

Abstract
Precisely controlled development of the somatosensory system is essential for detecting pain, itch, temperature, mechanical touch, and body position. To investigate the protein-level changes that occur during somatosensory development, we performed single-cell mass cytometry on dorsal root ganglia from C57/BL6 mice of both sexes, with litter replicates collected daily from E11.5 to P4. Measuring nearly 3 million cells, we quantified 30 molecularly distinct somatosensory glial and 41 distinct neuronal states across all time points. Analysis of differentiation trajectories revealed rare cells that coexpress two or more Trk receptors and overexpress stem cell markers, suggesting that these neurotrophic factor receptors play a role in cell fate specification. Comparison to previous RNA-based studies identified substantial differences between many protein/mRNA pairs, demonstrating the importance of protein-level measurements to identify functional cell states. Overall, this study demonstrates that mass cytometry is a high-throughput, scalable platform to rapidly phenotype somatosensory tissues.
Introduction
Somatosensory neurons residing in the dorsal root ganglia (DRG) transmit diverse sensory stimuli to the central nervous system (CNS), including mechanical pressure, changes in limb position, temperature, pain, and itch. Previous studies have identified up to 13 subpopulations of mature sensory neurons in the peripheral nervous system (PNS) by the first month of development in mice, and up to 18 subpopulations in adulthood1–5. While somatosensory neurons are relatively well characterized at maturity, many fundamental questions with respect to their development remain unresolved. In particular, intermediate progenitor cell types of the DRG remain poorly characterized, and the molecular profiles that control cell type specification have not been defined. Identifying the molecular trajectories and cell fate decisions that control DRG development promises to improve our understanding of sensory disorders with developmental components such as congenital insensitivity to pain with anhidrosis (CIPA) and autism spectrum disorder (ASD)6–8.
Previous efforts to monitor the diversification and maturation of somatosensory neurons have primarily relied either on microscopy, which detects a small number of proteins simultaneously, or single-cell RNA sequencing (scRNA-seq), which detects a large number of transcripts simultaneously. scRNA-seq and the related technique, single-nuclei RNA sequencing (snRNA-seq), have been applied to characterize the molecular diversity of cell types in a wide range of adult and developing neural tissues9–13, including the dorsal root ganglion2,3,14–23, but no study to date has measured every day of development across embryonic and postnatal timepoints. Such temporal resolution is essential if we are to determine precise lineages of both abundant and rare cell types that are responsible for somatosensory perception. In this study, we leverage the relatively high throughput (1×106 cells/hour) single-cell analysis technique, mass cytometry, to exhaustively profile the composition of the DRG at every day of development from embryonic day E11.5 to postnatal day P4.
Mass cytometry is a flow cytometry variant that uses rare earth metal isotope-labeled antibodies and other affinity reagents to quantify the abundance of proteins and other biomolecules at the single-cell level24. Commercially available reagents permit over 40 molecular markers to be measured and quantified simultaneously in each cell, including cell surface receptors and intracellular signaling molecules25, transcription factors26, cell cycle status and proliferation state27, and cell viability28. Mass cytometry has been used previously to characterize glioma cells and microglia in neural tissues29–31, but until now it has not been applied to neurons or other glial cell types in the CNS or PNS.
To investigate DRG development with mass cytometry, we developed a 41-antibody panel including key transcription factors, neurotrophic factor receptors, and other protein markers known to play a critical role in the specification and maturation of DRG cell types. We applied this panel to measure single-cell DRG samples from E11.5, shortly after the DRG have coalesced from migratory neural crest cells, to P4, when somatosensory neurons have innervated their peripheral targets and have begun to mature into distinct functional types1. With this approach, we identified and quantified the abundance of 30 molecularly distinct somatosensory glia and 41 somatosensory neuron subtypes across embryonic and postnatal development in the DRG.
The 41 somatosensory neuron subtypes we identify here show complementary overlap with postnatal DRG neurons previously identified by scRNA-seq2,5,19. However, a time course comparison reveals that mRNA transcript abundance does not accurately predict protein abundance, which is the best representation of a cell’s functional state. Collectively, the findings presented in this study demonstrate for the first time that mass cytometry is a high-throughput, scalable platform for single-cell analysis of neural tissues such as the DRG.
Results
DRG cell type characterization by protein expression
To identify and characterize cell types in the DRG by their protein expression signatures, we first adapted mass cytometry methods for neural tissues. This involved optimizing tissue dissection and cell dissociation techniques (Methods) and developing a 41-antibody staining panel for specific neuronal and glial subtypes (Supplementary Table 1)1. Each antibody was conjugated to a unique rare earth metal isotope32, and then titrated to identify its optimal staining concentration using known-positive and known-negative control cells (Extended Data Fig. 1a,b). Mouse DRGs were collected at daily time points from E11.5 to P4 in order to provide a continuous molecular expression profile (Fig. 1a, Supplementary Table 2). For embryonic time points, DRGs from a single litter were pooled for cell dissociation and mass cytometry analysis. For postnatal timepoints, the pups were first separated by sex, and then DRGs were combined to generate a pooled male and female sample from each litter. At least two biological replicates (i.e. pooled litters) were used for each developmental time point (Supplementary Table 2). After dissection, the pooled DRGs were dissociated into a single-cell suspension33 and then briefly incubated with cisplatin as a non-cell permeant viability stain28, followed by paraformaldehyde (PFA) fixation and storage at −80°C (Fig. 1a).
Fig. 1. Characterization of DRG cell types from E11.5 to P4 by mass cytometry.
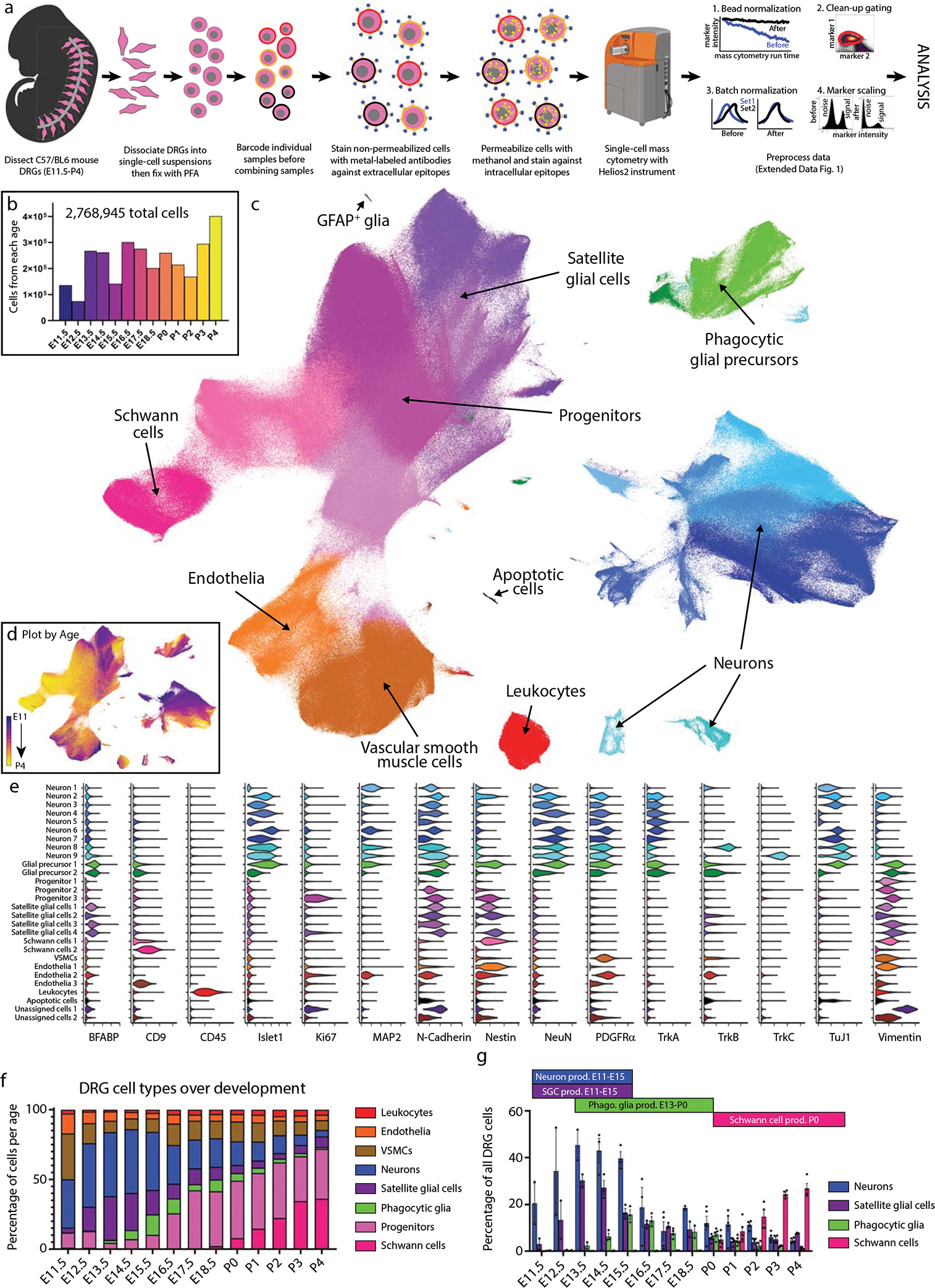
a) Neural mass cytometry workflow. Created with BioRender.com. b) Total number of clean-up gated and preprocessed cells analyzed at each day of DRG development. These show the sum of 2–4 separately analyzed litters (biological replicates) at each time point, for 43 samples in total. Bars are colored by time point. c) UMAP of all DRG cells after clean-up gating, colored by primary Leiden clustering and labeled by presumptive cell type according to protein expression profiles. d) UMAP from (c), colored by age. e) Violin plots of protein expression for clusters from c). f) The proportional abundance of major cell type classes across DRG development, created by combining the Leiden clusters from c) into 1) leukocytes: CD45+; 2) endothelia: CD31+, CD133+; 3) smooth muscle: p75NTR+, PDGFRα+, TrkB+, neuronal marker negative; 4) neurons: Islet1+, MAP2+, NeuN+, PGP9.5+, TuJ1+; 5) satellite glial cells: BFABP+, Sox10+, Vimentin+; 6) phagocytic glial precursors: BFABP+, Sox10+, Vimentin+, plus a mixture of neuronal markers such as Islet1+, MAP2+, NeuN+, PGP9.5+, TuJ1+; 7) neural progenitors: Ki67+, Nestin+, Sox10+, Vimentin+; and 8) Schwann cells: CD9+, cMet+, OligO4+. g) Changes in neuronal and glial abundance during periods of neuronal and SGC expansion, glial phagocytosis, and postnatal Schwann cell proliferation. Abbreviations: “phago.” stands for phagocytosis and “prod.” stands for production. Data are presented as mean values +/− SEM with biologically independent samples consisting of pool litters including both sexes with n=2 for E11.5-E13.5, and E18.5, n=3 for E14.5-E16.5, and n=4 for E18.5. Postnatal ages were biologically independent samples consisting of sex separated litters with n=2 of female and n=2 of male for P2-P4 and n=2 of female and n=3 of male for P0,P1. Number of pups per litter for each sample may be found in Supplementary Table 2.
After all samples were collected, they were thawed and barcode labeled34, followed by pooling into barcode sets(Supplementary Table 2) for uniform staining and subsequent mass cytometry analysis (Fig. 1a). Preliminary inspection revealed that five samples had a low percentage of Islet1+ cells and two additional samples had low overall cell numbers, so additional litters were collected for these timepoints and run as a third barcode set (Supplementary Table 3). The resulting 4,974,302 events from three barcode sets were preprocessed by: 1) bead normalization (Extended Data Fig. 2a)35, 2) debarcoding36, 3) clean-up gating to remove dead cells, aggregates, and debris (Extended Data Fig. 2b; www.cytobank.org), 4) batch normalization37, 5) marker scaling (Extended Data Fig. 2c) and 6) removal of low complexity cells (Extended Data Fig. 3a–f). After these preprocessing and clean-up steps, we were left with 2,768,945 high-quality, viable, singlet DRG cells for analysis (Fig. 1b; Supplementary Table 3).
To identify broad classes of DRG cell types, we performed Leiden clustering38 on the full ~2.8 million cell dataset and visualized the resulting clusters, or developmental age, on a 2D Uniform Manifold Approximation and Projection (UMAP) layout39,40 (Fig. 1c,d; Extended Data Fig. 3g–j). Cell type assignment for each cluster was determined by protein expression profiles matching neurons (23.09%), stem cells and glial progenitors (28.18%), satellite glial cells (12.21%), Schwann cells (11.87%), vascular smooth muscle cells (VSMCs) (11.51%), endothelial cells (5.17%), and leukocytes (3.07%) (Fig. 1e). Because glial precursors can act as “non-professional” phagocytes41, we assigned the identity of putative phagocytosing glial precursors (4.9%) to cells that express glial precursor markers along with additional markers of their presumptive phagocytosed cargo (Fig. 1e). In addition to clustering analysis, we also prepared a 2-dimensional gating hierarchy as an alternative method to identify key cell types (Extended Data Fig. 2d).
To investigate how the abundances of all DRG cell types change across development, we grouped the cell populations identified by Leiden clustering (Fig. 1c) by cell class and calculated their abundance and standard error of the mean for sample replicates at each time point (Fig. 1f and Extended Data Fig. 3k,l). Generally, leukocytes, endothelia, and VSMCs showed consistent abundance across development. Consistent with previous studies, we observe that the relative abundance of neurons increases until E16.5 due to initial proliferation and migration waves, and then diminishes due to apoptosis and concurrent increase in glial progenitors (Fig. 1f)42–47. The proportion of neurons, satellite glial cells, putative phagocytic glial precursors, and Schwann cells across the time course correspond to the expected windows of neuronal proliferation and glial subtype expansion indicated in Fig. 1g.
Comparison of DRG mass cytometry with immunohistochemistry
Neuronal cell types have not previously been characterized by mass cytometry, so we sought to validate our results by comparison with immunohistochemistry (IHC), assessing the total abundance of neurons observed with each technique, plus the relative abundance of TrkA, TrkB, TrkC, and Ret-expressing neurons. These neurotrophic factor receptors are essential for somatosensory growth and survival, but also delineate the broad neuronal cell types (Fig. 2a). First, we performed IHC on DRGs from E11.5, E12.5, E13.5, E14.5, and P0 using Islet1 as a global marker for somatosensory neurons, assessed the percentage of Islet1+ cells out of total DAPI+ cells by IHC, and compared this to the percentage of Islet1+ cells observed by mass cytometry at the same ages, excluding non-DRG cells (blood, endothelia, VSMCs, Schwann cells) (Fig. 2b). In both our mass cytometry and IHC analysis, the percentage of Islet1+ cells was relatively consistent across E11.5 to E14.5, and then dropped approximately 50% by P0, which is due to both neuron death and expansion of non-neuronal cell types43,46,47.
Fig. 2. Comparison of DRG analysis by mass cytometry and IHC.
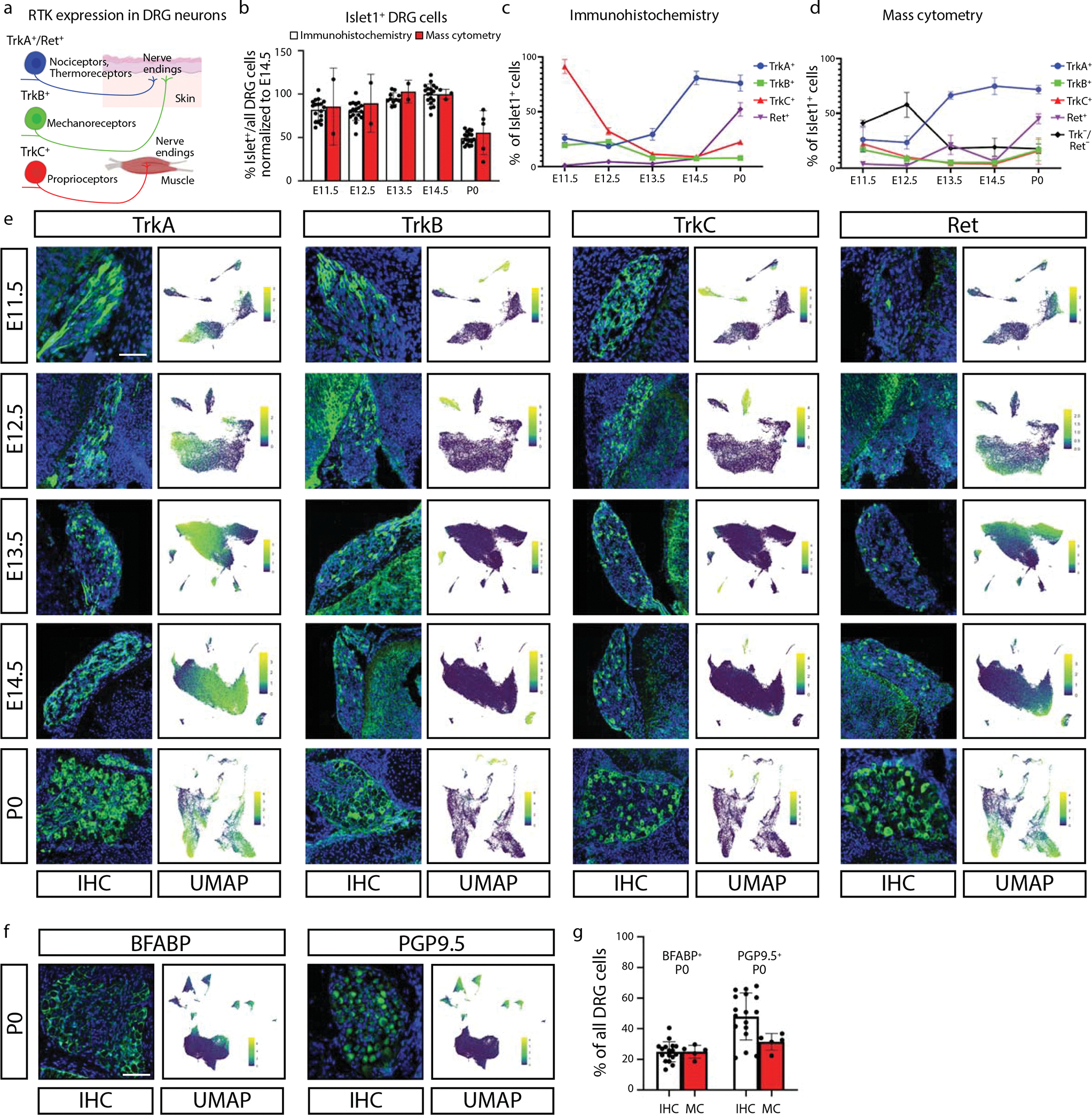
a) Schematic illustration of somatosensory neuron subtypes divided by TrkA+, TrkB+, TrkC+, and Ret+ expression during development. Created with BioRender.com. b) Proportion of Islet1+ cells out of all DRG cells, by IHC and mass cytometry. All ages are normalized to E14.5, the peak of Islet1+ cell abundance by IHC. Data are presented as mean values +/− SEM with biologically independent samples as in Fig. 1g except just E11.5-E14.5 and P0 for the mass cytometry and 1 or 2 sections from the L4 DRG of 3 mice (sexes unknown) from 3 litters, thus 17 datapoints for IHC at each age. c,d) Proportion of TrkA+, TrkB+, TrkC+, and/or Ret+ neurons across matching timepoints between IHC (c) and mass cytometry (d), respectively. Mass cytometry also identifies the number of neurons that express Islet1 but none of TrkA, TrB, TrkC, or Ret. Data are presented as mean values +/− SEM with the same samples in Fig. 2b. e) Representative IHC of lower lumbar (L3-L6) DRGs stained for TrkA, TrkB, TrkC, or Ret; at ages E11.5, E12.5, E13.5, E14.5, and P0 quantified in (c,d). IHC images are paired with mass cytometry UMAP layouts from neurons of the corresponding age, colored by protein expression for each RTK. Scale bar, 100μm. f) Representative IHC of P0 DRGs for BFABP and PGP9.5 quantified in (g), and UMAP layouts of all P0 DRG cells colored by expression of these two markers. Scale bar, 100μm. g) Relative abundance of BFABP+ and PGP9.5+ DRG cells at P0 by IHC or mass cytometry. Data are presented as mean values +/− SEM with the same samples as in Fig. 2b except just P0.
We next examined specific neuron subtypes by performing IHC on E11.5, E12.5, E13.5, E14.5, and P0 DRGs with antibodies against the receptor tyrosine kinases (RTKs) TrkA, TrkB, TrkC, and Ret, to distinguish the major classes of somatosensory neurons1. To compare cell abundance and staining intensity between mass cytometry and IHC, the microscopy images are shown side-by-side with UMAP plots from the same respective time points, subset for Islet1+ somatosensory neurons (Fig. 2c–e and Extended Data Fig. 4a–c). To quantify the abundance of somatosensory neuron subtypes observed by IHC, we counted the percentage of RTK+;DAPI+ double positive cells (Fig. 2c). To quantify the abundance of somatosensory neuron subtypes observed by mass cytometry the percentage of cells positive for each RTK was counted and then normalized to the percentage of Islet1+ cells from the same developmental age (Fig. 2d).
The patterns of cell abundance across this developmental window were similar for TrkA+, TrkB+, and Ret+ neurons, but TrkC+ neurons are more abundant in IHC than mass cytometry at E11.5-E12.5 (Fig. 2c,d). One potential explanation for this discrepancy is that only cell surface TrkC is detected by MC, while IHC detects total TrkC.
We also compared IHC and mass cytometry with the glial marker BFABP and the neuronal marker PGP9.5 at P0 (Fig. 2f,g). At P0, the relative percentages of BFABP+ cells are comparable between techniques, although the proportion of PGP9.5-expressing cells is slightly lower by mass cytometry (Fig. 2f,g). This difference is likely due to the expansion of glial Schwann cell precursors in proximal nerve roots that can be spatially excluded by IHC but not when dissected for mass cytometry.
Glial cell subtypes and their developmental trajectories
To further investigate how glia mature in the DRG, we selected all cell clusters that expressed glial markers Sox10, Vimentin, BFABP, CD9, cMet, OligO4, and GFAP; and then performed an additional round of Leiden clustering on this subset (Fig. 3a, Extended Data Fig. 5a–d), identifying four distinct glial cell types: 1) Schwann cells, 2) satellite glial cells (SGCs), 3) unspecified glial progenitors, and 4) putative phagocytic glial precursors. We then performed URD pseudotime analysis48, which uses timepoint-biased random walk iterations to identify the most likely cellular trajectory from a manually selected “root” cell type to manually selected “tip” cell types. We selected all E11.5 cells in the glial subset as the root, and each of the three mature cell types as tips (Fig. 3b,c and Extended Data Fig. 5e–i). For finer cluster-level resolution, we performed an additional tertiary round of subclustering on the SGCs, Schwann cells, and phagocytic glial precursors producing 30 molecularly distinct clusters (Fig. 3d,e,h,i,l,m).
Fig. 3. Glial subtypes show distinct developmental trajectories.
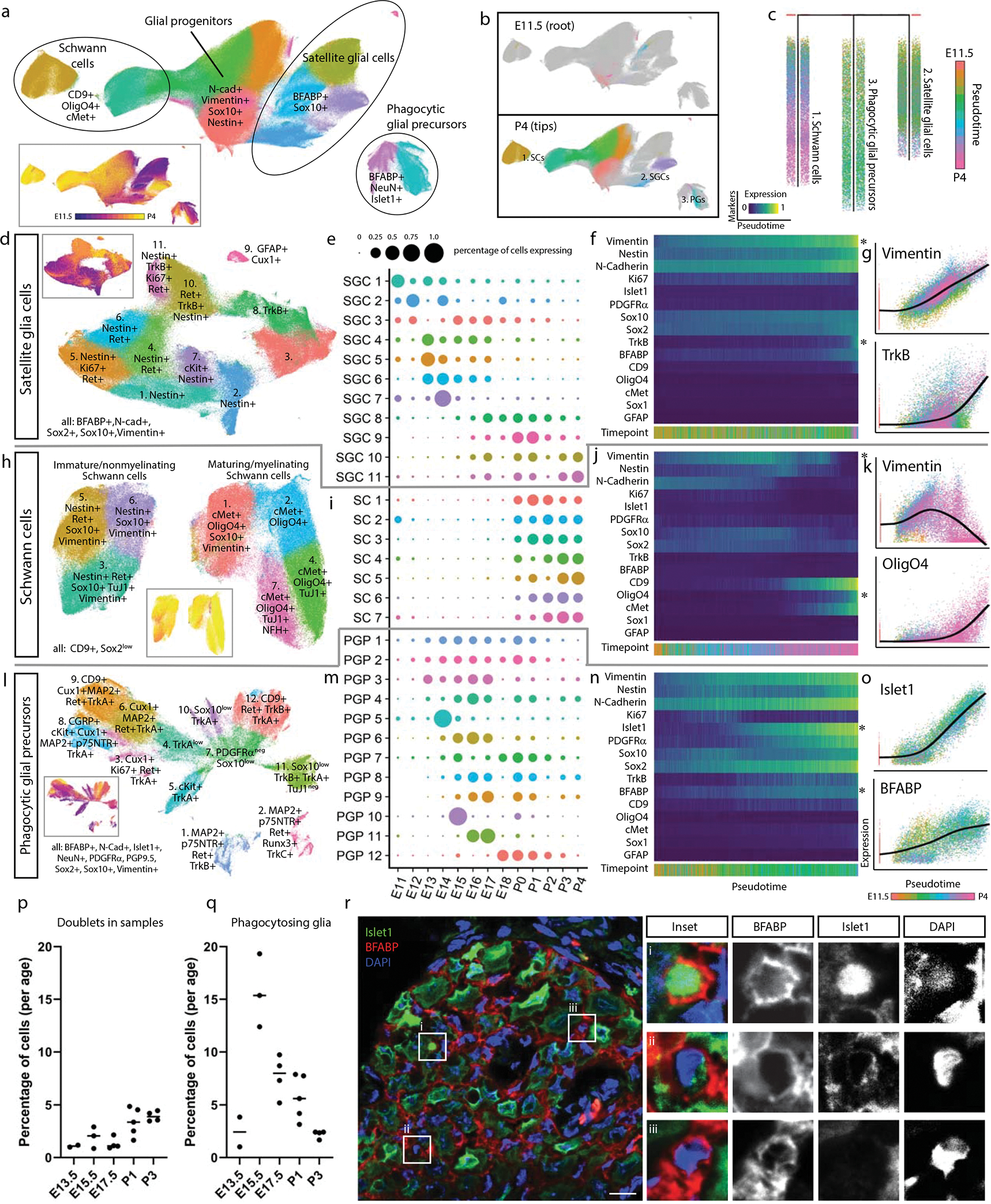
a) Leiden clustering and UMAP of E11.5-P4 DRG glia and glial precursors, performed on all cells from the clusters in Fig. 1c that expressed glial markers BFABP, CD9, cMet, GFAP, OligO4, Sox10, and/or Vimentin, labeled by glial types and key markers. Inset: colored by age. b) E11.5 or P4 cells only, colored by Leiden clustering, overlaid on the full UMAP from (a), colored gray. c) URD pseudotime hierarchy for satellite glial cells, Schwann cells, and putative phagocytic glial precursors, produced with E11.5 root (pooled) and P4 tips (individual clusters) from (b), colored by pseudotime. d-g) Satellite glial cell clusters were extracted from the glial dataset for an additional secondary round of Leiden clustering and UMAP, with SGC subtypes labeled by their characteristic marker expression (d). Inset is colored by age. Relative abundances for each SGC subtype are plotted across sample collection ages (e), and changes in protein expression are plotted across the SGC pseudotime trajectory (f). Expression markers denoted by asterisks are also plotted by scatterplot (g) to illustrate the cells underlying the pseudotime heatmap in (f). Similar analysis was performed for Schwann cells (h-k) and putative phagocytic glial precursors (l-o). p) Percentage of doublets counted in the samples from ages E13.5, E15.5, E17.5, P1, and P3 from manual inspection under brightfield analysis with hemocytometer. Data are biologically independent samples as in Fig. 1g for the ages used here. q) Percentage of phagocytic glia by mass cytometry after all cleanup gating. Data are biologically independent samples as in Fig. 1g for E13.5, E15.5, E17.5, P1, and P3. r) Representative immunohistochemistry of E15.5 L4/L5 DRGs with anti-Islet1 (neuronal marker) and anti-BFABP (satellite glial marker). Similar results were obtained across 3 or more DRGs per litter and 3 separate litters. Scale bar, 20μm. Insets i-iii show three phagocytic events as previously described41.
Glial precursors arrive in the DRG and start ensheathing somatosensory neuron cell bodies and phagocytosing dying neurons by E13.541,49–52. The non-myelinating, presumptive SGCs we observe here all express BFABP, N-cadherin, Sox2, Sox10, and Vimentin (Fig. 3d–g); and subcluster into eleven populations, four of which are retained at P4 (Fig. 3d). The early embryonic clusters express Nestin and a mix of Ki67, Ret, and c-Kit; while the postnatal SGCs upregulate TrkB (Fig. 3d–g). GFAP is a commonly used marker for SGCs, but only a small percentage of glial cells expressed GFAP, first appearing as ~2% of all BFABP+ glia at E16.5, and increasing to ~17% of BFABP+ glia by P4 (Fig. 3d,e).
Schwann cells (CD9+ and Sox2low) appear around birth and rapidly expand into two distinct classes: immature/non-myelinating (Nestin, Sox10, Vimentin) and maturing/myelinating (cMet, OligO4, and one subcluster still expressing Sox10 and Vimentin, suggesting a less mature state) (Fig. 3h). Each of these groups contain a subset of TuJ1low cells, which is likely not expressed by these cells but rather comes from axons wrapped by these Schwann cells and not removed by dissociation (Fig. 3h–k). The neurofilament NFH is also present in a subset of the maturing/myelinating cells. These subtypes do not simply transition from the immature/non-myelinating (clusters 2, 4, and 5) to maturing/myelinating island (clusters 1, 3, 6, and 7), suggesting that the distinct islands are likely delineating between non-myelinating and myelinating pools of the same populations rather than a difference in developmental lineages or maturation state (Fig. 3h,i).
Putative phagocytic glial precursors co-express non-myelinating glial markers (BFABP, Vimentin, and Sox10) but also contain neuronal markers (e.g. Islet1, NeuN, TuJ1) (Fig. 3l–o). Glial precursors have been shown to phagocytose dying neurons in the embryonic DRG before macrophages infiltrate and become the primary phagocytosing cell type41. We identified these putative phagocytic cells and performed the following analyses to rule out false positives associated with doublets and aberrant association with debris: 1) DNA intercalator gating, 2) “Barcode negative” filtering, 3) Abundance comparisons, 4) Expression intensity comparisons, and 5) Hemocytometer doublet analysis (Fig. 3p,q, Extended Data Fig. 5j). What are these glial precursors likely phagocytosing? Their subclustering is largely driven by the four RTKs that define somatosensory neuron types, TrkA, TrkB, TrkC, and Ret; but other neuronal proteins are also present, including nuclear transcription factors that indicate entire cell engulfment (Fig. 3l, Extended Data Fig. 5k,l). These putative phagocytosis events are observed across embryonic and postnatal development, but peak during the late embryonic stage (Fig. 3l,m). To validate these results, we performed immunohistochemistry on E15.5 DRGs with anti-Islet1 and anti-BFABP antibodies and DNA-label DAPI to demonstrate phagocytosis as previously observed by others41. Immature glial precursors were identified phagocytosing neuronal fragments (Fig. 3r).
Somatosensory neuron subtypes
To further investigate neuronal subtypes in the DRG, we extracted all clusters that expressed TuJ1, NeuN, Islet1, PGP9.5, and/or MAP2, and performed an additional round of clustering on this 637,744-cell subset. Specific subclusters were identified for removal as errant non-neurons, including putative phagocytic glia and spinal cord contamination (Extended Data Fig. 6a). After these cleanup steps, the remaining 533,488 cells were clustered again, dividing into three primary groups: TrkA+/Ret+, TrkB+, and TrkC+ (Extended Data Fig. 6b–h). These three groups were separated for a final round of subclustering to identify, as completely as possible, the somatosensory neuronal subtypes present across the DRG time course (Fig. 4a–d, Extended Data Fig. 7a–v). From E11.5 to P4, we identified 41 neuronal cell types and/or cell states, each molecularly distinguished by at least one of the 41 markers measured, and include transient intermediates as well as all known somatosensory neuron subtypes such as mechano-noxious heat peptidergic neurons (PEP), itch-mechano-heat non-peptidergic neurons (NP), cold-sensing neurons (which remain as a subpopulation within NP at P4, see Extended Data Fig. 6i), C-low-threshold mechanoreceptors (C-LTMR) (TH, see Fig. 4f–n), A-low-threshold mechanoreceptors (NF1, NF2, and NF3), and proprioceptors (NF4) (Fig. 4d and Extended Data Fig. 6b–h). These previously reported subtypes are labeled “E&E” in Fig. 4d, in reference to the review by Emery and Ernfors where they were described5.
Fig. 4. Distinct neuron subtypes emerge across development.
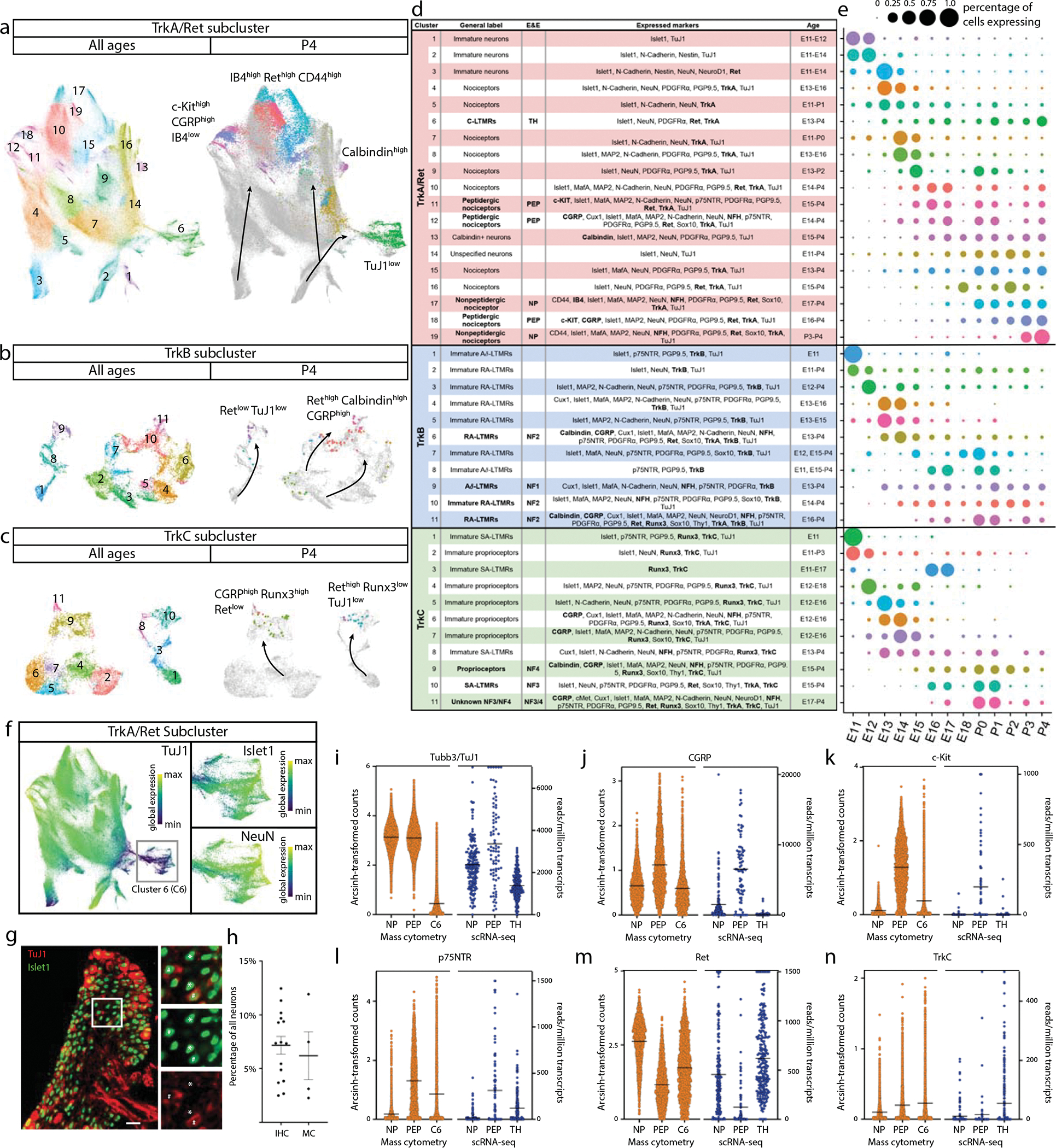
a-c) Clusters from Fig. 1c expressing only neuronal markers (Islet1, MAP2, NeuN, PGP9.5, and/or TuJ1) were extracted for a secondary round of analysis (Extended Data Fig. 6), producing three groups of somatosensory neurons characterized by TrkA/Ret, TrkB, or TrkC expression. Each group was extracted for tertiary analysis on TrkA+/Ret+ (a), TrkB+ (b), and TrkC+ (c) neurons. Cells from all ages are shown in the left UMAP, with clusters numbered by age of appearance. Black arrows denote population shifts across development from E11.5 to P4. In the right UMAP, all cells except P4 are colored gray to highlight the final time point populations. d) Table of all 41 neuron populations, listed in order of emergence. Cell types with protein expression that match designations from the recent Emery and Ernfors review are labeled5. Proteins expressed at a higher level relative to other neuronal populations are noted. Bolded proteins were used for subtype identification. The age range where each subtype was 1% or greater of all neurons in the TrkA/Ret, TrkB, or TrkC group is indicated. e) Relative abundance for neuronal subtypes, from E11.5-P4. f) TrkA/Ret UMAP from (a) colored by TuJ1 expression. Inset panels show cluster 6. g) Representative IHC of P3 DRGs stained for Islet1 (Alexafluor488, green) and TuJ1 (Alexafluor568, red) quantified in (h). Scale bar, 50 μm. Inset highlighting Islet1+ cells with corresponding high and low expression of TuJ1. Asterisk (*) denotes Islet1+;TuJ1low neurons, pound sign (#) denotes Islet1+;TuJ1+ neurons. h) Percentage of TuJ1low neurons observed by mass cytometry (MC, left) and immunohistochemistry (IHC, right) as shown in (g) at P3. For mass cytometry, two litters, sex separated were assessed (2 litters, 4 samples). For IHC, L4/L5 DRGs were stained from 5 pups, 2–3 females and males per litter, from 3 litters and counted for TuJ1 expression (15 DRGs in total). i) On the left, protein expression of TuJ1 from mass cytometry from P4 mice (in orange) for nonpeptidergic nociceptors (NP), peptidergic nociceptors (PEP), and Cluster 6 neurons (C6). On the right, gene expression of TUBB3 (TuJ1) from the scRNA-seq dataset from Usoskin et al., 2015 for the same nociceptive populations and TH+ C-LTMRs (TH) from P42-P56 mice2 (in blue).. Data are presented as mean values. j-n) Comparison of protein and mRNA transcript expression from the same datasets for CGRP, c-Kit, p75NTR, Ret, and TrkC, respectively. Data are presented as mean values.
Separately clustering 493,544 TrkA+/Ret+ neurons, 21,652 TrkB+ neurons, and 18,292 TrkC+ neurons revealed 19, 11, and 11 distinct somatosensory subtypes and/or cell states, respectively, labeled by their order of appearance during development (Fig. 4e). The TrkA+/Ret+ subtypes appear to emerge from three immature populations at E11.5 (clusters 1, 2, and 3) and diverge into four distinct subtypes: peptidergic nociceptors (c-Kit+, CGRP+, IB4low; clusters 11,12, and 18), nonpeptidergic nociceptors (IB4+, Ret+, CD44+; TrkA/Ret clusters 17 and 19), Calbindin+ neurons (TrkA/Ret cluster 13), and TH+ C-LTMRs (TuJ1low; TrkA/Ret cluster 6, see Fig. 4f–n) (Fig. 4a,d). The TrkB+ neurons separated into two distinct groups: Aδ low-threshold mechanoreceptors (Aδ-LTMR) (TrkB+, Retlow) and rapidly adapting low-threshold mechanoreceptors (RA-LTMR) (TrkB+, Ret+, Calbindin+, CGRP+). RA-LTMR clusters 2, 7, and 10 do not express the mature markers CGRP or Calbindin even at P4, indicating that these neurons do not mature until after P4 (Fig. 4b,d). TrkC+ neurons separate into two distinct groups: slowly adapting low-threshold mechanoreceptor (SA-LTMR) (TrkC+, Ret+) and proprioceptors (TrkC+, Runx3+). Interestingly, a small population of mostly postnatal TrkC+ neurons (TrkC cluster 11) express both Ret and Runx3 (Fig. 4c,d), indicating that these cells may undergo a period of cell fate plasticity during innervation of their final tissue targets, between SA-LTMR and proprioceptors.
We were initially surprised to find neuronal clusters with low TuJ1 expression, as TuJ1 is a widely accepted marker of somatosensory neurons (Fig. 4a–c)53–55. TrkB+ and TrkC+ TuJ1low clusters were identified by their differential expression of Ret and other key markers (Aδ-LTMR - TrkB+, Retlow, Calbindin−, CGRP−; SA-LTMR - TrkC+, Ret+, Runx3−). However, no markers were uniquely expressed in the TrkA+/Ret+ TuJ1low cluster 6 (Fig. 4f and Extended Data Fig. 6j–m). We verified that TuJ1low neurons exist in P3 L4/L5 DRG cryosections stained with TuJ1 and Islet1. These two neuronal markers were coexpressed in the majority of cells examined (Fig. 4g), but 7.16% (±0.8% SEM) of the Islet1+ cells had faint or no TuJ1 signal, compared to 6.2% (±1.9% SEM) by mass cytometry (Fig. 4h). In a publicly available scRNA-seq dataset on DRG neurons2, we identified a single TuJ1low population, C-LTMRs (Fig. 4i), which exhibited a similar expression pattern (CGRP, c-Kit, p75NTR, Ret, and TrkC) to our TrkA+/Ret+ TuJ1low cluster 6 (Fig. 4j–n and Extended Data Fig. 6n), and therefore, we labeled cluster 6 as TH+ C-LTMRs.
Developmental trajectories of somatosensory neurons
The TrkA+/Ret+ and TrkB+/TrkC+ lineages are thought to be distinct as separate migration waves of specific progenitors, with a common progenitor in the neural crest1. Because this common progenitor is present prior to our earliest collection, we performed URD pseudotime analysis48 on each lineage separately, with unique roots for TrkA+/Ret+ (TrkA/Ret clusters 1, 2, and 3 at E11.5) vs. TrkB+/TrkC+ (TrkB clusters 1 and 2, TrkC clusters 1, 2, and 3, all at E11.5)(Fig. 4a–d and Extended Data Fig. 8a–f). Viewed together, these trajectories represent the maturation of 14 somatosensory cell types: mechanoreceptors (segments 1,4,7,9,14, and 17), proprioceptors (segments 10, 20, and 23), Th+ C-LTRMs (segment 6) peptidergic nociceptors (segments 11, 13, 15, 19, and 21) and non-peptidergic nociceptors (segments 12, 16, 18, and 22) (Fig. 5a and Extended Data Fig. 8e–f). As expected, these somatosensory subtype trajectories are distinguished by Trk paralog expression, as well as markers such as Ret, IB4, CGRP, and c-Kit. (Fig. 5b and Extended Data Fig. 8g–t).
Fig. 5. Pseudotime analysis of neuronal differentiation.
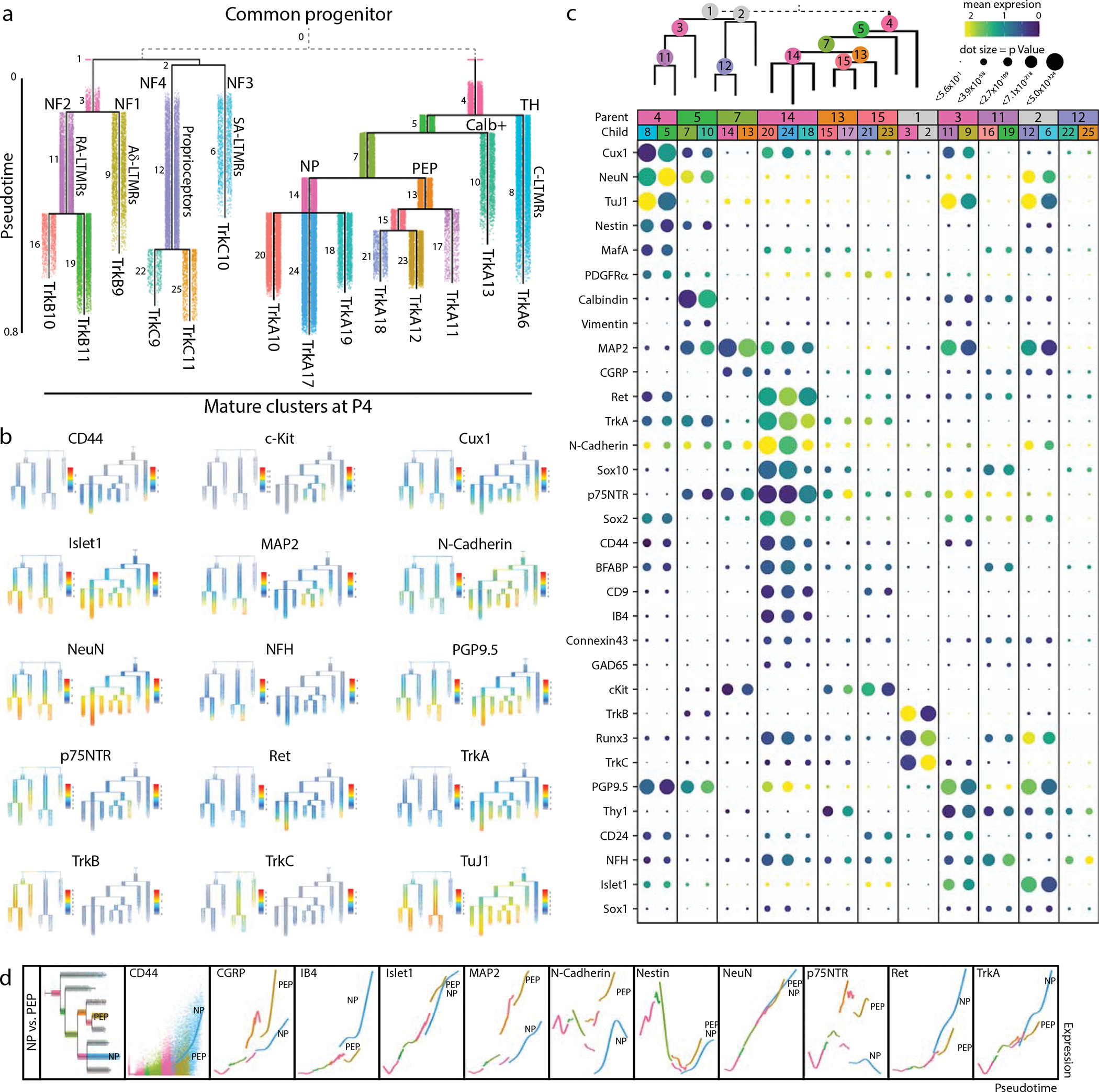
a) URD pseudotime analysis of TrkB/TrkC neurons (39,944 cells) and TrkA/Ret neurons (64,997 cells downsampled from 493,544 total). These were run separately (gray dashed line), each with all E11.5 cells designated as root, and all mature P4 clusters designated as tips (Extended Data Fig. 8). Cells are colored by their dendrogram segment, which are numbered by median pseudotime, from youngest to oldest. Key cell fate bifurcations are labeled at branch points. b) URD dendrograms from (a) colored by protein expression for key markers of somatosensory neurons and subtypes. c) Proteins with the highest URD branch point divergence, ranked by p Value of the two-sample t-test between protein expression in the cell populations of each child segment. For branch points with more than two child segments, pairwise comparisons were made, and the most highly divergent p Value was used for rank ordering. Circle size indicates these p Values (log transformed) and circle color indicates marker expression in each child segment. d) Trajectory plots showing protein expression by pseudotime for nonpeptidergic and peptidergic nociceptors. Each URD segment (colored in the grayed URD in the top left box in e) is plotted with all cells from that segment overlaid with a smoothed spline (e.g. CD44). For clarity the dots representing the cells on both trajectories were removed from the subsequent markers.
To investigate cell fate decisions at URD branch points, we identified the most significant differences in protein expression between child segments at each branch point in the TrkBTrkC and TrkARet URDs (Fig. 5c). As expected, TrkB and TrkC are the key markers that divide TrkB+ RA-LTMR and Aδ-LTMR from the TrkC+ SA-LTMR and proprioceptors, along with Runx3 (Fig. 5b,c). Divergence between RA-LTMR and Aδ-LTMR trajectories (from parent segment 1 into child segments 7 and 9, see Fig. 5a) corresponds to changes in expression of Thy1, Cux1, Islet1, CD24, Calbindin, p75NTR, and NFH, among others (Fig. 5b,c). NeuN, Runx3, and N-Cadherin similarly show the largest shift in expression between SA-LTMRs and proprioceptors (Fig. 5b,c). In the TrkARet URD branchpoints, the split between peptidergic and non-peptidergic nociceptors corresponded to expression differences in MAP2, N-Cadherin, p75NTR, and c-Kit, suggesting that peptidergic nociceptors mature earlier than non-peptidergic nociceptors (Fig. 5b,c).
We observed differences in the timing and expression level of neuronal maturation markers across both pseudotime dendrograms. For example, MAP2 increases across all somatosensory populations except for the presumptive non-peptidergic nociceptors, indicating that maturation for this subtype is delayed until after P4 (Fig. 5b,c). The TNFR family member p75NTR, a pro-growth signal in somatosensory development, is expressed broadly in both TrkB+ and TrkC+ types, but only strongly in a single TrkA+ branch, a presumptive peptidergic (CGRP+,c-Kit+) cell type (Fig. 5b,c)47. CD44 signaling is involved in nociception and we observe early expression of CD44 in non-peptidergic nociceptors as well as the TuJ1low TH cluster56 (Fig. 5b,c). Direct comparison of the peptidergic (PEP) and nonpeptidergic (NP) trajectories revealed an increase in CGRP, c-Kit, MAP2, N-Cadherin, and p75NTR for PEP, and elevated CD44, IB4, Ret and TrkA for NP (Fig. 5d).
Multi-Trk+ neurons with elevated pro-growth and stem markers
Since Trk receptors delineate somatosensory neuron subtypes and link a neuron’s survival to proper innervation of targets1, we investigated whether neurons could transiently express two or more Trks simultaneously before committing to a specific cell fate. This analysis revealed a small pool of multi-Trk+ neurons that express two Trk receptors simultaneously (TrkA+;TrkB+, TrkA+;TrkC+, or TrkB+;TrkC+, respectively) (Extended Data Fig. 9a–c). We also identified multi-Trk+ neurons in publicly available scRNA-seq datasets2,19 (Extended Data Fig. 9d,e), although these were not highlighted or commented on in the publication of these datasets.
The abundance of multi-Trk+ neurons begins to increase at E15.5 and peaks at P0 (Fig. 6a,c, Extended Data Fig. 9f). To confirm the presence of these multi-Trk+ neurons, we performed immunohistochemical analysis before, during, and after this peak, at E15.5, P0, and P4, respectively (Fig. 6b,d,e). This analysis suggests that P0 is a peak of abundance for all 3 types of multi-Trk+ neurons within this time course (Fig. 6a–e).
Fig. 6. Elevated expression of stem cell and pro-growth markers in multi-Trk+ neurons.
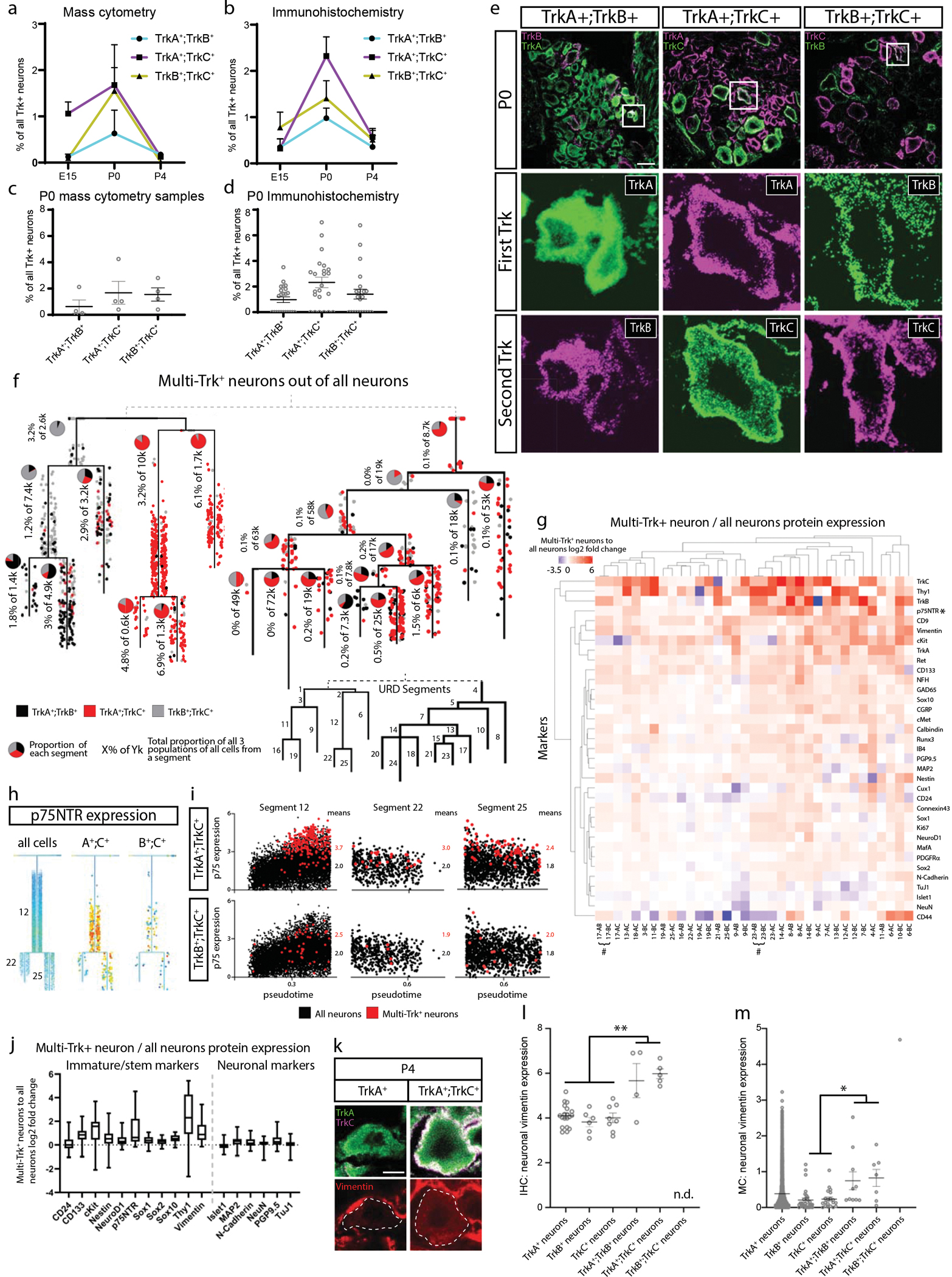
a) Proportion of multi-Trk+ neurons out of all Trk+ neurons (determined by thresholding expression values; TrkA>2, TrkB>3, and TrkC>2) at E15.5, P0, and P4 by mass cytometry. Data are presented as mean values +/− SEM with sample size as in Fig. 1g for the relevant age. b) Proportion of multi-Trk+ neurons out of all Trk+ neurons at E15.5, P0, and P4 by IHC of individual L4/L5 DRG cryosections. Data are presented as mean values +/− SEM with 3 litters with multiple DRGs analyzed per age for a total of 10, 23, and 14 DRGs at E15.5, P0, and P4, respectively. c) Relative proportion of each multi-Trk population in each P0 mass cytometry sample from (a). Data are presented as mean values +/− SEM with sample size as in Fig. 1g for only P0. d) Relative proportion of each multi-Trk population in individual L4/L5 DRG cryosections. Data are presented as mean values +/− SEM with 3 litters with 7–8 DRGs analyzed per litter, 23 DRGs in total. e) Representative IHC images of P0 L4/L5 DRGs with anti-TrkA, anti-TrkB, and anti-TrkC quantified in (b,d). Insets show a multi-Trk+ expressing neuron. Scale bar, 100 μm. f) All 1,480 multi-Trk+ neurons mapped onto the URD dendrogram from Fig. 5a by Nearest Neighbor URD Trajectory Tool (NNUTT) (Methods). In each segment, the relative proportions of each type of multi-Trk+ cells are indicated by pie charts, and the total proportion for all multi-Trk+ neurons relative to total neurons is indicated as a percentage. g) Log2 fold-change of protein expression in the multi-Trk+ neurons vs. total neurons in each URD dendrogram segment (Fig. 5a). Pound sign (#) denotes paired segments where all multi-Trk+ neurons were, in fact, tripleTrk+ neurons producing identical expression profiles in both segments. h) URD dendrograms of the proprioceptor population colored by p75NTR expression for the full dataset and two multi-Trk+ expressing neuron types. i) Every cell in segments 12, 22, and 25, respectively, plotted by pseudotime value and p75NTR expression. All neurons colored in black with the multi-Trk+ neurons are overlaid in red. j) Log2 fold change multi-Trk+ to all neuron comparison of marker expressions for immature/stem markers (left) and for neuronal markers (right) for all segments. Data are presented as mean values with the minimum value shown as the lower whisker, maximal value shown as the upper whisker, and the box bottom and top defines the first and third quartiles, respectively. Data are the marker expression comparisons for each URD segment, thus n=35. k) Representative immunohistochemistry of P4 L4/L5 DRG with anti-TrkA, anti-TrkC, and anti-vimentin. Dotted lines outline the indicated DRG neuron. Scale bar, 5μm. l) Quantification of vimentin immunohistochemical pixel intensity for single-Trk+ and multi-Trk+ neurons measured in FIJI. Three P4 litters with 3 L4/L5 DRGs (9 total) per litter were analyzed. Only 4 TrkA+;TrkB+ neurons, 5 TrkA+;TrkC+, and 0 TrkB+;TrkC+ neurons were detected in these DRGs. n.d. = “not detected”. Statistical analysis: one-way ANOVA using Tukey’s multiple comparison test and p Value **<0.005. Exact p Values are for all comparisons are in Extended Data Figure 9i. Data are presented as mean values +/− SEM. m) Quantification of vimentin expression from the mass cytometry dataset for single-Trk+ and multi-Trk+ neurons only from P4 samples. As in (a), multiTrk+ neurons were determined by thresholding expression values; TrkA>2, TrkB>3, and TrkC>2. Statistical analysis: one-way ANOVA using Tukey’s multiple comparison test and p Value *<0.05. Exact p Values are for all comparisons are in Extended Data Figure 9j. All neurons from P4 samples (n=2 biologically independent samples of pooled female and male litters) were included: 6034 TrkA+ neurons, 31 TrkB+ neurons, 20 TrkC+ neurons, 10 TrkA+;TrkB+ neurons, 7 TrkA+;TrkC+ neurons, and 1 TrkB+;TrkC+ neuron. Data are presented as mean values +/− SEM.
To assess whether the multi-Trk+ neurons were concentrated at cell fate decision points, we mapped all multi-Trk+ neurons from our analysis onto the URD dendrograms by Euclidean distance, to estimate their trajectory and pseudotime position (Fig. 6f and Extended Data Fig. 9g,h). Interestingly, the incidence of these multi-Trk+ neurons increases over pseudotime for many lineages (Fig. 6f and Extended Data Fig. 9g,h), and they exhibit characteristic shifts in the expression of pro-growth and stem cell markers when compared to single-Trk+ neurons from the same URD segment (Fig. 6g).
p75NTR, a receptor that promotes progressive developmental signaling in somatosensory neurons47,57–60, was expressed in TrkB+ and TrkC+ neurons from E11.5 to P4 and in peptidergic nociceptors, and not at all nonpeptidergic nociceptors (Fig. 4d and Extended Data Fig. 6f,h). In multi-Trk+ neurons however, expression of the pro-growth p75NTR was elevated in many URD segments, such as TrkA+;TrkC+ neurons in the proprioceptor trajectory (Fig. 6h,i). Multi-Trk+ neurons also showed increased expression of stem markers but remained largely unchanged for other marker types (Fig. 6j). IHC staining for vimentin in all three types of multi-Trk+ neurons confirms elevated vimentin compared to single-Trk+ neurons (Fig. 6k–m) These rare cells could represent a state of increased plasticity before cell fate decisions are made. Alternatively, they may be neurons with delayed cell fate, or a residual pool of immature cells that is destined to act as neural stem cells in the adult DRG.
Comparison of DRG mass cytometry with scRNA-seq
To determine the degree of correlation between our protein-based mass cytometry measurements and mRNA abundances, we compared our results to an scRNA-seq study with closely overlapping time points: E11.5, E12.5, E15.5, P0, and P519. Our antibody panel has 36 proteins with directly comparable cognate mRNAs, but the other markers are not directly comparable, such as the lectin IB4 and cleaved Caspase 3. Because mRNA and protein levels are not strictly related in a linear or predictable manner (see analysis below), we first compared the normalized mean expressions and the percentage of cells expressing these protein-mRNA cognate pairs. This comparison was performed at each matching time point, plus the close time points P4 (protein) and P5 (RNA) (Fig. 7a). In several cases, protein expression closely tracked RNA expression, but for the majority of protein-mRNA cognate pairs, there were large differences in the timing of relative trends in expression levels. For example, the transcription factors Cux1, Islet1, MafA, NeuN, Sox1, Sox2, and Sox10 all exhibit protein expression delayed by at least 5 days relative to RNA expression, and we see similar patterns for BFABP, GAD65, PGP9.5, TrkA, cMet, CD133, c-Kit, MAP2, and GFAP. In rare cases, we even see relative trends in protein expression levels occur before those for RNA, such as for Thy1 and CD31.
Fig. 7. Comparison of DRG analysis by mass cytometry and scRNA-seq.
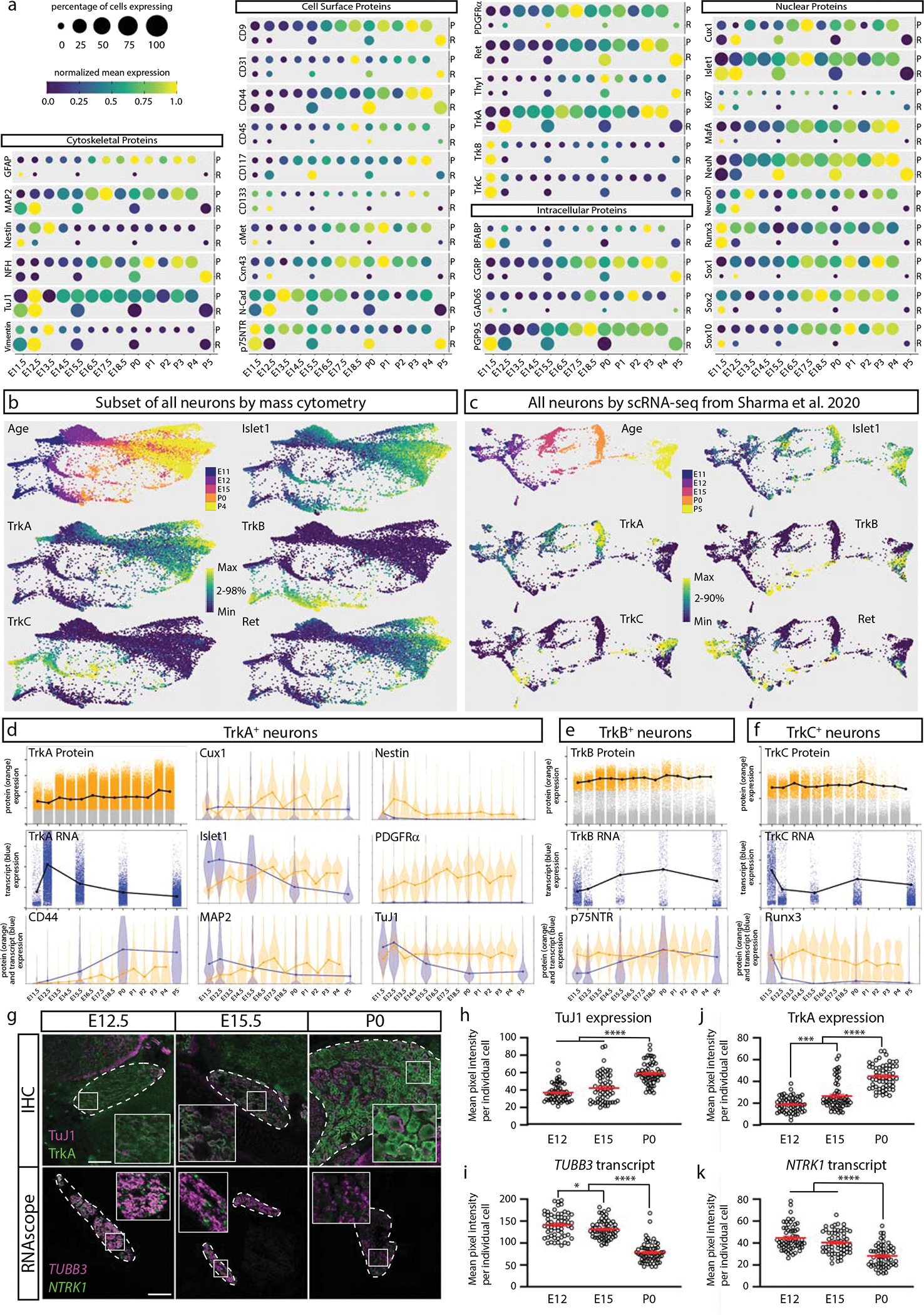
a) Protein abundance measured by mass cytometry (E11.5 to P4) and mRNA transcript abundance measured by scRNA-seq (E11.5, E12.5, E15.5, P0, P5; Sharma et al., 202019). Circle size indicates the percentage of cells with expression above marker-specific thresholds. For mass cytometry, the threshold was set at greater than the 99th percentile expression level of low complexity cells. For scRNA-seq, the threshold was set at zero; all non-zero values were included. Circle color indicates mean intensity of expression within the positive-expressing cells. Protein/mRNA pairs are grouped by protein subcellular localization. P=protein (mass cytometry measurements) and R=mRNA (Sharma et al., 2020 scRNA-seq measurements19). b,c) FLOW-MAP layouts from (a) mass cytometry (25,000 cells downsampled from all neuronal cells, Fig. 4a–c), and (b) scRNA-seq (full 32,169 cell dataset19), colored by age and marker expression level. d-f) Comparison of protein and mRNA expression levels in TrkA+, TrkB+, or TrkC+ neurons. Positive-expressing cells for each RTK were separated from non-expressers (gray) by thresholding for mass cytometry (orange) and scRNA-seq (blue) as described above. Violin plot overlays compare the normalized protein and mRNA abundance in each Trk-expressing population. g) Comparison of immunohistochemistry and RNAscope for TuJ1/TUBB3 and TrkA/NTRK1 of L4/L5 DRG at E12.5, E15.5, and P0. IHC scale bar = 100μm and RNAscope scale bar = 200μm. h-k) Quantification of the IHC (h,j) and RNAscope (i,k) in (g) for TuJ1/TUBB3 and TrkA/NTRK1. Twenty cells from 1 or more DRGs per litter with 3 litters (60 total DRG neurons) were traced per age per technique and pixel intensity measured in FIJI for each marker. Statistical analysis: one-way ANOVA using Tukey’s multiple comparison test and p Values *<0.05, ***<0.001, and ****<0.0001. Exact p Values for (h) E12 v. E15 p=0.0745, E12 v. P0 p<0.0001, E15 v. P0 p<0.0001; (i) E12 v. E15 p=0.0492, E12 v. P0 p<0.0001, E15 v. P0 p<0.0001; (j) E12 v. E15 p=0.0003, E12 v. P0 p<0.0001, E15 v. P0 p<0.0001; (k) E12 v. E15 p=0.0794, E12 v. P0 p<0.0001, E15 v. P0 p<0.0001. Data are presented as mean values +/− SEM.
We next compared mass cytometry and scRNA-seq DRG measurements across development at the population level. Due to the larger gaps in developmental time, FLOW-MAP was applied to promote connections between the most similar cells from adjacent time points61. Along with the 32,169 scRNA-seq neurons, we analyzed approximately 25,000 mass cytometry neurons, evenly downsampled to approximately 5000 cells per time point (Extended Data Fig. 10a). By mass cytometry, three distinct trajectories correspond to TrkA/Ret (top), TrkB (bottom), and TrkC (middle) (Fig 7b); and by scRNA-seq, two distinct trajectories correspond to TrkA (top) and TrkB, TrkC, and Ret (bottom) (Fig. 7c). Islet1 expression increases similarly with age along each trajectory for both datasets.
Because mRNA and protein levels are not well correlated, 1-to-1 comparison of mass cytometry and scRNA-seq cell clusters was not feasible. Instead, we used the general and widely accepted TrkA+, TrkB+, and TrkC+ populations (Extended data Fig. 10b) to compare RNA and protein expression in a more cell-type specific context.(Fig. 7d–f). As in the bulk comparison, we observe markers where RNA and protein levels track closely together, markers where RNA expression precedes the protein (Islet1 in all cell types, or CD44 in TrkA+ neurons), markers where the RNA levels swiftly diminish but the protein expression remains high (TuJ1 in all cell types, or Runx3 in TrkC+ neurons), and even markers where protein expression precedes the RNA levels (e.g. p75NTR in TrkB+ neurons). In agreement with these findings, we observed the same discrepancy between protein and mRNA levels in the two cognate pairs we selected for validation by IHC and RNAscope: TuJ1/TUBB3 and TrkA/NTRK1 (Fig. 7d).
Discussion
Seventy one neural cell types were identified in this study by their protein expression profiles, using an antibody panel with canonical markers of somatosensory development1 (Fig. 8a). Our panel provided sufficient intersectional coverage to identify cell types even when lacking canonical markers. For example C-LTMRs could be identified by low TuJ1 expression, even without their canonical marker tyrosine hydroxylase. The high-throughput nature of our dataset, with over 550,000 neurons, revealed rare neurons that coexpressed multiple Trk receptors and overexpressed stem cell markers. Comparison between our mass cytometry results and previous single-cell RNAseq data highlighted the complex relationship between transcripts and proteins. Thus, this study illustrates that mass cytometry is a high-throughput, scalable platform for developing neural tissues.
Fig. 8. Somatosensory maturation and development in the DRG.
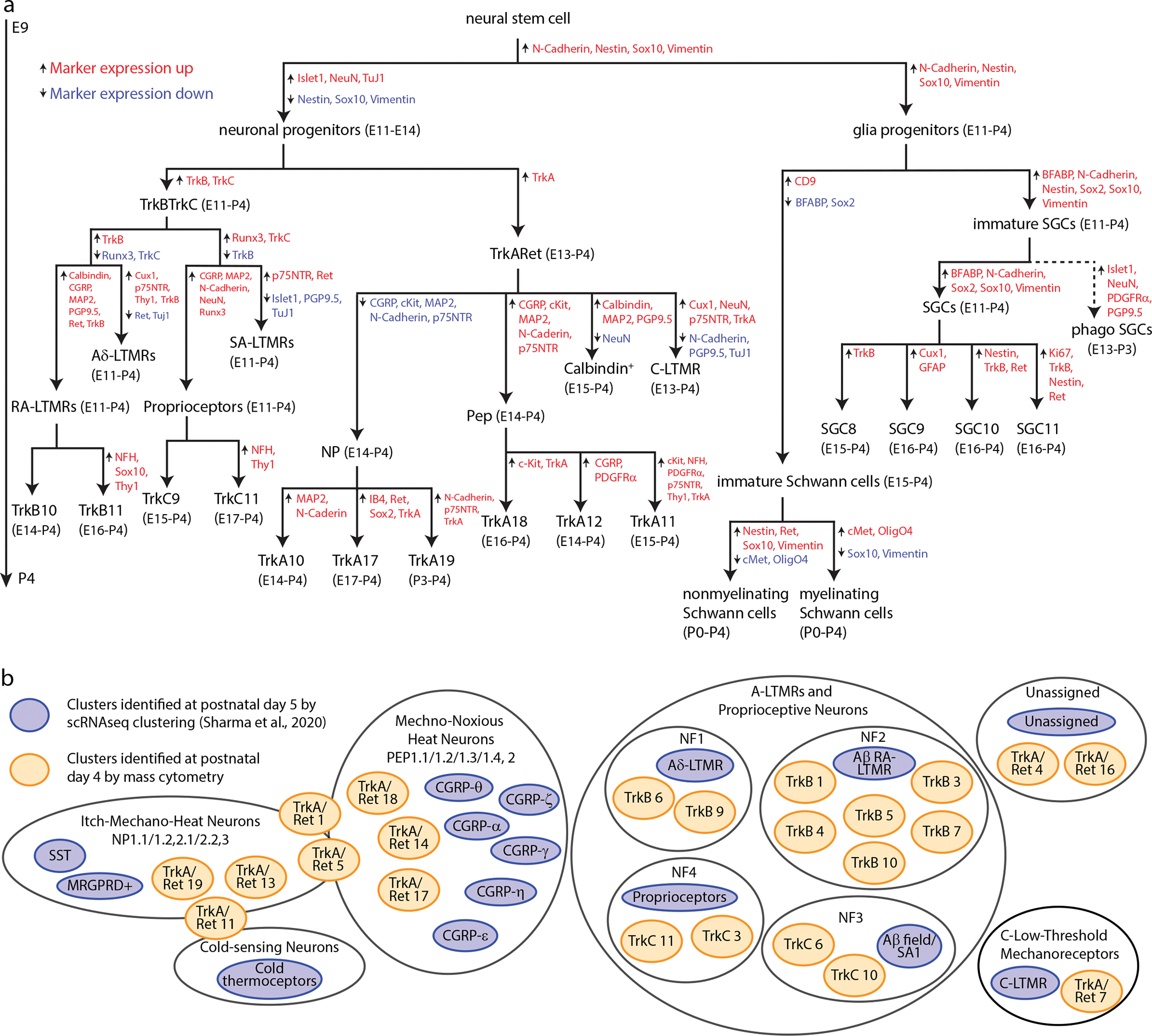
a) Expression level overlap from analysis in Fig. 7 between the cell types identified by mass cytometry at P4 (orange) and scRNA-seq at P519 (blue). b) Summary diagram of DRG cell populations identified by mass cytometry analysis. Marker expression changes between segments are indicated by upward arrows with red text denoting marker expression increases and downward arrows with blue text denoting marker expression decreases. The age range where each population comprises at least 1% of that cell type (e.g. a neuronal cell type is at least 1% of all neurons, not all cells) is indicated beside each population label.
An unexpected result of this study was the observation of putative phagocytic events, consistent with previous reports41. We were not intending to investigate this phenomenon, so did not include glial subtype markers or markers of phagocytosis such as CD6862, Jedi-1, and MEGF1041 in our antibody staining panel. Based on our gating strategy (Extended Data Fig. 2b), it is unlikely that the majority of putative phagocytic events result from cell doublets, larger aggregates, or incomplete dissociation of debris sticking to cells (Extended Data Fig. 5). Following additional validation, future studies could investigate whether glial subtypes have different patterns of phagocytic “food” over the course of development, and better characterize the molecular nature of these phagocytic events. This approach could provide additional insight into how the somatosensory nervous system is sculpted by axonal pruning and phagocytosis of whole cells and track the switch from phagocytosis by glial precursor cells to phagocytosis by macrophages later in development.
As illustrated in the discussion of putative phagocytic glia, an important caveat for single-cell analyses of neural tissue is that cell processes such as axons, dendrites, glial feet, and ensheathing membranes can be ripped away during tissue dissociation. For cell identification of neurons, the loss of pre-synaptic markers is more problematic than post-synaptic markers, because the latter can also be detected in synapses on the cell body. Many axonal and dendritic proteins are synthesized in the cell body and can be detected here too, although at lower levels. Glial process shearing can result in neurons appearing to express ensheathing membrane markers from glial cells, although neuronal identity can be confirmed by the lack of glial nuclear proteins. In this case, the wrapping membranes can be viewed as an additional level of characterization for the neuron’s phenotype and maturity.
Another caveat for single-cell analysis of neural tissue is the potential for biased loss of specific cell types during dissociation and processing. This could be due to more fragile cell types lysing during cell dissociation, or stickier cell types adhering to the inner walls of storage and processing tubes. Another potential source of biased cell loss is incomplete tissue dissociation, because the cells present in non-dissociated tissue are filtered out with a 40-micron strainer and lost to analysis. To validate the abundance levels of specific cell types, IHC can be applied to tissue slices (Fig. 2). Additionally, the metal-labeled antibodies described in this study can also be used for imaging mass cytometry (IMC)63 or multiplexed ion beam imaging (MIBI)64.
The multi-Trk+ neurons we identified in this study may represent transient cell states that can react to neurotrophic factors and switch cell fates if necessary, possibly related to previous work that showed cell state-specific differentiation outcomes from the same neurotrophin stimulus65. A signaling mass cytometry approach that combines antibodies for cell identity markers with antibodies for cell signaling molecules such as phospho-specific antibodies could facilitate a mechanistic investigation of these downstream pathways and cell states in every somatosensory cell type simultaneously, to determine how the internal cell signaling response to neurotrophins such as NGF, NT-3, BDNF, and GDNF differs between cell types and contributes to cell fate and survival decisions. Another aspect of cell state, receptor internalization, could be added to this analysis by staining with one antibody before permeabilization, and the same antibody with a different metal label after permeabilization, to investigate the distinct roles of surface vs. internalized receptors66.
Expression profiles from scRNA-seq have previously been used to define DRG cell types, but mRNA expression cannot directly predict protein expression and functional cell states. For example, translational control, protein degradation, and incomplete trafficking or internalization of surface proteins may result in high levels of mRNA but no protein present. On the other hand, mRNA degradation may result in high levels of long-lived proteins, but no mRNA remaining present. The discrepancy between RNA and protein is thought to be largest during dynamic cell transitions67,68, such as DRG development. We therefore sought to determine how well scRNA-seq could predict protein expression and functional cell states in the DRG19. In direct comparison, many protein-mRNA cognate pairs showed poor agreement across time points, highlighting the value of protein-level measurements by mass cytometry to identify functional cell states in the DRG. The mechanisms that control these observed differences are likely to be different for each protein-mRNA cognate pair, and could change considerably across developmental stages and between cell types. This could be investigated further by parallel measurement of split samples with scRNA-seq and mass cytometry, and by performing CITE-SEQ to simultaneously detect protein and RNA at the single-cell level, although this technique is limited to cell surface proteins69.
While the cell types defined by clustering of mass cytometry or scRNA-seq datasets are not expected to correspond perfectly, we were still interested to compare their similarities and differences. Because Ginty and colleagues found that early DRG time points were transcriptionally unspecialized19, we decided to focus on their P5 cell type clusters, and manually compare these with our P4 mass cytometry-based clusters. The two methods identified similar cell types, but interestingly, both methods found subpopulations not present in the other study (Fig. 8b). For example, while the scRNA-seq identified six CGRP subtypes, mass cytometry discerned at least three. Alternatively, while scRNA-seq identified a single cluster of Aβ RA-LTMRs, mass cytometry identified six molecularly distinct Aβ RA-LTMRs at P4. These similar (yet complementary) results were obtained by mass cytometry with a panel of just 41 markers, compared to the transcriptome-level measurements by scRNA-seq. While proper antibody selection and panel design is critical for successful identification of specific cell types by mass cytometry, these results suggest that the relatively limited number of measurement parameters is not an impediment to deep cell profiling, while the high sample throughput of mass cytometry facilitates replicate analysis and characterization of rare populations such as the multi-Trks.
The neural mass cytometry platform developed in this manuscript and the resulting cell atlas of somatosensory development will facilitate future mechanistic studies to characterize the effects of specific genetic or pharmacological perturbations on DRG development. Additionally, further optimization of tissue dissection and cell dissociation techniques for adult DRG tissues could expand our roadmap of embryonic and postnatal development to include DRG maturation and adult states, an important piece of the puzzle for studies linking animal behavior to cell population-level changes. Collectively, our mass cytometry analyses of the developing DRG demonstrate replicable ground truths associated with somatosensory development and provide a platform for future studies to ask fundamental developmental questions with enhanced speed and resolution.
Methods
Animals
All animal experiments were carried out in compliance with policies of the Association for Assessment of Laboratory Animal Care and approved by the University of Virginia Animal Care and Use Committee (Deppmann protocol no. 3795). Mice aged embryonic day 11.5 (E11.5) to postnatal day 4 (P4) were harvested from C57/BL6 females (Jackson Labs, 000664) bred in house. For timed pregnancies, animals were mated overnight and separated after 16 hours. Animals were housed on a 12-hour light/dark cycle with food and water ad libitum at 21°C with 45–50% humidity. For embryonic time points, pregnant females from single overnight (harem set up between 5–6pm and split between 7–8am the following morning) timed matings were used to ensure accurate embryo age.
Validation of antibodies
After conjugation to a specific metal isotope, each antibody was titrated using a variety of cell samples and counterstains. Antibodies that generated signal in DNA intercalator-positive cells that also correlated with one or more positive counterstains, but were absent in cells with a negative counterstain, were considered to be specific and reliable. Optimal concentrations for the discernment of relative protein expression were determined by titration to identify the concentration with the greatest separation between signal in positive controls compared to signal in negative controls while minimizing background. Optimal staining concentrations for each antibody are listed in Supplementary Table 1.
Metal conjugation of antibodies
Purified antibodies were conjugated to metals (listed in Supplementary Table 1) for mass cytometry analysis using MaxPAR antibody conjugation kits (Fluidigm) according to the manufacturer’s instructions. After labeling, antibodies were diluted at least 1:2 to a final concentration ranging from 0.05–0.4 mg/mL in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH) for long-term storage at 4°C.
Dissection
Spinal cords were removed from mice aged embryonic day 11.5 (E11.5) to postnatal day 4 (P4) and placed in 35-mm Petri dishes containing Dulbecco’s phosphate-buffered saline (PBS; Thermo Fisher Scientific, 14190) on ice. DRG were plucked either off the isolated spinal cord (E11.5-E15.5) or from within the ossified vertebrae (E16.5-P4), depending on age. All DRG were collected upon dissection, including the sacral, lumbar, thoracic, and cervical ganglia. Total numbers of animals and cells analyzed are listed in Supplementary Table 2.
Single-cell dissociation
After dissection, all the DRG from a whole litter (6+ pups) are transferred to a 15 ml conical filled with cold DMEM/F12. Excess media is then removed, followed by the addition of 5 ml of Enzyme Solution 1. For tissues E15.5 to P4, Enzyme Solution 1 is composed of 5 mg/ml bovine serum albumin (BSA; Sigma-Aldrich, A9418), 2 mg/ml Collagenase Type 2 (Worthington, LS004176), 0.2 mg/ml DNAse-I (Sigma-Aldrich, 11284932001), and 0.2 mg/ml hyaluronidase (Sigma-Aldrich, H3884) in DMEM/F12. For E11.5 to E14.5 tissues, Enzyme Solution 1 was prepared as above, and then diluted 1:10 in DMEM/F12. After 20 minutes of incubation in Enzyme Solution 1 at 37°C, this was then removed and replaced with 5 ml of Enzyme Solution 2 (Trypsin in DMEM/F12) for tissue from E15.5 to P4. Again, E11.5 to E14.5 was treated with Enzyme Solution 2 diluted 1:10 in DMEM/F12. The tissue was incubated for 15 minutes in Enzyme Solution 2 at 37°C before the solution was removed, leaving a residual volume of approximately 750 μl. Serial dissociation was performed with 4 fire polished pipettes with decreasing pore diameter, with approximately 10 triturations per pipette.
Our comparisons to IHC and scRNA-seq indicate that generally we were able to retain known cell types through our workflow and analysis. We also were not able to collect tissue older than P4 with enough efficiency and yield for mass cytometry analysis, due to the abundance of myelin and debris. Alternative dissociation methods or post-dissociation cleanup may make mass cytometry analysis possible for DRG past P4, including adult tissue.
Optimizing neural dissociation and antibody validation for mass cytometry of neural tissues
This study was begun without optimized protocols for cell dissociation of dissected samples, and without an optimized antibody panel for neural cell types. Without a working antibody panel, it was challenging to validate and optimize our dissection and cell dissociations, but without high quality dissociated samples, it was challenging to validate and optimize our antibodies. To solve this “chicken and egg” problem, we adopted a bootstrapping approach, starting with a few antibodies that worked relatively well (TuJ1, Sox2), and using these to test and optimize cell dissociation techniques for samples from various time points. These improved quality samples were then used to test more antibodies, and in this iterative manner, over many back-and-forth iterations, we were able to build up our antibody panel and improve our cell dissociation techniques to identify all the DRG cell types discussed in this study.
Cisplatin staining and fixation of cells
Final cell suspensions were mixed with 100 μl of 2× cisplatin solution (10 μM in PBS; Sigma Aldrich, P4394) and incubated for 30 sec before quenching with 1.3 ml of PBS containing 0.5% BSA. After centrifugation at 300 × g for 3 minutes at 4°C, resulting cell pellets were washed once with PBS with 0.5% BSA before fixation in 1 ml of 1.6% paraformaldehyde solution (Electron Microscopy Services, CAS 30525–89-4) in PBS for 10 minutes at room temperature. After centrifugation at 600 × g for 3 minutes at 4°C, cells were washed once with PBS before final resuspension in 1 ml of cell staining medium (CSM; 0.5% BSA, 0.02% NaN3 in PBS) and then the cell suspension was passed through a 75 μm sieve and 45 μm sieve (Thermo Fisher Scientific, 50871316 and 50871319) with a P1000 micropipette. Then the cells were stored at −80°C until all samples were collected.
Cell counts and visual inspection by light microscopy
Fixed cells were thawed and then visually inspected in bright field mode at 4X, 10X, and 20X on an EVOS AMF4300 microscope (Thermo Fisher Scientific, Waltham, MA) to determine if excessive amounts of cell clumping or debris were present in samples. Samples that passed visual inspection were counted using a Bio-Rad TC20 Automated Cell Counter (Hercules, CA) to determine cell number (Supplementary Table 2).
Sample barcoding, staining, and intercalation
Cells were thawed on ice, pelleted by centrifugation at 600 × g for 3 minutes at 4°C, and the supernatant was discarded. After washing once with CSM, cells were resuspended in 0.5 ml of cold saponin solution (0.02% in PBS) containing one of 20 specific combinations of 1 mM isothiocyanobenzyl EDTA-chelated palladium metals to barcode samples, as previously described34,36. After incubation at room temperature for 15 minutes on a shaker at 800 rpm, tubes were centrifuged at 600 × g for 3 minutes at 4°C, the supernatant was discarded, and the cell pellet was washed three times with CSM. At this point, individual samples were pooled into a total of three barcoded sets for antibody staining.
For staining of surface epitopes, cells were blocked in CSM containing 10% (v/v) normal donkey serum (Millipore, S30–100ML) for 30 minutes at room temperature. Next, primary antibodies indicated as “surface” in Supplementary Table 1 were diluted in CSM and added to cells (100 μl staining volume per 1 × 106 cells), which were incubated on a shaker at 800 rpm for 30 minutes at room temperature. After incubation, tubes were centrifuged at 600 × g for 3 minutes at 4°C, the supernatant was discarded, and the cell pellet was washed three times with CSM. For intracellular staining, cells were permeabilized by adding ice-cold 100% methanol to fill the tube and incubating on ice for 10 minutes with vortexing every 2 minutes. Next, tubes were centrifuged at 600 × g for 3 minutes at 4°C, and the supernatant was discarded. After washing cells once with CSM, primary antibodies listed as “intracellular” in Supplementary Table 1 were diluted in CSM and added to cells on a shaker at 800 rpm for 1 hour at room temperature. After incubation, tubes were centrifuged at 600 × g for 3 minutes at 4°C, the supernatant was discarded, and cells were washed three times with CSM.
After primary antibody staining, cells were incubated in 1.6% PFA containing 0.1 μM Cell-ID Intercalator-Ir (201192, Fluidigm) for 15 minutes at room temperature on a shaker at 800 rpm, or overnight at 4°C without shaking. After intercalation, cells were washed once with CSM, once with water, once with 0.05% Tween-20 (in water), and again with water. Cells were pelleted by centrifugation at 600 × g for 3 minutes at 4°C, then kept on ice until run on the mass cytometer.
Mass cytometry
Immediately before analysis on a Helios CyTOF 2 System (Fluidigm Corporation), cells were resuspended in water (approximately 1 mL per 1 × 106 cells) containing 1:20 EQ Four Element Calibration Beads (Fluidigm) and passed through a 40-μm nylon mesh filter. Cells were analyzed in multiple runs at a rate of 500 cells per second or less. Data was collected on a Fluidigm CyTOF 2 - Helios using CyTOF Software version 6.7.1014.
Normalization and debarcoding
To control for variations in signal sensitivity across individual runs on the mass cytometer, raw .FCS data files were first normalized using EQ Four Element Calibration Beads (https://github.com/nolanlab/bead-normalization)35. Next, normalized .FCS files from each run were concatenated for each sample set. Concatenated .FCS files were then debarcoded using software to deconvolute the 6-choose-3 Pd combinatorial barcode34,36, permitting identification of individual samples (https://github.com/zunderlab/single-cell-debarcoder)36. A new parameter was added to the FCS files: bc_neg, which is the sum of the 3 Pd measurements expected to be zero based on the cell barcode deconvolution assignment. High values for this bc_neg parameter indicate that the cell event in question is likely to contain two or more cells, and this was used for an additional clean-up gating step below.
Isolation of quality, single-cell events
To isolate single cells from fragments/debris and clumps of multiple cells, the normalized and debarcoded .FCS files described above were uploaded to Cytobank (https://community.cytobank.org) and clean-up gating was performed according to the strategy illustrated in Extended Data Fig. 2b. First, a secondary debarcoding clean-up process was performed by gating out events with a high bc_neg values, followed by gating out events with a low barcode separation distance and/or high Mahalanobis distance. Next, singlets and quality events were isolated by comparing the ion count length, center, and width parameters. Then, cells that were alive at the time of fixation were distinguished from debris and dissociation-damaged or destroyed cells with a gate using DNA-intercalator and cisplatin viability dye. Six unused channels (120Sn, 127I, 133Cs, 138Ba, 140Ce, and 208Pb) were identified in Cytobank to contain some background levels and events high in these channels were then gated out. Finally, the 4th sample set exhibited an elevated background (in nearly all channels, NeuN shown) in the last third of the runtime. These events were gated out with a time gate.
Batch correction
While sets 1 and 2 were exposed to a single master mix of antibodies, they were run independently after antibody staining. Further, set 3 was run later. To account for possible batch-dependent effects, batch correction was performed on universal samples made by combining excess cells from all samples after isolating single-cell events as described above. To correct signal intensities of individual markers for batch effects, debarcoded .FCS files were subjected to the batch adjustment process described in Schuyler et al. 2019 (https://github.com/CUHIMSR/CytofBatchAdjust)37. Specifically, signal intensities for the following antibodies were corrected at the 50th percentile because they yielded approximately Gaussian distributions and produced mean signals with variance greater than 1% for a universal sample included in each barcoded set: TuJ1, cMet, Connexin43, Sox2, CD9, CD117, Nestin, Sox1, CD24, NFH, CD133, CGRP, NeuN, Sox10, Ki67, Vimentin, Thy1.2, TrkC, Runx3, N-Cadherin, GAD65, Calbindin, MAP2, TrkA, MafA, Islet1, PDGFRa, Ret, Cux1, BFABP, Cleaved Caspase 3, TrkB, PGP9.5, p75NTR, IB4. Mean signals for OligO4, CD31, and CD45 had 2%–3% variance and normal distributions with truncated lower tails. Batch correction of CD31 and CD45 markers at 65th and OligO4 85th percentiles was determined to be effective at reducing the variance of mean signal. As the variance of mean signal for GFAP or CD44 was less than 1%, these markers were not subjected to batch correction.
Leiden clustering
Cells were partitioned with Leiden clustering (version number 0.7.0, https://github.com/vtraag/leidenalg)38 to identify molecularly defined cell types, with the nearest neighbors parameter set to 100. In some cases, this resulted in a memory error on 240GB High Mem compute nodes, and nearest neighbors = 15 was used instead. To assess if clusters were homogenous and unimodal, we inspected violin plots of marker expression for each cluster. To improve the homogeneity of cell populations, cell types of interest were subjected to multiple rounds of Leiden clustering. For the first round of clustering (primary clustering) and all subsequent rounds of clustering (secondary, tertiary, etc.), all 41 expression markers were used for Leiden clustering analysis. Cell cluster identities were annotated by comparison to previously reported expression profiles of DRG cell types.
UMAP dimensionality reduction
41-dimensional mass cytometry datasets (including all antibody markers), were embedded into 2 dimensions by uniform manifold approximation and projection (UMAP, version number 0.2.6.0, https://github.com/lmcinnes/umap/archive/0.2.4.tar.gz) with the following parameters: nearest neighbors = 15, metric = euclidean, local connectivity = 1, components = 2, epochs = 100039.
Identification of developmental cell trajectories with URD
Code for the URD algorithm (version number 1.1.1, https://github.com/farrellja/URD)48, originally designed to run on scRNA-seq datasets, was modified to interoperate with mass cytometry. Code including modified functions and a script to run the full analysis is included (see Code Availability). Datasets were downsampled proportionally (to ~65,000 cells) to accommodate the computational demands of URD except for the TrkB;TrkC dataset in Fig.7 which was smaller (39,944 cells) than the computational limits encountered. The URD parameters used were floodPseudotime n=500, minimum.cells.flooded = 2, and knn = 100. The sigma parameter was determined individually for each dataset with global auto-detection via Destiny in the URD pipeline.
NNUTT (Nearest Neighbor Urd Trajectory Tool) for analysis of rare cell populations
NNUTT was created to position a selected subset of cells on a full URD dendrogram. The code is available on GitHub at https://github.com/zunderlab/Keeler-et-al.-DRG-Development-Manuscript. The cells in this subset could either 1) be included in the approximately 60,000 cells used to generate the URD dendrogram, 2) come from the larger non-downsampled original dataset, or 3) come from a completely different dataset, measured separately. This tool simply maps each cell to its nearest neighbor in the 60,000 cell URD set by expression marker euclidean distance. Subsequent analyses can then be performed to identify which dendrogram segments are best represented by each mapped subset, and how protein expression in the mapped subsets compares to the original dendrogram cells for each segment.
All multi-Trk+ cells on the TrkB/TrkC dendrogram come from the original URD analysis (no downsampling was used for URD construction), but the majority of multi-Trk+ cells on the TrkA/Ret dendrogram were not in the original URD analysis (64,997 downsampled from 493,544 for URD construction). Segments were excluded from analysis with fewer than 10 cells and less than 0.1% proportion of multi-Trk-expressor/all neurons, or less than 5 cells regardless of proportion.
Tissue processing for IHC
Embryonic and postnatal mice were euthanized by decapitation. The lower lumbar spinal columns were dissected and fixed in 4% PFA overnight before cyroprotection in 30% sucrose in PBS for 2 days, all at 4°C. The tissue was subsequently embedded in OCT (VWR #25608–930) and then cryosectioned into 10-μm sections. The DRG from lower lumbar segments were analyzed at E11.5 and E12.5, immediately above the lower limb buds. The L4 DRG was analyzed at E13.5, E14.5, and P0, using the last rib as a landmark for T13.
Immunostaining
Mounted sections were warmed to room temperature and washed with 1× PBS three times for 5 minutes each. Antigen retrieval was performed for all antibodies by microwave boiling slides/sections in sodium citrate buffer (10 mM sodium citrate, pH 6.0). Sections were cooled to room temperature, sodium citrate buffer was replaced, and sections were microwaved until boiling again. Sections were then rinsed three times with 1× PBS and incubated with blocking solution (0.2% Triton X-100, 3% normal donkey serum) for 1 hour at room temperature. Sections were incubated with primary antibodies diluted as detailed below in blocking solution overnight at 4°C. Sections were washed with 1× PBS three times for 5 minutes each, incubated with secondary antibodies for 1 hour at room temperature protected from light, and then washed with 1× PBS three times for 5 minutes each. Sections were mounted in Fluoromount-G with DAPI (SouthernBiotech). Primary antibodies used in this study: Goat Anti-TrkA (R&D, AF1056, 1:200 or 0.2mg/mL, RRID:AB_2283049), Rabbit Anti-TrkA (Millipore-Sigma, 06–574, 1:50, RRID:AB_310180) Goat Anti-TrkB (R&D, AF1494, 1:100 or 0.2mg/mL, RRID:AB_2155264), Mouse anti-TrkB (R&D, MAB397, 1:50, RRID:AB_2298820) Goat Anti-TrkC (R&D, AF1404, 1:500 or 1:1000, RRID:AB_2155412), Mouse Anti-Beta III Tubulin (TuJ1) (Covance, MMS-435P, 1:500 or 1:1000, RRID:AB_2313773), Mouse Anti-Islet1/2 (DSHB, 39.4D5, 1:100, RRID:AB_528173), Rabbit Anti-Islet1/2 (Abcam, ab275990, 1:100, RRID:AB_10866454) Goat Anti-Ret (Neuromics, GT15002, 1:1000, RRID:AB_1622006), Rabbit Anti-Sox10 (Gift from S. Kucenas, 1:5,000), Rabbit Anti-BFABP (Gift from C. Birchmeier and T. Müller, 1:10,000, Kurtz et al., 1994), Rabbit Anti-BFABP (Abcam, ab281734, 1:250, RRID: unknown), Rat Anti-Vimentin (Biolegend, 699302, 1:250, RRID:AB_2716137). Secondary antibodies used in this study: Alexafluor 488 Donkey anti-mouse (ThermoFisher, A-21202, 1:500, RRID:AB_141607), Alexafluor 633 Donkey anti-goat (ThermoFisher, A-21082, 1:500, RRID:AB_141493), Alexafluor 568 Donkey anti-rabbit (ThermoFisher, A10042, 1:500, RRID:AB_2534017).
Cell count quantification
DRGs were sectioned into fifths (five representative sets) and each section was collected and stained with the indicated marker. All tissue was imaged on the laser scanning confocal Zeiss 780 NLO at 20× resolution in z-stacks at 3-μm intervals for manual quantification in Fiji70. Cells expressing each marker were counted and compared to counts of all cells determined from counting DAPI stained nuclei. To determine the percentage of Islet1+ and RTK+ cells observed by mass cytometry at these time points, we set a threshold value for each protein determined by the 99th percentile of expression of the low complexity cells as a measure of background: Islet1 > 0.75, TrkA > 0.9, TrkB > 3, TrkC > 2, and Ret > 1.9.
scRNA-seq data mining
Pre-processed and annotated scRNA-seq data from Sharma et al., 2020 was downloaded from the data browser provided with their publication (https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Sharma2019/all)19. As our analysis was focused on DRG development, we excluded the adult mouse timepoint (P40) from our analyses. For a per-feature comparison with our mass cytometry dataset, we selected the transcripts (from within those that had passed QC thresholds) that corresponded to our protein markers. The scRNA-seq dataset (External resource Table 1) from Usoskin et al., 2015 was downloaded from the additional supporting data for the manuscript (http://linnarssonlab.org/drg/)2.
Comparison of mRNA vs. protein expression
To compare scRNA-seq and mass cytometry, neurons from both analyses were first selected by thresholding for positive neuronal marker expression. For the scRNA-seq data, expression above zero was considered as ‘expressing’. For mass cytometry data, which typically exhibits low background for each marker similar to any antibody-based technique, thresholds for labelling cells as ‘expressing’ were determined by the 99th percentile marker expression of the low complexity cluster 6 (see Extended Data Fig. 3). For both datasets, expression values following respective preprocessing were per-feature range-normalized to fall between 0 and 1.
Visualization of high-dimensional data with FLOW-MAP
To incorporate time as a variable in inferring developmental trajectories of cell populations, graph structures incorporating the indicated time points from mass cytometry and scRNA-seq datasets were generated with FLOW-MAP26,61. FLOW-MAP output (.graphml files) was visualized with Gephi software (http://www.gephi.org)71 and force-directed layout was performed with the ForceAtlas2 algorithm72. For visualization, node size was adjusted to indicate cell type abundance, and the “prevent overlap” option was selected in Gephi to ensure all graph indices remained visible in the layout.
Population sorting and mRNA/protein comparison
For Trk+ population selection and comparison between scRNA-seq and mass cytometry datasets, both were thresholded on positive expression for TrkA, TrkB, or TrkC. For protein expression, we established TrkA, TrkB, or TrkC expression value by the 99th percentile of expression of the low complexity cells as a measure of background: TrkA > 0.9, TrkB > 3, and TrkC > 2, respectively. For RNA expression, we included any expression value above 0 for each Trk transcript. Cells were sorted in silico based on these per-marker expression thresholds to extract “positive” cells for the respective markers at each timepoint.
In situ Hybridization Assay by RNAscope
E12, E15, P0, and P4 DRG tissues were dissected from C57BL/6J mice and perfused with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) for 24 h at 4°C. Tissues were then incubated in 30% sucrose solution at 4°C for 3 days. Fixed tissues were embedded in optimal cutting temperature compound, frozen on dry ice, and stored at −80°C. Frozen tissues were cryosectioned at 15-μm thickness, collected onto positively charged slides (Shandon Superfrost Plus, Thermo Fisher Scientific, Waltham, MA; no: 6776214), and air dried overnight in the dark. The following day, slides were twice washed for 2 min in PBS, then incubated for 10 min in H2O2 (RNAscope H2O2 and Protease Reagents Kit, Advanced Cell Diagnostics, Newark, CA; no: 322381). Slides were then twice washed for 2 min with distilled water to remove H2O2, before incubation with protease IV solution (RNAscope H2O2 and Protease Reagents Kit, Advanced Cell Diagnostics; no: 322381) for 30 min at 40°C in a HybEZ II oven (Advanced Cell Diagnostics; no: 321710/321720), and two further 2 min washes in distilled water. Tissues were then incubated in probe master mix for 2 h at 40°C (Probe1: Mm-Ntrk1, no: 435791; Probe2: Mm-TUBB3-C2, no: 423391-C2; Advanced Cell Diagnostics), and then washed twice in 1 x wash buffer (RNAscope Wash Buffer Reagents, Advanced Cell Diagnostics; no: 310091). Following amplification by sequential incubations with AMP1, 2, and 3 solutions (RNAscope Multiplex Fluorescent Detection Kit v2, Advanced Cell Diagnostics; no: 323110) for 30 min at 40°C (separated with two, 2 min, washes with wash buffer between solutions), slides were incubated for 15 min at 40°C with horseradish peroxidase (HRP)-Channel 1 (HRP-C1) (RNAscope Multiplex Fluorescent Detection Kit v2, Advanced Cell Diagnostics; no: 323110), before a further two washes for 2 min in wash buffer. Slides were then incubated in a fluorescent dye for 30 min at 40°C (1:750 dilution; Tyramide Signal Amplification (TSA) Cyanine 3, Akoya, Marlborough MA; no: TS000202) before two 2 min washes with wash buffer and blocking with HRP blocker for 15 min at 40°C. The same HRP steps were repeated for channel 2 by applying a second fluorescent dye (1:750 dilution; TSA Fluorescein, Akoya; no: TS000200). Finally, 10–20 ml DAPI was applied at room temperature to stain the nuclei (Fluoromount-G, SouthernBiotech, Birmingham, AL; no: 0100–20), and then slides were sealed with coverslips.
Statistics & Reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. Data distribution was assumed to be normal but this was not formally tested.
Data availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by Eli Zunder (ezunder@virginia.edu). The unprocessed single-cell mass cytometry datasets are accessible at FlowRepository (https://flowrepository.org/) [ID: FR-FCM-Z4S9]. The debarcoded sample FCS files and clean-up gates used for pre-processing are available at Cytobank (https://community.cytobank.org/cytobank/experiments/102249). Pre-processed and annotated scRNA-seq data from Sharma et al., 2020 are available at https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Sharma2019/all19. The scRNA-seq dataset (External resource Table 1) from Usoskin et al., 2015 are available in the additional supporting data for the manuscript (http://linnarssonlab.org/drg/)2.
Code availability
The code used to perform analysis of mass cytometry data was adapted from standard R and Python packages, including UMAP, LeidenAlg, and URD. The code is available on GitHub at https://github.com/zunderlab/Keeler-et-al.-DRG-Development-Manuscript. More detailed information is available upon request.
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Eli Zunder (ezunder@virginia.edu).
Extended Data
Extended Data Fig. 1. Validation of antibodies for mass cytometry.

All antibodies validated and included in the DRG mass cytometry antibody panel. a) Each antibody was titrated across a range of concentrations (e.g. 9μg/ml to 0.01μg/ml). Known-positive and known-negative control cell samples were tailored for each antibody. Sometimes these were separate samples, and sometimes the known-positive and known-negative cells coexisted in a single sample, distinguishable by a separate antibody counterstain. Optimal staining concentrations for each antibody were determined by identifying the largest difference in signal intensity between known-positive and known-negative cells. b) Biaxials scatterplots for each antibody except for anti-Islet1 (y-axis) in the panel at each age by Islet1 (x-axis), demonstrating positive and negative staining across the DRG developmental time course. Samples were selected from Set 1. For postnatal ages the female sample from Set 1 was used. Full data available in Data Availability.
Extended Data Fig. 2. Pre-processing of DRG samples for mass cytometry.
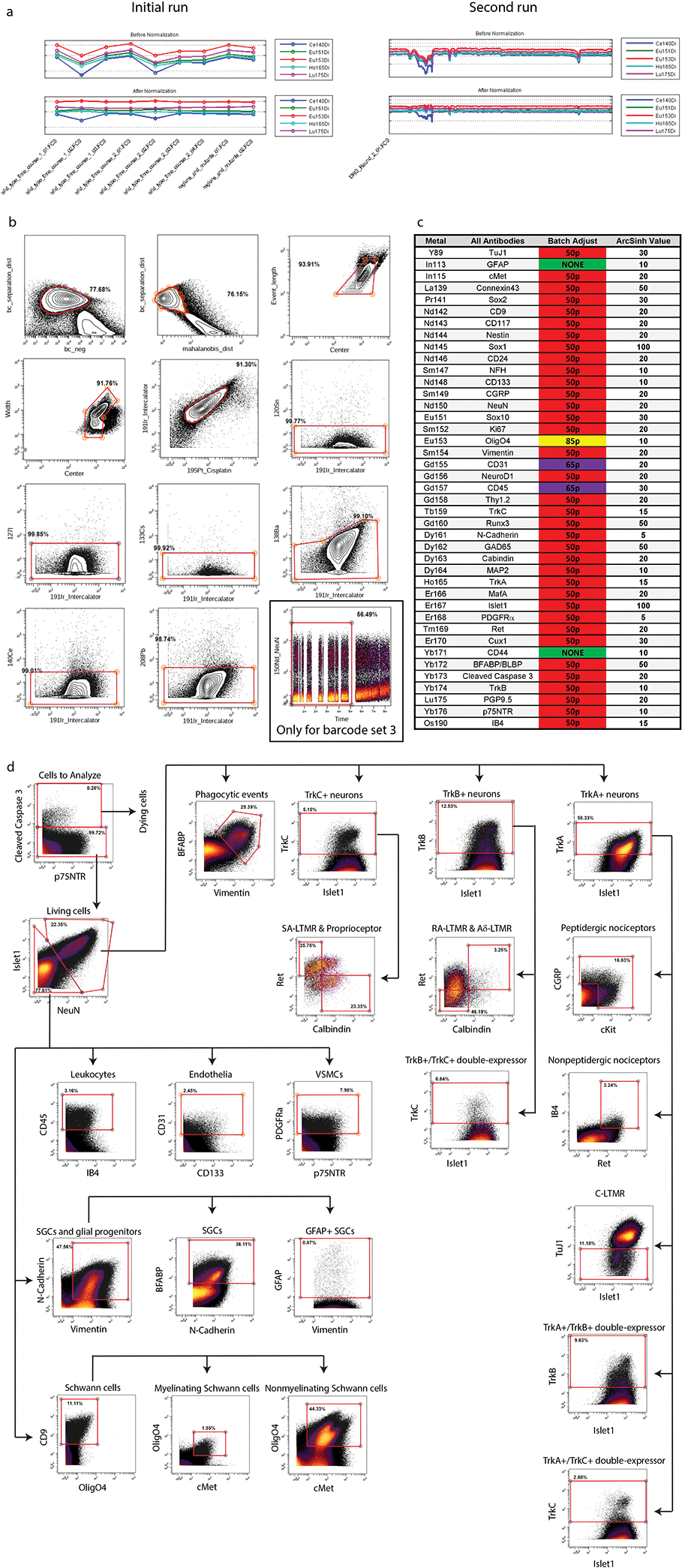
a) Calibration bead normalization of the raw mass cytometry data (stored as .fcs files) using the Matlab software described in Finck. et al., 201335. b) Clean up gating done with Cytobank (cytobank.org) to remove low quality events from the dataset. Biaxial gates as follows (from left to right, top to bottom): 1) barcode_separation x barcode_negative and 2) barcode_separation x malahoidis_distance removes events that cannot be confidently separated by barcode label; 3) event_length x center and 4) width x center remove events that fall outside of normal Gaussian parameter distribution – these events are often enriched for cell doublets; 5) intercalator x cisplatin removes both non-cell events (e.g. cellular debris) and dying cells; 6–11) unused metals x intercalator removes high background events. A twelfth clean up gate was required for samples from barcode Set 3 to remove a runtime-dependent increase in background in a subset of channels: time x NeuN. c) Batch correction was run to normalize signal strength between runs (Schuyler et al., 2019)37. Each barcode set included a “universal” sample consisting of excess samples from across the DRG time course. These excess cells were pooled together, and then aliquoted and stored at −80°C, to be included with each mass cytometry run as an unvarying control. After all samples were run, the universal samples between barcode sets were batch corrected to be as similar as possible on a per-marker basis, and then the batch adjustment process corrected the rest of the samples in that barcode set based on its corresponding universal sample. Arcsinh transformation values were manually adjusted to provide the greatest contrast between background and physiological values. d) A gating hierarchy of major populations found by high dimensional analysis of the whole time course. This gating hierarchy recapitulates the delineation of general cell types identified through high dimensional analysis with UMAP and leiden clustering.
Extended Data Fig. 3. High dimensional analysis of the entire somatosensory time course data set.
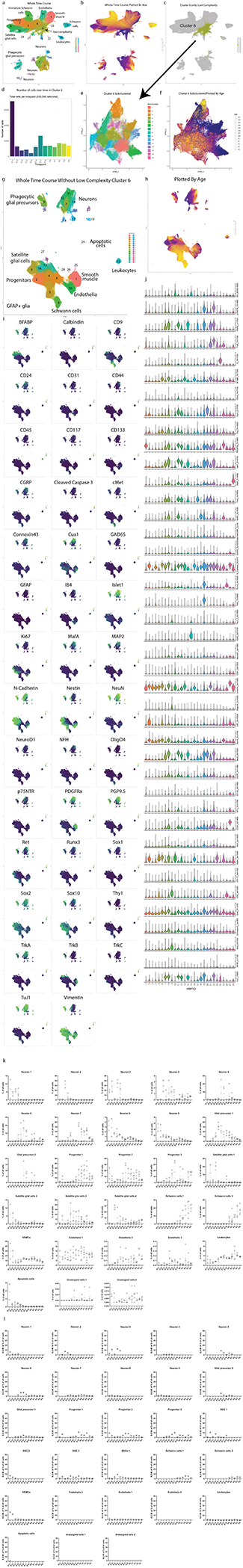
a) UMAP embedding of all ‘Cells to Analyze’ (cleanup gating applied) from the whole DRG time course. b) UMAP plot from (a) colored by age. c) UMAP plot grayed out except for low complexity Cluster 6. d) Cells from all ages in Cluster 6, with predominant contribution from E11.5. e) UMAP embedding of Cluster 6 after extraction and secondary Leiden clustering. f) UMAP plot from (e), colored by age. g) Unmodified UMAP embedding from Fig. 1c. UMAP layout was rotated and white space removed for improved visualization. h) UMAP plot from (g) colored by age. i) UMAP plot from (g) colored by expression level for every marker in the DRG antibody panel. j) Violin plots of all markers for all clusters; Fig. 1e is truncated to show just the most salient markers for the general populations. k) Analysis of the variability of each cluster by each sample at each age. The number of cells assigned to each cluster in each sample at each age was determined and the relative abundance of each cluster per sample per age calculated. Line and error bars denote the standard error of the mean. l) The standard error of the mean of the percentage of all cells for each cluster is shown, but only during the ages where the average abundance of that cluster comprised 1% or more of the cells at that age. Note, three clusters (Endothelia 3, Unassigned cells 1, and Unassigned cells 2) never reached that threshold.
Extended Data Fig. 4. Extraction of single age neuron sets for comparison to IHC.
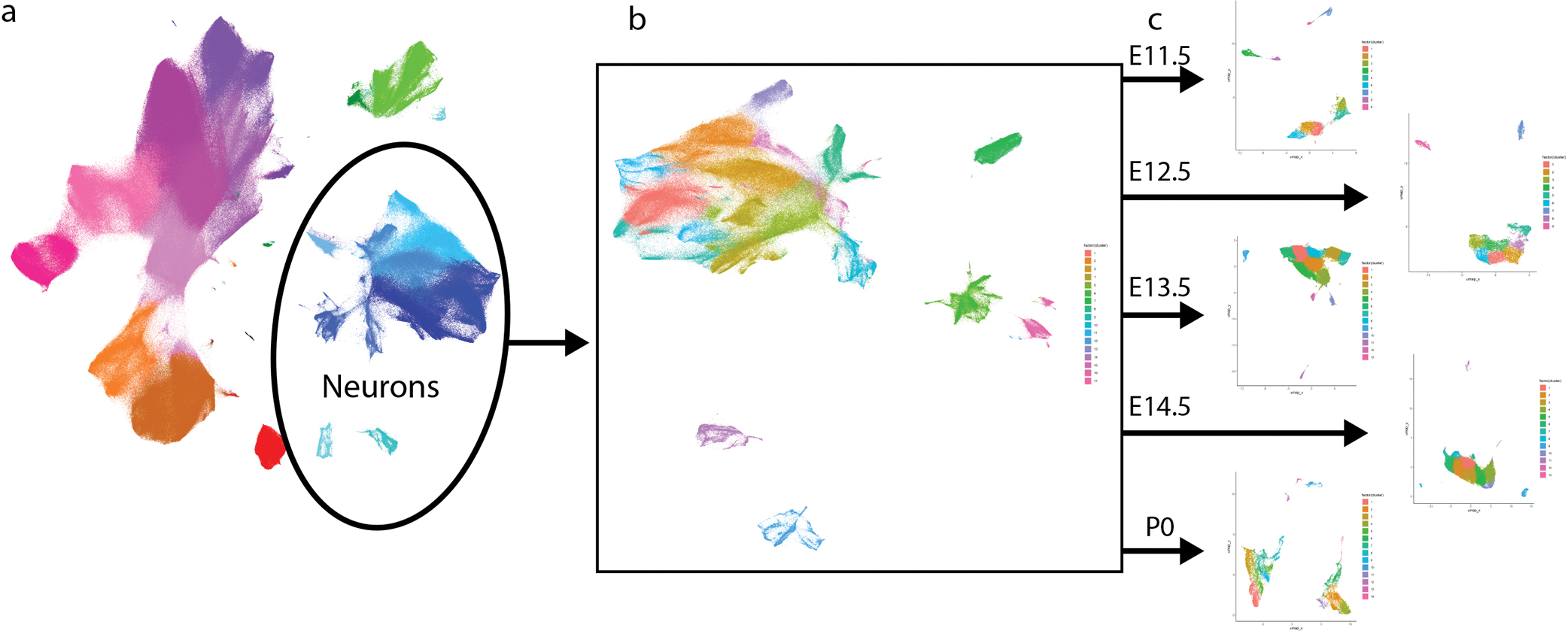
a) Neuronal clusters were extracted from the DRG time course (Fig. 1c), b) and then subjected to secondary Leiden clustering and UMAP embedding. c) Then, individual ages matched to IHC samples were further extracted and subjected to tertiary Leiden clustering and UMAP embedding.
Extended Data Fig. 5. High dimensional analysis of all glial cells and precursors.
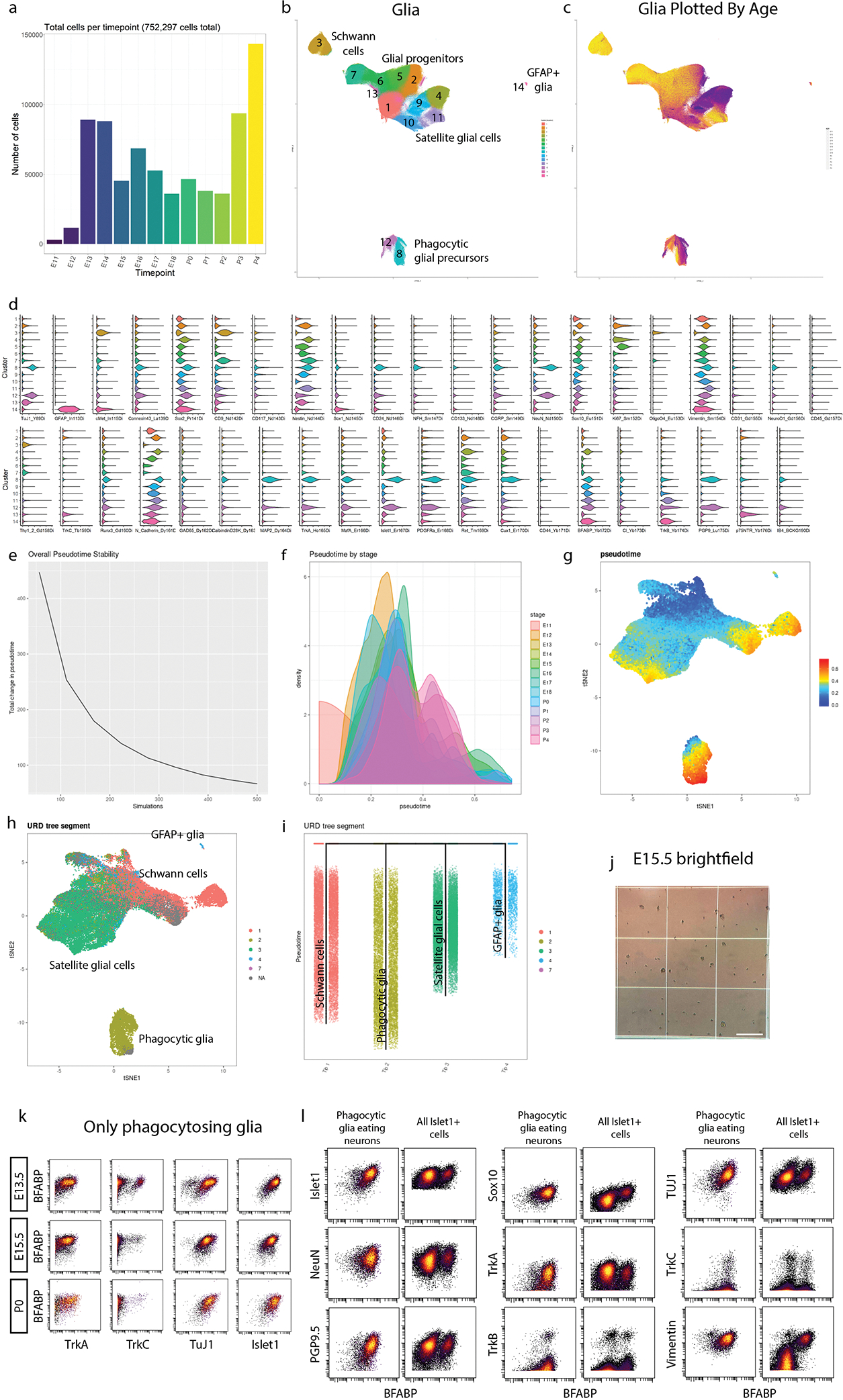
a) Cell number per age for all glia and glial precursors. b) Unmodified UMAP embedding from Fig. 3a. White space was trimmed for ease of visualization. c) UMAP plot from (b) colored by age. d) Violin plot of all markers for the glial clusters (from Fig. 3a) e) During pseudotime calculation, several simulations are run, allowing pseudotime to be calculated from these iterations. For ideal pseudotime stability (e.g. decreased change in cell pseudotime with increasing runs) we assessed the number of runs required to approach an asymptote. We determined 500 simulations was sufficient to reach a stability asymptote. f) We next assessed the distribution of pseudotime by real age (E11.5 to P4). There is a general progression across pseudotime with age with overlap between stages, as expected. g) UMAP plot colored by pseudotime value for all 63,796 downsampled cells included in this analysis. h) UMAP plot from (g) colored by URD segment. i) URD dendrogram of the 4 general populations colored by segment. j) Representative brightfield image of E15.5 sample on a hemocytometer quantified in Fig. 3p. Scale bar, 500μm.k) Biaxial scatterplots from only putative phagocytic glia showing expression of 4 neuronal markers (TrkA,TrkC, TuJ1, and Islet1), by satellite glial cell marker BFABP at E13.5, E15.5, and P0. l) Biaxial scatterplots comparing marker expression for several neuronally expressed markers (Islet1, NeuN, PGP9.5, TuJ1, TrkA, TrkB, and TrkC) and glial markers (Sox10, TrkB, and Vimentin) by BFABP between only putative phagocytic glia and all Islet1+ cells (neurons and putative phagocytic glia) from all ages.
Extended Data Fig. 6. High dimensional analysis of all neurons.

a) Leiden clustering (LC) and UMAP embedding of neurons extracted from the whole time course, labeled by cell type. Circles indicate the 3 main neuronal subtypes by RTK expression: TrkA+;Ret+, TrkB+, and TrkC+, respectively. b) LC and UMAP of the TrkA+;Ret+ neurons, extracted from a). Three clusters that did not exhibit neuronal markers were removed from the dataset before a final round of LC and UMAP (plot on right). c) LC and UMAP of the TrkB+ neurons extracted from a). d) LC and UMAP of the TrkC+ neurons extracted from a). Putative phagocytic glia expressing TrkC+ could not be removed from TrkC+ neurons in previous analytic iterations, but this could be done at this resolution resulting in a “cleaned” TrkC+ neuronal clustering and UMAP (plot on right). e) TrkA+;Ret+, TrkB+, and TrkC+ UMAP plots colored by age. f) Violin plots of all markers for TrkA+;Ret+, TrkB+, and TrkC+ neurons. g) Key markers in our panel that allow identification of somatosensory DRG populations5. h) UMAP plots colored by expression for all panel markers for TrkA+;Ret+, TrkB+, and TrkC+ neurons. i) TrpM8 transcript data from Usoskin et al., 2015 showing that TrpM8-expressing neurons are a subset of peptidergic nociceptors. j) UMAP plot of TH+ cells that were extracted from Fig. 4a (Cluster 6) and reclustered. k) UMAP plot of the TH+ cells from P4 overlaid on the grayed out UMAP plot from j). These were the cells used in the comparison to the Usoskin et al., 2015 transcript data in Fig. 4 i–n. l) UMAP plot from j) colored by age. m) Violin plot of marker expression of all clusters from j). n) Comparison of TH+ C-LTMR transcripts expression to protein expression for all markers in the mass cytometry panel2. In both cases, transcript or protein expression in C-LTMRs was normalized to all nociceptors.
Extended Data Fig. 7. Analysis of inter-replicate variability for TrkA+, TrkB+, and TrkC+ neuron subclusters.
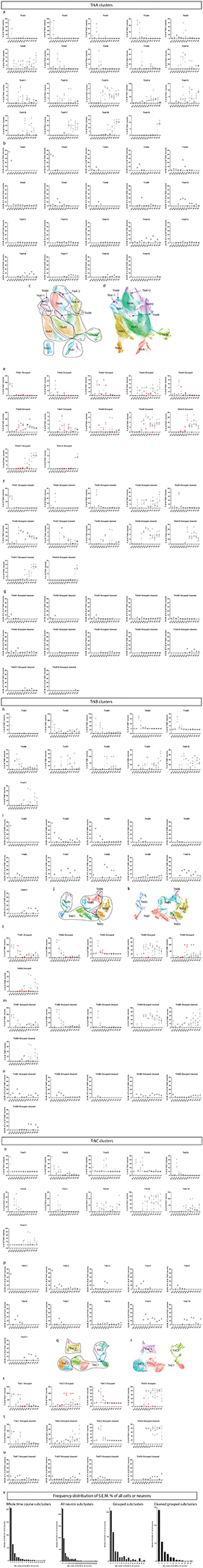
a) Analysis of the variability of each cluster by each sample for each age for the TrkA+/Ret+ neuron subcluster. The number of cells assigned to each cluster in each sample at each age was determined and the relative abundance of each compared to all neurons in the TrkA+/Ret+ cluster was calculated. b) The standard error of the mean of the percentage of all neurons from the TrkA+/Ret+ dataset for each TrkA+/Ret+ cluster is shown, but only during the ages where the average proportion of that cluster comprised 1% or more of the cells at that age. c) UMAP where TrkA+/Ret+ subclusters have been grouped together by similar expression of markers into a more general cell type. d) UMAP where TrkA+/Ret+ subclusters were recolored by general groups expressing similar markers as denoted in (c). e) The relative abundance for the general groups created in (d) are shown with the samples deemed ‘poorer quality’ (see Supplementary Table 3) indicated as a red circle. f) The relative abundance for the general groups created in (d) shown without the ‘poorer quality’ samples. g) The standard error of the mean of the percentage of all neurons from the grouped TrkA+/Ret+ dataset with the ‘poorer quality’ samples data removed for each TrkA+/Ret+ cluster is shown, but only during the ages where the average proportion of that cluster comprised 1% or more of the cells at that age. h-n) The same analysis as (a-g) except for TrkB+ neurons. (o-u) The same analysis as (a-g) except for TrkC+ neurons. v) Frequency distribution of the S.E.M % of either all cells or neurons for 1) the whole time course (from Extended Data Fig. 3k,l), 2) all neuron subclusters, 3) subclusters grouped by general cell types, and 4) these general cell types with the ‘poorer quality’ samples removed.
Extended Data Fig. 8. URD pseudotime analysis of all neurons.
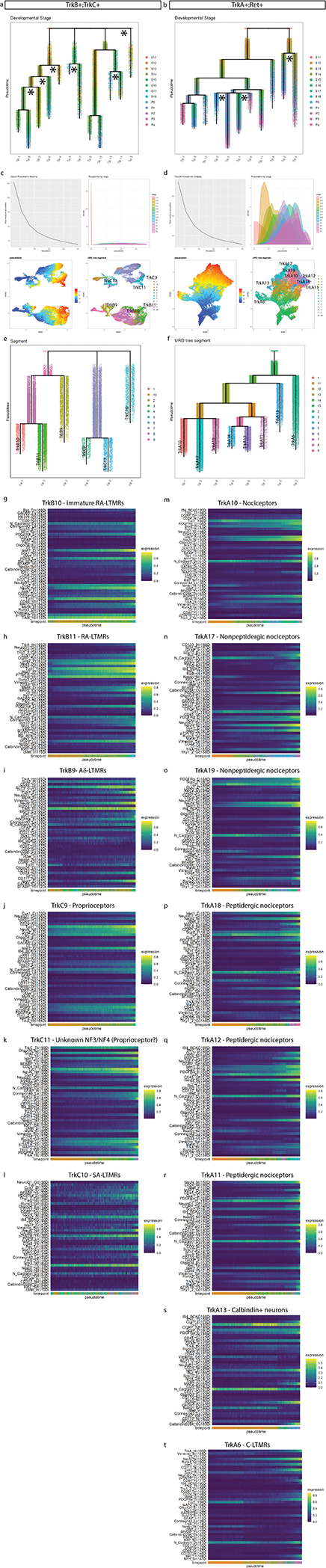
We initially defined tips as clusters present with ≥ 1% at P4 and all clusters at E11.5 as the root. However, this produced excessively branched pseudotime dendrograms for both a) TrkB+;TrkC+ and b) TrkA+;Ret+ where intermediate and more mature cell states were paired as tips. For instance, TrkA+/Ret+ clusters 5 and 13 are presumptively immature nonpeptidergic nociceptors. The presence of these immature cell types at P4 is expected as cell populations mature over development. By removing presumptive intermediates as tips, we were able to produce the most appropriate molecular trajectory across pseudotime, such as Fig.5 a. c-f) Supplemental URD analysis, same as in Extended Data Fig. 5: c,d) pseudotime stability to calculate simulation number, pseudotime by stage, UMAP of pseudotime value for all 39,944 (TrkB+;TrkC+) not downsampled and 64,997 (TrkA+/Ret+) downsampled cells included in this analysis, UMAP colored by URD segment, for TrkB+;TrkC+ and TrkA+/Ret+ datasets, respectively, and e,f) URD dendrogram colored by URD segment for TrkB+;TrkC+ and TrkA+/Ret+ datasets, respectively. g-t) Heatmaps for all URD tips from TrkB+;TrkC+ and TrkA+/Ret+ datasets, respectively, for all markers.
Extended Data Fig. 9. Multi-Trk+ neurons exhibit altered protein expression.
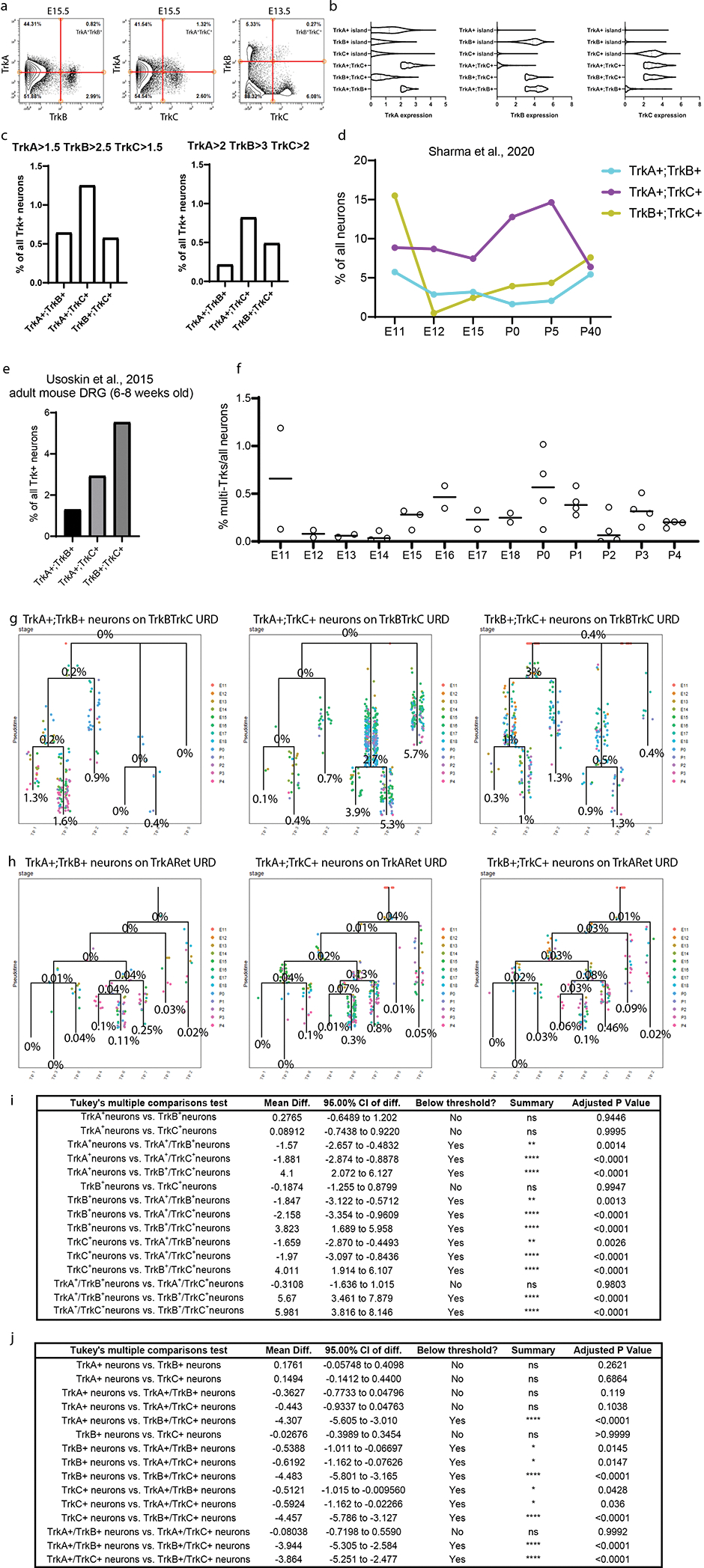
a) Biaxial scatterplots showing the neurons that express at least two Trks for all 3 combinations: TrkA;TrkB, TrkA;TrkC, and TrkB;TrkC. b) Violin plots of Trk expression for all single-Trk+ neurons (Fig. 4 a–c) compared to multi-Trk+ neurons. c) Proportion of TrkA+;TrkB+, TrkA+;TrkC+, and TrkB+;TrkC+ populations at different threshold values: left, TrkA>1.5, TrkB>2.5, TrkC>1.5 and right, TrkA>2, TrkB>3, TrkC>2. d) Proportion of TrkA+;TrkB+, TrkA+;TrkC+, and TrkB+;TrkC+ populations at E11, E12, E15, P0, P5, and P40 from Sharma et al., 2020 scRNA-seq. All cells with transcript expression values >0 were designated as expressing the given transcript. e) Proportion of TrkA+;TrkB+, TrkA+;TrkC+, and TrkB+;TrkC+ populations at P42–56 from Usoskin et al., 2015. Transcript expression was determined by thresholding as in Usoskin et al., 2015: briefly, the three cells with the highest expression for a given transcript were averaged and then multiplied by 5%. Cells with transcript expression greater than this threshold are designated as expressors. f) Proportion of all multi-Trk+ neurons combined at each age across the whole time course from the mass cytometry dataset. g,h) URD dendrograms from Fig. 5a without the URD dataset cells. Instead, each multi-Trk+ neuron type are mapped on the URDs, colored by the age of each cell, over the TrkB;TrkC URD (d) and the TrkA;Ret URD (e). Multi-Trk+ neuron numbers were counted and compared to the number of cells within each TrkB;TrkC URD segment (d). However, the TrkA+ and Ret+ neurons were downsampled (64,997 out of 492,982 neurons). Thus we multiplied the number of neurons in each segment of the TrkA;Ret URD by the downsampling coefficient (7.584689) before determining the proportion of multi-Trk+ neurons over the URD (e). i,j) All p Values for Fig. 6l and m, respectively.
Extended Data Fig. 10. Mass cytometry and scRNA-seq comparison.
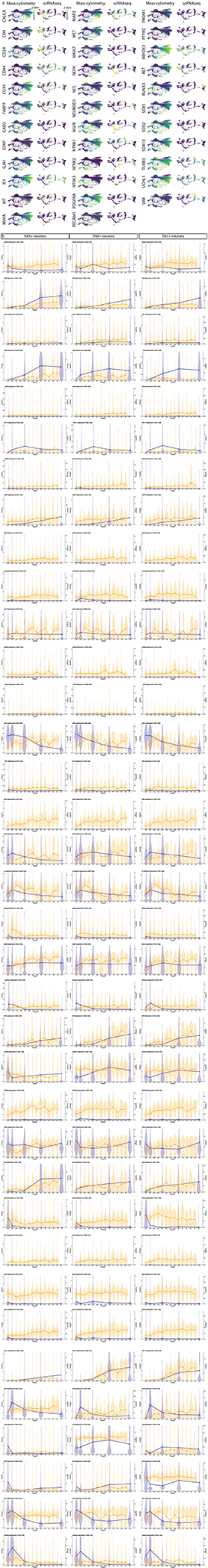
a) FLOW-MAP plots from Fig. 7b–c, colored by each protein marker in the DRG antibody panel for comparison between the mass cytometry and the scRNA-seq19. In cases where expression was not detected in the scRNA-seq dataset, the FLOW-MAPs are colored a single intermediate color (e.g. as in GAD2, GFAP, etc). Markers are named with the gene name. b) Graphed expression levels for all markers in the protein panel for TrkA+, TrkB+, and TrkC+ populations compared between mass cytometry and scRNA-seq.
Supplementary Material
Supplementary Table 1 Antibody panel tailored for dorsal root ganglia development. Each antibody or reagent used in the mass cytometry staining panel is listed with antigen, conjugated metal, manufacturer, product number, antibody clone, and concentration. Antibodies recognizing surface proteins were stained prior to methanol permeabilization in a single master mix and intracellular proteins, including nuclear proteins, were stained in a single master mix post-permeabilization, denoted by surface or intracellular stain protocol, respectively.
Supplementary Table 2 Sample data. Every sample included in this study is reported. For each sample, this includes the age of the mice, the date the cells were collected, the genotype, number of animals in the sample, sex for postnatal samples (all prenatal litters were not sex separated), the total cell count, and which mass cytometry run “set” the sample belonged to. Color indicates barcode set (blue = barcode set 1, green = barcode set 2, red = barcode set 3).
Supplementary Table 3 Efficiency across data clean-up. Data event counts for each sample across the 3 mass cytometry barcode sets for the full time course tracked from mass cytometry reads through preprocessing gating clean-up. Data events were first analyzed for the first two barcode sets, each comprising one replicate sample set of a complete time course from E11.5 to P4. These samples are first gated by barcode stringency to ensure that samples without a clear and clean barcode label are removed. Then data events outside of normal parameters for a mass cytometry read are gated out. The remaining events are gated by the viability dye cisplatin and DNA intercalator Iridium to remove damaged cells or non-single cells. Finally, empty isotope channels are gated to remove events with excessive background to produce a population of cleaned data entitled ‘Cells to Analyze’. An Islet1+ gate enabled an estimate on the percentage of neurons at each age. Using these initial numbers we determined a handful of ages to collect additional samples (barcode set 3). Barcode set 3 was also analyzed across gating clean-up.
Acknowledgments
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number F32NS103770 to A.B.K., and R01NS111220 to E.R.Z. and C.D.D.. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Further support was provided by the 3 Cavaliers Pilot research program (to C.D.D. and E.R.Z.). We thank Nikhil Sharma and David Ginty for consultation about data formatting and analysis steps from their scRNA-seq dataset. We thank Ali Güler, Amrita Pathak, Sushanth Kumar, Ekaterina Stepanova, Sonia Chandra, and Micah Hunter-Chang for feedback and critical evaluation of the data and manuscript. We thank the University of Virginia Flow Cytometry Core, RRID: SCR_017829 for technical assistance. The authors acknowledge Research Computing at The University of Virginia for providing computational resources and technical support that have contributed to the results reported within this publication. URL: https://rc.virginia.edu. We acknowledge the Keck Center for Cellular Imaging for the usage of the Zeiss 880/980 multiphoton Airy scan microscopy system (PI- AP; NIH-OD025156).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Bibliography
- 1.Lallemend F & Ernfors P Molecular interactions underlying the specification of sensory neurons. Trends Neurosci. 35, 373–381 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Usoskin D et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Li C-L et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 26, 83–102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li C, Wang S, Chen Y & Zhang X Somatosensory Neuron Typing with High-Coverage Single-Cell RNA Sequencing and Functional Analysis. Neurosci. Bull. 34, 200–207 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emery EC & Ernfors P Dorsal root ganglion neuron types and their functional specialization. in The oxford handbook of the neurobiology of pain (ed. Wood JN) (Oxford University Press, 2018). doi: 10.1093/oxfordhb/9780190860509.013.4. [DOI] [Google Scholar]
- 6.Orefice LL et al. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of asds. Cell 166, 299–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orefice LL et al. Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 178, 867–886.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Özkaya AK et al. A case of congenital insensitivity to pain with anhidrosis. Turk Pediatri Ars 49, 177–179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darmanis S et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA 112, 7285–7290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tasic B et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habib N et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiu IM et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. eLife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finno CJ et al. Single-Cell RNA-seq Reveals Profound Alterations in Mechanosensitive Dorsal Root Ganglion Neurons with Vitamin E Deficiency. iScience 21, 720–735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hockley JRF et al. Single-cell RNAseq reveals seven classes of colonic sensory neuron. Gut 68, 633–644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faure L et al. Single cell RNA sequencing identifies early diversity of sensory neurons forming via bi-potential intermediates. Nat. Commun. 11, 4175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma N et al. The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kupari J et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat. Commun. 12, 1510 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K et al. Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain. Cell Res. 31, 904–918 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H et al. Distinct subtypes of proprioceptive dorsal root ganglion neurons regulate adaptive proprioception in mice. Nat. Commun. 12, 1026 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ & Davidson S Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. eLife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandura DR et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81, 6813–6822 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Bendall SC et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zunder ER, Lujan E, Goltsev Y, Wernig M & Nolan GP A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell 16, 323–337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Behbehani GK, Bendall SC, Clutter MR, Fantl WJ & Nolan GP Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A 81, 552–566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fienberg HG, Simonds EF, Fantl WJ, Nolan GP & Bodenmiller B A platinum-based covalent viability reagent for single-cell mass cytometry. Cytometry A 81, 467–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mrdjen D et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 380–395.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Friebel E et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Ajami B et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 21, 541–551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ornatsky OI et al. Study of cell antigens and intracellular DNA by identification of element-containing labels and metallointercalators using inductively coupled plasma mass spectrometry. Anal. Chem. 80, 2539–2547 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Wheeler MA et al. TNF-α/TNFR1 signaling is required for the development and function of primary nociceptors. Neuron 82, 587–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zunder ER et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 10, 316–333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finck R et al. Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fread KI, Strickland WD, Nolan GP & Zunder ER An updated debarcoding tool for mass cytometry with cell type-specific and cell sample-specific stringency adjustment. Pac. Symp. Biocomput. 22, 588–598 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Schuyler RP et al. Minimizing batch effects in mass cytometry data. Front. Immunol. 10, 2367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv (2018). [Google Scholar]
- 40.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Wu H-H et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat. Neurosci. 12, 1534–1541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crowley C et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76, 1001–1011 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Fariñas I, Yoshida CK, Backus C & Reichardt LF Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron 17, 1065–1078 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White FA et al. Synchronous onset of NGF and TrkA survival dependence in developing dorsal root ganglia. J. Neurosci. 16, 4662–4672 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ & Snider WD Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J. Neurosci. 18, 1428–1439 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel TD, Jackman A, Rice FL, Kucera J & Snider WD Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron 25, 345–357 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Cheng I, Jin L, Rose LC & Deppmann CD Temporally restricted death and the role of p75NTR as a survival receptor in the developing sensory nervous system. Dev. Neurobiol. 78, 701–717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farrell JA et al. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Britsch S et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fariñas I, Cano-Jaimez M, Bellmunt E & Soriano M Regulation of neurogenesis by neurotrophins in developing spinal sensory ganglia. Brain Res. Bull. 57, 809–816 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Maro GS et al. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat. Neurosci. 7, 930–938 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Taylor MK, Yeager K & Morrison SJ Physiological Notch signaling promotes gliogenesis in the developing peripheral and central nervous systems. Development 134, 2435–2447 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo W et al. Developmental expression of P2X5 receptors in the mouse prenatal central and peripheral nervous systems. Purinergic Signal. 9, 239–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levin E, Andreadaki A, Gobrecht P, Bosse F & Fischer D Nociceptive DRG neurons express muscle lim protein upon axonal injury. Sci. Rep. 7, 643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avraham O et al. Satellite glial cells promote regenerative growth in sensory neurons. Nat. Commun. 11, 4891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrari LF, Khomula EV, Araldi D & Levine JD CD44 Signaling Mediates High Molecular Weight Hyaluronan-Induced Antihyperalgesia. J. Neurosci. 38, 308–321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee KF et al. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69, 737–749 (1992). [DOI] [PubMed] [Google Scholar]
- 58.Fan G, Jaenisch R & Kucera J A role for p75 receptor in neurotrophin-3 functioning during the development of limb proprioception. Neuroscience 90, 259–268 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Murray SS, Bartlett PF & Cheema SS Differential loss of spinal sensory but not motor neurons in the p75NTR knockout mouse. Neurosci. Lett. 267, 45–48 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Chen Z et al. p75 is required for the establishment of postnatal sensory neuron diversity by potentiating ret signaling. Cell Rep. 21, 707–720 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ko ME et al. FLOW-MAP: a graph-based, force-directed layout algorithm for trajectory mapping in single-cell time course datasets. Nature Protocols (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.da Silva RP & Gordon S Phagocytosis stimulates alternative glycosylation of macrosialin (mouse CD68), a macrophage-specific endosomal protein. Biochem. J. 338 ( Pt 3), 687–694 (1999). [PMC free article] [PubMed] [Google Scholar]
- 63.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo W et al. A hierarchical NGF signaling cascade controls Ret-dependent and Ret-independent events during development of nonpeptidergic DRG neurons. Neuron 54, 739–754 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Kuruvilla R et al. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118, 243–255 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, Beyer A & Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Reimegård J et al. A combined approach for single-cell mRNA and intracellular protein expression analysis. Commun. Biol. 4, 624 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bastian M, Heymann S & Jacomy M Gephi: An Open Source Software for Exploring and Manipulating Networks. Third Int. AAAI Conf. Weblogs Soc. Media 361–362 (2009). [Google Scholar]
- 72.Jacomy M, Venturini T, Heymann S & Bastian M ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 9, e98679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Antibody panel tailored for dorsal root ganglia development. Each antibody or reagent used in the mass cytometry staining panel is listed with antigen, conjugated metal, manufacturer, product number, antibody clone, and concentration. Antibodies recognizing surface proteins were stained prior to methanol permeabilization in a single master mix and intracellular proteins, including nuclear proteins, were stained in a single master mix post-permeabilization, denoted by surface or intracellular stain protocol, respectively.
Supplementary Table 2 Sample data. Every sample included in this study is reported. For each sample, this includes the age of the mice, the date the cells were collected, the genotype, number of animals in the sample, sex for postnatal samples (all prenatal litters were not sex separated), the total cell count, and which mass cytometry run “set” the sample belonged to. Color indicates barcode set (blue = barcode set 1, green = barcode set 2, red = barcode set 3).
Supplementary Table 3 Efficiency across data clean-up. Data event counts for each sample across the 3 mass cytometry barcode sets for the full time course tracked from mass cytometry reads through preprocessing gating clean-up. Data events were first analyzed for the first two barcode sets, each comprising one replicate sample set of a complete time course from E11.5 to P4. These samples are first gated by barcode stringency to ensure that samples without a clear and clean barcode label are removed. Then data events outside of normal parameters for a mass cytometry read are gated out. The remaining events are gated by the viability dye cisplatin and DNA intercalator Iridium to remove damaged cells or non-single cells. Finally, empty isotope channels are gated to remove events with excessive background to produce a population of cleaned data entitled ‘Cells to Analyze’. An Islet1+ gate enabled an estimate on the percentage of neurons at each age. Using these initial numbers we determined a handful of ages to collect additional samples (barcode set 3). Barcode set 3 was also analyzed across gating clean-up.
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by Eli Zunder (ezunder@virginia.edu). The unprocessed single-cell mass cytometry datasets are accessible at FlowRepository (https://flowrepository.org/) [ID: FR-FCM-Z4S9]. The debarcoded sample FCS files and clean-up gates used for pre-processing are available at Cytobank (https://community.cytobank.org/cytobank/experiments/102249). Pre-processed and annotated scRNA-seq data from Sharma et al., 2020 are available at https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Sharma2019/all19. The scRNA-seq dataset (External resource Table 1) from Usoskin et al., 2015 are available in the additional supporting data for the manuscript (http://linnarssonlab.org/drg/)2.