

Abstract
The rapid advancement of tumor immunotherapies poses challenges for the tools used in cancer immunology research, highlighting the need for highly effective biomarkers and reproducible experimental models. Current immunotherapy biomarkers encompass surface protein markers such as PD‐L1, genetic features such as microsatellite instability, tumor‐infiltrating lymphocytes, and biomarkers in liquid biopsy such as circulating tumor DNAs. Experimental models, ranging from 3D in vitro cultures (spheroids, submerged models, air–liquid interface models, organ‐on‐a‐chips) to advanced 3D bioprinting techniques, have emerged as valuable platforms for cancer immunology investigations and immunotherapy biomarker research. By preserving native immune components or coculturing with exogenous immune cells, these models replicate the tumor microenvironment in vitro. Animal models like syngeneic models, genetically engineered models, and patient‐derived xenografts provide opportunities to study in vivo tumor‐immune interactions. Humanized animal models further enable the simulation of the human‐specific tumor microenvironment. Here, we provide a comprehensive overview of the advantages, limitations, and prospects of different biomarkers and experimental models, specifically focusing on the role of biomarkers in predicting immunotherapy outcomes and the ability of experimental models to replicate the tumor microenvironment. By integrating cutting‐edge biomarkers and experimental models, this review serves as a valuable resource for accessing the forefront of cancer immunology investigation.
Keywords: biomarker, cancer immunotherapy, experimental model, lab‐on‐a‐chip devices, three‐dimensional model
The rapid progress in tumor immunotherapies poses challenges for cancer immunology research tools, particularly in terms of the need for high‐efficacy biomarkers and the limited reproducibility of experimental models. Biomarkers currently used in research can be categorized into surface protein markers, genetic features, tumor‐infiltrating lymphocytes, and liquid biopsy biomarkers. Experimental models, such as 3D in vitro cultures and animal models, have emerged as valuable platforms for biomarker research and cancer immunology investigations. 3D in vitro cultures, including spheroid models, submerged models, air–liquid interface models, and organ‐on‐a‐chip systems, offer the ability to replicate tumor‐immune interactions by preserving native immune components or through coculturing with immune cells. Animal models, including syngeneic models, genetically engineered models, and patient‐derived xenografts, offer opportunities to study tumor‐immune interactions in an in vivo setting. Furthermore, humanized animal models provide a means to simulate the human‐specific tumor microenvironment, enabling a more accurate representation of the complex immune responses observed in patients. In summary, the development of high‐efficacy biomarkers and the utilization of advanced experimental models, such as 3D in vitro cultures and animal models, offer promising avenues for studying tumor immunology and advancing cancer immunotherapies.
1. INTRODUCTION
Significant progress in cancer immunotherapy, including immune checkpoint blockers (ICBs), 1 tumor vaccines, 2 and adoptive cell therapies (ACT), 3 has led to an approximate 10% increase in the 5‐year overall survival (OS) rate across diverse cancer types, such as lymphoma, melanoma, and non‐small cell lung cancer (NSCLC). 4 , 5 , 6 Immunotherapy aims to manipulate the immune system to recognize and eliminate cancer cells. 1 Rapid development of tumor immunotherapies further pose challenges for the tools utilized in cancer immunology investigations, particularly concerning the availability of highly effective biomarkers. 2 Currently, guidelines recommend predictive markers are PD‐L1 expression in tumor cells and immune cells 7 and microsatellite instability (MSI). 8 However, challenges arise due to insufficient evidence for response prediction based on PD‐L1 expression in early‐stage tumors 9 and the limited application of MSI. 10 Other potential prediction biomarkers including tumor‐infiltrating lymphocytes (TILs) 11 and tumor mutation burden (TMB) 12 are constantly emerging. The emergence of experimental models that faithfully replicate the in vivo antitumor immune response has become crucial in biomarker research and cancer immunology investigations. 13 , 14
For the discovery of reliable immunotherapy biomarkers, 3D in vitro models that can reproduce the tumor microenvironment (TME) and tumor‐immune interactions are necessary. 15 TME, consisting of immune cells, stroma cells, blood, and lymphatic vessels embedded in a noncellular extracellular matrix (ECM), plays a crucial role in tumor progression, therapeutic response, and patient outcomes. 16 Though current tumor models such as two‐dimensional (2D) culture, 17 three‐dimensional (3D) culture, 18 and patient‐derived xenograft (PDX) 19 display high reproducibility in terms of tumor cell properties, they have limited capacity to mimic personalized TME. To simulate in vivo tumor‐immune interactions, 2D cultures can be cocultured with various exogenously added heterogeneous cells. 20 , 21 , 22 However, these reconstituted cells often do not originate from the native tumor, and the flat monolayer configuration in 2D cultures fails to replicate the complex 3D morphological structures. Compared with 3D cultures, the biology of oncogenes and tumor suppressors in 2D cultures may be less faithful to their in vivo counterparts. 21 , 23
Since the first in vitro 3D culture of human normal tissue was developed in 1975, 24 the 3D tumor culture has achieved significant development (Figure 1A). Spheroids, defined by tumor multicellular spherical colonies suspended in three dimensions, were described in 1992 on breast cancer, creating the first 3D in vitro tumor model. 25 In 2011, the first cancer organoid, characterized by 3D tumor cultures retaining histological and genetic features of the primary tumor, was established on colorectal cancer (CRC). 26 The first cancer‐on‐a‐chip, involving 3D tumor models based on microfluidic systems, was successfully established in 2012. 27 Over the past decade, there has been a growing interest in in vitro 3D tumor‐immune coculture systems. 28 Noteworthy, verifications of tumor‐immune interactions encompass both aspects of tumor cells and immune cells (Figure 1B). Additionally, animal models including humanized mouse models provide a valuable platform for evaluating immunotherapies and investigating in vivo TME. 29 Early in the 1970s, syngeneic mouse model, which involves injecting murine‐derived tumor cell lines into immunocompetent mice, has been constructed for melanoma. 30 Genetically engineered mouse models (GEMMs), introduced in 1974, enable spontaneous tumor formation in genetically engineered mice. 31 In 1984, PDXs emerged as the first animal models that directly preserve patient‐derived tumor cells. 32 In the 21st century, humanized mouse models allow the reconstruction of the human immune system in immunodeficient mice. 33
FIGURE 1.
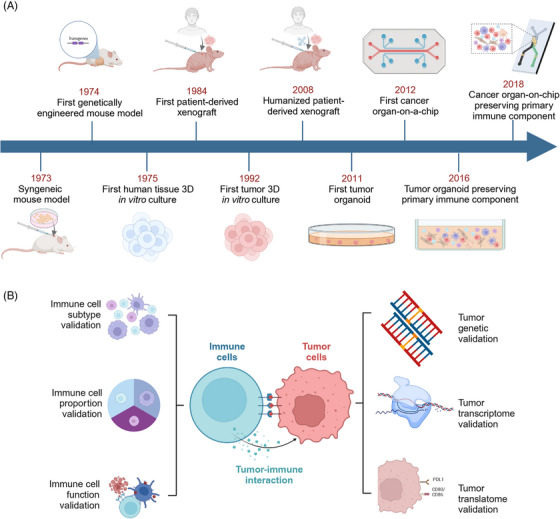
Development and validation: Experimental models for cancer immunology investigations. (A) Early in the 1970s, in vitro models such as spheroid and animal models such as syngeneic models have been utilized in cancer immunology investigations. In the recent decade, experimental models preserving original tumor components emerged, facilitating the research on tumor microenvironment and tumor immunotherapy. (B) Validation of a 3D in vitro tumor‐immune coculture system can be completed through two aspects: tumor components and immune components. Validations of tumor components include three different levels: genetic (DNA mutation), transcriptomic (RNA profiling), and translatomic (surface protein expression). Validations on immune components are composed of immune cell subtype, immune cell proportion, and immune cell function. Figure was created with BioRender.com.
Biomarkers and experimental models are interconnected and complementary in cancer immunology investigation. 13 In this review, we first provide a comprehensive summary of a range of classical and emerging biomarkers. We further clarify their relationship with the underlying tumor immunology mechanisms and various immunotherapies. Subsequently, we classify experimental models into two categories: in vivo and in vitro, and discuss the architecture, features, and applications of each model in the context of tumor immunology and immunotherapy. Special attention is given to exploring different approaches for TME reconstruction. Furthermore, we summarize the role of biomarkers and experimental models in cancer immunology investigation and outline future directions.
2. BIOMARKERS FOR TUMOR IMMUNOLOGY INVESTIGATION
Based on their mechanisms, potential biomarkers associated with tumor immunology and immunotherapy can be categorized based on their mechanisms: (1) genetic markers, (2) surface protein markers, (3) TILs, and (4) markers in liquid biopsy (Figure 2 and Table 1).
FIGURE 2.
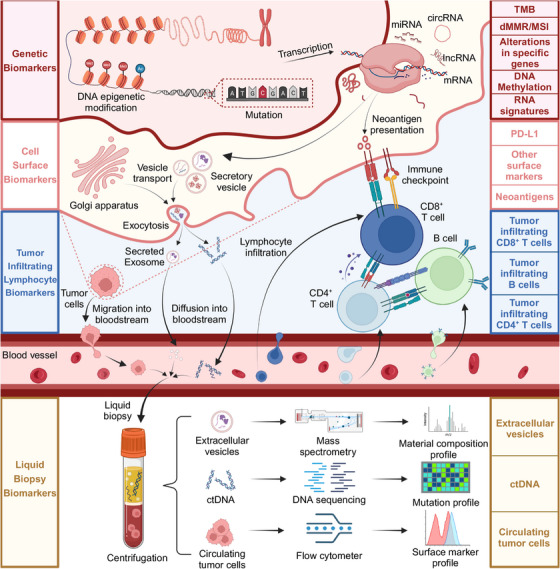
Biomarkers used in cancer immunology investigations. Various potential biomarkers associated with tumor immunology and immunotherapy have been categorized based on their mechanisms: (1) Genetic markers, including tumor mutation burden, mismatch repair system deficiency, and high microsatellite instability. Next‐generation sequencing technologies can be employed to detect these genetic markers. (2) Surface markers, including PD‐L1, some other inhibitory receptors, and tumor neoantigens. Immunohistochemistry of tumor tissues can be utilized to examine the expression of these surface markers. (3) Cytological markers, including tumor‐infiltrating lymphocytes such as tumor‐infiltrating lymphocytes and exhaustion cells, can be examined by RNA sequencing and immune‐related gene panel scoring. (4) Liquid biopsy markers, including circulating tumor DNA which exists in peripheral blood and can be accessible by blood sampling and analyses. Figure was created with BioRender.com.
TABLE 1.
Different biomarkers utilized in cancer immunology investigations.
| Category | Biomarkers | Measurement | Cancer types | Immunotherapies |
|---|---|---|---|---|
| Surface proteins | PD‐L1 | IHC | NSCLC, 34 RCC, 35 Melanoma, 35 etc. | Anti‐PD‐1 35 |
| LAG‐3 | Flow cytometer, IHC | NSCLC 36 | Anti‐PD‐1 36 | |
| Tumor neoantigen | Epitope discovery, NGS | Melanoma, 37 Sarcoma 38 | Anti‐PD‐1, 38 anti‐CTLA‐4, 37 ACT 39 | |
| Genetic features | TMB | NGS | NSCLC, 7 UC, 40 HNSCC, 41 etc. | Anti‐PD‐1, 7 anti‐CTLA‐4 42 |
| dMMR/MSI‐H | NGS | CRC, 43 NSCLC, 44 GC, 45 etc. | Anti‐PD‐1, 43 anti‐CTLA‐4 46 | |
| DNA methylation | Sulfite sequencing | CRC, 47 EC, 48 Melanoma, 49 etc. | Anti‐PD‐1, 50 anti‐CTLA‐4 49 | |
| MHC–TCR axis mutation | Targeted sequencing | NSCLC, 51 Melanoma 52 | Anti‐PD‐1, 52 anti‐CTLA‐4 52 | |
| RNA signatures | RNA sequencing | BC, 53 NSCLC, 54 CRC, 55 etc. | Anti‐PD‐1, 56 CAR‐T 55 | |
| TILs | CD8+ T cells | Flow cytometer | NSCLC, 57 RCC, 58 UC, 59 etc. | Anti‐PD‐1 59 |
| CD4+ T cells | Flow cytometer | Melanoma, 60 BC, 61 GC, 62 etc. | Anti‐PD‐1, 63 anti‐CTLA‐4 60 | |
| Exhausted T cells | Flow cytometer | NSCLC, 64 Melanoma 65 | Anti‐PD‐1 64 | |
| B cells | Flow cytometer | Melanoma, 66 RCC, 67 BC 68 | Anti‐PD‐1 66 | |
| Liquid biopsy | ctDNA | NGS, sulfite sequencing | NSCLC, 69 UC, 69 GC, 69 etc. | Anti‐PD‐1, 69 anti‐CTLA‐4 69 |
| CTC | Flow cytometer | NSCLC, 70 OC, 71 Melanoma 72 | Anti‐PD‐1, 70 anti‐CTLA‐4 72 | |
| tdEV | MS, NGS | NSCLC, 73 Melanoma 74 | Anti‐PD‐1 74 |
Abbreviations: ACT, adoptive cell therapy; BC, breast cancer; CAR‐T, chimeric antigen receptor T cell immunotherapy; CRC, colorectal cancer; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; dMMR, mismatch repair system deficiency; EC, esophagus carcinoma; GC, gastric cancer; HNSCC, head and neck squamous cell carcinoma; IHC, immunohistochemistry; MHC, major histocompatibility complex; MS, mass spectrometry.; MSI‐H, high microsatellite instability; NGS, next‐generation sequencing; NSCLC, non‐small cell lung cancer; RCC, renal cell carcinoma; tdEV, tumor‐derived extracellular vesicle; TMB, tumor mutation burden; UC, urothelial cancer.
2.1. Surface protein markers
Cancer progression is intricately linked to the immune evasion mechanisms involving immunosuppressive molecule expression. 75 , 76 Immune checkpoints, which are expressed on the immune cell surface, are crucial in preventing autoimmunity. 77 , 78 , 79 However, the excessive expression of immune checkpoints leads to immune function suppression. 75 , 76 Therefore, ICB therapy effectively hinders tumor growth by obstructing immune checkpoints and enhancing antitumor immune activity. 76 , 80 , 81 ICB development starting from PD‐1/PD‐L1 has brought revolutionary impacts on cancer therapy. 80 , 81 , 82 Nevertheless, only a proportion of patients exhibited disease remission, highlighting the need for individualized ICB utilizing biomarkers. 83 One approach is to focus on surface protein markers that can be directly detected using immunohistochemistry (IHC). 2
2.1.1. PD‐L1
PD‐L1 (B7‐H1) exhibits high expression across multiple tumor types and interacts with PD‐1, a crucial immunoregulatory protein found on various immune cell types, thereby facilitating immune evasion by tumors. 84 ICB employing PD‐1 antibodies specifically target PD‐1, mitigating the immunosuppressive control on T cells and enabling their engagement in tumor cell eradication. 85 PD‐L1 has been established as the primary biomarker for anti‐PD‐1 treatment, as evidenced by its inclusion in the prescribing information for pembrolizumab. 86 , 87 , 88
Notably, PD‐L1 expression can be influenced by several regulatory mechanisms involving transcriptional and translational levels, which may hinder its clinical applications. 89 , 90 , 91 , 92 For instance, the JAK–STAT–IRF1 axis severs as a key transcriptional regulator of interferon‐gamma (IFN‐γ) induced PD‐L1 expression, 93 while translational regulators include PI3K–AKT–mTOR pathway by oncogene activation. 94 Investigations have revealed mutations in PD‐L1 regulatory pathways that correlate with an unfavorable prognosis after ICB. 51 , 95 , 96 Research into acquired resistance to anti‐PD‐1 ICB uncovered genetic alterations within the interferon and antigen presentation pathways, which have now emerged as crucial biomarkers for predicting relapse following ICB. 97
It is yet to be investigated how PD‐L1 baseline expression affects tumor progress in the early stages of the disease. A recent study in 2022 demonstrated that all patients of untreated stage II or III triple‐negative breast cancer (TNBC) exhibited improved levels of pathological complete responses, regardless of PD‐L1 expression. 9 In contrast, in the KEYNOTE‐355 trial, pembrolizumab plus chemotherapy led to prolonged event‐free survival among metastatic TNBC patients with a high PD‐L1 expression level. 98 Similarly, atezolizumab therapy efficiency was only associated with high PD‐L1 level in late‐stage metastatic patients instead of early‐stage patients. 99 , 100 In conclusion, there is only insufficient evidence for the prediction efficiency of PD‐L1 expression in nonmetastatic tumors.
2.1.2. Other surface markers
Novel immunotherapy targets and immune biomarkers are of high interest. CTLA‐4 is a transmembrane protein expressed in activated CD4+ and CD8+ T cells, which suppresses effector T cells as an early IC in immune priming. 101 Biomarker studies of anti‐CTLA4 therapies focused on the diversity of peripheral blood lymphocytes (PBLs) rather than tumor cells. 102 In various tumor types, increased expression of the T‐cell costimulatory molecule (ICOS) on PBLs and TILs has been observed after CTLA4 blockade, suggesting that ICOS on immune cells may serve as a potential biomarker for anti‐CTLA‐4 therapy. 103
Other targets and surface markers include LAG3, TIM3, B7H3, NR2F6, TIGIT, VISTA, and BTLA. 2 , 104 High expression levels of TIM‐3 on TILs have been negatively correlated with OS. 105 In NSCLC, coexpression of PD‐1, LAG‐3, and TIM‐3 after anti‐PD‐1 treatment was significantly associated with significant T cell suppression and shorter OS. 36 In a study across several cancer types, multiplexed IHC demonstrated a higher prediction accuracy for PD‐1 blockage when compared with other biomarkers including PD‐L1 expression, TMB, and gene expression signatures. 106 Analyses for immunotherapy‐treated NSCLC cases revealed CD56 and CD4 expression in the CD45+ compartment to be an efficient biomarker for several clinical outcomes. 107 As for the tumor compartment, the CD44 expression in the tumor cells serves as a novel prognostic factor for extended PFS and OS under anti‐PD‐1 treatment. 108 Despite associations, these emerging surface markers still lack robustness for clinical use.
2.1.3. Tumor neoantigen
Neoantigens, as unique proteins expressed exclusively in tumor cells targeted by T cells in the immune system, have the potential to serve as ideal biomarkers for ICB including anti‐PD‐1 38 and anti‐CTLA‐4. 37 During tumor development, nonsynonymous mutations occur, leading to alterations in the amino acid coding sequence and the production of abnormal proteins specific to the tumor. These abnormal proteins can activate the immune system, triggering an immune response against the tumor. 109 The presence of neoantigens with high affinity for major histocompatibility complex (MHC) increases the likelihood of an effective immune response. 110 However, assessing the “quality” of neoantigens, which refers to their ability to induce immune recognition and activation, remains a challenge. 111 The current measurement approach, known as epitope discovery, can be achieved through two main approaches. The first method involves detecting mutations in the exons and subsequent candidate screening using MHC binding assays. 112 Alternatively, neoantigens can be directly obtained by TCR sequencing of tumor‐reactive T cells. 38 Currently, neoantigens are primarily used in conjunction with other biomarkers, such as TMB. The direct utilization of neoantigens as biomarkers requires the development of assays and algorithms capable of accurately detecting both the quantity and quality of neoantigens within a tumor. 113
2.1.4. Conclusions for cell surface markers
Surface markers, including PD‐L1 and other immune checkpoints as well as neoantigens, play a crucial role in cancer immunology investigations. 86 , 88 , 105 PD‐L1 serves as the only biomarker approved by clinical guidelines for ICB, but challenges remain due to its complex regulatory mechanisms and sufficient evidence in nonmetastatic cancer. 9 , 87 , 97 Besides PD‐L1, other surface markers including neoantigens have also shown potential as biomarkers for immunotherapy but require further validations. 36
2.2. Genetic features
The advancements in sequencing technologies, including polymerase chain reaction and next‐generation sequencing (NGS), have provided a crucial foundation for the research and application of genetic biomarkers. 114 , 115 , 116 Commonly used genetic markers include defective mismatch repair (dMMR)/MSI‐H (high microsatellite instability) and TMB, both associated with mutational loads. The abundance of mutations increases the likelihood of self‐neoantigens being immunogenic, leading to the activation of T cell responses. 117 Similar genetic markers include somatic copy number variation which has a larger‐scale impact on genome structure and has been reported to have prognosis predictive value in CRC. 118 , 119
2.2.1. Tumor mutation burden
TMB refers to the total number of mutations detected per megabase and serves as a prominent biomarker for ICB. 120 , 121 , 122 This assertion is based on the assumption that an elevated presence of mutant proteins will generate immunoreactive neoantigens, enhancing immunogenicity. 122 , 123 However, recognizable tumor neoantigens can occur even in a low mutation setting, and a high number of mutations does not guarantee the presence of immunogenic neo‐antigens. 124 Meanwhile, the complex microenvironment influences T cell‐mediated tumor killing and may compromise the inflammatory microenvironment . 125 Therefore, cancer immunology investigations concerning TMB must be considered along with multiple other factors including microenvironment features and specific mutation panels.
Accumulating evidence has suggested that associations between TMB and immunotherapy differ between different cancer types. 12 , 114 An observation study covering 32 cancer types compared the predictive efficiency of TMB‐H on ICB treatment. 12 TMB‐H demonstrated significantly better survival in tumors whose neoantigen loads positively correlated with CD8+ T cell levels, including lung cancer and melanoma. However, for cancer where neoantigen did not positively correlate with CD8+ T cell levels, such as breast and prostate cancer, TMB‐H tumors failed to achieve better outcomes. 12 A possible explanation is that the predictive efficacy of TMB‐H predominantly relies on the basal immune cell infiltration level.
Different tumor types depend on different mutational events during development. Thus, TMB across tumors cannot be defined by one universal mutational signature. 117 TMB characteristics should be more accurately identified for a certain cancer type or a given immunotherapy. 126 , 127 Signatures linked to external mutagens such as ultraviolet radiation and smoking are more prevalent in melanoma and lung cancer. In contrast, signatures associated with deficiencies in DNA repair genes (MRC1, POLE) are more prominent in endometrial, colorectal, and esophagogastric cancers. 128
2.2.2. Mismatch repair system and MSI
Microsatellites encompass brief recurring sequences of one to six nucleotides dispersed across the genome and are notably susceptible to DNA mismatch during replication. 129 , 130 , 131 In normal tissues, the crucial mismatch repair (MMR) system typically rectifies these errors in DNA replication or recombination. 132 , 133 As a result of MMR gene mutations, there is an exponential increase in mutation probability in microsatellite genome regions, causing high‐frequency MSI. 134 , 135 , 136 dMMR/MSI‐H has been considered a key prognosis influencing factor for CRC. 43 For ICB, dMMR/MSI‐H tumors have been reported to benefit from PD‐1 antibody treatment. 44 , 137 In 2017, pembrolizumab was approved by the United States Food and Drug Administration for treating relapsed or refractory solid tumors with MSI‐H and dMMR, marking the first approval of a biomarker that is agnostic to tumor‐type. 45 , 137 , 138
Notably, the impact of indel mutations varies according to the microsatellite location. 139 When indel occurs within noncoding segments, little effect can be observed. However, indel mutations in regulatory, splicing, or protein‐coding regions contribute to frameshifts which likely yield immunogenic neoantigens. 139 About 83.3% of the MSI‐H tumors demonstrated a high level of TMB, while 16% of the TMB‐H tumors are MSI‐H, suggesting that MSI‐H may serve as a contributing factor of TMB‐H. 140 However, as evidence does exist for the ICB treatment of certain hypermutator cancer types, especially for CRC patients with a high level of MSI, 141 , 142 there is still uncertainty about what threshold should be applied for treating different cancer types with ICBs. 121 , 143 , 144
2.2.3. DNA methylation
Associations exist between epigenetic characteristics and other biomarkers, suggesting intricated underlying mechanisms. In NSCLC, direct links between methylome alterations and TMB were observed. 145 In breast cancer, NEFM promoter hypomethylation was reported to be associated with increased immune infiltration and plays a role in TME reshaping. 146 Besides, methylations serve as targets for epigenetic therapy. Thus, identifying epigenetic biomarkers helps the combined application of epigenetic therapy and immunotherapy. 147
Various cancer subtypes exist with distinct progression and immunologic patterns based on the epigenetic landscape. For example, CRC can be categorized into MSI, chromosomal instability, and CpG island methylator phenotype (CIMP), in which CIMP is characterized by hypermethylation of CpG sites around certain gene promoter regions. 47 By calculating the scores of a series of key gene phenotypes within the CIMP subtype, predictions can be made regarding the therapeutic prognosis of patients. 47 , 148 , 149 Besides, ICB response prediction models utilizing DNA methylation profiles have emerged for several cancer types including melanoma, 150 esophagus carcinoma, 48 NSCLC, 151 glioma, 152 and bladder cancer. 153
However, despite the potential of DNA methylation being a reliable biomarker in various cancers, fundamental exploration is still needed. One key gap lies in the fact that in vivo methylation exists in a balance between generation and removal, while measurements only reflect stable levels instead of turnover rates. Therefore, dynamic analysis of tumor DNA methylation in tumor immunity is crucial for a comprehensive understanding. 154
2.2.4. Genomic alterations in specific genes
A variety of alterations in specific genes including the MHC–TCR axis and PD‐L1/CD274 gene serve as biomarkers individually. MHC diversity affects neoantigen presentation, while TCR repertoire determines antigen recognition. 52 , 155 Higher MHC heterozygosity or TCR clonality indicates the presence of tumor‐reactive T‐cells and correlates with improved survival during ICB treatment. 155 , 156 Mutation in POLE/POLD1 results in the heightened hydrophobicity of TCR‐contact residues and thus enhances T‐cell recognition and interaction. 157 , 158 However, the relationship between TCR clonality and ICB response differs depending on different checkpoint blockages and this phenomenon is partly explained by different associations between TCR mutation, neoantigen load, and TILs. 159 , 160 Apart from MHC–TCR axis, various genomic alterations can independently influence immunotherapy response, such as PD‐L1/CD274 gene amplification and B2M mutations. 88 , 161 , 162 , 163
2.2.5. RNA signatures
With the progress of sequencing techniques and comprehensive databases, identifications of RNA signatures using transcriptomic RNA data have become a prevalent practice. 164 Examples of transcriptomic signatures include MHC class II (HLA‐DR) expression in melanoma, 56 prognostic hypoxia‐immune genes in TNBC, 53 and ferroptosis signatures in breast cancer. 165 Furthermore, mRNA post‐transcriptional modifications have been reported to be associated with tumor immunology. 166 , 167 , 168 Additionally, noncoding RNA (ncRNA) has gained significant attention as a novel biomarker, 169 , 170 , 171 , 172 including lncRNA, 173 circRNA, 54 and miRNA. 55
2.2.6. Conclusions for genetic features
The TMB, dMMR/MSI‐H, DNA methylation, alterations in specific genes, and RNA signatures have shown significant implications in cancer immunology. 47 , 120 , 138 However, TMB and MSI may not apply to all cancer types and the specific threshold for different cancer types remains unclear. 12 , 114 DNA methylation patterns can influence immune responses and tumor progression but still need further elucidations. 154
2.3. Tumor‐infiltrating lymphocytes
Interaction, activation, and costimulation of lymphocytes are essential for a successful antitumor immune response, including CD8+ T cells, CD4+ T cells, and B cells. 11 The presence and proportion of different TIL subgroups, as well as their functional stage, differentiation process, and composition structure, have a fundamental impact on tumor immunotherapy. For instance, the T cell‐inflamed gene expression profile (GEP) has been reported to correlate with a good ICB prognosis in several cancer types. 118 , 174 , 175 This GEP contains IFN‐γ‐responsive genes related to antigen presentation, chemokine expression, and adaptive immunity and serves as a quantification of T cell‐inflamed microenvironment which can improve response to anti‐PD‐1 treatments. 176 Besides, the tertiary lymphoid structures (TLSs) as de novo lymphoid tissue resembling lymphoid organ structure serves as a promising prognostic predictor for improved post‐treatment survival. 66 , 67
2.3.1. CD8+ tumor‐infiltrating T cells
CD8+ T cells are characterized by their antitumor functions and are also referred to as cytotoxic T lymphocytes (CTLs) which serve as a producer for high levels of cytotoxic molecules (such as granzyme) and antitumor cytokines (such as tumor necrosis factor‐α, TNFα). Studies have demonstrated that CTLs are linked to favorable prognoses across diverse cancer types. Under physiological conditions, CTLs are transformed into memory subtypes to preserve long‐term protection capacity. Notably, memory CD8+ T cells are a heterogeneous group that can be further classified into central memory T (TCM), effector memory T (TEM), and other subgroups including stem cell‐like memory T (TSCM), and effector memory RA+ T (TEMRA) cells. 177 , 178 Despite naïve‐like features and limited direct effector functions, TCM have been reported to characterize ICB responders in naive tumors. 179 It has been demonstrated that treatment of TCM cells with ICB induces a cytolytic gene signature and an effector‐like phenotype. Increased expression of LAG3, BTLA4, and PD‐1 were observed in nonresponders. 180 TEM cells exhibit proinflammatory functions and serve as an independent prognostic factor of OS. 181 Recently, peripheral tissue‐resident memory (TRM) CTLs which normally stay in peripheral tissues and can be recruited for antitumor immune responses have attracted attention. 182 CD103+ TRM was associated with a favored prognosis, and there was also an increased TRM abundance in ICB responders. 183
2.3.2. CD4+ tumor‐infiltrating T cells
CD4+ T cells function with CD8+ T cells in antitumor immune response. 184 Among CD4 T cells, conventional helper CD4+ T cells (THC) use CD40L on the cell surface to interact with CD40 on dendritic cells (DCs) and help CD8+ T cells in the priming process. 184 Notably, only THC cells with functioning MHC‐I and MHC‐II (instead of a single MHC‐I) were able to eliminate tumors after ICB. 185 Additionally, the quality of T cell response is greatly influenced by the diversity and specificity of TCR. Studies have shown that the expansion of T cell clones can be observed in both ICB responders and nonresponders. 63 A higher TCR clonality and diversity correlated with improved response to ICB, as confirmed by multi‐variate regression models in a randomized controlled trial. 60
Unlike CD8+ T cells and previously mentioned CD4+ subgroups, CD4+ regulatory T (Treg) cells characterized by the high expression of CD25 and FOXP3 suppress CD8+ T cells by counteracting tumor immune response. 186 Treg cells have been associated with poorer survival in many kinds of solid tumors including breast, gastric, pancreatic, colon, and cervical cancers, 61 , 62 , 187 , 188 while Treg depletion contributes to the success of anti‐CTLA‐4 treatments. 189 Hyperprogressive disease (HPD) is a rare event in which a rapid type of tumor is caused by ICB. A recent study demonstrated that increased proliferation of Treg can be observed in HPD patients, and Treg cell depletion may serve as an HPD prevention before anti‐PD‐1 therapy. 190 Notably, recent studies have revealed diverse subgroups within Tregs. For instance, Treg subgroups expressing GZMB, LAG3, TIM3, or CCR8 exhibit significant immunosuppressive activity in CRC, 191 while CD30+OX40+ Tregs serve as negative Treg regulators, correlating with improved prognosis. 192 However, the significance of these diverse Treg subgroups as immunotherapy biomarkers remains to be explored.
2.3.3. Exhausted T cells
T cell exhaustion refers to the dysfunction of T cells caused by prolonged exposure to antigens. It is characterized by reduced cytolytic and proliferative ability, accompanied by elevated expression of various surface receptors such as PD‐1, CD103, CX3CR1, CD39, and TIM3. 178 , 193 Changing T cell phenotype is critical for switching between an inflammatory immune response that inhibits tumor growth or a regulatory state that promotes tumor growth. 178
Exhausted CD8+ T cells can be further classified into progenitor stem‐like exhausted (TPE) cells and terminally exhausted T (TEX) cells. 65 TPE cells possess expressions of transcription factor T cell factor 1 (TCF1) and are characterized by maintained tumor antigen‐specific immune response capacity. 194 This subgroup of exhausted T cells will eventually differentiate in the ultimate TEX stage with a decrease in TCF1 expression. 194 A single‐cell transcriptional analysis in 2018 have shown that PD‐1high CD8+ T cells serve as a predictive biomarker for anti‐PD‐1 therapy in NSCLC as anti‐PD‐1 therapy impact on TPE cells instead of TEX cells. 64 Several other studies also support this conclusion, showing that TPE cells increased their cytotoxic capacity while TEX cells did not show a response to ICB therapy. 65 , 194 , 195
2.3.4. Tumor‐infiltrating B cells
Studies have shown that TIL B cells were associated with an improved immunotherapy response as well as better survival. 66 , 196 , 197 For instance, melanoma and renal cell carcinoma (RCC) samples with a higher expression level of B cell gene panels had a higher response rate to ICB treatment. ICB responders exhibited an increased memory B cell proportion, an enhanced BCR diversity, and a larger B cell clonal expansion. 67 The coexistence of B cells and T cells facilitated the formation of TLS, served a curial role in TME formation in melanoma, and led to an improved prognosis after cancer immunotherapy. 66 Notably, ICOSL+ B cells, a small subgroup of TIL B cells, were found to serve as an antitumor immune response booster after neoadjuvant chemotherapy in breast cancer. 198 Another subgroup of TIL B cells, regulatory B (Breg) cells, have been reported to play an important regulatory role in cancer immunity. 199 , 200 , 201 , 202 However, evidence on the role of Breg in human in vivo studies is still limited. 202
2.3.5. Conclusions for TILs
TILs play a pivotal role in the tumor immune microenvironment, reflecting the complex interplay between the immune system and the tumor. 11 CD4+ T cells assist in activating other immune cells, while CD8+ T cells directly recognize and eliminate tumor cells. 179 , 184 Exhausted T cells are characterized by sustained antigen exposure and functional impairment. Among them, the TPE subgroup retains responsiveness to immunotherapy and partially explains the success of ICB. 64 Additionally, TIL B cells are attracting more and more attention as a key factor associated with an improved immunotherapy response. 67
2.4. Liquid biopsy
Liquid biopsy involves the analysis of circulating tumor material mostly in blood and has become one of the most powerful tools in the management of various kinds of cancer. 203 , 204 , 205 , 206 Compared with invasive tissue biopsy, noninvasive liquid biopsy provides a deeper understanding of cancer dynamics by utilizing frequent analysis of circulating biomarkers including circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), and extracellular vesicles (EVs). 207
2.4.1. Circulating tumor DNA
ctDNA is released from necrosis/apoptotic tumor cells into the bloodstream. 120 By identifying genetic mutations in ctDNA, it becomes possible to gain real‐time insights into the tumor states. 69 , 208 , 209 ctDNA pool provides a greater accuracy for determining TMB since it represents mutations from multiple tumor subclones. 210 , 211 Estimates of TMB based on blood samples (bTMB) exhibited significant concordance with tissue TMB (tTMB) and serves as a predictive biomarker in ICB response. 69 , 211 , 212 Similarly, estimations of MSI based on blood samples (bMSI) can be used for ICB response prediction as well. 213 , 214 , 215 While these findings recommended ctDNA‐based mutation estimations in patients whose tissue biopsy cannot be easily obtained, further studies concerning a broader range of cancer types and patients with low ctDNA levels are still needed. 208
Aside from TMB and MSI, longitudinal ctDNA tracings can provide insights into tumor dynamics and tumor immunity monitoring. 209 , 216 The CheckMate‐816 trial focusing on neoadjuvant ICB of NSCLC patients revealed a correlation between the elimination of specialized ctDNA panels and pathological complete response. 216 Post‐treatment ctDNA detection can also help identify patients with higher risks of cancer relapses, even only 3 days after operations. 217 , 218
Recently, DNA methylation in ctDNA has been shown to serve as novel diagnostic and prognostic biomarkers. 219 , 220 The aberrant DNA methylation status of ctDNA has emerged as a promising biomarker for the prediction of drug response in several cancer types including lung cancer, breast cancer, CRC, and prostate cancer. 219 , 221 Further research is needed to elucidate the functional significance of ctDNA methylation and its relationship with TME. Besides, exploring the dynamic of ctDNA methylation will also contribute to a deeper understanding of tumor‐immune interactions.
2.4.2. Circulating tumor cells
CTCs are tumor cells that have spread from primary or metastatic tumors to blood and serve as an intermediate stage in the metastasis process. 222 The tumor immunological characteristics of CTCs are closely associated with their ability to evade immune surveillance. 223 Despite thousands of tumor cells entering the bloodstream daily, only a small fraction of CTCs can be detected due to the loss of protective immunosuppressive microenvironment in the primary tumor. 223 CTCs can promote the formation of an immunosuppressive environment by downregulating MHC‐I molecules and upregulating immune checkpoints such as PD‐L1. 224 Therefore, higher levels of CTCs may indicate stronger immune suppression and lower immune cell activity. 225
Multiple studies have shown that CTC counts can serve as prognostic indicators for patients, despite their low detection rate. Baseline CTC counts have been employed as prognostic markers, and correlate with patients exhibiting either long or short OS. 70 , 226 , 227 For immunotherapies, several studies have revealed its predictive role. A study on metastatic or relapsed NSCLC immunotherapy candidates revealed a significant positive correlation between an extensive mutation burden and a higher number of CTCs. 228 Another study further demonstrated that CTCs PD‐L1 expression can efficiently predict ICB treatment outcomes advanced NSCLC. 229
In conclusion, the value of CTCs as biomarkers lies primarily in their quantity and biological characteristics. Currently, the relationship and molecular mechanisms between CTC formation and their immune evasion ability are not yet fully understood. Further research advancements in this area will facilitate the application of CTCs as biomarkers for cancer immunology and immunotherapy. Noteworthy, for CTC to be used for molecular diagnosis, the purity of CTCs is an important influencing factor on prediction efficiency. Therefore, the development of microfluid chip, nanotechnology, and 3D bio‐printing plays a crucial role in improving CTC capture efficiency and purity. 227 , 230
2.4.3. Extracellular vesicles
EVs, membrane‐bound vesicles secreted by various cell types, have been identified and isolated from different bodily fluids, providing a noninvasive approach to characterize the originating tumor cells. 231 Tumor‐derived EVs (tdEVs) carry a wide variety of tumor neoantigens and exhibit a distinct molecular signature that mirrors the genetic complexity. 232 tdEVs have emerged as potential mediators of cellular communication and modulators of TME, particularly in the establishment of immunosuppressive environments in distant metastatic sites. 233 , 234 TDEs have been identified as immunoregulatory factors that modulate various immune and stromal cells, including T cells, 235 Treg cells, 236 and cancer‐associated fibroblasts (CAFs). 237
tdEVs serve as carriers of diverse cargo, offering valuable insights into individualized tumor status. 238 tdEVs containing DNA enable the identification of genetic mutations, providing information about cancer‐specific alterations. 239 Additionally, tdEVs capable of predicting individualized treatment responses have been identified. 74 , 240 Significant differences in exosomal PD‐L1 levels before treatment have been observed between responder and nonresponder, suggesting that exosomal PD‐L1 is associated with anti‐PD‐1 responses. 74 However, due to its low content, even minimal contamination can lead to lowered efficacy, necessitating the development of accurate detection methods. 238
2.4.4. Conclusion for markers in liquid biopsy
Biomarkers in liquid biopsy, including ctDNA, CTCs, and tdEVs, have shown significant relevance in cancer immunology investigation due to their shared advantages of enabling noninvasive real‐time monitoring of tumor dynamics. 207 They provide an accessible approach for longitudinal assessment of cancer progression. 217 , 218 However, challenges remain due to the low abundance of these biomarkers in the bloodstream. 238 The development of biomedical engineering technologies, particularly microfluidic chips, holds promise for advancing this field by improving sensitivity, efficiency, and reliability in liquid biopsy. 227 , 230
3. IN VITRO PRECLINICAL MODEL FOR TUMOR IMMUNOLOGY INVESTIGATION
Rapid development of immunotherapy necessitates convenient, stable, and cost‐effective in vitro models for cancer immunology investigations. 18 Additionally, tumor immunology research requires effective preclinical models that faithfully reproduce the in vivo tumor, particularly the tumor‐immune interactions. 241 These dual demands have spurred the development of preclinical in vitro models effectively recapitulating the native TME. 242 Successful TME replication involves two key aspects: (1) reproduction of cellular components, including tumor components, immune cell subsets, and stromal cell subsets 243 , 244 ; (2) preservation of cellular functions, including the cytotoxicity of T and natural killing (NK) cells, antibody production by B cells, antigen presentation by myeloid cells, and ECM remodeling by CAFs. 28 , 242 Traditional 2D culture models have limitations in effectively replicating TME due to reasons including flat monolayer configurations and less representative distributions of oncogenes and tumor suppressors. 21 , 23 Compared with 2D models, 3D culture models create polarization of cells with distinct basal and apical poles through suspension, embedding, or advanced chip structures. Alterations in tissue microstructure result in modified distributions of oxygen, nutrients, and metabolites and further lead to optimized genomic and protein characteristics. 15 , 18 , 245 Additionally, by crosslinking biological materials in a manner that mimics in vivo tissue, 3D cultures create a solid ECM that closely resembles the properties of real TME. 246 This allows for the simulation of mechanical interactions between cells and the ECM, which are essential for various biological processes such as tumor growth, adhesion, migration, and immune infiltration. 247
3.1. 3D in vitro culture
3D tumor‐immune coculture systems can be divided into several subtypes based on construction architectures. The construction approach determines the specific spatial distribution of cancer and immune cells, thus having a fundamental impact on the characteristics of the coculture system (Figure 3A).
FIGURE 3.
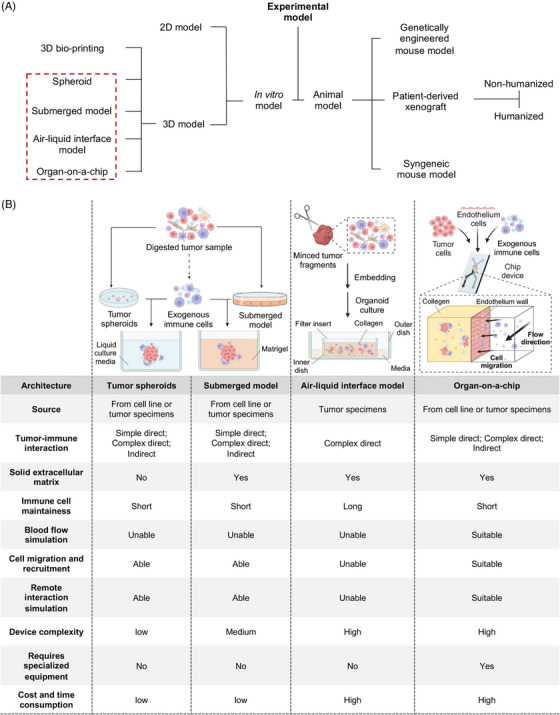
Comprehensive comparison of 3D tumor‐immune coculture system with different construction approaches. (A) Experimental models can be classified into in vitro models and animal models. In vitro models can be further divided into 2D models and 3D models which encompass spheroid, submerged model, air–liquid interface (ALI) model, and 3D bio‐printing based model. For animal models, they can be separated into syngeneic models, genetically engineered models, and patient‐derived xenografts. (B) Tumor immune microenvironment can be generated in in vitro 3D tumor‐immune coculture by four construction approaches. (1) Left: for tumor spheroid and submerged model, tumor‐immune interaction can be simulated by adding exogenous immune cells. (2) Middle: in the ALI model, minced tumor tissue fragments containing both tumor and immune cells are embedded in collagen. (3) Right: for the microfluid chip, tumor cell spheroids are mixed with collagen and injected into the central compartment while immune cells are circulating. Figure was created with BioRender.com.
3.1.1. Spheroid
Spheroids are collections of cells growing in three dimensions while suspended with or without an ECM. 18 A more complex model, such as a submerged model or microfluid chip, can be built based on tumor spheroid. 28 , 248
A variety of tumor types have completed the construction of 3D spheroid models, such as lung cancer, 243 prostate cancer, 249 and head/neck squamous cell carcinoma (HNSCC). 250 Generally, tumor spheroid exclusively preserves tumor epithelium cells. But with gentle digestion, spheroid can retain the immune cell components within the primary tumor for a period and preserve responsiveness towards immunotherapy. 243
Spheroids are used for testing immunotherapies by adding exogenous immune components, mainly to evaluate the effectiveness of therapeutic antibodies and conduct drug screening for improving immune cell infiltration and antitumor effects against the targets of spheroids. 249 , 251 , 252 Immune cells that can be added include CTLs, 249 , 253 Vδ2T cells, 251 and NK cells. 252
Overall, the spheroid model is a critical step forward in moving from 2D to 3D culture with the lowest cost and build difficulty. Despite the lack of retention of immune components, it is still one of the most widely used 3D tumor‐immune coculture models.
3.1.2. Submerged model
Submerged model (Figure 3B) cultures tumor spheroids with exogenously added stromal or immune cells in Matrigel submerged in a culture medium. 254 It is widely used and has been built for different cancer types, including breast cancer, 255 , 256 lung cancer, 257 , 258 ovarian cancer, 259 colon cancer, 241 , 257 pancreatic cancer, 260 , 261 and gastric cancer. 248 In the submerged model, native stroma cells and immune components cannot be preserved conventionally. Exogeneous cell types include stromal cells like CAFs, 258 and immune cells like CTLs, 261 , 262 tumor‐associated macrophages (TAMs), 261 and PBLs. 258 , 263
If organoids can be constructed suitably and a short time after tumor sample collection, part of the original immune cell components and even the original immunosuppressive environment in the tumor can be preserved. 241 , 259 To better preserve original tumor components, the time interval between sample collection and organoid construction must be possibly short (often less than one day), and a softer digestion method should be used during organoid construction. 259 Submerged models utilizing patient tumor samples from high‐grade serous ovarian cancer were employed to investigate the efficacy of dual‐specific anti‐PD‐1/PD‐L1 antibodies. 259 By adding ICBs and IL‐2, activation of T cells and NK cells can be observed. 259
Submerged models have provided insights into generating and screening highly cytotoxic and tumor‐selective lymphocytes. 257 , 264 Tumor‐reactive T cells with high cytotoxicity and specificity from peripheral blood can be efficiently enriched in coculture systems of autologous patient‐derived organoids (PDOs) from patients with dMMR CRC and NSCLC. 257 Submerged models can also reproduce characteristic responses of immunotherapy and have been widely used as a model for immunotherapy testing, including ICB, 241 , 256 Bacille Calmette‐Guerin (BCG) immunotherapy, 263 High‐affinity neoantigens, 265 iRGD peptide, 253 Bi‐mab antibody, 266 Chimeric antigen receptor T cell immunotherapy (CAR‐T), 255 , 267 , 268 cibisatamab, 269 and Vδ2T. 270 For instance, the cytotoxicity of peripheral blood mononuclear cells (PBMCs) activated with BCG vaccine were validated in 3D coculture systems involving HNSCC cell line FaDu. 263
3.1.3. Air–liquid interface model
In the ALI model, minced patient‐derived tumor tissues are directly embedded in the collagen matrix without digestion (Figure 3B). Collagen‐embedded tumor tissue is placed inside the inner plate where the culture medium can diffuse through the permeable plate wall from the outer plate. 271 The inner plate is in direct contact with the air to ensure adequate oxygen supply to the organoid. As the ALI model uses patient‐derived tumor tissue en bloc that includes endogenous stromal and immune cells, it has a strong advantage in simulating the native TME. 244 Tumors from multiple sources, for instance, skin, colon, pancreas, and lung can successfully prepare and form ALI models with high reproducibility. 242 , 244 IHC and fluorescence staining showed that ALI cultures not only effectively preserve stromal components such as fibroblasts and myofibroblasts but also retain a wide range of immune components including CD8+ and CD4+ T cells, B cells, NK cells, and PD‐1+ CD3+ T cells. Further single‐cell RNA sequencing (scRNAseq) revealed that 85% of T and B cells in ALI can be detected using VDJ enrichment assays, enabling the linkage of cell‐type identification and immune repertoires from the same cells. 242 Compared with the submerged model, the ALI model greatly improved lymphocyte lifespan (1 month without IL‐2, and 60 days after IL‐2 was added). 242 ALI model can also reproduce the TCR repertories of the original tumor and can be used for the tests of immunotherapy. 242 , 244
Among all constructs, the ALI model is the only one that does not require digestion and uses tissue en bloc directly for model construction. This construction mode directly preserves the original cellular components in the tumor tissue and greatly extends the survival time of the nonmalignant components. However, the culture method relying on fresh tumor tissue slices also limits the ability of ALI models to incorporate exogenous immune components from the external for TME reconstruction. Additionally, ALI models cannot be constructed using tumor cell lines other than fresh tissue.
3.1.4. Organ‐on‐a‐chip
Due to breakthroughs in biomedical engineering and material chemistry, the use of microfluidic chip technology in tumors has made great progress. 272 Tumor tissues are minced, digested, sieved to collect spheroids, mixed with collagen, and injected into the central gel compartment of the microfluid chip. 272 (Figure 3B) The culture medium is added to the fluid channel. Microfluidic chips have been widely used in the construction of in vitro 3D tumor‐immune models, 273 , 274 , 275 , 276 and have been used to test the efficacies of various immunotherapies such as ICB, 28 CAR‐T, 275 , 277 and Vδ2T. 270 Utilizing HeLa and NK‐92 cell line, rail‐based microfluidic design was integrated within a single 96‐well to achieve high‐throughput 3D coculture of cytotoxic lymphocytes with cancer cells. 275 Besides, combining the PDO with the spheroid‐based microfluid chip can also effectively preserve the original immune components in tumor samples. 28 The tumor organotypic slice model is a special type of organ‐on‐a‐chip. It allows for the investigation of native TME by directly utilizing undigested tumor tissue slices, such as exploring the role of astrocytes and microglia in immunosuppressive environment formation in glioblastoma. 278
Microfluidic chips can simulate complex TMEs including blood vessels, 279 and the blood–brain barrier (BBB). 276 In the vascular simulation, tumor cells, endothelial cells, and fibroblasts were cocultured in a certain proportion in the collagen‐embedded microfluidic chip. The cell mixture could spontaneously form vascular structures composed of endothelium cells. 273 , 279 To simulate the BBB, microfluidic chips can be utilized to coculture patient‐derived breast cancer cells, endothelial cells, and astrocytes. This in vitro model accurately replicates BBB structure and allows for the investigation of tumor‐immune interactions and their impact on tumor‐brain metastasis. 276 Microfluidic chips can also generate dynamic gradients of various substances. Therefore, it can be used as a cell migration model, 270 or a tumor‐lymph node remote interaction model. 274
Collectively, microfluidic chips are capable of simulating complex environments like blood circulation and cellular barriers. 273 , 276 However, due to its size limitation on spheroids, it only has a limited effect on the reconstruction of histological morphology.
3.1.5. Conclusions for different 3D in vitro model architectures
Different 3D in vitro culture architectures, including spheroids, submerged models, ALI models, and organ‐on‐a‐chip systems, offer unique advantages and applications in cancer immunology investigation (Figure 3B). Spheroids as the earliest tumor 3D in vitro culture represent a transition from 2D to 3D but lack the preservation of immune components within TME. 249 , 251 , 252 Submerged models provide a solid noncellular ECM and offer versatility in studying various aspects of cancer immunology. 241 , 259 ALI models exhibit the strongest capability to preserve the native TME but are limited to tissue culture and encounter difficulties incorporating exogenous immune components. 242 , 244 Organ‐on‐a‐chip systems excel in simulating complex environments and interactions. However, the limited sample size, high costs, and model complexity hinder their wider application. 273 , 274 , 275 , 276
3.2. 3D bio‐printing technology for in vitro model construction
The realm of tissue engineering witnessed the rise of 3D bio‐printing as a highly promising methodology for fabricating intricate biological structures. 280 As for tumor immunology, 3D bio‐printing is gaining prominence as a powerful tool due to its ability to preserve tumor cells in a near‐native state. It has found extensive applications in oncology research, providing new avenues for studying tumor immunology. 280 , 281
3.2.1. Approaches of 3D bio‐printing
The field of 3D bio‐printing relies on three key technological approaches: biomimicry, autonomous self‐assembly, and mini‐tissues. These enable the printed in vitro tumor model to exhibit the envisioned functions and structures, closely resembling primary tumors. 282 3D bio‐printing technologies used in tumor‐immune coculture include inkjet, laser‐assisted bio‐printing (LAB), and extrusion‐based bio‐printing (EBB).
Inkjet bio‐printers precisely deposit bio‐ink onto the targeted printing surface using thermal or acoustic mechanisms, ensuring a continuous flow or controlled droplet release from the nozzle. 283 , 284 With the ease of modification, low cost, and fast speed, it has been most widely used for cancer immunology studies. 285 LAB uses laser‐induced forward transfer, in which high‐pressure bubbles are generated by a high‐energy laser pulse in a thin biomaterial layer, ejecting it onto a specific area. 286 This precise process enables accurate biomaterial deposition, making LAB a promising technique for complex tissue fabrication in TME reconstructions. 287 LAB offers the advantages of accommodating a wide range of viscosity, ensuring high cell viability, and achieving high resolution. These advantages make it a valuable technique for creating functional tumor models such as exocrine pancreas spheroid models to study cancer initiation. 288 EBB merges a fluid‐dispensing system with an automated robotic system dedicated to extrusion and bio‐printing. 289 , 290 Its capability of creating porous constructs facilitates the engineering of vasculature in tumor models and the manipulation of cancer lymphatics. 291
3.2.2. 3D bio‐printing in tumor immunology investigations
Tumor‐immune cocultures enabled by 3D bio‐printing provide a new avenue for cancer immunotherapy research. 3D bio‐printed tumor models in collagen matrices containing immune cells enable the tracking of immune cell‐tumor interactions and facilitate simulated immunotherapy. 292 Besides, the ability to accurately measure T cell tumor infiltration demonstrates the potential of 3D bio‐printing as a valuable tool for preclinical characterization and selection of CAR‐T cells. Compared with 2D cocultures, the bio‐printed 3D neuroblastoma model showed high reproducibility and enabled the detection and quantification of CAR‐T cell tumor infiltration. 293 Additionally, the 3D bio‐printed tumor models provide a more physiologically relevant environment for studying tumor‐immune cell interactions. 294 For instance, 3D bio‐printing fabricates a miniature brain model by merging glioma cells with macrophages. This model unveiled the ability of glioma cells to attract macrophages and prompt their transitions to TAMs. 294 3D bio‐printing also allows for the creation of tumor models that replicate the microenvironment, enabling the study of cell fusion and the development of targeted therapies. 295 , 296
3.2.3. Conclusions for 3D bio‐printing
3D bio‐printing methods have shown great potential in generating valuable preclinical models for cancer immunotherapy and allow for the precise placement of tumor and immune cells. 280 , 281 Advances in bio‐printing techniques will be crucial in building more physiologically relevant models to study tumor‐immune interactions. 280
3.3. Reconstruction of tumor‐immune interactions in in vitro models
To apply 3D tumor‐immune coculture systems for TME and immunotherapy research, it is crucial to simulate in vivo tumor‐immune cell interactions. Tumor‐immune interactions can be divided into direct interaction relying on cell contacting and remote interaction relying on mediator secretion.
3.3.1. Simple direct interaction
Direct interaction refers to the interactions in which tumor cells and immune cells are in direct close contact, taking more account of the contact‐depending cytotoxic effects rather than remote interaction based on cytokine secretion or lymphocyte migration. TILs, primarily composed of T cells, are the most common mononuclear immune infiltrates observed in most patients. 11 Activated CTLs directly engage in immune killing by direct contact with tumor cells, thereby influencing tumor prognosis. 16 NK cells play a significant role in the treatment of hematological malignancies, while their cytotoxic effects on solid tumors remain controversial, possibly due to their weaker tumor‐infiltrating capacity. 297 Myeloid cells, such as TAMs, are a heterogeneous and plastic cell population within the tumor. TAM supports cancer progression and treatment resistance but can also mediate antitumor effects when responding to drugs that enhance phagocytic and oxidative functions. 298 , 299
Direct interaction is divided into simple and complex interactions. Simple interaction involves tumor cells and one type of lymphocyte, while complex interaction involves at least one other immunomodulatory cell. Simulations of different interaction patterns process different characteristics and have different corresponding construction architectures (Table 2). The simple direct interaction involves only tumor cells and a single type of lymphocyte. 256 , 262 , 268 It focuses on a specific immune cell and usually uses the constructs of tumor spheroid, submerged model, or microfluid chip. Coculture system has been widely used in preclinical testing and mechanism research of several types of novel immunotherapies such as ICB, 252 , 300 High‐affinity newsagents (HAN), 265 iRGD, 253 Bi‐mab Antibody, 266 CAR‐T, 255 , 267 , 268 cytokine‐induced killer cell (CIK), 249 and Vδ2T. 251 , 270
TABLE 2.
Comparison between different interaction simulations.
| Interaction patterns | Construction | Definition | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Simple direct interaction | Spheroid | Tumor and immune cells in direct contact. Involving only tumor cells and a single type of lymphocyte |
Focus on a specific immune cell type. Easy to construct. |
Unable to simulate complex TME or remote cell‐cell interaction | 252 , 255 , 266 |
| Submerged | 257 , 300 | ||||
| Microfluid chip | 275 | ||||
| Complex direct interaction | Spheroid | Tumor and immune cells in direct contact. Including at least two cellular components from the TME. | Able to simulate complex TME and preserve original tumor components. | Complex model construction and unable to simulate remote cell‐cell interaction | 243 |
| Submerged | 248 , 260 , 301 | ||||
| Microfluid chip | 28 , 273 , 279 | ||||
| ALI | 242 , 244 | ||||
| Remote interaction | Spheroid | Tumor and immune cells are cultured in different compartment. | Suitable for the simulation of remote interaction and immune cell migration. | Limited direct interaction | 302 |
| Submerged | 263 , 303 , 304 | ||||
| Microfluid chip | 274 , 277 |
Abbreviations: ALI, air–liquid interface model; TME, tumor microenvironment.
A coculture system that focuses on direct interaction is more suitable for generating and screening immune cells with tumor‐specific killing capacity as it does not include immunomodulatory components. 257 , 264 Cytotoxic T cells can be generated in a submerged model of colon cancer with dMMR, 257 and pancreatic cancer, 264 when T cells from PBMC were added into the coculture system with IL‐2 and IFN‐γ.
3.3.2. Complex direct interaction
Coculture systems focusing on complex direct interaction include at least one other immunomodulatory cell component. The coculture system can be constructed by adding additional immunomodulatory cell components and usually uses the constructs of a submerged model or microfluid chip. Cell components commonly used for complex coculture include various stroma cells and myeloid cells, including endothelium, 273 , 279 fibroblasts, 258 , 305 myeloid‐derived suppressor cells (MDSCs), 255 , 260 , 301 or macrophages. 260 , 261 Different conditional additives can be added into the medium to change the physiological state of cells in the model according to different tumor types and experimental purposes. 306 Such additives include R‐spondin, Noggin, Wnt3a, and other growth factors crucial for cell growth and differentiation. 241 , 260 , 307 This kind of coculture has been widely used in TME studies involving complex cell interactions, such as cancer–CTL interaction with TAMs 261 and CAFs. 305 Stroma components in TME can interact with tumor cells to form fine 3D structures and further influence the infiltration of lymphocytes into tumors. 258 By coculturing tumor cells, CTL, and MDSC or M2 macrophages, the addition of myeloid components generates an immunosuppressive environment, resulting in a significant decrease in the lethality of T cells. 248 , 261
The simulation of complex tumor‐immune interaction can also be established through a holistic approach, retaining the immune and stromal components of the original tumor. 28 , 241 , 242 Important cytokines such as R‐spondin, Noggin, epidermal growth factor, Prostaglandin 2, Gastrin, 241 and IL‐2, 259 should be added to the PDO culture system to better preserve native immune components (Table S1). ALI models have advantages in preserving immune components and have been widely used for PDO construction and immunotherapy testing. 242 In addition to ALI models, spheroid‐based submerged models, 241 and microfluid chips 28 can preserve original immune components under suitable culture conditions and operations. Thus, validation of the efficacies of immunotherapy 241 , 243 , 259 and testing of novel immunotherapies 28 , 244 can be carried out. Besides, a complex coculture system that preserves the original immune components can be used in the study of the tumor immunity process and mechanism. 28 , 259
3.3.3. Remote interaction
Remote interaction refers to the interplay in which tumor cells and immune cells are not in direct contact. For instance, T cell migration in response to chemokines and adhesion molecules plays a critical role in tumor immunity. This migration is facilitated by the activation of specific signaling pathways including chemokine receptor signaling and further contributes to the antitumor activities. B cells as the second population of tumor‐infiltrated immune cells, possess complex functions encompassing antibody production and immune function regulation. 308 The coculture model separates tumor cells from immune cells physically and requires lymphocytes to migrate to tumor cells or release cytokines or antibodies. Due to the advantages of creating cytokine gradients, the microfluid chip has been widely used in in vitro immune cell migration studies. 277 A separated submerged model, in which tumor cells and immune cells are separately cultured in different chambers and separated by a cell‐permeable membrane, can also be used for the study of cell migration. 270 , 302 , 304
A coculture system can be used to simulate remote interactions between tumor cells and immune cells. 263 , 274 , 303 For example, the coculture of tumor cells and endothelial cells using a chambered submerged model can be used to study the efficacies of BCG therapy on immune cell proliferation and cytokine secretion. 263 Besides, the chambered submerged model can be used as an in vitro simulation of B cell immunotherapy. 303 Through the utilization of organotypic slice cultures of breast cancer tissue and lymph node tissue, it has been observed that lymph node slices cocultured with tumor slices exhibit greater immunosuppression compared with those cocultured with healthy tissue. 274
3.3.4. Conclusions for tumor‐immune interaction reconstructions in vitro
The reconstruction of tumor‐immune interactions in 3D in vitro models involves both direct interactions and remote interactions. Direct interactions focus on T and NK cells. Coculture systems with different architectures can be employed for therapies that rely on direct cytotoxicity or to generate tumor‐reactive T lymphocytes. 257 , 264 Additionally, the inclusion of immunomodulatory cells such as MDSCs, TAMs, and CAFs contribute to the reconstruction of complex direct interactions. 255 , 260 , 301 Remote interactions encompass T cell migration as well as the secretion of cytokines and antibodies by lymphocytes, which play essential roles in tumor immunity. 308 Organ‐on‐a‐chip or chambered models can be utilized to investigate these aspects of tumor‐immune interactions. 277
3.4. Preservation of tumor immune microenvironment of in vitro models
The rapid development and extended use of 3D tumor‐immune coculture systems raised a question: whether the tumor‐immune coculture system can restore in vivo tumor immunity. Coculture systems need to be verified from two aspects: tumor cells and immune cells (Figure 1B). To date, most of the verifications have focused on the tumor cell, which can be mainly divided into genetic verification (mutation profile by NGS), 242 , 257 molecular biological verification (transcriptome profile by RNAseq), 309 cytological verification (surface protein profile by IHC), 267 and functional verification (drug sensitivity tests). 310 Nevertheless, there is still a lack of knowledge about the preservation of immune cells involved in in vitro tumor‐immune cocultures. Verifications on immune components involve two aspects: cell components and cell functions. For immune cell components, different immune cell subgroups with distinct characteristics can be evaluated based on surface markers using IHC and flow cytometry, which allows for assessing proportional changes of each subgroup. Immune cell functions can be evaluated through experiments measuring cytokine secretion, antibody secretion, and cytotoxicity. Alternatively, experimental immunotherapies can be conducted directly in in vitro models for efficacy observation. Table S2 summarizes the validation of the 3D tumor‐immune coculture system in different cancer types.
3.4.1. Validation of immune cell components
Several studies have shown that PDOs can retain a certain amount of native immune components for a period. 241 , 243 , 244 For the ALI model, IHC staining and other methods have confirmed that organoids can effectively retain a variety of nonimmune cell components including fibroblasts and a variety of lymphocyte components including TILs. 242 The longevity of lymphocytes can be extended to 60 days with the addition of IL‐2. 242 Further TCR repertories analysis revealed that TILs in PDOs could effectively reproduce TCR information of TILs in the primary tumor, where TCR components of exhausted T cells are best preserved. 242 Melanoma‐derived tumor spheroids embedded in a microfluid chip can also retain a variety of immune cell components from the primary tumors. 28 Spheroid‐based submerged model of human CRC retained a variety of cell components found in the original TME. 311
However, the proportion of immune cell subsets emerges as different between the original tumor and coculture system. The in vitro coculture model of low‐grade ovarian carcinoma containing PBMC was constructed by a magnetic field, and various lymphocyte components including NK, CD4+, CD8+, and Treg were retained. 311 However, the proportion of CD8+ T cells was significantly decreased in organoids, while the proportion of Treg was significantly increased. 311 In another submerged PDO model of human high‐grade serous ovarian cancer, scRNAseq showed that the myeloid component was significantly reduced, while the proportion of lymphocytes (including CD4+, CD8+, and NK) showed an overall upward trend. 259 In conclusion, a 3D tumor‐immune coculture system can retain the majority of immune cell types of the original tumor, but the proportion of cell types changes. Myeloid components are reported to generally decrease while the lymphoid components increase. 241 , 259 However, CD8+ T cells, B cells, and NK cells did not show a clear trend of change, and the underlying mechanisms of changes have also not been elucidated either.
3.4.2. Validation of immune cell function
The verification of immune cell function can be carried out from two perspectives: tumor immunity process reproducibility and immunotherapy efficacy. On the one hand, details of tumor immunity in vivo can be reproduced in the in vitro coculture model to explore the changes in cell composition and physiological state after immunotherapy. 28 , 259 After anti‐PD‐1 or anti‐CTLA‐4 treatment on human melanoma organoids, the expressions of CCL19 and CXCL13 were significantly up‐regulated, accompanied by significantly increased IFN‐γ, IL‐2, and TNFα secretion. 28 In 2021, Wan et al. 259 performed scRNAseq analysis on high‐degree ovarian cancer organoids before and after ICB treatment. ICB induced an increase in the number of certain immune cell populations, such as CD4+/CD8+ cells with high expression of CD107. Gene expression analysis further revealed increased cytotoxicity in T and NK cells and decreased exhaustion in T cells. 259
On the other hand, the immune function of the coculture system can be verified by analyzing the efficacies of immunotherapy in the in vitro coculture model. 28 , 242 , 243 Tumor spheroid from NSCLC patients could preserve original tumor immune components with reactivity to immunotherapy. 243 Spheroids from the different patients had different characteristics in tumor immune and responded differently to the same immunotherapy. 243 Spheroid‐based microfluid chip can retain original components including B cells, CD4+ T cells, CD8+ T cells, and myeloid components including MDSCs, DCs, and TAMs. PDOs can effectively reproduce the sensitivity (MC38) or resistance (Lewis lung carcinoma, B16F10) of the tumor to ICB therapy. 28 However, the stability and prediction accuracy of the in vitro coculture model for the response of a specific individual to certain immunotherapy remains unclear. It is necessary to conduct studies that compare the response of immunotherapy between the original tumors and the corresponding tumor organoids.
3.4.3. Conclusions on tumor immune microenvironment preservations
Several studies have demonstrated that PDOs can preserve various immune components and their functions. 28 , 241 However, several key challenges still exist. Further research is still needed to increase the complexity and reproducibility of immune cells in reconstructed TME. Additionally, the preservation of immune cell function in current models is still limited, hindering the study of the long‐term effects of immunotherapy. 242
3.5. Applications of in vitro tumor models in cancer immunology and immunotherapy
Applications of 3D in vitro tumor‐immune coculture systems focus on the mechanisms and key influence factors of tumor immunotherapy and tumor‐immune interactions (Figure 4). By controlling various factors such as immune cell populations, cytokines, and antibodies, models provide a controlled environment to assess the potential outcomes of immunotherapies. Moreover, in vitro models contribute to the development and optimization of immunotherapy by facilitating the screening of potential therapeutic targets and the evaluation of drug candidates. By changing key components of the TME, such as ECM, stromal cells, and immune cells, researchers can investigate the mechanisms underlying immune evasion and therapy resistance. Furthermore, by manipulating gene expression or using gene‐editing techniques, researchers can investigate the functional roles of specific genes in the immune response to cancer.
FIGURE 4.
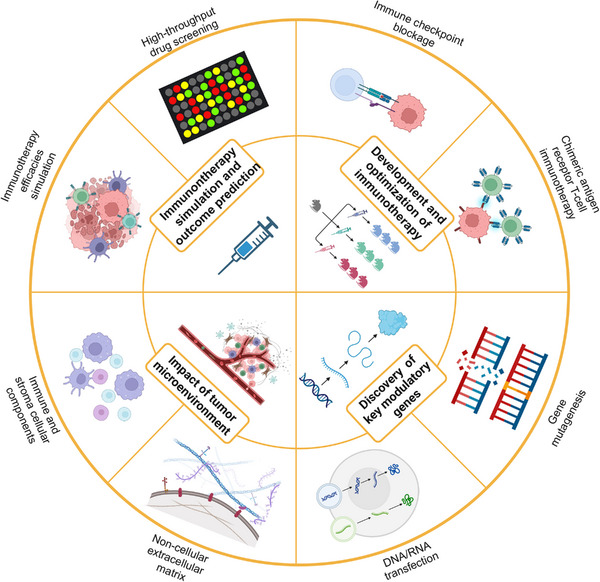
Application of 3D in vitro tumor‐immune coculture system on tumor immunology and tumor immunotherapy. Mechanisms and major influencing factors of tumor immunotherapy and tumor‐immune interactions are the main applications of the 3D in vitro tumor‐immune coculture system, which can be summarized in four aspects in the inner circle: immunotherapy simulation and prediction, immunotherapy optimization, tumor microenvironment factor analysis, and discovery of key modulatory genes. The eight parts of the outer circle are further subdivisions the four aspects of the inner circle. Figure was created with BioRender.com.
3.5.1. Immunotherapy simulation and outcome prediction
The efficacy of clinical immunotherapies has fostered an exponential interest in the tumor immune microenvironment, which in turn has engendered a pressing need for robust experimental systems modeling patient‐specific tumor‐immune interactions. The efficacies of different immunotherapies can be modeled in vitro by coculturing exogenous immune cells. 248 , 260 , 301 The in vitro coculture system based on exogenous immune cells makes it possible to develop a high‐throughput screening platform. 256 However, only adding a single kind of exogenous immune cells may not completely restore the complex interaction between tumor and nontumor components in the real TME.
The in vitro tumor immune cell coculture system based on a holistic approach can be used to simulate the ICB efficacies. 28 , 242 , 259 Compared with a coculture system depending on exogenous immune cells, the holistic coculture system has the following three main advantages. (1) The native model can effectively retain various stroma components, myeloid immune cells, and lymphocytes. 259 (2) The native model extends the duration for lymphocytes to remain active in the coculture system, enabling the study of medium‐ to long‐term ICB efficacies in vitro. 242 (3) Holistic coculture system simulates the response characteristics of tumors in vivo to ICB with higher recoverability. When PDOs from NSCLC, RCC, and melanoma were treated with anti‐PD‐1 therapy, the proportion of organoids with TIL activation was similar to that of clinical anti‐PD‐1 therapy. 242
PDO models have been utilized in personalized prediction for chemotherapy and chemoradiation. 312 , 313 Precision immunotherapy using PDOs not only necessitates preservation of tumor characteristics but also presents challenges in retaining patient‐specific TME. Models preserving native TME, such as ALI models and organotypic slice culture, represent significant opportunities. 28 In a study utilizing melanoma PDOs, the feasibility of PDOs as a personalized immunotherapy screening tool was demonstrated by comparing drug sensitivity from tumors with organoids. 314 Larger‐scale clinical validation studies, particularly parallel validation studies between patients and PDOs, are still needed.
3.5.2. Development and optimization of immunotherapy
The development of 3D tumor‐immune cell coculture has promoted the discovery of new molecular targets in tumor immunosuppressive environments. 252 , 300 In 2021, Sui et al. 300 determined that the DKK1 gene promotes the killing effect of CD8+ T cells through GSK3β/E2F1/T‐bet axis. In another study in 2021, scRNAseq analysis of the ALI model of high malignant ovarian cancer targeted BRD1 gene, which plays an important role in T cell and NK cell state transformation. 259 Other new molecular targets found or verified in 3D tumor immune coculture are summarized in Table S3.
The 3D in vitro culture of tumors preserves the surface antigen characteristics of tumor cells, making it an ideal preclinical testing platform for the application of CAR‐T therapy in solid tumors. 267 , 268 , 277 Combining CAR‐T, CRISPR, and microfluid technology, Preece et al. 277 tested the killing ability of Hepatitis B‐eTCR–/rTCR+‐CAR‐T on HepG2 cells in microfluid chips. Knockdown of eTCR upregulated the expression of rTCR and enhanced cell migration and cytotoxic killing effect on tumors. 277
In vitro 3D tumor‐immune coculture technology is also widely used in the testing of a variety of immunotherapies involving immune cell activation, recognition, and killing. Therapies that activate immune cells through direct stimulation include CIK cell, 249 BCG vaccine, 263 nanoparticles, 244 nanoformulated zoledronic acid, 270 zoledronate, 251 and so on. Additionally, by coculturing T cells with antigen‐presenting cells loaded with specific tumor antigens, antigen‐based immunotherapy can be tested. 265 , 266 After the bifunctional iRGD‐anti‐CD3 peptide is transmitted into T cells, CTLs targeting specific antigens are generated, and it has a strong killing and penetrating ability to the 3D culture of gastric cancer. 253 The amphiphilic antibody Bi‐mab enhances the killing ability of lymphocytes to breast cancer PDO by binding tumor cells and lymphocytes respectively. 266
3.5.3. Impact of TME on cancer immune response
3D tumor‐immune coculture, especially the coculture system involving more than one type of immunomodulatory component, can study the effects of microenvironment factors on tumor immunity. 258 , 261 , 262 The nature of ECM has an impact on tumor immunity and serves as a potential target for tumor immunotherapy. By changing the material and density of the matrix in the in vitro coculture system, tumor immunity in different hardness ECM can be simulated. 262 , 273 , 315 Oxygen concentration is altered in a coculture system for the hypoxic environment simulation. 273 , 305 In normal tissue, the oxygen partial pressure (PO2) is typically around 65 mmHg, ranging between 12.5 and 96 mmHg. For pathological conditions, cancer tissue usually exhibits lower PO2 levels around 10 mmHg and varies between 0 and 95 mmHg. 316 In comparison, organoids are typically constructed and cultured in CO2 cell incubators where PO2 is maintained at approximately 150 mmHg, indicating a significant difference in oxygen conditions. A hypoxic environment induces the production of endothelial components in the TME and ultimately leads to the decrease of lymphocyte killing ability, and even lymphocyte apoptosis. 273 Glucose restriction often occurs before and after eating. After T cell extraction, glucose restriction and re‐supply were carried out in vitro to simulate the change in glucose concentration. 315 After transient glucose restriction, the immunosuppressive characteristics of T cells decreased while the lethality of tumor organoids increased significantly. 315
For the cellular components in the TME, current research is mainly concentrated on fibroblasts, 258 macrophages, 261 endothelial cells/vascular epithelial cells, 273 , 279 and MDSC. 248 , 301 Notably, the addition of fibroblasts will change the cell composition and microenvironment architecture of the tumor immune microenvironment. 258 , 305 Fibroblasts form a marginal envelope around the coculture system as a microenvironment skeleton, preventing PBMC from infiltrating into the spheroid center. 258 Endothelial cells can spontaneously form vascular‐like structures in the coculture system, simulating the vascular environment in vitro. 279 Tumor cells form cell groups with different distributions and sizes according to their subtypes and further affect the migration and infiltration of immune cells. 279
3.5.4. Discovery and validation of key immune‐modulatory genes
A combination of in vitro tumor immune cell coculture and gene expression manipulation can directly examine the role of genes in tumor immunity. 248 , 276 , 301 Mutagenesis, RNA interference (RNAi), and other technologies specifically inhibit or knock out the expression of specific genes in cells and can be applied to the study of gene function. The glioma‐associated oncogene (GLI) related Akt–mTOR pathway directly affects the expression of PD‐L1 in the formation of the immunosuppressive environment of gastric cancer. 248 The knockout of HER2 inhibits the expression of the Akt–mTOR pathway and PD‐L1 gene and effectively increases the sensitivity of gastric cancer organoid tumors to anti‐PD‐1 therapy in vitro. 301 Breast cancer is one of the more common cancers that metastasize to the brain. By constructing a BBB in vitro in microfluidic chips, Mustafa et al. 276 found that the response of tumor cells to T cells was crucial to the development of brain metastasis, especially with GBP1 overexpression.
On the other hand, we can increase the expression of specific genes in cells by virus transfection and other methods and observe the effect of increased gene expression on tumor immunity. 304 , 307 Meng et al. 304 used gastric cancer cells transfected with CXCL10 expression and T cells induced with CXCR3 expression for the construction of a tumor‐immune coculture system and directly observed the dose‐dependent effect of T cell migration and infiltration and CXCL10–CXCR3 interaction intensity. Elmira Asl et al. 307 used a 1:1 ratio of tumor cell‐T cell coculture to induce Treg production in vitro. The miR124 indirectly inhibits the differentiation trend of Treg and weakens the generation of an immunosuppressive environment by mediating the decrease of STAT3 expression on the tumor surface. 307
3.5.5. Conclusions for in vitro tumor model applications
The use of spheroid and submerged models as immunotherapy testing platforms has been widely adopted due to their efficiency in immunotherapy simulation and outcome prediction. 248 , 260 , 301 Additionally, models preserving stromal and immune components, represented by the ALI model, replicate the native TME and thus simulate the ICB response of in vivo tumors with a higher recoverability. 28 , 242 , 259 Besides, high manipulability of in vitro models allows for the investigation of various TME factors. Organ‐on‐a‐chip has gained attention for its ability to faithfully reproduce complex TME structures. 273 , 276 , 279 Combined with gene editing techniques and RNAi, these models enable the exploration of gene functions within the TME. 248 , 276 , 301
4. IN VIVO MOUSE MODEL FOR TUMOR IMMUNOLOGY INVESTIGATION
With an increasing interest in the advancement of efficient immunotherapies, the creation of immune‐competent mouse models that accurately replicate human diseases appears to be a key challenge. 317 In the context of developing standard cytotoxic cancer therapies, xenotransplantation models serve as an industry gold standard in which human cancer cell transplantations are entailed into immunocompromised mice to evaluate the effectiveness and safety. 317 However, the development of immunotherapies further necessitates systems possessing a completely functional immune system characterized by heterogeneity and adaptability, enabling constant adaptation and evolution along with the tumor. 318 Consequently, a crucial criterion for assessing a preclinical mouse model is its ability to mimic human cancer progression, encompassing the faithful reproduction of cancer genomic heterogeneity, as well as the establishment of an intricate TME housing substantial populations of immune and stromal cells (Figure 5). 318
FIGURE 5.
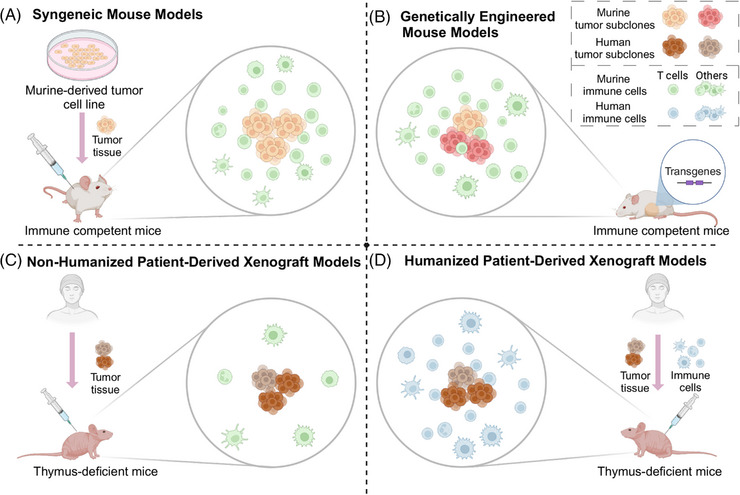
Comprehensive comparison of in vivo models in terms of tumor microenvironment reconstructions. (A) Syngeneic mouse models have the highest usability and the broadest applicability but are limited by factors including low tumor heterogeneity and the absence of tumor evolution processes. (B) Genetically engineered mouse models allow for the monitoring of the entire tumor development process, facilitating the study of gene contributions to tumor formation, but are still limited by the disparities between mice and human patients. (C) Patient‐derived xenografts maintain genomic diversity, tumor structure, and microenvironment of the human tumor. However, evaluating their utility requires careful consideration of host mice immunodeficiency levels. (D) Humanized patient‐derived xenografts facilitate the development of personalized immunotherapy, particularly in simulating the personalized tumor microenvironment. Figure was created with BioRender.com.
4.1. Syngeneic tumor model
Syngeneic tumor models are the most widely used choice in in vivo preclinical studies. 318 By utilizing inbred strains, tumor cell lines are isolated and expanded in vitro and subsequently transplanted to establish tumor‐bearing systems. 318 One of the key advantages is the high usability. By employing cell lines that can rapidly expand into consistent and large quantities, these models enable studies and databases that require substantial sample sizes, which can be challenging to achieve with genetically engineered models. 319 , 320 , 321 Moreover, syngeneic tumor models offer the opportunity to genetically manipulate the cells, allowing for the evaluation of specific biomarkers associated with immunotherapy sensitivity or resistance. 320 , 321 , 322 Additionally, researchers can assess the impact of various factors on the efficacy of immunotherapeutic approaches. 320 , 323 , 324 , 325
However, while syngeneic tumor models provide a stable biological nature of tumor grafts, they lack the genomic and microenvironmental heterogeneity that characterizes human tumors, including heterogeneity among different patients and heterogeneity within the same patient. 13 , 326 Fundamentally, this is due to the absence of the cancer stem cells, and the complex clonal evolution process from stem cells to the entire tumor. 327 To address this issue, one possible solution is to inject multiple lineages to generate tumors composed of multiple populations. For instance, when injecting SCLC into mouse models as a mixed population, the mesenchymal cells conferred metastatic capability to the neuroendocrine cells, thereby highlighting the significance of tumor cell heterogeneity in determining tumor properties. 328 On the other hand, syngeneic tumor models possess higher usability and efficiency compared with other models but lack the natural steps of tumor evolution. 329 To address this issue, tumor cells can be injected into the corresponding organ locations of mice with specific background diseases. 330 Additionally, the rapid growth rate of tumors in these models shortens the latency period, providing an inadequate time window to evaluate the efficacy of immunotherapy and making it impossible to study the early stages of cancer development. 331 , 332
4.2. Genetically engineered mouse model
GEMMs utilize transgenic mice with specific alleles, enabling the natural progression of malignancies in immunocompetent animals. 333 These models commonly employ tissue‐specific promoters to activate oncogene or induce deletion of tumor suppressor genes using recombinase enzymes. 334 , 335 By introducing these genomic alterations, GEMMs replicate the development of invasive cancers as well as precancerous lesions. 336 , 337 The extended period of tumor development in these models also provides a longer timeframe for immunotherapeutic interventions, which are important to elicit an effective antitumor immune response. 338 As GEMMs initiate the neoplastic conversion of healthy cells within the appropriate organ site, the stepwise evolution and advancement of cancer enable the establishment of a multifaceted TME, encompassing both immunosuppressive conditions and stromal vasculature. 333 Moreover, these models can be designed to mimic alterations in genes that impact the TME and, consequently, influence the immunotherapy efficacy. For example, by employing models in which tumor growth is induced by Pten deficiency, scientists can assess treatment strategies that augment the vulnerability of these tumors to immunotherapeutic interventions. 339
However, GEMMs have several limitations. The occurrence of deleterious mutations impacting multiple target cells at the organism or tissue level can lead to premature mortality in the model. 340 Furthermore, the tumor mutational burden observed in GEMMs may not precisely mirror that observed in the corresponding human cancer. 341 , 342 This is particularly crucial when assessing the efficacy of immunotherapies, as a higher mutational burden is a significant factor in evaluating the effectiveness of immune checkpoint blockade. 343 On the other hand, the incomplete penetrance of mutations in GEMMs leads to delayed cancer onset, causing nonsynchronous tumor occurrence among mice. 344 The development of nongermline GEMMs, conditional GEMMs, and advancements in genome editing technologies like CRISPR‐Cas9 have helped overcome these limitations. 29 , 335 , 345 As an illustration, when delivering potent tumor suppressors (Tp53 and Cdkn2a) targeted sgRNAs and Cas in GEMM liver, tumor formations were only observed when additional triggers existed, including Kras G12D mutation and CCl4 related inflammation. 346
4.3. Patient‐derived xenograft
PDXs are preclinical models created by injecting human tumor cells or implanting tumor tissue into immune‐deficient animal hosts. PDXs preserve the genomic heterogeneity, tumor architecture, and microenvironment factors of the primary tumor, making them valuable for evaluating therapeutic efficacy in vivo. 347 The success rate of establishing PDXs relies on various factors, including the animal species, cancer type, and specific implantation technique. 348 Metastatic tumors with aggressive behavior tend to have a higher engraftment success rate. Certain tumor types, such as colorectal or gastric cancer, have a higher success rate compared with others, like breast or kidney cancer. 348 , 349 , 350
Immunodeficiency levels in host mice are another critical consideration when assessing its applicability. Traditional athymic nude mice exhibit deficient thymic development and impaired T‐cell function but retain functional innate immune cells, including neutrophils, DCs, B cells, and NK cells. Thereby, they enable the representation of diverse aspects of the immune response. 351 To better replicate the original TME, researchers have developed humanized animal models. 352 , 353 These models involve the combination of reconstituted human hematopoietic systems with tumor samples. Immunodeficiency mice reconstituted with human CD34+ umbilical cord blood cells induce PDX regression when treated with anti‐PD‐1 therapy. 354 These responses were based on the combinations of human hematopoietic cells and allogeneic PDX, regardless of the degree of human leukocyte antigen (HLA) differentiation between hematopoietic stem cells (HSCs) and PDX. 354 Another approach to humanize mouse immune system is to use TILs from the same tumor. 355 Tumor cells and TILs from the same patient were transplanted sequentially into immune‐deficient mice and the demonstrations of antitumor activity were evaluated between responders and nonresponders. 355 Alternatively, modifying the host mice is another strategy to enhance the expansion of human immune cells. 356 , 357 As an exemplification, NSG‐SGM3 mice transgenically expressing human stem cell factors were found to have an improved human B cell development and function after human HSCs engraftment. 356 However, the impact of these modifications on the antitumor response in reconstituted mice remains unclear and requires further investigation.
4.4. Preservation of tumor immune microenvironment in in vivo models
To establish an effective testing platform for tumor immunotherapy in mouse models, it is crucial to reconstruct the TME that resembles the human tumor setting. GEMMs provide a valuable tool for studying tumor development in immunocompetent animals, allowing for the investigation of the TME. 29 However, the use of GEMMs for testing immunotherapy is limited by the potential cross‐reactivity between murine and human targets, particularly when agents require antigen presentation by human MHC class I. 355 To address this limitation, GEMMs incorporating human MHC class I and MHC class II have been developed to assess peptide‐specific T‐cell responses relevant to human antitumor immunity. 358 Additionally, GEMM models have been created to express target antigens, bridging the gap between human and mouse tumor antigens, and enabling a more accurate evaluation of antigen‐specific immunotherapies. 333 However, differences in antigen‐presenting mechanisms between humans and mice still exist, presenting potential challenges for future applications.
Another approach for reconstructing the TME in mouse models is using humanized models. Humanized models involve the engraftment of human immune cells into immunodeficient mice, allowing for the presence of both human tumor cells and human immune components. Humanized models can be generated by introducing PBMCs. 359 However, despite the feasibility of using PBMCs in autologous tumor‐bearing PDXs, challenges arise regarding the viability of these cells, the need for sequential blood draws from the patients, and the strong graft‐versus‐host reaction. 360 To address this, CD34+ HSCs or other hematopoietic progenitors could be used instead. 354 Besides, reconstructing the TME in PDX using autologous TILs is an emerging and promising approach. This strategy allows for the preservation of native immune components, thereby maintaining their complexity and functionality. 355
In addition, efforts have also been made to incorporate other TME components into mouse models. For example, the inclusion of stromal cells, such as CAFs, endothelial cells, and ECM components, can enhance the fidelity of the TME. 361 These models allow for the investigation of tumor‐stroma interactions, angiogenesis, and the influence of the ECM on tumor behavior. 362
4.5. Applications of in vivo tumor models in cancer immunology and immunotherapy
In recent years, there have been significant advances in cancer immunology and immunotherapy, which have revolutionized cancer treatment. In vivo mouse models play a pivotal role in evaluating the effectiveness of various immunotherapeutic strategies, including ICB and ACT. These models allow researchers to assess the tumor response to immunotherapy, measure immune cell infiltration, and investigate the mechanisms underlying treatment resistance. Furthermore, cancer immunology and TME have gained significant attention in recent years. In vivo tumor models provide a platform to study the complex interactions between cancer cells, immune cells, and stromal components within the TME. These models enable the exploration of key factors influencing tumor progression, immune evasion, and the development of novel immunomodulatory interventions.
4.5.1. Immunotherapy simulation, prediction, and optimization
Syngeneic mouse models are suitable for high‐throughput studies. The TISMO database encompassed 1518 RNA‐seq samples from 68 syngeneic mouse tumor models across 19 cancer types (832 were from ICB studies), providing great convenience for analyzing ICB response and resistance biomarkers. 319 Additionally, syngeneic mouse models can be used for biomarker research through genetical manipulations. For instance, PET radiotracers targeting CD4 and CD8 were developed and tested in syngeneic mouse models to find that CD4+ or CD8+ TILs can serve as anti‐PD‐1 therapy biomarkers. 322
GEMMs utilize transgenic mice with specific gene alterations, allowing for monitoring the natural progression of tumors. This characteristic provides a longer growth period and enables the study of long‐term effects of immunotherapy. 338 , 363 For adverse effects, GEMMs with slower tumor kinetics allow for immune‐related adverse events (irAE) development. Prolonged Treg depletion in Foxp3‐DTR mice served as biomarkers for the antitumor responses and irAE severity in ipilimumab/nivolumab‐treated patients. 363
PDX models based on humanized mice have rapidly developed and offer unique advantages in simulating personalized TME for immunotherapy investigations. 347 Moreover, the utilization of patient‐derived TILs allows for the study of ACT in humanized mouse models. 355 Tumor cells and TILs from the same patient were transplanted sequentially into immune‐deficient mice. TILs derived from ACT responders demonstrate antitumor activity, whereas TILs from nonresponders did not. 355
4.5.2. Cancer immunology and TME investigation
By utilizing syngeneic mouse models, researchers can explore various factors influencing the process of tumor immunity. 320 By utilizing three different syngeneic mouse models, distinct TME with different ICB response was constructed, demonstrating that increased TIL levels were related to high ICB sensitivity. 320 GEMMs have emerged as powerful tools for studying cancer progression, including the development of precursor lesions and the impact of various factors on tumor initiation and growth. 333 , 337 As an example, the concurrent expression of Trp53 R172H and Kras G12D in mouse pancreatic tissue resulted in the cooperative formation of invasive and highly metastatic carcinoma of pancreatic ductal adenocarcinoma. 337
PDX models involve the transplantation of patient tumor tissues into immunodeficient mice, allowing for the growth of tumors that retain the patient's original tumor characteristics and TME components. Therefore, PDX models provide a platform to study the interactions between tumor cells and the immune system in a personalized context. 347 Notably, CTCs have been employed to create animal models called cell line‐derived tumor xenografts (CDXs), which offer a distinct opportunity to assess the genomic characteristics of metastatic tumors. 364 CDX models were created by enriching CTCs from four SCLC patients and injecting them into animals. Genomic analysis of CDX models revealed preservation of the original mutational profile, and they also exhibited similar therapeutic responses as observed in patients. 365 Nevertheless, several limitations exist for this approach including technical challenges in isolating and expanding CTCs, the absence of noncancerous components, and the distinguishment of different metastatic sites. 222
4.5.3. Conclusions for in vivo tumor model applications
Compared with in vitro tumor models, in vivo mouse models’ advantage is possessing a functional immune system. However, it is important to acknowledge that a single type of model can only replicate an aspect of the in vivo TME. Therefore, developing new preclinical models or using multiple experimental models is necessary to complement each other's strengths and provide a more comprehensive understanding of cancer immunology. 318
5. DISCUSSION
Biomarkers serve as a crucial link between the unobservable immune status and the observable therapy response. In clinical applications, there is a growing demand for biomarkers that enable effective prediction and real‐time reflection of tumor status. 2 While PD‐L1 and MSI have been recommended by current clinical guidelines, 88 , 138 their efficacy remains limited. 97 , 144 In recent years, several novel biomarker candidates have emerged, including tumor neoantigens and biomarkers detected by liquid biopsy. 38 , 69 Tumor neoantigens, which are directly associated with immune recognition and T cell‐dependent cytotoxicity, have demonstrated effectiveness in various immunotherapies. 37 , 38 However, a significant challenge lies in accurately defining high‐quality neoantigens, which can be addressed through the application of emerging AI techniques that predict the relationships between protein structure and immunogenicity. 366 Biomarkers from liquid biopsy provide a noninvasive means of real‐time monitoring. 69 However, due to the low signal‐to‐noise ratio, it is crucial to employ noise reduction, decontamination, and feature extraction techniques. This necessitates the use of detection methods and prediction approaches with high sensitivity and specificity. 227 , 230
Biomarkers can reflect key aspects of the complex tumor immune processes, making them widely applicable in mechanism studies. For instance, surface markers and TILs can be used to evaluate the in vitro constructed TME, thereby aiding in the creation of a model that accurately reflects the real TME. 242 Compared with 2D models, 3D models offer a more comprehensive representation of the TME and can currently incorporate various tumor‐immune interactions. 28 , 241 , 242 However, challenges remain, including the need for improved accuracy in TME reconstruction, the alterations in cell type proportions over time, and the preservation of cell functionality. 242 In terms of animal models, the latest advancement involves reconstructing the human immune system in immune‐deficient mice. 359 However, due to insufficient TME reconstruction, long cultivation time, and low success rate, the clinical application of animal models is still limited. 318
Despite their widespread application, current experimental models are still far from fully replicating TME. Emerging technologies present an opportunity for the development of novel experimental models. 244 , 279 , 346 For instance, the ALI models allow direct tumor tissue culture, thereby avoiding cell damage during tissue processing and better preserving the diverse cell types within the model. 242 , 244 Organ‐on‐a‐chip utilizes microfluidic technologies to create 3D models that closely mimic the blood flow environment. 279 Tissue slice culture facilitates the simulation of interactions between two whole organs rather than isolated cells. 274 3D bio‐printing implements the precise construction of the TME. 280 Additionally, the CRISPR‐Cas9 gene editing technology has revolutionized the construction of humanized mouse models, making the process more convenient and efficient. 346
In conclusion, advancements in cancer immunology hold great potential for the development of efficient, accurate, and real‐time biomarkers, as well as experimental models that faithfully replicate the TME. With biomarkers acting as the bridge and experimental models serving as the platform, we will enter a new era of truly efficient and personalized cancer immunotherapy.
AUTHOR CONTRIBUTIONS
Hengyi Xu, Ziqi Jia, Jianzhong Su, and Jiaqi Liu contributed to the initial idea and conceptualization of the review article. Hengyi Xu, Ziqi Jia, Fengshuo Liu, and Jiayi Li were responsible for writing the initial draft of the review article. Hengyi Xu, Jiayi Li, Yiwen Jiang, Yufan Yang, and Yansong Huang prepared the figures. Pengming Pu, Pengrui Tang, Tongxuan Shang, Yufan Yang, and Yongxin Zhou prepared the tables. Hengyi Xu, Ziqi Jia, Fengshuo Liu, and Yufan Yang prepared the revision. Jiaqi Liu and Jianzhong Su reviewed the manuscript. All authors listed have made a substantial contribution to the work. All authors have read and approved the article.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
Supporting information
Supporting information
ACKNOWLEDGMENTS
I would like to express my sincere gratitude to the authors of the previous articles for providing valuable evidence and materials for this review. All figures including the graphic abstract were created by BioRender.com. This research was funded in part by National Natural Science Foundation of China (82272938 to Jiaqi Liu), Beijing Nova Program (20220484059 to Jiaqi Liu), the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐1‐014 to Jiaqi Liu), the Beijing Hope Run Special Fund (LC2020B05 to Jiaqi Liu), and the Beijing Science and Technology Innovation Foundation for University or College students (2022zglcss06074 to Hengyi Xu).
Xu H, Jia Z, Liu F, et al. Biomarkers and experimental models for cancer immunology investigation. MedComm. 2023;4:e437. 10.1002/mco2.437
Contributor Information
Jianzhong Su, Email: sujz@wmu.edu.cn.
Jiaqi Liu, Email: j.liu@cicams.ac.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Harlin H, Meng Y, Peterson AC, et al. Chemokine Expression in Melanoma Metastases associated with CD8+ T‐Cell recruitment. Cancer Res. 2009;69(7):3077‐3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang DR, Wu XL, Sun YL. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non‐response. Signal Transduct Target Ther. 2022;7(1):331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang M, Zhang C, Jiang X. CAR‐T: a potential gene carrier targeting solid tumor immune microenvironment. Signal Transduct Target Ther. 2021;6(1):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen R, Manochakian R, James L, et al. Emerging therapeutic agents for advanced non‐small cell lung cancer. J Hematol Oncol. 2020;13(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schadendorf D, van Akkooi ACJ, Berking C, et al. Melanoma. Lancet. 2018;392(10151):971‐984. [DOI] [PubMed] [Google Scholar]
- 6. Goldstein JS, Nastoupil LJ, Han X, Jemal A, Ward E, Flowers CR. Disparities in survival by insurance status in follicular lymphoma. Blood. 2018;132(11):1159‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375(19):1823. [DOI] [PubMed] [Google Scholar]
- 8. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science. 2017;357(6349):409‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmid P, Cortes J, Dent R, et al. Event‐free survival with pembrolizumab in early triple‐negative breast cancer. N Engl J Med. 2022;386(6):556‐567. [DOI] [PubMed] [Google Scholar]
- 10. Ren XY, Song Y, Wang J, Chen LY, Wu HW. Mismatch repair deficiency and microsatellite instability in triple‐negative breast cancer: a retrospective study of 440 patients. Front Oncol. 2021;11:570‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor‐infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18(4):842‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGrail DJ, Pilié PG, Rashid NU, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franklin MR, Platero S, Saini KS, Curigliano G, Anderson S. Immuno‐oncology trends: preclinical models, biomarkers, and clinical development. J Immunother Cancer. 2022;10(1):e003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu W, Zhou Y, Duan W, et al. Glutathione peroxidase 4‐dependent glutathione high‐consumption drives acquired platinum chemoresistance in lung cancer‐derived brain metastasis. Clin Transl Med. 2021;11(9):e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delarue M, Joanny JF, Jülicher F, Prost J. Stress distributions and cell flows in a growing cell aggregate. Interface Focus. 2014;4(6):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meijer TG, Naipal KA, Jager A, van Gent DC. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci OA. 2017;3(2):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boucherit N, Gorvel L, Olive D. 3D tumor models and their use for the testing of immunotherapies. Front Immunol. 2020;11:603640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida GJ. Applications of patient‐derived tumor xenograft models and tumor organoids. J Hematol Oncol. 2020;13(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sung PJ, Rama N, Imbach J, et al. Cancer‐associated fibroblasts produce netrin‐1 to control cancer cell plasticity. Cancer Res. 2019;79(14):3651‐3661. [DOI] [PubMed] [Google Scholar]
- 21. Riedl A, Schlederer M, Pudelko K, Stadler M, Dolznig H. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J Cell Sci. 2017;130(1):203‐218. [DOI] [PubMed] [Google Scholar]
- 22. Faget J, Biota C, Bachelot T, et al. Early detection of tumor cells by innate immune cells leads to Treg recruitment through CCL22 production by tumor cells. Cancer Res. 2011;71(19):6143‐6152. [DOI] [PubMed] [Google Scholar]
- 23. Han K, Pierce SE, Li A, Spees K, Bassik MC. CRISPR screens in cancer spheroids identify 3D growth‐specific vulnerabilities. Nature. 2020;580(7801):136‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rheinwald J, Green H. Serial cultivation of starins of human epidermal keratinocytes in defined clonal and serum‐free culture. J Invest Dermatol. 1975;6:331‐342. [Google Scholar]
- 25. Petersen OW, Rønnov‐Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89(19):9064‐9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato T, Stange DE, Ferrante M, et al. Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141(5):1762‐1772. [DOI] [PubMed] [Google Scholar]
- 27. Zervantonakis IK, Hughes‐Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three‐dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci USA. 2012;109(34):13515‐13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jenkins RW, Aref AR, Lizotte PH, et al. Ex vivo profiling of PD‐1 blockade using organotypic tumor spheroids. Cancer Discov. 2018;8(2):196‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zitvogel L, Pitt JM, Daillère R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16(12):759‐773. [DOI] [PubMed] [Google Scholar]
- 30. Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol. 1973;242(118):148‐149. [DOI] [PubMed] [Google Scholar]
- 31. Jaenisch R, Mintz B. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci USA. 1974;71(4):1250‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fiebig HH, Schuchhardt C, Henss H, Fiedler L, Löhr GW. Comparison of tumor response in nude mice and in the patients. Behring Inst Mitt. 1984(74):343‐352. [PubMed] [Google Scholar]
- 33. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59‐73. [DOI] [PubMed] [Google Scholar]
- 34. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Datar I, Sanmamed MF, Wang J, et al. Expression analysis and significance of PD‐1, LAG‐3, and TIM‐3 in human non‐small cell lung cancer using spatially resolved and multiparametric single‐cell analysis. Clin Cancer Res. 2019;25(15):4663‐4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kvistborg P, Philips D, Kelderman S, et al. Anti‐CTLA‐4 therapy broadens the melanoma‐reactive CD8+ T cell response. Sci Transl Med. 2014;6(254):254ra128. [DOI] [PubMed] [Google Scholar]
- 38. Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515(7528):577‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kvistborg P, Shu CJ, Heemskerk B, et al. TIL therapy broadens the tumor‐reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology. 2012;1(4):409‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Powles T, Durán I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum‐treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2018;391(10122):748‐757. [DOI] [PubMed] [Google Scholar]
- 41. Ferris RL, Blumenschein G, Jr. , Fayette J, et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science. 2015;350(6257):207‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sclafani F. PD‐1 inhibition in metastatic dMMR/MSI‐H colorectal cancer. Lancet Oncol. 2017;18(9):1141‐1142. [DOI] [PubMed] [Google Scholar]
- 44. Olivares‐Hernández A, Del Barco Morillo E, Parra Pérez C, et al. Influence of DNA mismatch repair (MMR) system in survival and response to immune checkpoint inhibitors (ICIs) in non‐small cell lung cancer (NSCLC): retrospective analysis. Biomedicines. 2022;10(2):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fucà G, Cohen R, Lonardi S, et al. Ascites and resistance to immune checkpoint inhibition in dMMR/MSI‐H metastatic colorectal and gastric cancers. J Immunother Cancer. 2022;10(2):e004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gim G, Kim Y, Park Y, et al. Response to nivolumab and ipilimumab in microsatellite instability‐high (MSI‐H) cervical carcinoma with acquired resistance to pembrolizumab: a case report and literature review. Oncologist. 2022;27(7):525‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yiu AJ, Yiu CY. Biomarkers in colorectal cancer. Anticancer Res. 2016;36(3):1093‐1102. [PubMed] [Google Scholar]
- 48. Zheng Y, Gao Q, Su X, et al. Genome‐wide DNA methylation and gene expression profiling characterizes molecular subtypes of esophagus squamous cell carcinoma for predicting patient survival and immunotherapy efficacy. Cancers (Basel). 2022;14(20):4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goltz D, Gevensleben H, Vogt TJ, et al. CTLA4 methylation predicts response to anti‐PD‐1 and anti‐CTLA‐4 immunotherapy in melanoma patients. JCI Insight. 2018;3(13):e96793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Starzer AM, Berghoff AS, Hamacher R, et al. Tumor DNA methylation profiles correlate with response to anti‐PD‐1 immune checkpoint inhibitor monotherapy in sarcoma patients. J Immunother Cancer. 2021;9(3):e001458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348(6230):124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang X, Weng X, Yang Y, et al. A combined hypoxia and immune gene signature for predicting survival and risk stratification in triple‐negative breast cancer. Aging (Albany NY). 2021;13(15):19486‐19509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J, Zhao X, Wang Y, et al. circRNA‐002178 act as a ceRNA to promote PDL1/PD1 expression in lung adenocarcinoma. Cell Death Dis. 2020;11(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang Q, Xia J, Wang L, et al. miR‐153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol. 2018;11(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson DB, Estrada MV, Salgado R, et al. Melanoma‐specific MHC‐II expression represents a tumour‐autonomous phenotype and predicts response to anti‐PD‐1/PD‐L1 therapy. Nat Commun. 2016;7:10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837‐1846. [DOI] [PubMed] [Google Scholar]
- 58. McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24(6):749‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hogan SA, Courtier A, Cheng PF, et al. Peripheral blood TCR repertoire profiling may facilitate patient stratification for immunotherapy against melanoma. Cancer Immunol Res. 2019;7(1):77‐85. [DOI] [PubMed] [Google Scholar]
- 61. Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high‐risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24(34):5373‐5380. [DOI] [PubMed] [Google Scholar]
- 62. Perrone G, Ruffini PA, Catalano V, et al. Intratumoural FOXP3‐positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer. 2008;44(13):1875‐1882. [DOI] [PubMed] [Google Scholar]
- 63. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med. 2019;25(8):1251‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thommen DS, Koelzer VH, Herzig P, et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med. 2018;24(7):994‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561‐565. [DOI] [PubMed] [Google Scholar]
- 67. Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim SS, Shen S, Miyauchi S, et al. B cells improve overall survival in HPV‐associated squamous cell carcinomas and are activated by radiation and PD‐1 blockade. Clin Cancer Res. 2020;26(13):3345‐3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang Q, Luo J, Wu S, et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov. 2020;10(12):1842‐1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lindsay CR, Faugeroux V, Michiels S, et al. A prospective examination of circulating tumor cell profiles in non‐small‐cell lung cancer molecular subgroups. Ann Oncol. 2017;28(7):1523‐1531. [DOI] [PubMed] [Google Scholar]
- 71. Luoma AM, Suo S, Wang Y, et al. Tissue‐resident memory and circulating T cells are early responders to pre‐surgical cancer immunotherapy. Cell. 2022;185(16):2918‐2935.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Forschner A, Battke F, Hadaschik D, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA‐4 and PD‐1 antibody therapy in metastatic melanoma ‐ results of a prospective biomarker study. J Immunother Cancer. 2019;7(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shimada Y, Matsubayashi J, Kudo Y, et al. Serum‐derived exosomal PD‐L1 expression to predict anti‐PD‐1 response and in patients with non‐small cell lung cancer. Sci Rep. 2021;11(1):7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen G, Huang AC, Zhang W, et al. Exosomal PD‐L1 contributes to immunosuppression and is associated with anti‐PD‐1 response. Nature. 2018;560(7718):382‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Abril‐Rodriguez G, Ribas A. SnapShot: Immune Checkpoint Inhibitors. Cancer Cell. 2017;31(6):848‐848.e1. [DOI] [PubMed] [Google Scholar]
- 76. Kroemer G, Zitvogel L. Immune checkpoint inhibitors. J Exp Med. 2021;218(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38(3):326‐333. [DOI] [PubMed] [Google Scholar]
- 78. Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J Exp Clin Cancer Res. 2018;37(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123(11):1904‐1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD‐1 and CTLA‐4. Mol Cancer. 2019;18(1):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rotte A. Combination of CTLA‐4 and PD‐1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD‐1, PD‐L1 and CTLA‐4: breakthroughs or backups. Nat Immunol. 2019;20(11):1425‐1434. [DOI] [PubMed] [Google Scholar]
- 83. Xu‐Monette ZY, Zhou J, Young KH. PD‐1 expression and clinical PD‐1 blockade in B‐cell lymphomas. Blood. 2018;131(1):68‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verma V, Shrimali RK, Ahmad S, et al. PD‐1 blockade in subprimed CD8 cells induces dysfunctional PD‐1(+)CD38(hi) cells and anti‐PD‐1 resistance. Nat Immunol. 2019;20(9):1231‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Garcia‐Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep. 2017;19(6):1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Powles T, Morrison L. Biomarker challenges for immune checkpoint inhibitors in urothelial carcinoma. Nat Rev Urol. 2018;15(10):585‐587. [DOI] [PubMed] [Google Scholar]
- 88. Zaretsky JM, Garcia‐Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med. 2016;375(9):819‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD‐L1 expression through 3'‐UTR disruption in multiple cancers. Nature. 2016;534(7607):402‐406. [DOI] [PubMed] [Google Scholar]
- 90. Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD‐L1 mRNA. Immunity. 2017;47(6):1083‐1099.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cheng X, Zhang B, Guo F, Wu H, Jin X. Deubiquitination of FBP1 by USP7 blocks FBP1‐DNMT1 interaction and decreases the sensitivity of pancreatic cancer cells to PARP inhibitors. Mol Oncol. 2022;16(7):1591‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD‐L1 expression in cancers and associated opportunities for novel small‐molecule therapeutics. Nat Rev Clin Oncol. 2022;19(5):287‐305. [DOI] [PubMed] [Google Scholar]
- 93. Doroshow DB, Bhalla S, Beasley MB, et al. PD‐L1 as a biomarker of response to immune‐checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345‐362. [DOI] [PubMed] [Google Scholar]
- 94. Lastwika KJ, Wilson W 3rd, Li QK, et al. Control of PD‐L1 expression by oncogenic activation of the AKT‐mTOR pathway in non‐small cell lung cancer. Cancer Res. 2016;76(2):227‐238. [DOI] [PubMed] [Google Scholar]
- 95. Lyu Q, Lin A, Cao M, Xu A, Luo P, Zhang J. Alterations in TP53 are a potential biomarker of bladder cancer patients who benefit from immune checkpoint inhibition. Cancer Control. 2020;27(1):1073274820976665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang F, Zhao Q, Wang YN, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5(10):1504‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hirashima T, Kanai T, Suzuki H, et al. The levels of interferon‐gamma release as a biomarker for non‐small‐cell lung cancer patients receiving immune checkpoint inhibitors. Anticancer Res. 2019;39(11):6231‐6240. [DOI] [PubMed] [Google Scholar]
- 98. Cortes J, Cescon DW, Rugo HS, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple‐negative breast cancer (KEYNOTE‐355): a randomised, placebo‐controlled, double‐blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817‐1828. [DOI] [PubMed] [Google Scholar]
- 99. Mittendorf EA, Zhang H, Barrios CH, et al. Neoadjuvant atezolizumab in combination with sequential nab‐paclitaxel and anthracycline‐based chemotherapy versus placebo and chemotherapy in patients with early‐stage triple‐negative breast cancer (IMpassion031): a randomised, double‐blind, phase 3 trial. Lancet. 2020;396(10257):1090‐1100. [DOI] [PubMed] [Google Scholar]
- 100. Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab‐paclitaxel as first‐line treatment for unresectable, locally advanced or metastatic triple‐negative breast cancer (IMpassion130): updated efficacy results from a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2020;21(1):44‐59. [DOI] [PubMed] [Google Scholar]
- 101. Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti‐CTLA‐4 and anti‐PD‐1 checkpoint blockade. Cell. 2017;170(6):1120‐1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Robert L, Harview C, Emerson R, et al. Distinct immunological mechanisms of CTLA‐4 and PD‐1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology. 2014;3:e29244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shi LZ, Goswami S, Fu T, et al. Blockade of CTLA‐4 and PD‐1 enhances adoptive T‐cell therapy efficacy in an ICOS‐mediated manner. Cancer Immunol Res. 2019;7(11):1803‐1812. [DOI] [PubMed] [Google Scholar]
- 104. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhao L, Cheng S, Fan L, Zhang B, Xu S. TIM‐3: an update on immunotherapy. Int Immunopharmacol. 2021;99:107933. [DOI] [PubMed] [Google Scholar]
- 106. Lu S, Stein JE, Rimm DL, et al. Comparison of biomarker modalities for predicting response to PD‐1/PD‐L1 checkpoint blockade: a systematic review and meta‐analysis. JAMA Oncol. 2019;5(8):1195‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zugazagoitia J, Gupta S, Liu Y, et al. Biomarkers associated with beneficial PD‐1 checkpoint blockade in Non‐Small Cell Lung Cancer (NSCLC) identified using high‐plex digital spatial profiling. Clin Cancer Res. 2020;26(16):4360‐4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Moutafi MK, Molero M, Martinez Morilla S, et al. Spatially resolved proteomic profiling identifies tumor cell CD44 as a biomarker associated with sensitivity to PD‐1 axis blockade in advanced non‐small‐cell lung cancer. J Immunother Cancer. 2022;10(8):e004757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA‐binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2(6):522‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wan JCM, Massie C, Garcia‐Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223‐238. [DOI] [PubMed] [Google Scholar]
- 112. Robbins PF, Lu YC, El‐Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor‐reactive T cells. Nat Med. 2013;19(6):747‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lo AA, Wallace A, Oreper D, et al. Indication‐specific tumor evolution and its impact on neoantigen targeting and biomarkers for individualized cancer immunotherapies. J Immunother Cancer. 2021;9(10):e003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8(1):e000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60(9):1192‐1199. [DOI] [PubMed] [Google Scholar]
- 116. Woodward ER, Green K, Burghel GJ, et al. 30 year experience of index case identification and outcomes of cascade testing in high‐risk breast and colorectal cancer predisposition genes. Eur J Hum Genet. 2022;30(4):413‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Jardim DL, Goodman A, de Melo Gagliato D, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. 2021;39(2):154‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lopez‐Beltran A, López‐Rios F, Montironi R, Wildsmith S, Eckstein M. Immune checkpoint inhibitors in urothelial carcinoma: recommendations for practical approaches to PD‐L1 and other potential predictive biomarker testing. Cancers (Basel). 2021;13(6):1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Domingo E, Freeman‐Mills L, Rayner E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1(3):207‐216. [DOI] [PubMed] [Google Scholar]
- 120. Weber S, van der Leest P, Donker HC, et al. Dynamic changes of circulating tumor DNA predict clinical outcome in patients with advanced non‐small‐cell lung cancer treated with immune checkpoint inhibitors. JCO Precis Oncol. 2021;5:1540‐1553. [DOI] [PubMed] [Google Scholar]
- 121. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069‐1086. [DOI] [PubMed] [Google Scholar]
- 124. Goodman AM, Castro A, Pyke RM, et al. MHC‐I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 2020;12(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science. 2016;352(6286):658‐660. [DOI] [PubMed] [Google Scholar]
- 126. Vokes NI, Liu D, Ricciuti B, et al. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non‐small‐cell lung cancer. JCO Precis Oncol. 2019;3:PO1900171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Stenzinger A, Endris V, Budczies J, et al. Harmonization and standardization of panel‐based tumor mutational burden measurement: Real‐world results and recommendations of the quality in pathology study. J Thorac Oncol. 2020;15(7):1177‐1189. [DOI] [PubMed] [Google Scholar]
- 128. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lobo J, Rodrigues Â, Guimarães R, et al. Detailed characterization of immune cell infiltrate and expression of immune checkpoint molecules PD‐L1/CTLA‐4 and MMR proteins in testicular germ cell tumors disclose novel disease biomarkers. Cancers (Basel). 2019;11(10):1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Guo X, Li S, Tong H, et al. Case report: complete response to antiangiogenesis and immune checkpoint blockade in an unresectable MMR‐deficient leiomyosarcoma harboring biallelic loss of PTEN. Front Oncol. 2022;12:802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sahin IH, Goyal S, Pumpalova Y, et al. Mismatch repair (MMR) gene alteration and BRAF V600E mutation are potential predictive biomarkers of immune checkpoint inhibitors in MMR‐deficient colorectal cancer. Oncologist. 2021;26(8):668‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Oliveira AF, Bretes L, Furtado I. Review of PD‐1/PD‐L1 inhibitors in metastatic dMMR/MSI‐H colorectal cancer. Front Oncol. 2019;9:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhou C, Jiang T, Xiao Y, et al. Good tumor response to chemoradioimmunotherapy in dMMR/MSI‐H advanced colorectal cancer: a case series. Front Immunol. 2021;12:784336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lower SS, McGurk MP, Clark AG, Barbash DA. Satellite DNA evolution: old ideas, new approaches. Curr Opin Genet Dev. 2018;49:70‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pećina‐Šlaus N, Kafka A, Salamon I, Bukovac A. Mismatch repair pathway, genome stability and cancer. Front Mol Biosci. 2020;7:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Luchini C, Bibeau F, Ligtenberg MJL, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD‐1/PD‐L1 expression and tumour mutational burden: a systematic review‐based approach. Ann Oncol. 2019;30(8):1232‐1243. [DOI] [PubMed] [Google Scholar]
- 137. Sahin IH, Akce M, Alese O, et al. Immune checkpoint inhibitors for the treatment of MSI‐H/MMR‐D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer. 2019;121(10):809‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site ‐ when a biomarker defines the indication. N Engl J Med. 2017;377(15):1409‐1412. [DOI] [PubMed] [Google Scholar]
- 139. Kloor M, von Knebel Doeberitz M. The immune biology of microsatellite‐unstable cancer. Trends Cancer. 2016;2(3):121‐133. [DOI] [PubMed] [Google Scholar]
- 140. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017:PO1700073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): an open‐label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Le DT, Uram JN, Wang H, et al. PD‐1 Blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: is more always better? Clin Cancer Res. 2021;27(5):1236‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Addeo A, Banna GL, Weiss GJ. Tumor mutation burden‐from hopes to doubts. JAMA Oncol. 2019;5(7):934‐935. [DOI] [PubMed] [Google Scholar]
- 145. Cai L, Bai H, Duan J, et al. Epigenetic alterations are associated with tumor mutation burden in non‐small cell lung cancer. J Immunother Cancer. 2019;7(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Li D, Zhao W, Zhang X, Lv H, Li C, Sun L. NEFM DNA methylation correlates with immune infiltration and survival in breast cancer. Clin Epigenetics. 2021;13(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cheng Y, Zhang T, Xu Q. Therapeutic advances in non‐small cell lung cancer: focus on clinical development of targeted therapy and immunotherapy. MedComm. 2021;2(4):692‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hamidi S, Nakaya Y, Nagai H, et al. Mesenchymal‐epithelial transition regulates initiation of pluripotency exit before gastrulation. Development. 2020;147(3):dev184960. [DOI] [PubMed] [Google Scholar]
- 149. Villalba M, Evans SR, Vidal‐Vanaclocha F, Calvo A. Role of TGF‐β in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370(1):29‐39. [DOI] [PubMed] [Google Scholar]
- 150. Yan J, Wu X, Zhu Y, Cang S. Genome‐wide DNA methylation profile analysis identifies an individualized predictive signature for melanoma immune response. J Cancer Res Clin Oncol. 2023;149(1):343‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang R, Chen C, Dong X, et al. Independent validation of early‐stage non‐small cell lung cancer prognostic scores incorporating epigenetic and transcriptional biomarkers with gene‐gene interactions and main effects. Chest. 2020;158(2):808‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yang G, Shan D, Zhao R, Li G. Metabolism‐associated DNA methylation signature stratifies lower‐grade glioma patients and predicts response to immunotherapy. Front Cell Dev Biol. 2022;10:902298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ye F, Liang Y, Hu J, et al. DNA methylation modification map to predict tumor molecular subtypes and efficacy of immunotherapy in bladder cancer. Front Cell Dev Biol. 2021;9:760369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590‐607. [DOI] [PubMed] [Google Scholar]
- 155. Postow MA, Manuel M, Wong P, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Linnemann C, Mezzadra R, Schumacher TN. TCR repertoires of intratumoral T‐cell subsets. Immunol Rev. 2014;257(1):72‐82. [DOI] [PubMed] [Google Scholar]
- 157. Ma X, Riaz N, Samstein RM, et al. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade‐dependent antitumor immunity. Nat Genet. 2022;54(7):996‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ma X, Dong L, Liu X, Ou K, Yang L. POLE/POLD1 mutation and tumor immunotherapy. J Exp Clin Cancer Res. 2022;41(1):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Yusko E, Vignali M, Wilson RK, et al. Association of tumor microenvironment T‐cell repertoire and mutational load with clinical outcome after sequential checkpoint blockade in melanoma. Cancer Immunol Res. 2019;7(3):458‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hopkins AC, Yarchoan M, Durham JN, et al. T cell receptor repertoire features associated with survival in immunotherapy‐treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3(13):e122092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ansell SM, Lesokhin AM, Borrello I, et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Goodman AM, Piccioni D, Kato S, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4(9):1237‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Roemer MG, Advani RH, Ligon AH, et al. PD‐L1 and PD‐L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34(23):2690‐2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol. 2016;17(12):e542‐e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang D, Wei G, Ma J, et al. Identification of the prognostic value of ferroptosis‐related gene signature in breast cancer patients. BMC Cancer. 2021;21(1):645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Chong W, Shang L, Liu J, et al. m(6)A regulator‐based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics. 2021;11(5):2201‐2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang H, Hu X, Huang M, et al. Mettl3‐mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Han D, Liu J, Chen C, et al. Anti‐tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Iqbal MA, Arora S, Prakasam G, Calin GA, Syed MA. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol Aspects Med. 2019;70:3‐20. [DOI] [PubMed] [Google Scholar]
- 170. Zhang Q, Wang W, Zhou Q, et al. Roles of circRNAs in the tumour microenvironment. Mol Cancer. 2020;19(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mohapatra S, Pioppini C, Ozpolat B, Calin GA. Non‐coding RNAs regulation of macrophage polarization in cancer. Mol Cancer. 2021;20(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Li J, Xue Y, Amin MT, et al. ncRNA‐eQTL: a database to systematically evaluate the effects of SNPs on non‐coding RNA expression across cancer types. Nucleic Acids Res. 2020;48(D1):D956‐d963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Liu Z, Liu L, Weng S, et al. Machine learning‐based integration develops an immune‐derived lncRNA signature for improving outcomes in colorectal cancer. Nat Commun. 2022;13(1):816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Cristescu R, Mogg R, Ayers M, et al. Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade‐based immunotherapy. Science. 2018;362(6411):eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Haddad RI, Seiwert TY, Chow LQM, et al. Influence of tumor mutational burden, inflammatory gene expression profile, and PD‐L1 expression on response to pembrolizumab in head and neck squamous cell carcinoma. J Immunother Cancer. 2022;10(2):e003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Ayers M, Lunceford J, Nebozhyn M, et al. IFN‐γ‐related mRNA profile predicts clinical response to PD‐1 blockade. J Clin Invest. 2017;127(8):2930‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Egelston CA, Avalos C, Tu TY, et al. Human breast tumor‐infiltrating CD8(+) T cells retain polyfunctionality despite PD‐1 expression. Nat Commun. 2018;9(1):4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wu TD, Madireddi S, de Almeida PE, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579(7798):274‐278. [DOI] [PubMed] [Google Scholar]
- 180. Araujo BdLV, Borch A, Hansen M, et al. Common phenotypic dynamics of tumor‐infiltrating lymphocytes across different histologies upon checkpoint inhibition: impact on clinical outcome. Cytotherapy. 2020;22(4):204‐213. [DOI] [PubMed] [Google Scholar]
- 181. Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654‐2666. [DOI] [PubMed] [Google Scholar]
- 182. Zaid A, Hor JL, Christo SN, et al. Chemokine receptor‐dependent control of skin tissue‐resident memory T cell formation. J Immunol. 2017;199(7):2451‐2459. [DOI] [PubMed] [Google Scholar]
- 183. Gide TN, Quek C, Menzies AM, et al. Distinct immune cell populations define response to anti‐PD‐1 monotherapy and anti‐PD‐1/anti‐CTLA‐4 combined therapy. Cancer Cell. 2019;35(2):238‐255.e6. [DOI] [PubMed] [Google Scholar]
- 184. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635‐647. [DOI] [PubMed] [Google Scholar]
- 185. Alspach E, Lussier DM, Miceli AP, et al. MHC‐II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co‐signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun. 2018;95:77‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Jordanova ES, Gorter A, Ayachi O, et al. Human leukocyte antigen class I, MHC class I chain‐related molecule A, and CD8+/regulatory T‐cell ratio: which variable determines survival of cervical cancer patients? Clin Cancer Res. 2008;14(7):2028‐2035. [DOI] [PubMed] [Google Scholar]
- 188. Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T‐cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137(4):1270‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Arce Vargas F, Furness AJS, Litchfield K, et al. Fc effector function contributes to the activity of human anti‐CTLA‐4 antibodies. Cancer Cell. 2018;33(4):649‐663.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kamada T, Togashi Y, Tay C, et al. PD‐1(+) regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA. 2019;116(20):9999‐10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Jiang Z, Zhu H, Wang P, et al. Different subpopulations of regulatory T cells in human autoimmune disease, transplantation, and tumor immunity. MedComm. 2022;3(2):e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Lam JH, Hong M, Koo SL, et al. CD30(+)OX40(+) Treg is associated with improved overall survival in colorectal cancer. Cancer Immunol Immunother. 2021;70(8):2353‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492‐499. [DOI] [PubMed] [Google Scholar]
- 194. Siddiqui I, Schaeuble K, Chennupati V, et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195‐211.e10. [DOI] [PubMed] [Google Scholar]
- 195. Yamauchi T, Hoki T, Oba T, et al. CX3CR1‐CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI Insight. 2020;5(8):e133920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Nelson MA, Ngamcherdtrakul W, Luoh SW, Yantasee W. Prognostic and therapeutic role of tumor‐infiltrating lymphocyte subtypes in breast cancer. Cancer Metastasis Rev. 2021;40(2):519‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Petitprez F, de Reyniès A, Keung EZ, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556‐560. [DOI] [PubMed] [Google Scholar]
- 198. Lu Y, Zhao Q, Liao JY, et al. Complement signals determine opposite effects of B cells in chemotherapy‐induced immunity. Cell. 2020;180(6):1081‐1097.e24. [DOI] [PubMed] [Google Scholar]
- 199. Lindner S, Dahlke K, Sontheimer K, et al. Interleukin 21‐induced granzyme B‐expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013;73(8):2468‐2479. [DOI] [PubMed] [Google Scholar]
- 200. Shen P, Roch T, Lampropoulou V, et al. IL‐35‐producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Schioppa T, Moore R, Thompson RG, et al. B regulatory cells and the tumor‐promoting actions of TNF‐α during squamous carcinogenesis. Proc Natl Acad Sci USA. 2011;108(26):10662‐10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour‐infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022;22(7):414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Sivapalan L, Murray JC, Canzoniero JV, et al. Liquid biopsy approaches to capture tumor evolution and clinical outcomes during cancer immunotherapy. J Immunother Cancer. 2023;11(1):e005924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Zhou H, Zhu L, Song J, et al. Liquid biopsy at the frontier of detection, prognosis and progression monitoring in colorectal cancer. Mol Cancer. 2022;21(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Tay TKY, Tan PH. Liquid biopsy in breast cancer: a focused review. Arch Pathol Lab Med. 2021;145(6):678‐686. [DOI] [PubMed] [Google Scholar]
- 206. Zhang Z, Wu H, Chong W, Shang L, Jing C, Li L. Liquid biopsy in gastric cancer: predictive and prognostic biomarkers. Cell Death Dis. 2022;13(10):903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Li W, Liu JB, Hou LK, et al. Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring. Mol Cancer. 2022;21(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Duffy MJ, Crown J. Use of circulating tumour DNA (ctDNA) for measurement of therapy predictive biomarkers in patients with cancer. J Pers Med. 2022;12(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Powles T, Assaf ZJ, Davarpanah N, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595(7867):432‐437. [DOI] [PubMed] [Google Scholar]
- 210. Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first‐line treatment of metastatic non‐small cell lung cancer: the MYSTIC Phase 3 randomized clinical trial. JAMA Oncol. 2020;6(5):661‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med. 2019;25(9):1415‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Gandara DR, Paul SM, Kowanetz M, et al. Blood‐based tumor mutational burden as a predictor of clinical benefit in non‐small‐cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441‐1448. [DOI] [PubMed] [Google Scholar]
- 213. Georgiadis A, Durham JN, Keefer LA, et al. Noninvasive detection of microsatellite instability and high tumor mutation burden in cancer patients treated with PD‐1 blockade. Clin Cancer Res. 2019;25(23):7024‐7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Willis J, Lefterova MI, Artyomenko A, et al. Validation of microsatellite instability detection using a comprehensive plasma‐based genotyping panel. Clin Cancer Res. 2019;25(23):7035‐7045. [DOI] [PubMed] [Google Scholar]
- 215. Kasi PM, Klempner SJ, Starr JS, et al. Clinical utility of microsatellite instability (MSI‐H) identified on liquid biopsy in advanced gastrointestinal cancers (aGI). J Clin Oncol. 2022;40(4_suppl):56‐56. [Google Scholar]
- 216. Forde PM, Spicer J, Lu S, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386(21):1973‐1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Zviran A, Schulman RC, Shah M, et al. Genome‐wide cell‐free DNA mutational integration enables ultra‐sensitive cancer monitoring. Nat Med. 2020;26(7):1114‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Chen K, Zhao H, Shi Y, et al. Perioperative dynamic changes in circulating tumor DNA in patients with lung cancer (DYNAMIC). Clin Cancer Res. 2019;25(23):7058‐7067. [DOI] [PubMed] [Google Scholar]
- 219. Oliver J, Garcia‐Aranda M, Chaves P, et al. Emerging noninvasive methylation biomarkers of cancer prognosis and drug response prediction. Semin Cancer Biol. 2022;83:584‐595. [DOI] [PubMed] [Google Scholar]
- 220. Locke WJ, Guanzon D, Ma C, et al. DNA methylation cancer biomarkers: translation to the clinic. Front Genet. 2019;10:1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Panagopoulou M, Karaglani M, Balgkouranidou I, et al. Circulating cell‐free DNA in breast cancer: size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene. 2019;38(18):3387‐3401. [DOI] [PubMed] [Google Scholar]
- 222. Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. 2016;35(10):1216‐1224. [DOI] [PubMed] [Google Scholar]
- 223. Zhong X, Zhang H, Zhu Y, et al. Circulating tumor cells in cancer patients: developments and clinical applications for immunotherapy. Mol Cancer. 2020;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. de Kruijf EM, Sajet A, van Nes JG, et al. HLA‐E and HLA‐G expression in classical HLA class I‐negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol. 2010;185(12):7452‐7459. [DOI] [PubMed] [Google Scholar]
- 225. Santos MF, Mannam VK, Craft BS, et al. Comparative analysis of innate immune system function in metastatic breast, colorectal, and prostate cancer patients with circulating tumor cells. Exp Mol Pathol. 2014;96(3):367‐374. [DOI] [PubMed] [Google Scholar]
- 226. Zhou J, Dong F, Cui F, Xu R, Tang X. The role of circulating tumor cells in evaluation of prognosis and treatment response in advanced non‐small‐cell lung cancer. Cancer Chemother Pharmacol. 2017;79(4):825‐833. [DOI] [PubMed] [Google Scholar]
- 227. Lim M, Kim CJ, Sunkara V, Kim MH, Cho YK. Liquid biopsy in lung cancer: clinical applications of circulating biomarkers (CTCs and ctDNA). Micromachines (Basel). 2018;9(3):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Monterisi S, Castello A, Toschi L, et al. Preliminary data on circulating tumor cells in metastatic NSCLC patients candidate to immunotherapy. Am J Nucl Med Mol Imaging. 2019;9(6):282‐295. [PMC free article] [PubMed] [Google Scholar]
- 229. Zhou Q, Liu X, Li J, et al. Circulating tumor cells PD‐L1 expression detection and correlation of therapeutic efficacy of immune checkpoint inhibition in advanced non‐small‐cell lung cancer. Thorac Cancer. 2023;14(5):470‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wei X, Chen K, Cai B, et al. An acoustic droplet‐induced enzyme responsive platform for the capture and on‐demand release of single circulating tumor cells. ACS Appl Mater Interfaces. 2019;11(44):41118‐41126. [DOI] [PubMed] [Google Scholar]
- 231. Kang H, Kim J, Park J. Methods to isolate extracellular vesicles for diagnosis. Micro Nano Syst Lett. 2017;5(1):15. [Google Scholar]
- 232. Mathew M, Zade M, Mezghani N, Patel R, Wang Y, Momen‐Heravi F. Extracellular vesicles as biomarkers in cancer immunotherapy. Cancers (Basel). 2020;12(10):2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Zhou W, Fong MY, Min Y, et al. Cancer‐secreted miR‐105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25(4):501‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shen Y, Xue C, Li X, et al. Effects of gastric cancer cell‐derived exosomes on the immune regulation of mesenchymal stem cells by the NF‐kB signaling pathway. Stem Cells Dev. 2019;28(7):464‐476. [DOI] [PubMed] [Google Scholar]
- 236. Yen EY, Miaw SC, Yu JS, Lai IR. Exosomal TGF‐β1 is correlated with lymphatic metastasis of gastric cancers. Am J Cancer Res. 2017;7(11):2199‐2208. [PMC free article] [PubMed] [Google Scholar]
- 237. Wang J, Guan X, Zhang Y, et al. Exosomal miR‐27a derived from gastric cancer cells regulates the transformation of fibroblasts into cancer‐associated fibroblasts. Cell Physiol Biochem. 2018;49(3):869‐883. [DOI] [PubMed] [Google Scholar]
- 238. Zhu L, Sun HT, Wang S, et al. Isolation and characterization of exosomes for cancer research. J Hematol Oncol. 2020;13(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Thakur BK, Zhang H, Becker A, et al. Double‐stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Yuwen DL, Sheng BB, Liu J, Wenyu W, Shu YQ. MiR‐146a‐5p level in serum exosomes predicts therapeutic effect of cisplatin in non‐small cell lung cancer. Eur Rev Med Pharmacol Sci. 2017;21(11):2650‐2658. [PubMed] [Google Scholar]
- 241. Hong HK, Yun NH, Jeong YL, et al. Establishment of patient‐derived organotypic tumor spheroid models for tumor microenvironment modeling. Cancer Med. 2021;10(16):5589‐5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Neal JT, Li X, Zhu J, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972‐1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Kamer I, Bab‐Dinitz E, Zadok O, et al. Immunotherapy response modeling by ex‐vivo organ culture for lung cancer. Cancer Immunol Immunother. 2021;70(8):2223‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Yin Q, Yu W, Grzeskowiak CL, et al. Nanoparticle‐enabled innate immune stimulation activates endogenous tumor‐infiltrating T cells with broad antigen specificities. Proc Natl Acad Sci USA. 2021;118(21):e2016168118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Delarue M, Montel F, Vignjevic D, Prost J, Joanny JF, Cappello G. Compressive stress inhibits proliferation in tumor spheroids through a volume limitation. Biophys J. 2014;107(8):1821‐1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7(3):211‐224. [DOI] [PubMed] [Google Scholar]
- 247. Pampaloni F, Reynaud EG, Stelzer EHK. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8(10):839‐845. [DOI] [PubMed] [Google Scholar]
- 248. Koh V, Chakrabarti J, Torvund M, et al. Hedgehog transcriptional effector GLI mediates mTOR‐Induced PD‐L1 expression in gastric cancer organoids. Cancer Lett. 2021;518:59‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Wang Z, Li Y, Wang Y, et al. Targeting prostate cancer stem‐like cells by an immunotherapeutic platform based on immunogenic peptide‐sensitized dendritic cells‐cytokine‐induced killer cells. Stem Cell Res Ther. 2020;11(1):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Weil S, Memmer S, Lechner A, et al. Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front Immunol. 2017;8:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Varesano S, Zocchi MR, Poggi A. Zoledronate triggers Vδ2 T cells to destroy and kill spheroids of colon carcinoma: quantitative image analysis of three‐dimensional cultures. Front Immunol. 2018;9:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Maas RJ, Hoogstad‐van Evert JS, Van der Meer JM, et al. TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM‐1/TIGIT/CD96 checkpoint molecules in ovarian cancer. Oncoimmunology. 2020;9(1):1843247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Zhou S, Meng F, Du S, et al. Bifunctional iRGD‐anti‐CD3 enhances antitumor potency of T cells by facilitating tumor infiltration and T‐cell activation. J Immunother Cancer. 2021;9(5):e001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Seino T, Kawasaki S, Shimokawa M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22(3):454‐467. [DOI] [PubMed] [Google Scholar]
- 255. Luangwattananun P, Junking M, Sujjitjoon J, et al. Fourth‐generation chimeric antigen receptor T cells targeting folate receptor alpha antigen expressed on breast cancer cells for adoptive T cell therapy. Breast Cancer Res Treat. 2021;186(1):25‐36. [DOI] [PubMed] [Google Scholar]
- 256. Gopal S, Kwon SJ, Ku B, Lee DW, Kim J, Dordick JS. 3D tumor spheroid microarray for high‐throughput, high‐content natural killer cell‐mediated cytotoxicity. Commun Biol. 2021;4(1):893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Dijkstra KK, Cattaneo CM, Weeber F, et al. Generation of tumor‐reactive T cells by co‐culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174(6):1586‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Koeck S, Kern J, Zwierzina M, et al. The influence of stromal cells and tumor‐microenvironment‐derived cytokines and chemokines on CD3(+)CD8(+) tumor infiltrating lymphocyte subpopulations. Oncoimmunology. 2017;6(6):e1323617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Wan C, Keany MP, Dong H, et al. Enhanced efficacy of simultaneous PD‐1 and PD‐L1 immune checkpoint blockade in high‐grade serous ovarian cancer. Cancer Res. 2021;81(1):158‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Holokai L, Chakrabarti J, Lundy J, et al. Murine‐ and human‐derived autologous organoid/immune cell co‐cultures as pre‐clinical models of pancreatic ductal adenocarcinoma. Cancers (Basel). 2020;12(12):3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Chandrakesan P, Panneerselvam J, May R, et al. DCLK1‐isoform2 alternative splice variant promotes pancreatic tumor immunosuppressive m2‐macrophage polarization. Mol Cancer Ther. 2020;19(7):1539‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Zhao R, Zhou X, Khan ES, et al. Targeting the microtubule‐network rescues CTL killing efficiency in dense 3d matrices. Front Immunol. 2021;12:729820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Sánchez‐Rodríguez C, Cruces KP, Riestra Ayora J, Martín‐Sanz E, Sanz‐Fernández R. BCG immune activation reduces growth and angiogenesis in an in vitro model of head and neck squamous cell carcinoma. Vaccine. 2017;35(47):6395‐6403. [DOI] [PubMed] [Google Scholar]
- 264. Meng Q, Xie S, Gray GK, et al. Empirical identification and validation of tumor‐targeting T cell receptors from circulation using autologous pancreatic tumor organoids. J Immunother Cancer. 2021;9(11):e003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Liu T, Tan J, Wu M, et al. High‐affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39(+)CD8(+) T cells. Gut. 2021;70(10):1965‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Warwas KM, Meyer M, Gonçalves M, et al. Co‐stimulatory bispecific antibodies induce enhanced T cell activation and tumor cell killing in breast cancer models. Front Immunol. 2021;12:719116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Zou F, Tan J, Liu T, et al. The CD39(+) HBV surface protein‐targeted CAR‐T and personalized tumor‐reactive CD8(+) T cells exhibit potent anti‐HCC activity. Mol Ther. 2021;29(5):1794‐1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Supimon K, Sangsuwannukul T, Sujjitjoon J, et al. Anti‐mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci Rep. 2021;11(1):6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Gonzalez‐Exposito R, Semiannikova M, Griffiths B, et al. CEA expression heterogeneity and plasticity confer resistance to the CEA‐targeting bispecific immunotherapy antibody cibisatamab (CEA‐TCB) in patient‐derived colorectal cancer organoids. J Immunother Cancer. 2019;7(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Di Mascolo D, Varesano S, Benelli R, et al. Nanoformulated zoledronic acid boosts the Vδ2 T cell immunotherapeutic potential in colorectal cancer. Cancers (Basel). 2019;12(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt‐dependent stem cell niche. Nat Med. 2019;15(6):701‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Sontheimer‐Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs‐on‐chips. Nat Rev Cancer. 2019;19(2):65‐81. [DOI] [PubMed] [Google Scholar]
- 273. Kim S, Park J, Kim J, Jeon JS. Microfluidic Tumor Vasculature Model to Recapitulate an Endothelial Immune Barrier Expressing FasL. ACS Biomater Sci Eng. 2021;7(3):1230‐1241. [DOI] [PubMed] [Google Scholar]
- 274. Shim S, Belanger MC, Harris AR, Munson JM, Pompano RR. Two‐way communication between ex vivo tissues on a microfluidic chip: application to tumor‐lymph node interaction. Lab Chip. 2019;19(6):1013‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Park D, Son K, Hwang Y, et al. High‐throughput microfluidic 3D cytotoxicity assay for cancer immunotherapy (CACI‐IMPACT Platform). Front Immunol. 2019;10:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Mustafa DAM, Pedrosa R, Smid M, et al. T lymphocytes facilitate brain metastasis of breast cancer by inducing Guanylate‐Binding Protein 1 expression. Acta Neuropathol. 2018;135(4):581‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Preece R, Pavesi A, Gkazi SA, et al. CRISPR‐mediated base conversion allows discriminatory depletion of endogenous T cell receptors for enhanced synthetic immunity. Mol Ther Methods Clin Dev. 2020;19:149‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Henrik Heiland D, Ravi VM, Behringer SP, et al. Tumor‐associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat Commun. 2019;10(1):2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Song J, Choi H, Koh SK, et al. High‐throughput 3D in vitro tumor vasculature model for real‐time monitoring of immune cell infiltration and cytotoxicity. Front Immunol. 2021;12:733317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Matai I, Kaur G, Seyedsalehi A, McClinton A, Laurencin CT. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials. 2020;226:119536. [DOI] [PubMed] [Google Scholar]
- 281. Dey M, Ozbolat IT. 3D bioprinting of cells, tissues and organs. Sci Rep. 2020;10(1):14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nat Biotechnol. 2014;32(8):773‐785. [DOI] [PubMed] [Google Scholar]
- 283. Hong N, Yang GH, Lee J, Kim G. 3D bioprinting and its in vivo applications. J Biomed Mater Res B Appl Biomater. 2018;106(1):444‐459. [DOI] [PubMed] [Google Scholar]
- 284. Li X, Liu B, Pei B, et al. Inkjet bioprinting of biomaterials. Chem Rev. 2020;120(19):10793‐10833. [DOI] [PubMed] [Google Scholar]
- 285. Chen K, Jiang E, Wei X, et al. The acoustic droplet printing of functional tumor microenvironments. Lab Chip. 2021;21(8):1604‐1612. [DOI] [PubMed] [Google Scholar]
- 286. Zhu W, Ma X, Gou M, Mei D, Zhang K, Chen S. 3D printing of functional biomaterials for tissue engineering. Curr Opin Biotechnol. 2016;40:103‐112. [DOI] [PubMed] [Google Scholar]
- 287. Yang H, Yang KH, Narayan RJ, Ma S. Laser‐based bioprinting for multilayer cell patterning in tissue engineering and cancer research. Essays Biochem. 2021;65(3):409‐416. [DOI] [PubMed] [Google Scholar]
- 288. Hakobyan D, Médina C, Dusserre N, et al. Laser‐assisted 3D bioprinting of exocrine pancreas spheroid models for cancer initiation study. Biofabrication. 2020;12(3):035001. [DOI] [PubMed] [Google Scholar]
- 289. Ozbolat IT, Hospodiuk M. Current advances and future perspectives in extrusion‐based bioprinting. Biomaterials. 2016;76:321‐343. [DOI] [PubMed] [Google Scholar]
- 290. Mironov V, Boland T, Trusk T, Forgacs G, Markwald RR. Organ printing: computer‐aided jet‐based 3D tissue engineering. Trends Biotechnol. 2003;21(4):157‐161. [DOI] [PubMed] [Google Scholar]
- 291. Flores‐Torres S, Jiang T, Kort‐Mascort J, et al. Constructing 3D in vitro models of heterocellular solid tumors and stromal tissues using extrusion‐based bioprinting. ACS Biomater Sci Eng. 2023;9(2):542‐561. [DOI] [PubMed] [Google Scholar]
- 292. Mazzaglia C, Sheng Y, Rodrigues LN, Lei IM, Shields JD, Huang YYS. Deployable extrusion bioprinting of compartmental tumoroids with cancer associated fibroblasts for immune cell interactions. Biofabrication. 2023;15(2). [DOI] [PubMed] [Google Scholar]
- 293. Grunewald L, Lam T, Andersch L, et al. A reproducible bioprinted 3D tumor model serves as a preselection tool for CAR T cell therapy optimization. Front Immunol. 2021;12:689697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Heinrich MA, Bansal R, Lammers T, Zhang YS, Michel Schiffelers R, Prakash J. 3D‐bioprinted mini‐brain: a glioblastoma model to study cellular interactions and therapeutics. Adv Mater. 2019;31(14):e1806590. [DOI] [PubMed] [Google Scholar]
- 295. Dai X, Shao Y, Tian X, et al. Fusion between glioma stem cells and mesenchymal stem cells promotes malignant progression in 3D‐bioprinted models. ACS Appl Mater Interfaces. 2022;14(31):35344‐35356. [DOI] [PubMed] [Google Scholar]
- 296. Choi YM, Lee H, Ann M, Song M, Rheey J, Jang J. 3D bioprinted vascularized lung cancer organoid models with underlying disease capable of more precise drug evaluation. Biofabrication. 2023;15(3). [DOI] [PubMed] [Google Scholar]
- 297. Guillerey C. NK Cells in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1273:69‐90. [DOI] [PubMed] [Google Scholar]
- 298. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36‐50. [DOI] [PubMed] [Google Scholar]
- 299. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13):6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Sui Q, Liu D, Jiang W, et al. Dickkopf 1 impairs the tumor response to PD‐1 blockade by inactivating CD8+ T cells in deficient mismatch repair colorectal cancer. J Immunother Cancer. 2021;9(3):e001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Chakrabarti J, Koh V, Steele N, et al. Disruption of Her2‐induced PD‐L1 inhibits tumor cell immune evasion in patient‐derived gastric cancer organoids. Cancers (Basel). 2021;13(24):6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Sherman H, Gitschier HJ, Rossi AE. A novel three‐dimensional immune oncology model for high‐throughput testing of tumoricidal activity. Front Immunol. 2018;9:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Hennenberg EM, Eyking A, Reis H, Cario E. MDR1A deficiency restrains tumor growth in murine colitis‐associated carcinogenesis. PLoS One. 2017;12(7):e0180834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Meng Q, Zhang Y, Hu LG. Targeting autophagy facilitates T lymphocyte migration by inducing the expression of CXCL10 in gastric cancer cell lines. Front Oncol. 2020;10:886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Zhou Z, Van der Jeught K, Fang Y, et al. An organoid‐based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T‐cell‐mediated cytotoxicity. Nat Biomed Eng. 2021;5(11):1320‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262‐265. [DOI] [PubMed] [Google Scholar]
- 307. Roshani Asl E , Rasmi Y, Baradaran B. MicroRNA‐124‐3p suppresses PD‐L1 expression and inhibits tumorigenesis of colorectal cancer cells via modulating STAT3 signaling. J Cell Physiol. 2021;236(10):7071‐7087. [DOI] [PubMed] [Google Scholar]
- 308. Downs‐Canner SM, Meier J, Vincent BG, Serody JS. B Cell Function in the Tumor Microenvironment. Annu Rev Immunol. 2022;40:169‐193. [DOI] [PubMed] [Google Scholar]
- 309. Wang R, Mao Y, Wang W, et al. Systematic evaluation of colorectal cancer organoid system by single‐cell RNA‐Seq analysis. Genome Biol. 2022;23(1):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Driehuis E, Kretzschmar K, Clevers H. Establishment of patient‐derived cancer organoids for drug‐screening applications. Nat Protoc. 2020;15(10):3380‐3409. [DOI] [PubMed] [Google Scholar]
- 311. Natânia de Souza‐Araújo C, Rodrigues Tonetti C, Cardoso MR, et al. Three‐dimensional cell culture based on magnetic fields to assemble low‐grade ovarian carcinoma cell aggregates containing lymphocytes. Cells. 2020;9(3):635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Ganesh K, Wu C, O'Rourke KP, et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat Med. 2019;25(10):1607‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Pasch CA, Favreau PF, Yueh AE, et al. Patient‐derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clin Cancer Res. 2019;25(17):5376‐5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Votanopoulos KI, Forsythe S, Sivakumar H, et al. Model of patient‐specific immune‐enhanced organoids for immunotherapy screening: feasibility study. Ann Surg Oncol. 2020;27(6):1956‐1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Klein Geltink RI, Edwards‐Hicks J, Apostolova P, et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat Metab. 2020;2(8):703‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9(8):1221‐1235. [DOI] [PubMed] [Google Scholar]
- 317. DeVita VT, Jr. , Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643‐8653. [DOI] [PubMed] [Google Scholar]
- 318. Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018;8(11):1358‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Zeng Z, Wong CJ, Yang L, et al. TISMO: syngeneic mouse tumor database to model tumor immunity and immunotherapy response. Nucleic Acids Res. 2022;50(D1):D1391‐d1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Lal JC, Townsend MG, Mehta AK, et al. Comparing syngeneic and autochthonous models of breast cancer to identify tumor immune components that correlate with response to immunotherapy in breast cancer. Breast Cancer Res. 2021;23(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Zeng Z, Gu SS, Wong CJ, et al. Machine learning on syngeneic mouse tumor profiles to model clinical immunotherapy response. Sci Adv. 2022;8(41):eabm8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Kristensen LK, Fröhlich C, Christensen C, et al. CD4(+) and CD8a(+) PET imaging predicts response to novel PD‐1 checkpoint inhibitor: studies of Sym021 in syngeneic mouse cancer models. Theranostics. 2019;9(26):8221‐8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Sato Y, Fu Y, Liu H, Lee MY, Shaw MH. Tumor‐immune profiling of CT‐26 and Colon 26 syngeneic mouse models reveals mechanism of anti‐PD‐1 response. BMC Cancer. 2021;21(1):1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Han MG, Jang BS, Kang MH, Na D, Kim IA. PI3Kγδ inhibitor plus radiation enhances the antitumour immune effect of PD‐1 blockade in syngenic murine breast cancer and humanised patient‐derived xenograft model. Eur J Cancer. 2021;157:450‐463. [DOI] [PubMed] [Google Scholar]
- 325. Mosely SI, Prime JE, Sainson RC, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5(1):29‐41. [DOI] [PubMed] [Google Scholar]
- 326. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 327. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822‐829. [DOI] [PubMed] [Google Scholar]
- 328. Calbo J, van Montfort E, Proost N, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19(2):244‐256. [DOI] [PubMed] [Google Scholar]
- 329. Greenberg NM, DeMayo F, Finegold MJ, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA. 1995;92(8):3439‐3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Bonnotte B, Gough M, Phan V, et al. Intradermal injection, as opposed to subcutaneous injection, enhances immunogenicity and suppresses tumorigenicity of tumor cells. Cancer Res. 2003;63(9):2145‐2149. [PubMed] [Google Scholar]
- 331. Madan RA, Gulley JL, Fojo T, Dahut WL. Therapeutic cancer vaccines in prostate cancer: the paradox of improved survival without changes in time to progression. Oncologist. 2010;15(9):969‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Gulley JL, Drake CG. Immunotherapy for prostate cancer: recent advances, lessons learned, and areas for further research. Clin Cancer Res. 2011;17(12):3884‐3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med. 2017;9(2):137‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v‐Ha‐ras and MMTV/c‐myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49(4):465‐475. [DOI] [PubMed] [Google Scholar]
- 335. Heyer J, Kwong LN, Lowe SW, Chin L. Non‐germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10(7):470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Kaplan‐Lefko PJ, Chen TM, Ittmann MM, et al. Pathobiology of autochthonous prostate cancer in a pre‐clinical transgenic mouse model. Prostate. 2003;55(3):219‐237. [DOI] [PubMed] [Google Scholar]
- 337. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469‐483. [DOI] [PubMed] [Google Scholar]
- 338. Liu J, Blake SJ, Yong MC, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6(12):1382‐1399. [DOI] [PubMed] [Google Scholar]
- 339. Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov. 2016;6(2):202‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163(1):39‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Yen J, White RM, Wedge DC, et al. The genetic heterogeneity and mutational burden of engineered melanomas in zebrafish models. Genome Biol. 2013;14(10):R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. McFadden DG, Politi K, Bhutkar A, et al. Mutational landscape of EGFR‐, MYC‐, and Kras‐driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci USA. 2016;113(42):E6409‐e6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Sharpless NE, DePinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006/09/01 2006;5(9):741‐754. [DOI] [PubMed] [Google Scholar]
- 345. Kaltenbacher T, Löprich J, Maresch R, et al. CRISPR somatic genome engineering and cancer modeling in the mouse pancreas and liver. Nat Protoc. 2022;17(4):1142‐1188. [DOI] [PubMed] [Google Scholar]
- 346. Weber J, Öllinger R, Friedrich M, et al. CRISPR/Cas9 somatic multiplex‐mutagenesis for high‐throughput functional cancer genomics in mice. Proc Natl Acad Sci USA. 2015;112(45):13982‐13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Hidalgo M, Amant F, Biankin AV, et al. Patient‐derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Okada S, Vaeteewoottacharn K, Kariya R. Application of highly immunocompromised mice for the establishment of patient‐derived xenograft (PDX) models. Cells. 2019;8(8):889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Murayama T, Gotoh N. Patient‐derived xenograft models of breast cancer and their application. Cells. 2019;8(6):621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Namekawa T, Ikeda K, Horie‐Inoue K, Inoue S. Application of prostate cancer models for preclinical study: advantages and limitations of cell lines, patient‐derived xenografts, and three‐dimensional culture of patient‐derived cells. Cells. 2019;8(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Hoffman RM. Patient‐derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer. 2015;15(8):451‐452. [DOI] [PubMed] [Google Scholar]
- 352. Du X, Liu M, Su J, et al. Uncoupling therapeutic from immunotherapy‐related adverse effects for safer and effective anti‐CTLA‐4 antibodies in CTLA4 humanized mice. Cell Res. 2018;28(4):433‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Mhaidly R, Verhoeyen E. Humanized mice are precious tools for preclinical evaluation of CAR T and CAR NK cell therapies. Cancers (Basel). 2020;12(7):1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Drake AC, Chen Q, Chen J. Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol. 2012;9(3):215‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Jespersen H, Lindberg MF, Donia M, et al. Clinical responses to adoptive T‐cell transfer can be modeled in an autologous immune‐humanized mouse model. Nat Commun. 2017;8(1):707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD‐scid IL2Rγ(null) mice transgenically expressing human stem cell factor, granulocyte‐macrophage colony‐stimulating factor and interleukin‐3. Immun Inflamm Dis. 2016;4(4):427‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Saito Y, Ellegast JM, Rafiei A, et al. Peripheral blood CD34(+) cells efficiently engraft human cytokine knock‐in mice. Blood. 2016;128(14):1829‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Pajot A, Michel ML, Fazilleau N, et al. A mouse model of human adaptive immune functions: HLA‐A2.1‐/HLA‐DR1‐transgenic H‐2 class I‐/class II‐knockout mice. Eur J Immunol. 2004;34(11):3060‐3069. [DOI] [PubMed] [Google Scholar]
- 359. Chuprin J, Buettner H, Seedhom MO, et al. Humanized mouse models for immuno‐oncology research. Nat Rev Clin Oncol. 2023/03/01 2023;20(3):192‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. King MA, Covassin L, Brehm MA, et al. Human peripheral blood leucocyte non‐obese diabetic‐severe combined immunodeficiency interleukin‐2 receptor gamma chain gene mouse model of xenogeneic graft‐versus‐host‐like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157(1):104‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Wang RM, Johnson TD, He J, et al. Humanized mouse model for assessing the human immune response to xenogeneic and allogeneic decellularized biomaterials. Biomaterials. 2017;129:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Li Y, Kumacheva E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci Adv. 2018;4(4):eaas8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Liu J, Blake SJ, Harjunpää H, et al. Assessing immune‐related adverse events of efficacious combination immunotherapies in preclinical models of cancer. Cancer Res. 2016;76(18):5288‐5301. [DOI] [PubMed] [Google Scholar]
- 364. Lin D, Shen L, Luo M, et al. Circulating tumor cells: biology and clinical significance. Signal Transduct Target Ther. 2021;6(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Hodgkinson CL, Morrow CJ, Li Y, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small‐cell lung cancer. Nat Med. 2014;20(8):897‐903. [DOI] [PubMed] [Google Scholar]
- 366. Lei Y, Li S, Liu Z, et al. A deep‐learning framework for multi‐level peptide‐protein interaction prediction. Nat Commun. 2021;12(1):5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Not applicable.