

Abstract
Efficient encapsulation and sustained release of small hydrophilic molecules from traditional hydrogel systems have been challenging due to the large mesh size of 3D networks and high water content. Furthermore, the encapsulated molecules are prone to early release from the hydrogel prior to use, resulting in a short shelf life of the formulation. Here, we present a hydration-induced void-containing hydrogel (HVH) based on hyperbranched polyglycerol-poly(propylene oxide)-hyperbranched polyglycerol (HPG-PPG-HPG) as a robust and efficient delivery system for small hydrophilic molecules. Specifically, after the HPG-PPG-HPG is incubated overnight at 4 °C in the drug solution, it is hydrated into a hydrogel containing micron-sized voids, which could encapsulate hydrophilic drugs and achieve 100% drug encapsulation efficiency. In addition, the voids are surrounded by a densely packed polymer matrix, which restricts drug transport to achieve sustained drug release. The hydrogel/drug formulation can be stored for several months without changing the drug encapsulation and release properties. HVH hydrogels are injectable due to shear thinning properties. In rats, a single injection of the HPG-PPG-HPG hydrogel containing 8 μg of tetrodotoxin (TTX) produced sciatic nerve block lasting up to 10 hours without any TTX-related systemic toxicity nor local toxicity to nerves and muscles.
Keywords: injectable hydrogel, local anesthetics, tetrodotoxin, pain treatment
Graphical Abstract
Small hydrophilic drugs can be encapsulated in hydrogels with micron-sized voids via a simple hydration process. The resulting formulation remains stable for several months. The hydrogels exhibit shear thinning properties, making them injectable. In rats, a single injection of this formulation containing 8 μg of tetrodotoxin effectively blocks the sciatic nerve for 10 hours without systemic toxicity.
1. Introduction
In recent decades, hydrogels have been widely used as drug delivery vehicles due to their excellent biocompatibility, controllable biodegradability, tunable mechanical strength and porous structure, and ability to deliver the drug to a desired location compared with other types of biomaterials[1–4]. Large biomolecules, such as proteins and peptides as well as hydrophobic drugs, can be efficiently encapsulated within hydrogels by physical trapping or covalent bonding[5–6]. However, in the case of small hydrophilic molecules, physical encapsulation is challenging due to the large mesh size of the three-dimensional (3D) network (5–100 nm) and weak hydrophobic interactions between the drug and the hydrogel matrix[4]. Although covalent binding is more stable than physical interaction, high water content increases the rate of hydrolysis of covalent bonds, resulting in rapid release of covalently bonded hydrophilic small molecules from hydrogels.
Some reported drug delivery systems can encapsulate and obtain sustained release of small hydrophilic molecules, such as liposomes[7–8], nanoparticles[9], polymer-drug conjugates[10], and peptides assembly[11], but they suffer from low encapsulation efficiency, drug waste, and cumbersome manufacturing processes. Moreover, these drug delivery systems are prone to early release of the encapsulated small hydrophilic molecules. During storage, this can result in depletion of the vehicles’ drug content; concerning in vivo, this can manifest as burst release and systemic toxicity, which is especially problematic with ultra-potent drugs[12–15].
Given the advantages of hydrogels in biomedical engineering applications, if the above challenges of small hydrophilic molecule delivery are addressed, hydrogels could have an extensive clinical impact on the local delivery of small hydrophilic molecules.
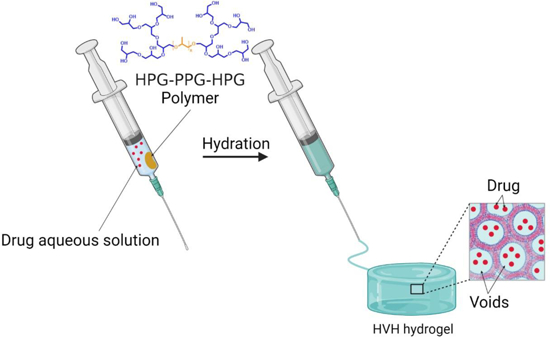
Void-containing hydrogels refer to hydrogels with a heterogeneous internal structure in which the polymer phase is densely packed and the aqueous phase appears as domains (e.g. voids)[16]. We hypothesize that, similar to liposomes, the voids provide space to encapsulate small hydrophilic molecules, while the densely packed polymer phase provides a barrier to limit the diffusion of the encapsulated drug to prevent leakage. Here, we present a hydration-induced void-containing hydrogel (HVH) based on a symmetrical triblock copolymer hyperbranched polyglycerol-poly(propylene oxide)-hyperbranched polyglycerol (HPG-PPG-HPG). Specifically, HPG-PPG-HPG was hydrated in an aqueous drug solution to form a void-containing hydrogel. While forming the hydrogel, the drug is simultaneously encapsulated within the voids (Scheme 1). The resulting hydrogel formulation has excellent physical stability and can be stored in a closed environment for several months without depleting the drug content of the formulation. Furthermore, the formulation is injectable, allowing easy and invasive administration to the site of interest.
Scheme 1.
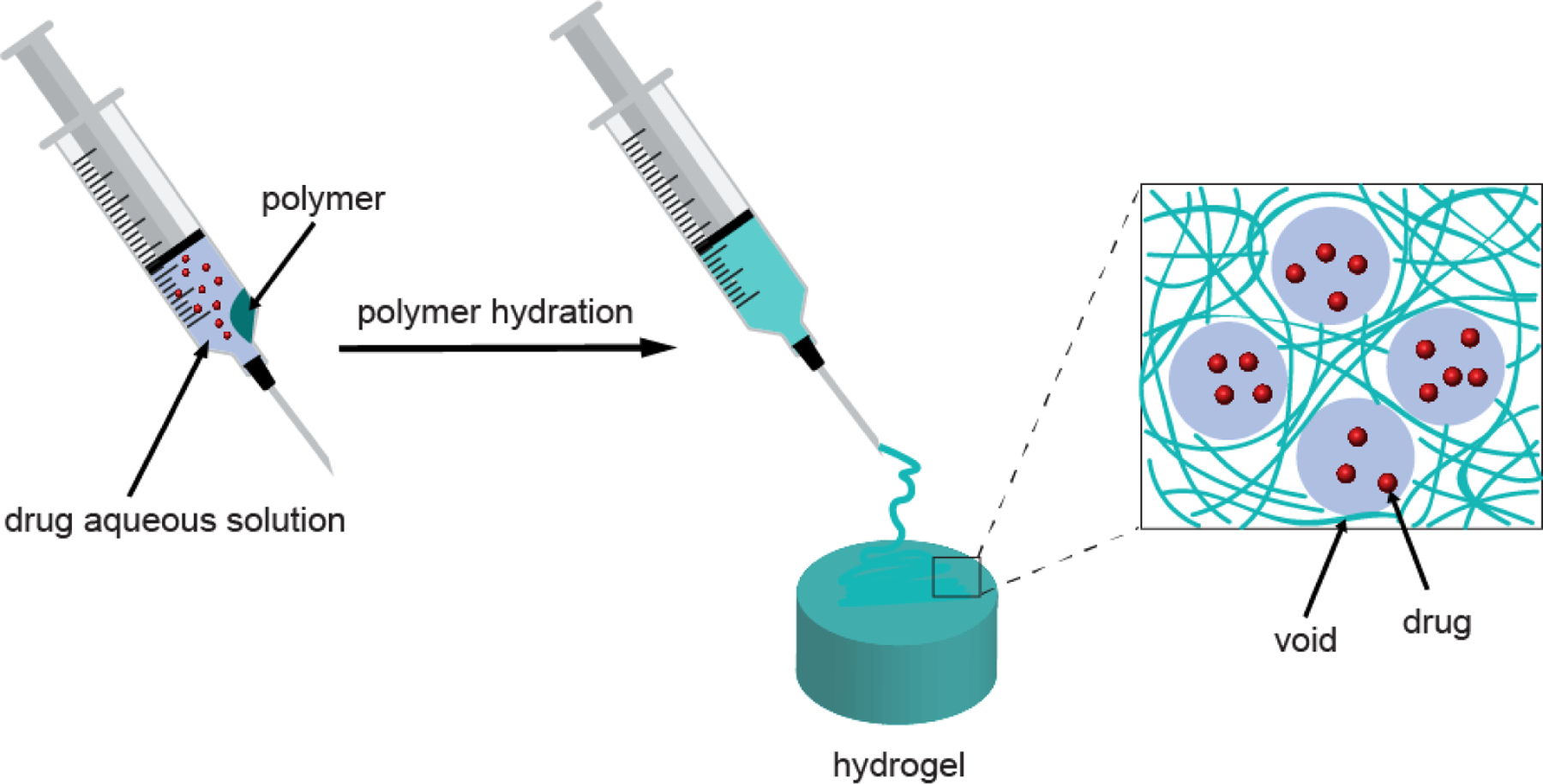
Schematic diagram of the preparation process of HVH hydrogels and drug encapsulation based on HPG-PPG-HPG copolymers
The performance of the HVH hydrogel in encapsulation and sustained release for small hydrophilic molecules was assessed using tetrodotoxin (TTX). TTX is one of the most challenging compounds to encapsulate because it is small (molecular weight = 319.27) and very hydrophilic (log P = ‒5.9)[17]. It is expected that if a drug delivery system can obtain encapsulation and sustained release of TTX, it will be well suited to many other small hydrophilic molecules. TTX is a potent non-protein specific blocker of voltage-gated sodium (NaV) channels. The ultra-high effectiveness in nerve blocks, harmlessness to local tissues, lack of affinity to cardiac NaV channels, and its inability to penetrate the blood-brain barrier make this toxin an attractive candidate for the design of local anesthetics[9, 12–13, 18–19]. However, a principal reason why TTX has not yet achieved clinical application is its systemic toxicity[20]. The most critical milestone of moving TTX into clinical practice is the precise control of TTX dosage to enhance its therapeutic effect but avoid its systemic toxicity. Therefore, a drug delivery system that can safely use TTX to treat pain will have the potential to fill an unmet need in pain control and help address the opioid crisis.
2. Results & Discussion
2.1. Synthesis and characterization of HPG-PPG-HPG
HPG-PPG-HPG was synthesized by a one-pot hydroxyl group initiated anionic ring-opening polymerization (AROP) of the glycidol using PPG (Mn = 2000, 4000 Da) as the initiator and potassium methylate (KOCH3) as the deprotonating agent (Figure 1a). All reactants and by-products (such as pure HPG) can be easily removed by dialysis against water and methanol to achieve high purity[15]. The chemical structure of the synthesized HPG-PPG-HPG was confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR) and Carbon-13 nuclear magnetic resonance (13C NMR) (Figure 1b). Specifically, the peak at 1.1 ppm represents the CH3 group of PPG, and the peak at 3.5 ppm represents the CH2 group of PPG. The CH2 and CH groups of HPG exhibit a broad resonance between 3.3 and 3.9 ppm. The OH group of HPG exhibit a broad resonance between 4.4 and 4.8 ppm. The signals of 13C-NMR spectra can be assigned as: (i) linear 1,3-unit (L13): CH2OH carbon at 61.5 ppm, CH2 carbon at 69.9 ppm, and CH carbon at 80.3 ppm; (ii) linear 1,4-unit (L14): both CH2 carbons at 73.3 ppm, CHOH carbon at 69.0 ppm; (iii) terminal unit (T): CH2OH carbon at 63.5 ppm, CHOH carbon at 70.9 ppm, and the CH2 carbon at about 71.2 ppm; (iv) dendritic unit (D): CH carbon at 78.5 ppm, one CH2 carbon at 72.0 ppm, and the other at about 71.2 overlapping with a CH2 carbon of a terminal unit.
Figure 1.
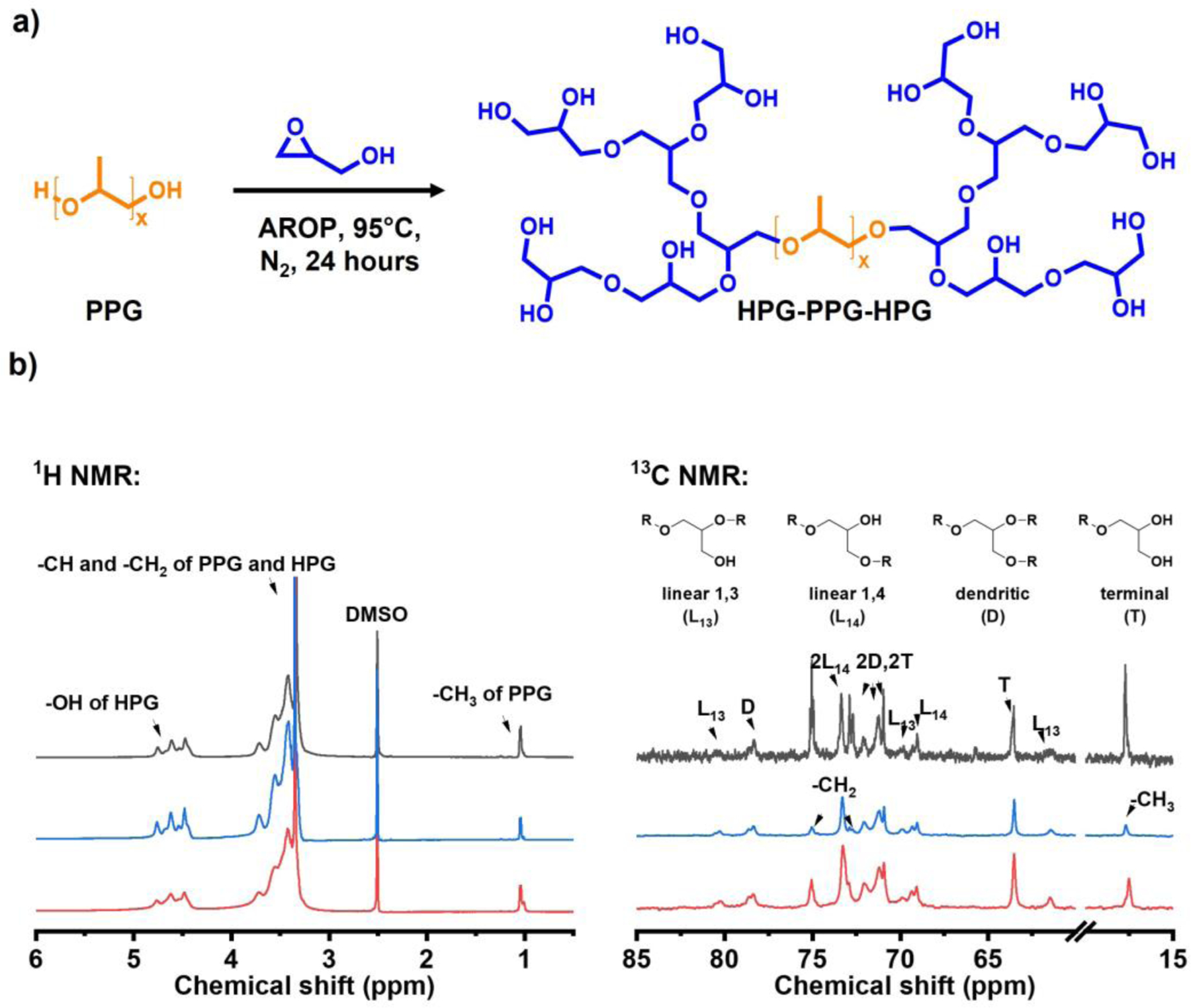
Synthesis and characterization of HPG-PPG-HPG copolymers. (a) Synthesis route of HPG-PPG-HPG copolymers, (b) 1H and inverse gated 13C NMR spectrums of the HPG-PPG-HPG copolymers. Black: HPG2656-PPG2000-HPG2656, blue: HPG695-PPG2000-HPG695, red: HPG608-PPG4000-HPG608.
Based on the integrated intensity analysis of 13C NMR spectrum, the HPG molecular weight was calculated as previously reported[15]. The synthesized HPG-PPG-HPG copolymers are denoted as HPG2656-PPG2000-HPG2656, HPG695-PPG2000-HPG695, HPG608-PPG4000-HPG608, where the numbers represent the molecular weight of each block (Table 1).
Table 1.
Characterization of HPG-PPG-HPG copolymers
| Copolymers | Mn of PPG (Da) | PPG Mass (g) | Glycidol Mass (g) | Yield (%) |
|---|---|---|---|---|
| HPG2656-PPG2000-HPG2656a | 2000 | 5 | 22.2 | 57.9 |
| HPG695-PPG2000-HPG695 | 2000 | 5 | 16.5 | 32.1 |
| HPG608-PPG4000-HPG608 | 4000 | 9.2 | 10.0 | 31.5 |
The molecular weight of HPG was determined by inverse gated 13C NMR spectrum.
2.2. Fabrication and characterization HVH hydrogels
To prepare hydrogels, 30 mg of HPG695-PPG2000-HPG695 and 0.2 mL of deionized water containing FS were placed in a syringe, then incubated at 4 °C overnight until the polymer was fully hydrated. After hydration, the mixture was warmed to room temperature and the hydrogel formed. The hydrogel was highly self-integrating, exhibited good stability, and was injectable at room temperature (Figure 2a).
Figure 2.
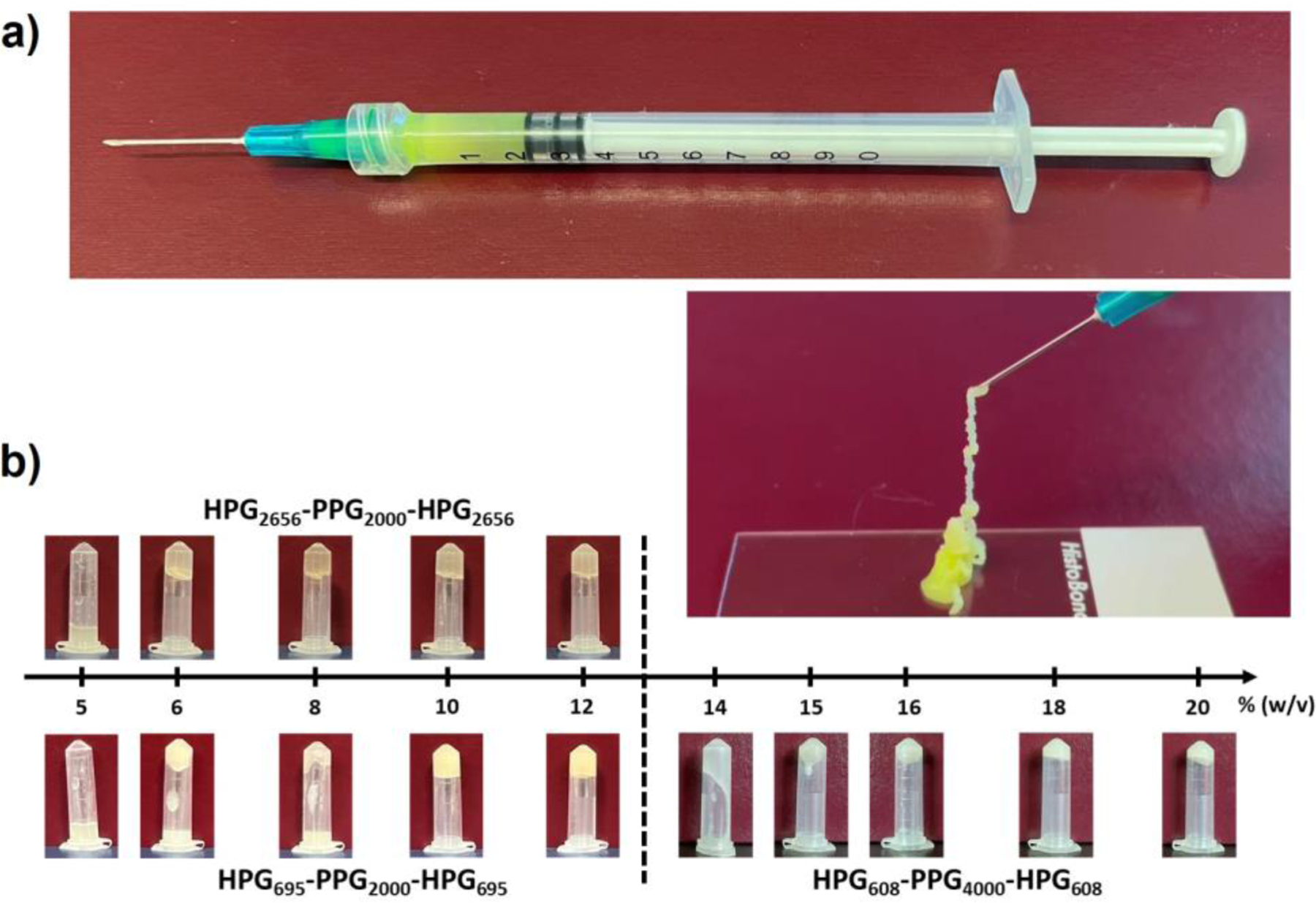
Characterization of HVH hydrogels. (a) Photographs of the hydrogel prepared by hydration of 15 (w/v) % concentration of HPG695-PPG2000-HPG695 copolymer overnight at 4 °C (FS was added as a colorful dye). The prepared hydrogel can be injected through a 23G needle. (b) Photographs of hydrogels after inverting the vials for 24 hours.
Polymer composition determines the capability of the HPG-PPG-HPG copolymers to form HVH hydrogels. HPG2656-PPG2000-HPG2656, HPG695-PPG2000-HPG695, and HPG608-PPG4000-HPG608 copolymers were hydrated in deionized water at different concentrations at overnight 4 °C (Table S1). The critical gelation concentrations (CGCs) of HPG-PPG-HPG copolymers were determined by the vial inversion method. The CGCs for HPG2656-PPG2000-HPG2656, HPG695-PPG2000-HPG695, and HPG608-PPG4000-HPG608 copolymers were 8%, 10%, and 18% (w/v), respectively, as determined from 24-hour vial inversion (Figure 2b). The hydration capacity of copolymers decreased in the sequence of HPG2656-PPG2000-HPG2656, HPG695-PPG2000-HPG695, and HPG608-PPG4000-HPG608 (Figure S1). The results showed an inverse relationship between the CGC and the hydration capacity of copolymer. A higher hydration capacity of a copolymer is considered a favorable property of hydrogels as it allows for gel formation at lower concentrations, resulting in a lower dose of polymer needed for administration into the human body.
Polymer composition determines the morphology of the HVH hydrogels. The morphologies of the hydrated polymers at three different concentrations were observed with the optical microscope. All tested samples had microscopically visible voids ranging in diameter from 10 μm to 300 μm (Figure 3a). The void size (diameter) is inversely proportional to the molecular weight of the HPG block, but is independent of polymer concentration (Figure 3b, Table S2, S3). Specifically, the void sizes of HPG2656-PPG2000-HPG2656, HPG695-PPG2000-HPG695, and HPG608-PPG4000-HPG608 samples at their highest concentration tested are 252.22 ± 67.55 μm, 39.17 ± 10.40 μm, and 15.96 ± 3.86 μm, respectively.
Figure 3.
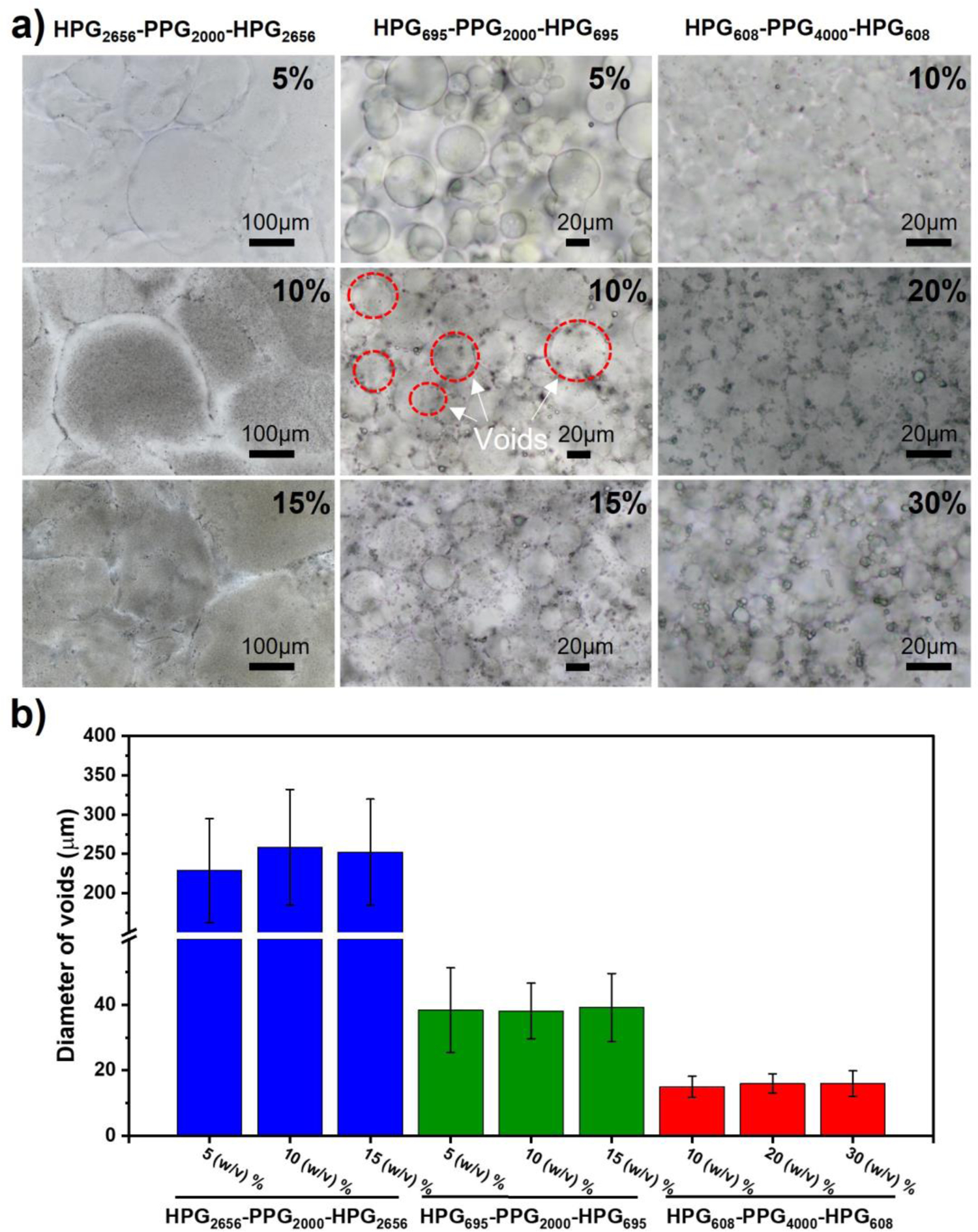
Morphology of HVH hydrogels. (a) Optical microscopy images of samples prepared by hydration of different concentrations of HPG-PPG-HPG in DI water overnight at 4 °C. (b) Void sizes of HPG-PPG-HPG hydrogels. Data are the means with SDs (n = 20 for HPG2656-PPG2000-HPG2656 hydrogels, n = 120 for HPG608-PPG4000-HPG608 and HPG695-PPG2000-HPG695 hydrogels).
The void walls of the hydrogels were visualized using DiI staining of the hydrophobic polymer shells, and their thickness was quantified from confocal images using ImageJ software. The measured void wall thicknesses were 0.56 ± 0.13 μm, 4.54 ± 2.75 μm, and 0.63 ± 0.12 μm for the HPG608-PPG4000-HPG608, HPG2656-PPG2000-HPG2656, and HPG695-PPG2000-HPG695 hydrogels, respectively. Notably, the measured thickness of the void walls in the HPG608-PPG4000-HPG608 hydrogel was significantly thinner than that of the other two hydrogels (Figure S2, Table S4, S5).
The rheological properties of HVH hydrogels were examined. Specifically, the modulus was measured at different frequency to present the gelation properties of the hydrogels, the complex viscosity was measured at different frequencies to test the shear strain resistance of the hydrogels, and the cyclic dynamic modulus were measured to illustrate the response of hydrogels to compression and stretching.
In the frequency spectrum measurement, HPG2656-PPG2000-HPG2656 samples with the concentrations of 5%, 10%, and 15%, HPG695-PPG2000-HPG695 samples with the concentrations of 10% and 15%, and HPG608-PPG4000-HPG608 samples with the concentrations of 15%, 20%, and 30%, exhibited a higher storage modulus (G′) than loss modulus (G′′) under the lower frequency sweep at a constant strain of 0.1%, indicating that these samples possess hydrogel characteristics (Figure 4a–c).
Figure 4.
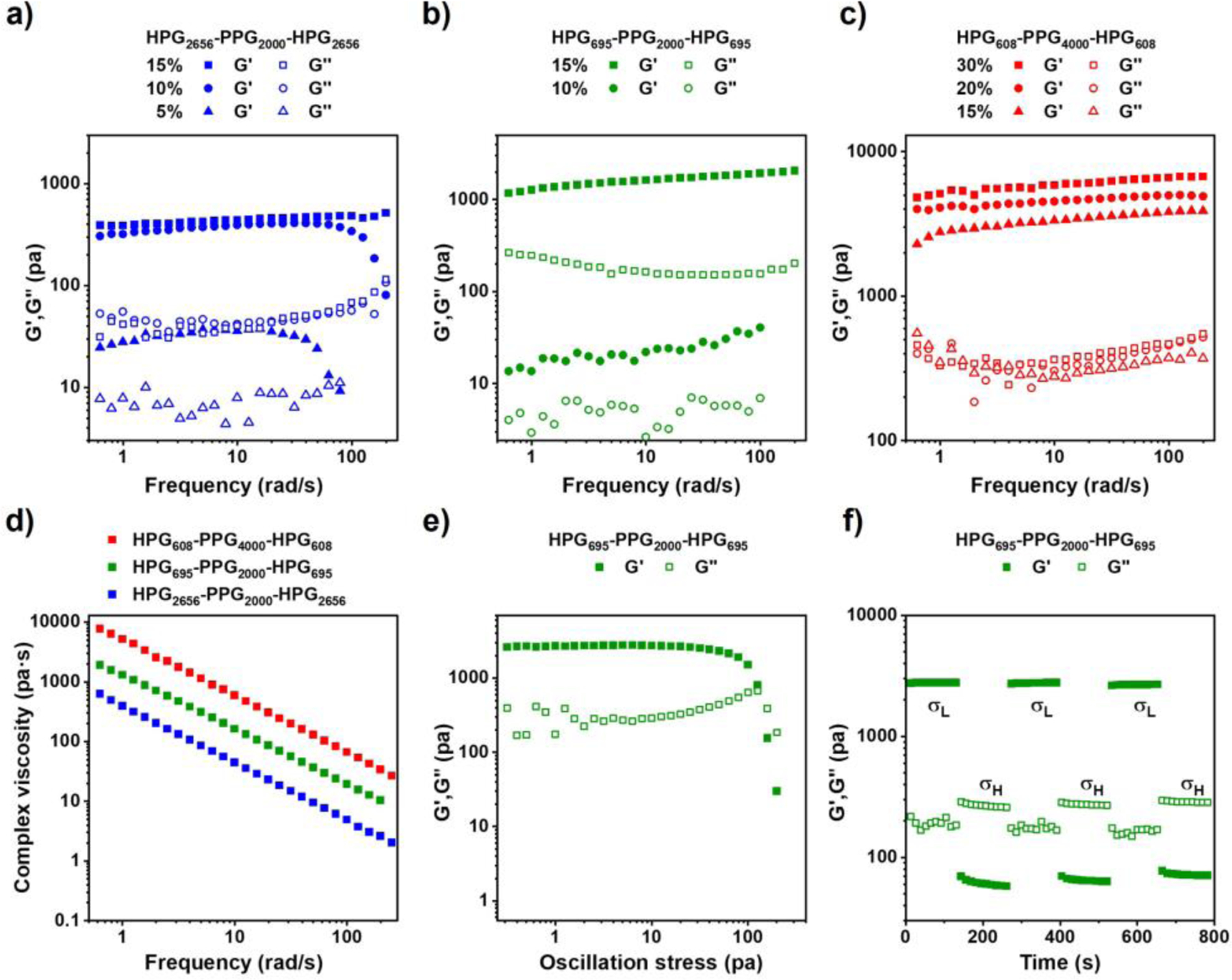
Characterization of the rheological properties of HPG-PPG-HPG hydrogels. Dynamic moduli as a function of frequency for a) HPG2656-PPG2000-HPG2656 hydrogels, b) HPG695-PPG2000-HPG695 hydrogels, and c) HPG608-PPG4000-HPG608 hydrogels. A constant strain 0.1% was applied. d) Dynamic viscosity as a function of frequency for the 30 (w/v) % HPG608-PPG4000-HPG608 hydrogel, 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel, and 15 (w/v) % HPG2656-PPG2000-HPG2656 hydrogel. e) Dynamic modulus as a function of oscillation stress for the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel. f) Dynamic modulus as a function of time with an imposed step change in shear stress between 10 Pa and 200 Pa for the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel.
All tested samples show the shear-thinning property, as shown by their decreased complex viscosity when a constant strain was applied and increasing the angular frequency (Figure 4d). To mimic the change from a statically stored gel in a syringe to the injectable state of the gel, a shear force was applied to the hydrogel while maintaining a constant frequency at 1 rad/s. It shows a typical spectrum of HPG695-PPG2000-HPG695 hydrogel at a concentration of 15 (w/v) % (Figure 4e). When the shear stress increased to its yield point, the hydrogel lost its mechanical integrity, where the storage modulus (G′) was lower than the loss modulus (G′′), and the hydrogel exhibited shear-thinning property. Furthermore, cyclic high and low stress were applied to the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel. At a low-stress level, the gel remained stable with G′ higher than G″. At a higher-stress level, the gel changed into a liquid-like state with G′ lower than G″. Upon removal of the high stress, the material immediately regained its gel property, demonstrating its ability to reside at the target site and release the drug continuously after injection (Figure 4f). These results explain the injectability of hydrogels from 23 G needles and their rapid recovery of gel properties after injection.
2.3. Mechanism of HVH hydrogel formation
As shown in Figure 5a, the mechanism of HVH hydrogels and voids was proposed. After synthesizing the HPG-PPG-HPG polymer, it was washed and emulsified with water using a vortex. The vortex process created HPG-PPG-HPG polymersomes with aqueous pockets through a water-in-oil-in-water (w/o/w) emulsion, as previously reported[15]. Upon centrifugation, the polymersomes underwent precipitation and packed together to form a hydrogel. More specifically, under centrifugal pressure, the polymersomes came into close contact with each other, and the polymersome shells merged together, ultimately forming a 3D network that maintained the aqueous pockets, thus converting the polymersomes into hydrogel precursors. The 3D network of hydrogel precursors was retained during lyophilization, allowing for the reformation of the hydrogel and voids upon rehydration. Notably, no significant difference in the HVH hydrogel morphology and void sizes (Table S2, S3) was observed between the hydrogel precursors (Figure 5b) and rehydrated hydrogels (Figure 3), which supports the proposed mechanism.
Figure 5.
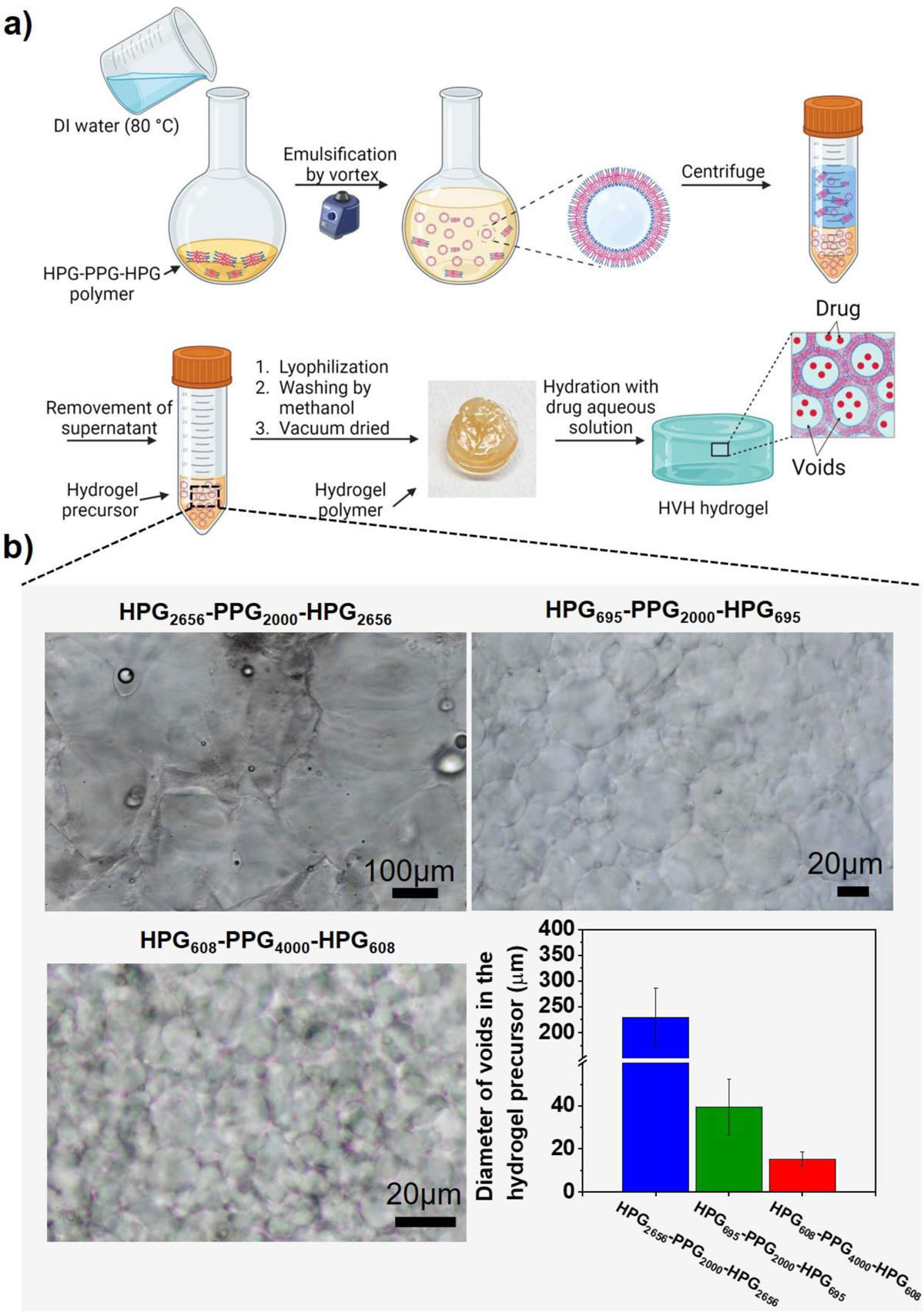
Study on the mechanism of formation of HVH hydrogels. (a) The proposed mechanism of the formation of HVH hydrogels. (b) Optical microscopy images and the diameters of voids in the hydrogel precursor formed after centrifuging the mixture of polymersome and water and before lyophilization. Data are the means with SDs (n = 20 for HPG2656-PPG2000-HPG2656 hydrogels, n = 120 for HPG608-PPG4000-HPG608 and HPG695-PPG2000-HPG695 hydrogels).
2.4. Drug encapsulation and release from HVH hydrogels
Using fluorescein sodium (FS, molecular weight = 376.27, logP = 2.6) and tetrodotoxin (TTX, molecular weight = 319.27, logP = –5.9) as model molecules, the drug encapsulation and release characteristics of HVH hydrogels were evaluated. Specifically, 6 μg of FS or 10 μg of TTX was dissolved in 0.3 mL of phosphate-buffered saline (PBS, pH 7.4) and mixed with a set amount of HPG-PPG-HPG polymer. The mixture was incubated overnight at 4 °C to fully hydrate the polymer. Confocal images of the formed FS-containing hydrogels showed that the voids of the HVH hydrogels were green (FS stained water green) and surrounded by red polymer shells (DiI was used as a hydrophobic dye and it stained polymer red) (Figure 6a, S3). The 3D structures of the hydrogels were also constructed from the confocal microscope images (Figure S4). This observation confirmed that the FS molecules were encapsulated inside the voids of the HVH hydrogels.
Figure 6.
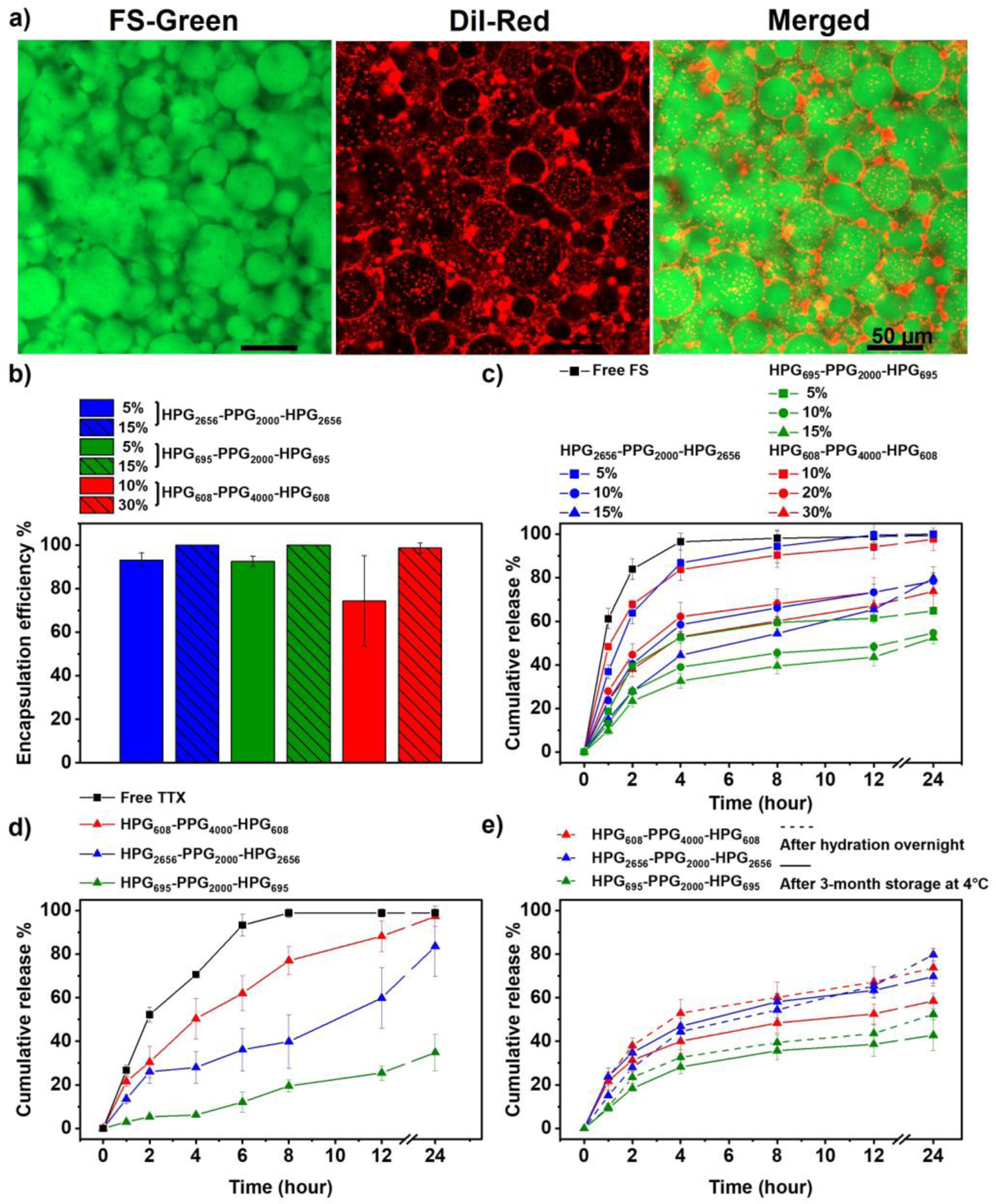
Characterization of drug encapsulation and release characteristics from HPG-PPG-HPG hydrogels. (a) Confocal microscope images of the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel, green: hydrophilic dye (FS), red: hydrophobic dye (DiI), scale bars: 50 μm. (b) Encapsulation efficiency of FS in hydrogels. (c) In vitro cumulative release of FS from hydrogels. (d) In vitro cumulative release of TTX from the 30 (w/v) % HPG608-PPG4000-HPG608 hydrogel, the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel, and the 15 (w/v) % HPG2656-PPG2000-HPG2656 hydrogel. (e) The in vitro cumulative release of FS was compared before and after hydrogel storage at 4°C for 3 months. Hydrogels tested included the 30 (w/v) % HPG608-PPG4000-HPG608 hydrogel, the 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel, and the 15 (w/v) % HPG2656-PPG2000-HPG2656 hydrogel. Data are the means with SDs (n = 3).
As illustrated by the mechanism of formation of the hydrogels and voids, the water-soluble drug is encapsulated in the HVH hydrogels by diffusion of an aqueous drug solution through the void walls during the hydration process. As a result, the encapsulation efficiency of water-soluble drugs by HVH hydrogel is determined by the polymer concentration. When the polymer concentration is higher than the CGC, all water is absorbed by the hydrogel, thus, the encapsulation efficiency is close to 100% (Figure 6b). The drug loading capacity is determined by the drug concentration in the aqueous drug solution and is limited by the solubility of drugs.
The encapsulated FS or TTX was sustainably released from the HVH hydrogels at a release rate much lower than that of the free drug control. The release profiles of FS from HVH hydrogels were analyzed using three models: the first-order model, Korsmeyer-Peppas model, and Higuchi model (Table S6). The first-order model did not fit the FS release data well (e.g. R2 < 0.5). As the first-order model is applicable for predicting drug release from homogeneous systems such as PEG-based and chitosan-based hydrogels[21–22], the unsatisfactory fitting of this model with the release data of HVH hydrogel is consistent with the non-homogeneous nature of the HVH hydrogel structure due to the presence of voids. Both the Korsmeyer-Peppas model and the Higuchi model yielded good fits to the FS release data (e.g. R2 > 0.8), indicating that the release of FS from the HVH hydrogel follows quasi-Fickian and Fickian diffusion, respectively. This indicates that the FS release from the HVH hydrogel is a diffusion-controlled process and is independent on polymer swelling or changes in hydrogel structure[23]. The HVH hydrogels have a stable structure as evidenced by the consistent size of voids during a 24-hour incubation in DI water (Figure S5). This finding suggests that there were no significant changes in the hydrogel structure during the incubation period, thereby supporting the diffusion-driven drug release mechanism.
The drug release rate from HVH hydrogels is influenced by both the void size of the hydrogel and the hydration capacity of the polymer. With the similar hydration capacity of the polymer, a smaller void size requires the drug to pass through more void walls before being released from the hydrogel (Scheme 2), resulting in a slower drug release rate. On the other hand, in hydrogels with the similar void sizes, the hydration capacity of the polymer determines the drug release. A higher hydration capacity the polymer results in increased water retention within the hydrogel, which leads to a slower drug release rate. Among the hydrogels tested, the HPG695-PPG2000-HPG695 hydrogels exhibit the slowest drug release rate (Figure 6c, d). This is due to their similar hydration capacity to the HPG2656-PPG2000-HPG2656 hydrogels but smaller void size, as well as their similar void size but higher hydration capacity compared to the HPG608-PPG4000-HPG608 hydrogels. All tested hydrogels maintained their drug encapsulation and release characteristics over time, as it turned out that their drug release profiles did not change significantly after three months of storage in syringes at 4 °C (Figure 6e, S6). Notably, there was no sign of any phase separation in hydrogels during storage.
Scheme 2.
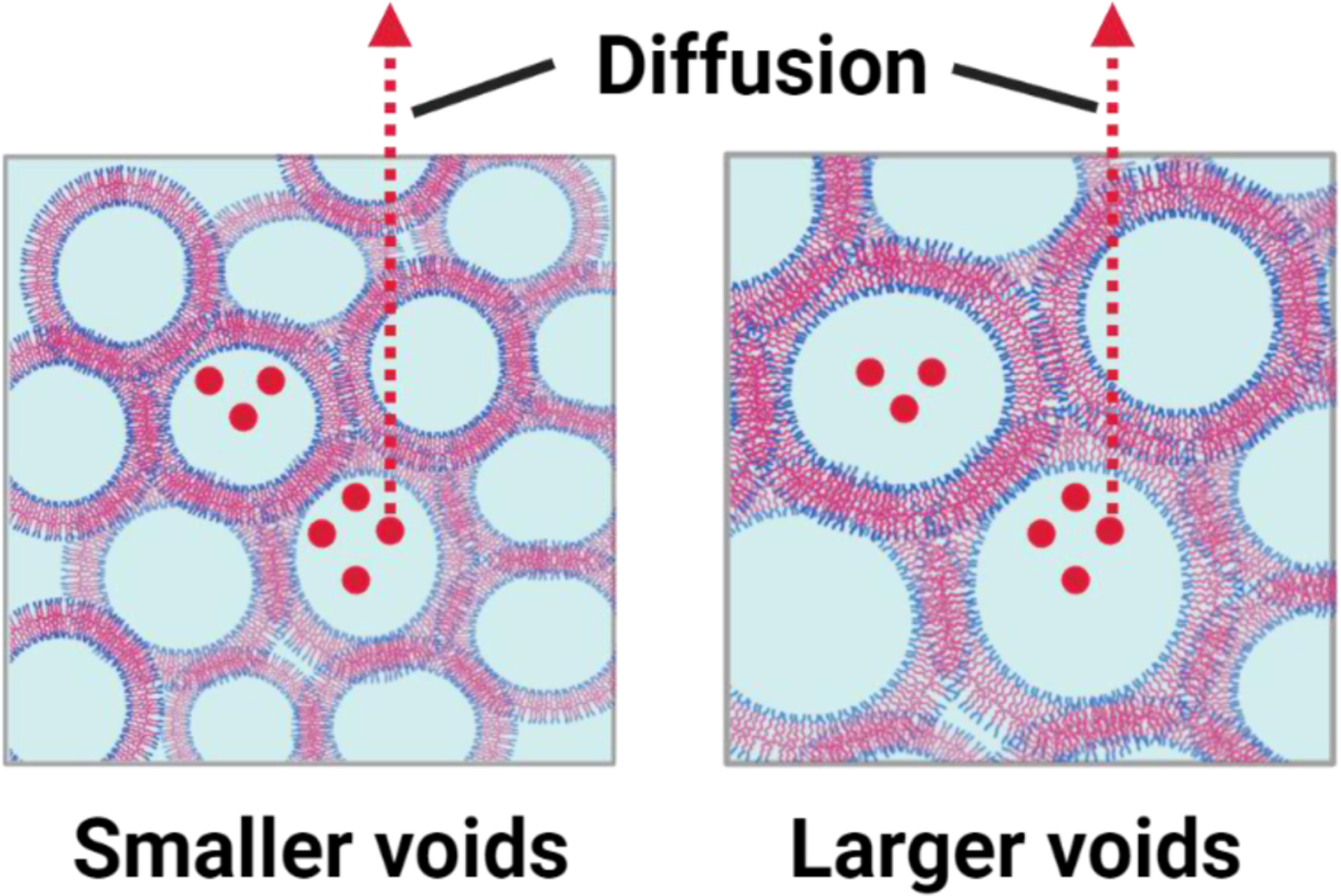
The drug release mechanism from the HVH hydrogels. As the void size decreases, the drug needs to diffuse through more void walls before being released from the HVH hydrogel.
2.5. Rat sciatic nerve blockade in vivo
TTX is a potent local anesthetic that is expected to fill the unmet pain treatment needs. However, its clinical application is hindered by its systemic toxicity that an overdose of TTX can lead to skeletal muscle paralysis and even death[11]. Due to its small molecular weight and hydrophilicity, TTX is one of the most challenging small hydrophilic molecules for encapsulation and controlled release. Here, we tested the ability of HVH hydrogels for TTX encapsulation and sustained release by evaluating their effects on the tolerated dose of TTX in rats and the effectiveness of TTX in blocking the rat sciatic nerve.
8 μg of TTX was dissolved in 0.1 mL of PBS. A specified amount of HPG-PPG-HPG was added into the prepared TTX aqueous solution. The mixture was hydrated overnight at 4 °C to prepare the HVH/TTX formulation. Male Sprague-Dawley rats (4 in each group) were injected with prepared formulation and the control solution (equal volume of PBS containing 2 μg, 3 μg, or 4 μg TTX alone) at the left sciatic nerve (Figure 7a). After injection, rats underwent neurobehavioral testing at set intervals to determine the duration of functional deficits in both hind paws. Specifically, the sensory nerve blockade was accessed by a modified hotplate test and the motor nerve blockade was accessed by a weight-bearing test. Measure thermal latency and maximum weight bearing on the injected leg to determine the duration of nerve blockade and on the un-injected leg to determine the systemic toxicity of TTX.
Figure 7.
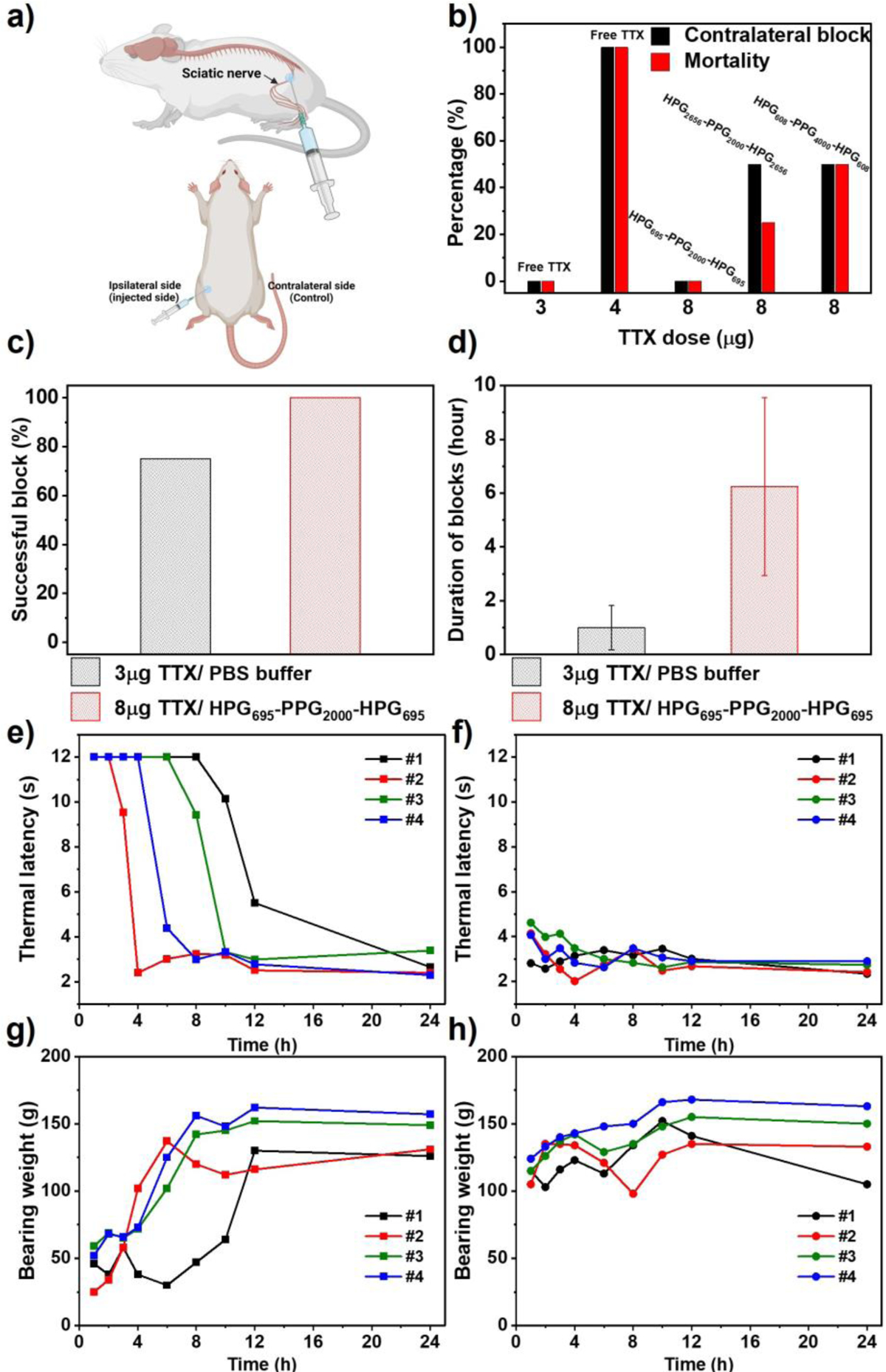
Neurobehavioral assessments of rats that received a single sciatic nerve injection of 0.1 mL PBS containing 8 μg TTX and HPG-PPG-HPG, or a single injection of 0.1 mL PBS containing 3 μg or 4 μg TTX alone. (a) Schematic illustrating the injected side and the contralateral side as control. (b) Frequency of nerve block in the uninjected (contralateral) leg and animal mortality. The hydrogels tested were 30 (w/v) % HPG608-PPG4000-HPG608, 15 (w/v) % HPG695-PPG2000-HPG695, and 15 (w/v) % HPG2656-PPG2000-HPG2656. Comparison between injections of 0.1 mL of PBS containing 3 μg free TTX and 0.1 mL of HPG695-PPG2000-HPG695 15 (w/v) % hydrogel containing 8 μg of TTX on (c) the frequency of successful blocks, and (d) the average duration of sensory nerve blocks. Data are the means with SDs (n = 4). (e) Ipsilateral and (f) contralateral paw withdrawal latency (PWL) to thermal stimulation in 4 rats injected with 0.1 mL of 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel containing 8 μg of TTX. (g) Ipsilateral and (h) contralateral paw leg bearing weight of 4 rats injected with 0.1 mL of HPG695-PPG2000-HPG695 15 (w/v) % hydrogel containing 8 μg of TTX.
Results demonstrated that 100% of rats (4 of 4) injected with 4 μg of TTX alone developed contralateral deficits (e.g. a thermal latency above 7 seconds in the un-injected limb suggests the systemic distribution of TTX), seizures and respiratory distress, and finally died (Figure 7b). In contrast, none of the rats injected with the 15 (w/v) % HPG695-PPG2000-HPG695/TTX formulation containing 8 μg of TTX developed contralateral deficits or died, demonstrating that injection of this formulation did not cause any TTX-related systemic toxicity. Notably, the HPG695-PPG2000-HPG695/TTX formulation was directly injected into rats after the hydration procedure without additional process, indicating that the HPG695-PPG2000-HPG695 hydrogel effectively encapsulated and greatly reduced the basal TTX release.
50% of rats (2 of 4) injected with the 15 (w/v) % HPG2656-PPG2000-HPG2656 hydrogel and the 30 (w/v) % HPG608-PPG4000-HPG608 hydrogel containing 8 μg of TTX developed contralateral deficits and some of them died. These results are consistent with the in vitro drug release profiles, i.e. the HPG695-PPG2000-HPG695 hydrogel showed the slowest drug release rate among the tested hydrogels.
TTX tolerance also depends on the hydrogel injected volume. When injected with 0.3 mL of the HPG695-PPG2000-HPG695/TTX hydrogel containing 8 μg of TTX, some rats developed contralateral deficits (3 of 4) or died (1 of 4) (Figure S7, S8). Similar trends were also seen in the HPG2656-PPG2000-HPG2656 hydrogel and the HPG608-PPG4000-HPG608 hydrogel. The percentage of rats developing TTX-related systemic toxicity increased when the injection volume was increased from 0.1 mL to 0.3 mL while the polymer concentration and TTX dose were kept constant (Figure S7, S8). The volume effect may be attributed to the higher surface area of the larger material volume, which increases the TTX release rate. To address concerns about possible systemic toxicity resulting from a larger polymer dose, rats were injected with 0.3 mL of 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel without TTX loading. The results showed that the injection of hydrogel alone didn’t induce any systemic toxicity in the rats (Figure S9). Additionally, in order to understand the drug release mechanism upon contact with tissue, the residue of the 15(w/v) % HPG695-PPG2000-HPG695 hydrogel was collected from the site of injection 4 days after injection. Microscopic examination of the collected residue revealed the presence of voids and no significant change in the hydrogel structure (Figure S10). These results suggest that the drug release mechanism from the hydrogel after injection is similar to that observed in vitro, which is a drug diffusion-driven process and is not related to the structural changes in the hydrogel.
In addition to safety, the HVH hydrogel/TTX formulation is an effective and reliable injectable local anesthetic compared to free TTX. Injection of 0.1 mL of PBS containing 3 μg of free TTX successfully blocked the ipsilateral sciatic nerve in 3 out of 4 rats, e.g. 75% of success rate (Figure 7c). The duration of nerve block of 3 rats was 1 ± 0.82 hours (Figure 7d). In contrast, injection of 0.1 mL of 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel containing 8 μg TTX induced reliable ipsilateral nerve blocks in all rats, e.g. 100% of success rate. In addition, the nerve block duration in 4 rats was 6.25 ± 3.30 hours, ranging from 3 to10 hours, and the injections did not cause TTX-related systemic toxicity (Figure 7e, f). A single injection of the HVH hydrogel/TTX formulation blocked the rat sciatic nerves as effectively as a single injection of the clinically used pain reliever Exparel (bupivacaine liposome injectable suspension)[24]. In all injected rats, motor blockade recovered synchronously with sensory blockade (Figure 7g, h).
2.6. Tissue reaction
Rats injected with 0.1 mL and 0.3 mL of TTX/15(w/v) % HPG695-PPG2000-HPG695 hydrogel were euthanized 4 and 14 days after injection (n = 4 at each time point). The sciatic nerve and surrounding tissues were harvested, sectioned, and stained for histologic evaluation. Muscle tissues were processed for hematoxylin–eosin (H&E) staining. The myotoxicity and inflammation were quantified using a previously reported scoring system[25]. Microscopic examination did not reveal myotoxicity or severe inflammation in rats at 4 or 14 days post-injection (Table 2, Figure 8a–d). Epon-embedded sections of the sciatic nerve were stained with toluidine blue. The formulation was found not to cause nerve damage (Figure 8e–h). The benign tissue response to the HVH hydrogel/TTX formulation is consistent with the excellent biocompatibility of high molecular weight PPG (2000 Da or greater)[10, 26] and HPG[27].
Table 2.
Myotoxicity and inflammation scores
| Group | Inflammation Score | Myotoxicity Score | |
|---|---|---|---|
| 8 μg TTX/ 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel | 0.1 mL, 4 days | 0.5 (0‒1) | 0 (0‒0) |
| 0.1 mL, 14 days | 0.5 (0‒1) | 0 (0‒0) | |
| 0.3 mL, 4 days | 0.5 (0‒2) | 0 (0‒0) | |
| 0.3 mL, 14 days | 0.5 (0‒1) | 0 (0‒0) | |
Inflammation score range: 0–4; myotoxicity score range: 0–6. Data for inflammation and myotoxicity are medians and range.
Figure 8.
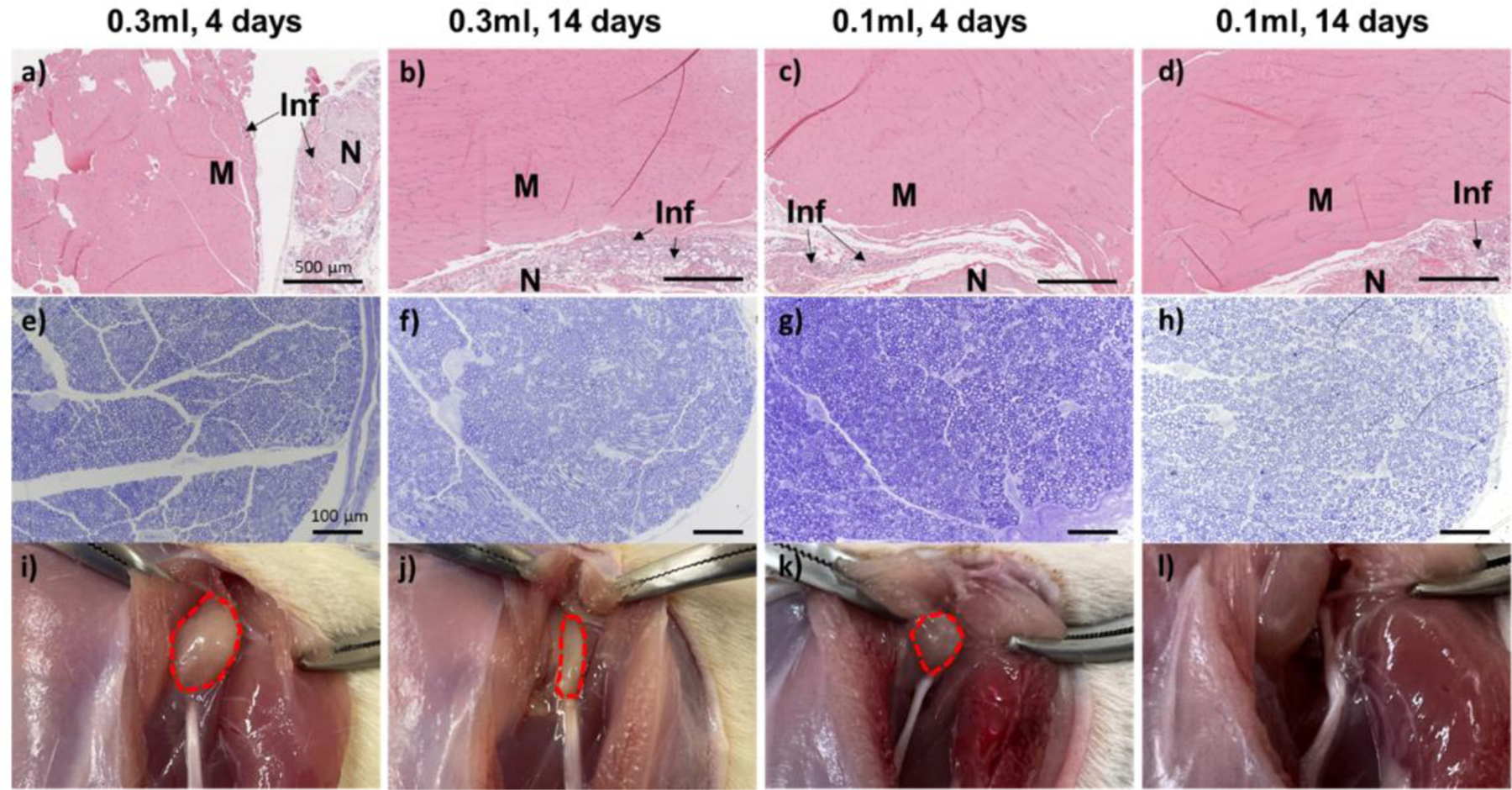
Tissue reaction to 0.1 mL or 0.3 mL of 15 (w/v) % HPG695-PPG2000-HPG695 hydrogel containing 8 μg of TTX at 4 days and 14 days post-injection. (a‒d) Representative H&E-stained sections of the muscle at the injection site. M: Muscle; N: Nerve region; Inf: inflammation. (e‒h) Representative toluidine blue-stained sections of the sciatic nerve from the injection site. (i‒l) Representative photographs of the injection site after dissection. Material residues are outlined dashed red.
The hydrogel residues at the injection site were photographed at the time of dissection. The volume of the hydrogel residue decreased significantly over time (Figure 8i–l). Notably, no visible hydrogel residue was observed 14 days after injection of 0.1mL TTX/ 15(w/v) % HPG695-PPG2000-HPG695 hydrogel (Figure 8l). The rapid clearance of the injected hydrogel helps avoid local tissue toxicity and allows subsequent injections.
3. Discussion
This work presents a void-containing hydrogel that can physically encapsulate small hydrophilic molecules and achieve sustained release. This new finding changes the generally accepted understanding that hydrogels are less effective as drug carriers for small hydrophilic molecules than poorly water-soluble drugs due to the loose packing of 3D networks and high water content[4, 28].
The HVH hydrogel is an advantageous and easy-to-implement approach to deliver small hydrophilic molecules. Due to the weak interaction between hydrophilic molecules and drug carriers, the current encapsulation and controlled release technologies (e.g. liposomes[8, 24, 29], polymersomes[30], and traditional hydrogels[31]) have shortcomings in delivering small hydrophilic molecules, such as low encapsulation efficiency, undesirable drug leakage during storage, and post-administration initial quick release. These shortcomings may lead to drug waste, drug overdose, and limited efficacy. In addition, the procedures for preparing these formulations are cumbersome and hence relatively costly[7–8]. The HVH hydrogel is a simple, rapid, single-step, low-cost drug delivery system for small hydrophilic molecules, yet achieves 100% drug encapsulation and diminishes drug leakage during storage. The simple procedure also makes it highly repeatable, minimizing the batch-to-batch variation (good batch-to-batch reproducibility). In addition, the hydration procedure is a mild process, without the use of high temperature, light, and organic solvents that can denature drugs during the formulation preparation.
TTX is a potent non-protein specific blocker of voltage-gated sodium (NaV) channels. The ultra-high effectiveness in nerve blocks, harmlessness to local tissues, lack of affinity with the heart muscle NaV channels, and the inability to penetrate the blood-brain barrier make this toxin an attractive candidate for the design of local anesthetics. However, the principal reason why TTX has not yet achieved clinical application is its systemic toxicity. The final and most critical milestone in TTX clinical practice is maintaining TTX concentrations above therapeutic levels to block nerves, but below toxic levels to avoid its systemic toxicity. HVH hydrogel is an excellent carrier for TTX to meet this requirement, as it allows injection of 8 μg of TTX, a 2-fold increase in tolerated dose compared to TTX alone, without causing TTX-related systemic toxicity in rats, and the injection produced reliable sciatic nerve block lasting more than 3 hours in all rats tested.
As a biomedical material, the degradability and metabolic pathway of the hydrogel are crucial factors to consider when justifying its clinical applications. The HVH hydrogel, being a physical hydrogel, gradually degrades as the HPG-PPG-HPG is absorbed and metabolized by the body. This is evidenced by the reduction in volume of the hydrogel residue over time, as shown in Figure 8i–l. HPG and PPG blocks were selected because of their excellent biocompatibility[10, 26–27]. PPG can be excreted via the kidneys or metabolized into downstream intermediates (lactate, CO2) in the human body[32]. HPG, although not biodegradable[33], can be eliminated via kidney without any organ accumulation[34], and has been proven to possess even better biocompatibility compared to polyethylene glycol (PEG) or hetastarch[35], thereby making the HPG-PPG-HPG safe for use in biomedical applications.
The composition of HPG-PPG-HPG significantly affects the hydrogel properties, including void structure, viscosity, and mechanical strength. Hydrogel properties affect the encapsulation and controlled release kinetics of small hydrophilic molecules. Specifically, the results demonstrate that, while other parameters are held constant, the void size increases as the percentage of HPG in the polymer increases. Moreover, it should be noted that the injection volume of the HVH hydrogel significantly affects the tolerated dose of TTX in rats with a more localized and targeted drug delivery at the injection site. The composition-structure-property-function relationships in polymer and hydrogel, and the volume effect indicate that there is still large room for optimization for the HVH hydrogel to encapsulate and achieve sustained release of small hydrophilic molecules.
4. Conclusion
HVH hydrogels based on HPG-PPG-HPG copolymers was developed. The HVH hydrogel has been shown to be a simple, efficient, and easy-to-implement delivery system for small hydrophilic molecules, which can achieve 100% drug encapsulation efficiency and sustained drug release. Compared with existing delivery systems for small hydrophilic molecules, the HVH hydrogel has the advantages of long shelf life, no premature drug release, no drug waste, and mild process conditions. In rats, the HVH hydrogel/TTX formulation increased the tolerated dose of TTX by 2-fold compared to TTX alone. A single injection of the HVH hydrogel/TTX formulation produced sciatic nerve blocks lasting for 3–10 hours without causing TTX-related toxicity and toxicity to local tissues. The HVH hydrogel is expected to be a clinically useful drug delivery system, especially for water-soluble drugs for disease treatment.
5. Methods
Materials.
Poly(propylene glycol) (PPG, Mn = 2000), potassium methoxide (KOCH3, 95%), glycidol (96%), phosphate buffer saline (PBS, pH 7.4, 0.15M, 138 mM NaCl, 2.7 mM KCl), fluorescein sodium salt, dimethyl sulphoxide-[D6] (≥ 99.8%) were purchased from VWR International Ltd (Radnor, PA). Poly(propylene glycol) (PPG, Mn = 4000) were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Tetrodotoxin was purchased from Abcam (Cambridge, MA, USA). TTX ELISA kits were purchased from Creative Diagnostics Company (Shirley, NY, USA).
Synthesis of HPG-PPG-HPG copolymers.
A predetermined amount of dry PPG4000 or PPG2000 polymer was added into a nitrogen-protected flask in a 95 °C oil bath, and a predetermined amount of KOCH3 was added (see details in Table.1). The mole ratio of the hydroxyl groups of PPG diol to KOCH3 was 1 : 0.45. The system was stirred (500 rpm) under vacuum for 1 hour. The flask was then placed in a vacuum oven at 80 °C for 24 hours. The flask was refilled with nitrogen and placed in a 95 °C oil bath. Then, a set amount of glycidol was added by a syringe pump with a speed of 1 mL/h. After adding glycidol, the reaction was continued for another 24 hours, The reaction mixture was washed three times with 15–40 mL of 80 °C hot water, and the collected samples were lyophilized. To further remove the free HPG, the dried samples were washed three times with 30 mL of methanol. The samples were dried under the vacuum and stored for further use. The HPG-PPG-HPG copolymers were rubber-like solid. The purity of the copolymers is over 90%.
Nuclear magnetic resonance (NMR) spectroscopy.
The 1H NMR and inverse gated 13C NMR spectra were recorded on a Bruker AVANCE Spectrometer 500. The polymers were hydrated with excess water and the mixture was vigorously sonicated. After lyophilization, DMSO‒d6 was used as solvent to dissolve the polymer, and the polymer concentration was 100 mg/mL. Analysis of 1H NMR and inverse gated 13C NMR spectra and calculation of molecular weights followed previously established methods[15, 36].
Preparation of hydrogels.
A predetermined amount of the copolymer and deionized water or aqueous solution (see details in Table S1) were placed in a syringe or test tube and incubated overnight at 4 °C until the polymer was fully hydrated.
Measurement of critical gelation concentration (CGC).
The CGC of copolymers was measured by the vial inversion method. After the polymer was hydrated in DI water with different concentrations in the test tube, the test tube was inverted for 24 hours and observed for any visible flow. Samples with no visible flow were considered to form a hydrogel, and the lowest polymer concentration to form a hydrogel was deemed as CGC.
Rheological test.
The rheological properties of the hydrogels were measured using a DHR-2 rheometer (TA instruments, New Castle, DE, United States). A parallel plate with a diameter of 25 mm was used for all the tests. The gap distance between the plates was 1 mm. The hydrogels were placed onto the plate through a syringe. A constant strain of 0.1% was applied to the frequency spectrum measurements. For dynamic modulus measurements, a constant 1 rad/s angular speed was used. The high stress and low stress used in the cyclic dynamic modulus measurement were 200 Pa and 10 Pa, respectively. Self-recovery of the modulus was validated after 3 cycles of high and low stress.
Measurement of polymer hydration capacity.
To determine the hydration capacity of the HPG-PPG-HPG copolymers. 100 mg of polymer was weighed in a 2 mL centrifuge tube and hydrated with an excess amount of DI water (1.5 mL) overnight. The mixture was then centrifuged (12000 rpm, 5 min) and the supernatant was removed. The hydration capacity was calculated with the following equation.
Optical microscope imaging and confocal imaging.
The hydrogel was injected onto a glass chip, covered with a cover slip, and then observed using an optical microscope (Ts2R, Nikon). For confocal imaging, 10 μg of DiI stain (1 mg/mL in DMSO, red, hydrophobic dye for staining the hydrophobic polymer phase) was added in 1 mL of PBS (e.g. 10 μg/mL), followed by a set amount of polymer added into the DiI stain solution and incubated at 4°C until fully hydrated. This process helps distribute the DiI stain evenly throughout the polymer. The hydrogel was lyophilized to remove water and DMSO. The lyophilized sample was added to 1 mL of PBS containing 10 μg of FS (10 μg/mL, green, hydrophilic dye for staining the water phase), and the mixture was incubated at 4 °C overnight. The hydrogels were observed under a confocal laser scanning microscopy (Leica TCS SP2 AOBS) using a 20× objective at room temperature.
The sizes of voids were measured by ImageJ Software (n = 20 for HPG2656-PPG2000-HPG2656 hydrogels, n = 120 for HPG608-PPG4000-HPG608 and HPG695-PPG2000-HPG695 hydrogels). The thickness of the void walls was also measured by Image J Software (n = 60).
Determination of drug encapsulation efficiency.
6 μg of FS was dissolved in 0.3 mL of PBS (20 μg/mL) and the solution was mixed with the predetermined amount of polymer in a 1 mL syringe. The mixture was incubated overnight at 4 °C to ensure complete hydration of the polymer. Measurements were performed by directly injecting the mixture through a 23 G needle into the bottom of a 5 mL sample vial containing 5 mL of PBS buffer pre-warmed at 37 °C. The vial was gently stirred to diffuse the unencapsulated drug into the supernatant. Then, 1 mL of the supernatant was filtered through a 0.22-μm pore size disposable syringe filter. The FS concentration in the solution was measured at 492 nm with an absorbance microplate reader (EMax Plus, Molecular Devices). The drug encapsulation efficiency can be calculated as the equation below.
Determination of in vitro drug release.
20 μg of FS or 10 μg of TTX was dissolved in 1 mL of PBS and mixed with the predetermined amount of polymer in a 5 mL syringe. The mixture was incubated overnight at 4°C to ensure the complete hydration of the polymer. The release of FS or TTX from each formulation was measured using a diffusion system at 37 °C. Specifically, transwell membrane inserts (0.4 μm pore size, Costar, Cambridge, MA) and 24-well culture plates were employed as the donor and acceptor chambers, respectively. Each formulation (~ 300 μL) was directly injected onto filter inserts through a 23 G needle. At each time point (30 min, 1 h, 2 h, 4 h, (6 h added for TTX release experiment), 8 h, 12 h, 24 h), 1 mL aliquots of the PBS receiving medium were sampled, and the filter inserts were sequentially transferred into new wells containing 1 mL prewarmed and fresh PBS. The FS concentration in the collected sample was measured at 492 nm with an absorbance microplate reader (EMax Plus, Molecular Devices). The concentration of TTX in the collected sample was measured with an ELISA kit. Experiments were performed in triplicate. The cumulative drug release percentage at different time points was calculated using the equation below:
Animal studies.
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Alabama (Protocol ID: 19–11-2992). Adult male Sprague-Dawley rats weighing 250–350 g (Charles River Laboratories, Wilmington, MA, USA) were group-housed under a 12 h/12 h light/dark cycle. Prior to injection, rats were briefly anesthetized with isoflurane through a facemask. The injection was conducted in the left leg by introducing a 23 G × 3/4” needle posteromedially into the greater trochanter, pointing in an anteromedial direction. Once in contact with bone, 0.1 mL or 0.3 mL of the test formulation was injected. Neurobehavioral testing was performed at predetermined intervals after injection. The right leg served as a control for systemic toxicity.
Sensory nerve blockade was assessed by a modified hotplate test. Briefly, the time that rats left their hindpaws on a hot plate (Model 39D Hot Plate Analgesia Meter, IITC Inc., Woodland Hills, CA) at 56 °C was measured by a stopwatch. The time is referred to as thermal latency. The paw was removed from the hotplate after 12 seconds to avoid injury to the rat. Measurements were repeated 3 times at each time point, and the average was used for data analysis. Motor function was tested by measuring extensor postural thrust. Briefly, the rat was placed on top of a digital balance and allowed to bear its weight on one hindpaw at a time. The maximum weight that the rat can bear without its ankle touching the balance was measured.
The duration of sensory block is the time it takes for the thermal latency to return to 7 seconds (the baseline thermal latency is approximately 2 seconds; therefore 7 seconds represents the midpoint between maximal latency, set as the 12-second cutoff, and the baseline). The duration of motor block was defined as the time to return 50% of weight bearing.
Tissue harvesting and histology.
Rats were sacrificed at 4 days (acute inflammation) or 14 days (chronic inflammation) after the injection, and the sciatic nerve was harvested together with surrounding tissues. Muscle samples were fixed in 10% neutral buffered formalin and processed for histology (hematoxylin–eosin stained slides) using standard techniques. Muscle samples were stained with Hematoxylin and eosin, and then scored for inflammation (0–4 points) and myotoxicity (0–6 points)[25].
To evaluate the neurotoxicity, the sciatic nerve samples were processed and fixed in Karnovsky’s KII Solution (2.5% glutaraldehyde, 2.0% paraformaldehyde, 0.025% calcium chloride in 0.1 M cacodylate buffer, pH 7.4). Samples were treated with osmium tetroxide for post-fixation for 2 hours and were subsequently dehydrated in graded ethanol solutions for 10 min each (30%, 60%, 90%, 100%, 100%, 100%). Then, the nerves were infiltrated with ethanol/Epon resin mixtures (1 : 1, 1 : 2, 0 : 1). After being sectioned by a diamond knife through ultramicrotome, nerve sections of 800 nm were stained with toluidine blue, followed by light microscopy. Slides were analyzed by an observer blinded to the nature of individual samples.
Statistical Analysis.
Data are presented as means ± SDs (n = 3 in drug encapsulation and release kinetics, n = 4 in neurobehavioral studies). The statistical differences between groups of void size and thickness of the void walls were tested by one-way analysis of variance (ANOVA) for multiple comparisons. Three post-hoc tests were used, including Bonferroni test, Scheffe test, and Tukey test. Statistical significance was defined as a p < 0.05. The inflammation scores and myotoxicity scores were reported as medians (n = 4 per experimental group). Statistical analysis were performed with Origin 2021 software (Origin Lab Corp. Northampton, MA).
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM144388. This research was partially supported by the University of Alabama’s Small Grant Program Award (GR14972). U.W. acknowledges support for her pain research program at the University of Alabama at Birmingham through the William A. Lell, M.D. Paul N. Samuelson, M.D. Endowed Professorship in Anesthesiology and in part from NCI, NIH (R01CA246708).
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Footnotes
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. U.W. reports research grants from the US National Institutes of Health. She serves on the External Consultant Board for the “NIH Preclinical Screening Platform for Pain”, a novel pre-clinical pain therapy screening platform that has been launched at the National Institute for Neurological Disorders and Stroke in the U.S. In her capacity as a special government employee of the US Food and Drug Administration (FDA), she has served as a voting member of the FDA Anesthetic and Analgesic Drug Products Advisory Committee. In the past 3 years she has received compensation for serving on advisory boards or for consulting activities for Aphrodite Health Inc., Wilmington, DE, Avenue Therapeutics Inc., New York, NY, Bayer Aktiengesellschaft, Leverkusen, Germany, and Biohaven Pharmaceuticals, New Haven, CT, all unrelated to the submitted work
Contributor Information
Qi Li, Department of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA.
Xiaosi Li, Department of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA.
Elizabeth Bury, Department of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA.
Amanda Koh, Department of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA.
Kimberly Lackey, Department of Biological Sciences, University of Alabama, Tuscaloosa, AL 35487, USA.
Ursula Wesselmann, Department of Anesthesiology and Perioperative Medicine, Division of Pain Medicine, and Department of Neurology, Consortium for Neuroengineering and Brain-Computer Interfaces, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Tony Yaksh, Department of Anesthesiology, University of California at San Diego, La Jolla, CA 92093, USA.
Chao Zhao, Department of Chemical and Biological Engineering, Center for Convergent Biosciences and Medicine, Alabama Life Research Institute, University of Alabama, Tuscaloosa, AL 35487, USA.
References
- [1].Vigata M, Meinert C, Hutmacher D, Bock N, Pharmaceutics 2020, 12, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abasalizadeh F, Moghaddam SV, Alizadeh E, Akbari E, Kashani E, Fazljou SMB, Torbati M, Akbarzadeh A, J. Biol. Eng 2020, 14, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoare TR, Kohane DS, Polymer 2008, 49, 1993. [Google Scholar]
- [4].Li J, Mooney DJ, Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vermonden T, Censi R, Hennink WE, Chem. Rev 2012, 112, 2853. [DOI] [PubMed] [Google Scholar]
- [6].Larrañeta E, Stewart S, Ervine M, Al-Kasasbeh R, Donnelly RF, J. Funct. Biomater 2018, 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shomorony A, Santamaria CM, Zhao C, Rwei AY, Mehta M, Zurakowski D, Kohane DS, Anesth. Analg 2019, 129, 709. [DOI] [PubMed] [Google Scholar]
- [8].Zhan C, Wang W, Santamaria C, Wang B, Rwei A, Timko BP, Kohane DS, Nano Lett 2017, 17, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu Q, Santamaria CM, Wei T, Zhao C, Ji T, Yang T, Shomorony A, Wang BY, Kohane DS, Nano Lett 2018, 18, 32. [DOI] [PubMed] [Google Scholar]
- [10].Zhao C, Liu A, Santamaria CM, Shomorony A, Ji T, Wei T, Gordon A, Elofsson H, Mehta M, Yang R, Kohane DS, Nat. Commun 2019, 10, 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ji T, Li Y, Deng X, Rwei AY, Offen A, Hall S, Zhang W, Zhao C, Mehta M, Kohane DS, Nat. Biomed. Eng 2021, 5, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kohane DS, Smith SE, Louis DN, Colombo G, Ghoroghchian P, Hunfeld NGM, Berde CB, Langer R, Pain 2003, 104, 415. [DOI] [PubMed] [Google Scholar]
- [13].Li X, Zhao Y, Zhao C, iScience 2021, 24, 102810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li X, Li Q, Zhao C, ACS Omega 2021, 6, 13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li X, Li Q, Song S, Stevens AO, Broemmel Z, He Y, Wesselmann U, Yaksh T, Zhao C, Adv. Ther 2023, 6, 2200199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Garske DS, Schmidt-Bleek K, Ellinghaus A, Dienelt A, Gu L, Mooney DJ, Duda GN, Cipitria A, Tissue Eng. Part A 2020, 26, 852. [DOI] [PubMed] [Google Scholar]
- [17].Li Q, Li X, Zhao C, Front. Bioeng. Biotechnol 2020, 8, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ji T, Ding Y, Zhao Y, Wang J, Qin H, Liu X, Lang J, Zhao R, Zhang Y, Shi J, Tao N, Qin Z, Nie G, Adv. Mater 2015, 27, 1865. [DOI] [PubMed] [Google Scholar]
- [19].González-Cano R, Ruiz-Cantero MC, Santos-Caballero M, Gómez-Navas C, Tejada MÁ, Nieto FR, Toxins 2021, 13, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hagen NA, Fisher KM, Lapointe B, du Souich P, Chary S, Moulin D, Sellers E, Ngoc AH, J. Pain Symptom Manag 2007, 34, 171. [DOI] [PubMed] [Google Scholar]
- [21].Sökmen N, Uysal S, Ayhan F, ÖZYAZICI M, Ayhan H, Hacettepe J Biol. Chem 2009, 37, 337. [Google Scholar]
- [22].Mahmood S, Almurisi SH, Al-Japairai K, Hilles AR, Alelwani W, Bannunah AM, Alshammari F, Alheibshy F, Gels 2021, 7, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dash S, Murthy PN, Nath L, Chowdhury P, Acta Pol. Pharm 2010, 67, 217. [PubMed] [Google Scholar]
- [24].Rwei AY, Sherburne RT, Zurakowski D, Wang B, Kohane DS, Anesth. Analg 2018, 126, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McAlvin JB, Padera RF, Shankarappa SA, Reznor G, Kwon AH, Chiang HH, Yang JS, Kohane DS, Biomaterials 2014, 35, 4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bingham E, Cohrssen B, Powell CH, Chem. Health Saf 2001, 8, 45. [Google Scholar]
- [27].Deng Y, Saucier-Sawyer JK, Hoimes CJ, Zhang J, Seo Y-E, Andrejecsk JW, Saltzman WM, Biomaterials 2014, 35, 6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McKenzie M, Betts D, Suh A, Bui K, Kim LD, Cho H, Molecules 2015, 20, 20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhan C, Santamaria CM, Wang W, McAlvin JB, Kohane DS, Biomaterials 2018, 181, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Du F, Bobbala S, Yi S, Scott EA, Control J. Release 2018, 282, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhao C, Li X, Li L, Cheng G, Gong X, Zheng J, Langmuir 2013, 29, 1517. [DOI] [PubMed] [Google Scholar]
- [32].Fowles JR, Banton MI, Pottenger LH, Crit. Rev. Toxicol 2013, 43, 363. [DOI] [PubMed] [Google Scholar]
- [33].Jafari M, Abolmaali SS, Najafi H, Tamaddon AM, Int. J. Pharm 2020, 576, 118959. [DOI] [PubMed] [Google Scholar]
- [34].Imran ul-haq M, Lai BFL, Chapanian R, Kizhakkedathu JN, Biomaterials 2012, 33, 9135. [DOI] [PubMed] [Google Scholar]
- [35].Kainthan RK, Janzen J, Levin E, Devine DV, Brooks DE, Biomacromolecules 2006, 7, 703. [DOI] [PubMed] [Google Scholar]
- [36].Sunder A, Hanselmann R, Frey H, Mulhaupt R, Macromolecules 1999, 32, 4240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.