

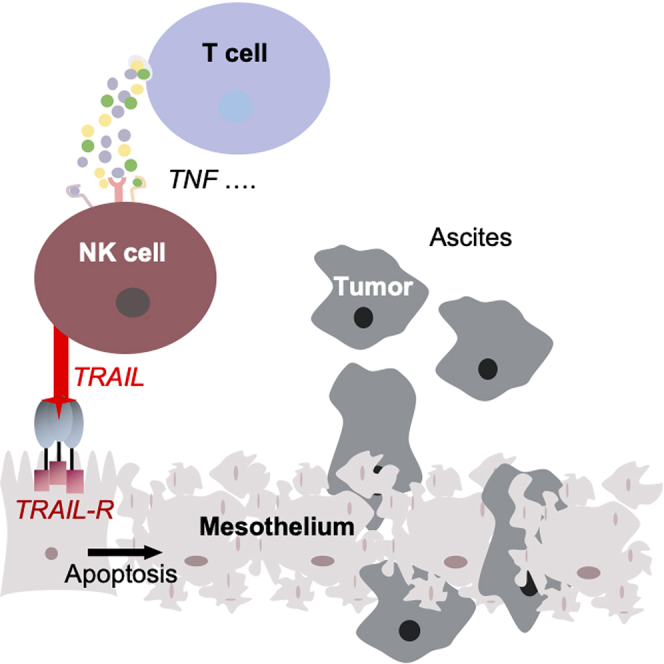
Summary
A crucial requirement for metastasis formation in ovarian high-grade serous carcinoma (HGSC) is the disruption of the protective peritoneal mesothelium. Using co-culture systems of primary human cells, we discovered that tumor-associated NK cells induce TRAIL-dependent apoptosis in mesothelial cells via death receptors DR4 and DR5 upon encounter with activated T cells. Upregulation of TRAIL expression in NK cells concomitant with enhanced cytotoxicity toward mesothelial cells was driven predominantly by T-cell-derived TNFα, as shown by affinity proteomics-based analysis of the T cell secretome in conjunction with functional studies. Consistent with these findings, we detected apoptotic mesothelial cells in the peritoneal fluid of HGSC patients. In contrast to mesothelial cells, HGSC cells express negligible levels of both DR4 and DR5 and are TRAIL resistant, indicating cell-type-selective killing by NK cells. Our data point to a cooperative action of T and NK in breaching the mesothelial barrier in HGSC patients.
Subject areas: Microenvironment, Immunology, Cancer
Graphical abstract
Highlights
-
•
Cancer-associated NK cells induce TRAIL-dependent apoptosis in mesothelial cells
-
•
Induction of apoptosis by NK cells is mainly driven by T-cell-derived TNFα
-
•
NK-cell-mediated mesothelial cell death promotes ovarian cancer cell invasion
-
•
Low TRAIL receptor levels on cancer cells contribute to their TRAIL resistance
Microenvironment; Immunology; Cancer
Introduction
Extended peritoneal metastasis is one of the central clinical hallmarks of high-grade serous ovarian carcinoma (HGSC) contributing to an overall poor prognosis of the disease. A prerequisite for metastatic spread is the invasion of cancer cells through the mesothelium covering peritoneal organs. It is generally believed that the mesothelial lining acts as a barrier against tumor cell invasion.1 This is supported by the presence of lesions in the mesothelial cell layer at metastatic sites.2,3 It is hypothesized that formation of such gaps precedes adherence of tumor cells from the malignant ascites to the submesothelial basement membrane, followed by its disruption.
The mechanisms underlying the cellular interactions in the tumor microenvironment (TME) that affect the integrity of the mesothelial cell layer and thereby enable tumor invasion, are only partly understood. Several factors are assumed to be involved, including changes in the mesothelial cell architecture due to mesothelial-mesenchymal transition (MMT) induced by ascites-derived mediators,4,5 induction of mesothelial cell senescence,6 and mesothelial clearance by myosin-dependent mechanical forces exerted by tumor cells.7 Apart from this, gastrointestinal and ovarian tumor cells are able to induce apoptosis in human peritoneal mesothelial cells (HPMC) by different mechanisms including Fas/FasL interaction,8 activation of the TGFβ signaling pathway9,10 or release of extracellular vesicles.11,12,13 Mesothelial injury caused by increased cell death has also been observed in the context of inflammation after long-term peritoneal dialysis or acute peritonitis.14,15,16,17,18 In explanation, a Fas/FasL dependent action was postulated, while other cytotoxic ligands like tumor necrosis factor α (TNFα) and tumor necrosis factor related apoptosis inducing ligand (TRAIL) did not appear to be relevant.14 Since acute peritonitis is accompanied by a sudden increase of peritoneal leukocytes, it was suggested that immune cells may play a role in regulating HPMC survival.14
In view of a general apparent susceptibility of mesothelial cells toward apoptosis in the context of pathological conditions including inflammation, we asked (i) if tumor-associated lymphocytes (TAL) exhibit selective cytotoxicity directed against HPMC in HGSC patients, (ii) whether this phenomenon persists in the presence of immunosuppressive ascites mimicking the HGSC TME, and may (iii) support peritoneal tumor invasion and metastasis as a consequence.
Results
Activated tumor-associated NK cells induce apoptosis in HPMC from HGSC patients
A monolayer of omental HPMC was subjected to co-culture with TAL isolated from malignant ascites for 6 h at an HPMC:TAL ratio of 1:10 (determined as optimal conditions in preliminary experiments) prior to annexin V/PI staining (schematic overview see Figure 1A). Intriguingly, TAL from different patients were generally able to attack HPMC, but apoptosis levels were significantly enhanced when TAL were activated by α-CD3 Ab stimulation prior to co-culture with HPMC (Figure 1B). This effect was maintained in a completely autologous setting using matched pairs of TAL and HPMC from the same patient (Figure 1B, blue dots; example dot plots of flow cytometric evaluation are given in Figure 1C). To address the question, which immune cells were responsible for the HPMC killing observed, TAL from ascites were fractionated by magnetic activated cell sorting (MACS) sorting into CD3+ T cells and CD3−CD56+ NK cells prior to co-culture with HPMC. As depicted in Figure 1D, neither CD3+ T cells subjected to α-CD3 Ab activation nor NK cells alone induced apoptosis in HPMC, whereas mixtures (1:1) of both cellular fractions yielded effects similar to those observed with unfractionated TAL (Figure 1B).
Figure 1.

T cell-activated NK cells induce HPMC apoptosis
(A) Schematic overview of the experimental setting. (A) Experimental setup. TAL derived from HGSC patient ascites were cultured +/− α-CD3 Ab stimulation prior to the co-culture with patient-derived HPMC. Cells were analyzed for HPMC apoptosis (annexin V/PI), as well as NK/T cell degranulation (CD335+/CD107a+, CD3+/CD107a+) by flow cytometry.
(B) Apoptotic HPMC after co-culture with TAL are shown as percentage of annexin V+ cells by gating on CD45−cells to exclude TAL contaminations (n = 11 patients, including n = 3 matched pairs of TAL and HPMC from the same patient as indicated by blue dots). The background apoptosis was measured from HPMC alone.
(C) Exemplary annexin V/PI dot plots gated on HPMC after co-culture with unstimulated and α-CD3 Ab stimulated TAL.
(D) Apoptosis induction in HPMC after co-culture with isolated CD3+ T cell, CD3− NK cells, and T/NK cell mix at 1:1 ratio (n = 4 different patients).
(E and F) Determination of NK and T cell degranulation by flow cytometry in response to HPMC. The percentage of degranulating NK cells (CD335+/CD107a+) (E) and T cells (CD3+/CD107a+) (F) in TAL co-cultured with HPMC is depicted (n = 4 different patients, including n = 3 autologous pairs as indicated by blue dots). The background degranulation was measured for TAL in the absence of HPMC.
(G) Representative FACS blots showing degranulating NK cells (CD335+/CD107a+) and T cells (CD3+/CD107a+) in HPMC co-cultures with TAL (+/− α-CD3 Ab stimulation). Background staining of TAL alone (+/− α-CD3 Ab stimulation) is given.
(H) The amount of degranulating NK cells (CD335+/CD107a+) is depicted after HPMC co-culture with isolated NK cells compared to NK/T cell mix (n = 5 different patients).
Horizontal bars indicate the mean and vertical error bars represent the standard deviation in B, D, E, F, and H. ∗ FDR < 0.05; ∗∗ FDR < 0.01; ∗∗∗ FDR < 0.001; determined by paired t test and Benjamini-Hochberg adjustment (ns: not significant).
To shed light onto the underlying killing mechanism, we next investigated the contribution of granzyme B (GrB)/perforin degranulation, in terms of HPMC killing by TAL subpopulations including T and NK cells. Increased expression of the degranulation marker CD107a was observed on CD335+ NK cells (within the α-CD3-activated TAL population) in response to HPMC co-culture (Figure 1E), whereas no induction of CD107a was observed on CD3+ T cells under those conditions (Figures 1F and 1G shows examples of flow cytometric evaluation of CD107a expression). Positive controls for degranulation of CD3+ T cells (in response to PMA/ionomycin stimulation) and of NK cells (in response to K562 cells) are presented in Figure S1. In accordance with the annexin V/PI measurement in Figure 1D, isolated T and NK cell fractions alone did not yield significant degranulation following HPMC co-culture, whereas CD107a+ NK cells were detected when NK cells were mixed with α-CD3 Ab-activated T cells prior to the co-culture (Figure 1H). Based on these data, we conclude that ascites-associated NK cells can acquire cytolytic activity targeting HPMC, but that a prior interaction with activated T cells is a prerequisite for HPMC killing. We further demonstrated that the pro-apoptotic activity of activated NK cells was clearly related to increased GrB/perforin degranulation.
TRAIL-dependent killing of HPMC by NK cells in contrast to GrB/perforin pathway is active in ascitic fluid
We next addressed the question if the cytolytic activity of NK cells toward HPMC persists in the presence of immunosuppressive ascitic fluid. To that end we compared induction of apoptosis in HPMC by α-CD3-activated TAL in the presence of cell-free malignant ascites or 5% AB serum. As depicted in Figure 2A, the fraction of apoptotic HPMC was not significantly altered in the presence of ascites. GrB/perforin degranulation of activated NK cells in response to HPMC was, however, almost completely blocked, when TALs were pre-cultured in cell-free ascites during activation with α-CD3 Ab (Figure 2B). Based on these data we concluded that T cell activated NK cells are generally able to kill HPMC in ascites, but that cytotoxic mechanisms other than the GrB/perforin pathway might be more relevant in the presence of ascites.
Figure 2.

Ascites blocks NK cell degranulation but not TRAIL-dependent HPMC apoptosis
(A and B) TAL were pre-cultured in RPMI/5% AB media or 100% ascites pool (+/− α-CD3 Ab stimulation) prior to co-culture with HPMC. (A) Apoptosis of HPMC is shown as percentage of annexin V+ cells after gating on CD45−cells (n = 7 patients). (B) Degranulating NK cells in response to HPMC were measured via flow cytometry and depicted as percentage of CD335+/CD107a+ cells (n = 6 patients).
(C and D) TAL were pretreated with α-TRAIL blocking Ab prior to HPMC co-culture. An irrelevant mouse IgG was included as control. Experiments were conducted with TAL cultured in RPMI/5% AB media (n = 11 patients) (C) and 100% ascites pool (n = 5 patients) (D). The amount of Annexin V+ HPMC was determined as described previously.
(E) Schematic representation of co-culture experiments applying T cell CM for NK cell activation. CM were collected from CD3+, CD3+/CD4+, and CD3+/CD8+ T cell subsets cultured in media +/− α-CD3 Ab stimulation. Purified NK cells were then stimulated with T cell CM prior to HPMC co-cultures.
(F) The amount of apoptotic HPMC induced by NK cells activated with CM of CD3+ T cells was compared with CM of CD3+/CD4+ and CD3+/CD8+ T cells (n = 4 matched pairs of different patients).
(G) Analysis of TRAIL signaling was performed by applying an α-TRAIL blocking Ab to NK cells stimulated with CD3+ T cell CM (α-CD3 Ab activated) prior to the co-culture with HPMC (n = 5 patients). An irrelevant mouse IgG was included as control.
The mean is shown by horizontal bars or boxes; vertical error bars represent the standard deviation in A–D, F, and G. ∗ FDR < 0.05; ∗∗ FDR < 0.01; ∗∗∗ FDR < 0.001; determined by paired t test and Benjamini-Hochberg adjustment (ns, not significant).
Since HPMC from the omentum of HGSC patients strongly express Fas when cultured in serum-containing medium or under ascites conditions (Figure S2A), we investigated a Fas/FasL-dependent mechanism for induction of apoptosis. HPMC were found to be generally prone to apoptosis induced by crosslinked sFasL, an effect blocked completely by anti-FasL blocking antibody, but HPMC were clearly less responsive as compared to the Fas-sensitive cell line Jurkat (Figures S2B and S2C). Furthermore, despite a strong expression of FasL on ascites-derived TAL (Figure S2D), induction of apoptosis by TAL does not seem to be Fas/FasL-dependent, as a validated neutralizing α-FasL antibody failed to prevent TAL-induced apoptosis in HPMC (Figure S2E). Additionally, direct cytotoxic activity of the NK cell-derived cytokines TNFα and IFNγ on HPMC does not appear essential, as suggested by the inefficacy of supplementing HPMC culture with the recombinant cytokines (Figure S3).
We next investigated a potential impact of TRAIL-dependent cytotoxicity by using a pathway-blocking α-TRAIL Ab. As depicted in Figure 2C, TAL-mediated induction of apoptosis was blocked in HPMC, at least partially, by this intervention, whereas an isotype-matched control mouse IgG was ineffective. Importantly, the specific blockade of HPMC apoptosis by α-TRAIL Ab was independent of the presence of ascites (Figure 2D). Beyond this, HPMC showed an increased sensitivity for killing by crosslinked rh-TRAIL in the presence of ascites versus serum-containing medium, which in both cases was selectively blocked by α-TRAIL Ab (Figures S4A and S4B). These observations demonstrate a likely relevance of a TRAIL-dependent mode of killing by NK cells in the HGSC TME.
T- NK cell crosstalk via soluble factors enhances TRAIL-mediated cytotoxicity against HPMC
Identifying TRAIL as a key mechanism by which NK cells kill HPMC in the presence of ascites raised the question as to how NK cells are activated by T cells in an immunosuppressive environment. To investigate the relevance of a cell contact-dependent crosstalk between T and NK cell subsets, we used conditioned media (CM) from T cells after stimulation with α-CD3 Ab to activate NK cells prior to HPMC encounter as outlined schematically in Figure 2E. Additionally, we examined the influence of different T cell subsets on the activation of NK cell cytotoxicity. Our findings indicate that CM of activated T cells was sufficient to induce NK cell cytotoxicity against HPMC as detected by annexin V staining, and point to a prominent role of soluble factors rather than cell-contact dependent signaling (Figure 2F). Furthermore, the CM from either activated CD4+ or CD8+ T cells stimulate the killing potential of ascites-associated NK cells in equal measure, indicating a prevalence of commonly expressed mediators as effector molecules (Figure 2F). In accordance with the data obtained from direct T-NK cell co-cultures, the cytolytic activity of isolated NK cell fractions stimulated with CM of activated T cells was blocked by α-TRAIL Ab (Figure 2G).
These observations suggested that activation of NK cell cytolytic function by soluble T cell-derived factors would trigger TRAIL-dependent killing mechanisms directed against HPMC. Indeed, when using the CM of T cells activated by α-CD3 Ab to stimulate NK cells, we clearly detected increased TRAIL expression on these NK cells as compared to NK cells exposed to CM from untreated T cells (Figure 3A), which also correlated with enhanced HPMC apoptosis (Figure 3B). We therefore focused on the identification of mediators in the activated T cell secretome responsible for the induction of TRAIL expression in NK cells. To this end, we applied affinity proteomics (Proximity Extension Assay; PEA) to investigate the secretome of CD3+ ascites-derived T cells in the presence and absence of α-CD3 Ab stimulation, followed by comparative bioinformatic analysis (Tables S1 and S2). As depicted in Figure 3C, activation of ascites-derived T cells strongly associated with an increased secretion of cytokines linked to NK cell activation and TRAIL according to the GeneCards database (n = 43; FDR < 0.05; Table S3). These include diverse members of the TNF ligand (e.g., TNFα, CD40LG, CD70, FASL, TNFSF8, TNFSF9, TNFSF10/TRAIL) and receptor superfamilies (e.g., FAS, TNFRSF10A/TRAILR1, TNFRSF10B/TRAILR2, TNFRSF4, TNFRSF8) together with cytokines assigned to different T cell subsets such as IL-2, IFNγ, IL-4, IL-10, and IL-17A. Among these, T cell-secreted factors related to death cell receptor pathways like GrA/GrB, TRAIL, and FasL (Figure 3C) were not further pursued, since activated T cells on their own were unable to trigger apoptosis in mesothelial cells as shown in Figures 1D and 1F. Apart from this, an implication of the CD40LG/CD40 as well as CD70/CD27 signaling axis in NK cell killing was ruled out by the failure of agonistic α-CD40 and α-CD27 Ab to stimulate NK cell cytotoxicity against HPMC targets (Figure S5). Thus, we further concentrated on prominent cytokines known to be involved in NK cell expansion and stimulation of NK cell cytotoxicity, including IFNγ, IL-2, and TNFα. IL-21 was not part of the PEA panel employed, but was included in the investigated factors, as published transcriptomic analysis of activated T cells suggest it as a candidate for NK cell activation (RNA-Seq datasets at GEO: GSE112899; http://www.ncbi.nlm.nih.gov/geo GSE112899).19 Furthermore, we confirmed its induction in our experimental setting by RT-qPCR (Figure S6A), and expression of TNF, IL2, IL21, and IFNG genes was validated in ascites-derived TAT (Figure S6B).
Figure 3.

NK cells are activated by T cell-derived soluble mediators to conduct a TRAIL-dependent cytotoxicity against HPMC
(A) TRAIL expression on CD335+ ascites-derived NK cells was measured after treatment with CM from unstimulated and α-CD3 Ab stimulated T cells by flow cytometry (n = 13 different patients). The geometric MFI was calculated after subtracting the isotype control.
(B) Corresponding to the TRAIL expression on NK cells induced by the CM of T cells (+/− α-CD3 Ab stimulation), HPMC apoptosis induced by these activated NK cells was also analyzed by annexin V/PI staining (n = 13 different patients).
(C) CD3 activation of ascites-derived T cells induce the secretion of cytokines fitting the terms “NK cell activation” and “TRAIL” in the Genecards database. Protein signals were determined by PEA-based affinity proteomics. Results are expressed as the fold change of T cells activated with α-CD3 Ab relative to untreated T cells (n = 5 matched pairs of different patients). Boxplots show the median (line), upper and lower quartiles (box), range (whiskers) and outliers (circles).
(D) Upregulation of TRAIL expression on NK cells upon stimulation with rh-TNFα and rh-IL-21 together with rh-IL-2 (n = 5 experiments) and combinations of all 3 cytokines (n = 3 experiments) was analyzed by flow cytometry.
(E) Co-culture of HPMC with NK cells previously activated with rh-TNFα (n = 6 patients) and rh-IL-21 (n = 4 patients) combined with rh-IL-2 were conducted and HPMC apoptosis induction was measured by flow cytometry.
(F) A TRAIL-dependent apoptosis induction in HPMC by NK cells activated with rh-IL-2/IL-21/TNFα was confirmed by adding α-TRAIL blocking Ab (n = 6 patients).
(G) To evaluate the contribution of TNFα and IL-21 on NK cell activation resulting in HPMC apoptosis, CM of T cells (α-CD3 Ab stimulated) were incubated with blocking Ab against IL-21 and TNFα (Infliximab) prior to NK cell treatment. The effect on HPMC apoptosis induction was analyzed in co-culture experiments (n = 5 experiments).
Horizontal bars or boxes indicate the mean and vertical error bars represent the standard deviation in panels A, B, and D–G. p values were determined by two-sided, paired t test and Benjamini-Hochberg adjustment: ∗ FDR < 0.05; ∗∗ FDR < 0.01; ∗∗∗ FDR < 0.001; ∗∗∗∗ FDR <0.0001.
Since IL-2 and IFNγ have also been linked to TRAIL regulation in NK cells,20,21,22 we first investigated their relevance for induction of TRAIL-mediated NK cell cytotoxicity. Stimulation of NK cells with rh-IFNγ combined with rh-IL-2 only slightly increased TRAIL expression in NK cells, as well as TRAIL-mediated cytotoxicity against HPMC. IL-2 alone was completely ineffective (Figures S6C and S6D), leading to the assumption that these cytokines do not play a pivotal role in the process. In contrast, enhanced secretion of IL-21 and TNFα from stimulated T cells appeared to be related to the activation of cytotoxic NK cell activity. Evidence for this arose from gain-of-function and loss-of-function experiments using recombinant cytokines and neutralizing antibodies. As depicted in Figure 3D, rh-TNFα combined with rh-IL-2 resulted in significantly increased TRAIL expression by NK cells, which was not seen with rh-IL-2 + rh-IL-21. Moreover, NK cell stimulation by rh-IL-2 + rh-IL-21 or rh-IL-2 + rh-TNFα, led to a strong induction of HPMC apoptosis (Figure 3E), which was at least partially dependent on TRAIL as shown by inhibition through a α-TRAIL blocking antibody of NK cell cytotoxicity induced by rh-IL-2 + rh-IL-21 + rh-TNFα (Figure 3F). Of note, rh-IL-21 did not significantly increase the effect of rh-IL-2 + rh-TNFα on TRAIL expression or NK cell mediated killing efficiency (Figures 3D and 3E). The stimulatory potential of CM from activated T cells on NK cell-mediated HPMC apoptosis was at least partially blocked by a neutralizing antibody against TNFα (infliximab) (Figure 3G), substantiating the relevance of T cell-derived TNFα for NK cell function in the TME. This blocking effect was not replicated when using an α-IL-21 neutralizing antibody, arguing for a minor role for IL-21 (Figure 3G).
TRAIL death receptor expression differs between HPMC and tumor cells
To address the question as to why HPMC are sensitive to NK cell attack, we first analyzed the expression level of MHC class I molecules on HPMC. MHC I determines the “immunological self” of healthy cells by interaction with inhibitory receptors on NK cells (killer cell immunoglobulin-like receptors; KIRs).23 As depicted in Figure 4A, the expression of HLA-A, B-, and C- genes was significantly lower in HPMC compared to immune cells (e.g., TAT), approaching the low levels detected in tumor cells. This low expression of MHC I molecules likely renders both HPMC and tumor cells vulnerable to NK cell-mediated clearance.
Figure 4.

TRAIL sensitivity of HPMC is linked to low expression of MHC class I and high expression of DR5 death receptor
(A and B) Expression of MHC class I genes (HLA-A, HLA-B, HLA-C) (A) and TRAIL receptor genes (TNFRSF10A, TNFRSF10B, TNFRSF10C, TNFRSF10D). (B) In HPMC (from omentum) compared to tumor cells and TAT (from ascites) from HGSC patients according to RNA-Seq datasets as described previously.24 TPM values are depicted from n = 5 HPMC, n = 34 tumor cells and n = 6 TAT of different patients.
(C) The expression of TRAIL receptors (DR4, DR5, DcR1, DcR2) on ex vivo tumor cells (n = 12 patients) and cultured HPMC from HGSC patients (n = 8 patients) and control HPMC originated from patients with benign gynecological diseases (Ctrl HPMC, n = 6 patients) was further validated on protein-level via flow cytometry. The geometric MFI minus the isotype control is depicted.
(D) Overlay of representative histograms showing the TRAIL receptor expression (DR4, DR5, DcR1, DcR2) in comparison to the isotype controls for HPMC and tumor cells from HGSC patients are presented.
The mean is shown by horizontal bars and vertical error bars represent the standard deviation. ∗ FDR < 0.05; ∗∗ FDR < 0.01; ∗∗∗ FDR < 0.001; ∗∗∗∗ FDR < 0.0001 determined by unpaired t test and Benjamini-Hochberg adjustment (ns: not significant).
We next addressed the potential relevance of the stress-induced ligands MICA and MICB (MHC class I polypeptide-related sequence A and B), which interact with the activating receptor KLRK1/NKG2D (killer cell lectin like receptor K1) on NK cells. In HPMC, we found MICA but not MICB mRNA to be expressed at high levels (Figure 7A). Surface expression of MICA/B in HPMC was found to be unaffected by cell-free ascites as evaluated by flow cytometry (Figures 7B and 7C). The corresponding receptor NKG2D was stably expressed on NK cells also in the presence of ascites (Figure 7D). Addition of a blocking NKG2D antibody resulted in a slight, but significant decrease in HPMC apoptosis induced by activated TAL (Figure 7E). In light of the more pronounced effects of blocking TRAIL signaling, however, we concluded that the MICA/B—NKG2D axis may not be a central pathway contributing to HPMC apoptosis.
Figure 7.

Immunohistochemical detection of apoptotis in metastatic omentum and in mesothelial cells from ascites
Matched pairs of omental tissue and ascites were retrieved from n = 6 HGSC patients including one patient (OC_67) with early micrometastatic lesions which is depicted in the following. (A-E) Paraffin sections from omentum specimens with early metastasis. (A) Sections subjected to HE staining. (B) Sections stained for caspase 3 to detect apoptotic cells (asterisks). (C) Tumor cells identified by staining for cytokeratin MNF 116. (D) Mesothelial cells were not detectable by calretinin staining, most likely due to early desquamation of apoptotic mesothelium. (E) Staining for αSMA was applied to identify the caspase 3+ apoptotic cells as fibroblasts. (F–K) Paraffin sections of ascites cells stained for caspase 3 (F–H) and calretinin (I–K). Apoptotic mesothelial cells (arrows) were identified as caspase 3+/calretinin+ cells with typical mesothelial morphology. Scale bar, 100 μm in all panels. Magnification: 200x in (A–E) and 400x in (F–K).
A prerequisite for TRAIL-mediated cell death is the presence of TRAIL death receptors on the surface of the target cell. We therefore analyzed the expression of TNFRSF10A (DR4, TRAIL R1) and TNFRSF10B (DR5, TRAIL R2), as well as the decoy receptors TNFRSF10C (DcR1, TRAIL-R3) and TNFRSF10D (DcR2, TRAIL-R4) on the mRNA (RNA-Seq data) and protein (flow cytometry) level in both HPMC and tumor cells. Of note, a strong expression of TNFRSF10B (DR5) was selectively detected in HPMC, whereas TNFRSF10A (DR4) and TNFRSF10C (DcR1) were only weakly expressed by both cell types. TNFRSF10D (DcR2) expression was also slightly enhanced in HPMC relative to tumor cells (Figure 4B). To assure maintenance of the observed phenotype in the cultured HPMC used in this study, we additionally compared death cell receptor expression in ex vivo and cultured HPMC. Despite a general decline of expression levels in cultured HPMC, the observed expression pattern was maintained in culture, irrespective of the presence of serum or ascites (Figure S8).
Flow cytometric analysis further confirmed a strong and selective surface expression of DR5 and to a lesser extent of DR4 in HPMC as compared to tumor cells, (Figures 4C and 4D), while low expression of the decoy receptor DcR2 was found in both. For DcR1, the RNA-Seq and flow cytometry data were inconsistent, the latter showing a high surface expression in HPMC (Figures 4B and 4C). TRAIL receptor expression on HPMC from patients with benign disease (Ctrl HPMC) did not reveal substantial differences, with HPMC from tumor patients trending toward enhanced DR4 and DcR1 expression (Figure 4C). Taken together, these data suggest that the high expression of DR5 (and to a lesser extent of DR4) in HPMC may provide an explanation for the susceptibility of HPMC to TRAIL-dependent apoptosis.
T cell-activated NK cells fail to kill primary HGSC tumor cells by TRAIL
Motivated by the low expression of both death receptors DR4 and DR5 on primary HGSC tumor cells, we postulated that, in contrast to HPMC, tumor cells may be less sensitive to TRAIL-dependent apoptosis by T cell-activated NK cells. To test this hypothesis for primary HGSC cells, we subjected them to the experimental settings used previously for the measurement of TRAIL-dependent apoptosis in HPMC. Induction of apoptosis through co-cultured α-CD3-Ab-activated TAL was less pronounced in HGSC cells as compared to HPMC despite a higher background in the untreated HGSC cells (Figure 5A: 26.62% mean increase for tumor cells; Figure 2C: 51.07% mean increase for HPMC). Importantly, in contrast to HPMC, TAL-mediated apoptosis of tumor cells was not affected by a blocking α-TRAIL Ab (Figure 5A), consistent with the low levels of DR4/5 death cell receptor expression by HGSC cells. These data support the conclusion that in the HGSC TME, other than HPMC, tumor cells are resistant to TRAIL-dependent killing.
Figure 5.

TAL-derived TRAIL does not mediate apoptosis of control HPMC from patients with benign gynecological diseases or tumor cells from HGSC patients
(A) Apoptosis induction in ex vivo tumor cells derived from ascites of HGSC patients was analyzed after encounter with TAL according to the protocol for HPMC co-cultures. TAL were pretreated with α-TRAIL blocking Ab prior to tumor co-culture where indicated. An irrelevant mouse IgG was included as control. Experiments were conducted with TAL previously cultured in 100% ascites pool (+/− α-CD3 Ab treatment) (n = 5). Apoptotic tumor cells were evaluated by annexin V staining of CD45−cells.
(B) Co-culture experiments were performed as in panel A using HPMC (Ctrl HPMC, n = 4) derived from ascites of patients with benign gynecological diseases instead of tumor cells. The mean is shown by horizontal bars and vertical error bars represent the standard deviation. ∗ FDR <0.05; determined by paired t test and Benjamini-Hochberg adjustment (ns: not significant).
Another important question concerns the conditions under which mesothelial cells become a target of TRAIL-dependent NK cell cytotoxicity in the TME. Similar to omental HPMC from HGSC patients, HPMC from peritoneal fluid of patients with benign gynecological disease (Ctrl HMPC) exhibited detectable levels of apoptotic cells when co-cultured with activated TAL in the presence of ascites-containing media, albeit to a far lesser extent (Figure 5B: 23.6% mean increase for Ctrl HPMC; Figure 2C: 51.07% mean increase for HPMC). As observed for tumor cells, TAL-induced apoptosis of Ctrl HPMC was not blocked by α-TRAIL Ab (Figure 5B), which argues against a TRAIL-dependent mode-of-action in this case. Both omental HPMC and Ctrl HPMC strongly express TRAIL death receptor DR5, but elevated DR4 expression is restricted to omental HPMC (Figure 4C), suggesting that an interplay between DR4 and DR5 may be crucial for TRAIL-mediated apoptosis.
TRAIL-related HMPC apoptosis is linked to tumor cell invasion
We next addressed a potential functional link between tumor cell invasion and mesothelial injury by TAL-mediated apoptosis. To this end, we established a three-dimensional in vitro culture system to measure the trans-mesothelial invasion of primary tumor cells into an underlying extracellular matrix (ECM) consisting of collagen I, which represents the major constituent of the peritoneal ECM. For proof-of-principle, we investigated the impact of rh-TRAIL-induced HPMC apoptosis in tumor cell invasion under conditions of ascites exposure. As shown in Figure 6A, HPMC pre-cultured in ascites built up an intact monolayer on Transwell inserts coated with a collagen I gel. Treatment with rh-TRAIL significantly increased the fraction of apoptotic cells as detected by Caspase 3/7 staining (Figure 6A), as well as reduced the integrity of a Ct-orange-stained HPMC monolayer (Figure S9). Furthermore, significantly increased matrix invasion by a primary OCMI tumor cell line (OC_58) toward FCS as a chemoattractant was observed when the HPMC monolayer was disrupted by TRAIL-mediated apoptosis (Figure 6B for an example of microscopic evaluation; Figure 6C). This finding was confirmed using a primary tumor cell line from another patient (OC_37). While OC_58 invasion was FCS-chemoattractant-dependent, rh-TRAIL enabled a high, but statistically not significant, OC_37 tumor cell invasion even in the absence of FCS (Figure 6D), which we attribute to increased invasive potential of tumor cells from this particular patient. Importantly, increased trans-mesothelial invasion toward a chemotactic gradient (FCS) was confirmed for primary tumor cells from 3 different patients (OC_37, OC_58, and OC_91; represented by different symbols in Figure 6E) in an experimental setting with an HPMC monolayer pre-cultured with ascites-derived TALs. In agreement with the data on TRAIL-mediated HPMC apoptosis (Figure 1E), the tumor invasion was more pronounced when TAL were stimulated with α-CD3 Ab to provide T cell-dependent NK cell activation toward full cytotoxic potential (Figure 6E).
Figure 6.

TRAIL-induced disruption of the mesothelial barrier promotes tumor cell invasion
(A–D) The invasion of primary OCMI tumor cell lines in collagen matrix through an HPMC monolayer was analyzed. Where indicated, the HPMC monolayer was pretreated with rh-TRAIL. (A) The loss of integrity of the HPMC monolayer was visualized by light microscopy and by fluorescence staining of caspase 3/7+ apoptotic HPMC and a representative picture is depicted. Trans-mesothelial invasion of OCMI tumor cells OC_58 (B and C) and OC_37 (D) was evaluated using fluorescence microscopy. The mean of invading cells was calculated for each experiment (n = 4–6 experiments using HPMC form different patients indicated by different colors) and is shown here relative to the untreated control with FCS as chemoattractant. (C) Representative fluorescent microscopic pictures of invaded tumor cells OCMI OC_58 are shown for untreated as well as rh-TRAIL-treated HPMC, both with and without FCS chemoattractant.
(E) Invasion of three primary OCMI tumor cell lines (OC-58: n = 4, blue dots; OC_37: n = 3, pink squares; OC_91: n = 3, orange triangles) through an HPMC monolayer pre-cultured with TAL (+/− α-CD3 Ab stimulation) to induce HPMC apoptosis.
The mean is shown by horizontal bars and vertical error bars represent the standard deviation in C–E. ∗ FDR <0.05; ∗∗ FDR <0.01; determined by paired t test and Benjamini-Hochberg adjustment (ns: not significant). Scale bars, 100μm.
To place these in vitro findings into a clinical context, we analyzed omentum specimens from HGSC patients by immunohistochemistry. Mesothelial cells which form the lining of the omentum were identified by calretinin expression, and apoptotic cells were detected by co-staining for active caspase 3. Unambiguous detection of caspase 3+ mesothelial cells at micrometastatic sites in omentum is challenging due to the scarcity of micrometastases and the early desquamation of apoptotic mesothelium at such sites.2,3 However, in one of six patients analyzed, we detected a loss of intact mesothelium with high numbers of underlying apoptotic caspase 3+/aSMA+ cells (Figure 7; patient OC_67). These findings suggest that apoptosis induction is not limited to mesothelial cells, but may also impact fibroblasts in the vicinity of micrometastases. Furthermore, we found apoptotic caspase-3+ mesothelial cells in matched ascites samples from the same patient, either as free-floating single cells (Figures 7F and 7I) or associated with spheroids (Figures 7G, 7J, 7H, and 7K). Taken together, these observations are consistent with the hypothesis that early steps of metastasis require disruption of the mesothelium through the induction of apoptosis.
In view of these data, we conclude that an intact HPMC monolayer functions as a barrier preventing tumor invasion. This protective function may be impaired by mesothelial cell apoptosis induced through TRAIL-mediated cytolytic activity of T cell-activated NK cells in the TME. Primary HGSC tumor cells, on the other hand, appear largely protected from TRAIL-mediated NK cell attack, potentially shifting the latter cell type toward a pro-metastatic role in the TME.
Discussion
Mesothelial cells are a central part of the HGSC TME and affect multiple stages of tumor progression through their interaction with tumor and other host cells.25 Due to their epithelial nature, mesothelial cells build a protective layer preventing peritoneal implantation of tumor cells. In the present study, have uncovered a novel mechanism by which tumor-associated immune cells, in particular NK cells, may compromise the integrity of the mesothelium by TRAIL-dependent apoptosis induction. Intriguingly, histological examination of HGSC specimens revealed the presence of detached apoptotic mesothelial cells in ascites, and a loss of intact mesothelium close to omental micrometastases. The later observation is consistent with previously published observations,2,3 and is consistent with a possible clinical relevance of mesothelial cell clearance for peritoneal tumor cell dissemination.
Malignant ascites of HGSC patients is rich in immune cells, predominantly tumor-associated macrophages (TAM), T and NK cell subsets,26 which can be actively recruited by peritoneal mesothelial cells secreting inflammatory cytokine IL-6 and chemokine CCL2.24,27 We have identified NK cells as an effector population which develop their full cytotoxic potential against HPMC in concert with activated T cells. As part of the innate immune system, NK cells are involved in anti-viral and anti-tumor immunity through various effector mechanisms, including (i) the activation of death cell receptor pathways via FasL and TRAIL, (ii) the release of lytic granules loaded with perforin and granzymes, and (iii) the secretion of cytokines (e.g., TNFα and IFNγ) with cytotoxic or regulative functions.28,29 Although HPMC from HGSC patients were susceptible to Fas/FasL-dependent killing induced by crosslinked sFasL, as described earlier for patients subjected to peritoneal dialysis,14 our data suggest that this mode-of-action might not account for the cytotoxic activity of ascites-derived NK cells despite high FasL expression on NK cells and Fas expression on HPMC. The reason for this dysfunctional Fas/FasL-interaction is not known, but different mechanisms are conceivable, including Fas receptor function (e.g., Fas downregulation, inactivating Fas mutations, transcriptional dysregulation) and impaired Fas-mediated signal transduction (e.g., by Fas-inhibitory proteins such as Fas-associated phosphatase-1.30,31 Furthermore, our data suggest that the GrB/perforin pathway does not play an important role in NK cell-mediated HPMC killing due to blockade by ascites compounds. In contrast, TRAIL-dependent killing of HPMC by NK cells, as demonstrated by blocking with α-TRAIL Ab, was maintained in the presence of ascites, indicating a potential relevance of this mechanism in the HGSC TME. Nevertheless, involvement of other, as yet unidentified, mechanisms contributing to NK-cell-induced mesothelial cell apoptosis cannot be excluded.
The TNF superfamily member TRAIL is frequently expressed by activated NK cells,32,33 whereas non-activated peripheral NK cells, in contrast to other immune cells of lymphoid or myeloid origin, do not express TRAIL.34 In our experimental system, the cytolytic potential of NK cells against HPMC was associated with an upregulation of TRAIL, which was triggered by soluble factors secreted by activated T cells. Multiple cytokines are implicated in the functional maturation and maintenance of the cytotoxic state of NK cells,35 whereas only a subset, including IL-2, IL-12, IL-15, and interferon class I molecules, are known to trigger surface expression of TRAIL on NK cells.20,21,32,36,37 Among the factors expressed by activated T cells, TNFα and, to a lesser extent, IL-21 and IFNγ, turned out to promote NK cell activation by upregulating TRAIL expression and TRAIL-dependent killing of HPMC. These cytokines have been unequivocally linked to the induction of TRAIL expression in NK cells. In liver NK cells, for example, IFNγ was reported to act as an autocrine regulator of TRAIL expression,22 contradicting an earlier study.36 Likewise, TNFα acts as positive TRAIL regulator in human fibroblasts,38 but has a negative impact on TRAIL expression in human neutrophils,39 suggesting that the cellular context may determine the impact of TNFα on IL-21 expression. IL-21, has not been implicated in the regulation of TRAIL expression, but is known to promote the differentiation and expansion of cytotoxic NK cells in concert with other cytokines.40 Our data suggest that ascites-derived NK cells with an impaired activation profile require additional stimuli, such as TNFα, IL-21, IFNγ or other unidentified factors derived from T cells, to reach full TRAIL-dependent cytotoxic potential against HPMC in the HGSC TME.
NK cell cytotoxic activity and the recognition of potential target cells is further dependent on the balance between activating and inactivating receptors expressed on NK cells.41 As proposed by the missing-self hypothesis, healthy cells are normally protected from NK-mediated killing through recognition of MHC I by KIRs/CD158 family members, C-type lectin receptors (CD94/NK group 2 member A, NKG2A), and leukocyte immunoglobulin-like receptor-1 (LILRB1/ILT2) expressed on the surface of NK cells.23 Indeed, a downregulation of HLA-A, HLA-B, and HLA-C gene expression was observed on HPMC in cancer patients and correlates with NK cell-mediated clearance. In line with these findings, HPMC also express stress-induced ligands, including MICA and MICB, serving as danger signals via the activating receptor NKG2D on NK cells. Importantly, the expression of NKG2D is not downregulated on ascites-derived NK cells as demonstrated by our previous data.42 We further showed that MICA/B-NKG2D interaction alone may not be sufficient for the induction of HPMC apoptosis by NK cells. However, MICA/B-NKG2D may act in concert with other mechanisms to promote pro-apoptotic signaling by NK cells.
Receptors interacting with TRAIL comprise two death receptors, DR4 and DR5, as well as two decoy receptors DcR1 and DcR2. By engagement with the death receptors DR4 and DR5, TRAIL forms a pro-apoptotic death-inducing signaling complex (DISC), leading to activation of the extrinsic apoptosis pathway via caspase 8 in the target cell.43,44 By contrast, the decoy receptors DcR1 and DcR2 lack functional death domains and protect from TRAIL-induced apoptosis.45 The expression of death receptors in relation to decoy receptors further determines the susceptibility of the target cell to TRAIL.46 As shown in the present study, omental HPMC from HGSC patients express high levels of DR5, and to a lesser degree DR4, which coincides with TRAIL sensitivity. TRAIL has been initially regarded as a promising therapeutic option for cancer treatment, as it was believed to selectively induce apoptosis in cancer cells with little toxicity toward normal cells due to high level of DR4 and DR5 expression and low expression of decoy receptors.47,48 This view has been revised, as a number of normal, untransformed cell types, including ovarian epithelial cells,49 were found to respond to TRAIL-mediated killing, sometimes concomitant with a low expression of decoy receptors.50 Furthermore, clinical efficacy of TRAIL-based therapies against ovarian cancer have been limited by an apparent TRAIL resistance of tumor cells.51 Our data confirm the resistance of primary ascites-derived HGSC cells to TRAIL-induced cell death through low expression of DR4 and DR5, which was strikingly different from patient HPMC. Loss or posttranslational modifications of DR4 and DR5 surface expression have been linked to TRAIL resistance in previous studies.52,53 Other factors favoring TRAIL resistance may implicate an activation of non-apoptotic, pro-tumorigenic pathways, e.g., by upregulation of c-FLIP, Bcl-2, or XIAP.54,55,56 Moreover, malignant ascites protects ovarian tumor cells from TRAIL-induced cell death, probably by activation of focal adhesion kinase and Akt.57
To better understand why HPMC, in contrast to ovarian cancer cells, become prone to NK cell lysis in the TME, we compared HPMC from omentum of HGSC patients with those from a benign gynecological background and found substantial differences. HPMC from a benign origin were less efficiently killed by ascites-derived NK cells, and moreover, were TRAIL-resistant. This was associated with lower DR4 expression, yet unchanged high DR5 expression. The relevance of this observation remains uncertain, as the role of DR4 or DR5 in TRAIL-mediated apoptosis appears to be context-dependent. TRAIL-mediated cell death, for example, has been attributed mainly to DR458,59 or DR5,60 but DR5 has also been reported to trigger pro-survival signaling.61 Only limited data on TRAIL-sensitivity in untransformed HPMC are available to this date but our observation is consistent with a study by Catalan et al. showing that HPMC death in peritoneal dialysis patients is caused by Fas/FasL interaction rather than TRAIL.14 Based on these observations, we propose that HPMC may be re-educated to become TRAIL-sensitive probably by factors of the TME, such as TNFα, IFNγ, or IL-6 known to modulate TRAIL receptor expression.38,62,63,64 Understanding the role of ascites-derived factors in sensitizing mesothelial and/or tumor cells to TRAIL-dependent killing by NK cells will be an interesting subject of upcoming studies that may pave the way to the development of new strategies to interfere with peritoneal metastasis.
Limitations of the study
There are nevertheless two major limitations of our study with respect to the clinical relevance of the identified interaction of T, NK, and mesothelial cells in promoting tumor cell invasion. First, our data are based on in vitro studies using primary host and tumor cells isolated from HGSC patients, and second, the induction of tumor cell invasion requires α-CD3 Ab activation of TAL which is unlikely to occur in vivo. Albeit our observations from immunohistological analyses of clinical specimens support the in vitro data by demonstrating the presence of apoptosis in mesothelial cells in the ascites, functional in vivo validation is required. Unfortunately, easily accessible tumor models that adequately recapitulate critical oncogenic features of HGSC as well as the human TME are still lacking. Currently available tumor models include xenografts with artificial mixed mouse-human compositions, which are problematic to study multifaceted interactions of tumor, immune, and other host cells. Genetically engineered mice recapitulating the genomics and the TME of human HGSC are available,65,66,67 but due to multiple genetic alterations these mouse models require complex breeding programs. Therefore, their application is beyond the scope of the present study but may represent useful tools for future investigations.
Furthermore, it is challenging to ultimately prove mechanistically that NK cells, in concert with T cells, are involved in mesothelial cell death in patients. However, several independent observations indicate that ascites may be the main route by which T cell-activated NK cells attack the mesothelium. First, NK cells expressing activating NKG2D receptor and also T cells are highly abundant in the ascites, as shown by our own previously published data.42 Second, we could confirm the expression of cytokines involved in the regulation of TRAIL-mediated NK cell cytotoxicity also in ex vivo ascites-derived T cells from patients. Third, a basal HPMC apoptosis is induced by ascites TAL without α-CD3 Ab activation, even though its extent was not sufficient to affect tumor invasion in our experimental setup. This may be attributable to the use of ex vivo TAL which require additional artificial stimuli, such as α-CD3 Ab. It is also conceivable that the local environment in vivo provides additional stimuli not present in the experimental setting.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human CD45-APC | Biolegend | Cat# 304011; RRID:AB_314399 |
| Mouse anti-human CD3-APC | Biolegend | Cat# 300412; RRID:AB_314066 |
| Mouse anti-human CD3 (Clone Okt3) | Biolegend | Cat# 317302; RRID:AB_571927 |
| Mouse anti-human CD4-PE-Cy7 | Southern Biotech | Cat# 9522-17; RRID:AB_2796857 |
| Mouse anti-human CD8 APC | Miltenyi Biotec | Cat# 130-113-154; RRID:AB_2725982 |
| Mouse anti-human CD335-eFluor 450 | eBioscience | Cat# 48-3359-42; RRID:AB_2574055 |
| Mouse anti-human CD56-PE | Miltenyi Biotec | Cat# 130-113-874; RRID:AB_2726367 |
| Mouse anti-human CD19-FITC | Miltenyi Biotec | Cat# 130-091-328; RRID:AB_244222 |
| Mouse anti-human EpCAM-PE | Miltenyi Biotec | Cat# 130-091-253; RRID:AB_871665 |
| Mouse anti-human FAP-PE | R&D Systems | Cat# FAP3715P; RRID:AB_2884010 |
| Mouse anti-human CD140a PE (clone 16A1) | Invitrogen | Cat# A15785; RRID:AB_2534564 |
| Mouse anti-human Vimentin FITC | Miltenyi Biotec | Cat# 130-116-663; RRID:AB_2727645 |
| Mouse anti-human Cytokeratin APC | Miltenyi Biotec | Cat# 130-112-933; RRID:AB_2651499 |
| Mouse anti-human NKG2D-FITC (clone 1D11) | Biolegend | Cat# 320820; RRID:AB_2562864 |
| Mouse anti-human NKG2D (clone 1D11) | Biolegend | Cat# 320802; RRID:AB_492956 |
| Mouse anti-human TRAIL-PE | eBioscience | Cat# 12-9927-42; RRID:AB_10557246 |
| Mouse anti-human TRAIL | R&D Systems | Cat# MAB375; RRID:AB_2256258 |
| Mouse anti-human TRAIL-R1-APC | Biolegend | Cat# 307207; RRID:AB_2256112 |
| Mouse anti-human TRAIL-R2-PE | Biolegend | Cat# 307405; RRID:AB_314677 |
| Mouse anti-human TRAIL-R3-PE | Miltenyi Biotec | Cat# 130-124-904; RRID:AB_2876902 |
| Mouse anti-human TRAIL-R4-APC | Antibodies.com | Cat# A121841; RRID:AB_2749664 |
| Mouse anti-human MICA/B.Fluor 647 clone 6D4 | Biolegend | Cat# 320914; RRID:AB_2266419 |
| Mouse anti-human HLA-ABC-PE (clone W6/32) | Biolegend | Cat# 311405; RRID:AB_314874 |
| Mouse anti-human FasL (clone #100419) | R&D Systems | Cat# MAB126; RRID:AB_2246667 |
| Mouse anti-human FasL-PE | Miltenyi Biotec | Cat# 130-118-491; RRID:AB_2751526 |
| Mouse anti-human CD40 (clone G28.5) | Bio X Cell InVivoMab | Cat# BE0189; RRID:AB_10950314 |
| Mouse anti-human CD107a-PE | eBioscience | Cat# 12-1079-42; RRID:AB_10853326 |
| Mouse anti-human CD95-APC | Miltenyi Biotec | Cat# 130-117-813; RRID:AB_2751526 |
| Mouse anti-human CD28 | Miltenyi Biotec | Cat# 130-093-386; RRID:AB_1036117 |
| Mouse anti-human IL-21 | MABTECH | Cat# 3540-0N-500 |
| Mouse anti-His-Tag (clone #AD.1.1.10) | R&D Systems | Cat# MAB050; RRID:AB_357353 |
| Mouse monoclonal anti-human calretinin (clone Calret 1) | DAKO | Cat# M 7245; RRID:AB_2068519 |
| Rabbit polyclonal anti-human cleaved caspase-3 | Cell Signaling | Cat# 9661; RRID:AB_2341188 |
| Mouse monoclonal anti-human cytokeratin (clone MNF116) | DAKO | Cat# M 0821; RRID:AB_2858276 |
| Mouse monoclonal anti-human alpha-smooth muscle actin (aSMA) (clone ASM-1) | Progen | Cat# 61001; RRID:AB_2920672 |
| Human CD27 antibody (Varlilumab biosimilar) | Biozol | Cat# BYT-ORB746677; RRID:AB_2921569 |
| Chimeric human-murin anti-human-TNFα (Infliximab, Remsima) | Celltrion Healthcare | kind gift from Christian Bauer, Clinic for Gastroenterology, Endocrinology, Infectiology and Metabolism, Philipps University Marburg, Germany |
| mouse IgG | Jackson Immuno Research | Cat# 015-000-002; RRID:AB_2337187 |
| Mouse IgG isotype control (Clone MOPC-2) | Biolegend | Cat# 400102; RRID:AB_2891079 |
| Mouse IgG isotype control FITC | Miltenyi Biotec | Cat# 130-113-833; RRID:AB_2733684 |
| Mouse IgG isotype control APC | Miltenyi Biotec | Cat# 130-113-269; RRID:AB_2733442 |
| Mouse IgG isotype control eFluor 450 | eBioscience | Cat# 48-4714-82; RRID:AB_1271992 |
| Mouse IgG sotype control PE-Cy7 | eBioscience | Cat# 25-4714-42; RRID:AB_1548705 |
| Mouse IgG isotype control PE | BD Biosciences | Cat# 555574; RRID:AB_395953 |
| Biological samples | ||
| HGSC patient-derived ascites and omental metastatic tissue samples | Marburg University Hospital | N/A |
| Leucoreduction system (LRS) chambers of healthy adult volunteers | Marburg University Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| rh-TNFα | PeproTech | Cat# 3000-01A-10UG |
| rh-IFNγ | Biomol | Cat# 51564.100 |
| rh-IL-2 | ImmunoTools | Cat# 11340025 |
| rh-IL-21 | PeproTech | Cat# 200-21 |
| rh-TRAIL (SuperKiller TRAIL) | Enzo Life Sciences | Cat# ALX-201-115 |
| sFasL | Biolegend | Cat# 589402 |
| Monensin (Golgi-Stop) | BD Biosciences | Cat# 554724 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma Aldrich | Cat# P1585 |
| Ionomycin | Sigma Aldrich | Cat# I9657 |
| Anti-APC microbeads | Miltenyi Biotec | Cat# 130-090-855 |
| CD14 Microbeads, human | Miltenyi Biotec | Cat# 130-050-201 |
| Collagen I gel (rat tail) | Ibidi | Cat# 50201 |
| CTgreen | Invitrogen | Cat# C2925 |
| CTorange | Invitrogen | Cat# C2927 |
| Critical commercial assays | ||
| FITC annexin V Apoptosis Detection Kit I | BD Bioscience | Cat# 556547 |
| CellEvent TM Caspase-3/7 Green | Invitrogen | Cat# C10723 |
| Dako REAL EnVision HRP Rabbit/Mouse polymer | DAKO | Cat# K5007 |
| Olink Explore 3072 | Olink | N/A |
| Deposited data | ||
| E-MTAB-4162 | EBI ArrayExpress | |
| E-MTAB-10611 | EBI ArrayExpress | |
| complete PEA affinity proteomics dataset | This paper, Table S1 | |
| Experimental models: Cell lines | ||
| Jurkat | kind gift from Miriam Frech, Department of Internal Medicine and Hematology, Oncology and Immunology, Philipps University Marburg, Germany) | |
| K562 | kind gift of Elke Pogge von Strandmann, Institute for Tumor Immunology, Center for Tumor Biology and Immunology, Philipps University, Marburg, Germany | |
| Oligonucleotides | ||
| RPL27, AAAGCTGTCATCGTGAAGAAC and GCTGTC ACTTTGCGGGGGTAG | N/A | N/A |
| IL-21, TGTGAATGACTTGGTCCCTGAA and CTGCATTTGTGGAAGGTGGTTTCC | N/A | n/A |
| Software and algorithms | ||
| Diva Software | BD Biosciences | |
| FlowJo™ v10.8 Software | BD Life Sciences | |
| ImageJ Software | ImageJ | |
| GraphPad Prism 8 & 9 Software | GraphPad | |
Resource availability
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Rolf Müller (rolf.mueller@uni-marburg.de).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon request with a material transfer agreement.
Data and code availability
-
•
RNA-Seq data were deposited at EBI ArrayExpress (accession numbers E-MTAB-4162, E-MTAB-10611). The complete PEA affinity proteomics dataset is included as Table S1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Patient samples and healthy donors
As approved by the local ethics committee (Philipps University Marburg: reference number 205/10), samples of the malignant ascites and omentum tissue with metastatic lesions were obtained from first line HGSC patients without prior treatment preceding the surgery at the Marburg University Hospital. As a control, HPMC were propagated from peritoneal lavage of female patients with benign gynecological disease obtained during hysterectomy. Written consent was given by each patient according to the Declaration of Helsinki. The patient characteristics are summarized in . In addition, peripheral lymphocytes were also retrieved from leucoreduction system (LRS) chambers of healthy adult volunteers which were kindly provided by the Center for Transfusion Medicine and Hemotherapy at the University Hospital Gieβen and Marburg. Blood lymphocytes enriched by ficoll gradient centrifugation were only applied for generation of T cell conditioned media (CM) as described in the method details section.
Method details
Peritoneal cell types from malignant ascites
Peritoneal cells, including tumor-associated macrophages (TAM), tumor-associated lymphocytes (TAL) and tumor cells were isolated from the malignant ascites following our established standard protocol.26,68 This includes a first separation of mononuclear cells by applying Lymphocyte Separation Medium 1077 (PromoCell, Heidelberg, Germany) and density gradient centrifugation. Tumor spheroids were separated by size exclusion using 30 μm and 100 μm cell strainer. The whole TAL fraction was isolated from the remaining peritoneal cells composing of less than 3.5% EpCAM+ cells by depletion of CD14+ TAM via magnetic activated cell sorting (MACS) following the standard protocols of the supplier (Miltenyi Biotec, Bergisch Gladbach, Germany). CD14− TAL were cryopreserved for the purification of T and NK cells at a later point. To isolate CD3-/CD56+ NK cells for functional assays, a negative MACS sorting strategy was chosen to avoid unspecific NK cell activation by direct labeling. Therefore, the CD14-depleted TAL fraction was stained with APC-labelled anti-human CD3 antibody (Biolegend, San Diego, CA, USA) followed by incubation with anti-APC microbeads (Miltenyi Biotec). After MACS separation, CD3+ T cells were eluted from the columns, whereas the flow through contained the CD3−cell fraction, mainly consisting of CD3-/CD56+ NK cells. The purity of the fractions was determined via flow cytometry. A purity of >95% was achieved for CD3+ T cell fractions, and >80% for the NK cell fraction with CD19+ B cells as major contaminating cell population. The viability of TAL subpopulations was >80% as determined by trypan blue staining.
To further divide the CD3+ T cell fraction into CD3+/CD4+ T helper cells and CD3+/CD8+ cytotoxic T-cells, the preselected CD14−lymphocytes were stained with APC-labelled anti-human CD8 antibody (Miltenyi Biotec), incubated with anti-APC microbeads and isolated via MACS. CD3+/CD8+ cytotoxic T-cells were eluted from the column, whereas the flow through was further applied to a CD3-positive MACS selection as described above to isolate CD3+/CD4+ T-Helper cells from CD3− NK and B cells.
Apart from cellular fractions, cell-free ascites was cryopreserved at −80°C and an ascites pool of n = 10 HGSC patients were used for co-culture experiments where indicated.
HPMC from patients
The isolation of HPMC from omentum tissue of HGSC patients was performed according to Sommerfeld et al.24 First, macroscopically tumor-free omentum tissue was minced and digested with trypsin (0.05% Trypsin/0.02% EDTA, Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 30 min at 37°C, 5% CO2. HPMC-enriched fractions obtained after filtration through a 100 μm cell strainer and centrifugation (10 min, 300x g) were either used directly (ex vivo cells) or initially cultured in RPMI 1640/10% FCS/1% sodium pyruvate/1% Pen-Strep (RPMI1640, Life Technologies, Darmstadt, Germany). The purity of the HPMC primary culture was determined by flow cytometric analysis of CD45+ leukocyte and EpCAM+ tumor cell contaminations. In case of <95% purity, an additional MACS depletion of CD45+ and EpCAM+ cells was performed. As exemplified in Figure S10, HPMC were routinely examined for their typical cobblestone-like morphology by microscopy (Figure S10A), high expression of the mesothelial markers cytokeratin and vimentin (flow cytometry) and reduced staining for the fibroblast-selective markers CD140a and FAP (Figure S10B) as described previously.24 Additionally, RNA-Seq data24 were used to verify the mesothelial origin of isolated cells (Figure S10C). Primary HPMC were further propagated in OCMI medium69 for a maximum of five passages with a viability of >90%. Depending on the experimental setting, the OCMI medium composition was modified by adding 50% ascites pool or removing EGF (for trans-mesothelial invasion assay) where indicated.
In order to obtain HPMC from patients with benign gynecological disease, mononuclear cells were enriched from peritoneal lavage by Ficoll gradient prior to MACS depletion of CD14+ macrophage contaminations. CD14-depleted peritoneal fractions containing floating HPMC were cultivated in OCMI medium until confluency. Purity was determined by flow cytometry using mesothelial markers (vimentin, cytokeratin 8), fibroblast-specific markers (FAP) and leukocyte marker CD45.
Primary tumor cell culture
Emanating from ascites tumor spheroids, permanent primary tumor cell cultures (OCMI tumor cells) were established from patients OC_37, OC_58 and OC_91, following the description of Ince et al.70 with slight modifications as delineated previously.69 The benefit of this culture method is to retain the original tumor characteristics and propagate the tumor cells long-term without the induction of cell culture crisis and genetic alterations. Patient-derived OCMI cell lines were tested for mycoplasma contaminations prior to their experimental use.
HPMC/tumor cell co-culture experiments with TAL or purified T/NK cells
Co-culture experiments comprising of HPMC and either TAL or purified T or NK cells were performed in 96-well plates based on published protocols8 with some modifications as follows. Prior to the co-culture, an HPMC monolayer was generated by cultivating 4 × 104 HPMC per 96-well in OCMI/5% FCS or OCMI/50% ascites pool media for two days. The lymphocytes were harvested after initial stimulation (see above) and resuspended in OCMI/5% FCS media. The lymphocytes were applied to the HPMC monolayer in a TAL:HPMC ratio of 10:1. The co-cultivation was performed for 6 h at 37°C, 5% CO2.
In single experiments, 5 × 104 primary tumor cells isolated from ascites of HGSC patients (viability of >80%) were tested instead of HPMC by applying the same experimental setting for TAL co-cultures.
Susceptibility to TRAIL-mediated killing of HPMC was tested by incubating HPMC with 100 ng/mL rh-TRAIL (SuperKiller TRAIL, Enzo Life Sciences, Lörrach, Germany) for 18 h according to the suggestions of the manufacturer. Furthermore, for TRAIL-blocking analysis, the lymphocytes were pre-treated with 10 μg/mL human α-TRAIL antibody (R&D Systems, Minnesota, USA), mouse IgG (Jackson Immuno Research, Cambridgeshire, United Kingdom) as isotype control and PBS as solvent control for 1 h at 37°C prior to the co-culture with HPMC. A possible contribution of NKG2D expression on NK cells to HPMC apoptosis induction was determined using lymphocytes pre-treated with 10 μg/mL human α-NKG2D antibody (Clone 1D11, BioLegend) or an appropriate IgG control (Clone MOPC-21, BioLegend).
To evaluate the impact of CD40−and CD27-mediated signaling on NK cell activation, purified NK cells were stimulated with 10 μg/mL agonistic α-CD40 antibody (mouse, clone G28.5, Bio X Cell InVivoMab, Lebanon, NH) and α-CD27 antibody (human, Biozol, Eching, Germany) for 30 min at 37°C prior to HPMC co-culture based on published protocols.71,72 Mouse and human IgG (both from Biolegend) were included as controls.
To analyze Fas/FasL-mediated killing of HPMC, soluble sFasL (1-100 ng/mL, Biolegend) was crosslinked with α-His-Tag antibody (10 μg/mL, Clone #AD.1.1.10, R&D Systems) for 1 h at 37°C, 5% CO2 according to the supplier’s information. The crosslinked FasL was added to the HPMC monolayer and incubated for 24 h. The Fas-sensitive cell line Jurkat (kind gift of Miriam Frech, Department of Internal Medicine and Hematology, Oncology and Immunology, Philipps University Marburg, Germany) was used as a positive control for Fas/FasL-mediated killing. To specifically block Fas/FasL-mediated killing of HPMC by TAL, TAL were pre-treated with α-FasL antibody (500 ng/mL as suggested by the supplier, R&D Systems) for 1 h and were then added to HPMC. After co-cultivation flow cytometric analysis were performed to determine NK cell and T cell degranulation, as well as HPMC apoptosis induction, as described in the following sections.
For evaluation of direct cytotoxicity of NK cell-derived cytokines, HPMC were incubated with 100 ng/mL rh-TNFα and 20 ng/mL rh-IFNγ for 6h, 37°C, 5% CO2 prior to apoptosis detection.
Treatment of TAL and T/NK cell co-cultures
As indicated in individual experimental settings, either whole ascites-derived TAL, purified T cells or NK cells, as well as NK/T cell co-cultures were cultivated for 2 days in the presence or absence of α-CD3 antibody for T cell stimulation prior to the co-culture with HPMC. Therefore, a 24-well plate was pre-coated with 0.5 μg/mL α-CD3 antibody (Clone Okt3, Biolegend) in PBS for 2 h at 37°C. The wells were washed twice with PBS and once with the corresponding cultivation media (either RPMI 1640/5% AB-serum (Sigma Aldrich, Taufkirchen, Germany)/1% sodium pyruvate or 100% ascites pool), a total of 2 × 106 TAL per 24-well were then added and incubated for 2 days at 37°C, 5% CO2. In single experiments, costimulatory α-CD28 antibody (2 μg/mL; Miltenyi Biotec) was added to TAL cultures. In case of NK/T co-cultures, the purified fractions were mixed in a ratio of 1:1.
In individual experiments, NK cells were treated with the following cytokines – alone or in combination – for two days: 10 ng/mL rh-TNFα (PeproTech, Hamburg, Germany), 10 ng/mL rh-IFNγ (Biomol), 20 ng/mL rh-IL-2 (ImmunoTools, Friesoythe, Germany), and 10 ng/mL rh-IL-21 (PeproTech). Instead of T cell co-cultures, NK cells were also stimulated with conditioned media (CM) of CD3+ T cells (or CD4+ and CD8+ T cell subsets) diluted 1:1 in RPMI 1640/5% AB serum/1% sodium pyruvate. CM of T cells were collected after two days cultivation with or without α-CD3 antibody stimulation followed by two centrifugation steps (10 min, 300x g and 10 min, 14,000x g). The CM was preserved at −20°C until used. For blocking experiments, CM of T cells stimulated with α-CD3 antibody were pre-treated with 10 μg/mL neutralizing α-hIL-21 antibody (MABTECH, Nacka Strand Schweden) and α-hTNFα antibody (Infliximab, Remsima, Celltrion Healthcare, Bad Homburg vor der Höhe, Germany; a kind gift from Christian Bauer, Clinic for Gastroenterology, Endocrinology, Infectiology and Metabolism, Philipps University Marburg, Germany) for 1 h at 37°C prior to their use for NK cell activation.
CD107a degranulation assay
To detect specific degranulation of TAL or purified NK and T cell subsets in response to HPMC, 5 μL CD107a-PE (eBioscience, Frankfurt, Germany) and 20 nM Monensin (Golgi-Stop, BD Biosciences, Heidelberg, Germany) were added during HPMC/TAL co-cultures in 96-well plates for 6 h. Unstimulated TAL, NK or T cells alone served as negative controls. As a positive control for NK cell degranulation, NK cells or TAL were incubated together with the myeloid cell line K562 in an NK cell/TAL to K562 ratio of 10:1 for 6 h. Stimulation of TAL or isolated T cells with phorbol 12-myristate 13-acetate (PMA, 1 μg/mL) and ionomycin (15 ng/mL, both from Sigma Aldrich) for 6 h was included as positive control for T cell-specific degranulation. Afterward the TAL (or isolated cell subsets) were harvested using EDTA (20 mM in PBS), washed and resuspended in staining buffer (PBS with 1% FCS). The following antibodies were added for 30 min at 4°C for detection of cell-type specific degranulation: CD3-APC, CD335-eFluor 450 (eBioscience) and CD4-PE-Cy7 (Southern Biotech, Birmingham, Alabama, USA). Isotype controls were obtained from Miltenyi Biotec, eBioscience and BD Bioscience BD. Flow cytometric analysis was performed on a FACS Canto II instrument using Diva Software (BD Biosciences) and FlowJo v10.8 Software (BD Life Sciences). The percentage of degranulating CD107a+ cells was calculated in CD335+ and CD3+ subsets.
Detection of apoptosis by annexin V staining
After co-culture with lymphocytes, HPMC (or primary tumor cells in individual experiments) were harvested with trypsin for flow cytometric detection of apoptosis by annexin V/propidium iodide (PI) staining. To exclude contaminating lymphocytes from the analysis, HPMC were counterstained with CD45-APC (BioLegend) for 30 min at 4°C. Cells were then washed once in 1x annexin V binding buffer (FITC annexin V Apoptosis Detection Kit I, BD Bioscience) before 5 μL annexin V-FITC and 5 μL PI were added and incubated for 15 min in the dark at room temperature. After adding 250 μL 1x annexin V binding buffer, the cells were measured immediately. HPMC as well as lymphocytes alone were included as negative controls to enable correct gating of CD45 negative HPMC by excluding contaminating lymphocytes. The amount of apoptotic cells within the CD45 negative population was calculated based on the total numbers of annexin V+ CD45−cells including PI- (early apoptosis) and PI + cells (late apoptosis).
Flow cytometric analysis of surface receptor expression
In order to analyze the purity and phenotype of peritoneal cell fractions, cells were stained following the standard staining protocol for surface staining (as described above) applying the following antibodies: CD3-APC, CD4-PE-Cy7, CD335-eFluor 450, CD45-APC, CD56-PE (Miltenyi Biotec), CD19-FITC (Miltenyi Biotec), EpCAM-PE (Miltenyi Biotec), CD140a-PE (Invitrogen) and FAP-PE (both R&D Systems). For phenotyping of HPMC, additional intracellular staining with Vimentin-FITC (Miltenyi Biotec) and Cytokeratin-APC (Miltenyi Biotech) was applied. TRAIL and NKG2D surface expression on NK cells were analyzed using TRAIL-PE (eBioscience) and NKG2D-FITC (Clone 1D11, Biolegend) combined with CD335-eFluor 450. The expression of TRAIL-R1 (DR4), TRAIL-R2 (DR5), TRAIL-R3 (DcR1), TRAIL-R4 (DcR2) and MICA/B on HPMC was measured after staining with antibodies TRAIL-R1-APC (CD261, BioLegend), TRAIL-R2-PE (CD262, BioLegend), TRAIL-R3-PE (CD263, Miltenyi Biotec), TRAIL-R4-APC (CD264, antibodies.com) and MICA/B-Fluor 647 (Clone 6D4, Biolegend). HLA-ABC-PE (Clone W6/32, Biolegend) was used for evaluation of MHC class I expression on HPMC, tumor and lymphocytes. Staining of Fas ligand on lymphocyte subsets was performed for 10 min at 4°C with antibody FasL (CD178)-PE (Miltenyi Biotec) and Fas receptor (CD95) expression on HPMC was detected with CD95-APC (Miltenyi Biotec).
The corresponding isotype control antibodies were purchased from BD Biosciences, Miltenyi Biotec, Biolegend and eBioscience. Flow cytometry was performed as described above and results were calculated as percentage of positive cells. Geometric mean fluorescence intensities (MFI) are given after subtracting the isotype control.
Trans-mesothelial tumor cell invasion analysis
To determine the impact on TRAIL-mediated apoptosis induction in HPMC on tumor invasion, a 3D trans-mesothelial tumor invasion assay was established based on a transwell system. Therefore, transwell inserts (24-well inserts, 8.0 μm pore size, BD Biosciences) were coated with 10 μL collagen I gel (rat tail, Ibidi, Gräfelfing, Germany) at a concentration of 150 μg/mL in OCMI media (without EGF). The gel was allowed to polymerize for 3 h at 37°C, 5% CO2 and was then equilibrated in 100 μL OCMI media (without EGF) for 1 h. HPMC previously cultivated in OCMI/50% ascites pool for 3–4 days, were labeled with CT-orange (Invitrogen) and plated on top of the collagen I gel in a density of 8 × 104 cells in 300 μL OCMI media (without EGF). In individual experiments, 100 ng/mL rh-TRAIL was added to the HPMC for 18 h before tumor cells were applied. Alternatively, TAL (derived from 100% ascites pool cultivation ± α-CD3 antibody stimulation for 2 days) were added to the HPMC monolayer on collagen I gel in a 10-fold concentration and incubated for 24 h. The induction of apoptosis in the mesothelial layer was monitored by caspase3/7 staining using 10 μM CellEvent TM Caspase-3/7 Green (Invitrogen) according to the instructions of the supplier. After incubation for 30 min, apoptotic cells were documented by microscopy (Leica DMI3000B microscope; Leica, Wetzlar, Germany).
Primary tumor cells (OCMI cells) from patients OC_37, OC_58 and OC_91 were used for invasion assay. Tumor cells were washed serum-free using serum-free OCMI media (without EGF), stained with CTgreen (Invitrogen) and added at a concentration of 4 × 105 cells/insert in 300 μL serum free OCMI media. Complete OCMI media (without EGF) containing 5% FCS was used as chemoattractant in the bottom well. The tumor cells were allowed to invade for 2 h at 37°C, 5% CO2. Remaining cells were removed from the inside of the inserts, invaded cells were fixated with methanol, filters were cut out and fixed on to microscope slides for microscopic evaluation on a Leica DMI3000B microscope. Invaded tumor cells were counted in 6 visual fields per filter using the ImageJ software.
RT-qPCR
Isolation of RNA and cDNA transcription from T-cells, as well as RT-qPCR analyses were performed as described previously.73 RPL27 was applied for normalization. The following primers were used: RPL27, AAAGCTGTCATCGTGAAGAAC and GCTGTC ACTTTGCGGGGGTAG; and IL-21, TGTGAATGACTTGGTCCCTGAA and CTGCATTTGTGGAAGGTGGTTTCC. For raw data evaluation the Cy0 method was applied.74
Immunohistochemistry
For immunohistochemistry, heat-induced epitope retrieval was performed with EDTA according to the manufacturer’s protocol of the respective primary antibody. Staining was performed on a DAKO autostainer Link 48. After blocking endogenous peroxidase, sections were incubated for 45 min with mouse monoclonal α-calretinin antibody (1:100; Dako M 7245, clone DAK Calret 1; DAKO Waldbronn, Germany), rabbit polyclonal α-cleaved caspase-3 antibody (1:200; Cell Signaling #9661), mouse monoclonal α-human cytokeratin antibody (1:100; Dako M 0821, clone MNF116) or mouse monoclonal α-alpha-smooth muscle actin (aSMA) antibody (1:200; Progen 61001, clone ASM-1; Progen, Heidelberg, Germany). Sections were washed and incubated with Dako REAL EnVision HRP Rabbit/Mouse polymer, which reacts with DAB-Chromogen, according to the manufacturer's protocol.
Affinity-based proteomics
Ascites-derived CD3+ T cells from 5 patients were stimulated with or without a-CD3 activation for 2 days at 37°C and 5% CO2. Supernatant from stimulation cultures was harvested as described above and tested for NK cell activation and induction of NK-mediated HPMC killing in functional assays. Basal medium (RPMI 1640/5% AB serum/1% sodium pyruvate) was included as background control for medium derived proteins. Olink analysis was performed using the Olink Explore 3072 platform at the Core Facility Translational Proteomics at the Medical Department of Philipps-University Marburg (UMR), following the Olink standard protocol (v1.5, 2022–12.21). All samples were randomized and plated on a 96-well plate. Samples were processed in one batch. Next-Generation Sequencing (NGS) of the generated libraries was performed at the Genomics Core Facility of the Department of Medicine at UMR. Olink Explore uses Proximity Extension Assays (PEA) technology75 and has been optimized for high-throughput analysis with NGS readout.76 In brief, the PEA immunoassay uses two matched antibodies per target, binding simultaneously to different epitopes on the protein. The antibodies are covalently labeled with complementary oligonucleotide probes that only hybridize when the correct pair of matching antibodies is in close proximity. The resulting short dsDNA sequences contain an assay specific barcode and are pre-amplified by a first PCR. In a second PCR, additional DNA tags providing information on the respective sample are added to the barcodes. This coding allows for the parallel measurement of ∼3000 proteins.
Protein levels are expressed as Normalized Protein eXpression (NPX; Olink-provided arbitrary unit in log2 scale). For mediators present in more than one panel (CXCL8, IDO1, IL6, LMOD1, SCRIB, TNF) mean NPX values were used in subsequent bioinformatic analyses (Table S1; complete dataset). aCD3-induced proteins were identified by determining the difference of NPX values (ΔNPX) for samples with and without CD3 stimulation. Fold change (FC) values (2ΔNPX) were calculated as the median of n = 5 biological replicates. False discovery rates (FDR) were determined by applying the Benjamini-Hochberg method to nominal p values determined by unpaired t test of FC values (Table S2). Identification of potential T cell secreted mediators promoting NK-cell-induced TRAIL-dependent apoptosis was achieved by filtering the hits for proteins retrieved by the search term “NK cell AND TRAIL” in the genecards.org database (Table S3).
RNA sequencing
RNA-Seq datasets for omentum-derived ex vivo HPMC as well as for tumor cells and tumor-associated T cells (TAT) from ascites of HGSC patients were retrieved from Sommerfeld et al.24 and used for Figures 4A and 4B; Figures S6B, S7A, S8, and S10C; Tables S5 and S6. RNA-Seq data were processed as described previously26,68 using Ensembl 96.77
Quantification and statistical analysis
Comparative data were statistically analyzed by paired or unpaired Student’s t test (two-sided, unequal variance), as indicated in the figure legends. Post-hoc analysis of multiple comparisons (if n > 2) was performed by the Benjamini–Hochberg method. Results were expressed as follows: ∗FDR <0.05; ∗∗FDR <0.01; ∗∗∗FDR <0.001; ∗∗∗∗FDR <0.0001. Boxplots were constructed using the Matplotlib boxplot function with Python. Boxplots show medians (horizontal line), upper and lower quartiles (box), range (whiskers) and outliers (open circles).
Acknowledgments
We are grateful to T. Plaum-Allmeroth, A. Allmeroth, V. Wischmann, and Manasi J. Benurwar for excellent technical assistance and to Christian Bauer (Clinic for Gastroenterology, Endocrinology, Infectiology and Metabolism, Philipps University Marburg, Germany) for kindly providing the α-hTNFα antibody Infliximab. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) - 416910386 - GRK 2573.
Author contributions
A.M.S, C.S., and I.K. carried out most of the experimental work and collected and analyzed the data. C.K. performed the histological analyses of clinical specimens. L.S. prepared primary human mesothelial cells and cooperated in coculture experiments with tumor cell spheroids. V.M.N. and J.G. carried out affinity proteomics and processed the data; F.F. and R.M. performed bioinformatic analyses. T.W., M.H., and E.P.S. contributed conceptual and methodological details. S.M.B., R.M., and S.R. conceived the study and oversaw the project; S.R. and R.M. wrote the paper. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108401.
Contributor Information
Rolf Müller, Email: rolf.mueller@uni-marburg.de.
Silke Reinartz, Email: silke.reinartz@uni-marburg.de.
Supplemental information
Sample 1–5 = ascites-derived T cells of 5 different patients; a = untreated; b = activated with aCD3 Ab; Sample M = medium control, Control 1 + 2 = pooled plasma samples (internal quality control).
Table S5. RNASeq data of TRAIL receptor, HLA, MICA/B expression in ex vivo HPMC, ascites-derived Tumor (ascTU) and TAT, related to STAR METHODS
References
- 1.Sodek K.L., Murphy K.J., Brown T.J., Ringuette M.J. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev. 2012;31:397–414. doi: 10.1007/s10555-012-9351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niedbala M.J., Crickard K., Bernacki R.J. Interactions of human ovarian tumor cells with human mesothelial cells grown on extracellular matrix. Exp. Cell Res. 1985;160:499–513. doi: 10.1016/0014-4827(85)90197-1. [DOI] [PubMed] [Google Scholar]
- 3.Kenny H.A., Nieman K.M., Mitra A.K., Lengyel E. The first line of intra-abdominal metastatic attack: breaching the mesothelial cell layer. Cancer Discov. 2011;1:100–102. doi: 10.1158/2159-8290.CD-11-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandoval P., Jiménez-Heffernan J.A., Rynne-Vidal Á., Pérez-Lozano M.L., Gilsanz Á., Ruiz-Carpio V., Reyes R., García-Bordas J., Stamatakis K., Dotor J., et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013;231:517–531. doi: 10.1002/path.4281. [DOI] [PubMed] [Google Scholar]
- 5.Mutsaers S.E., Birnie K., Lansley S., Herrick S.E., Lim C.-B., Prêle C.M. Mesothelial cells in tissue repair and fibrosis. Front. Pharmacol. 2015;6:113. doi: 10.3389/fphar.2015.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mikuła-Pietrasik J., Uruski P., Sosińska P., Maksin K., Piotrowska-Kempisty H., Kucińska M., Murias M., Szubert S., Woźniak A., Szpurek D., et al. Senescent peritoneal mesothelium creates a niche for ovarian cancer metastases. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwanicki M.P., Davidowitz R.A., Ng M.R., Besser A., Muranen T., Merritt M., Danuser G., Ince T.A., Brugge J.S., Brugge J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011;1:144–157. doi: 10.1158/2159-8274.CD-11-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heath R.M., Jayne D.G., O'Leary R., Morrison E.E., Guillou P.J. Tumour-induced apoptosis in human mesothelial cells: a mechanism of peritoneal invasion by Fas Ligand/Fas interaction. Br. J. Cancer. 2004;90:1437–1442. doi: 10.1038/sj.bjc.6601635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Na D., Lv Z.-D., Liu F.-N., Xu Y., Jiang C.-G., Sun Z., Miao Z.-F., Li F., Xu H.-M. Gastric cancer cell supernatant causes apoptosis and fibrosis in the peritoneal tissues and results in an environment favorable to peritoneal metastases, in vitro and in vivo. BMC Gastroenterol. 2012;12:34. doi: 10.1186/1471-230X-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu P., Wang J., Mao X., Xu H., Zhu Z. PDCD4 regulates apoptosis in human peritoneal mesothelial cells and promotes gastric cancer peritoneal metastasis. Histol. Histopathol. 2021;36:447–457. doi: 10.14670/HH-18-305. [DOI] [PubMed] [Google Scholar]
- 11.Deng G., Qu J., Zhang Y., Che X., Cheng Y., Fan Y., Zhang S., Na D., Liu Y., Qu X. Gastric cancer-derived exosomes promote peritoneal metastasis by destroying the mesothelial barrier. FEBS Lett. 2017;591:2167–2179. doi: 10.1002/1873-3468.12722. [DOI] [PubMed] [Google Scholar]
- 12.Yokoi A., Yoshioka Y., Yamamoto Y., Ishikawa M., Ikeda S.-I., Kato T., Kiyono T., Takeshita F., Kajiyama H., Kikkawa F., Ochiya T. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 2017;8 doi: 10.1038/ncomms14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu M., Zhang N., He S., Lu X. Exosomal miR-106a derived from gastric cancer promotes peritoneal metastasis via direct regulation of Smad7. Cell Cycle. 2020;19:1200–1221. doi: 10.1080/15384101.2020.1749467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catalan M.P., Subirá D., Reyero A., Selgas R., Ortiz-Gonzalez A., Egido J., Ortiz A. Regulation of apoptosis by lethal cytokines in human mesothelial cells. Kidney Int. 2003;64:321–330. doi: 10.1046/j.1523-1755.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen J.-Y., Chi C.-W., Chen H.-L., Wan C.-P., Yang W.-C., Yang A.-H. TNF-alpha renders human peritoneal mesothelial cells sensitive to anti-Fas antibody-induced apoptosis. Nephrol. Dial. Transplant. 2003;18:1741–1747. doi: 10.1093/ndt/gfg275. [DOI] [PubMed] [Google Scholar]
- 16.Santamaría B., Benito-Martin A., Ucero A.C., Aroeira L.S., Reyero A., Vicent M.J., Orzáez M., Celdrán A., Esteban J., Selgas R., et al. A nanoconjugate Apaf-1 inhibitor protects mesothelial cells from cytokine-induced injury. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krediet R.T., Struijk D.G. Peritoneal changes in patients on long-term peritoneal dialysis. Nat. Rev. Nephrol. 2013;9:419–429. doi: 10.1038/nrneph.2013.99. [DOI] [PubMed] [Google Scholar]
- 18.Simon F., Tapia P., Armisen R., Echeverria C., Gatica S., Vallejos A., Pacheco A., Sanhueza M.E., Alvo M., Segovia E., Torres R. Human Peritoneal Mesothelial Cell Death Induced by High-Glucose Hypertonic Solution Involves Ca2+ and Na+ Ions and Oxidative Stress with the Participation of PKC/NOX2 and PI3K/Akt Pathways. Front. Physiol. 2017;8:379. doi: 10.3389/fphys.2017.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sousa I.G., Simi K.C.R., do Almo M.M., Bezerra M.A.G., Doose G., Raiol T., Stadler P.F., Hoffmann S., Maranhão A.Q., Brigido M.M. Gene expression profile of human T cells following a single stimulation of peripheral blood mononuclear cells with anti-CD3 antibodies. BMC Genom. 2019;20:593. doi: 10.1186/s12864-019-5967-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnsen A.C., Haux J., Steinkjer B., Nonstad U., Egeberg K., Sundan A., Ashkenazi A., Espevik T. Regulation of APO-2 ligand/trail expression in NK cells-involvement in NK cell-mediated cytotoxicity. Cytokine. 1999;11:664–672. doi: 10.1006/cyto.1999.0489. [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N., Yamaguchi N., Nakayama M., Takeda K., Akiba H., Tsutsui H., Okamura H., Nakanishi K., Okumura K., Yagita H. Expression and Function of TNF-Related Apoptosis-Inducing Ligand on Murine Activated NK Cells. J. Immunol. 1999;163:1906–1913. [PubMed] [Google Scholar]
- 22.Takeda K., Hayakawa Y., Smyth M.J., Kayagaki N., Yamaguchi N., Kakuta S., Iwakura Y., Yagita H., Okumura K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001;7:94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 23.Sordo-Bahamonde C., Lorenzo-Herrero S., Payer Á.R., Gonzalez S., López-Soto A. Mechanisms of Apoptosis Resistance to NK Cell-Mediated Cytotoxicity in Cancer. Int. J. Mol. Sci. 2020;21:3726. doi: 10.3390/ijms21103726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sommerfeld L., Finkernagel F., Jansen J.M., Wagner U., Nist A., Stiewe T., Müller-Brüsselbach S., Sokol A.M., Graumann J., Reinartz S., Müller R. The multicellular signalling network of ovarian cancer metastases. Clin. Transl. Med. 2021;11:e633. doi: 10.1002/ctm2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng A., Wei Y., Zhao Y., Zhang T., Ma X. The role of cancer-associated mesothelial cells in the progression and therapy of ovarian cancer. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.1013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Worzfeld T., Finkernagel F., Reinartz S., Konzer A., Adhikary T., Nist A., Stiewe T., Wagner U., Looso M., Graumann J., Müller R. Proteotranscriptomics Reveal Signaling Networks in the Ovarian Cancer Microenvironment. Mol. Cell. Proteomics. 2018;17:270–289. doi: 10.1074/mcp.RA117.000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hausmann M.J., Rogachev B., Weiler M., Chaimovitz C., Douvdevani A. Accessory role of human peritoneal mesothelial cells in antigen presentation and T-cell growth. Kidney Int. 2000;57:476–486. doi: 10.1046/j.1523-1755.2000.00867.x. [DOI] [PubMed] [Google Scholar]
- 28.Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 29.Prager I., Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019;105:1319–1329. doi: 10.1002/JLB.MR0718-269R. [DOI] [PubMed] [Google Scholar]
- 30.O'Connell J., Bennett M.W., O'Sullivan G.C., Collins J.K., Shanahan F. The Fas counterattack: a molecular mechanism of tumor immune privilege. Mol. Med. 1997;3:294–300. [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y., Kanki H., Hachiya T., Ohyama T., Irie S., Tang G., Mukai J., Sato T. Negative regulation of Fas-mediated apoptosis by FAP-1 in human cancer cells. Int. J. Cancer. 2000;87:473–479. doi: 10.1002/1097-0215(20000815)87:4<473::aid-ijc3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Sato K., Hida S., Takayanagi H., Yokochi T., Kayagaki N., Takeda K., Yagita H., Okumura K., Tanaka N., Taniguchi T., Ogasawara K. Antiviral response by natural killer cells throughTRAIL gene induction by IFN-α/β. Eur. J. Immunol. 2001;31:3138–3146. doi: 10.1002/1521-4141(200111)31:11<3138::aid-immu3138>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K., Cretney E., Hayakawa Y., Ota T., Akiba H., Ogasawara K., Yagita H., Kinoshita K., Okumura K., Smyth M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood. 2005;105:2082–2089. doi: 10.1182/blood-2004-08-3262. [DOI] [PubMed] [Google Scholar]
- 34.von Karstedt S., Montinaro A., Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer. 2017;17:352–366. doi: 10.1038/nrc.2017.28. [DOI] [PubMed] [Google Scholar]
- 35.Wu Y., Tian Z., Wei H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017;8:930. doi: 10.3389/fimmu.2017.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kayagaki N., Yamaguchi N., Nakayama M., Eto H., Okumura K., Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999;189:1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smyth M.J., Cretney E., Takeda K., Wiltrout R.H., Sedger L.M., Kayagaki N., Yagita H., Okumura K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001;193:661–670. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sedger L.M., Shows D.M., Blanton R.A., Peschon J.J., Goodwin R.G., Cosman D., Wiley S.R. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 1999;163:920–926. [PubMed] [Google Scholar]
- 39.Kamohara H., Matsuyama W., Shimozato O., Abe K., Galligan C., Hashimoto S.-I., Matsushima K., Yoshimura T. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor expression in human neutrophils. Immunology. 2004;111:186–194. doi: 10.1111/j.0019-2805.2003.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almishri W., Santodomingo-Garzon T., Le T., Stack D., Mody C.H., Swain M.G. TNFα Augments Cytokine-Induced NK Cell IFNγ Production through TNFR2. J. Innate Immun. 2016;8:617–629. doi: 10.1159/000448077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vyas M., Reinartz S., Hoffmann N., Reiners K.S., Lieber S., Jansen J.M., Wagner U., Müller R., von Strandmann E.P. Soluble NKG2D ligands in the ovarian cancer microenvironment are associated with an adverse clinical outcome and decreased memory effector T cells independent of NKG2D downregulation. OncoImmunology. 2017;6 doi: 10.1080/2162402X.2017.1339854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan G., O'Rourke K., Chinnaiyan A.M., Gentz R., Ebner R., Ni J., Dixit V.M. The receptor for the cytotoxic ligand TRAIL. Science (New York, N.Y.) 1997;276:111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 44.Sheridan J.P., Marsters S.A., Pitti R.M., Gurney A., Skubatch M., Baldwin D., Ramakrishnan L., Gray C.L., Baker K., Wood W.I., et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science (New York, N.Y.) 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 45.Degli-Esposti M.A., Dougall W.C., Smolak P.J., Waugh J.Y., Smith C.A., Goodwin R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 46.Höfle J., Trenkner T., Kleist N., Schwane V., Vollmers S., Barcelona B., Niehrs A., Fittje P., Huynh-Tran V.H., Sauter J., et al. Engagement of TRAIL triggers degranulation and IFNγ production in human natural killer cells. EMBO Rep. 2022;23 doi: 10.15252/embr.202154133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koornstra J.J., Kleibeuker J.H., van Geelen C.M.M., Rijcken F.E.M., Hollema H., de Vries E.G.E., de Jong S. Expression of TRAIL (TNF-related apoptosis-inducing ligand) and its receptors in normal colonic mucosa, adenomas, and carcinomas. J. Pathol. 2003;200:327–335. doi: 10.1002/path.1364. [DOI] [PubMed] [Google Scholar]
- 48.Daniels R.A., Turley H., Kimberley F.C., Liu X.S., Mongkolsapaya J., Ch'En P., Xu X.N., Jin B.Q., Pezzella F., Screaton G.R. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005;15:430–438. doi: 10.1038/sj.cr.7290311. [DOI] [PubMed] [Google Scholar]
- 49.Lane D., Cartier A., L'Espérance S., Côté M., Rancourt C., Piché A. Differential induction of apoptosis by tumor necrosis factor-related apoptosis-inducing ligand in human ovarian carcinoma cells. Gynecol. Oncol. 2004;93:594–604. doi: 10.1016/j.ygyno.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 50.Nesterov A., Ivashchenko Y., Kraft A.S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) triggers apoptosis in normal prostate epithelial cells. Oncogene. 2002;21:1135–1140. doi: 10.1038/sj.onc.1205151. [DOI] [PubMed] [Google Scholar]
- 51.Khaider N.G., Lane D., Matte I., Rancourt C., Piché A. Targeted ovarian cancer treatment: the TRAILs of resistance. Am. J. Cancer Res. 2012;2:75–92. [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner K.W., Punnoose E.A., Januario T., Lawrence D.A., Pitti R.M., Lancaster K., Lee D., von Goetz M., Totpal K., et al. Yee S.F., Totpal K. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007;13:1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y., Zhang B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol. Cancer Res. 2008;6:1861–1871. doi: 10.1158/1541-7786.MCR-08-0313. [DOI] [PubMed] [Google Scholar]
- 54.Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J.L., Schröter M., Burns K., Mattmann C., et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 55.Liston P., Fong W.G., Korneluk R.G. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- 56.Takeda K., Stagg J., Yagita H., Okumura K., Smyth M.J. Targeting death-inducing receptors in cancer therapy. Oncogene. 2007;26:3745–3757. doi: 10.1038/sj.onc.1210374. [DOI] [PubMed] [Google Scholar]
- 57.Lane D., Goncharenko-Khaider N., Rancourt C., Piché A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene. 2010;29:3519–3531. doi: 10.1038/onc.2010.107. [DOI] [PubMed] [Google Scholar]
- 58.MacFarlane M., Inoue S., Kohlhaas S.L., Majid A., Harper N., Kennedy D.B.J., Dyer M.J.S., Cohen G.M. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005;12:773–782. doi: 10.1038/sj.cdd.4401649. [DOI] [PubMed] [Google Scholar]
- 59.Lemke J., Noack A., Adam D., Tchikov V., Bertsch U., Röder C., Schütze S., Wajant H., Kalthoff H., Trauzold A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J. Mol. Med. (Berl) 2010;88:729–740. doi: 10.1007/s00109-010-0619-0. [DOI] [PubMed] [Google Scholar]
- 60.Kelley R.F., Totpal K., Lindstrom S.H., Mathieu M., Billeci K., Deforge L., Pai R., Hymowitz S.G., Ashkenazi A. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J. Biol. Chem. 2005;280:2205–2212. doi: 10.1074/jbc.M410660200. [DOI] [PubMed] [Google Scholar]
- 61.Shlyakhtina Y., Pavet V., Gronemeyer H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J.-J., Knudsen S., Mazin W., Dahlgaard J., Zhang B. A 71-gene signature of TRAIL sensitivity in cancer cells. Mol. Cancer Ther. 2012;11:34–44. doi: 10.1158/1535-7163.MCT-11-0620. [DOI] [PubMed] [Google Scholar]
- 63.Yabe M., Ishibashi K., Onagi A., Tanji R., Honda-Takinami R., Koguchi T., Matsuoka K., Hoshi S., Hata J., Kataoka M., et al. Suppression of SOCS3 enhances TRAIL-induced cell growth inhibition through the upregulation of DR4 expression in renal cell carcinoma cells. Oncotarget. 2018;9:31697–31708. doi: 10.18632/oncotarget.25851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sano E., Kazaana A., Tadakuma H., Takei T., Yoshimura S., Hanashima Y., Ozawa Y., Yoshino A., Suzuki Y., Ueda T. Interleukin-6 sensitizes TNF-α and TRAIL/Apo2L dependent cell death through upregulation of death receptors in human cancer cells. Biochim. Biophys. Acta. Mol. Cell Res. 2021;1868 doi: 10.1016/j.bbamcr.2021.119037. [DOI] [PubMed] [Google Scholar]
- 65.Kim O., Park E.Y., Klinkebiel D.L., Pack S.D., Shin Y.-H., Abdullaev Z., Emerson R.E., Coffey D.M., Kwon S.Y., Creighton C.J., et al. In vivo modeling of metastatic human high-grade serous ovarian cancer in mice. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maniati E., Berlato C., Gopinathan G., Heath O., Kotantaki P., Lakhani A., McDermott J., Pegrum C., Delaine-Smith R.M., Pearce O.M.T., et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020;30:525–540.e7. doi: 10.1016/j.celrep.2019.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iyer S., Zhang S., Yucel S., Horn H., Smith S.G., Reinhardt F., Hoefsmit E., Assatova B., Casado J., Meinsohn M.-C., et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov. 2021;11:384–407. doi: 10.1158/2159-8290.CD-20-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reinartz S., Finkernagel F., Adhikary T., Rohnalter V., Schumann T., Schober Y., Nockher W.A., Nist A., Stiewe T., Jansen J.M., et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016;17:108. doi: 10.1186/s13059-016-0956-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reinartz S., Lieber S., Pesek J., Brandt D.T., Asafova A., Finkernagel F., Watzer B., Nockher W.A., Nist A., Stiewe T., et al. Cell type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol. Oncol. 2019;13:185–201. doi: 10.1002/1878-0261.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ince T.A., Sousa A.D., Jones M.A., Harrell J.C., Agoston E.S., Krohn M., Selfors L.M., Liu W., Chen K., Yong M., et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015;6:7419. doi: 10.1038/ncomms8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankert K.C., Oxley K.L., Smith S.M., Graham J.P., de Boer M., Thewissen M., Simons P.J., Bishop G.A. Induction of an altered CD40 signaling complex by an antagonistic human monoclonal antibody to CD40. J. Immunol. 2015;194:4319–4327. doi: 10.4049/jimmunol.1402903. [DOI] [PubMed] [Google Scholar]
- 72.Ahrends T., Spanjaard A., Pilzecker B., Bąbała N., Bovens A., Xiao Y., Jacobs H., Borst J. CD4+ T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity. 2017;47:848–861.e5. doi: 10.1016/j.immuni.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 73.Unger A., Finkernagel F., Hoffmann N., Neuhaus F., Joos B., Nist A., Stiewe T., Visekruna A., Wagner U., Reinartz S., et al. Chromatin Binding of c-REL and p65 Is Not Limiting for Macrophage IL12B Transcription During Immediate Suppression by Ovarian Carcinoma Ascites. Front. Immunol. 2018;9:1425. doi: 10.3389/fimmu.2018.01425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guescini M., Sisti D., Rocchi M.B.L., Stocchi L., Stocchi V. A new real-time PCR method to overcome significant quantitative inaccuracy due to slight amplification inhibition. BMC Bioinf. 2008;9:326. doi: 10.1186/1471-2105-9-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Assarsson E., Lundberg M., Holmquist G., Björkesten J., Thorsen S.B., Ekman D., Eriksson A., Rennel Dickens E., Ohlsson S., Edfeldt G., et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9 doi: 10.1371/journal.pone.0095192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wik L., Nordberg N., Broberg J., Björkesten J., Assarsson E., Henriksson S., Grundberg I., Pettersson E., Westerberg C., Liljeroth E., et al. Proximity Extension Assay in Combination with Next-Generation Sequencing for High-throughput Proteome-wide Analysis. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., et al. Ensembl 2020. Nucleic Acids Res. 2020;48:D682–D688. doi: 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample 1–5 = ascites-derived T cells of 5 different patients; a = untreated; b = activated with aCD3 Ab; Sample M = medium control, Control 1 + 2 = pooled plasma samples (internal quality control).
Table S5. RNASeq data of TRAIL receptor, HLA, MICA/B expression in ex vivo HPMC, ascites-derived Tumor (ascTU) and TAT, related to STAR METHODS
Data Availability Statement
-
•
RNA-Seq data were deposited at EBI ArrayExpress (accession numbers E-MTAB-4162, E-MTAB-10611). The complete PEA affinity proteomics dataset is included as Table S1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.