mL was sufficient for a twofold reduction in the frequency of granzyme B+ OT-I Tmem). In contrast, when we reactivated OT-I Tmem with either N4 or Q4 peptide, we found that a 10-fold lower concentration of TGF-β was sufficient to reduce the frequency of granzyme B+ OT-I Tmem twofold (0.16 and 0.09 ng/mL of TGF-β for N4 and Q4, respectively). These data strongly suggest that lower affinity responders are particularly susceptible to losing cytotoxic function, which is an important consideration for antitumor responses. This potent suppression of cytotoxic function of low-affinity CD8 T cells then also begs the question how low-affinity T cells could possibly contribute to pathogen clearance. A previous study reported a potential decrease in total TGF-β in blood following infection with Listeria monocytogenes (LM) (28) and we similarly observed a decrease of total TGF-β in the spleen following infection with LM from 10.1 ng/g tissue during homeostasis, and decreased to 4.5 ng/g tissue 3 d following infection with LM (SI Appendix, Fig. S1A). Of note, active TGF-β is often only a fraction of total TGF-β (13). Such an infection-associated decrease in TGF-β may be critical to allow for low-affinity CD8 Tmem to exert cytotoxic function. It is also worthwhile to consider that such a decrease in active TGF-β represents a window of opportunity for self-reactive T cells to acquire cytotoxic function. An association of viral infection and an autoimmune response was first suggested 40 y ago with autoreactive antibodies (33), but has since been demonstrated for T cells as well (34). This is typically thought to be the result of molecular mimicry between viral and self-antigen, which could be facilitated during a decline in active TGF-β availability (10).
Since infections also elicit cytokine-driven activation of CD8 Tmem (35–37), we examined how bystander-activated CD8 Tmem are affected by TGF-β signals. Interestingly, TGF-β had a similar effect on CD8 Tmem reactivated with IL-12, 15, and 18: The reduction in gzmB expression was comparable to CD3/CD28 cross-linking (1.3 ng/mL was sufficient for a 2× reduction in the frequency of granzyme B+ T cells) with a high concentration of proinflammatory cytokines, but the susceptibility to TGF-β-mediated inhibition of cytotoxicity increased as we decreased cytokine concentrations. Overall, these data highlight the importance of the strength of the activating signal in regard to the ability of TGF-β to inhibit cytotoxic function.
Based on studies that relied on priming on naive T cells or used T cell clones, it is often assumed that TGF-β concurrently inhibits IFN-γ and cytotoxic function (25). However, across all experimental conditions, we consistently observed that the impact of TGF-β signals on IFN-γ expression by reactivated CD8 Tmem was rather limited. This distinct effect of TGF-β on granzyme B and IFN-γ expression in reactivated CD8 Tmem is curious, particularly in context of the tumor microenvironment with presumably abundant active TGF-β. Our data indicate that TGF-β can inhibit direct cytotoxicity by reactivated CD8 Tmem, but IFN-γ could still allow for myeloid cell-mediated tumor killing (38, 39). In context of an infection, this selective disabling of cytotoxicity could limit pathology while still allowing for IFN-γ-mediated protective effects and continued recruitment of immune cells (40).
In our initial set of experiments, we provided reactivation and TGF-β signals at the same time, but we considered that CD8 Tmem may receive activating signals before or after a TGF-β signal (for example, reactivation in a lymph node followed by TGF-β exposure in the tissue, or vice versa). Since our ex vivo experimental system allowed us to have temporal control of the sequence of signaling events (TGF-β exposure before, together with or after the activation event), we explored these different scenarios. We found that receiving TGF-β signals after reactivation still efficiently reduced cytotoxicity and, similarly, brief exposure to TGF-β prior to an activation event was sufficient to reduce cytotoxic effector function. In our system, this suppressive effect lasts for 24 h, but this observation of course begs the question of how long the decrease in cytotoxic function may last in vivo. Defining the duration of suppression will be important in follow-up studies and is relevant in context of the association between viral infection and autoimmune responses, as well as antitumor responses. Based on these data, we speculated that TGF-β may alter chromatin accessibility.
However, we did not observe changes in chromatin accessibility to perforin or granzyme loci, while transcript abundance was significantly decreased in the presence of TGF-β.
Previous studies suggested that SMAD transcription factors can bind to stimulation-induced transcription factors of the AP-1 (41, 42) and IRF (43) families. Thus, potential cooperative or antagonistic activities could affect the overall state of regulatory elements leading to changes in transcription of these loci without substantial changes in chromatin accessibility. A previous study reported acetylation of histone H3 lysine 9 (H3K9) proximal to perforin and gzmB promoter and first exon regions in memory CD8 T cells, but not naive T cells (44). Thus, permissive histones seem to also play a role in controlling transcription in memory T cells.
Interestingly, we detected epigenetic changes for several chemokine receptors, including CCR6, CXCR3, CXCR4, and CCR8. These data suggest that at least some of the TGF-β-mediated changes are epigenetic in nature. We observed a TGF-β-mediated increase in CXCR3 expression in context of CD8 T Tmem reactivation, while a recent study reported that deletion of TGF-βRI driven by CD8a-cre enhanced CXCR3 expression on CD8 T cells (45). A possible explanation for this difference is due the timing of deletion as noted in other TGF-β studies in regard to T cell activation and differentiation (10, 11). We were particularly interested in CCR8 expression, which has often been observed on intratumoral regulatory T cells (46). Thus, TGF-β could potentially push reactivated CD8 Tmem to colocalize with these Tregs in tumors thereby ensuring continued control over their effector function. It could also be a critical signal to route CD8 Tmem to the skin, which is a physiological target site for CCR8+ T cells (47). Of note, induction of CCR8 expression was TGF-β dose-dependent and even low doses of 0.04 to 0.06 ng/mL were sufficient to elicit expression in about 50% of OT-I T cells reactivated with N4 or Q4, respectively. TGF-β also increased expression of the adhesion receptor ninjurin-1 (Ninj1), which is involved in T cell crawling in blood vessels (48), metalloproteinase 1 (Timp1) and the metalloprotease Meltrin β (ADAM19) and the chemokine CCL20, which orchestrates interactions with CCR6-expressing immune cells subsets (including Tregs, Th17 and dendritic cells) (49, 50). In addition to gene expression changes related to cell motility and trafficking, GO analysis also revealed changes related to cell metabolism (SI Appendix, Fig. S5C).
We were surprised by the large number of regulatory elements that changed during reactivation (almost 26,000). About 10% of these elements were affected by TGF-β 24 h after reactivation, indicating that TGF-β signals are not merely a specific suppressor of effector function, but rather a modifier CD8 Tmem function, which is highly relevant in regard to blocking TGF-β signaling for therapeutic purposes. Targeting TGF-β for therapeutic purposes, specifically to alter immune responses, is of great clinical interest, but the pleiotropic properties of TGF-β across different cell types have complicated these efforts (1, 51). Advances in the design of biologic therapeutics now allow for a more specific targeting of cells to block or activate receptor function (51), but our data highlight that even for CD8 Tmem, inhibition of TGF-β signals does not simply equal increased functionality: For example, complete blocking of TGF-β may preclude CCR8 expression and prevent trafficking to sites in which ligands (including CCL1 and CCL8) are expressed (46, 52). This includes trafficking to the skin, which would presumably interfere with targeting melanomas (53), but also trafficking to the CCL8+ hypoxic regions of solid tumors (54).
Overall, our data indicate that TGF-β should not be considered a suppressor of effector function for CD8 Tmem, but rather a modifier of CD8 Tmem function in the context of reactivation. Our data support the notion that TGF-β does not affect all CD8 Tmem equally since the functional consequences of a TGF-β signal are shaped by the strength of the reactivation signal. Finally, our data also highlight that TGF-β signals can exert their function regardless if they are received before or after the reactivating event, which is an important consideration for interpreting studies that assess CD8 Tmem function in situ.
Methods
Mice.
Mouse protocols and experimentation conducted at the Fred Hutchinson Cancer Center were approved by and in compliance with the ethical regulations of the Fred Hutchinson Cancer Center’s Institutional Animal Care and Use Committee. All animals were maintained in specific pathogen-free facilities and infected in modified pathogen-free facilities. Experimental groups were nonblinded and animals were randomly assigned to experimental groups. We purchased C57BL/6J mice from the Jackson Laboratory; OT-I mice were maintained on CD45.1 congenic backgrounds. To generate OT-I memory mice, we adoptively transferred 1 × 104 OT-I T cells in sterile 1× PBS i.v. per C57BL/6J recipient, and subsequently infected recipients i.v. with 1 × 106 PFU OVA-expressing vesicular stomatitis virus (VSV-OVA) (55) or 4 × 103 CFU OVA-expressing Listeria monocytogenes (LM-OVA) as previously described (56). We allowed ≥60 d to pass after initial VSV or ≥30 d LM infections before using OT-I memory T cells for experiments.
T Cell Isolation and Ex Vivo Stimulation.
To enrich bulk T cells from single-cell suspensions, we used negative T cell isolation MACS (STEMCELL Technologies, Canada). We plated 0.5–1 ×106 T cells per well in 96-well V-bottom tissue culture plates. We cultured cells in RP10 media (RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin–streptomycin, 1 mM sodium pyruvate, 0.05 mM β-mercaptoethanol, and 1 mM HEPES). T cells were activated with rIL-12, rIL-15, and rIL-18 (BioLegend) (at specified concentrations), or plate-bound anti-CD3 and anti-CD28 antibodies, or N4, Q4, or with media alone with our without recombinant mouse TGF-β1 (Biolegend Cat # 763104). Cells were incubated at 37 °C, 5% CO2 for up to 24 h. T cells were activated in the presence of GolgiPlug (BD Biosciences) (1:1,000 dilution) for the final 4 h of stimulation, prior to intracellular cytokine staining (ICS). The methods for our experiments with deidentified human PBMCs are outlined in the legend of SI Appendix, Fig. S2.
Flow Cytometry.
We conducted all flow staining for mouse and human T cells on ice and at room temperature, respectively. All mouse and human flow panel reagent information, stain conditions, and gating are included in SI Appendix, Figs. S7–S9 and Tables S1–S3. We conducted LIVE/DEAD fixable aqua (AViD) staining in 1× PBS. For surface staining, we utilized FACSWash (1× PBS supplemented with 2% FBS and 0.2% sodium azide) as the stain diluent. We fixed cells with the FOXP3 fixation/permeabilization buffer kit (Thermo Fisher) and conducted intranuclear stains using the FOXP3 permeabilization buffer (Thermo Fisher) as diluent. For ICS panels, we fixed cells with Cytofix/Cytoperm (BD Biosciences) and conducted intracellular stains using Perm/Wash buffer (BD Biosciences) as diluent. We resuspended cells in FACSWash and acquired events on a FACSSymphony, which we analyzed using FlowJo v10 (BD Biosciences). We conducted statistical testing using Prism v8 (GraphPad).
ELISA for TGF-β.
Female C57BL/6J mice were infected with 4 × 103 CFU LM-OVA. Spleens were weighed and then mechanically dissociated in 500 μL buffer (1× PBS supplemented with 0.05% Tween) with scissors in a microcentrifuge tube. To separate debris, samples were centrifuged at 1,000 g for 10 min at 4 °C, and the supernatants were stored at −80 °C until assay. Total TGF-β levels were determined by acid activation of the latent TGF-β1 in the sample using the sample activation kit 1 (DY010) (R&D Systems).
RNA-sequencing.
Bulk RNA-seq was performed on 500 sort-purified OT-I T cells derived from OT-I memory mice after culture in conditions of 2 h no stimulation, 2 h stimulation with 100 ng/mL TGF-β, 24 h stimulation with 100 nM N4, and 24 h stimulation with 100 nM N4 and 100 ng/mL TGF-β. Twenty-four-hour stimulation stain control was performed to ensure T cell activation occurred consistently with prior experiments (SI Appendix, Fig. S7A). In total, 28 samples were sequenced, and each condition was represented by a total of seven biological replicates (combined from two independent experiments). Cells were prepared for RNA sequencing, and data were overall analyzed as previously described (57) aside from using the GRCm38 reference genome.
ATAC Sequencing.
ATAC-seq was performed on pools of 40,000 to 50,000 sort-purified OT-I T cells pooled from 2 to 3 mice. DNA was purified as previously described (58). Fastq files were used to map to the mm10 genome using the ENCODE ATAC-seq pipeline (59), with default parameters, except bam files used for peak calling were randomly downsampled to a maximum of 50 million mapped reads. Peaks with a MACS2 (60) computed q value of less than 0.0001 in at least one replicate were merged with bedtools (61) function intersect and processed to uniform peaks of 500 bp width with the functions getPeaks and resize from R package chromVAR (62). Reads overlapping peaks were enumerated with getCounts function from chromVAR and normalized and log2-transformed with voom from R package limma (63, 64). Peaks with 3 or more normalized counts per million mapped reads at least one replicate were included to define a global peak set of 99,317 peaks. Differentially accessible peaks were identified in pairwise comparisons based on FDR-adjusted P values of less than 0.01, fold change of at least 1.5, and with an average of 3 normalized counts per million mapped reads using R package limma. Motif-associated variability in ATAC-seq signal was computed with R package chromVAR using homer motif definitions from R package chromVARmotifs (https://github.com/GreenleafLab/chromVARmotifs). Genome-wide visualization of ATAC-seq coverage was computed with deeptools (65) function coveragebam, using scale factors computed based on the number of reads within the total peak set.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank members of the Prlic lab for critical discussion. We thank the Flow Cytometry Shared Resources of the FHCRC and the Genomics Core Lab of the Benaroya Research Institute for sequencing. This work was supported by NIH grants R01AI123323 (to M.P.) and R01AI151021 to (J.S.-B.). Figs. 1A and 4A and SI Appendix, Fig. S2A use templates from BioRender.com.
Author contributions
A.T., A.K., and M.P. designed research; A.T. and A.K. performed research; S.K.O. contributed new reagents/analytic tools; A.T., A.K., J.S.-B., and M.P. analyzed data; and A.T., A.K., S.K.O., J.S.-B., and M.P. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Sequencing data are available on GEO under access # GSE246933 (66). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Akhurst R. J., Hata A., Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug. Discov. 11, 790–811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nixon B. G., Gao S., Wang X., Li M. O., TGFbeta control of immune responses in cancer: A holistic immuno-oncology perspective. Nat. Rev. Immunol. 23, 346–362 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travis M. A., Sheppard D., TGF-beta activation and function in immunity. Annu. Rev. Immunol. 32, 51–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanjabi S., Oh S. A., Li M. O., Regulation of the immune response by TGF-beta: From conception to autoimmunity and infection. Cold Spring Harb. Perspect. Biol. 9, a022236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorelik L., Flavell R. A., Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12, 171–181 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Lucas P. J., Kim S. J., Melby S. J., Gress R. E., Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J. Exp. Med. 191, 1187–1196 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishigame H., Mosaheb M. M., Sanjabi S., Flavell R. A., Truncated form of TGF-betaRII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice J. Immunol. 190, 6340–6350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marie J. C., Liggitt D., Rudensky A. Y., Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25, 441–454 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Li M. O., Sanjabi S., Flavell R. A., Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Zhang N., Bevan M. J., TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 13, 667–673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma C., Zhang N., Transforming growth factor-beta signaling is constantly shaping memory T-cell population. Proc. Natl. Acad. Sci. U.S.A. 112, 11013–11017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith K. A., The quantal theory of immunity. Cell Res. 16, 11–19 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Grainger D. J., Mosedale D. E., Metcalfe J. C., TGF-beta in blood: A complex problem. Cytokine Growth Factor Rev. 11, 133–145 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Abe M., et al. , An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276–284 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Grainger D. J., Mosedale D. E., Metcalfe J. C., Weissberg P. L., Kemp P. R., Active and acid-activatable TGF-beta in human sera, platelets and plasma. Clin. Chim. Acta 235, 11–31 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Disis M. L. N., et al. , Safety and outcomes of a plasmid DNA vaccine encoding the ERBB2 intracellular domain in patients with advanced-stage ERBB2-positive breast cancer: A phase 1 nonrandomized clinical trial. JAMA Oncol. 9, 71–78 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J., et al. , Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 15, 28 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Q., et al. , The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 185, 4049–4066.e25 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Freeman B. E., Hammarlund E., Raue H. P., Slifka M. K., Regulation of innate CD8+ T-cell activation mediated by cytokines Proc. Natl. Acad. Sci. U.S.A. 109, 9971–9976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman B. E., Meyer C., Slifka M. K., Anti-inflammatory cytokines directly inhibit innate but not adaptive CD8+ T cell functions J. Virol. 88, 7474–7484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanders K. C., et al. , Quantitation of TGF-beta proteins in mouse tissues shows reciprocal changes in TGF-beta1 and TGF-beta3 in normal vs neoplastic mammary epithelium. Oncotarget 7, 38164–38179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zehn D., Lee S. Y., Bevan M. J., Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiedemann G. M., et al. , Deconvoluting global cytokine signaling networks in natural killer cells. Nat. Immunol. 22, 627–638 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmierer B., Hill C. S., TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Thomas D. A., Massague J., TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Hu Y., et al. , TGF-beta regulates the stem-like state of PD-1+ TCF-1+ virus-specific CD8 T cells during chronic infection. J. Exp. Med. 219, e20211574 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C., et al. , TGF-beta promotes stem-like T cells via enforcing their lymphoid tissue retention. J. Exp. Med. 219, e20211538 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjabi S., Mosaheb M. M., Flavell R. A., Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 31, 131–144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prlic M., Williams M. A., Bevan M. J., Requirements for CD8 T-cell priming, memory generation and maintenance. Curr. Opin. Immunol. 19, 315–319 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Zhang N., Bevan M. J., CD8(+) T cells: Foot soldiers of the immune system. Immunity 35, 161–168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C. H., et al. , Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 197, 1689–1699 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwata A., et al. , Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 18, 563–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujinami R. S., Oldstone M. B., Wroblewska Z., Frankel M. E., Koprowski H., Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. U.S.A. 80, 2346–2350 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cusick M. F., Libbey J. E., Fujinami R. S., Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 42, 102–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu T., et al. , Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. 3, 701–708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurice N. J., Taber A. K., Prlic M., The ugly duckling turned to swan: A change in perception of bystander-activated memory CD8 T cells. J. Immunol. 206, 455–462 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim T. S., Shin E. C., The activation of bystander CD8(+) T cells and their roles in viral infection. Exp. Mol. Med. 51, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaCasse C. J., et al. , Th-1 lymphocytes induce dendritic cell tumor killing activity by an IFN-gamma-dependent mechanism. J. Immunol. 187, 6310–6317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mumberg D., et al. , CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. U.S.A. 96, 8633–8638 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schenkel J. M., Fraser K. A., Vezys V., Masopust D., Sensing and alarm function of resident memory CD8(+) T cells. Nat. Immunol. 14, 509–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Feng X. H., Derynck R., Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394, 909–913 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Liberati N. T., et al. , Smads bind directly to the Jun family of AP-1 transcription factors. Proc. Natl. Acad. Sci. U.S.A. 96, 4844–4849 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamiya T., et al. , Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J. Immunol. 191, 2360–2371 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Araki Y., Fann M., Wersto R., Weng N. P., Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J. Immunol. 180, 8102–8108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gunderson A. J., et al. , TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 11, 1749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moser B., Chemokine receptor-targeted therapies: Special case for CCR8. Cancers (Basel) 14, 511 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaerli P., et al. , A skin-selective homing mechanism for human immune surveillance T cells. J. Exp. Med. 199, 1265–1275 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Odoardi F., et al. , T cells become licensed in the lung to enter the central nervous system. Nature 488, 675–679 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Yamazaki T., et al. , CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 181, 8391–8401 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Borgne M., et al. , Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity 24, 191–201 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Chen S. Y., Mamai O., Akhurst R. J., TGFbeta: Signaling blockade for cancer immunotherapy. Annu. Rev. Cancer Biol. 6, 123–146 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Islam S. A., et al. , Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat. Immunol. 12, 167–177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ebert L. M., Meuter S., Moser B., Homing and function of human skin gammadelta T cells and NK cells: Relevance for tumor surveillance. J. Immunol. 176, 4331–4336 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Sattiraju A., et al. , Hypoxic niches attract and sequester tumor-associated macrophages and cytotoxic T cells and reprogram them for immunosuppression. Immunity 56, 1825–1843.e6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turner D. L., Cauley L. S., Khanna K. M., Lefrancois L., Persistent antigen presentation after acute vesicular stomatitis virus infection. J. Virol. 81, 2039–2046 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zehn D., Roepke S., Weakly K., Bevan M. J., Prlic M., Inflammation and TCR signal strength determine the breadth of the T cell response in a bim-dependent manner. J. Immunol. 192, 200–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mair F., et al. , Extricating human tumour immune alterations from tissue inflammation. Nature 605, 728–735 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J., ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y., et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schep A. N., Wu B., Buenrostro J. D., Greenleaf W. J., chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Law C. W., Chen Y., Shi W., Smyth G. K., voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritchie M. E., et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramirez F., Dundar F., Diehl S., Gruning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taber A., et al. , TGF-β broadly modifies rather than specifically suppresses reactivated memory CD8 T cells in a dose-dependent manner. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246933. Deposited 2 November 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Sequencing data are available on GEO under access # GSE246933 (66). All other data are included in the manuscript and/or SI Appendix.