

Abstract
Tricyclic pyrone (TP) molecules have shown protection of MC65 neuroblastoma cells death induced by amyloid-β proteins through SβC gene, a decrease of amyloid-β peptide levels, and improvement of motor functions and memory in Alzheimer’s disease mouse and rat models. Mechanistic studies suggest TP molecules modulate N-methyl-D-aspartate receptor. A short synthesis of chiral TP analogs was sought using a Pd(0)-catalyzed displacement of TP allylic acetate intermediate with sodium azide or substituted benzylamines. A three-step sequence of reactions by the treatment of 2-{(5aS,7S)-3-methyl-1-oxo-1,5a,6,7,8,9-hexahydropyrano[4,3-b]chromen-7-yl}allyl acetate (9) with (Ph3P)4Pd and sodium azide, followed by reduction with Zn-NH4OCHO and coupling with 3-fluoro-4-hydroxybenzaldehyde and NaCNBH3 was found to give TP coupling molecule, (5aS,7S)-7-(1-(3-fluoro-4-hydroxybenzylamino)prop-2-en-2-yl)-3-methyl-6,7,8,9-tetrahydropyrano[4,3-b]chromen-1(5aH)-one (2), in a good yield. An alternative shorter pathway – a two-step sequence of reactions – by the displacement of 9 by 4-(t-butyldimethylsilyloxy)-3-fluoro-benzylamine with a catalytic amount of (Ph3P)4Pd in THF followed by removal of the silyl ether protecting group gave 2, albeit in a lower chemical yield. The described syntheses should provide general procedures for the synthesis of a library of TP molecules for the discovery of anti-Alzheimer drugs.
Keywords: NMDAR (N-methyl-D-aspartate receptor) modulators, Palladium-catalyzed displacement of allylic acetates by amines, Tricyclic pyrone molecules
Graphical Abstract

Introduction
Alzheimer’s disease (AD), a most common form of dementia in the elderly, inflicts millions of people.[1] Intra- and extra-cellular oligomers of amyloid-β (Aβ), derived from transmembrane amyloid-β protein precursor (APP) glycoproteins, and tau proteins have been hypothesized to initiate a cascade of molecular changes leading to synaptic dysfunction, inflammation, and neuronal death in the AD brain.[2] Following the amyloid cascade hypothesis, most AD therapeutics under development target Aβ peptide including vaccines, and to lesser extents, tau proteins, and N-methyl-D-aspartate (NMDA) receptors.[3] Current treatments include cholinesterase inhibitors, memantine,[1,3] and Aβ monoclonal antibody lecanemab,[3d] which improve the quality of life, but do not cure the disease. We screened a library of tricyclic pyrone molecules using MC65 cell lines for their neural protective properties against Aβ oligomers induced cell death.[4–7] Among the tested tricyclic pyrone molecules, CP2 (code name) and TP70 (or compound 1) are the most active molecules (Scheme 1).[6,7] Tricyclic pyrone molecules were initially synthesized based on the structure of pyripyropene A, a natural product obtained from the culture medium of Aspergillus fumigatus.[8] It was found that CP2 and TP70 oppositely modulate hippocampal excitatory postsynaptic potentials (EPSPs) and plasticity.[7] Similar to memantine, CP2 inhibits NMDAR (N-methyl-D-aspartate receptor)-mediated EPSPs, whereas TP70 enhances synaptic NMDAR-mediated EPSPs and restores AβO (amyloid-β oligomer)-impaired long-term potentiation (LTP). Hence, synthesis of TP70 analogs, such as molecule 2 (Scheme 1) was carried out.
Scheme 1.
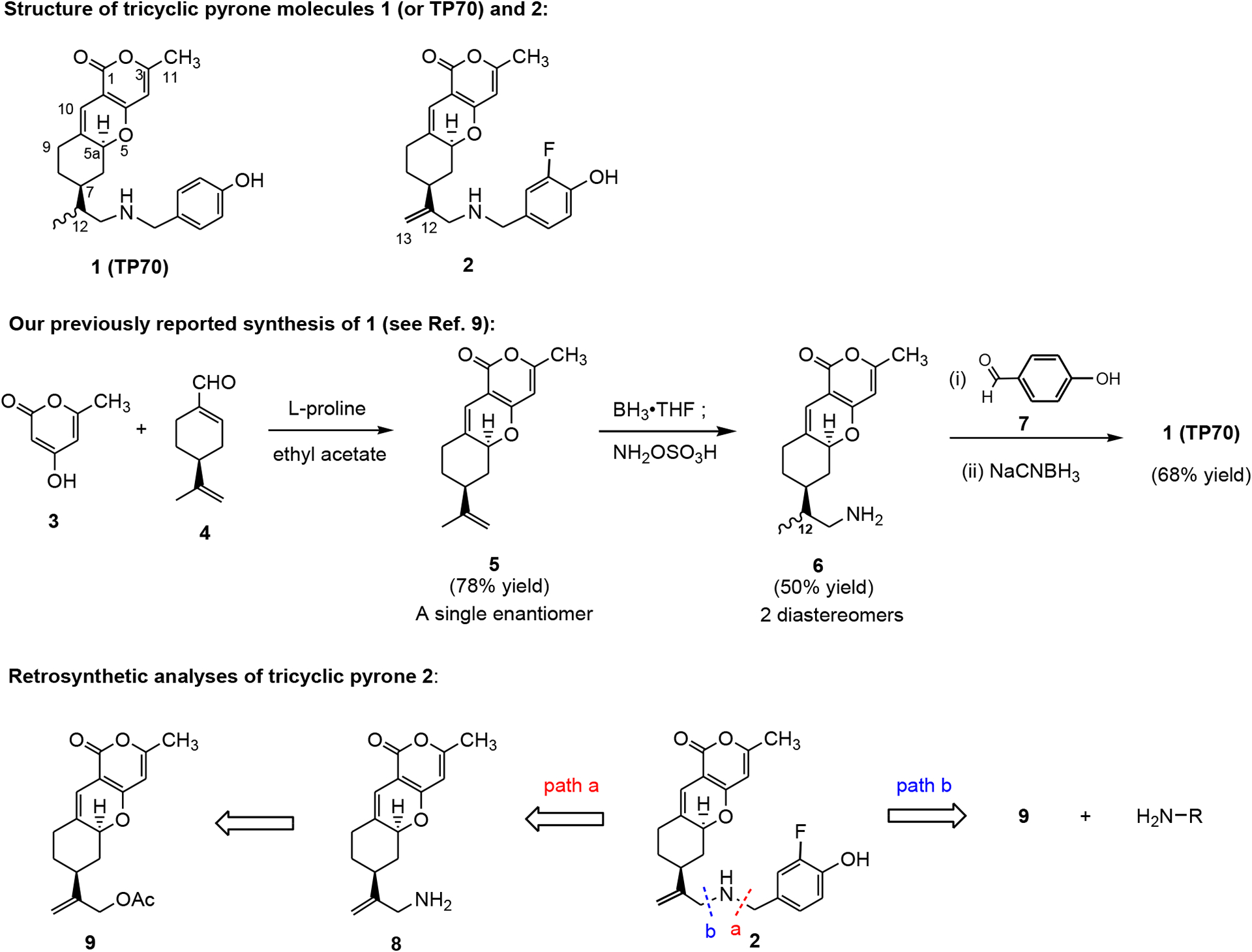
Structures of TP70 (1) and new analog 2, previously reported synthesis of TP70 (1),[9] and retrosynthetic analyses of 2.
Previously, a reductive amination reaction was used for the synthesis of various tricyclic pyrone molecules - such as 1 - starting from amine 6 and various aldehydes, such as 7 (Scheme 1).[9] However, in the preparation of amine 6 via a hydroboration/amination procedure of optically pure tricyclic pyrone 5, two chiral stereoisomers at C12, were produced, which are inseparable by silica gel column chromatography. We report herein new and facile approaches for the synthesis of tricyclic pyrone amine derivative 2, eliminating the stereochemistry issue at C12, by two different routes and their retrosyntheses depicted in Scheme 1. A fluorine incorporated into the aromatic ring of 2 is due to its hydrogen-bond acceptor character, improving absorption, distribution, metabolism, and excretion property in biological systems.[10] Similar to the synthesis of TP70, tricyclic pyrone molecule 2 could be obtained by the coupling of an aldehyde and amine 8. Amine 8 would derive from allylic acetate 9, as illustrated in path a of the retrosynthetic analysis. In addition, molecule 2 might be achieved by a direct displacement of 9 with various amines, depicted in path b of the retrosynthetic analysis. The latter method would allow a short synthesis of a library of analogs of TP70.
Results and Discussion
The synthesis started from tricyclic pyrone acetate 9, which obtained by following a previously reported allylic oxidation of 5 using palladium acetate, 1,4-benzoquinone and diethyl malonate in acetic acid (Scheme 2).[5,11] Modification in the work-up procedure allowed a slightly increase of chemical yield of 9 to 68% yield. Displacement of the acetate group of 9 with sodium azide and a catalytic amount of tetrakis(triphenylphosphine)palladium[12] followed by reduction of the azido group with activated zinc and ammonium formate afforded allylic amine 8 in a 48% overall yield. Similar to amine 6, allylic amine 8 serves as a key intermediate for the synthesis of various tricyclic pyrone molecules.[6,9] For example, coupling of 8 with 3-fluoro-4-hydroxybenzaldehyde (11) under a reductive amination reaction condition gave tricylic pyrone 2 in a 55% yield.
Scheme 2.

Synthesis of tricyclic pyrone 2 via a reductive amination process.
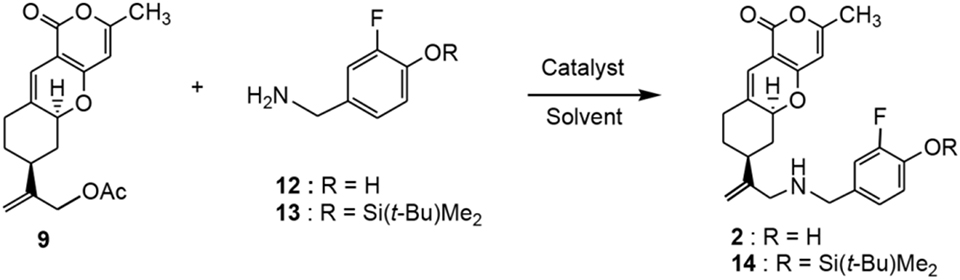
An alternative and shorter route for the synthesis of 2 was sought via a direct displacement[13a] of allylic acetate 9 with primary amines using palladium catalysts. The reported palladium-catalyzed displacement of naphthylmethyl acetates with morpholine, using N,N’-dimethylpyropyleneurea (DMPU) as a solvent, gave moderate to good yields of the products.[13a] However, the yields are solvent dependent along with limited acetate substrates. In addition to displacement of allylic acetates, direct allylic aminations of Pd(II)-allyl complexes or alkenes were restricted to the use of highly stabilized anion deriving from N-fluoro-dibenzenesulfonimide and KF.[13b,c] Hence, we studied the displacement reactions of allylic acetate 9 with 3-fluoro-4-(aminomethyl)phenol (12) or 3-fluoro-4-(t-butyldimethylsilyloxy)benzylamine (13) catalyzed by different palladium complexes in the presence of a base, and in different solvents such as DMF, THF or DMPU. Results of the displacement reactions are summarized in Table 1. The reaction of 9 and 2 equiv. of amine 12 in the presence of 3 mol% of Pd(PPh3)4 or tris(dibenzylideneacetone)-dipalladium(0)•chloroform [Pd2(dba)3•CHCl3] and 3 eq. of diisopropylethylamine (DIPEA) or cesium carbonate, respectively, in DMF gave no detectable of the desired displacement product 2. Only starting materials were recovered, see Entries 1 and 2 of Table 1. Presumably, the phenolic group of 12 might hinder the displacement reaction. Therefore, t-butyldimethylsilyl (TBS) protected benzylamine 13 was used in the displacement reactions. When 3 mol% of Pd2(dba)3•CHCl3 and 5 mol% of 1,2-bis(diphenylphosphine)ethane (DPPE) along with cesium carbonate in THF at 50 °C, reaction of 9 and 13 provided a trace amount (~2%) of the desired product 14, Entry 3. Likewise, under similar reaction conditions but using 3 mol% of Pd(PPh3)4 and DIPEA in THF at 60 °C also gave a trace amount (~3%) of 14, Entry 4. An attempt to use a Pd(II) complex, 4,7-diphenyl-1,10-phenanthroline palladium bis(trifluoroacetate) [(DPP)Pd(OCOCF3)2][14] in DMPU[13a] at 80 °C without DPPE failed as well to obtain the desired product 14, Entry 5. Since the presence of DPPE and DMPU did not afford any desired product, the use of Pd(PPh3)4, in the absence of DPPE, under increment temperatures from 50 – 80 °C were conducted, Entries 7 – 10. Interestingly, at 50 °C for 20 h, a 3% yield of 14 was isolated, Entry 7, while reactions at 60 and 70 °C for 12 and 18 h, respectively, provided 8% and 11% yield of 14, Entries 8 and 9. Fortuitously, an increase of the amount of Pd(PPh3)4 to 0.1 equiv. and at 80 °C in THF for 2 days furnished a 20% yield of 14 along with 25% recovery of acetate 9, Entry 10. Notably, under similar reaction conditions, the use of amine 12 did not produce compound 2.
Table 1.
Reaction conditions and results of the Pd(0)-catalyzed displacement of tricyclic pyrone acetate 9 (1 equiv.) with aryl amine 12 or 13. Formation of compound 14. DIPEA: N,N-diisopropylethylamine; NR: no reaction; DPPE: 1,2-bis(diphenylphosphine)ethane.
| Entry | Amine (2 eq.) | Catalyst (0.03 eq.) | Ligand (eq.) | Base (3 eq.) | Solvent | Temp (°C) | Time (hour) | Yield (%)[a] |
|---|---|---|---|---|---|---|---|---|
| 1 | 12 | Pd(PPh3)4 | None | DIPEA | DMF | 25 | 48 | NR |
| 2 | 12 | Pd2(dba)3•CHCl3 | None | Cs2CO3 | DMF | 25 | 48 | NR |
| 3 | 13 | Pd2(dba)3•CHCl3 | DPPE (0.05) | Cs2CO3 | THF | 50 | 24 | 2 |
| 4 | 13 | Pd(PPh3)4 | DPPE (0.05) | DIPEA | THF | 60 | 24 | 3 |
| 5 | 13 | (DPP)Pd(TFA)2 | None | DIPEA | DMPU | 80 | 120 | 0 |
| 6 | 13 | Pd(PPh3)4 | None | DIPEA | DMPU | 80 | 120 | 0 |
| 7 | 13 | Pd(PPh3)4 | None | DIPEA | THF | 50 | 20 | 3 |
| 8 | 13 | Pd(PPh3)4 | None | DIPEA | THF | 60 | 12 | 8 |
| 9 | 13 | Pd(PPh3)4 | None | DIPEA | THF | 70 | 18 | 11 |
| 10 | 13 | Pd(PPh3)4[b] | None | DIPEA | THF | 80 | 48 | 20 |
Note:
Starting material 12 or 13 recovered in all Entries in varies amounts. The reported yields are isolated yields.
0.10 equiv. of Pd(PPh3)4 was used, and 47% of 9 was recovered. NR: no reaction.
Removal of the TBS protecting of 14 with n-Bu4NF in THF gave 2 in a 81% yield, whose spectral data is identical with that obtained from the coupling of amine 8 and aldehyde 11. Tricyclic pyrone amine 14 also unequivocally synthesized in a 67% yield by the coupling of amine 8 and aldehyde 15 in methanol followed by reduction with sodium cyanoborohydride. Benzylamine 13 was prepared by the silylation of 4-cyano-2-fluorophenol (16) with t-butyldimethylsilyl chloride and by hydrogenation with Pd/C and hydrogen in an 80% overall yield (Scheme 3). 4-(Aminomethyl)-2-fluorophenol (12) was obtained in a 55% yield from the hydrogenation of 16 with 5% Pd/C and 30 psi of hydrogen in methanol at 25 °C.
Scheme 3.

Synthesis of tricyclic pyrone 14 from the displacement reaction of tricyclic pyrone acetate 9 and aryl amine 13.
Conclusion
Facile displacement reactions of the allylic acetate group of a tricyclic pyrone molecule with sodium azide or substituted benzylamines catalyzed by Pd(PPh3)4 was found. Expectedly, the former displacement reaction gave a high yield of the tricyclic pyrone allylic azide, since the azide ion is a weak base and good nucleophile. The latter displacement reaction with substituted benzylamine appears to be a challenging one, since the primary benzylamine is a stronger base and weaker nucleophile comparing with the azide ion. Upon examination of several palladium catalysts, different bases and solvents, a catalytic amount of Pd(PPh3)4 and diisopropylethylamine in THF yielded the desired displacement product, albeit in a low yield. The finding highlights the difficulty in displacing moderately complex allylic acetate molecules with substituted benzylamines.
Experimental Section
General Methods.
Chemicals were purchased from Fisher Scientifics and Avantor VWR International. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra obtained from a Bruker Avance Neo 400-MHz NMR spectrometer, and were measured from a solution in CDCl3 unless otherwise stated. The chemical shift data reported in 1H NMR are given in units of δ relative to TMS (δ = 0) or CHCl3 (δ = 7.26 ppm). For 13C NMR spectra, the chemical shifts are recorded in ppm relative to CDCl3 (δ = 77.0 ppm). Low-resolution mass spectra were taken from a Waters Acquity TQD Ultra Performance LC/MS/MS system. High-resolution mass spectra obtained using a LCT Premier time of flight mass spectrometer (Waters Inc.). All solvents distilled over appropriated drying agent such as CaH2 for DMF, dichloromethane and acetonitrile, or Na/benzophenone for THF and diethyl ether. Flash column chromatography carried out on silica gel (400 mesh) for purification of organic products. All compounds were characterized by 1H and 13C NMR spectroscopies, and either low-resolution mass spectrometry for known compounds or high-resolution mass spectrometry for new molecules.
2-{(5aS,7S)-3-Methyl-1-oxo-1,5a,6,7,8,9-hexahydropyrano[4,3-b]chromen-7-yl}allyl acetate (9), an improved procedure.
To a solution of 3.0 g (11.8 mmol) of tricyclic pyrone 5,[5] 3.76 g (24.8 mmol) of 1,4-benzoquinone, and 0.373 g (2.3 mmol) of diethyl malonate in 75 mL of acetic acid under argon at 25 °C, was added 0.26 g (1.16 mmol) of palladium acetate. The mixture was heated to 75 °C and stirred for 2 h and then at 50 °C for 4 days. The reaction mixture was cooled to 25 °C, and acetic acid was removed under reduced pressure on a rotary evaporator. To the residue, 50 mL water and 100 mL ethyl acetate were added, and the resulting mixture was filtered through Celite to remove the gummy semisolid materials. The semisolid materials were washed with 100 mL water and 100 mL ethyl acetate. The filtrate was extracted three times with ethyl acetate (100 mL each) and the combined extracts were washed with aqueous sodium bicarbonate containing few drops of aqueous NaOH to remove acetic acid, and washed with water and brine. The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was diluted with 150 mL of ethyl acetate, washed with 100 mL of 5 % NaOH and then 50 mL of saturated aqueous solution of Na2S2O5, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and ethyl acetate (ratios increased from 4:1 to 3:2) as eluents. The fractions containing desired product 9 and 1,4-dihydroxybenzene were combined. This mixture was washed with 2% aqueous NaOH solution, saturated aqueous Na2S2O5 solution, and water, dried (MgSO4), concentrated to give 2.5 g (68% yield) of compound 9 as a light yellow oil: Rf = 0.30 (hexane/ethyl acetate 2:1); [α]D20 = +6.82 (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.08 (s, 1H, =CH), 5.70 (s, 1H, =CH), 5.08 (s, 1H, =CH2), 5.06 – 5.05 (m, 1H, CHO), 4.98 (s, 1H, =CH2), 4.54 (s, 2H, CH2OAc), 2.48 (dt, J = 14, 4 Hz, 1H), 2.25 – 2.23 (m, 2H), 2.17 (s, 3H, Me), 2.07 (s, 3H, CH3CO), 2.04 – 2.00 (m, 1H), 1.91 – 1.86 (m, 1H), 1.77 (dd, J = 12, 11 Hz, 1H), 1.29 (qd, J = 12.8, 4 Hz, 1H); 13C NMR (100 MHz, CDCl3) 171.0, 163.6, 162.8, 162.0, 146.5, 132.0, 113.0, 110.0, 100.0, 97.6, 79.3, 66.2, 40.1, 39.6, 32.5, 32.4, 21.3, 20.4. Spectral data are identical to those reported.[5]
(5aS,7S)-7-(3-Azido-prop-1-en-2-yl)-3-methyl-6,7,8,9-tetrahydropyrano[4,3-b]chromen-1(5aH)-one (10).
To 0.257 g (0.2 mmol) of Pd(PPh3)4 in a two-necked round-bottom flask under argon at 25 °C, was added a solution of 2.337 g (7.4 mmol) of tricyclic pyrone acetate 9 and 1.2 g (18.5 mmol) of sodium azide in degassed THF and water (1:1). The reaction mixture was stirred at 70 °C for 3 hours. After cooling to room temperature, most of the THF solvent was removed on a rotary evaporator. The residue was diluted with 30 mL of 1N HCl and extracted three times with ethyl acetate (40 mL each). The combined extract was washed with saturated aqueous sodium bicarbonate (40 mL) and brine (35 mL), dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate as eluents to give 1.703 g (77 % yield) of compound 10 as a white solid: Rf = 0.27 (hexane/ethyl acetate 2:1); m.p. 85 – 87 °C; [α]D20 = +5.48 (c = 0.36 in CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.09 (s, 1H, =CH), 5.70 (s, 1H, =CH), 5.12 – 5.01 (m, 1H, CHO), 5.08 (s, 1H, =CH2), 5.05 (s, 1H, =CH2), 3.76 (s, 2H, CH2N), 2.47 (ddd, J = 14.3, 4.1, 2.4 Hz, 1H), 2.26 – 2.18 (m, 2H), 2.17 (s, 3H, Me), 2.05 (tdd, J = 14.4, 4.5, 2.2 Hz, 1H), 1.91 – 1.84 (m, 1H), 1.74 (dd, J = 12, 11 Hz, 1H), 1.35 – 1.23 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 163.2, 162.5, 161.7, 145.6, 131.5, 114.0, 109.8, 99.72, 97.3, 78.9, 55.1, 39.8, 39.7, 32.2, 32.0, 20.1. MS (ESI): m/z = 299.562 ([M]+); HRMS-ESI: m/z [M + H]+ calcd for C16H18N3O3+: 300.1343, found: 300.1348.
(5aS,7S)-7-(1-Aminoprop-2-en-2-yl)-3-methyl-6,7,8,9-tetrahydropyrano[4,3-b]chromen-1(5aH)-one (8).
A three-necked flask containing 1.687 g (5.64 mmol) of azide 10 and 1.780 g (28.2 mmol) of ammonium formate was dried under vacuum and maintained under argon at 25 °C. To it, 30 mL of dried methanol and 1.474 g (22.56 mmol) of activated zinc dust were added. The reaction mixture was stirred at 25 °C for 12 hours, filtered through Celite, washed with ethyl acetate, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 100 mL of ethyl acetate and extracted three times with aqueous 1N HCl (20 mL each). The combined aqueous layers were basified with 1N NaOH to pH 12, and extracted three times with ethyl acetate (40 mL each). The combined organic layer was washed with water (50 mL) and brine (50 mL), dried (anhydrous Na2SO4), concentrated, and recrystallized using dichloromethane and n-pentane (in a ratio of 1:15) to afford 0.949 g (62% yield) of amine 8 as a light yellow oil: Rf = 0.22 (CH2Cl2/MeOH 5:1); [α]D20 = +7.27 (c = 0.33 in CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.11 (s, 1H, =CH), 5.72 (s, 1H, =CH), 5.10 (dd, J = 11.4, 4.8 Hz, 1H, CHO), 5.00 (s, 1H, =CH2), 4.88 (s, 1H, =CH2), 3.30 (s, 2H, CH2N), 2.50 – 2.45 (m, 1H), 2.30 – 2.20 (m, 2H), 2.19 (s, 3H, Me), 2.14 – 2.00 (m, 1H), 1.87 (ddt, J = 13.2, 5.6, 2.9 Hz, 1H), 1.75 (dd, J = 12.7, 11.3 Hz, 1H), 1.44 – 1.15 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 163.0, 162.2, 161.3, 152.4, 131.7, 109.2, 107.9, 99.5, 97.0, 78.8, 45.2, 39.9, 39.8, 32.2, 32.1, 19.8. HRMS-ESI: m/z [M + H]+ calcd for C16H20NO3+: 274.1438, found: 274.1448.
(5aS,7S)-7-(1-(3-Fluoro-4-hydroxybenzylamino)prop-2-en-2-yl)-3-methyl-6,7,8,9-tetrahydropyrano[4,3-b]chromen-1(5aH)-one (2).
A 100-mL three-necked flask containing 0.347 g (1.27 mmol) tricyclic pyrone amine 8 and 0.5 g of 3Å molecular sieves was dried under vacuum and then maintained under argon at 25 °C. To it, 20 mL of dry methanol was added followed by a solution of 0.160 g (1.14 mmol) of 3-fluoro-4-hydroxy-benzaldehyde (11) in 15 mL of dry methanol. The reaction mixture was stirred at 25 °C for 12 hours. The reaction progress was monitored by analyzing the 1H NMR spectrum of an aliquot taken from the reaction mixture, which showed the presence of ~10% of aldehyde 11. Additional 40 mg (0.15 mmol) of amine 8 in 2 mL of dry methanol was added to the reaction mixture and stirred for one hour. To the reaction mixture, 25 μL of acetic acid and a solution of 0.118 g (1.9 mmol) of sodium cyanoborohydride in 5 mL of methanol were added, stirred at 25 °C for 2 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. The residue was dissolved in 40 mL of ethyl acetate and extracted five times with 1N HCl (30 mL each). The combined extracts were basified to pH 11 using 1N NaOH, and the resulting solution was extracted five times with dichloromethane (30 mL each). The combined dichloromethane extract was dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford 0.257 g (55% yield) of 2 as a viscous oil: Rf = 0.23 (CH2Cl2/MeOH 5:1); [α]D20 = +47.3 (c = 0.22 in MeOH) (for the HCl salt); 1H NMR (400 MHz, CDCl3) δ 7.04 (dd, J = 11.5, 1.8 Hz, 1H), 6.98 – 6.85 (m, 2H), 6.10 (s, 1H, =CH), 5.72 (s, 1H, =CH), 5.10 (dd, J = 11.3, 5.1 Hz, 1H, CHO), 5.02 (s, 1H, =CH2), 4.90 (s, 1H, =CH2), 3.68 (s, 2H, CH2N), 3.23 (s, 2H, CH2N), 2.48 (dt, J = 14.1, 3.4 Hz, 1H), 2.33 – 2.15 (m, 2H), 2.19 (s, 3H, Me), 2.14 – 2.00 (m, 1H), 1.92 – 1.70 (m, 2H), 1.37 – 1.26 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 163.6, 162.9, 161.9, 151.6 (d, J = 240 Hz, C-F), 149.8, 143.4, 132.1, 124.7, 118.1, 115.9 (d, J1,3 = 18 Hz, C-F coupling), 110.8, 109.8, 100.1, 97.6, 79.5 (CHO), 53.0, 52.7, 40.4, 40.2, 32.7, 32.6, 30.0, 20.4. HRMS-ESI: m/z [M + H]+ calcd for C23H25FNO4+: 398.1768, found: 398.1764.
A representative palladium-catalyzed displacement reaction. (5aS,7S)-7-{1-[3-Fluoro-4-(t-butyldimethylsilyloxy)benzylamino)prop-2-en-2-yl)-3-methyl-6,7,8,9-tetrahydropyrano[4,3-b]chromen-1(5aH)-one (14) from Pd(PPh3)4 catalyzed displacement reaction of acetate 9 and benzylamine 13.
A sealable tube containing 50 mg (0.158 mmol) tricyclic pyrone acetate 9, 60.5 mg (0.237 mmol) of amine 13, and 20 mg (15 μmol) of Pd(PPh3)4 was dried under vacuum and then maintained under argon at 25 °C. To it was added 2 mL of dried THF followed by 20 mg (0.156 mmol) of N,N-diisopropylethylamine (Hünig’s base), and the reaction tube was sealed and heated to 80 °C with stirring for 2 days. It was cooled to 25 °C, diluted with water, and extracted three times with dichloromethane. The combined extract was washed with water and brine, dried (MgSO4), concentrated and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate as eluents to give 16 mg (20% yield) of tricyclic pyrone 14 as a viscous oil and 12 mg (25% recovery) of 9. Compound 14: Rf = 0.40 (CH2Cl2/MeOH 5:1); [α]D20 = +87.9 (c = 0.38 in CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.04 (d, JC-F = 11.2 Hz, 1H, Ar C2-H), 6.91 (d, J = 8.8 Hz, 1H, Ar C6-H), 6.85 (dd, J = 10, 6.8 Hz, Ar C5-H), 6.10 (s, 1H, =CH), 5.71 (s, 1H, =CH), 5.12 – 5.06 (m, 1H, CHO), 5.01 (s, 1H, =CH2), 4.89 (s, 1H, =CH2), 3.68 (s, 2H, NCH2Ar), 3.22 (s, 2H, CH2N), 2.43 (d, J = 14.8 Hz, 1H), 2.25 – 2.20 (m, 1H), 2.19 (s, 3H, Me), 2.10 – 2.00 (m, 1H), 1.88 – 1.81 (m, 1H), 1.74 (q, J = 11 Hz, 1H), 1.34 – 1.24 (m, 2H), 1.00 (s, 9H, t-Bu), 0.18 (s, 6H, 2 Me); 13C NMR (100 MHz, CDCl3) δ 163.3, 162.5, 161.6, 154.1 (d, J = 242 Hz, C-F), 150.2, 134.4, 132.1, 123.7, 122.0, 116.0 (d, J1,3 = 18.6 Hz, C-F coupling), 110.1, 109.5, 99.8, 97.4, 79.3 (CHO), 52.8, 52.4, 40.2, 39.9, 32.4, 25.6 (3C, t-Bu), 20.1, 18.3, −4.7 (2C, Me2Si). HRMS-ESI: m/z [M + H]+ calcd for C29H39FNO4Si+: 512.2632, found: 512.2641.
Conversion of 14 to 2.
To a solution of 10 mg (0.019 mmol) of compound 14 in 1 mL of THF under argon at 25 °C, was added 0.1 mL (0.095 mmol) of n-Bu4NF (1.0 M in THF). The solution was stirred for 1 h, diluted with 10 mL of water, and extracted twice with CH2Cl2. The combined extracts were washed with brine, dried (anhydrous Na2SO4), and concentrated to give 6.8 mg (87% yield) of compound 2, whose spectral data were identical to those obtained from aforementioned coupling reaction of tricyclic pyrone amine 8 and benzaldehyde 11.
Coupling of amine 8 and aldehyde 15. Formation of compound 14.
A mixture of 39 mg (0.14 mmol) tricyclic pyrone amine 8 and 0.1 g of 3Å molecular sieves was dried under vacuum and then maintained under argon at 25 °C. To it, 2 mL of dry methanol was added followed by a solution of 33 mg (0.13 mmol) of 4-(t-butyldimethylsilyloxy)-3-fluorobenzaldehyde (15) in 1 mL of dry methanol. The reaction mixture was stirred at 25 °C for 12 hours and added 5 μL of acetic acid followed by the addition of a solution of 13 mg (0.21 mmol) of sodium cyanoborohydride in 1 mL of methanol. The mixture was stirred at 25 °C for 2 hours, filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The residue as subjected to column chromatographic purification on silica gel using a gradient mixture of hexane, ethyl acetate, and methanol gave 44 mg (67% yield) of 14, whose spectral data are identical to those obtained from the aforementioned displacement reaction.
4-(Aminomethyl)-2-fluorophenol (12).
To a solution of 1.0 g (7.3 mmol) of 3-fluoro-4-hydroxybenzonitrile (16) in 30 mL of methanol in a hydrogenation flask at 25 °C, was added 0.59 g (0.55 mmol) of 10% Pd/C, and the mixture was shaken on a Parr hydrogenator at 30 psi H2 for 2 h. The mixture was taken out from the hydrogenator, filtered through a pad of Celite, washed with methanol, and the filtrate was concentrated to dryness. The residue was purified by silica gel column chromatography using a gradient mixture of hexane, ethyl acetate and methanol as eluents to give 0.566 g (55% yield) of compound 12.[16] Since the amine is unstable, its HCl salt was prepared: m.p. 210–211 °C (as a HCl salt); MS (ESI; positive mode): m/z calcd for C7H9FNO+ (M+H)+: 142.1, found 142.2. 1H NMR (400 MHz, CDCl3) (as a HCl salt) δ 7.22 (dd, J = 11.6, 2 Hz, 1H), 7.12 (dd, J = 8.4, 2 Hz, 1H), 7.04 (t, J = 8.6 Hz, 1H), 4.09 (s, 2H); 13C NMR (100 MHz, CDCl3) (as a HCl salt) δ 150.9 (d, JC,F(1,2) = 239 Hz, C2), 143.9 (d, JC,F(1,3) = 12.5 Hz, C1), 125.7 (C5), 124.9 (d, JC,F(1,4) = 6.3 Hz, C4), 118.1 (C6), 116.8 (d, JC,F(1,3) = 19.1 Hz, C3), 42.3 (CH2N). The spectral data are in agreement with the assigned structure.
4-(tert-Butyldimethylsilyloxy)-3-fluorobenzaldehyde (15).
A mixture of 0.50 g (3.57 mmol) of 3-fluoro-4-hydroxybenzaldehyde, 0.32 g (4.64 mmol) of imidazole and 22 mg (0.18 mmol) of 4-(N,N-dimethylamino)pyridine (DMAP) was dried under vacuum and maintained under argon at 0 °C. To it were added 10 mL of dichloromethane and 0.81 g (5.4 mmol) of t-butyldimethylsilyl chloride. The solution was stirred at 25 °C for 12 h, filtered to remove the solids, and the solids were washed with ethyl acetate. The combined organic filtrate was washed with 10 mL of 1N HCl, 10 mL of 1% aqueous NaOH until pH ~10, and 20 mL each of water and brine. The organic layers was dried over MgSO4, concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate as eluents to give 0.798 mg (88% yield) of compound 15:[15] Rf = 0.42 (hexane/diethyl ether 3:1); MS (ESI; positive mode): m/z calcd for C13H20FO2Si (M+H)+: 255.3, found 255.1. 1H NMR (400 MHz, CDCl3) δ 9.86 (s, 1H, CHO), 7.61 – 7.54 (m, 2H, Ar), 7.05 – 7.03 (m, 1H, Ar), 1.02 (s, 9H, t-Bu), 0.24 (s, 6H, Me2); 13C NMR (100 MHz, CDCl3) δ 190.0 (CHO), 154,5 (d, JC-F(1,2) = 246 Hz, C3), 149.5 (d, JC-F(1,3) = 12 Hz, C4), 131.0 (C1), 127.5 (C2), 122.3 (C6), 116.6 (d, JC-F(1,4) = 19 Hz, C5), 25.5 (3C, t-Bu), 18.4 (C-Si), −4.7 (2C, Me). Spectral data are in agreement with the structure.
{4-[(t-Butyldimethylsilyl)oxy]-3-fluorophenyl}methanamine (13).
To a solution of 3.5 g (25.5 mmol) of 3-fluoro-4-hydroxybenzonitrile (16), 2.25 g (33.2 mmol) of imidazole and 0.156 g (1.27 mmol) of DMAP in 30 mL of dichloromethane under argon at 0 °C, was added 4.23 g (28 mmol) of t-butyldimethylsilyl chloride, and the solution was stirred at 25 °C for 16 h. The mixture was filtered and the solid was washed with 20 mL of dichloromethane. The filtrate was sequentially washed with 10 mL each of 1N HCl, 1M aqueous NaOH solution, water, and brine, dried (anhydrous Na2SO4), concentrated and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate to give 5.80 g (93% yield) of 4-[(t-butyldimethylsilyl)oxy]-3-fluorobenzonitrile.[17] Rf = 0.43 (hexane/diethyl ether 3:1); 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.33 (m, 2H), 6.99 (t, J = 8 Hz, 1H), 1.00 (s, 9H, Me3), 0.227 (s, 3H), 0.224 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.2 (d, JC,F = 246 Hz, C3), 148.1, 129.3, 123.0, 120.2, 118.0, 105.0 (C=N), 25.5 (3C, Me3), 18.3 (CSi), −4.73 (2C, Me2Si).
To a solution of 2.5 g (9.96 mmol) of the aforementioned benzonitrile in 30 mL of methanol in a hydrogenation flask at 25 °C was added 0.8 g (0.75 mmol) of 10% Pd/C, and the mixture was shaken on a Parr hydrogenator at 30 psi H2 for 4 h. The mixture was removed from the hydrogenator, filtered through a pad of Celite, washed with methanol, and the filtrate was concentrated to dryness. The residue was purified by silica gel column chromatography using a gradient mixture of hexane, ethyl acetate and methanol as eluents to give 2.18 g (86% yield) of compound 13: Rf = 0.31 (CH2Cl2/MeOH 9:1); MS (ESI; positive mode): m/z calcd for C13H23FNOSi (M+H)+: 256.4, found 256.2. 1H NMR (400 MHz, CDCl3) δ 7.0 – 6.75 (m, 3H, Ar-H), 3.78 (s, 2H, CH2N), 1.00 (s, 9H, t-Bu), 0.18 (s, 6H, Me2Si); 13C NMR (100 MHz, CDCl3) δ 154.1 (d, JC,F(1,2) = 243 Hz, C3), 141.9 (d, JC,F(1,3) = 12 Hz), 137.3 (d, JC,F(1,4) = 5 Hz), 122.4 (d, JC,F(1,3) = 48 Hz), 116.0 (d, JC,F(1,4) = 19 Hz), 115.0 (d, JC,F(1,5) = 19 Hz), 45.6 (CH2N), 25.8 (3C), 18.3 (CSi), −4.76 (2C). HRMS-ESI: m/z [M + H]+ calcd for C13H23FNOSi+: 256.1527, found: 256.1540.
Supplementary Material
Acknowledgement
We are grateful to the National Institutes of Health, National Institute of General Medical Sciences (R01 GM128659) for financial support in part of this research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Drs. Zongbo Tong and Deepak Barange for technical assistance.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information Summary
The 1H and 13C NMR spectra that support the findings of this study are available in the Supporting Information of this article.
References
- [1].a) Weller J, Budson A, F1000Research 2018, 7, 1161; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bhushan I, Kour M, Kour G, Gupta S, Sharma S, Yadav A, Ann. Biotechnol 2018, 1, 1002; [Google Scholar]; c) Castillo S, Hoie E, O’Brien KK, US Pharm. 2019, 44, 20–23. [Google Scholar]
- [2].Gulisano W, Maugeri D, Baltrons MA, Fa M, Amato A, Palmeri A, D’Adamio L, Grassi C, Devenand DP, Honig LS, Puzzo D, Arancio O, J. Alzheimer’s Dis 2018, 64, S611–S631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Rafii MS, Aisen PS, BMC Med. 2015, 13, 62; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sun X, Jin L, Ling P, Drug Discoveries Ther. 2012, 6, 285–290; [PubMed] [Google Scholar]; c) Cummings J, Lee G, Nahed P, Kambar MEZN, Zhong K, Fonseca J, Taghva K, Alzheimer’s Dement. 2022, 8, e12295; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S N. Engl. J. Med 2023, 388, 9–21. [DOI] [PubMed] [Google Scholar]
- [4].Hong H–S, Rana S, Barrigan L, Shi A, Zhang Y, Zhou F, Jin L–W, Hua DH, J. Neurochem 2009, 108, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rana S, Hong H–S, Barrigan L, Jin L–W, Hua DH, Bioorg. Med. Chem. Lett 2009, 19, 670–674. [DOI] [PubMed] [Google Scholar]
- [6].Pokhrel L, Maezawa I, Nguyen TDT, Chang K–O, Jin L–W, Hua DH, J. Med. Chem 2012, 55, 8969–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Maezawa I, Zou B, Di Lucente J, Cao WS, Pascual C, Weerasekara S, Zhang M, Xie XS, Hua DH, Jin L–W, J. Alzheimer’s Dis 2017, 58, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tomoda H, Tabata N, Nakata Y, Nishida H, Kaneko T, Obata R, Sunazuka T, Omura S, J. Org. Chem 1996, 61, 882–886. [Google Scholar]
- [9].Pokhrel L, Kim Y, Nguyen TDT, Prior AM, Lu J, Chang K–O, Hua DH, Bioorg. Med. Chem. Lett 2012, 22, 3480–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dalvit C, Invernizzi C, Vulpetti A, Chem. Eur. J 2014, 20, 11058–11068. [DOI] [PubMed] [Google Scholar]
- [11].McMurry JE, Kočotovský P, Tetrahedron Lett. 1984, 25, 4187–4190. [Google Scholar]
- [12].Murahashi S–I, Taniguchi Y, Imada Y, Tanigawa Y, J. Org. Chem 1989, 54, 3292–3303. [Google Scholar]
- [13].a) Toffano M, Legros J–Y, Fiaud J–C, Tetrahedron Lett. 1997, 38, 77–80. [Google Scholar]; b) Wang T–C, Rath NP, Mirica LM, Organometallics 2022, 41, 2067–2076. [Google Scholar]; c) Xiong T, Li Y, Mao L, Zhang Q, Zhang Q, Chem. Commun 2012, 48, 2246–2248. [DOI] [PubMed] [Google Scholar]
- [14].Brice JL, Meerdink JE, Stahl SS, Org. Lett 2004, 6, 1845–1848. [DOI] [PubMed] [Google Scholar]
- [15].Punchi Hewage AND, Yao H, Nammalwar B, Gnanasekaran KK, Lovell S, Bunce RA, Eschelman K, Phaniraj SM, Lee MM, Peterson BR, Battaile KP, Reitz AB, Rivera M, J. Am. Chem. Soc 2019, 141, 8171–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ma Z, Chandrashekhar VG, Zhou B, Alenad AM, Rockstroh N, Bartling S, Beller M, Jagadeesh RV, Chem. Sci 2022, 13, 10914–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Johansson JO, Hansen HC, Chiacchia FS, Wong NCW, World Intellectual Property Organization, WO2007016525 A2 2007-02-08.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.