

Abstract
Purpose
Using the model of experimental autoimmune uveitis (EAU) induced by immunization with a retinal antigen, two studies have reported contradictory results on disease development following oral antibiotic treatment (ABX). We showed that long-term ABX did not affect EAU, but another study showed that short-term ABX was protective. We therefore studied the effects of ABX on EAU, gut microbiota, and host immune responses as a function of treatment duration.
Methods
EAU-susceptible mice were treated orally with broad-spectrum antibiotics starting at least 10 weeks (long-term) or 1 week (short-term) before immunization until termination of the experiment. Gut microbiota were characterized by 16S amplicon sequencing, and host gut immune elements were studied phenotypically and functionally.
Results
Long-term ABX had no effect, whereas short-term ABX delayed EAU, as previously reported by us and others, respectively. Microbial sequencing revealed progressive reduction of gut microbiota that showed some differences in the two ABX groups. Interestingly, duration of ABX was associated with a gradual disappearance of the CD4+ and CD4+CD8+ subset of gut intraepithelial lymphocytes (IELs). This IEL subset is microbiota dependent and is absent in germ-free mice. Relative abundance of Lactobacillus reuteri correlated with the frequencies of CD4+CD8+ IELs. IELs suppressed antigen-specific activation of autoreactive T cells in culture.
Conclusions
Gut microbiota may play dual roles in uveitis development: They promote EAU development but also help maintain IEL populations that have regulatory function against autoreactive T cells. We propose that the progressive loss of this population during long-term ABX reverses the EAU-ameliorating effects of microbiota depletion.
Keywords: EAU, antibiotic treatment, intraepithelial lymphocytes, autoreactive T cells, Lactobacillus
Autoimmune uveitis in humans is an intraocular inflammatory disease believed to be driven by autoreactive T cells that can disrupt the neuroretina and cause blindness.1,2 Two animal models in susceptible B10.RIII mice have been useful to study the disease: (1) the induced model elicited by active immunization with peptide 161–180 of the interphotoreceptor retinoid binding protein (IRBP)2 and (2) the spontaneous uveitis model in R161H mice made transgenic for a CD4-restricted T-cell receptor (TCR) specific to this epitope,3 which allows the study of natural triggers that contribute to uveitis. This model led to the conclusion that gut commensals provide a signal in the intestine to activate the autoreactive T cells,4,5 based on data showing ameliorated disease in R161H mice treated with a broad-spectrum antibiotic cocktail (ABX) since before birth or reared under germ-free conditions.5 Together with data showing activation of the clonotypic retina-specific T cells in the gut environment, this suggested that gut microbiota provide a “mimic” antigen that activates the autoreactive T cells for pathogenicity. In contrast, wild-type (WT) littermates of these mice that were actively immunized for experimental autoimmune uveitis (EAU) with the IRBP161–180 peptide showed no reduction of disease, suggesting that when the antigen was provided by immunization there was no further role for gut microbiota in modulating disease.5
However, a subsequent study by another group showed that ABX started 1 week prior to the active immunization of WT mice for EAU inhibited the disease, a finding that appeared to be in conflict with our data.6 One major difference between the two studies was the duration of ABX prior to immunization. To reconcile these opposing observations, we set out to study the differences in disease, microbiota, and host immunological parameters between IRBP peptide–immunized WT mice receiving long-term versus short-term ABX.
We report that changes in both microbiota and gut immune parameters are differentially affected by the duration of ABX and are associated with distinct effects on EAU progression. ABX-induced changes in microbiota composition continue beyond the first week of treatment and correlate with progressive depletion of T-cell subsets within the intraepithelial lymphocyte (IEL) compartment that could be functionally relevant to uveitis. Based on this correlation and on the inhibitory function of IELs that we have observed in vitro, we propose that the change in the IEL compartment during long-term ABX results in loss of a regulatory mechanism that counteracts the EAU-ameliorating effect of microbiota depletion.
Methods
Mice
WT or Rag2−/−IRBP−/− R161H mice on the B10.RIII background were bred in-house and maintained under specific pathogen-free (SPF) conditions and fed standard facility chow ad libitum. The animal study protocol (NEI-688) was approved by the Institutional Animal Care and Use Committee.
Induction and Evaluation of EAU
B10.RIII WT mice, both females and males, between the ages of 10 and 15 weeks were immunized with 10 µg IRBP161–180 peptide emulsified in complete Freund's adjuvant (CFA). Development of uveitis was monitored by fundoscopic observation of anesthetized mice via a binocular microscope at the indicated days post-immunization. Scores were assigned by a masked observer on a scale of 0 to 4, taking into account the number, size, and appearance of inflammatory lesions as described in detail elsewhere.2
Antibiotic Treatment
WT B10.RIII mice were given a mixture of broad-spectrum antibiotics (ampicillin, 1 g/L; metronidazole, 1 g/L; neomycin, 1 g/L; vancomycin, 0.5 g/L5; and gentamicin, 0.4 g/L) in MediDrop Sucralose gel (ClearH2O, Westbrook, ME, USA), to minimize settling of the antibiotic suspension, for 1 week (short-term) or at least 10 weeks (long-term) prior to immunization for EAU. Antibiotic treatment was continued throughout the experiment. Treatment groups were kept in separate cages, and extra care was taken when handling the groups so as not to transfer microbiota, such as by changing gloves, reagents, and materials between groups during routine cage changing and fundoscopy monitoring.
Fecal DNA Extraction
Before and after the start of antibiotic treatment or EAU induction, fecal pellets were collected from each mouse and immediately frozen on dry ice and kept at –80°C until processing for bacterial DNA extraction in house or through the National Cancer Institute Microbiome Core (see 16S rRNA gene sequencing discussion below). For in-house DNA extraction, fecal pellets were subjected to lysozyme (Sigma-Aldrich, St. Louis, MO, USA), Invitrogen RNaseA (Thermo Fisher Scientific, Waltham, MA, USA), Achromopeptidase (Wako Biochemicals, Richmond, VA, USA), and proteinase K (QIAGEN, Germantown, MD, USA) digestions, and DNAs were extracted by the phenol:chloroform:isoamyl alcohol phase separation method.7
Calculation of 16S rRNA Gene Abundance
Quantitative polymerase chain reaction (qPCR) was performed using UniF340 (5′-ACTCCTACGGGAAGGGCAGCAGT-3′) and UniR514 (5′-ATTACCGCGGCTGCTGGC-3′) primers, which are specific for the eubacteria 16S rRNA gene8 and used here for normalization; EUB1114-F (5′-CGGCAACGAGCGCAACCC-3′) and EUB1221-R (5′-CCATTGTAGCACGTGTGTAGCC-3′), which are specific for the eubacteria 16S rRNA gene9; Sg-Lreu-F (5′-GAACGCAYTGGCCCAA-3′) and Sg-Lreu-R (5′-TCCATTGTGGCCGATCAGT-3′), which are specific for Lactobacillus reuteri9; and ITS1Fungal_F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2_R (5′-GCTGCGTTCTTCATCGATGC-3′), which are specific for internal transcribed spacer (ITS) genes for fungi, with Fast SYBR Green Supermix (Applied Biosystems, Bedford, MA, USA). Fecal DNA (2.5–5 ng) was amplified per qPCR reaction. CT values were normalized to respective CT values of control (untreated) mice (ΔCT = CTTxn – CTControl). The 2−∆CT method was used to calculate a qPCR factor relative to untreated controls (normalized to 1). Relative 16S rRNA gene abundance was calculated as total µg DNA per gram feces × qPCR factor.
16S rRNA Gene Sequencing and Analyses
In some experiments, fecal pellets were directly processed at the National Cancer Institute Microbiome Core using the MagAttract PowerMicrobiome DNA/RNA Kit (QIAGEN) on liquid-handling robots (epMotion5075 and epMotion 5073; Eppendorf, Enfield, CT, USA). Fecal DNAs were sequenced on a MiSeq platform (Illumina, San Diego, CA, USA) targeting the 16S V4 region (515F–806R). The 16S amplicon sequence variants were inferred using DADA2 1.10,10 and microbial diversity was analyzed and visualized with phyloseq 1.42.011 and other functions using R 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria). Part of the analysis was also performed on the Nephele cloud platform.12 The raw data generated for this study are available at National Center for Biotechnology Information Sequence Read Archive (SRA Accession: SRP450596).
Tissue Harvesting and T-Cell Subset Characterization by Flow Cytometry
At the time of sacrifice, peripheral tissues (submandibular lymph nodes, spleen, bone marrow, inguinal lymph node, subcutaneous, and mesenteric adipose tissues) and gut-associated lymphoid tissue (GALT), including mesenteric lymph nodes, Peyer's patches (PP), small and large intestine lamina propria (LP), and IELs, were collected and processed for fluorescence-activated cell sorting (FACS) analysis using the CytoFLEX LX (Beckman Coulter, Brea, CA, USA). Fluorochrome-conjugated antibodies and viability dyes used in this study were purchased from BD Biosciences (Franklin Lakes, NJ, USA), BioLegend (San Diego, CA, USA), eBioscience (San Diego, CA, USA), or Tonbo Biosciences (San Diego, CA, USA). FlowJo 10.6.1 was used for the t-distributed stochastic neighbor embedding (t-SNE) analyses. Surface markers (clone) included CD3 (17A2 or 145-2C11), CD4 (RM-4.5), TCRγδ (GL3), CD8a (53-6.7), TCRβ (H57-597), CD90.2 (30-H12), Ghost UV450, and ViaKrome 808 (Beckman Coulter). Intracellular markers included granzyme A (3G8.5) and granzyme B (GB11).
IEL Suppression Assay
IRBP-specific naïve CD4+ T cells were harvested from spleen and peripheral lymph nodes of Rag2−/−IRBP−/− R161H mice and enriched for naïve CD4+ cells (EasySep Mouse Naïve CD4+ T Cell Isolation Kit; Stem Cell Technologies, Vancouver, BC, Canada). Cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) in some experiments. IELs and PP cells were harvested from small intestines of B10.RIII WT mice and enriched for CD90.2+ T cells (EasySep Mouse CD90.2 Positive Selection Kit II; Stem Cell Technologies), and the CD90.2− fraction was collected as a source of antigen-presenting cells (APCs). Cell co-cultures were set up in various combinations of cells and stimulated with 100 ng/mL IRBP161–180 incubated at 37°C for 1 to 4 days and stained for FACS analysis. Culture supernatant was collected at 21 hours to measure IL-2 levels, following the manufacturer's protocol (R&D Systems, Minneapolis, MN, USA).
Cell Sorting and NanoString Analyses
Cells from the inguinal and mesenteric lymph nodes, small intestine LP, and IEL compartment were sorted for CD4 and CD8 populations by the National Eye Institute Flow Cytometry Core on FACSAria II and FACSAria Fusion (BD Biosciences). RNA was isolated using the RNeasy Kit (QIAGEN). Multiplex hybridization was performed using the NanoString platform at the National Cancer Institute Center for Cancer Research Genomics Core. Expression of 770 genes was profiled using an nCounter panel (NS_MM_CANCERIMM_C3400) and quantified with the nCounter Digital Analyzer (NanoString Technologies, Seattle, WA, USA). The data were normalized using nSolver 4. The log2 normalized data were used for pre-ranked gene set enrichment analysis (GSEA), against a custom annotated gene set based on the nSolver Immune Response Categories. The processed and raw gene expression data generated in this study are available at the National Center for Biotechnology Information Gene Expression Omnibus (GSE236922). Custom R scripts and BASH scripts were used to preprocess the data and generate visualizations. All of the scripts used for this research are documented in detail and are available at the National Eye Institute GitHub as open-source resources (https://github.com/NIH-NEI/abx-eau).
Statistical Analyses
Statistical analysis was performed using Prism (GraphPad, San Diego, CA, USA). Student's t-tests for paired samples, one-way ANOVA, and two-way ANOVA (with Tukey's multiple comparison test) were used to evaluate differences between groups of variables. Where not specified, statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Short-Term But Not Long-Term Antibiotic Treatment Delays Development of EAU Disease
B10.RIII WT mice were started on ABX for 1 week for short-term treatment or at least 10 weeks for long-term treatment before immunization with IRBP peptide to elicit EAU (Fig. 1A). Short-term ABX partially inhibited the development of EAU, whereas long-term ABX had no effect on disease kinetics compared to untreated controls (Figs. 1B, 1C), supporting the data in previous reports from our lab and the Lin Lab5,6 showing differential effects on EAU development depending on different durations of antibiotic treatment. However, disease scores in the short-term ABX group eventually reached those of the untreated or long-term ABX mice (Fig. 1B). Thus, short-term ABX was able to reduce susceptibility to EAU, but the effect was transient. This trend was consistently observed in three independent experiments. Because of the transient nature of the protective effect, we concentrated further efforts on examining the differences between long-term and short-term ABX, which might explain the delay in the disease kinetics, during the first 2 weeks after immunization (weeks 2–3 of short-term ABX).
Figure 1.
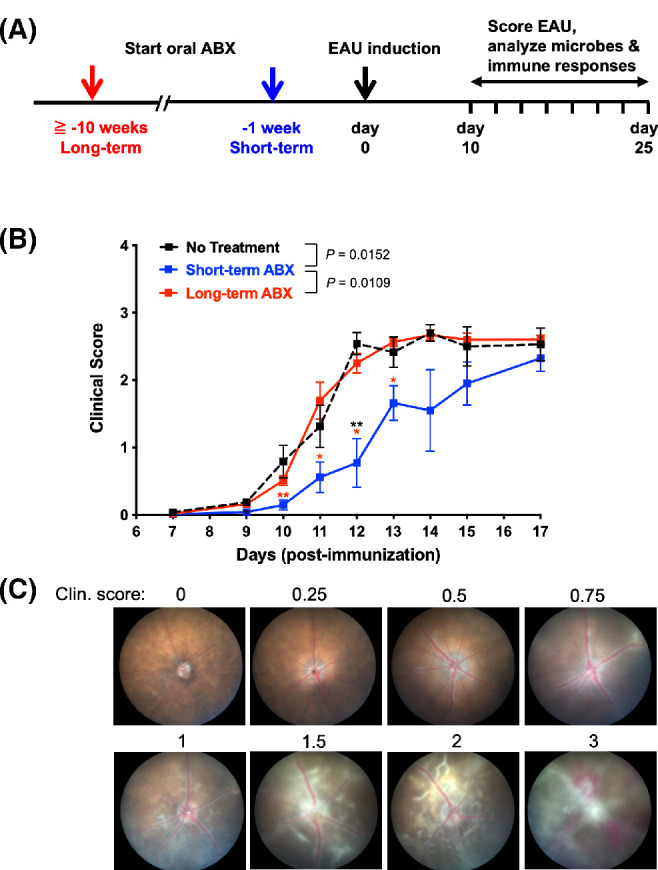
Short-term, but not long-term, antibiotic treatment delayed the development of EAU. WT B10.RIII mice were treated, or not, with antibiotics for at least 10 weeks (long-term) or 1 week (short-term) prior to immunization with 10 µg IRBP161–180 with CFA. (A) Experimental scheme of ABX treatment and EAU induction. (B) Disease was monitored weekly, and mean fundoscopy scores were plotted by treatment groups; data for no treatment (n = 16), long-term ABX (n = 25), and short-term ABX (n = 18) were combined from three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001 relative to no treatment (black symbol) or long-term ABX (red symbol) by Tukey's multiple comparison test on indicated time points. P values between treatment groups for the course of EAU are shown in the graph. (C) Representative fundus pictures for corresponding clinical scores.
Short-Term and Long-Term ABX Mice Differ in Gut Microbiota When Inductive Events of EAU Take Place
We sought to correlate the changes in microbial composition and gut-related immune parameters with the disease process. Fecal bacterial abundance determined by qPCR in ABX-treated mice quickly declined after initiation of treatment, such that 1 week after immunization (2 weeks after the start of ABX for the short-term group and >10 weeks ABX for the long-term group), fecal bacterial mass from both ABX groups became comparable (Fig. 2A). Sequences representing fungi were inversely proportional to the level of gut bacteria and were undetectable in feces of untreated mice (Fig. 2A), indicating that fungi expand to fill niches made available by depletion of bacteria due to ABX. A comparison of microbial richness in individual samples (Chao1 index) among the short-term ABX, long-term ABX, and no ABX groups at each time point supported the notion that most of the decrease in abundance of microbial composition had taken place within the first week of ABX (Fig. 2B). The Bray–Curtis beta diversity analysis, comparing microbial composition among the groups, revealed distinct clustering among the no-treatment, short-term ABX, and long-term ABX groups, with short-term ABX samples being more dissimilar from each other, likely reflecting ongoing changes in the bacterial composition (Fig. 2C). By visualizing the relative abundance of bacterial phyla over time, Bacteroidetes and Firmicutes progressively decreased as Proteobacteria and Tenericutes progressively increased; diversity reduced to only few dominant phyla, and the composition of short-term ABX flora gradually became closer to that of long-term ABX (Fig. 2D). Nevertheless, some differences in flora composition were still detectable during the second week of ABX, corresponding to the first week after immunization, when inductive events of EAU were taking place (Fig. 2D). During the second and third weeks after initiation of ABX, the gut microbiota composition of the short-term ABX group increasingly approached that of the long-term ABX group (Figs. 2A, 2B), with an uptick in disease scores during the third week of EAU (Fig. 1).
Figure 2.

Long-term versus short-term duration of ABX affected gut microbiome differently. WT B10.RIII mice were treated, or not, with antibiotics for at least 10 weeks (long-term) or 1 week (short-term) prior to immunization with 10 µg IRBP161–180 with CFA. Fecal pellets were collected weekly over time from unimmunized and immunized mice, and qPCR on microbes and 16S rRNA gene sequencing were performed. (A) The relative abundance of bacteria (16S rRNA gene, left) or fungi (ITS gene, right) is expressed as 2−ΔCT × DNA per gram feces. No treatment, n = 3 or 4; short-term ABX, n = 5 or 6; and long-term ABX, n = 4, per time point. *P < 0.05 and **P < 0.01 relative to no treatment (same as respective treatment colors) or between long-term ABX and short-term ABX (purple) on week -1. (B) Chao1 alpha-diversity among the groups over time. No treatment, n = 9 to 17; short-term ABX, n = 9 to 11; and long-term ABX, n = 8, per time point. ****P < 0.0001 relative to no treatment (same as respective treatment colors) by comparing median estimates across cohorts using the Kruskal–Wallis test and Dunn's post hoc tests for multiple comparisons. (C) Bray–Curtis beta-diversity from a representative two experiments. Time points 0, 1, and 2 weeks relative to immunization are compiled. NMDS, non-metric multidimensional scaling. (D) Bar plots of taxonomy by phylum.
Antibiotic Treatment Depletes CD4+ and CD4+CD8+ Cells in the IEL Compartment in a Time-Dependent Fashion
We examined the effects of ABX duration more broadly on lymphoid cell populations in the gut and other peripheral tissues by flow cytometry with visualization in t-SNE plots by treatment or population (Fig. 3A). ABX effects were pronounced within the IEL compartment and small intestine LP on CD4+ and CD4+CD8+ cells, which were depleted in the long-term ABX mice but not in the short-term ABX mice compared to the no-treatment group (Figs. 3A, 3B). Numbers of CD8+ IELs were not affected by ABX (data not shown). CD4+CD8+ IELs were progressively depleted over time (Figs. 3B–3D), reminiscent of what has been reported by others.9,13 This appears to be a predominant effect on IELs and not on lymphocytes from small intestine LP or peripheral lymph nodes (Fig. 3C). To investigate the dependence of IELs on microbiota in this model, we examined the IEL compartment in germ-free B10.RIII mice. CD4+ and CD4+CD8+ IEL frequency, but not CD8+ IELs, were strongly reduced in germ-free mice (Fig. 3E). These results support the notion that progressive depletion of CD4-expressing IELs by ABX could be secondary to the dependence of CD4+ and CD4+CD8+ IEL maintenance on the microbiota.
Figure 3.

CD4+CD8+ T cells in the IEL compartment are modulated by antibiotic treatment. Experimental mice were treated and immunized as previously described. Tissues from unimmunized and immunized groups were collected 7 days after immunization (2 weeks after short-term ABX) and processed for FACS analysis. (A) A total of 15,000 live lymphocytes from each sample were concatenated by tissue, and t-SNE analyses were done separately by tissue. The t-SNE plots of various tissues are shown by the experimental parameters: treatment (upper row) and T-cell populations (lower row). Plots reflect three mice per group from one representative experiment of three. (B) Representative FACS plots of CD4 and CD8 profiles in IELs on day 7 of EAU among live lymphocytes. (C) Kinetic changes in CD4+, CD4+CD8+ (double-positive [DP]), and CD8+ proportions by antibiotic treatment in various tissues (n = 3 or 4 unimmunized mice per time point). (D) Proportions of CD4+ and CD4+CD8+ (DP) IELs among live lymphocytes on day 7 of EAU. Data were combined from unimmunized mice and immunized mice under the same ABX, as they displayed no differences. (E) Percentages of CD4+, CD4+CD8+ (DP), and CD8+ IELs in specific pathogen-free (SPF) and germ-free (GF) mice. *P < 0.05, ***P < 0.001, ****P < 0.0001.
IELs Suppress Activation of IRBP-Specific T Cells In Vitro
Some IELs have been reported to have immunoregulatory function complementary to that of regulatory T cells (Tregs), and some may even arise from FoxP3+ Tregs.14 We therefore examined whether IELs could inhibit the activation of antigen-specific T cells when they are co-cultured in vitro (Fig. 4A). For this assay, we enriched naïve IRBP-specific CD4+ T cells from Rag2−/−IRBP−/− R161H mice as responders. CD90.2+ cells were isolated from either the PP or IEL compartment of WT mice as potential suppressors, and the CD90.2− fraction from PP cells was used as a source of APCs. Upon stimulation with IRBP peptide in the presence of CD90.2− APCs from PP, antigen-specific CD4+ T cells expressed early activation markers CD69 and CD25 (Fig. 4B, top plot). Inhibition of these markers was significant in the presence of suppressors from the IELs but not from PP (Fig. 4B, bottom vs. middle plots; Fig. 4C). This suppression occurred in a dose-dependent manner, with increasing ratios of IEL suppressors to responders but not with PP suppressors (Fig. 4D). Production of IL-2 in culture supernatant was correlated with the activation of retina-specific T cells. In co-cultures containing IEL suppressors, IL-2 levels were lower compared to those co-cultured with PP cells (Fig. 4E). Antigen-specific T-cell proliferation, assessed by CFSE dilutions at 4 days of co-culture, was inhibited in the presence of increasing ratios of IEL as suppressors (Fig. 4F). These results raise the possibility that IELs could exert a suppressive role on antigen-specific T cells and/or on other immune cells that pass through the gut on their way to the eye.15
Figure 4.

IELs suppressed activation and proliferation of IRBP-specific T cells in vitro. Responder cells from Rag2−/−IRBP−/− R161H mice were enriched for naïve CD4+ T cells. IELs and PP cells were isolated from B10.RIII-CD45.1 congenic mice and enriched for CD90.2+ cells (which served as potential suppressors); the CD90.2− fraction served as APCs. Cell co-cultures (suppressor-to-responder ratio of 1:1) were stimulated with 100 ng/mL IRBP161–180 for 18 to 21 hours, and cells were stained for FACS analysis. CD4 cells from Rag2−/−IRBP−/− R161H mice were gated by singlets, time, live lymphocytes, CD45.2+, CD90.2+, CD8−, and CD4+. (A) Experimental scheme of co-culture. (B) Representative FACS plots of CD69 early activation marker expression on antigen-specific CD4 responders co-cultured with APCs and respective CD90.2+ IELs or PP suppressors. (C) Compiled results of five independent experiments. The line connects the data points from the same experiment. *P < 0.05. (D) Dose-dependent suppression of CD69 expression by CD90.2+ IELs or PP suppressors on responder CD4+ T cells (0.125:1, 0.25:1, 0.5:1, and 1:1 suppressor-to-responder ratios) under IRBP peptide stimulation. One representative experiment of five is shown. (E) Dose-dependent inhibition of IL-2 production in the culture supernatant at 21 hours of co-culture. One representative experiment of three is shown. P < 0.05 by paired t-test. (F) CFSE dilution at 87 hours of co-culture. One representative experiment of two is shown.
IELs Express Genes With Cytotoxic or Regulatory Functions That Are Microbiota Dependent
To gain insight into the function of IELs in our model in vivo, we examined gene expression profiles of IEL subpopulations compared to CD4+ or CD8+ T cells from small intestine LP, mesenteric lymph nodes, and peripheral lymph nodes from unimmunized mice by NanoString. Hierarchical clustering of the top 50 immune-related genes that were most highly expressed in CD4+CD8+ IELs revealed that many of them were also expressed in CD4+ or CD8+ IELs, but CD4+CD8+ IELs were consistently the highest expressors. Among these genes, Gzma and Gzmb (encoding granzymes A and B, respectively), Cd244, Cd48, and Cd160 were prominent in IELs, suggesting that the inhibitory function of these cells could in part be attributed to cytotoxicity (Fig. 5A). Genes associated with T-cell activation and signaling, such as Cd6, Fyn, Zap70, and Spn, were also highly expressed in IELs (Fig. 5A). GSEA supported enrichment of pathways associated with cytotoxicity as being positively regulated in IEL populations compared to other intestinal or peripheral compartments (Fig. 5B). Flow cytometry staining for intracellular granzymes A and B confirmed protein expression of granzymes by IELs (Fig. 5C). Importantly, on day 7 of EAU, expression of granzymes in CD4+ and CD4+CD8+ IELs was still well maintained in the short-term ABX mice and comparable to that of the no-treatment mice. In contrast, granzyme expression was lost after long-term ABX (Fig. 5C), closely mimicking the situation in germ-free mice (Figs. 5D, 5E) and indicating dependence on microbiota not only for physical IEL maintenance (in CD4+ IELs) (Fig 3E) but also for their functional maintenance (Fig 5E). This included CD8+ IELs, whose numbers were maintained in germ-free mice (Fig. 3E), but more than half lost granzyme expression (Figs. 5D, 5E).
Figure 5.

IELs expressed high levels of granzymes A and B, dependent on microbiota. CD4+, CD4+CD8a+ (DP), and CD8a+CD90+ cells from peripheral lymph nodes (PLN) and mesenteric lymph nodes (MES), small intestine lamina propria (SI), and IEL compartments were FACS sorted from naïve B10.RIII mice, and their gene expression was analyzed by NanoString. (A) Heatmap clustering of the top 50 genes generated by Morpheus. (B) GSEA of the pre-ranked transcriptome using custom annotated gene sets based on the nSolver immune response categories. Positive gene set enrichments are shown as red dots (pos). Negative enrichments are shown as blue dots (neg). The size of the dots represents the P value significance of the gene set enrichments. (C) Representative flow plots of granzyme-producing IELs on day 7 of EAU. (D) Representative flow plots of granzyme-producing IELs in SPF and GF mice from three independent experiments. (E) Counts of granzyme A– and granzyme B–producing IELs from SPF and GF mice. **P < 0.01.
Maintenance of CD4+ and CD4+CD8+ IEL Compartments in ABX Mice Is Associated With Levels of Firmicutes and Lactobacilli
Firmicutes and Bacteroidetes are dominant phyla in untreated mice and show strong reduction within the first week of ABX (Fig. 2D). Lactobacillus reuteri (L. reuteri) has been associated with the induction and maintenance of CD4+CD8+ IELs in the gut epithelium in another model.9 ABX efficiently depleted the genus Lactobacillus as determined by 16S rRNA sequencing (Fig. 6A), as well as L. reuteri as determined by qPCR (Fig. 6B). Short-term ABX mice still had approximately 10 times more L. reuteri compared to long-term ABX at the 1-week time point and reached similar levels after 1 more week (Fig. 6B). Thus, physical reduction of CD4+ and CD4+CD8+ IELs appeared to lag 1 to 2 weeks behind depletion of L. reuteri.
Figure 6.

CD4+CD8+ T cells in the IEL compartment were microbiota dependent. (A) Relative abundance of the Lactobacillus genus extrapolated from 16S sequencing reads. Feces were collected at the time points of 0, 1, and 2 weeks of EAU (1, 2, and 3 weeks of ABX), and the compiled results are shown. *P < 0.05, **P < 0.01 by ordinary one-way ANOVA. (B) Quantification of L. reuteri by qPCR from fecal DNA (no treatment, n = 7; short-term ABX, n = 6; long-term ABX, n = 4). *P < 0.05 by two-way ANOVA relative to no treatment. (C) Linear regression analysis of CD4+, CD4+CD8+ (DP), or CD8+ IEL subsets and abundance of eubacteria, Bacteroidetes and Firmicutes phyla, or L. reuteri. P values shown in the graphs indicate the significant linear relationships between IEL subsets and bacterial abundance (DP, green; CD4, red). No significance was found in the CD8+ population.
To examine the correlation between gut bacteria and IEL subsets, we performed simple linear regression analyses of relative abundance of fecal bacteria and frequencies of CD4+ and CD4+CD8+ IELs. In support of published results and our data (Figs. 3C–3E), total bacterial abundance (eubacteria) showed a positive correlation with the frequencies of CD4+ and CD4+CD8+, but not CD8+, IELs (Fig. 6C). Importantly, a positive association between the frequency of CD4+CD8+ IELs and L. reuteri abundance was observed, but such an association was not observed with the frequencies of CD4+ IELs or CD8+ IELs (Fig. 6C). A positive association was also observed between the frequency of CD4+CD8+ IELs and total bacteria and the Firmicutes phylum, to which L. reuteri belongs, but not between the frequency of CD4+CD8+ IELs and the Bacteroidetes phylum (Fig. 6C). These results suggest that the abundance of L. reuteri in the gut affects the development and maintenance of CD4+CD8+ cells within the IEL compartment.
Taken together, transient protection from EAU with short-term ABX can be associated with the presence of microbial taxa, possibly including L. reuteri, that can support the maintenance of CD4+CD8+ IELs in the intestine. When these IELs are depleted by longer duration of ABX, the protective effects by beneficial commensals are no longer maintained, and the susceptibility to EAU of the short-term ABX mice becomes equivalent to that of the long-term ABX mice. It is therefore conceivable that commensal microbiota may play a dual role in EAU development by promoting disease and also by maintaining IELs that could have a regulatory function against autoreactive T cells.
Discussion
In this study, we reconcile seemingly contradictory reports regarding protection from EAU by broad-spectrum antibiotic treatment in the same mouse strain (B10.RIII) immunized for EAU with the same antigen (IRBP161–180).5,6 Here, we report that short-term ABX provided disease protection, albeit temporary, whereas prolonged ABX did not reduce disease. It is now recognized that gut microbiota can affect autoimmune disease in distal sites by many and diverse mechanisms, including antigenic, innate stimulatory, and metabolic effects.4,16 Our study demonstrates that, as short-term ABX becomes long term with continued treatment, dynamic and sequential changes occur in the microbiota and gut immune cells in the intestine that have a prominent influence on IELs. The data are suggestive of a sequential loss of two opposing influences during ABX: removal of disease-stimulatory microbiota, resulting in protection, followed by secondary depletion of regulatory IELs that were being maintained by them and consequent reversal of the protective effect. A limitation of our data is that, although they implicate IELs in the regulation or EAU by gut microbiota, they do not prove causality and do not exclude additional effects and interactions that play into the observed effects on disease. Indeed, it is also possible that the subsequent rise in fungal sequences, reflecting a transition toward ABX-resistant gut microorganisms, could have contributed over time to additional immune stimulation in the gut, compounding the loss of protection as short-term ABX continued and became long-term ABX. The effect on disease appears to lag behind the gut environment changes, as would be expected.
The protective effect of short-term ABX in our study was more transient than what had been reported by Nakamura et al.6 This can be attributed to differences in the conditions between the two laboratories, such as food and other environmental factors, that likely resulted in different gut flora in our respective facilities. A case in point is the enhancement of gut Tregs reported in the Nakamura et al.6 study, which we did not observe. Similarly, the kinetics and effectiveness of IEL depletion following ABX could also differ, contributing to the longer protection from EAU reported in the Nakamura et al.6 study.
Our data highlight one possible scenario, that of inhibitory IELs suppressing uveitogenic effector T cells. This raises the inevitable question regarding where and how these cells communicate. Although in the immunization-induced EAU model priming of pathogenic T cells occurs mainly in the lymph nodes that drain the sites of immunization, broad migration of lymphocytes to and from the gut has been observed under homeostatic conditions.17 In eye and brain autoimmunity, the migration of activated T cells through peripheral tissues is thought to be required for functional “licensing” of pathogenic T cells to cause pathology in the target tissue.18–21 Of note, uveitis-relevant immune cells that were labeled by photoconversion in the gut were found in eyes of C57BL/6 mice immunized for EAU, although the precise lineage has not been established.15 It is thus conceivable that primed uveitogenic T cells migrating through the gut may come into contact and be influenced by the gut environment, including resident immune cells in the gut epithelium.
CD4+ and CD4+CD8+ IELs in the small intestine differentiate from intestinal CD4+ T cells that migrate into the epithelial cell layer and take on regulatory functions. Conversion of FoxP3+CD4+ T cells into CD4+CD8+ IELs has been reported to occur, as LP Tregs migrate to the intestinal epithelium; downregulate expression of FoxP3, CD25, and ThPOK; and become CD4+CD8+ IELs in a microbiota-dependent manner.14,22 This finding supports the possible scenario that microbial antigens may control CD4+ T cell plasticity between FoxP3+ Treg cells and regulatory IELs.23 Nakamura et al.6 reported expansions of gut CD4+ Tregs after ABX. Such FoxP3+CD4+ Tregs in the gut were not expanded in our study, which might be due to differences in gut microbiota between our animal facilities. That said, we cannot rule out the possibility that FoxP3+ Tregs had been induced but were converted to IEL-like cells.
IELs are microbiota dependent and are strongly reduced or absent in germ-free mice (Fig. 3E).24 Recently, the presence of this population in SPF mice has been reported to be associated with L. reuteri in C57BL/6 mice.9 L. reuteri metabolizes dietary tryptophan into indole derivatives, which activate the aryl hydrocarbon receptor and promote differentiation of CD4+ and CD4+CD8+ IEL precursors. In agreement with these findings, we detected an association between L. reuteri abundance and the frequency of CD4+CD8+ IELs, as well as massive reduction in L. reuteri in mice on prolonged ABX, which correlated with progressive depletion of CD4+CD8+ IELs. It is conceivable that L. reuteri–dependent maintenance of IEL subsets and IEL regulation of EAU expression may be among the several pathways involved in reversal of the protective effect of microbiota with continued ABX. Of note, although it was reported that L. reuteri reduces the severity of experimental autoimmune encephalomyelitis (EAE) by reducing Th1/Th17 cells,25 tryptophan metabolites of L. reuteri unexpectedly enhanced EAE.26 The role of this bacterium in EAU and in autoimmunity in general will require further investigation.27
The data have implications on how we think of potentially modulating autoimmune eye disease by manipulation of gut microbiota, and they underline the importance of taking into consideration the duration of antibiotic treatment. The findings highlight the potential unintended consequences of the extended use of antibiotics in clinical settings for the immunomodulatory role of IELs in the microbiota-driven gut–eye axis. More comprehensive approaches will be needed to gain insights into the interaction of microbiome and its contribution to systemic and ocular immune responses by integrating microbiome and metabolome studies with those of the cellular and transcriptional signatures of inflammation.16,28
Acknowledgments
The authors thank Colm O'Huigin, Vishal Thovarai, and Wuxing Yuan from the Genetics and Microbiome Core at the National Cancer Institute, National Institutes of Health (Bethesda, MD, USA) for 16S rRNA gene sequencing and analyses; the National Cancer Institute CCR Genomics Core for NanoString; the National Eye Institute Flow Cytometry Core for cell sorting; and all of the members of the R.R. Caspi Lab for their help.
Supported by National Eye Institute, National Institutes of Health, intramural funding (Project #EY000184).
Disclosure: R. Salvador, None; R. Horai, None; A. Zhang, None; Y. Jittayasothorn, None; J. Tang, None; A. Gupta, None; V. Nagarajan, None; R.R. Caspi, None
References
- 1. Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. 2010; 120: 3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horai R, Caspi RR.. Retinal inflammation: uveitis/uveoretinitis. In: Pang I-H, Clark AF, eds. Animal Models for Retinal Diseases . Totowa, NJ: Humana Press; 2010: 207–225. [Google Scholar]
- 3. Horai R, Silver PB, Chen J, et al.. Breakdown of immune privilege and spontaneous autoimmunity in mice expressing a transgenic T cell receptor specific for a retinal autoantigen. J Autoimmun. 2013; 44: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horai R, Caspi RR. Microbiome and autoimmune uveitis. Front Immunol. 2019; 10: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horai R, Zarate-Blades CR, Dillenburg-Pilla P, et al.. Microbiota-dependent activation of an autoreactive T Cell receptor provokes autoimmunity in an immunologically privileged site. Immunity. 2015; 43: 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura YK, Metea C, Karstens L, et al.. Gut microbial alterations associated with protection from autoimmune uveitis. Invest Ophthalmol Vis Sci. 2016; 57: 3747–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atarashi K, Suda W, Luo C, et al.. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 2017; 358: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barman M, Unold D, Shifley K, et al.. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008; 76: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cervantes-Barragan L, Chai JN, Tianero MD, et al.. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science. 2017; 357: 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP.. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016; 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McMurdie PJ, Holmes S.. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013; 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weber N, Liou D, Dommer J, et al.. Nephele: a cloud platform for simplified, standardized and reproducible microbiome data analysis. Bioinformatics. 2018; 34: 1411–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cortez VS, Cervantes-Barragan L, Song C, et al.. CRTAM controls residency of gut CD4+CD8+ T cells in the steady state and maintenance of gut CD4+ Th17 during parasitic infection. J Exp Med. 2014; 211: 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sujino T, London M, Hoytema van Konijnenburg DP, et al.. Tissue adaptation of regulatory and intraepithelial CD4+ T cells controls gut inflammation. Science. 2016; 352: 1581–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakamura YK, Janowitz C, Metea C, et al.. Short chain fatty acids ameliorate immune-mediated uveitis partially by altering migration of lymphocytes from the intestine. Sci Rep. 2017; 7: 11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salvador R, Zhang A, Horai R, Caspi RR.. Microbiota as drivers and as therapeutic targets in ocular and tissue specific autoimmunity. Front Cell Dev Biol. 2020; 8: 606751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D.. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci USA. 2014; 111: 6696–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen J, Vistica BP, Takase H, et al.. A unique pattern of up- and down-regulation of chemokine receptor CXCR3 on inflammation-inducing Th1 cells. Eur J Immunol. 2004; 34: 2885–2894. [DOI] [PubMed] [Google Scholar]
- 19. Flugel A, Berkowicz T, Ritter T, et al.. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001; 14: 547–560. [DOI] [PubMed] [Google Scholar]
- 20. Odoardi F, Sie C, Streyl K, et al.. T cells become licensed in the lung to enter the central nervous system. Nature. 2012; 488: 675–679. [DOI] [PubMed] [Google Scholar]
- 21. Tan C, Wandu WS, Lee RS, et al.. Shedding new light on the process of “licensing” for pathogenicity by Th lymphocytes. J Immunol. 2017; 198: 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prakhar P, Alvarez-DelValle J, Keller H, et al.. The small intestine epithelium exempts Foxp3+ Tregs from their IL-2 requirement for homeostasis and effector function. JCI Insight. 2021; 6: e149656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDonald BD, Jabri B, Bendelac A.. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat Rev Immunol. 2018; 18: 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olivares-Villagomez D, Van Kaer L.. Intestinal intraepithelial lymphocytes: sentinels of the mucosal barrier. Trends Immunol. 2018; 39: 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He B, Hoang TK, Tian X, et al.. Lactobacillus reuteri reduces the severity of experimental autoimmune encephalomyelitis in mice by modulating gut microbiota. Front Immunol. 2019; 10: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montgomery TL, Eckstrom K, Lile KH, et al.. Lactobacillus reuteri tryptophan metabolism promotes host susceptibility to CNS autoimmunity. Microbiome. 2022; 10: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fine RL, Mubiru DL, Kriegel MA.. Friend or foe? Lactobacillus in the context of autoimmune disease. Adv Immunol. 2020; 146: 29–56. [DOI] [PubMed] [Google Scholar]
- 28. Hagan T, Cortese M, Rouphael N, et al.. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell. 2019; 178: 1313–1328.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]