

Abstract
Aerobic organisms contain antioxidant enzymes, such as superoxide dismutase (SOD) and catalase, to protect them from both direct and indirect effects of reactive oxygen species, such as O2·− and H2O2. Previous work by others has shown that Escherichia coli mutants lacking SOD not only are more susceptible to DNA damage and killing by H2O2 but also contain larger pools of intracellular free iron. The present study investigated if SOD-deficient E. coli cells are exposed to increased levels of hydroxyl radical (·OH) as a consequence of the reaction of H2O2 with this increased iron pool. When the parental E. coli strain AB1157 was exposed to H2O2 in the presence of an α-(4-pyridyl-1-oxide)-N-tert-butyl-nitrone (4-POBN)–ethanol spin-trapping system, the 4-POBN–·CH(CH3)OH spin adduct was detectable by electron paramagnetic resonance (EPR) spectroscopy, indicating ·OH production. When the isogenic E. coli mutant JI132, lacking both Fe- and Mn-containing SODs, was exposed to H2O2 in a similar manner, the magnitude of ·OH spin trapped was significantly greater than with the control strain. Preincubation of the bacteria with the iron chelator deferoxamine markedly inhibited the magnitude of ·OH spin trapped. Exogenous SOD failed to inhibit ·OH formation, indicating the need for intracellular SOD. Redox-active iron, defined as EPR-detectable ascorbyl radical, was greater in the SOD-deficient strain than in the control strain. These studies (i) extend recent data from others demonstrating increased levels of iron in E. coli SOD mutants and (ii) support the hypothesis that a resulting increase in ·OH formation generated by Fenton chemistry is responsible for the observed enhancement of DNA damage and the increased susceptibility to H2O2-mediated killing seen in these mutants lacking SOD.
Although the aerobic metabolism of bacteria optimally results in the near simultaneous four-electron reduction of O2 to H2O, a variable percentage of O2 reduction occurs initially via either one-electron reduction of O2 to superoxide (O2·−) or divalent reduction to H2O2 (7). At physiological pH, O2·− rapidly reacts with itself (dismutes) to form H2O2 (7). Pathogenic microorganisms are also exposed to exogenous O2·− and H2O2 generated by host neutrophils and other phagocytes (17).
O2·− and H2O2, in the presence of free iron, can form the hydroxyl radical (·OH), a highly reactive molecule that will react at diffusion-limited rates with various biomolecules, including lipids, proteins, and DNA (17).
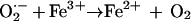 |
1 |
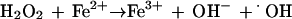 |
2 |
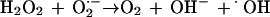 |
3 |
The reaction of H2O2 with reduced iron is the well-known Fenton reaction (reaction 2). In this scheme, O2·− enhances ·OH formation both by reducing Fe3+ to Fe2+ and by serving as a source of H2O2. Most bacteria, including Escherichia coli, contain superoxide dismutase (SOD) and catalase as means of eliminating O2·− and H2O2, respectively (16, 17). SOD catalyzes the dismutation of O2·− to H2O2, thus preventing the first reaction above.
Earlier studies established that exposure of E. coli to increasing H2O2 concentrations results in a bimodal dose-response curve (8). Low-dose (1 to 3 mM H2O2) killing is greatly enhanced in strains deficient in DNA repair systems. This led to the hypothesis that the low-dose component was due to iron-dependent ·OH formation on or near the DNA, presumably as a result of Fenton chemistry as described above. Additional studies, however, demonstrated that E. coli mutant strains lacking SOD activity are more susceptible to H2O2-mediated killing (9) than are wild-type strains. While this implies a role for O2·− in H2O2-dependent ·OH formation in vivo, the exact nature of that role has been unclear.
One hypothesis to explain the increased sensitivity of mutants lacking SOD to H2O2-mediated killing is that the absence of SOD results in increased levels of O2·−, required for the maintenance of iron in the ferrous form (reaction 1 above) (5, 9). However, most iron inside a cell is either bound to an enzyme or sequestered by an iron storage protein. Also, there are other cellular reductants more plentiful than O2·−, such as glutathione or NADH, which are capable of reducing free iron.
Liochev and Fridovich (15) have proposed that an increased flux of O2·− could lead to an increase in free iron by oxidatively attacking the [Fe-S]x clusters of dehydratases. Recently, Keyer et al. (11, 12) have presented evidence verifying that the increased fluxes of O2·− in an SOD-deficient E. coli strain lead to increased levels of free intracellular iron and that this is the result of O2·−-mediated release of iron from [Fe-S]x proteins such as aconitase. These studies reconcile the ambiguity mentioned earlier regarding the role of O2·− in this process.
Several studies have concluded that exposure to O2·− inactivates a number of enzymes in E. coli which contain Fe-S clusters through oxidation of these sites (6, 13–15).
Based on these findings, Keyer and Imlay (12) proposed but did not demonstrate that the increased susceptibility of SOD-deficient E. coli to H2O2-mediated killing resulted from increased production of ·OH due to the presence of catalytic iron. In the present study, we tested this proposal with electron paramagnetic resonance (EPR)-based techniques for detection of both ·OH generation and the presence of redox-active iron. We have subsequently confirmed both enhanced ·OH generation upon exposure to H2O2 and the presence of higher levels of redox-active iron in these cells.
(Part of this work was presented in abstract form at the 1997 meeting of the American Federation for Medical Research (Biomedicine 97), Washington, D.C., May 1997.)
MATERIALS AND METHODS
Bacterial strains.
This work utilized a well-characterized E. coli strain, JI132, lacking both FeSOD and MnSOD (SOD−) and previously constructed by transduction of E. coli AB1157 (SOD+) (9). We also employed the catalase-deficient mutant UM255 (CAT−), which was constructed by transduction of strain KL 16-99 (CAT+). Both strains were provided to us by Michael Gunther, National Institute of Environmental Health Sciences. Native-activity gels confirmed the continued absence of SOD and catalase from strains JI132 and UM255, respectively.
Bacterial growth.
Bacteria were streaked on agar plates and grown overnight at 37°C. Colonies were isolated, placed in media (1% [wt/vol] tryptone [Difco]–0.5% [wt/vol] yeast extract [Difco]–170 mM NaCl adjusted to pH 7.0 with NaOH), and grown for 4 to 6 h at 37°C. The bacteria were washed two times with cold chelated Hanks balanced salt solution (HBSS), and cell numbers were determined from the optical density at 600 nm, which had been previously correlated with known E. coli concentrations as determined by quantification of CFU. The bacteria were resuspended at a density of 2.5 × 1010/ml in chelated HBSS.
Spin trapping.
Spin-trapping experiments to detect formation of ·OH utilized a spin-trapping system containing 10 mM α-(4-pyridyl-1-oxide)-N-tert-butyl-nitrone (4-POBN; Aldrich, Milwaukee, Wis.) and 170 mM ethanol. In this spin-trapping system, ·OH reacts with ethanol, abstracting a hydrogen atom and yielding the α-hydroxyethyl radical [·CH(CH3)OH], which forms a stable spin adduct with 4-POBN (aN = 15.5 G, aH = 2.6 G) (18). This spin trap does not directly yield stable spin adducts with either O2·− or ·OH. As the α-hydroxyethyl radical cannot be formed by O2·−, the presence of the 4-POBN–·CH(CH3)OH spin adduct is direct evidence for the presence of ·OH. Background EPR signals were determined for each batch of spin trap and were at the noise level for the instrument settings used in these studies.
To minimize the effects of adventitious metals, all buffers were Chelex-treated and all samples contained a 100 μM concentration of the iron chelator diethylenetriaminepentaacetic acid (DTPA). In some experiments, bacteria were exposed to 2 mM deferoxamine mesylate (DFO) for 50 min at room temperature before H2O2 treatment. In all EPR studies, bacteria were added to a final concentration of 2.5 × 109/ml. All spin-trapping spectra are the result of seven signal-averaged scans and were obtained at room temperature with a model ESP 300 spectrometer (Bruker Instruments, Karlsruhe, Germany). The magnitude of the EPR signal observed is directly proportional to the amount of spin adduct in the sample. Instrument settings were as follows: microwave power, 20.0 mW; modulation frequency, 100 kHz; modulation amplitude, 0.941 G; and receiver gain, 8 × 104. The sweep rate for each scan was 80 G/84 s.
Ascorbate assay for reactive iron.
Bacteria (2.5 × 109/ml) were added to a system containing a 100 μM concentration of the metal chelator EDTA (Fisher) (recrystalized four times to remove adventitious metals) in 50 mM phosphate buffer at pH 7.0; 5 mM N-ethylmaleimide (NEM) was added to the bacteria 5 min before ascorbate exposure to inhibit ascorbate reductase activity (19). Upon addition of ascorbate (100 μM) to the samples, they were quickly transferred to a flat cell and placed in the cavity of the EPR spectrometer. The resulting spectra represent direct detection of the ascorbate free radical. The EPR signal intensity of the ascorbate radical can be related directly to the concentration of free iron in the sample; however, this free iron must first be converted to a standard catalytic form (e.g., chelated with EDTA) (2, 4). Ascorbate and NEM stock solutions were made fresh daily. Instrument settings for the detection of the ascorbate free radical were as follows: microwave power, 40 mW; modulation frequency, 100 kHz; modulation amplitude, 0.594 G; receiver gain, 2.5 × 105. The sweep rate for each scan was 10 G/42 s.
RESULTS
Previously we have demonstrated that exposure of various E. coli strains to O2·−/H2O2 leads to the formation of ·OH, as detected with a 4-POBN–ethanol spin-trapping system (1). Bacterium-associated iron appears to serve as the catalyst for ·OH production. Consistent with these earlier findings, exposure of an E. coli strain (AB1157) containing both MnSOD and FeSOD to 100 μM H2O2 in the presence of 4-POBN and ethanol resulted in the formation of the 4-POBN–·CH(CH3)OH spin adduct (aN = 15.5 G, aH = 2.6 G). This is indicative of ·OH formation (Fig. 1B). When 100 μM H2O2 was added to the 4-POBN–ethanol spin-trapping system in the absence of bacteria, the resulting EPR spectrum lacked any evidence of the formation of stable spin adducts (Fig. 1A). This indicates that the bacterium is required for the generation of ·OH. When strain AB1157 was preincubated with DFO (2 mM for 1 h) there was a marked reduction in the magnitude of ·OH spin trapped (Fig. 2A and B), indicating that ·OH production occurred due to the interaction of H2O2 with bacterium-associated redox-active iron.
FIG. 1.

4-POBN–ethanol (EtOH) spin trapping of ·OH in wild-type and SOD-deficient E. coli. The EPR spectrum was obtained 10 min after addition of 100 μM H2O2 to Chelex-treated HBSS containing 100 μM DTPA, 10 mM 4-POBN, and 170 mM ethanol (A), in the presence of wild-type E. coli (2.5 × 109/ml) (B), and in the presence of SOD-deficient E. coli (2.5 × 109/ml) (C).
FIG. 2.

Inhibition by DFO of 4-POBN–ethanol spin trapping of ·OH in wild-type and SOD-deficient E. coli. The EPR spectrum was obtained 10 min after addition of 100 μM H2O2 to Chelex-treated HBSS containing 100 μM DTPA, 10 mM 4-POBN, 170 mM ethanol, and wild-type E. coli (2.5 × 109/ml) (A) pretreated for 50 min with 2 mM DFO (B) or containing SOD-deficient E. coli (2.5 × 109/ml) (C) pretreated for 50 min with 2 mM DFO (D). DSFL, DFO.
When the isogenic E. coli mutant (JI132) lacking both FeSOD and MnSOD (SOD−) was exposed to H2O2 in a similar manner, the resulting 4-POBN–·CH(CH3)OH signal was fivefold greater than that seen with strain AB1157 (Fig. 1C). DFO treatment also led to a large decrease in the 4-POBN–·CH(CH3)OH signal seen with the JI132 strain (Fig. 2C and D), consistent with the hypothesis that the absence of SOD activity enhances formation of ·OH upon exposure of these organisms to H2O2. Furthermore, the addition of exogenous SOD immediately before H2O2 exposure failed to inhibit ·OH formation, consistent with the need for SOD to be present intracellularly during bacterial growth in order to produce its protective effect (data not shown).
To provide additional evidence that the observed increase in ·OH formation was due to the SOD-dependent control of intracellular iron and not to unexpected alterations in bacterial H2O2 metabolism, we performed the same H2O2 and DFO treatments with a control (KL 16-99) and a matching isogenic bacterium lacking catalase activity (UM255). When these bacteria were examined with the 4-POBN–ethanol spin-trapping system, we observed that the catalase-replete and catalase-deficient strains showed no difference in ·OH formation upon H2O2 exposure (Fig. 3A and C). Pretreatment of these bacteria with DFO also reduced the 4-POBN–·CH(CH3)OH signal to near background levels (Fig. 3C and D). These data suggest a role for noncatalase pathways in the removal of exogenous H2O2 in E. coli.
FIG. 3.

Inhibition by DFO of 4-POBN–ethanol spin trapping of ·OH in wild-type and catalase-deficient E. coli. The EPR spectrum was obtained 10 min after addition of 100 μM H2O2 to Chelex-treated HBSS containing 100 μM DTPA, 10 mM 4-POBN, 170 mM ethanol, and wild-type E. coli (2.5 × 109/ml) (A) pretreated for 50 min with 2 mM DFO (B) or containing catalase-deficient E. coli (2.5 × 109/ml) (C) pretreated for 50 min with 2 mM DFO (D). DSFL, DFO.
We next sought to quantify the redox-active iron in both the SOD+ and SOD− bacteria by determining the relative ability of each to form the EPR-detectable ascorbyl radical (Asc·−) (Fig. 4A). It is important to note that when ascorbate is dissolved in aqueous buffer, a small background Asc·− signal forms spontaneously, which serves as an excellent internal control. The intensity of this background signal is pH-dependent, so these experiments were carried out at pH 7.0, minimizing spontaneous Asc·− formation yet representing a pH appropriate for biological studies (3). When added to bacterial suspensions, ascorbate reacts with bacterium-associated Fe3+ to form Fe2+ and Asc·−, which is manifested as an increase in the background Asc·− signal. Asc·− formation in the presence of the SOD+ bacteria was similar to the signal in the absence of bacteria (Fig. 4B). In contrast, a stronger signal was seen with the SOD− bacteria (Fig. 4C). When the pure ascorbate (i.e., no cells) background signal is subtracted from those of both the SOD+ and SOD− bacteria, the resulting signal for the SOD+ bacteria is essentially at the level of noise (Fig. 4D). The signal for the SOD− bacteria is above background (Fig. 4E), indicating the presence of detectable redox-active iron with this strain. These data are consistent with the increase in total DFO-chelatable iron seen with the same SOD− and SOD+ strains of E. coli in the previous work of Keyer and Imlay (12). Since the Asc·− signal with the control (SOD+) strain was not above background, we are unable to calculate the magnitude of the increase in redox-active iron over control in the SOD− strain.
FIG. 4.

EPR visualization of ascorbate free radical. The EPR spectrum was obtained immediately after addition of ascorbate (200 μM) to a solution containing 200 μM EDTA and 5 mM NEM (A) in the presence of wild-type E. coli (2.5 × 109/ml) (B) or in the presence of SOD-deficient E. coli (2.5 × 109/ml) (C). (D and E) Results of subtraction of spectrum A from spectrum B and from spectrum C, respectively. bkg, background.
DISCUSSION
When E. coli organisms are exposed to H2O2, a bimodal dose-response curve is observed: mode one killing, seen at low H2O2 concentrations (1 to 3 mM), is caused by direct DNA damage; mode two killing, seen at higher H2O2 concentrations (>20 mM), is not well defined (8). Mutant bacteria deficient in either recombinational or excision repair pathways are extremely sensitive to mode one killing (9). In addition, mutant bacteria lacking SOD also show increased sensitivity to mode one killing, which implies a role for O2·− in this pathway (9).
Previous studies have proposed that the DNA damage seen following low-dose H2O2 exposure (1 to 3 mM) is a consequence of Fenton chemistry (reaction 2 above) occurring on or near DNA, generating a highly reactive species such as ·OH, which is then the effector of DNA damage (5, 9). The increased sensitivity seen in the SOD mutants was initially thought to be due to an O2·−-dependent enhancement of iron reduction, leading to increased ·OH formation. However, recent work by Keyer and Imlay (12) has shown that the SOD-deficient mutants have greatly increased levels of free iron, most likely due to the release of iron from O2·−-sensitive [Fe-S]x proteins such as aconitase. In bacteria devoid of SOD activity (i.e., strain JI132), and thus presumably under the influence of a higher steady-state level of O2·−, intracellular levels of free iron were sevenfold higher than that observed in the parental strain (AB1157) (12).
Based on these findings, Keyer and Imlay hypothesized that excess O2·− in these mutants enhances sensitivity to H2O2 by increasing the pool of free iron, resulting in enhanced production of DNA-damaging ·OH. In the present study, we have provided direct evidence in support of this hypothesis. We demonstrated a significant enhancement of spin trap-detectable ·OH formation upon exposure of the SOD-deficient E. coli strain JI132 to 100 μM H2O2 compared to that seen with the parental strain, AB1157. Pretreatment of the JI132 (SOD-deficient) bacteria with DFO greatly reduced the magnitude of ·OH generation, confirming that it arose as a consequence of Fenton chemistry, as iron bound to DFO is no longer available for this redox chemistry (10). At the levels used, and in the time frame of the present study, DFO does not remove tightly bound iron from proteins (12). Thus, these data are consistent with the bacteria that lack SOD containing a larger pool of redox-active free iron than wild-type bacteria.
Using an assay based on the oxidation of ascorbate to the ascorbate free radical, we found the concentration of ascorbate-reactive iron (i.e., Fe3+) to be below the limit of detection in the SOD+ samples. In normal bacteria, iron availability is tightly regulated. The demonstration of ascorbate-reactive iron in the bacteria lacking SOD can be interpreted as indicating an increase in the steady-state levels of catalytic iron. This catalytic iron would lead to increased ·OH formation, as observed in our spin-trapping experiments.
When the same H2O2 exposures were examined with an E. coli strain lacking catalase, no differences was observed between the parental and the mutant strains. While it is somewhat surprising that no differences were seen, there are some possible explanations. First, an absence of catalase does not necessarily equate with the inability to metabolize and/or remove H2O2. The bacteria could contain multiple enzymatic systems that facilitate removal of H2O2, or the levels of H2O2 added may have been too low for efficient removal by catalase. In any case, the enhanced ·OH formation seen in the SOD-deficient (but catalase-proficient) strain following H2O2 exposure implies that in the presence of significant levels of redox-active iron, catalase alone is not able to compensate and protect the cell. It follows that iron, not [H2O2], is potentially rate limiting in ·OH formation.
In summary, these studies extend recent data demonstrating increased levels of iron in E. coli strains lacking SOD. They further support the hypothesis that a resulting increase in ·OH formation is responsible for the enhanced DNA damage seen in these organisms following H2O2 exposure. Thus, SOD plays a critical role in bacterial resistance to H2O2-mediated damage by limiting release of iron from O2·−-susceptible bacterial enzymes (e.g., aconitase), which would in turn enhance ·OH production.
ACKNOWLEDGMENTS
This work was supported by a merit review grant from the Department of Veterans Affairs (B.E.B.), Public Health Services grants AI28412 (B.E.B.) and CAII081 (G.R.B.), and an American Heart Association Established Investigator award (B.E.B.).
We thank Michael Gunther, NIEHS, for providing the bacterial strains used in the present study and Sherry Flanagan for help in preparing the manuscript.
REFERENCES
- 1.Britigan B E, Ratcliffe H R, Buettner G R, Rosen G M. Binding of myeloperoxidase to bacteria: effect on hydroxyl radical formation and susceptibility to oxidant-mediated killing. Biochim Biophys Acta. 1996;1290:231–240. doi: 10.1016/0304-4165(96)00014-1. [DOI] [PubMed] [Google Scholar]
- 2.Buettner G R. Ascorbate oxidation: UV absorbance of ascorbate and ESR spectroscopy of the ascorbyl radical as assays for iron. Free Radical Res Commun. 1990;10:5–9. doi: 10.3109/10715769009145927. [DOI] [PubMed] [Google Scholar]
- 3.Buettner G R, Jurkiewicz B A. Ascorbate free radical as a marker of oxidative stress: an EPR study. Free Radical Biol Med. 1993;14:49–55. doi: 10.1016/0891-5849(93)90508-r. [DOI] [PubMed] [Google Scholar]
- 4.Buettner G R, Jurkiewicz B A. Catalytic metals, ascorbate, and free radicals: combinations to avoid. Radical Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 5.Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flint D H, Tuminello J F, Emptage M H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 7.Halliwell B, Gutteridge J M C. Free radicals in biology and medicine. Oxford, England: Clarendon Press; 1985. pp. 346–378. [Google Scholar]
- 8.Imlay J A, Linn S. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J Bacteriol. 1986;166:519–527. doi: 10.1128/jb.166.2.519-527.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imlay J A, Linn S. Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J Bacteriol. 1987;169:2967–2976. doi: 10.1128/jb.169.7.2967-2976.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imlay J A, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 11.Keyer K, Gort A S, Imlay J A. Superoxide and the production of oxidative DNA damage. J Bacteriol. 1995;177:6782–6790. doi: 10.1128/jb.177.23.6782-6790.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keyer K, Imlay J A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo C F, Mashino T, Fridovich I. α,β-Dihydroxyisovalerate dehydratase. J Biol Chem. 1987;262:4724–4727. [PubMed] [Google Scholar]
- 14.Liochev S I, Fridovich I. Modulation of the fumarases of Escherichia coli in response to oxidative stress. Arch Biochem Biophys. 1993;301:379–384. doi: 10.1006/abbi.1993.1159. [DOI] [PubMed] [Google Scholar]
- 15.Liochev S I, Fridovich I. The role of O2·− in the production of HO·: in vitro and in vivo. Free Radical Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 16.McCord J M, Keele B B, Jr, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci USA. 1971;68:499–506. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller R A, Britigan B E. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev. 1997;10:1–18. doi: 10.1128/cmr.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramos C L, Pou S, Britigan B E, Cohen M S, Rosen G M. Spin trapping evidence for myeloperoxidase-dependent hydroxyl radical formation by human neutrophils and monocytes. J Biol Chem. 1992;267:8307–8312. [PubMed] [Google Scholar]
- 19.Villalba J M, Canalejo A, Buron M I, Cordoba F, Navas P. Thiol groups are involved in NADH-ascorbate free radical reductase activity of rat liver plasma membrane. Biochem Biophys Res Commun. 1993;192:707–713. doi: 10.1006/bbrc.1993.1472. [DOI] [PubMed] [Google Scholar]