

Key Points
-
•
This study assessed safety and efficacy of defibrotide in preventing CAR-T–associated neurotoxicity in patients with large B-cell lymphoma.
-
•
Defibrotide treatment modestly reduced the rate of CAR-T–associated neurotoxicity/high-grade event duration vs that in historical data.
Visual Abstract
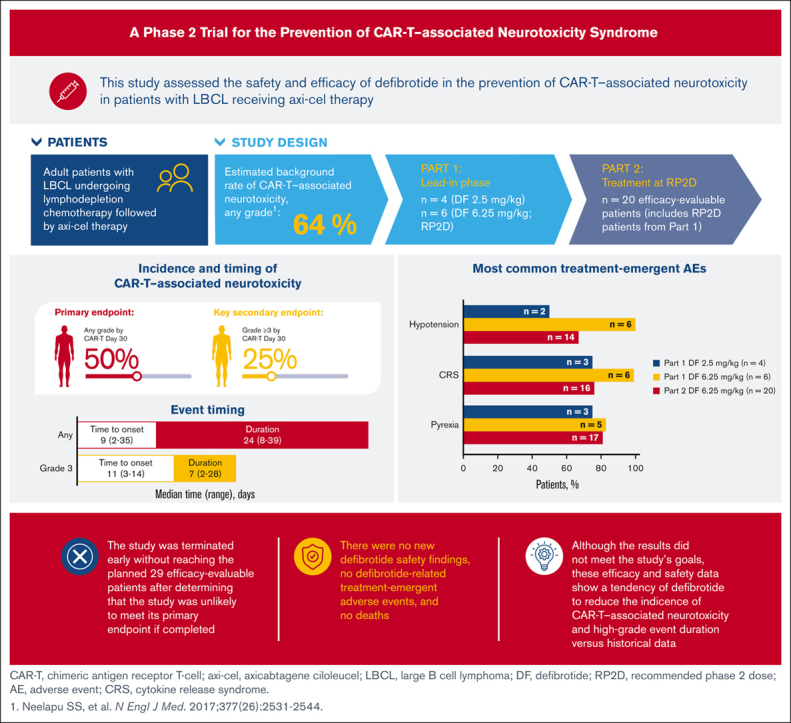
Abstract
Chimeric antigen receptor T-cell (CAR-T) therapy represents a major advance in cancer immunotherapy; however, it can be associated with life-threatening neurotoxicity linked to blood-brain barrier disruption and endothelial activation. Defibrotide was shown to reduce endothelial cell activation in vitro and is approved in the United States for treatment of veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) in patients with renal or pulmonary dysfunction after hematopoietic cell transplantation (HCT), and in the European Union for severe VOD/SOS after HCT in patients aged >1 month. Defibrotide may stabilize the endothelium during CAR-T therapy and reduce the rate of CAR-T–associated neurotoxicity. This phase 2 study evaluated the safety and efficacy of defibrotide for prevention of CAR-T–associated neurotoxicity in patients with relapsed/refractory large B-cell lymphoma receiving axicabtagene ciloleucel. Part 1 established the recommended phase 2 dose (RP2D; 6.25 mg/kg); 20 patients (from parts 1 and 2) receiving the RP2D were evaluable for efficacy. Rate of CAR-T–associated neurotoxicity by day 30 (primary end point) was ∼50%, lower than reported in the ZUMA-1 trial (64%). Median event duration of grade ≥3 neurotoxicity was 7 days. No unexpected defibrotide-related safety findings and defibrotide-related treatment-emergent adverse events or deaths were reported. Results showed modest reduction in rate of CAR-T–associated neurotoxicity and high-grade neurotoxicity event duration relative to historical data; however, reduction was unlikely to meet the primary end point, so the study was terminated early. Nevertheless, results contribute valuable data for potential therapeutic insight on the management of CAR-T–associated neurotoxicity. This trial was registered at www.clinicaltrials.gov as #NCT03954106.
Introduction
Chimeric antigen receptor T-cell (CAR-T) therapy is one 1 of the most significant advances in cancer immunotherapy. It has generated impressive outcomes in hematologic malignancies, including B-cell non-Hodgkin lymphoma, multiple myeloma, and B-cell acute lymphoblastic leukemia. To date, 6 different CAR-T products have been approved by the US Food and Drug Administration, including 4 products targeting CD19 for B-cell non-Hodgkin lymphoma and B-cell acute lymphoblastic leukemia and 2 products targeting the B-cell maturation antigen for multiple myeloma.1 CAR T cells are genetically modified to express CARs that can specifically recognize tumor antigens, activate, and multiply to use a strong antitumor effect via a cascade of cytokines. Despite outstanding efficacy results, CAR-T therapy benefits are offset by class-effect toxicities that can be life threatening, such as cytokine release syndrome (CRS) and neurotoxicity.2
Although the pathophysiology and management of CRS are well described and standardized using anti–interleukin-6 therapy (ie, tocilizumab3) with or without steroids, which yields rapid resolution of CRS signs and symptoms, our understanding of CAR-T neurotoxicity is less established and continues to evolve.4 The pathophysiology of CAR-T neurotoxicity is believed to involve disruption of the blood-brain barrier (BBB) after inflammation and increased levels of cytokines as initiating factors.5 The resulting endothelial cell activation is thought to then trigger BBB disruption and increase permeability and coagulopathy, as demonstrated in a prior mouse model.6 The angiopoietin (Ang; 1 and 2)–Tie2 axis plays a significant role in balancing the endothelium between quiescence and activation. During severe neurotoxicity after CD19-directed CAR-T therapy, levels of Ang-2 and the ratio of Ang-2 to Ang-1 are significantly increased, potentially representing endothelial injury and subsequent disruption of the BBB and capillary leakage.7 The presence of high levels of von Willebrand factor, high molecular weight von Willebrand factor multimers, and depleted ADAMTS 13 levels also reinforce the endothelial activation and coagulopathy during neurotoxicity.7 Neurotoxicity of any severity occurs in 18% or 70% of patients treated with CAR-T therapy.8, 9, 10, 11, 12, 13, 14, 15, 16, 17 Severe neurotoxicity has been observed in the pivotal studies of all 6 CAR-T products. Differences in the costimulatory domains of each product and target tumor antigen may affect, among other factors, neurotoxicity risk.7 Among the available products, rates and severities of neurotoxicity are generally higher with CD19 CARs, compared with those with B-cell maturation antigen CARs, and with CARs with a CD28 costimulatory domain, compared with those with a 4-1BB costimulatory domain. The CD19-CD28-3z CAR-T axicabtagene ciloleucel (axi-cel) had the highest rate of any grade or high-grade neurotoxicity in large B-cell lymphoma (LBCL), at 64% and 28%, respectively, in the pivotal ZUMA-1 trial.17
Defibrotide is approved in the United States for the treatment of adult and pediatric patients with hepatic veno-occlusive disease/sinusoidal obstruction syndrome with renal or pulmonary dysfunction after hematopoietic cell transplantation (HCT) and in the European Union for the treatment of severe veno-occlusive disease/sinusoidal obstruction syndrome after HCT in patients aged >1 month.18,19 The mechanism of action of defibrotide involves reducing endothelial cell activation and damage and restoring the thrombofibrinolytic balance.20 Defibrotide also protects endothelial cells from damage caused by chemotherapy-related toxicities. In vitro studies have demonstrated that defibrotide reduces endothelial cell activation and protects the endothelium via antithrombotic, profibrinolytic, anti-inflammatory, anti-ischemic, and antioxidant effects.20, 21, 22, 23 It is hypothesized that defibrotide administered before and during CAR-T therapy may contribute to the stabilization of endothelial cells, thereby reducing the rate of CAR-T–associated neurotoxicity and CRS without affecting the antilymphoma effect of CAR-T therapy. Defibrotide, therefore, may be able to prevent the onset or progression of endothelial cell damage,24 perhaps by reducing the cytokine surge or strengthening the cellular barrier. This study was conducted to assess the safety and efficacy of defibrotide in the prevention of CAR-T–associated neurotoxicity in patients with relapsed or refractory LBCL who are receiving axi-cel.
Methods
Study design
This phase 2, prospective, multicenter, open-label, single-arm study (clinically registered as #NCT03954106) assessed the safety and efficacy of defibrotide for the prevention of CAR-T–associated neurotoxicity in patients with relapsed or refractory LBCL who are receiving axi-cel. The study was conducted in 2 parts: a lead-in phase (part 1) to assess the safety of defibrotide at a low dose (2.5 mg/kg) and then at a higher final dose (6.25 mg/kg), with the intent of setting the 6.25 mg/kg dose level as the recommended phase 2 dose (RP2D), and a treatment-extension phase (part 2) at the RP2D level to further evaluate both safety and efficacy of defibrotide in preventing CAR-T–associated neurotoxicity (Figure 1). A standard 3 + 3 design was used in part 1 to evaluate defibrotide at the 2 dose levels in 3 or 6 patients who were eligible. Dose-limiting toxicity (DLT) from the start of the first dose to 7 days after the last dose of defibrotide was assessed during the study and was adjudicated by the safety assessment committee (SAC) for each dose level during part 1 to determine the RP2D. The determination of DLTs was made from treatment-emergent adverse events (TEAEs) considered by the SAC to have a causal relationship with defibrotide. Because all hemorrhagic events were considered TEAEs of defibrotide, the SAC focused on any grade of intracranial hemorrhage and any other hemorrhage of grade ≥2. Nonhemorrhagic TEAEs of grade ≥3 were also deemed possible DLTs in this clinical setting. In part 2, the study was expanded to enroll additional patients at the RP2D to obtain up to a total of 29 patients who were evaluable for efficacy, including those from part 1 who were treated at the RP2D. Patients were observed for CAR-T–associated neurotoxicity up to CAR-T day 30. AEs and serious AEs were assessed from the signing of informed consent to 30 days after the last dose of defibrotide, and lymphoma response was captured up to day 60 of CAR-T therapy. The SAC continued to assess and monitor safety data, including serious and grade ≥3 TEAEs throughout part 2 of the study.
Figure 1.

Study design. ASBMT, American Society for Blood and Marrow Transplantation; CTCAE, Common Terminology Criteria for Adverse Events; R/R, relapsed or refractory; SAE, serious adverse event.
Patients
Patients were eligible for enrollment in the study if they were aged ≥18 years and scheduled to receive lymphodepletion chemotherapy followed by CAR-T immunotherapy with axi-cel for the treatment of relapsed or refractory LBCL. Study exclusion criteria included primary central nervous system lymphoma, ongoing or planned dialysis, recent or planned use of any investigational anticancer agent within 3 weeks before the first dose of defibrotide, previous CAR-T therapy, hemodynamic instability, clinically significant bleeding or use of medications that increase bleeding risk (including but not limited to systemic heparin, low molecular weight heparin, heparin analogs, alteplase, streptokinase, urokinase, antithrombin III, oral anticoagulants including warfarin, and factor Xa inhibitors; heparin for routine IV line management was allowed), serious disease or comorbid medical condition, and pregnancy/lactation.
The protocol for this study was approved by the institutional review board, and the study was conducted in accordance with the Declaration of Helsinki and all relevant ethical guidelines. All patients provided written informed consent.
Treatment and dosing
Treatment schedule and dosing are shown in Figure 2. Defibrotide was administered IV for a total of 11 days as follows: once daily as a single infusion for 3 days on CAR-T days −5, −4, and −3 (study days 1, 2, and 3) immediately before lymphodepletion chemotherapy; no defibrotide on CAR-T days −2 and −1 (study days 4 and 5); and 4 times a day for 8 days starting on CAR-T day 0 (the day CAR-T infusion was administered) to day +7 (study days 6-13). Defibrotide was infused over 2 hours (±15 minutes) at 2.5 mg/kg per dose or 6.25 mg/kg per dose every 6 hours. A minimum of 2 doses of defibrotide must have been administered before axi-cel infusion on day 0. If administration of axi-cel was delayed for >2 days, the patient was not evaluable for efficacy.
Figure 2.

Study treatment schedule and doses. DF, defibrotide.
Study end points
The primary end point of the study was the rate of CAR-T–associated neurotoxicity (any grade, defined by Common Terminology Criteria for Adverse Events, version 5.0) by CAR-T day 30. Secondary efficacy end points included the rate of grade ≥3 CAR-T–associated neurotoxicity; rate of any grade CAR-T–associated neurotoxicity per the American Society for Transplantation and Cellular Therapy consensus grading system by CAR-T day 30; rate of any grade and grade ≥3 CRS per the American Society for Transplantation and Cellular Therapy criteria by CAR-T day 30; and use of high-dose steroids (defined as a dose of dexamethasone of ≥7.5 mg/d; considered equivalent to hydrocortisone 200 mg/d, methylprednisolone 40 mg/d, or prednisone 50 mg/d) by CAR-T day 30.25 Lymphoma response evaluation by Cheson criteria was captured by day +60 of CAR-T administration and was also a secondary end point.26 Safety end points included TEAEs and serious TEAEs that occurred up to 30 days after the last dose of defibrotide. Severity was classified using Common Terminology Criteria for Adverse Events version 5.0 for AEs. Exploratory end points included duration of hospital and intensive care unit stay; data for median duration of neurotoxicity, especially high grade, was also explored. Although samples were collected over the course of the study, translational sample analysis was not completed because the study was terminated early.
Statistical analysis
Data are summarized based on the descriptive statistics for continuous variables or by the number and percentage of patients for categorical variables. All analyses were completed using Statistical Analysis System version 9.3 or later (SAS Institute Inc, Cary, NC). A Simon optimal 2-stage test was used to test the rate of no CAR-T–associated neurotoxicity of any grade by day 30 after CAR-T therapy.27 This study design was used to avoid unnecessarily exposing patients to a potentially nonefficacious therapy with defibrotide. In stage 1, the planned accrual was 10 patients who were evaluable for efficacy. If ≥6 patients had CAR-T–associated neurotoxicity after CAR-T therapy, the study would be stopped; otherwise, 19 additional patients would be accrued for a total of 29 patients who were evaluable. Study design was based on the reported rate of CAR-T–associated neurotoxicity (any grade) of 64% found in the ZUMA-1 trial (NCT02348216), the pivotal multicenter phase 2 trial with axi-cel.17 A clinically relevant reduction of 50% in the rate of neurotoxicity (ie, 64%-32%) was anticipated for this study.
The sample size calculation for phase 2 of the study was based on an overall 1-sided type 1 error of 0.05 and a statistical power of ≥90%.27 The null hypothesis would be rejected if ≥15 patients (out of 29) had no CAR-T–associated neurotoxicity after CAR-T therapy, suggesting that the CAR-T–associated neurotoxicity rate for defibrotide is significantly <64%.
In this analysis, the overall sample size included patients from part 1 and part 2. Patients treated at the RP2D in part 1 were included in the analyses for efficacy and safety end points. Assuming that 10% of enrolled patients would be ineligible for the efficacy evaluation, the study planned to enroll a total of 32 patients (anticipated 29 who were evaluable for efficacy) to be treated at the RP2D. The efficacy-evaluable analysis cohort included all patients receiving ≥18 doses of defibrotide at the RP2D who either developed CAR-T–associated neurotoxicity on/before day 30 or completed the day 30 neurologic assessment, plus those who discontinued defibrotide before 18 doses because of CAR-T–associated neurotoxicity. Patients whose axi-cel therapy was delayed by >2 days from the original schedule were considered not evaluable for efficacy. The safety analysis cohort included all patients who received ≥1 dose of defibrotide.
Results
Patients
In total, 26 patients were screened, and 25 patients were enrolled at 4 United States sites (Figure 3). Of these, 10 patients participated in part 1 (4 patients received defibrotide at 2.5 mg/kg, and 6 patients received defibrotide at 6.25 mg/kg). One of the first 3 patients enrolled at the 2.5-mg/kg dose level discontinued defibrotide after serious grade 3 events of pleural effusion and pulmonary embolism (PE) determined by the investigator as not related to defibrotide. Defibrotide was discontinued when anticoagulation medication was initiated to treat PE because this type of medication was prohibited per protocol. This participant had to be replaced, resulting in a total of 4 patients being treated at this dose. There were no DLTs during part 1 of the study per the SAC, and 6.25 mg/kg was identified as the RP2D in part 1. A total of 15 patients were enrolled in part 2, contributing to a total of 21 patients (6 patients from part 1, and 15 enrolled from part 2) treated at the RP2D of 6.25 mg/kg. The efficacy-evaluable analysis cohort comprised 20 patients (6 patients from part 1 and 14 patients from part 2) for analysis of the primary and secondary efficacy end points; 1 patient enrolled in part 2 had a CAR-T infusion delay of >2 days and was not included in the efficacy-evaluable analysis. The first 10 patients at the RP2D (6 patients from part 1 and 4 patients from part 2) were used in stage 1 of the analysis. The safety analysis cohort consisted of 25 patients, 20 (80%) of whom completed defibrotide treatment; of the 5 patients who discontinued defibrotide, reasons for discontinuation included TEAEs (3 [12%]; 1 patient had a grade 3 immune effector cell–associated encephalopathy score and grade 2 depressed level of consciousness; 1 patient had serious TEAEs of CRS and grade 2 immune effector cell neurotoxicity, grade 2 intentional upper extremity tremor, grade 2 autonomic dysfunction, and grade 2 dysphasia; and 1 patient had a serious grade 3 event of febrile neutropenia that led to study-drug interruption; however, this participant did not resume defibrotide treatment), investigator decision (1 [4%]; 1 patient had grade 3 pleural effusion and PE in part 1 of the study and had to be replaced), and delay in axi-cel administration of >2 days (1 [4%]). None of the TEAEs leading to defibrotide discontinuation were assessed by the investigator as related to defibrotide treatment.
Figure 3.

Patient disposition.
For the overall patient population (combined part 1 and part 2 participants), the median age was 68 years (range, 31-78 years), 17 of 25 (68%) were male, and all (100%) were White, non-Hispanic (Table 1). Patients had received a median of 2 previous treatment regimens (range, 1-6), 21 (84%) patients were refractory to previous treatment, and 5 (20%) had received previous autologous stem cell transplantation; 1 patient was treatment refractory and had prior HCT. Patients at the RP2D received a median of 35 total defibrotide doses (range, 3-35), for a median defibrotide exposure of 11 days (range, 3-12 days).
Table 1.
Demographic and baseline disease characteristics (safety analysis cohort)
| Part 1 |
Patients treated at RP2D from part 1 and part 2 |
Overall (N = 25) | ||
|---|---|---|---|---|
| Defibrotide 2.5 mg/kg (n = 4) |
Defibrotide 6.25 mg/kg (n = 6) |
Defibrotide 6.25 mg/kg (n = 21) |
||
| Age, y | ||||
| Median | 63.5 | 72.0 | 69.0 | 68.0 |
| Min, max | 31, 73 | 52, 76 | 43, 78 | 31, 78 |
| Age group, y, n (%) | ||||
| 18-64 | 2 (50) | 2 (33) | 7 (33) | 9 (36) |
| ≥65 | 2 (50) | 4 (67) | 14 (67) | 16 (64) |
| Sex, n (%) | ||||
| Female | 1 (25) | 2 (33) | 7 (33) | 8 (32) |
| Male | 3 (75) | 4 (67) | 14 (67) | 17 (68) |
| White, non-Hispanic, n (%) | 4 (100) | 6 (100) | 21 (100) | 25 (100) |
| ECOG PS at baseline, n (%) | ||||
| 0 | 2 (50) | 0 | 5 (24) | 7 (28) |
| 1 | 2 (50) | 6 (100) | 16 (76) | 18 (72) |
| Number of previous treatment regimens | ||||
| Median | 2.0 | 3.5 | 3.0 | 2.0 |
| Min, max | 2, 2 | 2, 6 | 1, 6 | 1, 6 |
| Primary refractory disease, n (%) | ||||
| Yes | 3 (75) | 2 (33) | 11 (52) | 14 (56) |
| No | 1 (25) | 4 (67) | 10 (48) | 11 (44) |
| Refractory to previous treatment, n (%) | ||||
| Yes | 4 (100) | 6 (100) | 17 (81) | 21 (84) |
| No | 0 | 0 | 4 (19) | 4 (16) |
| Previous autologous stem cell transplantation, n (%) | ||||
| Yes | 0 | 2 (33) | 5 (24) | 5 (20) |
| No | 4 (100) | 4 (67) | 16 (76) | 20 (80) |
| Number of recurrences | ||||
| Median | 1.0 | 2.5 | 2.0 | 1.0 |
| Min, max | 1, 1 | 1, 5 | 0, 5 | 0, 5 |
| Defibrotide exposure, d | ||||
| Median | 11.0 | 11.5 | 11.0 | – |
| Min, max | 1, 12 | 11, 12 | 3, 12 | – |
| Total doses received | ||||
| Median | 34.0 | 35.0 | 35.0 | – |
| Min, max | 1, 35 | 35, 35 | 3, 35 | – |
| Daily dose, mg/kg per day | ||||
| Median | 7.320 | 19.190 | 19.760 | – |
| Min, max | 2.46, 7.89 | 18.26, 20.07 | 6.25, 20.37 | – |
ECOG PS, Eastern Cooperative Oncology Group performance status; Max, maximum; Min, minimum.
Efficacy
In stage 1 of the analysis, 5 (50%) of the first 10 patients who were treated with defibrotide at the RP2D did not have CAR-T–associated neurotoxicity of any grade by day 30; therefore, the stopping rule was not met, and additional patients were accrued. As enrollment continued, 5 of the 10 additional patients who were evaluable for efficacy did not have CAR-T–associated neurotoxicity of any grade by day 30 of CAR-T therapy. The predictive probability of meeting the primary efficacy end point if enrollment continued to the planned 29 patients who were evaluable for efficacy was calculated after that of these 20 patients evaluable for efficacy. It was determined that the study was unlikely to meet the primary efficacy end point after full enrollment; therefore, the study was terminated early.
The rate of no CAR-T–associated neurotoxicity of any grade was 51% (90% confidence interval, 36-66; P = .099; vs that in historical data from the ZUMA-1 trial) after adjusting for the stage 1 analysis; correspondingly, the incidence rate of neurotoxicity of any grade was 49%.17,28 Because the study was terminated early, efficacy results are considered descriptive, and P values are nominal. Overall, 50% of patients had any grade neurotoxicity, and 25% had grade ≥3 neurotoxicity (Table 2). Data showed a median time to neurotoxicity onset (ie, any grade) of 9 days; the median duration of grade ≥3 neurotoxicity was 7 days. High-dose steroids were used in 45% of patients, and the median length of hospitalization was 19 days (Table 3). The overall rate of no CRS of any grade was 15% by day 30 of CAR-T treatment.
Table 2.
Proportions and timing of CAR-T–associated neurotoxicity
| Neurotoxicity, CTCAE version 5.0 | Patients who were evaluable (n = 20) |
|---|---|
| Rating by CAR-T day 30 | |
| Any grade, n (%) | 10 (50)∗ |
| Grade ≥3, n (%) | 5 (25) |
| Median event duration, d (range) | |
| Any grade | 24 (8-39)† |
| Grade ≥3 | 7 (2-28) |
CTCAE, Common Terminology Criteria for Adverse Events.
Value is unadjusted for stage 1 analysis.
Event duration was calculated from the onset of the first CAR-T neurotoxicity event to the end date of the last CAR-T neurotoxicity event.
Table 3.
Use of high-dose steroids and hospitalization
| Parameter | Current RP2D evaluation (n = 20) |
|---|---|
| High-dose steroid use for neurotoxicity, n (%) | 9 (45) |
| Hospitalization, median, d (range) | 19 (9-42) |
Safety
There were no DLTs reported by the SAC during the study. One patient in part 1 who received 2.5 mg/kg had a grade 3 serious TEAE of PE and pleural effusion that was assessed by the investigator as not related to defibrotide.
All study participants had ≥1 TEAE. The most common TEAEs (≥20%) in the safety analysis population based on dose groups are shown in Table 4, and a summary based on the maximum severity is presented in supplemental Table 1. Most patients (76%) had ≥1 TEAE of grade ≥3. Approximately half of the patients (52%) had ≥1 serious TEAE, and there were 3 discontinuations because of TEAEs. Moreover, 6 patients experienced ≥1 bleeding events. For patients receiving the RP2D (defibrotide 6.25 mg/kg), the most common TEAEs were pyrexia (81%), CRS (76%), hypotension (67%), nausea (67%), diarrhea (57%), fatigue (57%), constipation (52%), tremor (52%), and neutropenia (48%). No TEAEs were assessed as related to defibrotide. Furthermore, timing of CAR-T–associated neurotoxicity, based on the severity, in 10 patients who were evaluable is shown in supplemental Figure 1.
Table 4.
TEAEs occurring in 20% or more patients (safety analysis cohort)
| Preferred term, n (%) | Part 1 |
Patients treated at RP2D from part 1 and part 2 |
|
|---|---|---|---|
| Defibrotide 2.5 mg/kg (n = 4) |
Defibrotide 6.25 mg/kg (n = 6) |
Defibrotide 6.25 mg/kg (n = 21) |
|
| Patients with ≥1 TEAE∗ | 4 (100) | 6 (100) | 21 (100) |
| Pyrexia | 3 (75) | 5 (83) | 17 (81) |
| CRS | 3 (75) | 6 (100) | 16 (76) |
| Hypotension | 2 (50) | 6 (100) | 14 (67) |
| Nausea | 3 (75) | 3 (50) | 14 (67) |
| Diarrhea | 3 (75) | 4 (67) | 12 (57) |
| Fatigue | 1 (25) | 2 (33) | 12 (57) |
| Constipation | 2 (50) | 3 (50) | 11 (52) |
| Tremor | 1 (25) | 4 (67) | 11 (52) |
| Chills | 2 (50) | 5 (83) | 10 (48) |
| Headache | 2 (50) | 1 (17) | 10 (48) |
| Neutropenia | 1 (25) | 5 (83) | 10 (48) |
| Neurotoxicity | 0 | 4 (67) | 9 (43) |
| Decreased appetite | 1 (25) | 2 (33) | 8 (38) |
| Confusional state | 2 (50) | 3 (50) | 7 (33) |
| Febrile neutropenia | 1 (25) | 5 (83) | 7 (33) |
| Hypoxia | 2 (50) | 3 (50) | 7 (33) |
| Anemia | 1 (25) | 4 (67) | 6 (29) |
| Dizziness | 1 (25) | 2 (33) | 6 (29) |
| Hypocalcemia | 1 (25) | 2 (33) | 6 (29) |
| Hypomagnesemia | 0 | 3 (50) | 6 (29) |
| Sinus tachycardia | 0 | 2 (33) | 6 (29) |
| Back pain | 0 | 2 (33) | 5 (24) |
| Hypophosphatemia | 2 (50) | 1 (17) | 5 (24) |
| Edema peripheral | 1 (25) | 1 (17) | 5 (24) |
| Oropharyngeal pain | 0 | 1 (17) | 5 (24) |
| Urinary incontinence | 1 (25) | 3 (50) | 5 (24) |
| Vomiting | 1 (25) | 3 (50) | 5 (24) |
Preferred terms are sorted in descending order of incident based on phase 2.
A total of 10 patients receiving the RP2D (48%) had ≥1 grade 3 TEAEs, most commonly neutropenia (29%), and febrile neutropenia, anemia, and neurotoxicity (each 24%). Moreover, 5 patients receiving the RP2D (24%) had ≥1 grade 4 TEAEs, including neutropenia (14%), and febrile neutropenia, thrombocytopenia, neutrophil count decreased, platelet count decreased, and tumor lysis syndrome (each 5%). Nine patients receiving the RP2D (43%) experienced ≥1 serious TEAE: CRS, neurotoxicity, and hypotension each in 2 patients (10%); all other serious events occurred in 1 patient each (5%). Three patients discontinued defibrotide because of TEAEs. Among the patients who discontinued defibrotide because of TEAEs, 1 patient receiving the RP2D had serious grade 3 neurotoxicity (grade 3 immune effector cell–associated encephalopathy score and grade 2 depressed level of consciousness); 1 patient receiving the RP2D had serious TEAEs of CRS and grade 2 AEs of immune effector cell neurotoxicity, upper extremity tremor, autonomic dysfunction, and dysphasia; and 1 patient receiving the RP2D had a serious grade 3 event of febrile neutropenia that led to study-drug interruption and did not resume defibrotide treatment. One patient who received 2.5 mg/kg in part 1 had serious grade 3 pleural effusion and PE, and this patient was replaced in the study. No TEAEs were related to defibrotide, and none were fatal. There were no deaths during the study.
Lymphoma response
Lymphoma response up to CAR-T day +60 was assessed in all 4 patients who received 2.5 mg/kg dosing in part 1, with 1 patient (25%) achieving complete response and 3 patients (75%) achieving partial response. Of the 21 patients who received the RP2D at 6.25 mg/kg, the overall response rate was 95% (n = 20), with 11 patients (52%) achieving complete response and 9 patients (43%) achieving partial response; 1 patient (5%) had progressive disease (supplemental Table 2).
Discussion
In this proof-of-concept study, 20 patients receiving the RP2D of defibrotide 6.25 mg/kg were evaluable for efficacy. Although the study did not meet the stopping rule and passed the interim analysis, the predictive probability that the study would achieve the primary end point at the final analysis if enrollment was completed was low, and hence, the study was terminated prematurely. There were, however, some interesting findings from the study. Study results showed a modest reduction in the rate of CAR-T–associated neurotoxicity and high-grade event duration relative to that in historical data (64%).17 The rate of no CAR-T–associated neurotoxicity was 51% (correspondingly, the incidence rate of neurotoxicity of any grade was 49%), and prophylactic therapy with defibrotide was well tolerated in participants undergoing CAR-T therapy. TEAEs were consistent with either the known safety profile of defibrotide or with complications associated with CAR-T therapy. There were no DLTs with defibrotide 2.5 or 6.25 mg/kg. In addition, there were no deaths during the study, no TEAEs were related to defibrotide, and no significant bleeding was associated with defibrotide.
Several observations can be made from the study results. The study provides additional information with regard to historical data and contributes to the small pool of published information available on neurotoxicity in patients receiving CAR-T therapy. Overall, the neurotoxicity events were lower in this study than those in ZUMA-1 (overall, 50% vs 64%, respectively; grade ≥3, 25% vs 28%, respectively).17 The median onset and event duration of neurotoxicity also showed interesting results. Our data showed a median onset of any grade neurotoxicity of 9 days and a median event duration of grade ≥3 neurotoxicity of 7 days, whereas the ZUMA-1 trial reported 5 and 26 days, respectively.17,29 The median length of hospitalization was slightly longer in this study than in ZUMA-1 (19 vs 13 days, respectively), and the use of high-dose steroids was higher than that in reference axi-cel data (45% vs 17%, respectively). This increase in steroid use may be related to changes in current clinical practice relative to the initial axi-cel trial. There is growing evidence showing that liberal corticosteroid use does not alter efficacy; therefore, there is, now, greater comfort with starting corticosteroids to treat neurotoxicity and CRS.4,30 Furthermore, 15% of patients did not develop any grade CRS by CAR-T day 30. Of 21 patients, only 2 patients who received the RP2D experienced serious TEAEs of CRS; of these 2 patients, 1 patient experienced serious TEAEs of CRS and neurotoxicity that led to discontinuation of defibrotide.
It is also worth noting that the length of hospital stay is often determined based on discharge barriers such as rehabilitation placement,31 so our observations may not directly reflect the safety and tolerability of CAR-T therapy. Moreover, lymphoma response evaluation was collected per the protocol, and all but 1 patient with progressive disease at CAR-T day 28 had a complete or partial lymphoma response, suggesting that defibrotide did not decrease the efficacy of CAR-T therapy. Given these limitations, the short duration of grade ≥3 neurotoxicity is the most relevant finding of this study.
Defibrotide is known to reduce endothelial cell activation and to protect the endothelium from inflammation, such as that observed in CAR-T–associated neurotoxicity.4,20, 21, 22, 23 The data from this study showed a modest defibrotide treatment effect that reduced CAR-T–associated neurotoxicity and high-grade event duration compared with historical data. The study conclusions and generalizability of these results are limited by the fact that the study was terminated early, given it was unlikely that the primary end point would be met upon completion of target enrollment. This limitation of the trial, along with the small sample size, likely contributed to the lack of statistical significance. Furthermore, clinical practice has changed since the ZUMA-1 pivotal trial, limiting comparability with our current study. A key difference was the use of levetiracetam and earlier use of corticosteroids and tocilizumab for the management of CRS and neurotoxicity, similar to ZUMA-1 cohorts 4 and 6.17,32,33
In conclusion, although accrual was terminated early because of futility, findings from this study are clinically relevant and may suggest a proof-of-concept for the use of defibrotide in the prevention of CAR-T–associated neurotoxicity. We note the lower numerical rate of all-grade neurotoxicity incidence and the shorter duration of grade ≥3 neurotoxicity compared with that in historical data of axi-cel in ZUMA-1. In addition, there was a delay in the onset of neurotoxicity vs what was observed in ZUMA-1. Although larger studies are required to validate the results of this study, these data contribute valuable knowledge related to therapy for CAR-T–associated neurotoxicity.
Conflict-of-interest disclosure: C.A.J. has served as a consultant for Kite/Gilead, Novartis, Bristol Myers Squibb (BMS)/Celgene, bluebird bio, Epizyme, Ipsen, Daiichi Sankyo, Instill Bio, ImmPACT Bio, Abintus Bio, and Caribou Bio, and has received research funding from Kite/Gilead and Pfizer. J.A. has served as a consultant for Regeneron and Juno/BMS. S.A. is an employee of Jazz Pharmaceuticals and holds stock and/or stock options in Jazz Pharmaceuticals. P.Z. was an employee of Jazz Pharmaceuticals at the time of the study; holds stock and/or stock options in Jazz Pharmaceuticals; and is a current employee of, and holds stock and/or stock options in, Incyte Corporation. W.W. is an employee of Jazz Pharmaceuticals and holds stock and/or stock options in Jazz Pharmaceuticals. V.A. is an employee of Jazz Pharmaceuticals and holds stock and/or stock options in Jazz Pharmaceuticals. J.A.Y. has served as a consultant and advisory board member for Sanofi, Kite/Gilead, Omeros, Kadmon, and Jazz Pharmaceuticals, and has received research funding from Gilead. A.C.R. declares no competing financial interests.
Acknowledgments
The authors thank all the study participants, and their families and caregivers.
Medical writing and editorial assistance were provided by Laura Ninger, and Laura Weber, of Lumanity Scientific Inc and financially supported by Jazz Pharmaceuticals and Trina Soluta and Caroline Davidson of CMC AFFINITY, a division of IPG Health Medical Communications, under direction of the authors and with funding from Jazz Pharmaceuticals, in accordance with Good Publication Practice (GPP 2022) guidelines.
Authorship
Contribution: C.A.J. and P.Z. designed the study, participated in the acquisition, analysis, and interpretation of the data, wrote and critically revised the manuscript, and gave final approval for the manuscript to be submitted; A.C.R., W.W., and J.A.Y. participated in the acquisition, analysis, and interpretation of the data, wrote and critically revised the manuscript, and gave final approval for the manuscript to be submitted; and J.A., S.A., and V.A. participated in the conduct of the trial, participated in the acquisition, analysis, and interpretation of the data, wrote and critically revised the manuscript, and gave final approval for the manuscript to be submitted.
Footnotes
All relevant data are provided within the manuscript. Jazz has established a process to review requests from qualified external researchers for data from Jazz-sponsored clinical trials in a responsible manner that includes protecting patient privacy, assuring data security and integrity, and furthering scientific and medical innovation. Qualified researchers may request acess through Jazz Pharmaceuticals’ data-sharing criteria at https://www.jazzpharma.com/science/clinical-trial-data-sharing/.
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.National Cancer Institute at the National Institutes of Health. CAR T Cells: Engineering Patients’ Immune Cells to Treat Their Cancers. 2022 https://www.cancer.gov/about-cancer/treatment/research/car-t-cells Accessed 27 July 2023. [Google Scholar]
- 2.Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T-Cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–444. doi: 10.1200/EDBK_238691. [DOI] [PubMed] [Google Scholar]
- 3.Actemra (tocilizumab). Prescribing information. Genentech, Inc. 2010. [Google Scholar]
- 4.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022;22(2):85–96. doi: 10.1038/s41577-021-00547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 7.Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7(12):1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson CA, Chavez JC, Sehgal AR, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23(1):91–103. doi: 10.1016/S1470-2045(21)00591-X. [DOI] [PubMed] [Google Scholar]
- 11.Schuster SJ, Tam CS, Borchmann P, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22(10):1403–1415. doi: 10.1016/S1470-2045(21)00375-2. [DOI] [PubMed] [Google Scholar]
- 12.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 13.Fowler NH, Dickinson M, Dreyling M, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. 2022;28(2):325–332. doi: 10.1038/s41591-021-01622-0. [DOI] [PubMed] [Google Scholar]
- 14.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munshi NC, Anderson LD, Jr., Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 16.Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324. doi: 10.1016/S0140-6736(21)00933-8. [DOI] [PubMed] [Google Scholar]
- 17.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Defitelio. Summary of Product Characteristics. Defitelio. 2019 [Google Scholar]
- 19.Defitelio. 80 mg/mL Summary of Product Characteristics EU. Gentium SRL. 2016 [Google Scholar]
- 20.Richardson PG, Palomo M, Kernan NA, Hildebrandt GC, Chao N, Carreras E. The importance of endothelial protection: the emerging role of defibrotide in reversing endothelial injury and its sequelae. Bone Marrow Transplant. 2021;56(12):2889–2896. doi: 10.1038/s41409-021-01383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127(13):1719–1727. doi: 10.1182/blood-2015-10-676114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palomo M, Diaz-Ricart M, Rovira M, Escolar G, Carreras E. Defibrotide prevents the activation of macrovascular and microvascular endothelia caused by soluble factors released to blood by autologous hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2011;17(4):497–506. doi: 10.1016/j.bbmt.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Pescador R, Capuzzi L, Mantovani M, Fulgenzi A, Ferrero ME. Defibrotide: properties and clinical use of an old/new drug. Vascul Pharmacol. 2013;59(1-2):1–10. doi: 10.1016/j.vph.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11:1973. doi: 10.3389/fimmu.2020.01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee DW, Santomasso BD, Locke FL, et al. ASBMT consensus grading for cytokine release syndrome and neurological toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheson BD, Ansell S, Schwartz L, et al. Refinement of the Lugano classification lymphoma response criteria in the era of immunomodulatory therapy. Blood. 2016;128(21):2489–2496. doi: 10.1182/blood-2016-05-718528. [DOI] [PubMed] [Google Scholar]
- 27.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10(1):1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 28.Koyama T, Chen H. Proper inference from Simon's two-stage designs. Stat Med. 2008;27(16):3145–3154. doi: 10.1002/sim.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.BLA Clinical Review Memorandum – YESCARTA. FDA. 2017 [Google Scholar]
- 30.Liu S, Deng B, Yin Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15. doi: 10.1038/s41408-020-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao EJ, Yeluru A, Manjunath L, et al. A long wait: barriers to discharge for long length of stay patients. Postgrad Med J. 2018;94(1116):546–550. doi: 10.1136/postgradmedj-2018-135815. [DOI] [PubMed] [Google Scholar]
- 32.Topp MS, van Meerten T, Houot R, et al. Earlier corticosteroid use for adverse event management in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. 2021;195(3):388–398. doi: 10.1111/bjh.17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oluwole OO, Bouabdallah K, Muñoz J, et al. Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. 2021;194(4):690–700. doi: 10.1111/bjh.17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.