

Abstract
Liver fibrosis is a wound-healing process characterized by excess formation of extracellular matrix (ECM) from activated hepatic stellate cells (HSCs). Previous studies show that both EZH2, an epigenetic regulator that catalyzes lysine 27 trimethylation on histone 3 (H3K27me3), and long non-coding RNA H19 are highly correlated with fibrogenesis. In the current study, we investigated the underlying mechanisms. Various models of liver fibrosis including Mdr2−/−, bile duct ligation (BDL) and CCl4 mice were adapted. We found that EZH2 was markedly upregulated and correlated with H19 and fibrotic markers expression in these models. Administration of EZH2 inhibitor 3-DZNeP caused significant protective effects in these models. Furthermore, treatment with 3-DZNeP or GSK126 significantly inhibited primary HSC activation and proliferation in TGF-β-treated HSCs and H19-overexpreesing LX2 cells in vivo. Using RNA-pull down assay combined with RNA immunoprecipitation, we demonstrated that H19 could directly bind to EZH2. Integrated analysis of RNA-sequencing (RNA-seq) and chromatin immunoprecipitation sequencing (ChIP-seq) further revealed that H19 regulated the reprogramming of EZH2-mediated H3K27me3 profiles, which epigenetically promoted several pathways favoring HSCs activation and proliferation, including epithelial-mesenchymal transition and Wnt/β-catenin signaling. In conclusion, highly expressed H19 in chronic liver diseases promotes fibrogenesis by reprogramming EZH2-mediated epigenetic regulation of HSCs activation. Targeting the H19-EZH2 interaction may serve as a novel therapeutic approach for liver fibrosis.
Keywords: liver fibrosis, H19, EZH2, epigenetic regulation, 3-DZNeP, quercetin
Introduction
Liver fibrosis is a reversible wound-healing response that is characterized by excessive deposition of extracellular matrix (ECM) and the formation of fibrous scar in the liver [1]. The etiology of liver fibrosis is typically triggered by repeated and chronic external stimuli, including but not limited to alcohol abuse, hepatitis B and C virus infections, biliary obstruction and overloaded lipids [2]. In response to these injuries, epithelial and endothelial cells are damaged first and release various physiological signaling substances to neighboring hepatic stellate cells (HSCs), thereby promoting the transformation of these cells into α-smooth muscle actin (α-SMA)-expressing and ECM-producing hyperproliferative myofibroblasts [3]. Despite great efforts have been made, liver fibrosis remains challenging to manage and can eventually progress to advanced stages, including cirrhosis, liver failure and cancers [4, 5]. Consequently, it is of great importance to uncover novel therapeutic targets of liver fibrosis and explore potential therapies.
Long noncoding RNAs (lncRNAs) are a class of transcripts with more than 200 nucleotides in length, which functions as RNAs but do not encode proteins [6]. Emerging studies have suggested that lncRNAs are unignorable regulators in the development of liver fibrosis [7]. Among these lncRNAs, lncRNA H19 (H19) is an imprinted maternally transcribed and evolutionary conserved lncRNA, which is highly expressed in skeletal muscle and liver during the fetal period but downregulated postnatally [8, 9]. In the past few years, a series of our studies highlighted that the aberrant expression of H19 was correlated with the progression of liver fibrotic diseases, especially cholangiopathies [10, 11]. In multidrug resistance 2 knockout (Mdr2−/−) mice, we found that cholangiocyte-derived exosomal H19 were transferred into adjacent hepatocytes and HSCs, which further promoted the progression of cholestatic liver fibrosis by regulating bile acid accumulation in hepatocytes and facilitating the activation and proliferation of HSCs [12, 13]. However, the underlying and detailed mechanism about how H19 directly regulated the transcription of pro-fibrotic genes in HSCs and thus influence hepatic fibrosis was still unclear.
LncRNAs extensively modulate gene expression at transcriptional, post-transcriptional and epigenetic levels by interacting with proteins, DNAs or mRNAs. Enhancer of zeste homolog 2 (EZH2), a key catalytic protein that belongs to polycomb repressive complex 2 (PRC2), is highly expressed in proliferating cells and epigenetically silences the expression of genes by catalyzing tri-methylation of histone H3 at lysine 27 (H3K27me3) [14]. In addition, mounting evidence has indicated that lncRNAs can functionally interact with EZH2 and therefore regulate disease progression such as oncogenesis by affecting signaling pathways including the Wnt/β-catenin and p53 classic pathways [15–17]. Recently, it has been revealed that H19 might share an intimate correlation with EZH2 and regulate its expression in various chronic diseases. By extension, Zhang’s group first identified that H19 promoted tumor progression through the interaction with EZH2, which further regulated the downstream β-Catenin/GSK3β/epithelial-mesenchymal transition (EMT) signaling whereases the knockdown of H19 significantly reversed this injurious effect [18]. Moreover, Zhen et al. suggested that H19 repressed the gene expression of large tumor suppressor 1 (LATS1) through recruiting the EZH2 protein into the nucleus and further regulated EZH2-induced H3K27me3 trimethylation in dental pulp stem cells [19]. Additionally, EZH2 is positively related to the severity of various chronic liver diseases and several inhibitors of EZH2 like 3-Deazaneplanocin A (DZNeP) and GSK126 have been reported to show therapeutic effects [20]. However, effective pro-fibrotic target pathways of EZH2-mediated epigenetic regulation and the potential interaction between H19 and EZH2 remain to be further elucidated.
In the current study, we highlighted the direct interaction between H19 and EZH2 using RNA pulldown and RNA immunoprecipitation (RIP) and further verified downstream signaling pathways in response to H19-EZH2-mediated shifts in H3K27me3 profiles during fibrogenesis, particularly cholestatic liver fibrosis in both BDL and Mdr2−/− mice. Collectively, our findings uncovered the synergistic interaction between the two distinct fibrogenic targets, overexpressed H19 and EZH2, in accelerating liver fibrosis and suggested a potential therapeutic strategy for treating relevant complications.
Materials and methods
Materials
3-DZNeP and GSK126 were purchased from Innochem Technology Co., Ltd. (Beijing, China). Quercetin was purchased from Yuanye Biotechnology (Shanghai, China). Carbon tetrachloride (CCl4) (A33986) was purchased from Innochem (Beijing, China). Collagenase from Clostridium histolyticum (C5138) and other materials for cell culture were all purchased from Sigma (St. Louis, MO, USA). AceQTM Universal SYBR qPCR Master Mix (Q511-02) and HiScript III RT SuperMix cDNA Reverse Transcription Kits (R323-01) were purchased from Vazyme Biotech (Nanjing, China). Antibodies against EZH2 (21800-1-AP), Collagen 1 (Col1a1) (67288-1-lg), Fibronectin (Fn1) (15613-1-AP), HuR (ab200342), β-Tubulin (p01l06) and β-ACTIN (66009-1-lg) were obtained from Proteintech Group (Rosemont, IL, USA). Antibodies against H3K27Me3 (9733 s), α-SMA (19245 S), c-Myc (18583 S), normal rabbit IgG (2729 S) were purchased from Cell Signaling Technology (Danvers, MA, USA) and adsorbed secondary antibody (Alexa Fluor Plus 488) (Ul287767) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Goat anti-mouse IgG-HRP (abs20039) and goat anti-rabbit IgG-HRP (abs20040) were obtained from Absin Bioscience (Shanghai, China). The Pierce Agarose ChIP Kit and ChIP Grade Protein A/G Plus agarose were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Animal studies
C57BL/6 J mice (7-week-old, female) were purchased from SPF Biotechnology (Beijing, China). Mdr2 knockout mice (Mdr2−/−) with C57BL/6 J background were purchased and bred from Shanghai Model Organisms Center (Shanghai, China). Mice were kept in a temperature-controlled room (22 ± 2 °C) and supplied with sterile water and normal chow. For Mdr2−/− mice, mice at different ages (60-day old and 100-day old) were used in this study (n = 6). For 3-DZNeP experiments, Mdr2−/− mice (50-day old) were treated with an EZH2 inhibitor 3-DZNeP at a dose of 1 mg/kg once a day for two weeks by intraperitoneal injection (n = 6). For CCl4 chronic injury mouse model, mice (7-week-old) were orally administrated with CCl4 (1 mL/kg) or sesame oil twice a week for 4 weeks or 8 weeks as previously described (n = 6) [21]. For 3-DZNeP experiments, mice were orally pre-treated with CCl4 for 4 weeks and then co-injected with 3-DZNeP intraperitoneally at a dose of 1 mg/kg once a week from week 5 to week 8 (n = 6). For the BDL mouse model, mice were subjected to BDL or sham operation at different time points, either day 3 or day 7 (n = 6). For 3-DZNeP experiments, BDL mice were treated with 3-DZNeP intraperitoneally at a dose of 1 mg/kg once a day from day 4 to day 7 (n = 6). At the end of different treatments, mice were sacrificed and collected serum, liver tissues and other organs for further experiments. Moreover, as there was no difference between the protein expression of control group in different ages or operation periods, we randomly picked control samples from different control groups for WB analysis. All animal studies and procedures were approved by the Institutional Animal Care and Use Committee of Beijing University of Chinese Medicine and carried out in compliance with all guidelines and regulations.
Cell culture and transfection
The human hepatic stellate cells LX2 were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). Primary mouse HSCs were isolated according to a gradient centrifugation method modified based on our previous report [22, 23]. Primary HSCs and LX2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution in a humidified incubator containing 5% CO2. For transfection experiments, control plasmid (CP), H19 plasmid, EZH2 plasmid obtained from Hanbio Biotechnology (Shanghai, China) were transfected into LX2 cells using Lipofectamine 2000. For quercetin intervention experiments, cells were transfected with H19 plasmid for 24 h and then treated with quercetin (20 μM or 40 μM) for another 24 h. For miR-675 transfection, miR-675 mimic and miR-675 inhibitor purchased from Hanbio Biotechnology (Shanghai, China) were transfected into LX2 cells using RNAFit (Hanbio Biotechnology, Shanghai, China).
RNA pulldown
To construct pH19-S1 (expressing S1-tagged H19), an annealed oligonucleotide fragment containing S1 aptamer sequence was inserted into the 3' end of human H19 full-length sequence through a linked sequence (GTAGAAA), and PCR was carried out to amplify the target fragment. The resulting PCR target gene product was inserted into pcDNA3.1 (Hanbio Biotechnology) opened with EcoR I and BamH I, which was also reported in previous studies [24]. All clones were checked by sequencing. The detailed sequence of the construction and other critical information has been supplied in the Supplementary Materials. HEK293 cells and LX2 were transfected with pH19-S1 or pH19 in a 15-cm plate scale and harvested 48 h later by manual scraping with ice-PBS and pelleted by centrifugation. After washing with ice-cold PBS, cell pellet was resuspended in ice-cold gentle lysis buffer GLB. The lysate was precleared by incubation at 4 °C with avidin agarose beads (Pierce, 20219), and was incubated with streptavidin beads at 4 °C overnight. Then the beads were collected and washed with 800 μL of ice-cold GLB buffer 4 times by rotating the tube at 4 °C for 5 min each. The purified RNPs were eluted from the beads in 20 μL of 2× SDS sample buffer (Sigma, 1610747) by heating at 100 °C for 10 min and resolved by 10% SDS-PAGE. The eluted proteins were then used for immunoblotting.
RNA immunoprecipitation (RIP)
For RNA immunoprecipitation (RIP) assays, H19OE LX2 cells were harvested and lysed. Protein A + G Sepharose beads were incubated with EZH2 antibody or rabbit normal IgG in IP buffer at 4 °C overnight. The next day, the beads were washed 3 times with IP buffer and kept on ice until used. The suspensions were incubated on ice for 20 min. After removing insoluble materials by centrifugation, lysates were precleared using protein A + G sepharose, followed by addition of yeast tRNA (Ambion) to a final concentration of 40 μg/mL. Cleared lysates were transferred to tubes containing antibody- or pre-immune IgG-coated beads, and IP was carried out by rotating the tubes at 4 °C, overnight. Following IP, the beads were washed 4 times with IP buffer and the RNA of immunoprecipitated group and Input was extracted and analyzed by RT-qPCR.
Chromatin immunoprecipitation (ChIP) sequencing
Cell samples were prepared for ChIP-Seq using a Thermo Pierce Agarose ChIP Kit. LX2 cells transfected with H19 overexpression plasmid or control plasmid were cross-linked using 1% formaldehyde. After glycine treatment, cells were harvested with ice-cold PBS containing halt cocktail, lysed using lysis buffer containing protease inhibitors and re-suspended in MNase digestion buffer containing Micrococcal Nuclease (ChIP grade). The digested chromatin supernatant was immunoprecipitated with H3K27Me3 antibody (9733s, CST) on a rocking platform overnight at 4 °C. Following overnight incubation, ChIP Grade Protein A/G Plus agaroses were added and washed. In the end, chromatin DNA was recovered and purified using DNA Clean-Up Column and DNA Column Elution Solution. ChIP-seq analysis was performed by Novogene (Novogene, Tianjin, China). Briefly, raw reads were quality trimmed by removing bases with low quality and pre-processing sequencing adapters were clipped using the fastp software. These reads were then mapped into the Human Reference Genome (hg19) using BWA MEM version 0.7.17-r1188. Resulting bam files were filtered to remove alignments that are not “proper-paired”, reads mapping to more than one location and other non-primary alignments using SAMtools (v.0.1.19). PCR duplicates were removed by the MarkDuplicates module of Picard (v.1.141).
Statistical analysis
All results were expressed as mean ± SEM and repeated at least three independent times. Data were performed by One-way ANOVA to compare the differences between multiple groups with GraphPad Prism 9.0 software (Graph-Pad, San Diego, CA, USA). Differences below were identified as statistically significant (*P < 0.05, **P < 0.01, and ***P < 0.001).
Results
Hepatic EZH2 levels are correlated to the severity of liver fibrosis in different mouse models
Mdr2−/−, BDL- and CCl4-induced mice are all widely applied mouse models for the study of liver fibrosis. Our previous studies reported that Mdr2 deficiency and BDL time-dependently induced hepatic H19 levels during disease progression and H19 is correlated with the severity of fibrotic liver injuries in patients with biliary atresia [12, 25]. To investigate whether EZH2 was also in a positive correlation with the development of hepatic fibrosis and H19 expression, we measured the expression of Ezh2 and various fibrotic genes in the above-mentioned mouse models. Hematoxylin and eosin (H&E) and Masson’s Trichrome staining indicated that Mdr2−/− mice at age 60 days have a higher level of inflammation infiltration and collagenous fibers in the liver, which become more severe at age 100 days (Fig. 1a). Similar progressive injuries were also found in BDL (3 days and 7 days)- and CCl4 (4 weeks and 8 weeks)-induced liver fibrosis (Fig. 1e, i). Notably, serum biochemistry assays also showed that the ALT and AST levels in serum were significantly increased in Mdr2−/−, BDL and CCl4 model mice (Supplementary Fig. S1a). Similar to histological examinations, immunofluorescence staining of α-SMA (red) showed a significant increase of α-SMA positive fibroblasts in these hepatic fibrosis models (Supplementary Fig. S1b). The gene expression levels of Ezh2, H19 and fibrotic genes (Acta2, Col1a1, Fn1 and Timp1) in the liver were all upregulated when compared with relative control groups, which were continually upregulated during disease progression (Fig. 1b, f, j). Moreover, as depicted in Fig. 1c, g, k, the protein expression of EZH2 as well as the fibrotic proteins such as α-SMA, Collagen 1 and Fibronectin were significantly upregulated in the livers of Mdr2−/−, BDL and CCl4 treated mice. EZH2 was widely demonstrated as a catalytic subunit of PRC2 and catalyzed histone H3K27me3 to silence the expression of target genes. In addition, an increased expression of H3K27me3 in those models of liver fibrosis when compared with the control mice was also identified. Collectively, these data indicated that the expression of EZH2 was upregulated and was in a positive correlation with the expression of H19 and fibrotic marker genes in liver fibrosis.
Fig. 1. Hepatic EZH2 levels are correlated to the severity of liver fibrosis in various mouse models.

a, e, i Representative images of H&E and Masson’s Trichrome staining in relative control, Mdr2-/-, BDL and CCl4-induced mice. Scale bar =100 μm. b, f, j Relative mRNA levels of H19, Ezh2, Acta2, Col1a1, Fn1 and Timp1 were determined by qPCR and normalized using Hprt1 as an internal control. c, g, k Representative immunoblots against EZH2, H3K27me3, α-SMA, Collagen 1, Fibronectin and β-ACTIN were shown. d, h, l The relative densities of EZH2/β-ACTIN, H3K27me3/β-ACTIN, α-SMA/β-ACTIN, Collagen 1/β-ACTIN and Fibronectin/β-ACTIN. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the relative control groups; One-way ANOVA with Tukey’s post-hoc tests (n = 6).
EZH2 is essential for HSCs activation in liver fibrosis
TGF-β is the master regulator of HSCs activation through both Smad (mothers against decapentaplegic homolog)-based and non-Smad-based signaling pathways [4]. As depicted in Fig. 2a, TGF-β significantly triggered HSCs activation, as indicated by remarkable upregulation of fibrotic genes as well as H19. EZH2 inhibition mediated by either 3-DZNeP or GSK126 significantly reduced the expression of these genes, indicating a decisive role of EZH2 in regulating HSCs activation (Fig. 2b). Meanwhile, the change of contraction force of HSCs after EZH2 inhibition was also explored by gel contraction analysis. As shown in Fig. 2c, both 3-DZNeP and GSK126 could inhibit collagen gel contraction under normal conditions. Moreover, once stimulated by TGF-β, collagen gels were significantly contracted, and were further reversed in the presence of different EZH2 inhibitors (Fig. 2d). Mouse primary HSCs were further used for verification. Interestingly, we found that as primary HSCs grew, the expression of H19, proliferative and profibrotic markers were all remarkably upregulated, which were also markedly reduced by 3-DZNeP and GSK126 (Supplementary Fig. S2a). Further, even with the simulation of TGF-β, these EZH2 inhibitors still markedly reduced the activation and proliferation of primary HSCs (Supplementary Fig. S2b).
Fig. 2. EZH2 inhibitors 3-DZNeP and GSK126 significantly abrogate TGF-β-mediated fibrotic response in HSCs.

LX2 cells were administered 3-DZNeP or GSK26 with or without TGF-β (5 ng/mL) treatment. a Relative mRNA levels of H19, ACTA2, COL1A1, FN1 and DESMIN were determined by qPCR and normalized using HPRT1 as an internal control. b Representative immunoblots and the relative density of EZH2/β-ACTIN, H3K27me3/β-ACTIN, α-SMA/β-ACTIN, Collagen 1/β-ACTIN and Fibronectin/β-ACTIN were shown. c Collagen gel containing HSC-derived fibroblasts were treated with 3-DZNeP or GSK26 at different concentrations. d Collagen gel containing HSC-derived fibroblasts were treated with 3-DZNeP or GSK26 with or without TGF-β (5 ng/mL) treatment. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the TGF-β group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
Given the key role of EZH2 in promoting HSCs activation in vitro, we further investigated the dependency of disease progression on EZH2 by pharmacological inhibition in vivo. 3-DZNeP was used since it showed better anti-fibrotic efficacy than GSK126 in vitro. For Mdr2−/− mice, 3-DZNeP was administered at a dose of 1.0 mg/kg once a day (from age day 50 to day 64) as depicted in Fig. 3a. Histological examination depicted that 3-DZNeP relieved liver fibrosis as evidenced by improved lobular inflammation and reduced collagen deposition in the liver (Fig. 3b). The upregulation of Acta2, Col1a1 and Fn1 were all significantly downregulated by EZH2 inhibition (Fig. 3c). Furthermore, the mRNA and protein levels of EZH2, H3K27me3 and fibrotic proteins (α-SMA, Collagen 1 and Fibronectin) were also remarkably inhibited by 3-DZNeP treatment in Mdr2−/− mice (Fig. 3c, d), indicating a protective effect of 3-DZNeP on fibrogenesis in cholangiopathies. In addition, BDL mice were treated with 3-DZNeP at a dose of 1.0 mg/kg once a day from day 4 to day 7 and CCl4 induced mice were administered with the same dose from week 5 to week 8 (Fig. 3e, i). Similar to the results observed in Mdr2−/− mice, EZH2 inhibition also exhibited a protective effect on BDL and CCl4 mice, which is supported by a most recently published study [20]. Specifically, 3-DZNeP improved collagen deposition when compared with corresponding model groups (Fig. 3f, j). Notably, the induced serum levels of ALT and AST in these three liver fibrosis models were markedly reduced by 3-DZNeP treatment (Supplementary Fig. S3a). Furthermore, both transcription and translation of EZH2, H3K27me3, α-SMA, Collagen 1 and Fibronectin were significantly downregulated by 3-DZNeP (Fig. 3g, h, k, l). Taken together, these results emergingly confirmed that EZH2 is essential for HSCs activation in liver fibrosis.
Fig. 3. The inhibition of EZH2 significantly alleviates hepatic fibrosis in vivo.

a, e, i Schematic diagram of in vivo experimental designs in control, Mdr2-/-, BDL and CCl4 treated mice. b, f, j Representative images of H&E and Masson’s Trichrome staining in control, Mdr2-/-, BDL and CCl4 treated mice. Scale bar = 100 μm. c, g, k Representative immunoblots against EZH2, H3K27me3, α-SMA, Collagen 1, Fibronectin and β-ACTIN were shown. d, h, l Relative mRNA levels of Acta2, Col1a1 and Fn1 were determined by qPCR and normalized using Hprt1 as an internal control. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, compared with the Mdr2-/-, BDL and CCl4 groups, separately; One-way ANOVA with Tukey’s post-hoc tests (n = 6).
H19 overexpression promotes HSCs activation by facilitating EZH2-mediated epigenetic regulation
LncRNAs are responsible for a plethora of cellular functions via tethering proteins into chromatin-modifying complexes. To specifically examine whether H19 could regulate EZH2 or vice versa and further control the fibrotic responses of HSC activation, we established H19 overexpressed (H19OE) LX2 cells and then measured the expression of EZH2 and fibrotic genes. As shown in Fig. 4a, an activated HSC-phenotype was observed in H19OE cells (the level of H19 was increased to 7 times than that in CP-transfected cells), as illustrated by increased expression of ACTA2, COL1A1 and FN1, while the expression of transforming growth factor-β (TGFB1) was not altered. We next measured the protein expression of EZH2, H3K27me3 and fibrotic proteins (α-SMA, Collagen 1 and Fibronectin) and found that they were all upregulated in H19OE LX2 cells, indicating that H19 could induce the expression of EZH2, histone trimethylation and HSC activation (Fig. 4b). Thus, we further checked whether EZH2 was able to regulate H19 expression as well and EZH2OE LX2 cells were constructed. In EZH2OE cells, the expression of H19 was almost unchanged while the downstream fibrotic genes including ACTA2, COL1A1 and FN1 was significantly upregulated. In consistent with mRNA expression results, the protein levels of H3K27me3, α-SMA, Collagen 1 and Fibronectin were all markedly upregulated, suggesting increased histone trimethylation and HSC activation induced by EZH2 overexpression (Fig. 4d).
Fig. 4. H19 overexpression promotes EZH2-mediated fibrotic gene expression.

a Relative mRNA levels of H19, ACTA2, COL1A1, FN1 and TGFB1 in H19-overexpressed LX2 cells were determined by qPCR and normalized using HPRT1 as an internal control. b Representative immunoblots against EZH2, H3K27me3, α-SMA, Collagen 1, Fibronectin and β-ACTIN in H19-overexpressed LX2 cells were shown. c Relative mRNA levels of EZH2, H19, ACTA2, COL1A1 and FN1 in EZH2-overexpressed LX2 cells were determined by qPCR and normalized using Hprt1 as an internal control. d Representative immunoblots against EZH2, H3K27me3, α-SMA, Collagen 1, Fibronectin and β-ACTIN in EZH2-overexpressed LX2 cells were shown. e The relative density of EZH2/β-ACTIN, H3K27me3/β-ACTIN, α-SMA/β-ACTIN, Collagen 1/β-ACTIN and Fibronectin/β-ACTIN. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
To further investigate whether the pro-fibrotic effects of H19 is depending on EZH2-mediated epigenetic regulation, H19OE LX2 cells were subjected to 3-DZNeP and GSK126 treatments. In agreement with our anticipation, 3-DZNeP and GSK126 promoted the transformation of H19OE-activated HSCs to a quiescent phenotype, with a significantly decreased expression of ACTA2, COL1A1, FN1, DESMIN, as well as TIMP1, when compared with the H19OE group. Paradoxically, when EZH2 was inhibited by DZNeP or GSK126, the gene expression of H19 was slightly downregulated, indicating that EZH2 might still influence the expression of H19 under certain circumstances (Fig. 5a). Similarly, H19 overexpression induced increased protein levels of EZH2, H3K27me3, α-SMA, Collagen 1 and Fibronectin, which were subsequently reversed by 3-DZNeP and GSK126-induced EZH2 inhibition (Fig. 5b). Inflammatory response has been considered as a key driver of the persistent fibrotic response. Consequently, we measured the mRNA expression of Il6 and found it was significantly decreased after 3-DZNeP or GSK126 treatment (Supplementary Fig. S4a). These findings suggested that EZH2 functioned as a positive character and downstream effector in H19-induced HSC activation.
Fig. 5. EZH2 inhibitors 3-DZNeP and GSK126 significantly inhibit H19-induced HSC activation.

H19 overexpressed LX2 cells or WT LX2 cells (transfected with control plasmid, referred to as CP) were administered with 3-DZNeP or GSK126. a Relative mRNA levels of H19, ACTA2, COL1A1, FN1, DESMIN and TIMP1 were determined by qPCR and normalized using HPRT1 as an internal control. b Representative immunoblots against EZH2, H3K27me3, α-SMA, Collagen 1, Fibronectin and β-ACTIN in EZH2-overexpressed LX2 cells and the relative densities of EZH2/β-ACTIN, H3K27me3/β-ACTIN, α-SMA/β-ACTIN, Collagen 1/β-ACTIN and Fibronectin/β-ACTIN were shown. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the CP group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H19OE group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
H19 promotes HSCs activation by directly interacting with EZH2 rather than acting as miR-675 precursor
Indeed, H19 has diverse operating patterns through interactions with both proteins and RNAs. Figure 6a is the schematic diagram of the hypothesis and verification of this part. Previous studies have demonstrated that H19 is the precursor of microRNA 675 (miR-675, located within the exon1 of H19) and plays essential regulatory roles in multiple biological processes based on the function of miR-675. For instance, H19 was found to promote tumorigenesis and invasion through the regulation of its integrated carcinogenic miR-675 [26]. To further determine whether H19 regulated pro-fibrotic process and HSC activation by producing miR-675, we transfected LX2 cells with miR-675 mimic and inhibitor and found that miR-675 had ignorable effects on the expression of ACTA2, FN1, COL1A1 and TGFB1 (Fig. 6b). Human antigen R (HuR), one of the best characterized RNA binding proteins, has been proved to stabilize H19 from post-transcriptional processing, so that decreases the cellular abundance of miR-675 [27]. Here, our results indicated that HuR inhibition induced by quercetin (QU) at both 20 μM and 40 μM, which markedly increased miR-675 levels, significantly prevented HSCs from activation and proliferation, as evidenced by decreased expression of ACTA2, FN1, COL1A1 and KI67 (Fig. 6c-e). These results suggested that the pro-fibrotic role of H19 was independent of miR-675 production.
Fig. 6. H19 can directly interact with EZH2.

a Schematic diagram of the relationship between HUR, H19 and miR-675. (b) LX2 cells were transfected with miR-675 mimics and miR-675 inhibitor. Relative mRNA levels of ACTA2, COL1A1, FN1 and TGFB1 were determined by qPCR and normalized using HPRT1 as an internal control. c–e LX2 cells were transfected H19 plasmid for 24 h and then treated with Quercetin (20 μM and 40 μM). Relative miRNA level of miR-675 was determined by qPCR and normalized using U6 as an internal control. Relative mRNA levels of H19, ACTA2, COL1A1 and FN1 were determined by qPCR and normalized using HPRT1 as an internal control. Relative mRNA levels of MKI67 were determined by qPCR and normalized using Hprt1 as an internal control. (f) Schematic diagram of the establishment of pH19-S1. g, h Interaction between the pH19 and EZH2 in LX2 cells was detected by RNA pulldown. HUR was used a positive control. (i) EZH2 protein was coimmunoprecipitated with H19 in RIP assays. IgG group: immunoprecipitation using IgG antibody; EZH2 group: immunoprecipitation using anti-EZH2 antibody. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H19OE group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
The interactions between lncRNAs and proteins have been increasingly found to be key aspects in many cellular processes. For instance, HOTAIR, one of the first identified lncRNAs, forms chromatin modification complexes through interacting with the PRC2 and regulates cancer progression [28]. Notably, a previous global screening profiling, using RIP and RNA sequencing, revealed that H19 was potentially presented in the EZH2 complex, yet without further verification [29]. Thus, we speculated that H19 could directly interacted with EZH2, which further led to cascade reactions of epigenetic regulation. We then designed a H19 full length sequence linked with S1 aptamer for RNA pull down assay (Fig. 6f), and revealed that pH19-S1 could directly interact with EZH2 in both 293 cells and LX2 cells (Fig. 6g, h). Subsequently, the RIP assay was conducted, and further verified the high enrichment of H19 in EZH2-RNA precipitates compared to the IgG control in LX2 cells, with over 300-fold higher levels observed. Meanwhile, HPRT1 RNAs were not retrieved, providing strong evidence of direct interaction between H19 and EZH2 (Fig. 6i). Collectively, these results indicated that H19 could strongly bind with EZH2 and further epigenetically regulate downstream pathways in the fibrotic liver.
H19 regulates proliferation, apoptosis, and EMT through EZH2-mediated epigenetic regulation in HSCs
To further elucidate the underlying mechanism of H19-regulated H3K27me3 reprogramming through EZH2 and following target signaling, we therefore performed both RNA-seq and ChIP-seq comparison of H19OE LX2 cells and CP LX2 cells were conducted for comprehensive analysis. The general volcano plot and heatmap of RNA-seq revealed differential transcripts between control and H19OE groups (Fig. 7a, b). Gene Ontology (GO) analysis indicated that differentially expressed genes (DEGs) were associated with immune regulation, cell cycle, and cell adhesion (Supplementary Fig. S5a). In ChIP-seq results, H19 overexpression led to an overall increased H3K27me3 distribution over the genome, which was mainly located at the promoter area (Fig. 7c, d). A total of 102 genes (referred as H19 target DEGs) were identified in both RNA-seq and CHIP-seq results, suggesting that these DEGs were epigenetically regulated by H19-EZH2 interaction (Fig. 7e). As shown in Fig. 7f, g, transcriptionally upregulated H19 target DEGs were significantly enriched in cell proliferation-related pathways (regulation of cell cycle, cell division, positive regulation of fibroblast proliferation) and H19 target DEGs enriched in apoptosis-related pathways (positive regulation of apoptosis process, apoptotic process), were transcriptionally suppressed and H3K27me3 upregulated. We also observed a significant upregulation of H19 target DEGs enriched in extracellular matrix-related pathways (cell adhesion, positive regulation of cell-substrate adhesion, ECM organization, regulation of cell-matrix adhesion pathways) and loss of H3K27me3 signatures on these genes (Fig. 7g). Matrix metallopeptidase 9 (MMP9) is a type IV collagenase that degrades ECM and TIMP2 is the endogenous inhibitor of matrixin and adamalysin endopeptidase activity. HIC ZBTB transcriptional repressor 2 (HIC2) was a new transcription activator of sirtuin 1 (SIRT1) that was proved to inhibit EMT through the TGF-β/Smad4 pathway [30]. In addition, loss of protein tyrosine phosphatase non-receptor type 23 (PTPN23) was also found to promote EMT in human digestive cancers [31]. From qPCR results in Fig. 7g, we can see that the expression levels of MMP9, TIMP2, HIC2 and PTPN23 were markedly downregulated in H19OE cells, while the expression of COL8A1 was significantly upregulated. In consistent with mRNA expression, in H19OE LX2 cells, CHIP-seq peaks located in COL8A1 and COL6A5 genes were significantly increased, while less peaks were identified in MMP9, TIMP2, HIC2 and PTNP23 genes in H19OE LX2 cells (Supplementary Fig. S5b). Taken together, these data revealed that H19-EZH2 interaction epigenetically upregulated genes associated with cell proliferation, extracellular matrix and inhibited apoptosis-related genes.
Fig. 7. H19 overexpression epigenetically regulates cell proliferation, apoptosis and EMT pathways.

The (a) volcano plots, and (b) the heatmap of DEGs (H19OE LX2 cells vs CP LX2 cells) are presented. c The ChIP-seq signal enrichment around the transcription start sites (TSSs) in control and H19 groups. d Peak distribution on the genomic function area in ChIP-seq analysis. e Overlapped genes of interests found in both RNA-seq and ChIP-seq data. f GO biological processes enrichment analysis of representative DEGs in RNA-seq and ChIP-seq data. The relative expression levels of DEGs enriched in proliferation and apoptosis pathways were shown as a heatmap. g The relative expression levels of DEGs enriched in EMT and cell adhesion pathways were shown as a heatmap. Relative mRNA levels of MMP9, TIMP2, COL8A1, HIC2 and PTPN23 were exhibited. Statistical significance: *P < 0.05, **P < 0.01, compared with the control group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
Pivotal cell signaling pathways responsible for H19-EZH2 interaction-mediated HSCs activation were further explored. As can be seen from Fig. 8a, some essential pathways that effectively regulated cell proliferation, cell apoptosis, EMT, energy metabolism as well as signaling transduction were enriched. Collectively, the affected pathways can be mainly divided into two parts: Wnt-related signaling pathways (Canonical Wnt signaling pathway and negative regulation of Wnt signaling pathway) and other classic signaling including phosphoinositide 3-kinase (PI3K), extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways (Regulation of PI3K signaling, positive regulation of ERK1 and ERK2 cascade and MAPK cascade). β-Catenin (CTNNB) is a key nuclear effector of canonical Wnt signaling that is responsible for the transduction of the signal to the nucleus and leads to the activation of various downstream signaling cascades including cell proliferation and EMT (Fig. 8b). WNT7A and WNT2B are both important Wnt ligands that can bind to their receptor frizzled (FZD) to preferentially induce non-canonical Wnt signaling by activating β-catenin [32, 33]. Previous studies revealed that transcription factor retinoic acid receptor beta (RARβ) can transcriptionally upregulate the expression of WNT2B and WNT7A, which further promoted the activation of Wnt/β-catenin signaling [34, 35]. Furthermore, LIM Homeobox 6 (LHX6) was a negative regulator of transcriptional expression of β-catenin by inhibiting the promoter region (-1161 bp to +27 bp) [36]. In the results of Fig. 8a and Supplementary Fig. S5c, we can see that the expression of CTNNB, WNT2B, WNT7A and RARβ were significantly upregulated while the expression of LXH6 was downregulated, which was also consistent with the H3K27 methylation levels of these genes. Taken together, these findings highly suggested that H19-EZH2 interaction facilitated Wnt/β-catenin signaling transduction and following cell proliferation and EMT processes which eventually promoted HSCs activation.
Fig. 8. H19 overexpression epigenetically enhances WNT signaling and promotes liver fibrosis.

a GO biological processes enrichment analysis of DEGs of interests. The relative expression levels of DEGs enriched in representative cell signaling pathways were shown as a heatmap. b Relative mRNA levels of CTNNB1, WNT7A, WNT2B, RARB and LHX6. c Schematic diagram of the proposed epigenetic mechanisms underlying the effects of H19-EZH2 on cell proliferation and EMT. Statistical significance: *P < 0.05, **P < 0.01, compared with the control group; One-way ANOVA with Tukey’s post-hoc tests (n = 3).
Discussion
Liver fibrosis is characterized by progressive accumulation of ECM and HSCs transdifferentiation and excessive proliferation are major contributors for persistent myofibroblast activation during fibrogenesis. Functioned as the most pivotal character in the pathogenesis of liver fibrosis, HSCs have received widespread attention, leading to numerous attempts aiming at inhibiting the activation of HSCs, such as PPARγ agonists, αv integrin inhibitors, Hedgehog inhibitors and more. However, there are still no approved therapies for liver fibrosis and it has been recognized as a pre-cancerous condition in more than 60% of HCC cases, which is in urgent need of novel treatment [37]. Dysregulation of the epigenome has always been found to drive aberrant gene expression and therefore promoted disease progression. EZH2 is an important epigenetic regulator that catalyzes H3K27me3, which was identified to regulate miscellaneous biological processes such as cancer, diabetic nephropathy, acute kidney injury and liver fibrosis [38]. Previous study has demonstrated that EZH2 was positively upregulated during HSCs activation and liver fibrosis progression and inhibition of EZH2 in CCl4 and BDL mice ameliorates disease development in vivo [20]. Our current study not only verified previous reports but also proved the anti-fibrosis effects of EZH2 inhibition in Mdr2−/− mice, a widely accepted primary sclerosis cholangitis mouse model, further highlighting the therapeutic potential of targeting EZH2 [39]. Recently, a potential communication between lncRNA networks and epigenetic regulation was suggested to play essential roles in the pathogenesis of various diseases. However, little is known about the specific molecular mechanism underlying. LncRNA H19 has been well-elucidated to promote fibrogenesis in cholangiopathies, such as primary sclerosis cholangitis and biliary atresia and has become a valuable diagnostic marker and therapeutic target, as reported by previous studies from both our and other groups. We have also identified that overexpression of H19 in response to bile acid stimulation promoted HSC activation and cholestatic liver fibrosis via exosomal transmission. However, whether H19 promoted liver fibrosis via epigenetic regulation remains elusive and has brought significant obstacles to the development of certain therapeutic strategies. Herein, we observed that H19 directly binds to EZH2, and thus reprogramming H3K27me3 signatures and regulating the transcription of genes related to HSC proliferation and ECM remodeling. Pharmacological EZH2 inhibition significantly attenuated H19OE-mediated HSC activation and improved fibrosis in Mdr2−/− mice, BDL mice and CCl4 treated mice.
To explore how H19 regulate EZH2-mediated histone modification, the potential crosstalk between H19 and EZH2 was investigated and the subsequent mechanism was studied. At first, we found a positive correlation between H19 expression and EZH2 activation, which is also consistent with the progression of fibrosis. Interestingly, EZH2 inhibition by 3-DZNeP reversely inhibited the expression of H19, which pointed out EZH2 might also regulate the expression of H19 and functioned as a negative feedback mechanism, consistent with the results in other research [40]. However, H19 overexpression only slightly induced the expression of EZH2 through an unidentified pathway, suggesting that regulating EZH2 expression is not the primary mechanism.
LncRNA H19 is the precursor miR-675, which has been widely identified as regulators in multiple biological processes such as tumorigenesis and atherosclerosis. However, we found that direct delivery of miR-675 mimic into LX2 cells could not promote HSC activation, suggesting that the epigenetic regulatory effects of H19 is not relying on producing miR-675. This speculation is further confirmed by HuR inhibition analysis. Since HuR binds to and stabilizes H19 from being processed into miR-675, HuR inhibition mediated by quercetin increased the production of miR-675 and abolished H19OE-induced HSC activation and proliferation (Fig. 6c-e). After excluding the role of miR-675 in promoting HSC development, we noticed that most of the epigenetic enzymes requires the guidance of certain lncRNAs or transcriptional factors to recruit them into the specific site on genomic DNA to achieve fully functionality. The interaction between lncRNAs and proteins, especially epigenetic regulators, have been widely reported. For instance, lncRNA Xist has been found to recruit EZH2 into the promoter region of the DKK1 protein on the X chromosome, which inhibited the expression of DKK1 and further promoted neuroblastoma carcinogenesis [41]. Ye et al. have found H19 was an EZH2-interacting lncRNA by performing RIP-seq in SH-SY5Y cells. However, further verification of the interaction between H19 and EZH2 and evidence regarding whether H19 can deliver specific H3K27me3 signatures were absent in this study [42]. Inspired by these reports, we performed RNA pulldown and RIP experiments and verified that H19 can directly bind with EZH2, which is probably the primary mechanism underlying H19-EZH2 interaction. Further studies are still required to comprehensively identify specific sequences on H19 and key residues on EZH2 responsible for the binding.
Following binding to EZH2, overexpressed H19 significantly reprogramed H3K27me3 profiles in HSCs. By combining the results of the ChIP-seq and RNA-seq analysis, we picked out significant H19 target DEGs for further enrichment analysis and verification. It is noteworthy that H19 target DEGs are mainly regulating cell proliferation, apoptosis, ECM remodeling and EMT pathways, indicating that H19 epigenetically regulated HSC cell fate and therefore promoted the progression of liver fibrosis. By applying bioinformatic analysis on detailed signaling pathways of integrated genes, we preliminarily identified that pathways including canonical Wnt signaling pathway, phosphatidylinositol 3-kinase signaling, positive regulation of ERK1 and ERK2 cascade and MAPK cascade were markedly upregulated, which were all widely-identified pathways regulating cell proliferation, survival and metabolism. β-Catenin (CTNNB) is a key nuclear effector of canonical Wnt signaling that is responsible for the transduction of the signal and downstream cascade activation. We found that H19OE significantly induced the expression of CTNNB and also markedly decreased the distribution of H3K27me3 peaks on its gene. In addition, we also observed an increased expression of Wnt ligands, the upregulation of RARB (encoding RARβ, a transcription factor that promotes expression of WNT2B and WNT7A) [34], and the downregulation of LHX6, a negative regulator of transcriptional expression of β-catenin [43]. Consequently, we speculated that H19 epigenetically promoted Wnt/β-catenin signaling pathway by activating RARβ and suppressing LHX6 by altering their DNA accessibility, which further activated downstream pathways concerning cell proliferation and EMT. Interestingly, a most recent study revealed that EZH2 inhibition in primary HSCs inhibited H3K27me3 enrichment on genes encoding bone morphogenetic protein and activin membrane bound inhibitor (BAMBI, a TGF-β1 pseudoreceptor) and cell cycle regulators cyclin dependent kinase inhibitor (CDKN1A), growth arrest and DNA damage inducible alpha (GADD45A), GADD45B and anti-inflammatory cytokine IL10, which may further promoted the TGF-β/SMAD signaling pathway and CDKN1A and GADD45 mediated growth arrest inhibition. These results are mainly deduced from the transcriptomic analysis of 3-DZNeP treated HSCs and are different from our observations [20]. Hence, although further evidence is required, we still speculate that H19OE could steer EZH2 away from delivering canonical H3K27me3 signatures but results in a highly specific histone modification and thus transcription pattern of certain target genes.
Conclusion
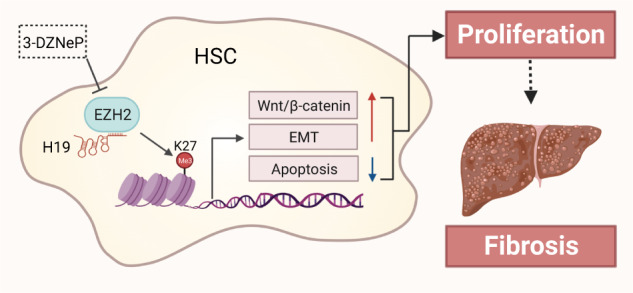
In summary, as illustrated in the graphical abstract, our study demonstrates that highly expressed H19 in various chronic liver diseases epigenetically reprograms H3K27me3 signatures over genes involved in cell proliferation and ECM remodeling pathways by directly binding to EZH2, thereby facilitating HSCs activation. Our findings uncover the mysterious mechanism underlying the pro-fibrotic role of H19 and shed novel light on the development of therapeutic approaches targeting H19-EZH2 interaction for the management of liver fibrosis.
Supplementary information
Acknowledgements
This work was supported by grants from Beijing Nova Program of Science & Technology (Z201100006820025 and Z211100002121167 to RPL); the National High-Level Talents Special Support Program to XJYL; National Natural Science Foundation of China (Grant No. 82274186 to XJYL, and 82274201 to RPL); Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (Grant No. ZYYCXTD-C-202006 to XJYL).
Author contributions
XJYL and FZ contributed equally to this work. XJYL and RPL conceived the original idea and supervised the study. FZ, XJYL and RPL prepared the manuscript and figures. YJL, XYX, JRQ, JL, GFF, RS, JZW, and QZ conducted all the experiments and performed data analysis. All authors have approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Xiao-jiao-yang Li, Fei Zhou
Contributor Information
Xiao-jiao-yang Li, Email: xiaojiaoyang.li@bucm.edu.cn.
Run-ping Liu, Email: liurunping@bucm.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01145-z.
References
- 1.Li Y, Wu J, Liu R, Zhang Y, Li X. Extracellular vesicles: catching the light of intercellular communication in fibrotic liver diseases. Theranostics. 2022;12:6955–71. doi: 10.7150/thno.77256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–36. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Liu R, Wu J, Li X. Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics. 2020;10:7993–8017. doi: 10.7150/thno.47826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parola M, Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Asp Med. 2019;65:37–55. doi: 10.1016/j.mam.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151–66.. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 6.Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220:e202009045. doi: 10.1083/jcb.202009045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci. 2013;56:876–85. doi: 10.1007/s11427-013-4553-6. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Qi M, Fei X, Wang X, Wang K. LncRNA H19: A novel oncogene in multiple cancers. Int J Biol Sci. 2021;17:3188–208.. doi: 10.7150/ijbs.62573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Liu R. Long non-coding RNA H19 in the liver-gut axis: A diagnostic marker and therapeutic target for liver diseases. Exp Mol Pathol. 2020;115:104472. doi: 10.1016/j.yexmp.2020.104472. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Hylemon PB, Zhou H. Long noncoding RNA H19: A key player in liver diseases. Hepatology. 2021;74:1652–9. doi: 10.1002/hep.31765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Liu R, Yang J, Sun L, Zhang L, Jiang Z, et al. The role of long noncoding RNA H19 in gender disparity of cholestatic liver injury in multidrug resistance 2 gene knockout mice. Hepatology. 2017;66:869–84.. doi: 10.1002/hep.29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu R, Li X, Zhu W, Wang Y, Zhao D, Wang X, et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology. 2019;70:1317–35.. doi: 10.1002/hep.30662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Liu R, Huang Z, Gurley EC, Wang X, Wang J, et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology. 2018;68:599–615. doi: 10.1002/hep.29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen Y, Hou Y, Yi X, Sun S, Guo J, He X, et al. EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics. 2021;11:1795–813.. doi: 10.7150/thno.48101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishimwe N, Wei P, Wang M, Zhang H, Wang L, Jing M, et al. Autophagy impairment through lysosome dysfunction by brucine induces immunogenic cell death (ICD) Am J Chin Med. 2020;48:1915–40.. doi: 10.1142/S0192415X20500962. [DOI] [PubMed] [Google Scholar]
- 16.Zhu P, Wang Y, Huang G, Ye B, Liu B, Wu J, et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23:631–9. doi: 10.1038/nsmb.3235. [DOI] [PubMed] [Google Scholar]
- 17.Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M, et al. MDM2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol Cell. 2016;61:68–83. doi: 10.1016/j.molcel.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DM, Lin ZY, Yang ZH, Wang YY, Wan D, Zhong JL, et al. IncRNA H19 promotes tongue squamous cell carcinoma progression through beta-catenin/GSK3beta/EMT signaling via association with EZH2. Am J Transl Res. 2017;9:3474–86.. [PMC free article] [PubMed] [Google Scholar]
- 19.Du Z, Shi X, Guan A. lncRNA H19 facilitates the proliferation and differentiation of human dental pulp stem cells via EZH2-dependent LATS1 methylation. Mol Ther Nucleic Acids. 2021;25:116–26.. doi: 10.1016/j.omtn.2021.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Xiang C, Zhong F, Zhang Y, Wang L, Zhao Y, et al. Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics. 2021;11:361–78.. doi: 10.7150/thno.46360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YJ, Liu RP, Ding MN, Zheng Q, Wu JZ, Xue XY, et al. Tetramethylpyrazine prevents liver fibrotic injury in mice by targeting hepatocyte-derived and mitochondrial DNA-enriched extracellular vesicles. Acta Pharmacol Sin. 2022;43:2026–41. doi: 10.1038/s41401-021-00843-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiskirchen R, Gressner AM. Isolation and culture of hepatic stellate cells. Methods Mol Med. 2005;117:99–113. doi: 10.1385/1-59259-940-0:099. [DOI] [PubMed] [Google Scholar]
- 23.Liu R, Li X, Huang Z, Zhao D, Ganesh BS, Lai G, et al. C/EBP homologous protein-induced loss of intestinal epithelial stemness contributes to bile duct ligation-induced cholestatic liver injury in mice. Hepatology. 2018;67:1441–57.. doi: 10.1002/hep.29540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. 2013;52:101–12. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Y, Liu R, Li X, Gurley EC, Hylemon PB, Lu Y, et al. Long noncoding RNA H19 contributes to cholangiocyte proliferation and cholestatic liver fibrosis in biliary atresia. Hepatology. 2019;70:1658–73.. doi: 10.1002/hep.30698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vennin C, Spruyt N, Dahmani F, Julien S, Bertucci F, Finetti P, et al. H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget. 2015;6:29209–23. doi: 10.18632/oncotarget.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung HK, Xiao L, Jaladanki KC, Wang JY. Regulation of paneth cell function by RNA-binding proteins and noncoding RNAs. Cells. 2021;10:2107. doi: 10.3390/cells10082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Xie Y, Li L, He Y, Zheng D, Yu P, et al. EZH2 RIP-seq identifies tissue-specific long non-coding RNAs. Curr Gene Ther. 2018;18:275–85.. doi: 10.2174/1566523218666181008125010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song JY, Lee SH, Kim MK, Jeon BN, Cho SY, Lee SH, et al. HIC2, a new transcription activator of SIRT1. FEBS Lett. 2019;593:1763–76.. doi: 10.1002/1873-3468.13456. [DOI] [PubMed] [Google Scholar]
- 31.van der Lely L, Hafliger J, Montalban-Arques A, Babler K, Schwarzfischer M, Sabev M, et al. Loss of PTPN23 promotes proliferation and epithelial-to-mesenchymal transition in human intestinal cancer cells. Inflamm Intest Dis. 2019;4:161–73.. doi: 10.1159/000502861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Han Q, Zhao H, Zhang J. Promotion of epithelial-mesenchymal transformation by hepatocellular carcinoma-educated macrophages through Wnt2b/beta-catenin/c-Myc signaling and reprogramming glycolysis. J Exp Clin Cancer Res. 2021;40:13. doi: 10.1186/s13046-020-01808-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu G, Wan N, Liu Q, Chen Y, Cui H, Wang Y, et al. Resolvin E1 attenuates pulmonary hypertension by suppressing Wnt7a/beta-catenin signaling. Hypertension. 2021;78:1914–26.. doi: 10.1161/HYPERTENSIONAHA.121.17809. [DOI] [PubMed] [Google Scholar]
- 34.Seko Y, Azuma N, Yokoi T, Kami D, Ishii R, Nishina S, et al. Anteroposterior patterning of gene expression in the human infant sclera: chondrogenic potential and Wnt signaling. Curr Eye Res. 2017;42:145–54.. doi: 10.3109/02713683.2016.1143015. [DOI] [PubMed] [Google Scholar]
- 35.Bouasker S, Patel N, Greenlees R, Wellesley D, Fares Taie L, Almontashiri NA, et al. Bi-allelic variants in WNT7B disrupt the development of multiple organs in humans. J Med Genet. 2022;60:294–300. [DOI] [PubMed]
- 36.Yang J, Han F, Liu W, Zhang M, Huang Y, Hao X, et al. LHX6, an independent prognostic factor, inhibits lung adenocarcinoma progression through transcriptional silencing of beta-catenin. J Cancer. 2017;8:2561–74.. doi: 10.7150/jca.19972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang J, Lan T, Li C, Ji X, Zheng L, Gou H, et al. Activation of Slit2-Robo1 signaling promotes liver fibrosis. J Hepatol. 2015;63:1413–20. doi: 10.1016/j.jhep.2015.07.033. [DOI] [PubMed] [Google Scholar]
- 38.Li T, Yu C, Zhuang S. Histone methyltransferase EZH2: a potential therapeutic target for kidney diseases. Front Physiol. 2021;12:640700. doi: 10.3389/fphys.2021.640700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 40.Chen MJ, Deng J, Chen C, Hu W, Yuan YC, Xia ZK. LncRNA H19 promotes epithelial mesenchymal transition and metastasis of esophageal cancer via STAT3/EZH2 axis. Int J Biochem Cell Biol. 2019;113:27–36. doi: 10.1016/j.biocel.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Zhu Y, Ni T, Lin J, Zhang C, Zheng L, Luo M. Long non-coding RNA H19, a negative regulator of microRNA-148b-3p, participates in hypoxia stress in human hepatic sinusoidal endothelial cells via NOX4 and eNOS/NO signaling. Biochimie. 2019;163:128–36.. doi: 10.1016/j.biochi.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Ye M, Xie L, Zhang J, Liu B, Liu X, He J, et al. Determination of long non-coding RNAs associated with EZH2 in neuroblastoma by RIP-seq, RNA-seq and ChIP-seq. Oncol Lett. 2020;20:1. doi: 10.3892/ol.2020.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Z, Xie L. LHX6 inhibits breast cancer cell proliferation and invasion via repression of the Wnt/beta-catenin signaling pathway. Mol Med Rep. 2015;12:4634–9. doi: 10.3892/mmr.2015.3997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.