

Significance
Almost all cases of HIV-1 infections are caused by the pandemic HIV-1 M. The nonpandemic HIV-1s N, O, and P do not spread much in humans for unknown reasons. Human cells express TRIM5α that restricts HIV-1. HIV-1 M evolved to escape this restriction by binding cyclophilin A (CYPA) to the viral core. Our data indicate that nonpandemic HIV-1s are sensitive to human TRIM5α. In these viruses, cyclophilin A binding cannot protect against TRIM5α because its trans/cis isomerase enzymatic activity is reduced. Our data suggest that subtle changes induced by CYPA in the capsid have a severe impact on viral infection.
Keywords: HIV-1, TRIM5, restriction factor, cyclophilin, capsid
Abstract
The rather few cases of humans infected by HIV-1 N, O, or P raise the question of their incomplete adaptation to humans. We hypothesized that early postentry restrictions may be relevant for the impaired spread of these HIVs. One of the best-characterized species-specific restriction factors is TRIM5α. HIV-1 M can escape human (hu) TRIM5α restriction by binding cyclophilin A (CYPA, also known as PPIA, peptidylprolyl isomerase A) to the so-called CYPA-binding loop of its capsid protein. How non-M HIV-1s interact with huTRIM5α is ill-defined. By testing full-length reporter viruses (Δ env) of HIV-1 N, O, P, and SIVgor (simian IV of gorillas), we found that in contrast to HIV-1 M, the nonpandemic HIVs and SIVgor showed restriction by huTRIM5α. Work to identify capsid residues that mediate susceptibility to huTRIM5α revealed that residue 88 in the capsid CYPA-binding loop was important for such differences. There, HIV-1 M uses alanine to resist, while non-M HIV-1s have either valine or methionine, which avail them for huTRIM5α. Capsid residue 88 determines the sensitivity to TRIM5α in an unknown way. Molecular simulations indicated that capsid residue 88 can affect trans-to-cis isomerization patterns on the capsids of the viruses we tested. These differential CYPA usages by pandemic and nonpandemic HIV-1 suggest that the enzymatic activity of CYPA on the viral core might be important for its protective function against huTRIM5α.
Human immunodeficiency virus 1 (HIV-1) is subdivided into four groups, the pandemic group M and the nonpandemic groups N, O, and P (1). HIV-1 N was identified in 20 patients and HIV-1 P in two patients, circulating mostly in Cameroon (https://www.hiv.lanl.gov/). About 100,000 people have been infected with HIV-1 group O found in West and Central Africa with the highest prevalence in Cameroon, Gabun, and Equatorial Guinea (2). HIV-1s are the evolutionary results of rare successful transmission events of simian IV (SIV) to humans. Group M and group N HIV-1s are derived from SIV of chimpanzees (SIVcpz), while group O and group P are the results of spillover of SIV of gorillas (SIVgor) (1, 3, 4). To halt retroviral replication, vertebrates use several cellular restriction factors (5–9). It is ill-defined whether the low prevalence of non-M HIVs is associated with the activity of antiviral factors such as the capsid-binding TRIM5α, which may limit their spread among humans.
TRIM proteins have a RING domain, a coiled-coil domain, and one to two B box domains (10). In addition, some TRIM proteins have a C-terminal PRYSPRY (B30.2) domain such as in the alpha isoform of TRIM5, the TRIM5α, and TRIM25, or a cyclophilin A (CYPA) domain such as in the TRIM-Cyclophilin A (TRIMCyp) fusion proteins (11). The PRYSPRY or CYPA domains of TRIM5 proteins can bind retroviral cores. This binding initiates TRIM5 oligomerization to form higher-order oligomers around the conical core to affect the core integrity and, thereby, impairs nuclear import and integration of the reverse-transcribed viral genome (11–13). The TRIM5α binding to the capsid can also result in intracellular signaling events for an extended viral restriction (11, 14). The antiretroviral activity of TRIM5 proteins has been shown to mainly be PRYSPRY or CYPA domain-dependent (11). CYPA presence at the C terminus of TRIM5 proteins is a result of the evolutionary retrotransposition of CYPA in some primates, like the Old World rhesus monkeys (Macaca mulatta) or the New World Aotus night/owl monkeys (15, 16). The antiretroviral activity of TRIM5α is thought to have formed a selective protective shield against the retroviral spread in vertebrate hosts (17, 18). As an example, human (hu) TRIM5α restricts infections by the horse lentivirus equine infectious anemia virus (EIAV) (19); in contrast, the pandemic HIV-1 M escapes the antiviral activity of huTRIM5α by displaying binding sites in the viral core for CYPA (20, 21). The viral core is composed of about 1,200 to 1,500 capsid proteins in mostly hexameric organizations (22). Cellular CYPA and TRIMCyp bind the so-called CYPA-binding loop of the capsid formed by residues 85 to 93 (23–26). The CYPA - core interaction during the early phase of infection in the viral target cell is essential for its protective function against huTRIM5α (20). CYPA is also packaged by HIV virions, with less defined roles (27, 28). In viral particles, a CYPA-capsid stoichiometry of 0.1 and in in vitro assembled capsid tubes, a stoichiometry of ~0.3 to 0.4 was described (29–31). CYPA can influence many early steps of HIV infection (32–34), but the molecular mechanisms are not fully understood and may involve a trans/cis isomerization activity of CYPA and altered dynamics of the core (35, 36). Here, we tested the antiviral property of huTRIM5α against non-M group HIVs but also SIVgor, identified viral determinants of sensitivity, and assessed the role of CYPA in the mechanism of TRIM5α to restrict HIVs.
Materials and methods
Cells.
CrFK and HEK293T cells were maintained in Dulbecco’s high-glucose modified Eagle’s medium (PAN Biotech, Aidenbach, Germany) with addition of 10% fetal bovine serum (PAN Biotech), 2 mM L-glutamine (PAN Biotech), 100 U/mL penicillin, and 100 μg/mL streptomycin (PAN Biotech). TRIM5α KO U87-MG and control cells were kindly donated by Michael Malim (37); CYPA knockdown (KD) and control U87-MG cells were cultured under 1 µg puromycin. Human macrophages were isolated from whole blood, obtained from the university hospital of Heinrich Heine University Düsseldorf (ethical approval study number 3180). Macrophages were maintained in RPMI 1640 containing 1,000 U/mL monocyte colony-stimulating factor.
Plasmids.
The replication-competent HIV-1 N DJO0131, HIV-1 O RBF206, HIV-1 P RBF168, SIVcpzPtt MB897 clones were kindly provided by Frank Kirchhoff. SIVgorCP2139 and SIVcpzPts clone TAN1.910 clones were obtained from NIH AIDS repository (38). The murine leukemia virus (MLV) packaging construct pHIT60, which encodes the gag-pol of Moloney MLV, was provided by Jonathan Stoye (39). Reporter viruses for HIV-1 N, HIV-1 O, HIV-1 P, and SIVgor were constructed as follows: Using fusion PCR, nanoluciferase gene was cloned in replacement of nef of HIV-1 N, HIV-1 O, HIV-1 P, and SIVgor. Two stop codons were introduced in the envelope genes of these viruses, as the glycoprotein of the vesicular stomatitis virus (VSV-G) was to be used for envelope. Capsid mutant reporter viruses of HIV-1 M, HIV-1 N, HIV-1 O, and HIV-1 P were generated with fusion PCR using specific primers including those with desired mutations. HIV-1 M constructs expressing the capsid of HIV-1 N or HIV-1 O or HIV-1 P were made by fusion PCR, their inserts were cloned in PmlI-MfeI digested pMDLg/pRRE vector to produce chimeric gag-pol of HIV-1 M with CA genes of other viruses: pMDLg/pRRE.HIV-1 N CA, pMDLg/pRRE.HIV-1 O CA, pMDLg/pRRE.HIV-1 P CA. Complete gag-pol constructs for HIV-1 N, HIV-1 O, and HIV-1 P were constructed using the HIV-1 M pMDLg/pRRE plasmid and replacing HIV-1 M gag-pol by digestion with PmlI and BspEI and insertion of gag-pols of interest, produced through a series of overlapping PCR reactions, to make pMDLg/pRRE.HIV-1 N, pMDLg/pRRE.HIV-1 O, pMDLg/pRRE.HIV-1 P, and pMDLg/pRRE.SIVgor. CYPA-binding loop capsid mutants were made through PCR with primers bearing specific CYPA-binding loop sequences and their inserts were respectively cloned in specific vectors to make HIV-1 M.N loop, HIV-1 M.O loop, and HIV-1 N.M loop. HIV-1 vector pSIN.PPT.CMV.Luc.IRES.GFP expresses firefly luciferase and GFP. psPAX2 was obtained from the NIH AIDS Reagent Program (Cat# 11348); pRSV-Rev, pMDLg/pRRE, and pMD.G (VSV-G) have been described (40). Using fusion PCR on TRIMCyp in pLNCX2 plasmid (41), fusion PCR was used to introduce the desired mutations in the CYPA domain; N66D, H69R, or N66D-H69R rhTRIMCyp-HA mutants were generated.
Transfection and Viral Particle Production.
The pMDLg/pRRE-based viral particle production was done using pMDLg/pRRE (800 ng), pSIN.PPT.CMV.Luc.IRES.GFP (800 ng), pRSV-Rev (400 ng), and pMDG.VSV-G (200 ng). For nanoluciferase-based reporter viruses (HIV-1 N, O, P, SIVgor, and SIVcpz), 106 HEK293T cells were seeded; the following day, these cells were transfected using 200 ng VSV-G and 2,000 ng of viral plasmids using polyjet (Tebubio GmbH, Offenbach, Germany). Forty-eight hrs posttransfection, viral particles were collected. Where needed, the reverse transcriptase activity of viruses was quantified using the previously described approach (42).
Generation of Stable Cells Expressing TRIM5 Proteins.
Viral particles produced in HEK293T cells using a pLNCX2.TRIMCyp construct, pHIT60, and pMD.G (VSV-G) were used to transduce CrFK cells for 3 d, followed by a selection under 400 μg/mL G418 (Biochrom GmbH, Berlin, Germany), for the expression of HA-tagged WT and mutant rhTRIMCyp proteins. Protein expression was confirmed with immunoblots. Cells expressing empty pLNCX2 vector were used for control.
Single Round Infection Assay.
A total of 10 × 104 CrFK cells or 5 × 103 U-87 MG cells were seeded into 96-well plates, and infection was performed the following day. For experiments involving cyclosporin A (CsA, Sigma-Aldrich, Germany), 1 to 10 µM of CsA or control DMSO was used to treat cells 2 h before infection. Cells were then infected with different reporter viral particles, and after 48 to 72 h, luciferase activity was measured. For infection with nanoluciferase-containing reporter viruses, cells were washed with phosphate-buffered saline three times before lysis and luciferase measurement, in addition to medium change 24 h following infection, to eliminate the effect of background nanoluciferase. Nanoluciferase activity was measured with the Nano-Glo Luciferase system (Promega, Mannheim, Germany), and firefly luciferase activity was measured with the Steady-Glo Luciferase system (Promega) on a MicroLumat Plus luminometer (Berthold Detection Systems, Pforzheim, Germany). Each experiment was performed in triplicates for at least three times.
Pulldown Assays and Immunoblots.
The GST-CYPA-based pulldown experiments followed a protocol previously described (43). To confirm the expression of primate TRIM5 proteins, cells were lysed using RIPA lysis buffer [25 mM Tris-HCl (pH 8.0), 137 mM NaCl, 1% NP-40, 1% glycerol, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 2 mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitor cocktail set III (Calbiochem, Darmstadt, Germany). The cell lysate was centrifuged at 14,800 rpm for 20 min at 4 °C. The protein supernatant was denatured using Roti-load sample buffer (Roth, Karlsruhe, Germany) and was used for SDS polyacrylamide gel electrophoresis (PAGE). The following primary antibodies were used: anti-HA (mouse MMS-101P, Covance, Münster, Germany, 1:7,500 dilution) for HA-tagged proteins, anti-human TRIM5α (rabbit monoclonal, # 143265; Cell Signaling Technology Europe BV, Frankfurt, Germany, 1:1,000 dilution), anti-CYPA (mouse monoclonal; Santa Cruz Biotechnology, 1:500 dilution), anti-GST (mouse SAB4200237-200 UL, Sigma-Aldrich, Germany,1:7,500 dilution). For viral proteins, viral supernatant was centrifuged through 20% sucrose gradient for a minimum of 4 h at 4 °C, 14,800 rpm. The supernatant was discarded, and the viral pellet was lysed using RIPA buffer for 5 min followed by denaturation at 95 °C in Roti-load sample buffer for 5 min and SDS PAGE using anti-p24 antibody (NIH).
Modeling of HIV-1 Capsid Variants.
The structural models of the HIV-1 M capsid and CYPA generated by TopModel (44, 45) were taken from ref. 43. The capsid-CYPA complex was formed by superimposing the structures of the components onto the proteins of the cryo-EM structure PDB ID 5FJB. Mutations at the CYPA-binding loop to HIV-1 N, HIV-1 O, and the HIV-1 M A88V, I91L, and H87P variants were introduced with SCWRL (46). HIV-1 N P87H and L91I monomers were modeled with (47). The capsid-CYPA complex was formed as above. PDB2PQR (48) was used to determine the protonation state at pH = 7.4 with the help of PROPKA 3 (49). The systems were packed using PACKMOL-Memgen (46) adding KCl in a concentration of 0.15 M and using a minimum distance between the protein(s) and the edges of the water box of 15.0 Å. The ff19SB force field (44) was used for the proteins; the OPC force field (50) with the corresponding Li/Merz ion parameters (49) was used for water and ions, respectively.
Thermalization, Density Adjustment, and Dihedral Modification For PMF (Potential of Mean-Force) Calculations.
Thermalization and density adjustment were carried out using pmemd from the Amber22 software package (51, 52) with a time step of 2 fs. The Langevin thermostat (53) and Berendsen barostat (44) were used for temperature and pressure control, respectively. For the treatment of long-range electrostatic interactions, the Particle Mesh Ewald method (54) was used with a direct-space nonbonded cutoff of 9.0 Å. The SHAKE algorithm was used to constrain bond lengths involving hydrogen atoms (55). The loop of each investigated variant either binding to CYPA or unbound was turned at the ω dihedral between G89 and P90 in 3° steps, which leads to 121 windows for each setup. The dihedral was modified to the target value during 10,000 steps of relaxing the system with the steepest decent algorithm followed by 10,000 steps with the conjugate gradient algorithm applying a force at the ω dihedral of 200 kcal mol–1 rad–2. During 10 ns, the system was heated to 300 K and 1 bar under NPT conditions.
Umbrella Sampling and PMF Calculations.
20 ns of umbrella sampling simulations in the NPT ensemble were performed for each window, using a harmonic restraining potential with a force constant of 200 kcal mol–1 rad–2 and writing out the ω dihedral every 50 simulation steps. The ω dihedral distribution was analyzed with the Weighted Histogram Analysis Method v2.0.9.1 (48). The periodicity was considered in the analysis, histogram limits were set to –0.5° and 360.5° for 722 bins in total, and the tolerance was set to 10–7 kcal mol–1. The histograms showed a median overlap of 25% between contiguous windows (SI Appendix, Figs. S6 and Table S1), well suited for PMF calculations (56).
The error along the PMF [G(x)] was estimated by block averaging; for each system, the data were separated into five parts of 4 ns each. The squared error in the estimate of the mean position of ω in window was calculated based on the block averages (for further details, see SI Appendix, Eq. S1) (57). From there, the error was propagated to derive the variance of PMFs taking into account as well as the used force constant (k), the sampling step size (Δω), and the starting position (ω0) as suggested by Zhu and Hummer (Eq. 1) (54).
| [1] |
From var[G(x)], the SD and the SEM were computed. For error propagation when subtracting values of windows n and m, e.g., for obtaining ΔG#t→c, SEM was calculated according to Eq. 2:
| [2] |
Individual states, e.g., cis (ω = 0°), trans (ω = 180°), and transition state (ω = 90°), were visually checked, and figures were prepared with PyMOL.
Results
Construction of Non-M HIV and SIVgor Reporter Viruses.
We constructed nanoluciferase-based reporter viruses for HIV-1 N, HIV-1 O, HIV-1 P, and SIVgor. Nanoluciferase was cloned in replacement of nef genes. In addition, two stop codons were inserted in the env genes. Production of VSV-G-pseudotyped viral particles was done through transfection of HEK293T cells, and such virions were tested on human HeLa cells, for infectivity by nanoluciferase measurement, 2 d after infection (data not shown). We also constructed pMDLg/pRRE-based gag-pol constructs for HIV-1 N, HIV-1 O, HIV-1 P, and SIVgor. Such constructs were made by replacing HIV-1 M gag-pol in pMDLg/pRRE with non-M or SIVgor gag-pol.
Non-M HIVs Are Inhibited by huTRIM5α in Human Cells.
To test whether the replication of nonpandemic HIVs is restricted by huTRIM5α, equal amounts of reporter viruses of non-M HIVs (HIV-1 N, HIV-1 O, and HIV-1 P) and SIVs (SIVcpzPtt, SIVcpzPts, and SIVgor) were used for infections of human wild-type (WT) U-87 MG and huTRIM5α knockout (KO) U-87 MG cells (37) (Fig. 1A). In addition, we included reporter viruses for HIV-1 M and for the EIAV; EIAV is known to be sensitive to huTRIM5α (19). While the infectivity of HIV-1 M was equal in WT and TRIM5 KO cells, infection by non-M HIVs but also SIVgor was enhanced when huTRIM5α was knocked down (Fig. 1B and SI Appendix, Fig. S1A). Infection by HIV-1 N increased to up to three folds in the huTRIM5α KO cells as compared to control cells, infection by HIV-1 O, HIV-1 P, and SIVgor reached 2.5-, 3.5-, and twofolds, respectively in the absence of huTRIM5α, suggesting inhibition of these viruses by huTRIM5α. EIAV showed 6.5-fold higher infectivity in the absence of huTRIM5α, compared to WT cells. In contrast, SIVcpzPtt and SIVcpzPts did not benefit from the absence of huTRIM5α (Fig. 1B), as described before (58). We also compared infection of WT U-87 MG cells with HIV-1 M vs. non-M HIV-1s virions produced by their respective gag-pol expression plasmids and confirmed a strongly reduced infectivity of HIV-1 N, O, and P compared to the infectivity of HIV-1 M (SI Appendix, Fig. S1B). The restriction of nonpandemic HIV-1s was seen over a wide range of virus input in WT U87-MG cells and in primary human macrophages (SI Appendix, Fig. S2).
Fig. 1.

TRIM5α depletion in human U-87 MG cells increases non-M HIV infection in a capsid CYPA-binding loop-dependent way. (A) Immunoblot of WT and TRIM5α KO U-87 MG cells. Anti-TRIM5 antibody was used to detect TRIM5α; anti-GAPH antibody was used to ensure equal protein loading. (B) WT or TRIM5α KO U-87 MG cells were infected with equal amounts of luciferase reporter viruses for HIV-1 M, HIV-1 N, HIV-1 O, HIV-1 P, SIVcpzPtt, SIVcpzPts, and EIAV. Two to three days later, luciferase activity was measured; infection of KO cells was normalized to infection of WT cells. (C) HIV-1 N, HIV-1 O, or HIV-1 P capsids were cloned in HIV-1 M gag-pol as replacements for its WT capsid. (D) HIV-1 M gag-pol with the capsid of either HIV-1 N, O, or P were tested in WT and TRIM5α KO U-87 MG cells. Two to three days after infection, luciferase activity was measured; infection of KO cells was normalized to infection of WT cells. (E) Protein sequence alignment of CYPA-binding loop (box) regions of capsids of HIV-1 M, N, O, and P and HIV-2. (F) Using their respective gag-pol constructs, the CYPA-binding loop from HIV-1 N or HIV-1 O was introduced in HIV-1 M gag-pol, and the HIV-1 M CYPA-binding loop was transferred to HIV-1 N and HIV-1 O gag-pol. These CYPA loop mutants were then used to infect WT or TRIM5α KO U-87 MG cells for 2 to 3 d, Luciferase activity was measured; infection of KO cells was normalized to infection of WT cells. All experiments were repeated for a minimum of three times.
Capsid and Its CYPA-Binding Loop Mediate Differences in Sensitivity to huTRIM5α.
Since TRIM5α is a capsid-binding factor (11), we opted to investigate on the mechanisms behind differences in sensitivity to huTRIM5α between HIV-1 M and nonpandemic HIVs. We transferred the capsid encoding sequence from HIV-1 N, HIV-1 O, HIV-1 P, to HIV-1 M in the gag-pol expression construct pMDLg/pRRE representing HIV-1 M sequence (Fig. 1C). In contrast to WT HIV-1 M, chimeric viruses with the capsid from non-M viruses were inhibited 2.5 to 8-fold in WT cells compared to huTRIM5α KO cells (Fig. 1D), confirming the role of viral capsid for restriction by huTRIM5α.
Reports have described that in human cells, the capsid-interacting cellular protein CYPA protects HIV-1 M against the antiviral activity of huTRIM5α (20, 21). In the capsid, CYPA binds to a loop between helix 4 and helix 5, termed CYPA-binding loop. Here, CYPA directly interacts with residues G89 and P90 (23), which are conserved in pandemic and nonpandemic HIVs. However, there are variations in other residues of the capsid CYPA-binding loop between these viruses (Fig. 1E and SI Appendix, Fig. S3). To explore the impact of loop variability, we swapped in the capsid the CYPA-binding loops between HIV-1 M, HIV-1 N, and HIV-1 O. Thus, we created HIV-1 M gag-pol constructs with a CYPA-binding loop of HIV-1 N (identical to HIV-1 P) or HIV-1 O. In reverse, we transferred the HIV-1 M CYPA-binding loop to HIV-1 N or HIV-1 O gag-pol constructs. Stunningly, viruses that had the CYPA-binding loop of nonpandemic HIVs showed a restriction in WT cells compared to TRIM5α KO cells, and nonpandemic viruses with a CYPA-binding loop of HIV-1 M escaped the restriction by TRIM5α (Fig. 1F). These findings demonstrated that the capsid CYPA-binding loops account for differences in infection levels of pandemic and nonpandemic HIVs in huTRIM5α expressing cells.
Capsid Residue at Position 88 Mediates HIV Sensitivity to huTRIM5α.
In the capsid CYPA-binding loop, the human CYPA protein binds strongly to residues G89 and P90 (23) but capsid residue A88 also locates in the active site groove of CYPA (59). We hypothesized that variability in residue 88 in the CYPA-binding loops could explain our observations regarding the differential sensitivity of HIV-1 M and the nonpandemic HIV-1s to TRIM5α. We infected both WT and TRIM5α KO cells with reporter viruses that had mutations in the CYPA-binding loop at position 88, HIV-1 M A88V, HIV-1 N V88A, HIV-1 O M88A, or HIV-1 P V88A and compared their infectivity to the corresponding WT viruses. For HIV-1 M, we included the well-characterized HIV-1 M G89V capsid mutant, which does not bind CYPA. Both mutations in HIV-1 M, A88V, and G89V were strongly inhibited in WT U-87 MG cells compared to WT virus (Fig. 2A). However, in TRIM5α KO cells infections by both capsid mutants were not reduced compared to WT HIV-1 M, with infection by the A88V mutant even 4.3-fold higher than infection by WT virus (Fig. 2B and SI Appendix, Fig. S4 A and B). Infections by capsid mutants of nonpandemic HIVs showed a contrasting pattern. In WT cells, the infectivity of HIV-1 N V88A was fivefold higher, of HIV-1 O M88A was 2.3 folds higher, and of HIV-1 P V88A was 2.1 folds higher than infections by their WT viruses (Fig. 2C). However, these capsid mutants of nonpandemic viruses had a similar infectivity to WT virus in TRIM5α KO cells (Fig. 2D). To further analyze whether other residues in the CYPA-binding loop could inhibit the protection in HIV-1 M or induce a resistance to TRIM5α in HIV-1 N, additional mutations were tested (SI Appendix, Fig. S5). As a positive control, we included the A92E change in HIV-1 M and the P92E mutation in HIV-1 N. As it was shown that the Ala 92 to Glu mutation disrupts the binding of huTRIM5α to HIV-1 M viral cores (20). In HIV-1 M, the mutations H87P, I91L, and A92E, in contrast to A88V or G89V did not enhance the sensitivity to TRIM5α (SI Appendix, Fig. S5A). In HIV-1 N, however, the reverse mutations P87H, L91I, or P92E, similar to V88A, induced a protection against TRIM5α (SI Appendix, Fig. S5B). Together, these findings show that residue 88 in the CYPA-binding loop of the capsid is a unique determinant of TRIM5α restriction in pandemic and nonpandemic HIV-1. Other CYPA-binding loop residues in nonpandemic HIV-1 may be additionally important.
Fig. 2.

Capsid residues 88 and 89 mediate susceptibility to huTRIM5α. (A and B) A88V and G89V capsid mutations for HIV-1 M, (C and D) V88A for HIV-1 N, M88A for HIV-1 O, and V88A mutation for HIV-1 P were introduced in their respective gag-pol constructs. WT and capsid mutants were tested in infections of WT or TRIM5α KO U-87 MG cells. Infection of mutant viruses was normalized to infection of corresponding WT virus. (E) GST-pulldown of CYPA (CYPA-GST) with capsid proteins of HIV-1 M, N, O, or P. GST: GST not fused to CYPA. Viral lysates and GST-tagged CYPA protein lysates were specifically incubated together with GST Sepharose beads. The eluate was subjected to immunoblotting to detect viral p24 (capsid) and GST-tagged CYPA (pulldown). Cell and viral lysates were also loaded as inputs for GST-tagged CYPA and p24, respectively. (F) Immunoblot of viral particles and corresponding virus producer cells. The level of CYPA packaged by virions was analyzed by anti-HA staining (for CYPA-HA), virus was confirmed by anti-p24 (capsid) antibody staining, and anti-tubulin was used to confirm equal amounts of cell lysates loaded. Cells were treated with cyclosporine A (CsA) from 0 to 2.5 µM. (G) The amount of packaged CYPA in relation to the used CsA dose was quantified using ImageJ.
Pandemic and Nonpandemic HIV-1 Capsids Bind CYPA.
With the observed differences in the capsid CYPA loop between M and non-M HIV-1s (Fig. 1F), we wanted to test whether all four viruses interact with CYPA similarly. Using a GST-tagged CYPA, we assessed CYPA-capsid interaction in pulldown experiments. Immunoblots of these precipitations found similar levels of capsid protein for all four HIV-1s (Fig. 2E). In addition, we found that CYPA is packaged in virions of pandemic and nonpandemic HIV-1s (Fig. 2F). To understand whether these viruses package similarly CYPA, we exposed them to increasing amounts of cyclosporine A (CsA), a drug that binds CYPA and prevents CYPA interaction with the capsid. The dose-dependent reduction of CYPA packaging showed a small difference between HIV-1 M and the nonpandemic HIV-1s. A CsA dose of 1 µM reduced virion-associated CYPA by around 70% in the context of non-M HIVs, as opposed to only 40% by HIV-1 M, possibly suggesting differences in CYPA-binding strength between capsids of these viruses (Fig. 2 F and G).
CYPA Decreases the Isomerization Barrier the Most for HIV-1 M WT and Variants that Are Protected against TRIM5α.
We performed PMF computations of the isomerization reaction catalyzed by CYPA to address the question if the binding of CYPA to HIV-1 capsid has different effects on the pandemic HIV-1 M vs HIV-1 N, HIV-1 O, HIV-1 P, or variants of HIV-1 types. HIV-1 P was not considered because the loop composition is as in HIV-1 N. The isomerization occurs between the trans and cis conformations of the peptide bond formed by residues G89 and P90 in the CYPA-binding loop of the capsid (36). The conformational change is characterized by the torsion angle ω (trans: ω = 180°, cis: ω = 0°; Fig. 3 A and B). The PMF is the free energy change during the isomerization evaluated along the reaction coordinate ω (Fig. 3C); the PMF describes an average over the conformations of the surrounding protein residues and solvent molecules such that the effect of other CYPA-binding loop residues that vary among different HIV-1 clades is considered.
Fig. 3.

CYPA binds to the CYPA-binding loop of the HIV-1 M capsid, where it can exert cis/trans isomerase activity at the G89-P90 ω dihedral (36). (A) Overview of the simulated complex. CYPA (shown in gray) binds to the CYPA binding loop (orange) on the surface of the HIV-1 capsid. Monomers are colored differently; the region in the black box is shown as a blowup in panel (B). (B) Close-up view of the CYPA binding loop. The ω dihedral between residues G89 and P90 (shown as sticks) is marked, evaluated as the angle between the normals on the planes formed by the atoms [Cα(G89), C(G89), N(P90)] and [C(G89), N(P90), Cα(P90)]. (C) Potential of mean force (free energy profile) along the ω dihedral of G89-P90 in HIV-1 M wild type with or without CYPA. The height of the energy barrier for the transition from the trans (180°) to the cis (0°) conformation is marked with a black line. The HIV-1 M capsid without CYPA (red triangle) has a higher isomerization barrier than HIV-1 M bound to CYPA (orange triangle). The height of the free energy difference between trans (180°) and cis (0°) conformation is marked with a gray line. The HIV-1 M capsid without CYPA (red circle) has a lower free energy difference than HIV-1 M bound to CYPA (orange circle). (D) Barrier heights (ΔG#t→c, triangles) and free energy differences (ΔGt,c, circles) from the potential of mean force computations for the cis/trans isomerization of the ω angle between G89 and P90 (SI Appendix, Fig. S4). The HIV-1 types and variants are sorted from Left to Right according to the decrease in ΔG#t→c in the presence of CYPA.
We computed PMFs of the isomerization in the presence and absence of CYPA and calculated the free energy difference ΔGt,c as the difference between the PMF values for the trans and cis conformations (SI Appendix, Fig. S6 and Table S2 and Fig. 3D) as well as the barrier height ΔG#t→c as the difference between the PMF values for the trans conformation and the maximal value at ω ≈ 90° (SI Appendix, Fig. S6 and Table S3 and Fig. 3D).
In the absence of CYPA, ΔGt,c ≈ 1.7 to 2.3 kcal mol–1 for the WT systems (SI Appendix, Fig. S6 and Table S2), indicating that the trans conformation of the peptide bond between residues G89 and P90 is preferred over the cis conformation by ~95:5 in line with NMR experiments (36). This indicates that the energetic description of the states at the minima is appropriate, confirming previous work (60).
In the presence of CYPA, the barrier height ΔG#t→c decreases in all WT systems by 1.7 to 2.9 kcal mol–1 compared to the absence of CYPA (Fig. 3D and SI Appendix, Fig. S6 and Table S3), indicating a faster isomerization when catalyzed by CYPA, as expected. The decrease of ΔG#t→c is the largest for HIV-1 M, indicating that the isomerization is most accelerated by CYPA for HIV-1 M, which is protected against TRIM5α (Fig. 1), and that it is ~5 to 10 fold faster for HIV-1 M than for the other WT systems, which are not protected (Fig. 1).
As to variants, HIV-1 M A88V is not protected, and its behavior differs from the WT in the presence of CYPA: The trans conformation is more favorable, and the isomerization barrier is comparable to that in solution (SI Appendix, Fig. S6 and Tables S2 and S3). This may be caused by V88 sticking into the hydrophobic surface of CYPA, which hampers the isomerization (SI Appendix, Fig. S7). Notably, variants HIV-1 N L91I and P87H, which become protected against TRIM5α in contrast to the WT (SI Appendix, Fig. S5B), show decreases in ΔG#t→c in the presence of CYPA that are as large as or comparable to that in HIV-1 M. The reverse mutation in HIV-1 M, I91L, keeps the variant protected (SI Appendix, Fig. S5A), and, analogously, the decrease in ΔG#t→c is the fourth largest.
Overall, the magnitude of the decrease in ΔG#t→c in the presence of CYPA allows for an almost perfect ordering of the investigated HIV-1 types and variants, with protected ones showing generally the largest decreases (Fig. 3D). The change in ΔGt,c would not allow such an ordering. Although the quantitative relation of barrier heights derived from PMFs to kinetics requires caution (61), this finding suggests that the kinetics of the trans/cis isomerization plays a decisive role in the protective effect of CYPA for HIV-1 M.
Rhesus TRIMCyp Inhibits Non-M HIVs.
TRIMCyp proteins halt retroviral infections through the binding of their CYPA domain to the capsid CYPA-binding loop, early during infection, this inhibition can be blocked by cell treatment with CsA (26). For instance, owl monkey TRIMCyp is active against HIV-1 but not HIV-2, and rhTRIMCyp from rhesus macaques (rh) inhibits HIV-2 but not HIV-1 M (18, 41). In rhTRIMCyp, CYPA differs from human CYPA by two residues, the CYPA in rhTRIMCyp has N66 and H69 while human CYPA has D66 and R69 (62). It was shown that HIV-1 M’s resistance and HIV-1 O’s inhibition by rhTRIMCyp were due to the presence of N66 and H69 in this CYPA domain (59). To further understand whether rhTRIMCyp or a variant in which we reversed the two residues to D66 and R69 has differential antiviral activity to pandemic and nonpandemic HIV-1, we generated cell lines expressing WT rhTRIMCyp or its CYPA mutants N66D, H69R, and N66D-H69H (Fig. 4A). We challenged such cells with HIV-1 M, HIV-1 N, HIV-1 O, HIV-1 P, and SIVgor. As expected, rhTRIMCyp did not reduce the infectivity of HIV-1 M but strongly inhibited HIV-1 O (58). In addition, all other viruses, HIV-1 N, HIV-1 P, and SIVgor, were also strongly inhibited in cells expressing rhTRIMCyp by or more than 90% (Fig. 4 B and C and SI Appendix, Fig. S8). TRIMCyp with a human identical CYPA domain (N66D H69R, DR) recognized pandemic and nonpandemic viruses and displayed strong antiviral activity, further indicating that human CYPA interacts with all tested viruses. Mutating only residue H69R (NR) in rhTRIMCyp caused a complete loss of antiviral activity against HIV-1 N and HIV-1 O, in addition to remaining inactive against HIV-1 M. However, HIV-1 P and SIVgor were still inhibited by around 50%. In contrast, mutating N66 to the human D in rhTRIMCyp (DH mutant) generated an antiviral protein that inhibited efficiently all viruses (Fig. 4 B and C and SI Appendix, Fig. S8). Previous data suggested that capsid residue 88 in the CYPA-binding loop mediates differential interaction of HIV-1 M and O with rhTRIMCyp (59). The mutations in residue 88 reversed the resistance of HIV-1 M and the sensitivity of HIV-1 N, HIV-1 O, and HIV-1 P to rhTRIMCyp (Fig. 4 B and C). The nonpandemic HIVs with capsid mutations at position 88 were still sensitive to antiviral activity of TRIMCyp.DR, as was HIV-1 M A88V (Fig. 4B). The HIV-1 M G89V was predictably not inhibited by any TRIMCyp protein, likely because the G89V mutation prevented the interaction of TRIMCyp (Fig. 4B). These data suggest that the human CYPA domain in TRIMCyp proteins and likely free CYPA can recognize the incoming viral cores of nonpandemic HIV-1s.
Fig. 4.

WT, N66D, H69R, or N66D-H69R mutations in the CYPA domain of rhTRIMCyp or capsid mutations at position 88 affect rhTRIMCyp antiviral activity in a virus-dependent way. (A) Immunoblot of CrFK cells expressing rhTRIMCyp or its mutants with N66D (rhTRIMCyp.N66D) or H69R (rhTRIMCyp.H69R) or N66D-H69R (rhTRIMCyp.N66D.H69R). (B and C) CrFK cells expressing WT or mutated rhTRIMCyps were infected by luciferase reporter viruses from HIV-1 M, N, O, or P or their capsid mutants at position 88. Two to three days after infection, cells were lysed, and luciferase activity was measured. Infectivity of each virus on control cells expressing the empty vector (vector) was used as the reference. NH: rhTRIMCyp with N66 and H69 in CYPA domain, DH: rhTRIMCyp with N66D mutation in CYPA, NR: rhTRIMCyp with H69R mutation in CYPA, DR: rhTRIMCyp with N66D and H69R mutations in CYPA. All experiments were done at least three times in triplicates.
Pandemic and Nonpandemic HIVs Show Different Dependencies on CYPA.
To test whether the depletion of CYPA affects pandemic and nonpandemic HIVs differently, a CYPA KD in U-87 MG cells and huTRIM5α KO U-87 MG using the CRISPR/Cas9 system was done (Fig. 5A). CYPA KD in WT cells was associated with a strong and significant inhibition of HIV-1 M infectivity by up to 86%, and while the TRIM5α KO generated infections comparable to WT cells, the cells that lost TRIM5α and additionally CYPA expression were unexpectedly less infectable than WT or TRIM5α KO cells, suggesting that CYPA function for HIV-1 M is beyond protection against TRIM5α (Fig. 5B). Non-M HIVs and SIVgor were also tested in such conditions. The CYPA KD affected the nonpandemic HIVs less than HIV-1 M and caused only 50% inhibition. In further contrast to HIV-1 M, cells with no TRIM5α and no CYPA were much better infectable than the TRIM5α KO cells by the nonpandemic HIV-1s (Fig. 5 C–E). Infection with SIVgor differed from all HIV-1s and was not affected by CYPA KD in WT or TRIM5α KO cells, demonstrating that the CYPA KD did not impair cell vitality (SI Appendix, Fig. S9A).
Fig. 5.
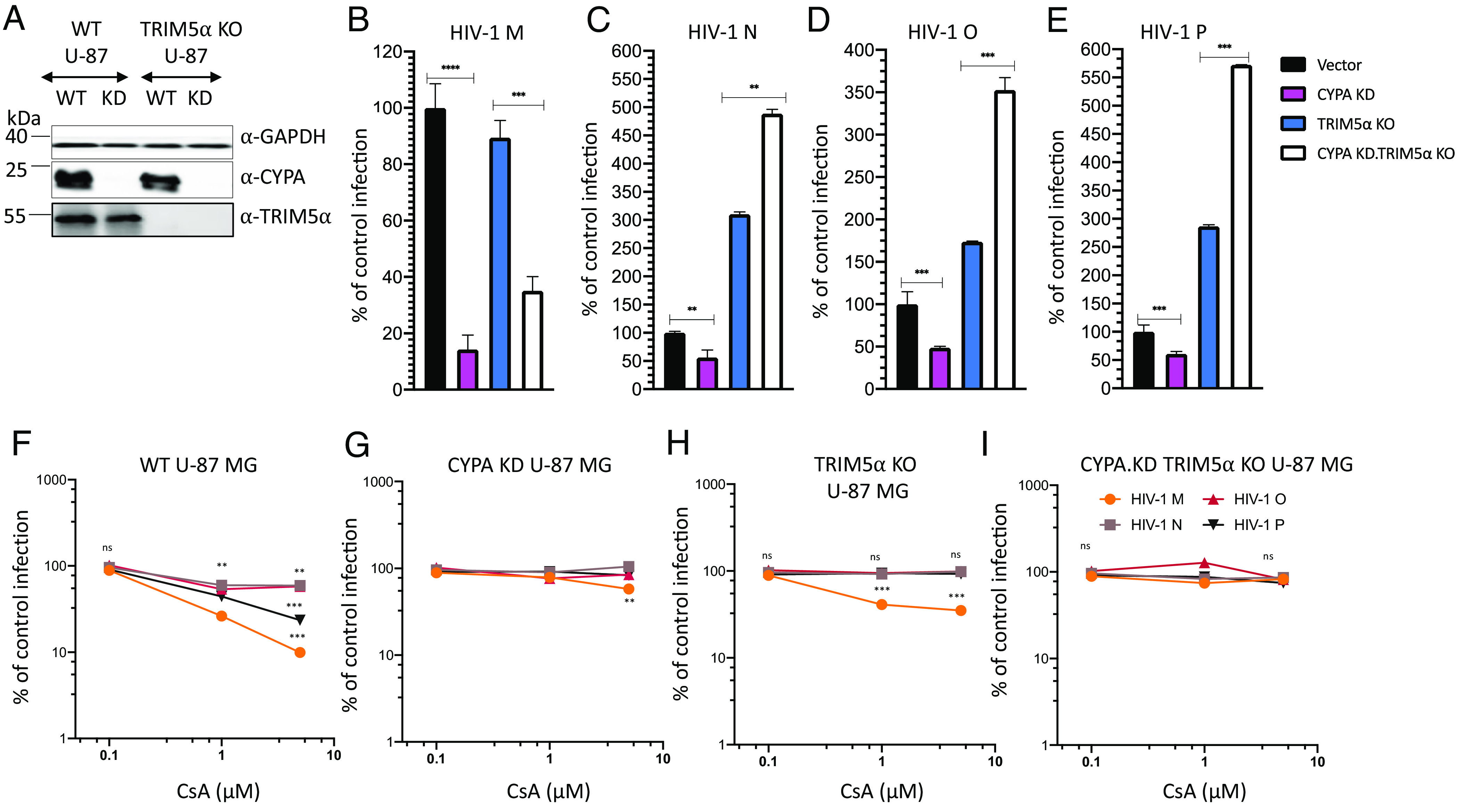
CYPA knockdown alone or CYPA and TRIM5α double depletion differentially affect infection by HIV-1 M, N, O, and P. (A) Immunoblot of CYPA KD in WT and in TRIM5α KO U-87 MG cells. CYPA expression was detected by anti-CYPA antibodies and GAPDH detection served as control for equal protein loading. (B–E) WT and TRIM5α, CYPA, and double KO U-87 MG cells were infected by HIV luciferase reporter viruses from different groups M, N, O, or P for 48 to 72 h, and luciferase activity was measured. Infection of mutated cells was normalized to infection of WT (vector) cells. (F–I) Infection of HIV-1 M, HIV-1 N, HIV-1 O or HIV-1 P, in the presence of increasing amounts of CsA, of (F) WT U-87 MG cells, (G) CYPA KD U-87 MG cells, (H) TRIM5α KO U-87 MG cells, and (I) CYPA KD - TRIM5α KO U-87 MG cells. Two to three days later, luciferase activity was assessed, and data analysis was done in comparison to control infection. All experiments were repeated at least three times independently with similar findings.
In an additional approach, we tested CsA treatment of cells in infection experiments. Increasing levels of CsA (0.1 µM to 10 µM) inhibited up to 91% of infection by HIV-1 M in WT U87 MG cells (Fig. 5F). In WT cells treated with 1 µM CsA, HIV-1 N was inhibited by 41%, HIV-1 O by 57%, and HIV-1 P by 66%, while HIV-1 M’s infectivity was decreased by 74%. Interestingly, high CsA concentrations had no further effect on infections by HIV-1 N and HIV-1 O but inhibited 77% of HIV-1 P (Fig. 5F). As expected, SIVgor did not react to CsA treatment of WT U-87 MG cells (SI Appendix, Fig. S9B). In CYPA-depleted cells, CsA treatment lost almost all its antiviral activity and only HIV-1 M showed some mild inhibition using 10 µM CsA (Fig. 5G). In TRIM5α KO cells, CsA treatment inhibited only HIV-1 M but had no effect on non-M HIVs, mirroring data obtained with CYPA KD (Fig. 5H). In CYPA and TRIM5α double depleted cells, CsA had no significant inhibitory activity against any virus tested, suggesting that CsA itself does inhibit these viruses (Fig. 5I and SI Appendix, Fig. S9B). Together, these data suggest that the nonpandemic HIV-1s have a weak and partial protection against TRIM5α by its CYPA binding.
Discussion
The reason(s) why non-M HIVs did not extensively spread in the human population remain(s) elusive. Some reports have pointed out differences in GAG capsid sequences but also differences in the functional activity of viral accessory proteins (63, 64). Here, we found that nonpandemic HIV-1s are subject to restriction by huTRIM5α. While it was shown that CYPA forms a protective layer around the viral core of HIV-1 M to prevent destruction by huTRIM5α (20, 21), the CYPA interaction with nonpandemic HIV-1 N, O, and P does not protect strongly against TRIM5α.
Our data demonstrate that the nonpandemic HIV-1s interact with and bind CYPA to a level that appears similar to the level of binding of HIV-1 M. Surprisingly, our findings show that the viral capsid CYPA binding loop determines the different sensitivities to TRIM5α of HIV-1 M and the nonpandemic HIV-1s. A recent report (65) has suggested capsid residues, such as residue at position 50, as important for differential sensitivity to TRIM5α by HIVs. Our findings suggest that more than one region of the capsid is involved in the regulation of TRIM5α activity. Thus, we postulate that not the binding of CYPA itself but the nature of CYPA capsid interaction is a regulator of TRIM5α sensitivity. Despite an identical GP motif in capsid CYPA-binding loops in pandemic and nonpandemic HIV-1s, these viruses engage CYPA differently, suggesting an important role from other residues of the CYPA-binding loop.
Moreover, a rescue of infection of non-M HIV-1s, when their capsids were specifically mutated at position 88 or when the CYPA domain of rhTRIMCyp was mutated at position 66 or 69, shows the importance of the capsid CYPA-binding loop for such restriction by rhTRIMCyp, as previously shown for HIV-1 M, HIV-1 O, and HIV-2 (18, 59). It is possible that the presence of valine (HIV-1 N and HIV-1 P capsid) or methionine for HIV-1 O at position 88 of the capsid, as opposed to alanine in the HIV-1 M, changes capsid conformation and allows availability of the loop to other host factors like TRIM5α, in addition to CYPA binding. This may restrict non-M HIVs by TRIM5α, but this hypothesis will need clarification.
Based on our study results, we postulated that the CYPA trans/cis isomerization activity on the capsid G89-P90 peptide bond (36, 61) differs between HIV-1 M and nonpandemic HIVs and that this difference changes the binding strength of TRIM5α in a way that may involve allostery (35). This hypothesis is supported by the PMF computations of the isomerization reaction. The capsid binding sites for TRIM5α are still ill-defined, although it is known that the binding involves the capsid CYPA-binding loop (13). In crystal structures, the loop of HIV-1 M is in the trans conformation in the presence of CYPA, whereas the loop in HIV-1 O is in the cis conformation in the presence of CYPA (61). It is not known which of the conformations is preferred under native conditions. However, TRIM5α should bind neither too strongly nor too weakly to the HIV-1 capsid to build an antiviral scaffold around the core (66). Considering that in G-P peptides the trans conformation prevails according to crystal structure (67) and NMR spectroscopy (68) analyses, our results lead us to speculate that a faster isomerization process might lead to reaching a higher proportion of cis conformation faster in the CYPA-binding loop, which might affect TRIM5α binding. This would be most relevant for HIV-1 M. In turn, and, in line with this, in the HIV-1 M A88V capsid mutant in the presence of CYPA, the isomerization barrier remains unchanged and almost as high as in the other HIV-1 WT in solution, which might explain why this variant does not show resistance against TRIM5α anymore. This would also explain why non-M HIV-1s with valine or methionine are also inhibited by TRIM5α. The magnitude of the decrease in the isomerization barrier in the presence of CYPA allows for an almost perfect ordering of all HIV-1 types and variants investigated in PMF computations, with protected ones showing generally the largest decreases, suggesting that the kinetics of the trans/cis isomerization plays a decisive role in the protective effect of CYPA for HIV-1 M.
The nonpandemic HIV-1s likely face several restrictions in human cells that slow a rapid adaptation. Ala88 is conserved in different HIV-1 M isolates and Val in the few cases of HIV-1 N and HIV-1 P. However, HIV-1 O isolates show more variability, and there are viruses that have at position 88 Val, Met, or Ile (SI Appendix, Fig. S3). SIVgor, the evolutionary ancestor virus of HIV-1 O and HIV-1 P has also Val at position 88 and shows similar restriction by huTRIM5α (SI Appendix, Fig. S1). SIVcpz is escaping huTRIM5α likely by its A88. Therefore, the generation of HIV-1 M was not restricted by huTRIM5α. It is, however, puzzling to understand why in the generation of HIV-1 N the A88 changed to V88. As we show in Fig. 2B and SI Appendix, Fig. S4, HIV-1 M with an A88V mutation has higher infectivity in cells lacking TRIM5α compared to WT virus. Thus, HIV-1 N may have evolved in an environment with reduced antiviral TRIM5α activity.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Wioletta Hörschken, Björn Wefers, and Yvonne Dickschen for excellent technical assistance. We thank Thomas Gramberg, Beatrice H. Hahn, Frank Kirchhoff, Michael Malim, Daniel Sauter, Heiner Schaal, Jonathan P. Stoye, Greg Towers, and Stephen R. Yant for reagents. The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: psPAX2 (Cat# 11348) from Didier Trono, SIVcpz TAN1.910 infectious molecular clone (Cat #11496) and SIVgorCP2139 (Cat # ARP-11722) from Jun Takehisa, Matthias H. Kraus and Beatrice H. Hahn. A.P.T. is supported by the German Academic Exchange Service (DAAD), and C.M. is supported by the Heinz-Ansmann Foundation for AIDS Research. This work was, in part, supported by Deutsche Forschungsgemeinschaft through GRK 2158/2 (project number 270650915) to H.G. H.G. is grateful for computational support and infrastructure provided by the “Zentrum für Informations- und Medientechnologie” at the Heinrich-Heine-University Düsseldorf and the computing time provided by the John von Neumann Institute for Computing to H.G. on the supercomputer JUWELS at Jülich Supercomputing Centre (user ID: VSK33).
Author contributions
A.P.T., H.G., and C.M. designed research; A.P.T., D.B., S.Z., and H.G. performed research; A.P.T., D.B., T.L., H.G., and C.M. analyzed data; and A.P.T., D.B., H.G., and C.M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix. All MD input structures, MD infiles, umbrella sampling files, and scripts that were used to analyze the umbrella sampling results are provided in the supporting repository: http://dx.doi.org/10.25838/d5p-52 (69). For molecular simulations, the AMBER22 package of molecular simulation codes was used. AMBER22 is available from here: http://ambermd.org/ (51, 52). The PMF calculations were performed with WHAM 2.9.9.1. WHAM is available from here: http://membrane.urmc.rochester.edu/?page_id=126 (57).
Supporting Information
References
- 1.Sharp P. M., Hahn B. H., Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 1, a006841 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush S., Tebit D. M., HIV-1 group O origin, evolution, pathogenesis, and treatment: Unraveling the complexity of an outlier 25 years later. AIDS Rev. 17, 147–158 (2015). [PubMed] [Google Scholar]
- 3.Bell S. M., Bedford T., Modern-day SIV viral diversity generated by extensive recombination and cross-species transmission. PLoS Pathog. 13, e1006466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Twizerimana A. P., Scheck R., Häussinger D., Münk C., Post-entry restriction factors of SIVcpz. Future Virol. 13, 727–745 (2018). [Google Scholar]
- 5.Harris R. S., Hultquist J. F., Evans D. T., The restriction factors of human immunodeficiency virus. J. Biol. Chem. 287, 40875–40883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malim M. H., Bieniasz P. D., HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2, a006940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staeheli P., Haller O., Human MX2/MxB: A potent interferon-induced postentry inhibitor of herpesviruses and HIV-1. J. Virol. 92, e00709-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boso G., Kozak C. A., Retroviral restriction factors and their viral targets: Restriction strategies and evolutionary adaptations. Microorganisms 8, 1965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cano-Ortiz L., Luedde T., Münk C., HIV-1 restriction by SERINC5. Med. Microbiol. Immunol. 212, 133–140 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reymond A., et al. , The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganser-Pornillos B. K., Pornillos O., Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 17, 546–556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kutluay S. B., Perez-Caballero D., Bieniasz P. D., Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 9, e1003214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., et al. , Structural insight into HIV-1 capsid recognition by rhesus TRIM5α. Proc. Natl. Acad. Sci. U.S.A. 109, 18372–18377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pertel T., et al. , TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 (2011). 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayah D. M., Sokolskaja E., Berthoux L., Luban J., Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430, 569–573 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Brennan G., Kozyrev Y., Hu S.-L., TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. U.S.A. 105, 3569–3574 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sebastian S., Luban J., TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2, 40 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakayama E. E., Shioda T., TRIM5α and species tropism of HIV/SIV. Front. Microbiol. 3, 1–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saenz D. T., Teo W., Olsen J. C., Poeschla E. M., Restriction of feline immunodeficiency virus by Ref1, Lv1, and primate TRIM5alpha proteins. J. Virol. 79, 15175–15188 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selyutina A., et al. , Cyclophilin A prevents HIV-1 restriction in lymphocytes by blocking human TRIM5α binding to the viral core. Cell Rep. 30, 3766–3777.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim K., et al. , Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 4, 2044–2051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganser B. K., Li S., Klishko V. Y., Finch J. T., Sundquist W. I., Assembly and analysis of conical models for the HIV-1 core. Science 1979, 80–83 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Yoo S., et al. , Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269, 780–795 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Caines M. E. C., et al. , Diverse HIV viruses are targeted by a conformationally dynamic antiviral. Nat. Struct. Mol. Biol. 19, 411–416 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamble T. R., et al. , Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Towers G. J., et al. , Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9, 1138–1143 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Toccafondi E., Lener D., Negroni M., HIV-1 capsid core: A bullet to the heart of the target cell. Front. Microbiol. 12, 755 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kootstra N. A., Münk C., Tonnu N., Landau N. R., Verma I. M., Abrogation of postentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc. Natl. Acad. Sci. U.S.A. 100, 1298–1303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke E. K., Yuan H. E. H., Luban J., Specific incorporation of cyclophilin a into HIV-1 virions. Nature 372, 359–362 (1994). [DOI] [PubMed] [Google Scholar]
- 30.Thali M., et al. , Functional association of cyclophilin A with HIV-1 virions. Nature 372, 363–365 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Peng W., et al. , Functional analysis of the secondary HIV-1 capsid binding site in the host protein cyclophilin A. Retrovirology 16, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambrose Z., et al. , Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J. Virol. 86, 4708–4714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J., Zhou J., Shah V. B., Aiken C., Whitby K., Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85, 542–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaller T., et al. , HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu M., et al. , Dynamic allostery governs cyclophilin A-HIV capsid interplay. Proc. Natl. Acad. Sci. U.S.A. 112, 14617–14622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosco D. A., Eisenmesser E. Z., Pochapsky S., Sundquist W. I., Kern D., Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 99, 5247–5252 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jimenez-Guardeño J. M., Apolonia L., Betancor G., Malim M. H., Immunoproteasome activation enables human TRIM5α restriction of HIV-1. Nat. Microbiol. 4, 933–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takehisa J., et al. , Generation of infectious molecular clones of simian immunodeficiency virus from fecal consensus sequences of wild chimpanzees. J. Virol. 81, 7463–7475 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bock M., Bishop K. N., Towers G., Stoye J. P., Use of a transient assay for studying the genetic determinants of Fv1 restriction. J. Virol. 74, 7422–7430 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dull T., et al. , A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson S. J., et al. , Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. U.S.A. 105, 3557–3562 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeire J., et al. , Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS One 7, e50859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Twizerimana A. P., et al. , Cell type-dependent escape of capsid inhibitors by simian immunodeficiency virus SIVcpz. J. Virol. 94, e01338-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li P., Song L. F., Merz K. M., Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 11, 1645–1657 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Mulnaes D., et al. , TopModel: Template-based protein structure prediction at low sequence identity using top-down consensus and deep neural networks. J. Chem. Theory Comput. 16, 1953–1967 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Krivov G. G., Shapovalov M. V., Dunbrack R. L., Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77, 778–795 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R., Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
- 49.Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H., PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 7, 525–537 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Schott-Verdugo S., Gohlke H., PACKMOL-memgen: A simple-to-use, generalized workflow for membrane-protein-lipid-bilayer system building. J. Chem. Inf. Model 59, 2522–2528 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Tian C., et al. , ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Case D. A., et al. , AMBER 2022 (University of California, San Francisco, 2022). [Google Scholar]
- 53.Izadi S., Anandakrishnan R., Onufriev A. V., Building water models: A different approach. J. Phys. Chem. Lett. 5, 3863–3871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu F., Hummer G., Convergence and error estimation in free energy calculations using the weighted histogram analysis method. J. Comput. Chem. 33, 453–465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quigley D., Probert M. I. J., Langevin dynamics in constant pressure extended systems. J. Chem. Phys. 120, 11432–11441 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Ryckaert J.-P., Ciccotti G., Berendsen H. J. C., Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977). [Google Scholar]
- 57.Grossfield A., WHAM: the weighted histogram analysis method (Version 2.0.10, Rochester, NY, 2017). [Google Scholar]
- 58.Kratovac Z., et al. , Primate lentivirus capsid sensitivity to TRIM5 proteins. J. Virol. 82, 6772–6777 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price A. J., et al. , Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat. Struct. Mol. Biol. 16, 1036–1042 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doshi U., Hamelberg D., Reoptimization of the AMBER force field parameters for peptide bond (omega) torsions using accelerated molecular dynamics. J. Phys. Chem. B 113, 16590–16595 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Howard B. R., Vajdos F. F., Li S., Sundquist W. I., Hill C. P., Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 10, 475–481 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Virgen C. A., Kratovac Z., Bieniasz P. D., Hatziioannou T., Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. U.S.A. 105, 3563–3568 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sauter D., et al. , Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6, 409–421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sauter D., Kirchhoff F., Key viral adaptations preceding the AIDS pandemic. Cell Host Microbe 25, 27–38 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Zuliani-Alvarez L., et al. , Evasion of cGAS and TRIM5 defines pandemic HIV. Nat. Microbiol. 7, 1762–1776 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu A., et al. , TRIM5α self-assembly and compartmentalization of the HIV-1 viral capsid. Nat. Commun. 11, 1307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gres A. T., et al. , X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 1979, 99–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang C., Ndassa Y., Summers M. F., Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 9, 537–543 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Becker D., Münk C., Gohlke H., MD simulation data for: “The cyclophilin A binding loop of the capsid regulates the human TRIM5α sensitivity of nonpandemic HIV-1”. HHU ResearchData. 10.25838/d5p-52. Deposited 1 November 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix. All MD input structures, MD infiles, umbrella sampling files, and scripts that were used to analyze the umbrella sampling results are provided in the supporting repository: http://dx.doi.org/10.25838/d5p-52 (69). For molecular simulations, the AMBER22 package of molecular simulation codes was used. AMBER22 is available from here: http://ambermd.org/ (51, 52). The PMF calculations were performed with WHAM 2.9.9.1. WHAM is available from here: http://membrane.urmc.rochester.edu/?page_id=126 (57).