

Abstract
Background and Objectives
SYNGAP1 variants are associated with rare developmental and epileptic encephalopathies (DEEs). Although SYNGAP1-related childhood phenotypes are well characterized, the adult phenotype remains ill-defined. We sought to investigate phenotypes and outcomes in adults with SYNGAP1 variants and epilepsy.
Methods
Patients 18 years or older with DEE carrying likely pathogenic and pathogenic (LP/P) SYNGAP1 variants were recruited through physicians' practices and patient organization groups. We used standardized questionnaires to evaluate current seizures, medication use, sleep, gastrointestinal symptoms, pain response, gait, social communication disorder and adaptive skills of patients. We also assessed caregiver burden.
Results
Fourteen unrelated adult patients (median: 21 years, range: 18–65 years) with SYNGAP1-DEE were identified, 11 with novel and 3 with known LP/P SYNGAP1 de novo variants. One patient with a partial exon 3 deletion had greater daily living skills and social skills than others with single-nucleotide variants. Ten of 14 (71%) patients had drug-resistant seizures, treated with a median of 2 antiseizure medications. All patients (100%) had abnormal pain processing. Sleep disturbances, social communication disorders, and aggressive/self-injurious behaviors were each reported in 86% of patients. Only half of adults could walk with minimal or no assistance. Toileting was normal in 29%, and 71% had constipation. No adult patients could read or understand verbal material at a sixth-grade level or higher. Aggressive/self-injurious behaviors were leading cause of caregiver burden. The oldest patient was aged 65 years; although nonambulant, she had walked independently when younger.
Discussion
Seventy-one percent of patients with SYNGAP1-DEEs continue to have seizures when adults. Nonseizure comorbidities, especially aggression and self-injurious behaviors, are major management challenges in adults with SYNGAP1-DEE. Only 50% of adults can ambulate with minimal or no assistance. Almost all adult patients depend on caregivers for many activities of daily living. Prompt diagnostic genetic testing of adults with DEE can inform clinical care and guide outcomes of precision therapies.
Introduction
The human SYNGAP1 gene on chromosome 6p21.3 encodes the synaptic RAS-GTPase-activating protein 1 (SynGAP), which regulates cell growth and synaptic plasticity.1,2 As a highly enriched protein in excitatory glutamatergic synapses, SynGAP is necessary for normal brain function by maintaining a balance between excitation and inhibition.3
SYNGAP1 variants were initially associated with nonsyndromic intellectual disability (ID) (OMIM#306684).4-7 The link between developmental and epileptic encephalopathies (DEEs) and SYNGAP1 was first observed in 2013, when 7 unrelated patients presented with epilepsy, delayed development in the first years of life, autism spectrum disorder (ASD), and varying degrees of ID.8 Most patients with SYNGAP1-related DEE harbor de novo heterozygous variants and present with varying degrees of clinical severity.
In patients with ID without epilepsy, SYNGAP1 pathogenic variants have a prevalence of roughly 1:1,000 to 1:10,000, comprising approximately 1% of all ID cases.6,7 Similarly, when SYNGAP1 variants were first linked to DEE, these variants comprised approximately 1% of patients with DEE.8 Currently, only 13 adult patients, from 9 different studies, have been reported in the literature (median: 25 years, range: 18–33 years).8-16
We evaluated an international group of adults with DEE and likely pathogenic or pathogenic (LP/P) SYNGAP1 variants. Specifically, we investigated seizures, adaptive skills, social communication skills, sleep, gait and gastrointestinal disturbances, pain tolerance, aggression, and behavior. Finally, we evaluated the functional dependence of adult patients and caregiver burden.
Methods
Study Population and Data Collection
We recruited patients aged 18 years or older with P/LP SYNGAP1 variants to participate in our study. Enrollment was performed between July 2021 and February 2022. Patients were recruited through the investigator's institutions, and through the SYNGAP1 Research Fund, across the following countries: Canada, the United States, Spain, and Germany. Study materials were translated from English to Spanish and German. The study was approved by the Research Ethics Board with the University Health Network (protocol 21-5009) in Toronto, Canada. Informed consent was obtained from the substitute decision makers of all patients.
Clinical data were collected using these validated assessments tools.
Severity of Clinical Outcomes
A modified version of the Severity Assessment17 tool was used to determine a composite score of disease severity in patients. The final severity assessment comprised 51 items that comprehensively describe seizures, treatment usage, gait, pain responsiveness, toileting, reflux, and abnormal sleep. Each item consisted of a Likert scale rating. Ratings of therapy effectiveness and the patient's overall condition were also included. A maximum score of 129/129 is indicative of the most severe clinical phenotype. Seizures were classified according to the 2017 International League Against Epilepsy guidelines.18
Social Communication Skills
The Social Communication Questionnaire (SCQ) (Lifetime version) was used to screen for the presence of communication deficits suggestive of ASD. The Lifetime version yields a Total Score that is interpreted with reference to cut-off scores. Scores above the cutoff of 15 suggest that the patient is likely to be on the autism spectrum and that a more extended evaluation should be undertaken.
Adaptive Behavioral Abilities
The Vineland Adaptive Behavior Scales 3 was used to determine adaptive behavior abilities in the following domains: communication (receptive, expressive, and written), daily living skills (personal, domestic, and community), socialization (interpersonal relationships, play/leisure, and coping skills), motor ability (fine and gross motor), and maladaptive behaviors. Raw scores were extracted from each domain, which were selected due to floor effects found in age-wise comparisons. An overall rating was given based on the patient's results compared with those of a norm sample, which is a representative group of patients the same age from across the United States.
Statistical Analyses
Descriptive statistics were used to summarize clinical features and characteristics of the patients in the study. The Kruskal-Wallis test was used to compare assessment scores with variant types (missense, nonsense, deletion, splice acceptor, and frameshift where applicable) and affected functional domains. Statistical significance was set to p < 0.05, and statistical values were not reported for nonstatistically significant findings. Analyses were conducted using R, and figures were created using GraphPad PRISM.
Genotyping
The pathogenicity of SYNGAP1 variants was interpreted according to the American College of Medical Genetics and Genomics guidelines.19 Each of the SYNGAP1 variants was also queried using the ClinVar database and GnomAD browser database.20,21
Data Availability
Deidentified data may be provided on reasonable request.
Results
Description of Participants
Fourteen unrelated adult patients (9 female patients) participated in the study, with a median age of 21 years (range: 18–65) (Table 1). Five patients had a family history of epilepsy. All patients came from nonconsanguineous families. A full table of genotypic and phenotypic data from previously reported patients in the literature and patients from this study is presented in eTable 1 (links.lww.com/NXG/A647).
Table 1.
Demographics of 14 Adult Patients With SYNGAP1 Variants and Their Genetic Profiles (n = 14)

| Patient # | Sex | Age (y) | ASD diagnosis (age of diagnosis) | Ethnicity | Family history | Exon # | cDNA | Protein consequence | Variant classification | Inheritance | ACMG interpretation | Variant previously reported? |
| 1 | F | 65 | Yes | White | No | Exon 8 | c.1329_1333delCAAGG | p.Lys444Glyfs*27 | Frameshift leading to truncation | Unknown | Pathogenic | No |
| 2 | M | 20 | Yes (2y) | White | No | Exon 8 | c.781_784delGACA | p.Asp261Metfs*3 | Frameshift leading to truncation | De novo | Pathogenic | No |
| 3 | F | 20 | No | White, Jewish | Yes, sister | Exon 15 | c.3295delT | p.Tyr1099Metfs* | Frameshift leading to truncation | De novo | Pathogenic | No |
| 4 | F | 48 | Yes | Other | No | Exon 8 | c.870del | p.Tyr291fs | Frameshift | De novo | Pathogenic | No |
| 5 | F | 24 | Yes (5y) | White | Yes, half sister | Exon 12 | c.2019delA | p.Thr674Profs*36 | Frameshift leading to truncation | De novo | Pathogenic | No |
| 6 | F | 19 | Yes | South Asian, Latino/Hispanic | Yes, maternal uncle | Exon 8 | c.1167_1168del | p.Gly391fs | Frameshift | De novo | Pathogenic | Jimenez-Gomez et al. (2019) |
| 7 | M | 23 | Yes | White | N/A | Exon 15 | c.3233_3236delTCAG | p.Val1078fs* | Frameshift leading to truncation | De novo | Pathogenic | No |
| 8 | F | 20 | Yes | Latino/Hispanic | Yes, grandmother | Exon 15 | c.2526dup | p.Met843Hisfs*7 | Frameshift leading to truncation | De novo | Likely pathogenic | No |
| 9 | F | 23 | Yes | White, Latino/Hispanic | Yes, cousin | Exon 19 | c.4006G>A | p.Glu1336Lys | Missense | De novo | Likely pathogenic | No |
| 10 | F | 20 | No | White | No | Exon 5 | c.403C>T | p.Arg135* | Nonsense | De novo | Pathogenic | Mignot et al. (2016) |
| 11 | F | 22 | Yes | Latino/Hispanic | No | Exon 11 | c.1861C>T | p.Arg621* | Nonsense | De novo | Pathogenic | Aguilera et al. (2021) Verma et al. (2020) |
| 12 | M | 20 | Yes | White | No | Exon 15 | c.3227delT | p.Leu1076* | Nonsense | De novo | Pathogenic | No |
| 13 | M | 18 | No | White | No | Exon 3 | c.190-15_206delins28 | — | Insertion/deletion (indel) | De novo | Pathogenic | No |
| 14 | M | 22 | Yes | White, Jewish | N/A | — | c.1532-1G>C | — | Splice acceptor leading to exon 10 skipping | De novo | Likely pathogenic | No |
Abbreviations: ASD = autism spectrum disorder; F=Female; M = Male; N/A = not available.
Sex, age, ASD diagnosis, family history of epilepsy, and ethnicity are listed when provided. Information on the genetic variant, molecular consequences, inheritance, and interpretation from a patient's genetic report is provided. The zygosity of all variants was heterozygous.
Genotypic Spectrum
All patients had LP/P SYNGAP1 variants. Eleven are new, previously unreported variants. Three variants have been previously reported in the literature.10,14,22,23 Eight of 14 patients had frameshift variants. Five of 8 frameshift variants led to protein truncation (nonsense). Another 3 patients had nonsense variants, one had a missense variant, and one had a splice acceptor variant leading to exon skipping. One patient had an indel in exon 3 of SYNGAP1. In all patients, the transcript analyzed was the longest one: NM006772.2. Together, these 14 patients harbored 11 novel (likely) pathogenic variants (Table 1).
Phenotypic Spectrum
Seizures
Overall, 10/14 (71%) patients had at least one type of seizure in the past 12 months. Seven of 14 (50%) patients had ongoing nonconvulsive seizures (including eyelid myoclonia with absence): yearly in one patient, monthly in another patient, weekly in 2 patients, and daily in 3 patients.
Four (29%) patients had convulsive seizures in the past 12 months. Of these patients, 3 had monthly convulsive seizures and 1 patient had daily convulsive seizures.
Three (21%) adult patients had prolonged convulsive seizures lasting over 5 minutes in the previous 12 months. One patient had seizures associated with hyperventilation.
Disruptive isolated epileptic spasms in the past 12 months were reported by 4 (26%) patients.
In this cohort of adult patients, there was a median lifetime of 4 antiseizure medications (ASMs) usage (range: 1–5+) and median current usage of 2 (range: 1–5+) ASMs. There was no reported current usage of ketogenic diet, vagus nerve stimulation (VNS), deep brain stimulation (DBS), or responsive neurostimulation (RNS) as treatment options, although 1 patient had used ketogenic diet in the past. Thirteen patients (93%) reported no use of rescue medications or hospital visits for prolonged seizures. Four patients reported no current usage of ASMs; however, only 2 of those patients were seizure free without ASMs.
Comorbidities
Constipation, pain responsiveness, sleep disturbances, disruptive daytime sleepiness, gait, toileting, and reflux were evaluated (Figure 1). Four of 14 patients (29%) had no constipation; 9 patients (64%) had constipation that was controlled either with or without medication over the previous 12 months. One patient had uncontrolled constipation that was reported to be a key component to the patient's quality of life.
Figure 1. Summary Graphs of Various Clinical Features in Adults With SYNGAP1.

Severity assessment results regarding: (A) constipation, (B) pain responsiveness, (C) sleep disturbances, (D) daytime sleepiness, (E) toileting, and (F) reflux (n = 14).
Reflux was absent in 9 (64%) patients; 5 (36%) patients had reflux which was controlled with daily medication in 4/5 patients. Toileting was normal in 4 (29%) patients, timed in 5 (36%), and 5 (36%) patients required the use of diapers.
Pain responsiveness was abnormal in all patients; 4 (29%) had delayed reactions to minor pain, 5 (36%) had delayed reactions to major pain, whereas 5 (36%) had no response to minor pain.
Sleep disturbances were present in 12 (86%) patients—only 2 (14%) patients had normal sleep with no issues. Daytime sleepiness varied across the cohort, with 11/14 (79%) displaying some form of sleepiness and fatigue that was disruptive during the day.
Scoliosis and Walking Abilities
Mild scoliosis not requiring treatment was reported in 8/14 (57%) patients, with no patients requiring braces or surgery (Figure 2). Walking abilities varied as 3 (21%) walked independently with minimal assistance and 4 (29%) walked community distances with minimal assistance. Two (14%) walked without assistance only on even surfaces. Another 2 patients (14%) could walk outdoors, but typically used wheelchairs. One patient walked only indoors, while another could only take some steps. The oldest patient (patient #1) was nonambulant, although she was able to walk independently when she was younger.
Figure 2. Walking Abilities of Adult Patients With SYNGAP1-DEE.

Autism Spectrum Disorder
Twelve of 14 (86%) patients scored above the threshold on the SCQ, indicating further evaluation for autism is required (eTable 2, links.lww.com/NXG/A648). In fact, 11 of these 12 patients already had a previous formal diagnosis of ASD, given by a physician or health care provider. Specific characteristics in this cohort included ritual-like behaviors (11/14, 79%) and specific interests with high intensity (11/14, 79%). Of relevance, 12/14 (86%) patients showed self-injurious behavior, e.g., head banging and biting oneself.
Adaptive Behavioral Abilities
Adaptive behavioral abilities were varied among SYNGAP1-DEE patients. Domain level scores are presented in eTable 2 (links.lww.com/NXG/A648). There were no statistically significant differences between clinical findings and genotypes, except for one patient, a 19-year-old man carrying an indel of SYNGAP1 exon 3 (c.190-15_206delins28). This patient demonstrated an elevated ability to perform daily living skills. He also exhibited stronger social skills and abilities to pursue relationships, compared with the rest of the cohort. Although his overall summary score for adaptive behaviors was moderately low for his age, all other patients in this cohort had lower scores compared with normative data. Nine patients (64%) were able to feed themselves with a fork and spoon. Twelve (86%) were cooperative in personal activities, such as undressing, dressing, and washing of the hands and face. Of the 9 patients who were able to dress themselves, none could use zippers (Table 2).
Table 2.
Comparison of Daily Living Abilities Between Pediatric Patients and Adult Patients With SYNGAP1 Variants
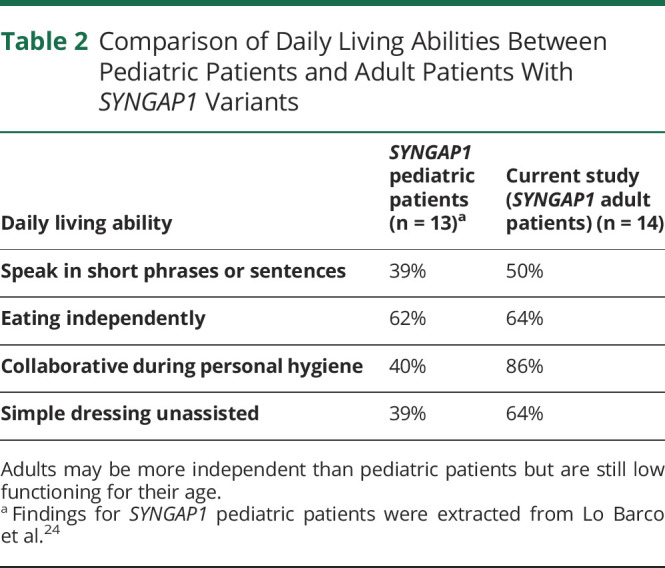
| Daily living ability | SYNGAP1 pediatric patients (n = 13)a | Current study (SYNGAP1 adult patients) (n = 14) |
| Speak in short phrases or sentences | 39% | 50% |
| Eating independently | 62% | 64% |
| Collaborative during personal hygiene | 40% | 86% |
| Simple dressing unassisted | 39% | 64% |
Adults may be more independent than pediatric patients but are still low functioning for their age.
Findings for SYNGAP1 pediatric patients were extracted from Lo Barco et al.24
With respect to gross and fine motor skills, 11 (79%) patients were able to sit unsupported for at least 10 minutes, 12 (86%) could walk upstairs, and 11 (79%) could walk downstairs. Seven (50%) could jump off the ground with both feet. However, no patient had the ability to manipulate very small objects.
Regarding language and learning abilities, 7 (50%) patients could talk using short phrases or sentences. However, all patients were responsive to caregivers, could recognize their own names, and respond to one-word actions. Eight patients (57%) could identify all letters of the alphabet, but only 2 (14%) could sometimes write at least 10 simple words from memory. Although 7 patients (50%) could read at least 10 words, only 5 (36%) could read simple sentences out loud and just 3 (21%) could read simple stories out loud. No adult patients were able to read or understand material at a sixth-grade level or higher.
Maladaptive Behaviors
Physical aggression, temper tantrums, and neediness were observed in 11 (79%) patients. Ten patients (71%) disobeyed those in authority and had eating problems, such as a refusal to eat or overeating. Loss of awareness regarding surroundings was identified by caregivers in 12 (86%) patients.
Negative findings included no lying and breaking rules/laws because of peer pressure, no harming animals, or interest in extreme violence. Furthermore, there were no reports about holding untrue beliefs or talks about auditory/visual hallucinations. One patient expressed feelings of helplessness/hopelessness, and another patient has threatened to hurt/kill someone in the past.
Longevity
This study features the oldest SYNGAP1-DEE patient currently reported in the literature, a 65-year-old White woman carrying a pathogenic frameshift variant (p.Lys444Glyfs*27) of unknown inheritance.
This patient experienced her first seizure at age 7 months. She developed absence seizures and generalized tonic-clonic seizures that were drug resistant. At age 18 years (several years before receiving the SYNGAP1 genetic diagnosis), she underwent a frontal lobe resection for the treatment of seizures. Unfortunately, the surgery was unsuccessful. She has never had the ketogenic diet, VNS, or DBS/RNS. By age 65 years, her caregiver reported daily absences with eyelid myoclonia induced by sounds and lights and daily isolated epileptic spasms that are disruptive to the patient and/or family. Other convulsive seizures had not occurred in over a year, and no prolonged seizures lasting more than 5 minutes were reported in the previous 6 months. The caregiver's impression of seizures in the past 12 months was of worsening seizures, with daily seizures that are disruptive to daily life.
This patient has moderate intellectual disability and has received a formal diagnosis of autism spectrum disorder. She is at present wheelchair-bound, has feeding/swallowing issues, and is unable to consume previously enjoyed foods due to choking hazards. Other clinical features of concern include uncontrolled constipation and toileting accidents. Daytime sleepiness is disruptive throughout most of the week, and the patient often arouses from sleep more than once per week. Overall, the caregiver reported a “really worse” patient condition compared with the first 10 years of life, particularly pertaining to motor ability. Some key maladaptive behaviors reported included a tendency to harm herself, frequent threats to hurt or kill someone, lose awareness of surrounding, and fixation on a specific topic.
Discussion
In this study, we present molecular and clinical information on 14 adults with SYNGAP1-DEE, the largest such cohort yet to be reported. Of the 14 adult patients included in this study, 11 had previously unreported likely pathogenic or pathogenic SYNGAP1 variants. One patient with an indel of SYNGAP1 exon 3 had stronger daily living skills and social skills including abilities to pursue relationships, compared with the rest of the cohort who had frameshift, missense, nonsense, and splice acceptor site variants affecting exons 3 to 19.
Half of the adult patients were free of convulsive seizures. Four patients were free of all types of seizures in the previous 12 months, and 2 of those 4 were off ASMs. In previous reports, only 19% of patients 7 years and older were seizure-free.16 On one end of the spectrum, 28% of adult patients in our study are seizure-free; on the other end, 21% of these adults still have prolonged convulsive seizures, which represent a significant morbidity.9,10
Abnormal pain responsiveness was observed in 100% of adults, making it more common than previously reported in children.16 Although it is possible that the pain threshold may change over time, methodological differences could also have led to this finding. For example, previous studies evaluated severely abnormal pain responsiveness (e.g., to broken bones),16 while we also asked about mild nociception abnormalities. The precise mechanism leading to abnormal nociception is unclear, but Syngap1 mouse models reveal that touch is weakly encoded in upper-lamina neurons in the somatosensory cortex, leading to improper sensory processing.24
Self-injury behaviors were previously observed in children with SYNGAP1.11,14,16 However, our findings showed a whopping 86% prevalence of self-injury in adults with this condition. Self-injury has been noted in patients with DEE as a whole, but it seems to be less prevalent compared with SYNGAP1-DEE. For example, only 28% of patients with Rett syndrome may have externalizing behavioral issues, such as self-injury.25,26 Similarly, one cohort of adult patients with Dravet syndrome saw self-injurious behaviors in 31% of patients.27 Self-injury and aggression are rarely observed in patients with CDKL5 variants.17 Finally, an evaluation of adults with KCNQ2-DEEs found that 31% of patients demonstrated self-injurious behaviors.28
It is unclear why most adults with SYNGAP1-DEE show self-injurious behavior. One potential mechanism could include a relationship to abnormal sensory perception of self-harming behavior as a form of self-stimulation. While ASMs are generally well tolerated in several forms of DEEs,29 one patient in our study showed some behavioral improvement after discontinuing an ASM. Similarly, one adult in the literature discontinued levetiracetam at age 13 years due to behavioral issues.12 Given the caregiver burden associated with neurobehavioural challenges in patients with DEEs, considerations for behavioral changes when selecting an ASM may be worthwhile.30,31
The prevalence of ASD diagnoses is greater in adults (79%) with SYNGAP1-DEE than previously reported for children (53%).16 The reasons for this discrepancy are unclear. Regardless, ASD emerges as a key finding in adults, aligning with other adults in the literature exhibiting autistic features, such as stereotypies.9,13,14 Other behavioral issues, such as aggression, temper tantrums, and obsessive behaviors, align with previous reports in both children and adults.11,14-16
The daily living abilities of adult SYNGAP1-DEE patients have not been previously explored. We found that all adults in our cohort had below average adaptive skills, at similar levels to pediatric SYNGAP1 patients.30 While direct comparisons cannot be made due to different assessment tools used in each study, Table 2 presents a brief summary of the daily living abilities of pediatric patients (range: 3.7–17.7 years) as reported by Lo Barco et al. and the adults of this study.30 When compared with the pediatric sample, 25% more adults were able to dress themselves with assistance, pointing toward possible improved daily living abilities across the life course. The proportion of patients able to feed themselves was similar in both pediatric and adult patients (∼60–70%), suggesting that these skills may be preserved into middle age. Of interest, a slightly higher proportion of adults could speak using short phrases or sentences compared with pediatric patients. This is in contrast to adults previously presented in the literature, where speech was either absent or, at most, of the ability of a 1-year-old.10,11,14
The exact reason for the improvement in daily living abilities compared with pediatric patients is unknown. However, in other DEEs, such as Dravet syndrome, seizure freedom has been associated with improved everyday executive functioning of children and young adults.32,33 Although direct comparisons cannot be made, it is possible that this may be the case in some adults with SYNGAP1. Longitudinal studies would be required to confirm this relationship.
Regarding sleep, our findings align with previous research observing sleep disturbances in SYNGAP1-DEE pediatric patients, particularly difficulties initiating and maintaining sleep.16,34 Other studies of adult cases in the literature report insomnia and night-time awakenings.11 As such, sleep disturbances and daytime sleepiness may be important features that warrant continuous monitoring and treatment as patients age. The reasons for sleep problems are unclear, but studies of adult mice have shown that SYNGAP1's abnormal interictal epileptiform discharges can increase during sleep and interrupt sleep architecture.35 It is possible that a similar pathophysiology underlies the sleep problems in adult patients.
As seen in other DEEs, walking may be worse in adults.36 In this cohort, only 50% of patients could walk with minimal or no assistance, and the oldest patient (age 65 years) is wheelchair-bound, although she had been able to walk when younger.
Most patients with SYNGAP1 LP/P variants diagnosed today are children. This is in part due to the recency of our knowledge of this gene as a cause of DEE. As such, when parents of newly diagnosed SYNGAP1-DEE children ask about longevity, there are no definitive answers. Here, we describe the natural history of the oldest patient with SYNGAP1-DEE reported so far, aged 65 years.
Although this is the largest cohort of adult patients with SYNGAP1-DEE yet studied, the sample size is small, and only 2 patients were older than 24 years. This might be in part due to the relatively recent recognition of SYNGAP1 gene variants as a cause of DEEs and adults not receiving up-to-date genetic testing.5 As with any caregiver reported outcome, findings may be subject to recall bias. This study did not track specific ASM usage. Studies are needed to examine the efficacy of drugs, as there is no standardized ASM management of SYNGAP1-DEE patients. Conclusions about adults with SYNGAP1-DEE will require large numbers and detailed longitudinal follow-up data, ideally in a prospective study.
This is the study of adult patients with SYNGAP1-DEE. The detailed characterization of the natural history of this condition, including seizures, communication skills, pain responsiveness, sleep, digestive issues, gait abnormalities, and other comorbidities, may help to identifying adults with DEE who so far lack genetic diagnosis. We also report the adaptive behavioral abilities of adults, allowing caregivers an opportunity to help plan for future care as SYNGAP1-DEE patients enter adulthood.
In this study, we also report the oldest SYNGAP1-DEE patient in the literature and the first view into possible longevity issues. Further studies in larger groups of adults are still necessary to have a more comprehensive view of the natural history of this condition. Encouraging genetic (re)testing of adults with undiagnosed epilepsies may contribute to these efforts.
Acknowledgment
The authors acknowledge the participating patients and families for their time, especially the SYNGAP1 Research Fund.
Glossary
- ASD
autism spectrum disorder
- ASMs
antiseizure medications
- DBS
deep brain stimulation
- DEEs
developmental and epileptic encephalopathies
- ID
intellectual disability
- LP/P
likely pathogenic or pathogenic
- RNS
responsive neurostimulation
- SCQ
Social Communication Questionnaire
- VNS
vagus nerve stimulation
Appendix. Authors
| Name | Location | Contribution |
| Marlene Rong, MSc | Institute of Medical Science, University of Toronto; Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Tim Benke, MD, PhD | Department of Pediatrics, Neurology, Pharmacology and Otolaryngology, University of Colorado School of Medicine and Children's Hospital Colorado, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design |
| Quratulain Zulfiqar Ali, MD | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design |
| Ángel Aledo-Serrano, MD, PhD | Epilepsy and Neurogenetics Program, Neurology Department, Ruber Internacional Hospital; Initiative for Neuroscience (INCE) Foundation, Madrid, Spain | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Allan Bayat, MD, PhD | Department of Drug Design and Pharmacology, University of Copenhagen; Department for Genetics and Personalized Medicine, Danish Epilepsy Centre, Dianalund; Institute for Regional Health Services, University of Southern Denmark, Odense, Denmark | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Alessandra Rossi, MD | Department of Epilepsy Genetics and Personalized Medicine, Danish Epilepsy Centre, Dianalund; Pediatric Clinic, IRCCS San Matteo Hospital Foundation, University of Pavia, Italy | Major role in the acquisition of data |
| Orrin Devinsky, MD | NYU Langone Epilepsy Center, NY | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Farah Qaiser, MSc | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Anum S. Ali, BScN, MSc | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Alfonso Fasano, MD, PhD, FAAN | Edmond J. Safra Program in Parkinson's Disease, Morton and Gloria Shulman Movement Disorders Clinic, Toronto Western Hospital; Division of Neurology, University of Toronto; Krembil Brain Institute, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Anne S. Bassett, MD, FRCPC | Clinical Genetics Research Program, Centre for Addiction and Mental Health; The Dalglish Family 22q Clinic, Toronto General Hospital, University Health Network; Department of Psychiatry, University of Toronto; Toronto Congenital Cardiac Centre for Adults, Division of Cardiology, Department of Medicine, and Department of Psychiatry, University Health Network; Toronto General Hospital Research Institute and Campbell Family Mental Health Research Institute, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Danielle M. Andrade, MD, MSc | Adult Genetic Epilepsy (AGE) Program, Krembil Neurosciences Institute, Toronto Western Hospital, University Health Network; Division of Neurology, Department of Medicine, University of Toronto, Ontario, Canada | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
MR received unrestricted educational funding from Biocodex. AB is funded by a BRIDGE - Translational Excellence Programme grant funded by the Novo Nordisk Foundation, grant agreement number: NNF20SA0064340. ASB holds the Dalglish Chair in 22q11.2 Deletion Syndrome at the University Health Network and University of Toronto. DMA received grant support from Ontario Brain Institute and McLaughlin Foundation for this study.
Disclosure
M. Rong reports no disclosures relevant to the manuscript; T.A. Benke receives grant support from NINDS, NIDCD, NIA, Simons Foundation and IRSF. He performs consultancy for AveXis, Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda, Taysha, CureGRIN, GRIN Therapeutics, Alcyone, Neurogene, and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department; Q. Zulfiqar Ali reports no disclosures relevant to the manuscript; A. Aledo‐Serrano reports no disclosures relevant to the manuscript; A. Bayat reports no disclosures relevant to the manuscript; A. Rossi reports no disclosures relevant to the manuscript; O. Devinsky receives grant support from NINDS, NIMH, MURI, CDC and NSF. He has equity and/or compensation from the following companies: Ajna Biosciences, Tilray, Receptor Life Sciences, Hitch Biosciences, Tevard Biosciences, Regel Biosciences, Script Biosciences, Actio Biosciences, Empatica, SilverSpike, and California Cannabis Enterprises (CCE). He has received consulting fees from Zogenix, Ultragenyx, BridgeBio, GeneMedicine and Marinus. He holds patents for the use of cannabidiol in treating neurologic disorders which are owned by GW Pharmaceuticals for which he waived financial interests. He holds other patents in molecular biology. He is the managing partner of the PhiFund Ventures; F. Qaiser reports no disclosures relevant to the manuscript; A. Ali reports no disclosures relevant to the manuscript; A. Fasano reports no disclosures relevant to the manuscript; A.S. Bassett reports no disclosures relevant to the manuscript; D.M. Andrade receives grant support from McLaughlin Foundation, UHN Foundation, Dravet Syndrome Foundation. She also received consulting fees from UCB, Biocodex, Paladin, Eisai. Finally, she receives royalties from UpToDate. Go to Neurology.org/NG for full disclosures.
References
- 1.Hae Kim J, Liao D, Lau LF, Huganir RL. SynGAP: A Synaptic RasGAP That Associates with the PSD-95/SAP90 Protein Family, Vol 20; 1998:683-691. [DOI] [PubMed] [Google Scholar]
- 2.Ligeti E, Welti S, Scheffzek K. Inhibition and termination of physiological responses by GTPase activating proteins. Physiol Rev. 2012;92(1):237-272. doi: 10.1152/physrev.00045.2010 [DOI] [PubMed] [Google Scholar]
- 3.Clement JP, Aceti M, Creson TK, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151(4):709-723. doi: 10.1016/j.cell.2012.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamdan FF, Gauthier J, Spiegelman D, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360(6):599-605. doi: 10.1056/NEJMoa0805392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berryer MH, Hamdan FF, Klitten LL, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34(2):385-394. doi: 10.1002/humu.22248 [DOI] [PubMed] [Google Scholar]
- 6.McRae JF, Clayton S, Fitzgerald TW, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433-438. doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223-228. doi: 10.1038/nature14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825-830. doi: 10.1038/ng.2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klitten LL, Møller RS, Nikanorova M, Silahtaroglu A, Hjalgrim H, Tommerup N. A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia. 2011;52(12):e190-e193. doi: 10.1111/j.1528-1167.2011.03304.x [DOI] [PubMed] [Google Scholar]
- 10.Mignot C, von Stülpnagel C, Nava C, et al. Genetic and neurodevelopmental spectrum of SYNGAP1 -associated intellectual disability and epilepsy. J Med Genet. 2016;53(8):511-522. doi: 10.1136/jmedgenet-2015-103451 [DOI] [PubMed] [Google Scholar]
- 11.Prchalova D, Havlovicova M, Sterbova K, Stranecky V, Hancarova M, Sedlacek Z. Analysis of 31-year-old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1-associated phenotype: case report. BMC Med Genet. 2017;18(1):62. doi: 10.1186/s12881-017-0425-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kluger G, von Stülpnagel-Steinbeis C, Arnold S, Eschermann K, Hartlieb T. Positive short-term effect of low-dose rosuvastatin in a patient with SYNGAP1-associated epilepsy. Neuropediatrics. 2019;50(4):266-267. doi: 10.1055/s-0039-1681066 [DOI] [PubMed] [Google Scholar]
- 13.Vidal S, Brandi N, Pacheco P, et al. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur J Paediatr Neurol. 2019;23(4):609-620. doi: 10.1016/j.ejpn.2019.04.006 [DOI] [PubMed] [Google Scholar]
- 14.Aguilera C, Gabau E, Ramirez-Mallafré A, et al. New genes involved in Angelman syndrome-like: expanding the genetic spectrum. PLoS ONE. 2021;16(10):e0258766. doi: 10.1371/journal.pone.0258766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook EH, Masaki JT, Guter SJ, Najjar F. Lovastatin treatment of a patient with a De Novo SYNGAP1 protein truncating variant. J Child Adolesc Psychopharmacol. 2019;29(4):321-322. doi: 10.1089/cap.2018.0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vlaskamp DRM, Shaw BJ, Burgess R, et al. SYNGAP1 encephalopathy: a distinctive generalized developmental and epileptic encephalopathy. Neurology. 2019;92(2):e96-e107. doi: 10.1212/WNL.0000000000006729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saldaris J, Weisenberg J, Pestana-Knight E, et al. Content validation of clinician-reported items for a severity measure for CDKL5 deficiency disorder. J Child Neurol. 2021;36(11):998-1006. doi: 10.1177/08830738211019576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522-530. doi: 10.1111/epi.13670 [DOI] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062-D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma V, Kumar MJV, Sharma K, et al. Pharmacological intervention in young adolescents rescues synaptic physiology and behavioural deficits in Syngap1+/− mice. Exp Brain Res. 2022;240(1):289-309. doi: 10.1007/s00221-021-06254-x [DOI] [PubMed] [Google Scholar]
- 23.Jimenez-Gomez A, Niu S, Andujar-Perez F, et al. Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J Neurodev Disord. 2019;11(1):18-18. doi: 10.1186/s11689-019-9276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaelson SD, Ozkan ED, Aceti M, et al. SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nat Neurosci. 2018;21(12):1-13. doi: 10.1038/s41593-018-0268-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchanan CB, Stallworth JL, Scott AE, et al. Behavioral profiles in Rett syndrome: data from the natural history study. Brain Dev. 2019;41(2):123-134. doi: 10.1016/j.braindev.2018.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cianfaglione R, Clarke A, Kerr M, Hastings RP, Oliver C, Felce D. Ageing in Rett syndrome. J Intellect Disabil Res. 2016;60(2):182-190. doi: 10.1111/jir.12228 [DOI] [PubMed] [Google Scholar]
- 27.Berkvens JJL, Veugen I, Veendrick-Meekes MJBM, et al. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. 2015;47:11-16. doi: 10.1016/j.yebeh.2015.04.057 [DOI] [PubMed] [Google Scholar]
- 28.Boets S, Johannesen KM, Destree A, et al. Adult phenotype of KCNQ2 encephalopathy. J Med Genet. 2022;59(6):528-535. doi: 10.1136/jmedgenet-2020-107449 [DOI] [PubMed] [Google Scholar]
- 29.Perucca P, Mula M. Antiepileptic drug effects on mood and behavior: molecular targets. Epilepsy Behav. 2013;26(3):440-449. doi: 10.1016/j.yebeh.2012.09.018 [DOI] [PubMed] [Google Scholar]
- 30.Lo Barco T, De Gaetano L, Santangelo E, et al. SYNGAP1-related developmental and epileptic encephalopathy: the impact on daily life. Epilepsy Behav. 2022;127:108500. doi: 10.1016/j.yebeh.2021.108500 [DOI] [PubMed] [Google Scholar]
- 31.Gallop K, Lloyd AJ, Olt J, Marshall J. Impact of developmental and epileptic encephalopathies on caregivers: a literature review. Epilepsy Behav. 2021;124:108324. doi: 10.1016/j.yebeh.2021.108324 [DOI] [PubMed] [Google Scholar]
- 32.Bishop KI, Isquith PK, Gioia GA, et al. Improved everyday executive functioning following profound reduction in seizure frequency with fenfluramine: analysis from a phase 3 long-term extension study in children/young adults with Dravet syndrome. Epilepsy Behav. 2021;121(Pt A):108024. doi: 10.1016/j.yebeh.2021.108024 [DOI] [PubMed] [Google Scholar]
- 33.Catarino CB, Liu JYW, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain J Neurol. 2011;134(Pt 10):2982-3010. doi: 10.1093/brain/awr129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith-Hicks C, Wright D, Kenny A, et al. Sleep abnormalities in the synaptopathies—SYNGAP1-related intellectual disability and Phelan–McDermid syndrome. Brain Sci. 2021;11(9):1229-1229. doi: 10.3390/brainsci11091229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Creson TK, Rojas C, Hwaun E, et al. Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. eLife. 2019;8:e46752. doi: 10.7554/eLife.46752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selvarajah A, Gorodetsky C, Marques P, et al. Progressive worsening of gait and motor abnormalities in older adults with Dravet syndrome. Neurology. 2022;98(22):e2204-e2210. doi: 10.1212/WNL.0000000000200341 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data may be provided on reasonable request.