

Abstract
Emerging evidence indicates that cancer cells can mimic characteristics of embryonic development, promoting their development and progression. Cancer cells share features with embryonic development, characterized by robust proliferation and differentiation regulated by signaling pathways such as Wnt, Notch, hedgehog, and Hippo signaling. In certain phase, these cells also mimic embryonic diapause and fertilized egg implantation to evade treatments or immune elimination and promote metastasis. Additionally, the upregulation of ATP‐binding cassette (ABC) transporters, including multidrug resistance protein 1 (MDR1), multidrug resistance‐associated protein 1 (MRP1), and breast cancer‐resistant protein (BCRP), in drug‐resistant cancer cells, analogous to their role in placental development, may facilitate chemotherapy efflux, further resulting in treatment resistance. In this review, we concentrate on the underlying mechanisms that contribute to tumor development and progression from the perspective of embryonic development, encompassing the dysregulation of developmental signaling pathways, the emergence of dormant cancer cells, immune microenvironment remodeling, and the hyperactivation of ABC transporters. Furthermore, we synthesize and emphasize the connections between cancer hallmarks and embryonic development, offering novel insights for the development of innovative cancer treatment strategies.
Keywords: cancer therapy, drug resistance, embryonic development, tumor development and progression
Various regulatory mechanisms are involved in the development of a fertilized egg into an embryo to ensure that it proceeds normally. Meanwhile, cancer cells also exhibit several embryo‐like characteristics that enhance multidrug resistance. (A) The level of corpus‐secreted progesterone can be mediated by the inner or outer environment and lead to embryonic diapause. (a) Cancer cells are able to transition to a dormant state after treatment. (B) Wnt, Notch, Hedgehog, and Hippo signaling pathways contribute to development by driving the proliferation and differentiation of embryonic cells. (b) Cancer stem cells are considered drug‐resistant cells associated with some embryonic developmental signaling pathways. (C) Preimplantation embryo was reported to drive the formation of immunosuppressive microenvironment to evade attack from maternal immune system. (c) The immune microenvironment components of cancer cells can confer resistance to immune checkpoint blockades (ICBs). (D) To export environmental toxins and provide a favorable condition for development, ABC transporters are usually overexpressed in the blood–embryo barrier. (d) ABC transporter overexpression in cancer cells results in the development of multidrug resistance.
1. BACKGROUND
Embryogenesis is a sophisticated process accompanied by rapid cell proliferation, differentiation, and material exchange in early or later development. 1 During this period, embryonic development‐related pathways, such as Wnt, Notch, hedgehog, Hippo, and transforming growth factor (TGF)‐β signaling, are continuously activated and coordinated with each other to meet the needs of embryonic development. 2 , 3 Specifically, the Wnt family regulates embryo polarity and patterning and the morphogenesis of several organs. 4 Endothelial‐to‐hematopoietic and epithelial‐to‐mesenchymal transitions (EMTs) can both be regulated by Notch signaling. Hedgehog signaling provides positional information and fate instruction to cells. 5 The Hippo signaling is critical for angiogenesis and vascular development. 6 In addition, transforming Growth Factor‐β (TGF‐β) signaling cascade contributes to organogenesis. 7
To cope with harsh conditions for embryonic development, hundreds of mammalian species utilize diapause, a period of suspended development, to avoid the adverse effects of the environment. 8 , 9 Moreover, fetuses can help to reprogram immune microenvironment to prevent immunological rejection during implantation. 10 In addition, to supply a large amount of energy and nutrients required during embryonic development, the exchange of substances between mammalian embryos and mothers must operate rapidly, simultaneously leading to the accumulation of environmental toxins. 11 , 12 But in fact, ABC transporters, a class of transmembrane proteins in placental barrier functions and significant reproductive processes, are overexpressed in the placenta and control the efflux of toxic substances. 13 Although the underlying mechanisms controlling embryonic features need further investigation, these characteristics provide an essential foundation for embryonic development.
A growing body of evidence suggests that cancer cells can mimic embryonic traits in development and progression called oncofetal reprogramming 14 , 15 , 16 (Figure 1). First, the dysregulation of embryonic development signaling cascades was frequently detected in cancer cells, which contributes to tumor development and progression. 17 , 18 , 19 , 20 Targeted therapies focused on developmental pathways have been developed, and many of them have become clinical treatment drugs for various types of cancers. 21 Meanwhile, the overexpression of ABC transporters plays a role in regulating the tumor immune microenvironment through the transport of various cytokines, thereby influencing antitumor immunity and the sensitivity to anticancer drugs. 22 The administration of ABC transporter inhibitors in combination with standard chemotherapeutics or immunotherapy could attenuate, at least in part, cancer resistance. 23 , 24 Moreover, clinical studies have demonstrated that a small fraction of tumor cells can survive after systemic treatment, exhibiting several characteristics akin to circulating tumor cells, although the underlying mechanism remains a mystery. 25 Several excellent studies have revealed that these residual tumor cells could transition into an embryonic diapause‐like state, referred to as drug‐tolerant persister cells, cancer stem cells (CSCs), or dormant cancer cells, which contribute to cancer relapse after a period of dormancy. 26 , 27 , 28 While there are no well‐established methods for eliminating these residual cancer cells, understanding how cells enter a quiescent state may provide new therapeutic approaches to cancer recurrence. In addition, the reprogramming of the immune microenvironment endows cancer cells with the ability to escape immune surveillance. 29
FIGURE 1.
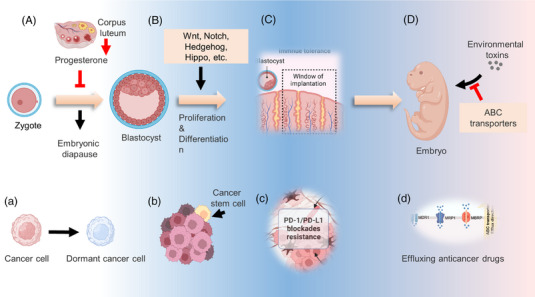
The similar characteristics between embryonic development and cancer development and progression. Various regulatory mechanisms are involved in the development of a fertilized egg into an embryo to ensure that it proceeds normally. Meanwhile, cancer cells also exhibit several embryo‐like characteristics that enhance multidrug resistance. (A) The level of corpus‐secreted progesterone can be mediated by the inner or outer environment and lead to embryonic diapause. (a) Cancer cells are able to transition to a dormant state after treatment. (B) Wnt, Notch, Hedgehog, and Hippo signaling pathways contribute to development by driving the proliferation and differentiation of embryonic cells. (b) Cancer stem cells are considered drug‐resistant cells associated with some embryonic developmental signaling pathways. (C) Preimplantation embryo was reported to drive the formation of immunosuppressive microenvironment to evade attack from maternal immune system. (c) The immune microenvironment components of cancer cells can confer resistance to immune checkpoint blockades (ICBs) such as programmed death‐ligand 1 (PD‐L1)/programmed cell death‐1 (PD‐1). (D) To export environmental toxins and provide a favorable condition for development, ABC transporters are usually overexpressed in the blood–embryo barrier. (d) ATP‐binding cassette (ABC) transporters overexpression in cancer cells results in the development of multidrug resistance.
To date, there has been far less emphasis on the systematic revisit of the specific traits that govern tumor development and progression. In this review, we examine in detail how cancer cells mimic developmental‐related features, including the dysregulation of development pathways, embryonic diapause‐like transition, reprogramming of the immune microenvironment, and overexpression of the ABC transporter. Furthermore, we also discuss potential strategies to reverse the tumor embryo‐like state in cancer therapy.
2. DYSREGULATION OF EMBRYONIC DEVELOPMENT SIGNALING PATHWAYS IN CANCER
Wnt, Notch, Hedgehog, Hippo, TGF‐β, and Fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) signaling are essential for embryonic development. However, abnormal regulation of these pathways usually correlates with the development and progression of numerous cancers. 30 , 31 , 32 , 33 Here, we review how cancer cells hijack these development‐related pathways.
2.1. Reactivation of Wnt signaling pathway
The first Wnt gene, Wnt1, which accounts for mammary tumorigenesis in mice, was discovered approximately 40 years ago. 34 At the turn of the century, most of the crucial elements in the Wnt pathway had been discovered. In the canonical Wnt cascade, the ligand‐bound Wnt receptor complexes can block the phosphorylation of β‐catenin. Therefore, the component of specific E3 ubiquitin ligase β‐TrCP can no longer recognize and deregulate β‐catenin. Stabilized β‐catenin binds to T‐cell factor (TCF) in the nucleus, promoting the transcription of Wnt downstream genes. Wnt signaling is crucial for embryonic development and tissue homeostasis in nature. It regulates stem cell self‐renewal and determines the cell fate in various organs, including the intestine and skin, which tumor suppressors tightly control via negative feedback loops or direct regulation. 35 , 36 , 37 However, persistent hyperactivation of the Wnt pathway is observed in various kinds of cancer cells 38 (Figure 2).
FIGURE 2.
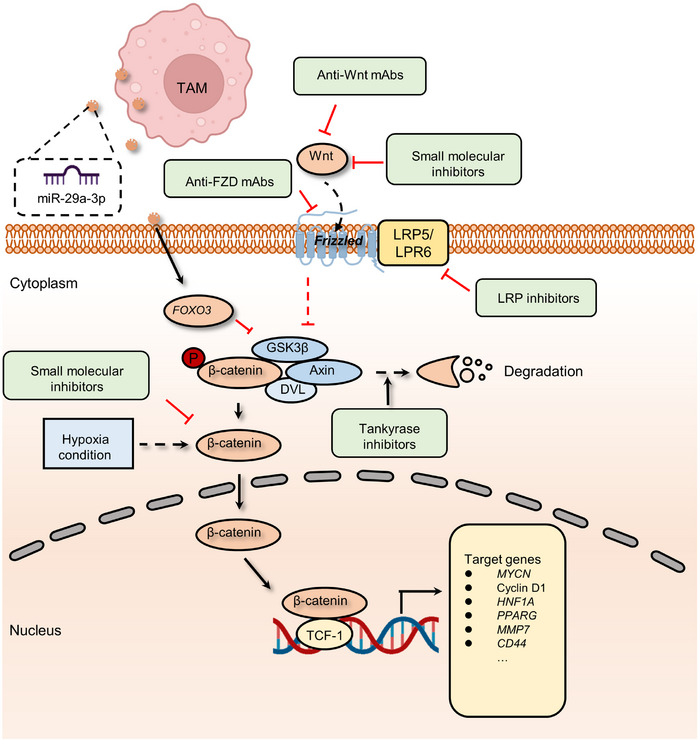
The mechanism of canonical Wnt signaling and related pharmacological inhibitors. The binding of Wnt proteins to Frizzled (Fzd) family receptors can inhibit the phosphorylation of β‐catenin mediated by the destruction complex (mainly including glycogen synthase kinase 3β (GSK3β), Axin, and Dishevelled protein (DVL)) and thereby avoiding degradation. Stable β‐catenin will be translocated into the nucleus and trigger target gene transcription by interacting with TCF‐1 and other factors. Wnt target gene expression, such as MYCN, endows resistance to cancer drugs on tumor cells. However, GSK3β could be inhibited by forkhead box O3 (FOXO3), which is activated by tumor‐associated macrophage (TAM)‐secreted exosomal miR‐29a‐3p. Hypoxic conditions also enhance the proliferation and function of myeloid‐derived suppressor cells (MDSCs) and lead to an immunosuppressive environment. Agents targeting diverse proteins were validated to impede the activation of Wnt pathway, including anti‐Wnt mAbs and small molecular inhibitors of Wnt ligands, low‐density lipoprotein (LDL)‐related protein (LRP) inhibitors, small molecular inhibitors targeting β‐catenin, and tankyrase inhibitors that promote β‐catenin degradation.
The role of the Wnt pathway in cancer has been extensively elucidated, and its functions primarily include promoting tumor proliferation, metastasis, stem cell maintenance, and drug resistance. 39 In gastric cancer, long non‐coding RNA (lncRNA) small nucleolar host gene 11 (SNHG11), upregulated in multiple cancer, induced glycogen synthase kinase 3β (GSK‐3β) ubiquitination activate the Wnt/β‐catenin pathway and contribute to cell proliferation, stemness, migration, invasion, and EMT. 40 In addition to the canonical Wnt/β‐catenin cascade mentioned above, currently, there are two different pathways believed to be activated when Wnt receptors are activated: the planar cell polarity (PCP) pathway and the Wnt/Ca2+ pathway. Moreover, the noncanonical Wnt pathways play crucial roles in embryonic development and can also be hijacked and activate the transcription of downstream target genes by tumor cells. 39 For example, frizzled family receptor 7 (FZD7), a receptor for Wnt signaling, is associated with aggressiveness in Stem‐A ovarian cancer by casein kinase 1ɛ‐mediated non‐canonical Wnt/PCP pathway. 41
With the continuous development of antitumor drugs, the role of the Wnt pathway in tumor treatment resistance is increasingly emerging. A body of research has shown that the activity of the Wnt/β‐catenin pathway can be upregulated in various cancers, 42 , 43 , 44 confer tumor cells the ability to survive in the insult from radiotherapy or chemotherapeutics, such as 5‐fluorouracil (5‐FU), oxaliplatin, temozolomide, and other agents. 45 , 46 , 47 For instance, chronic hypoxia increased the expression of hypoxia‐inducible factor 2α (HIF‐2α) and induced the resistance of breast cancer cells to paclitaxel (PTX). 48 More precisely, HIF‐2α overexpression increases the activation of Wnt signaling. Dickkopf‐1, a Wnt inhibitor, strikingly reverses the resistance to PTX. 46 In addition to chemotherapy, hyperactivation of the Wnt pathway is also associated with decreased sensitivity to radiation therapy. The miR‐301a in hypoxic glioma cell‐derived exosomes can directly target TCEAL7, which upregulates the activity of the Wnt pathway by boosting β‐catenin translocation from the cytoplasm into the nucleus and enhances radiation sensitivity. This resistance to radiotherapy can be reversed by inhibiting the activation of the Wnt/β‐catenin pathway. 49 Moreover, reactivation of Wnt signaling is also involved in resistance to immunotherapy by disrupting various components of tumor immunity. 50 , 51 , 52 In autochthonous mouse melanoma models, active β‐catenin signaling negatively regulates the antitumor T‐cell responses. 53 This study found that melanoma without T‐cell infiltration was highly linked to tumor‐intrinsic Wnt pathway activation. Meanwhile, active Wnt/β‐catenin cascade contributes to T‐cell exclusion and therefore results in resistance to antiprogrammed death‐ligand 1 (anti‐PD‐L1)/anticytotoxic T lymphocyte antigen 4 immunotherapy. 54 Tumor‐associated macrophage (TAM)‐derived exosomal miR‐29a‐3p enhances PD‐L1 expression in ovarian cancer, promoting tumor proliferation and immunosuppression. Examination of the underlying mechanism shows that miR‐29a directly targets FOXO3, which results in the inhibition of an antagonist of the Wnt pathway, termed GSK3β, to facilitate Wnt pathway activation and PD‐L1 expression. 55 , 56 Similarly, hypoxic conditions also enhance the expression of miR‐29a in glioblastoma (GBM), which can promote the proliferation of myeloid‐derived suppressor cells (MDSCs) and eventually lead to an immunosuppressive environment. 57 , 58
2.2. Hyperactivation of notch signaling pathway
Notch signaling has been known for over a century, and its roles in embryonic and organ development have been well studied. 59 , 60 , 61 When the classical Notch signaling cascade is activated, three cleavages occur. 62 In brief, after synthesis in the endoplasmic reticulum, the Notch extracellular domain (NEC) of the receptor can be transferred into the Golgi compartment and then processed by furin‐like convertase (S1 cleavage). At the cell surface, NEC and Notch transmembrane fragment are linked by disulfide bonds to form the heterodimeric Notch receptor, which interacts with its ligand on the juxtaposed cell. With ligand interaction exposed to disintegrin and metalloproteases (ADAM) metalloproteases (S2 cleavage), the C‐terminal cleavage domain of the receptor is further cleaved by the γ‐secretase complex (S3 cleavage). Finally, the liberated Notch intracellular domain (NICD) is translocated to the nucleus and forms a trimeric complex with CSL (also known as CBF1) and mastermind‐like protein (MAML), changing CSL function to initiate its related transcription of downstream targets. The Notch pathway participates in the developmental programs of most organs and tissues and often plays an iterative role during the progression of a particular cell lineage. 62 Although there are many conditions where the Notch pathway blocks differentiation and secures a pool of stem or progenitor cells (PCs), in some contexts, the Notch pathway can promote differentiated cell fate, for example, in the skin. 63 Notch pathway is also essential for the formation of lateral inhibition in some differentiation programs, such as in the inner ear development. 64
Notch signaling's dual function in cell fate decisions (blocking or promoting differentiation) under different conditions may endow cancer cells with the ability to promote the development of tumors as well as drug resistance 65 , 66 , 67 (Figure 3). For instance, FGF4 secreted by B‐cell lymphoma cell promotes the expression of Jag1 within endothelial cells, activating Notch2 in adjacent cancer cells. The juxtacrine pathway promotes Notch signaling activation in endothelial cells thereby inducing invasiveness and chemoresistance. 68 The above results are consistent with previous evidence about the oncogenic role of Jag1 in breast, colon, and liver cancers. 69 , 70 , 71 The upregulation of Notch receptors could also activate the Notch pathway. 72 , 73 Stromal‐derived exosomes containing noncoding transcripts and transposable elements can be delivered into breast cancer cells and activate the STAT1‐dependent antiviral pathway. 74 In turn, active antiviral signaling promotes Notch3 expression and Notch signaling‐dependent therapeutic resistance. 74 Other important stromal cells involved in upregulating the Notch pathway are cancer‐associated fibroblasts (CAFs). 75 Primary CAFs enhance the expression of chemokine (C‐C motif) ligand 2 (CCL2), which contributes to the stemness maintenance of breast cancer cells. Increased CCL2 expression is correlated with high expression of Notch1 and therefore confers Notch signaling‐induced CSC features in vitro and in vivo. 76 Another research revealed that downregulating CCL2 expression could significantly reduce carcinogenesis and Notch1 expression in a xenograft model containing both fibroblasts and breast cancer cells. 77 Taken together, the data demonstrate that cancer cells can directly or indirectly hijack Notch signaling to promote cancer progression.
FIGURE 3.
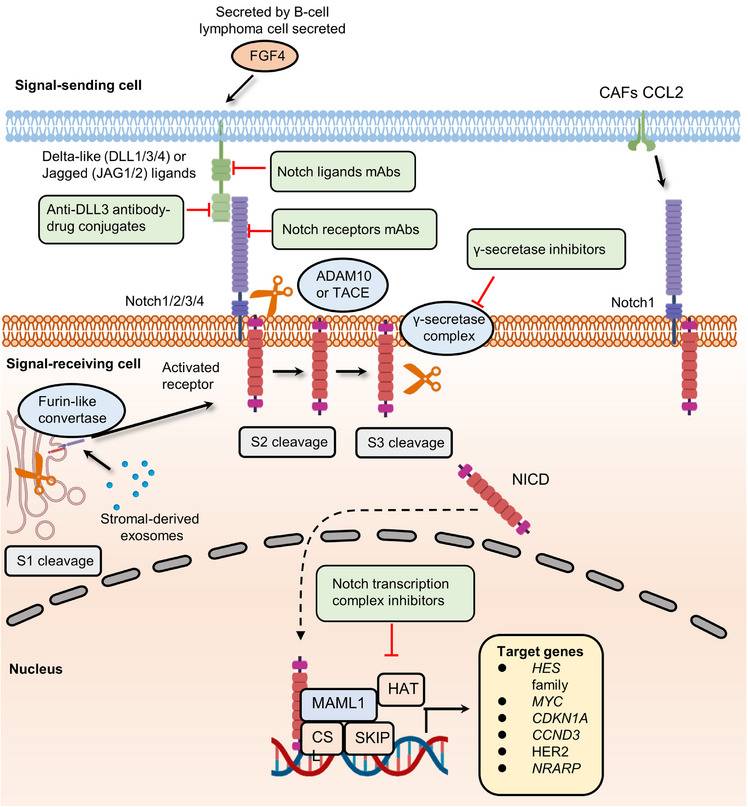
The canonical Notch signaling pathway and related pharmacological inhibitors that reverse Notch pathway‐induced cancer progression. The activation of Notch receptors needs to undergo three times cleavages. The first‐time cleavage, known as S1 cleavage, occurs in the Golgi apparatus and is mediated by a furin‐like convertase. Following S1 cleavage, the interaction between signal‐sending cell Notch ligands (Delta‐like ligands (DLL1, DLL3, and DLL4) and Jagged ligands (JAG1 and JAG2)) and signaling receiving cell Notch receptors (Notch1, Notch2, Notch3, and Notch4) on the cell surface can result in the cleavage of Notch receptors mediated by ADAM10 or ADAM17, termed (S2 cleavage). In the end, γ‐secretase‐mediated S3 cleavage leads to the release of NICD and the formation of Notch transcription complex. The expression of Notch pathway target genes promotes the development of cancer drug resistance. The Notch signaling receptor Notch1 was upregulated by overexpression of chemokine (C‐C motif) ligand 2 (CCL2) in CAFs, and FGF4 in adjacent tumor cells stimulated the expression of Jag1. Both regulatory axis contributed to the activation of the Notch pathway. Moreover, several agents were used to inhibit the activation of Notch signaling compressing Notch ligands mAbs, anti‐DLL3 antibody–drug conjugates, Notch receptors mAbs, γ‐secretase inhibitors, and Notch transcription complex inhibitors.
2.3. High‐level activation of hedgehog signaling
Hedgehog signaling is a vital pathway that determines cell location and fate in early embryonic development. 78 , 79 , 80 Following the development, the Hedgehog pathway participates in tissue homeostasis and wound healing. 81 , 82 , 83 The canonical mammalian Hedgehog signaling cascade can be activated by the interaction between Hedgehog ligands (Desert Hedgehog, Indian Hedgehog, and Sonic Hedgehog (SHH)) and Hedgehog receptors (Patched‐1 (PTCH1) and Patched‐2 (PTCH2)). 84 Their interplay results in the phosphorylation of Smoothened (SMO), the main effector of Hedgehog signaling, and the inhibition of multiprotein complexes containing GSK3β, protein kinase A (PKA), and suppressor of fused homolog (SUFU). The proteolysis of transcription factor glioma‐associated oncogene family zinc finger (Gli) is then blocked. Finally, Gli transcription factors translocate to the nucleus and activate transcription at start sites. During embryonic development and tissue homeostasis, Hedgehog signaling is typically modulated spatially and temporally. Given the important roles of Hh pathway in in the maintenance of stem PCs in many adult tissues, dysregulation of Hedgehog signaling can drive the development of several cancers, such as basal cell and colorectal carcinomas 85 , 86 , 87 (Figure 4).
FIGURE 4.
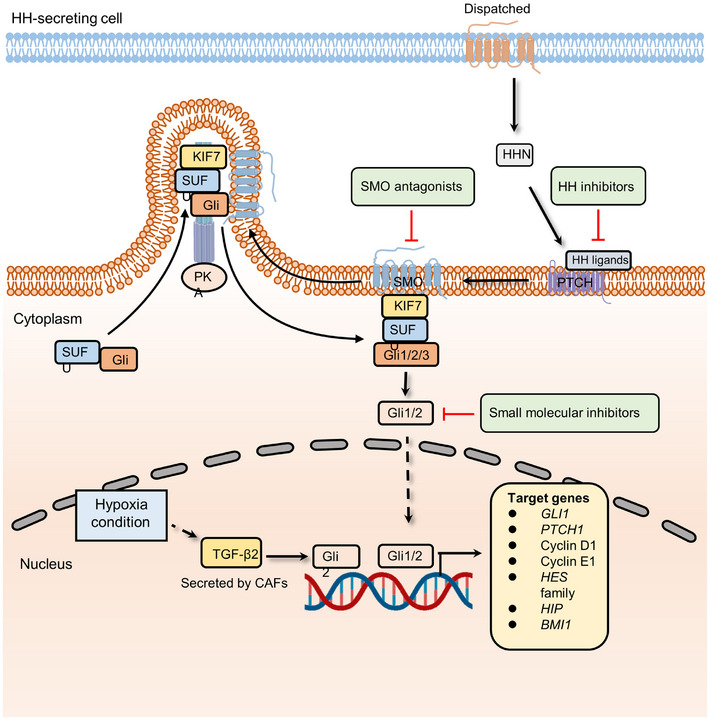
The canonical Hedgehog signaling pathway involved in cancer and associated therapeutic targets. The secretion of HHN regulated by Dispatched homolog can bind to PTCH receptor and hence releases SMO. Then, accumulated SMO, sequestrated kinesin family member 7 (KIF7), suppressor of fused homolog (SUFU) proteins, and Gli transcription factors form a multiprotein complex in cilia, which prevents Gli from inhibitory phosphorylation by PKA. Stable Gli is further released from SUFU complex and mediates the transcription of Hedgehog signaling pathway. CAF‐derived hypoxia stimulates the expression of TGF‐β2, which increases the level of Gli2 and activates Hedgehog signaling. Meanwhile, SMO antagonists, HH inhibitors, and small molecular inhibitors were proven to inhibit hedgehog pathway activation.
As the most common skin cancer in the western world, basal cell carcinoma (BCC) was first linked to BCC through the identification of germline mutations in Ptch1, which are responsible for Gorlin syndrome (also known as nevoid BCC syndrome or NBCCS). 88 Lineage tracing experiments showed that activating Smo oncogenes in interfollicular epidermal stem cells (IFE‐SCs), but not in hair follicle bulge stem cells, led to BCC development. This pinpointed IFE‐SCs as the source of BCC in mice. 89 Subsequent investigations showed that Smo activation in IFE‐SCs resulted in more aggressive tumor growth compared to Smo activation in PCs. This heightened growth was attributed to the greater capacity of SCs for symmetric self‐renewing divisions and their increased P53‐dependent resistance to cell death compared with PCs. 90 As expected, given the crucial role of the Hh signaling pathway in maintaining stemness, its role in tumor treatment resistance is continuously being unveiled. A multidimensional genomics analysis revealed that the active transcription factor serum response factor (SRF) could cause Gli1 transcriptional activity amplification in drug‐resistant BCCs. 87 The overexpression of Gli1 confers the activation of the Hedgehog pathway as well as drug resistance. 91 , 92 A recent study indicates that CAFs and hypoxia are involved in chemoresistance by upregulating the expression of Gli2 (a ligand of Hedgehog signaling). 86 In this study, researchers found that low‐oxygen conditions could induce CAFs to secrete TGF‐β2. High‐level TGF‐β2 augments the transcription of Gli2, promoting the occurrence of drug resistance. Moreover, a retinoic acid‐low (RA‐low) microenvironment plays a crucial role in bortezomib (BTZ) resistance. 93 Multiple myeloma cells secrete paracrine Hedgehog, which increases the expression of stromal CYP26, a cytochrome P450 monooxygenase, favoring the establishment of an RA‐low microenvironment. Inhibition of retinoid signaling blocks the differentiation of malignant hematopoietic cells and reduces BTZ sensitivity. 93 In addition, endothelial cells can promote the stem‐like phenotype of cancer cells by activating the Hedgehog pathway. 94 In glioma cells, the expression of stemness‐related genes, including Sox2, Olig2, Bmi1, and CD133, was upregulated when cocultured with endothelial cells. However, knockdown of Smo in endothelial cells abolished the stem‐like phenotype in glioma cells. To further unveil the mechanism of the activation of the Hedgehog signaling cascade in perivascular glioma cells, tissue specimens from glioma patients were examined, which indicated that some canonical development pathways, such as the Wnt pathway, could be the “intermediary” to promote Hedgehog signaling‐mediated drug resistance. In a study combining a 3D culture model and whole‐transcriptome analysis, Wnt and Hedgehog signaling components were found to be overexpressed in CRC. 95 Of note, Wnt signaling is negatively regulated by the canonical Gli‐dependent Hedgehog pathway in CRC. To further investigate the underlying mechanism, the expression of specific Hedgehog pathway genes was detected. The results showed that the activation is mainly driven by Gli‐independent and noncanonical Hedgehog signaling components, which are the positive regulators of the Wnt pathway. Noncanonical Hedgehog signaling cooperates with Wnt pathway to maintain the stemness of CSCs and develop resistance to antitumor drugs. 96
2.4. Aberrant regulation of hippo signaling cascade
Hippo signaling plays an important role in mediating organ development, tissue homeostasis, immune modulation, and wound healing. 97 , 98 , 99 , 100 At the turn of the 21st century, Hippo pathway was found to restrict the growth of Drosophila tissues. 101 In the mammalian canonical Hippo kinase cascade, the MST1/2–Salvador homolog 1 (SAV1) complex phosphorylates and activates LATS1/2–MOB domain kinase activator 1A/1B (MOB1A/1B) complexes. The activated LATS1/2–MOB1A/B complex then phosphorylates and inactivates YAP/TAZ, thereby inhibiting the transcription of downstream target genes 102 (Figure 5).
FIGURE 5.
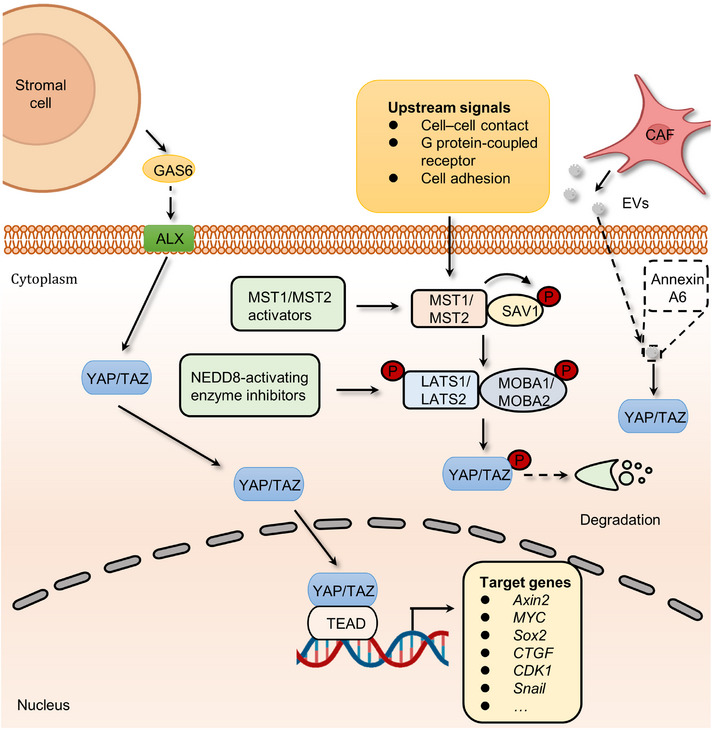
The mechanism by which cancer cells utilize the Hippo signaling pathway and associated therapeutic targets. The core proteins of Hippo pathway include MST1/2, LATS1/2, SAV1, MOB1A/1B, and YAP1/TAZ. Once activated by upstream signals, MST1/2 can phosphorylates SAV1, which subsequently activates LATS complex comprising LATS1/2 and MOB1A/1B. Then, the activated LATS complex phosphorylates YAP1/TAZ, thereby leading to the degradation of transcriptional factors of Hippo pathway. Otherwise, YAP1/TAZ can translocate to the nucleus and bind with TEA domain (TEAD) family members to promote the expression of Hippo signaling target genes. Stromal cells‐secreted GAS6, and CAF‐derived EVs (encompassing Annexin A6) mediate YAP/TAZ shuttling to the nucleus, which favors the expression of drug resistance‐related genes. MST1/2 activators and NEDD8 activating enzyme inhibitors were developed to prevent the transcription of Hippo pathway target genes, thereby increasing the degradation of YAP1/TAZ.
Analyses of various tumors and cancer cell lines, as well as data from The Cancer Genome Atlas with over 9000 tumors, emphasize the prominent role of YAP/TAZ in cancer, with the Hippo pathway being one of the frequently altered signaling pathways in human cancer. 103 Remarkably, the YAP1 and WWTR1 genes, which code for YAP and TAZ respectively, undergo amplification in approximately 14% of head and neck squamous cell carcinomas (HNSCCs), around 16% of lung squamous carcinomas, approximately 17% of cervical squamous cell carcinomas, and about 15% of esophageal squamous cell carcinomas. 103 As mentioned above, the Hippo pathway can promote the occurrence and development of tumors by regulating processes such as tumor cell migration and immune modulation. Initial investigations demonstrated that the ectopic expression of YAP, particularly nuclear‐localized mutants of YAP, exhibits robust prometastatic activity. This activity is contingent on TEAD binding, implying that interfering with this interaction might hold therapeutic promise in aggressive cancers. YAP further facilitates a metabolic shift in cancers with lymph node metastasis by stimulating the expression of genes that bolster fatty acid oxidation. 104 In addition, both YAP and TAZ have the ability to induce the expression of programmed cell death 1 ligand (PD‐L1), a ligand for the programmed cell death‐1 (PD‐1) receptor, which in turn creates an immunosuppressive microenvironment. 105 TAZ, for instance, stimulates the expression of PD‐L1 in lung and breast cancer cells, while YAP promotes PD‐L1 expression in melanomas and HNSCC cells. 106 Consequently, targeting YAP/TAZ presents an appealing strategy to reduce PD‐L1 levels and, as a result, enhance the efficacy of immunotherapy employing neutralizing antibodies against PD‐L1 and/or PD‐1. Meanwhile, YAP/TAZ have been found to participate in various Hippo pathway‐dependent drug resistance and show a central role in mediating resistance to cancer therapeutics. 107 , 108 , 109 For instance, extracellular vesicles (EVs) from CAFs contribute to the activation of focal adhesion kinase (FAK)‐YAP signaling and enhance drug resistance. 110 In this work, a comprehensive proteomic analysis of CAF‐EVs was performed, which identified a key effector, Annexin A6, involved in drug resistance. Annexin A6 secreted by CAFs activated FAK‐YAP by stabilizing β1 integrin at the cell surface of gastric cancer cells and in turn resulted in resistance to cisplatin. More recently another study also discovered that CAFs within gastric cancers expand resistance to 5‐FU by activating YAP/TAZ. 111 In addition to chemotherapy, the inactivation of Hippo signaling confers resistance to targeted therapies. For example, the links between the activation of YAP/TAZ and tyrosine kinase inhibitors (TKIs) resistance have been revealed. 112 , 113 Although the precise mechanism of how YAP/TAZ signaling develops resistance to TKIs is poorly understood, AXL, one of the YAP/TAZ transcriptional targets, seems to be a candidate to drive resistance. 114 , 115 AXL is a receptor tyrosine kinase that belongs to the Tyro3, Axl, and Mer (TAM) receptors and is activated by growth arrest‐specific protein 6 (GAS6). 116 , 117 At present, hyperactivation of GAS6/AXL signaling is considered as one of the hallmarks in many types of multidrug‐resistant cancer cells. 118 , 119 , 120 , 121 , 122 Interestingly, a study revealed that stromal cells in the tumor microenvironment (TME) could continuously express GAS6 to activate the AXL receptor of adjacent tumor cells, thus promoting cancer drug resistance. 123 The administration of AXL inhibitors, such as R428, abolishes GAS6/AXL signaling‐induced resistance to quizartinib. 119 Taken together, the above studies present concrete proof for the key roles of Hippo signaling in the development of cancer and drug resistance.
2.5. Abnormal activation of other signaling pathways
In addition to the above‐mentioned embryonic development‐related pathways, the TGF‐β superfamily and FGF/FGFR signaling cascades also contribute to cancer development and progression.
TGF‐β superfamily signaling, which contains over 30 ligands, including TGF‐βs, growth and differentiation factors, bone morphogenetic proteins, Nodal, and Activins, is required for the development and homeostasis of complex multicellular animals. 124 These members and their downstream pathway components are highly conserved during evolution and contribute to various cellular functions, such as migration, differentiation, growth, apoptosis, and adhesion. 124 To perform these complex biological functions, ligand dimers need to bind to and activate heterogeneous complexes of type I and type II receptors that phosphorylate intracellular mediators (Smads). Then, phosphorylated mediators form complexes with each other and other components to mediate the transcription of target genes in the nucleus. 125 Finally, the expression of effectors leads to related cellular responses during different embryonic developmental stages. For instance, TGF‐β family members can induce EMT, an essential process in the temporally distinct phases of heart development, by upregulating the expression of related markers, including Snail1/2, ZEB1/2, Twist, and ids. 125 TGF‐β signaling dysregulation, on the other hand, may drive cancer cell proliferation, metastasis, and drug resistance (Figure 6). For example, mutations in the SMAD gene are detected in 60% of pancreatic cancer patients. The coexistence of KRAS mutations and Smad mutations in pancreatic ductal adenocarcinoma (PDAC) patients plays a crucial role in driving early tumor formation and metastasis. 126 Through Smad signaling, TGF‐β1 suppresses the expression of miR‐198, a cellular methylguanine DNA methyltransferase regulator, in GBM to alter temozolomide sensitivity. 127 Meanwhile, CAF‐secreted exosomal miR‐423‐5p promotes chemotherapy resistance by regulating the TGF‐β pathway. 128 The TGF‐β pathway in prostate cancer cells is activated by miR‐423‐5p, which leads to the inhibition of GREM2, a differential screening‐selected gene aberrant in the neuroblastoma (DAN) family member, and enhances drug sensitivity. Of note, current studies reported the vital function of TGF‐β in attenuating TME response to PD‐L1 blockade. The role of the TGF‐β pathway in restraining antitumor immunity by restricting T‐cell infiltration was revealed by examining tumors from a large cohort of patients with metastatic urothelial cancer treated with atezolizumab, a PD‐L1 inhibitor, and using a mouse model to recapitulate the above findings. 129 Therapeutic cotreatment with TGF‐β blockage and anti‐PD‐L1 antibodies reverses TGF‐β signaling‐induced T‐cell exclusion in the center of tumors, which triggers robust antitumor immunity and tumor regression. Moreover, YM101, an anti‐TGF‐β/PD‐L1 bispecific antibody, was also found to reshape the immunosuppressive microenvironment induced by the TGF‐β/Smad pathway and promote the formation of “hot tumors”. 130 In addition, TGF‐β signaling fosters drug resistance and regulates stemness in various cancers. For example, compared to the reversible state induced by a shorter exposure, chronic TGF‐β exposure could drive stable EMT, tumor stemness, and chemoresistance in breast cancer cells. 131 Similarly, informative research showed that HIF‐1α and CAF‐secreted TGF‐β2 converge to enhance the expression of Gli2 in CSCs, promoting stemness and resistance to chemotherapy. 86
FIGURE 6.
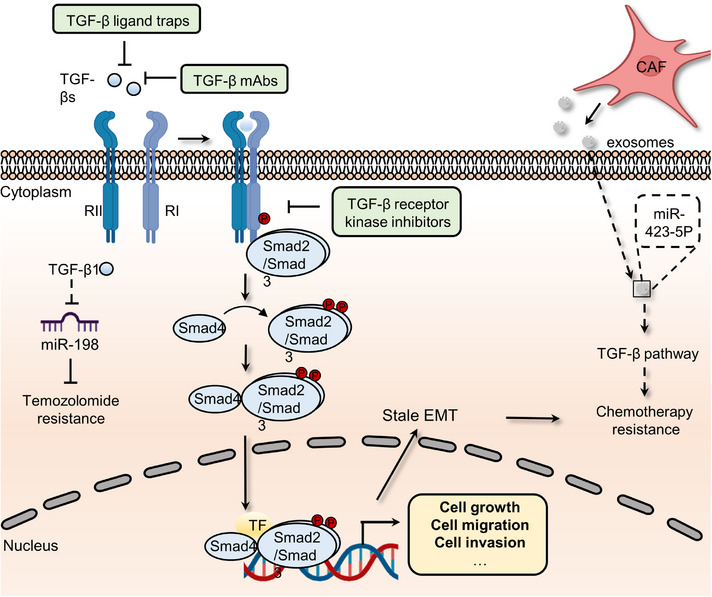
The roles of canonical TGF‐β signaling pathway in cancer and inhibitors that target TGF‐β pathway. The ligands, TGF‐βs, bind TGF‐β type II (RII) induce the phosphorylation of TGF‐β type I (RI) receptors on cell surface. Activated kinase activity of RI further phosphorylates Smad2 and Smad3, which form trimeric complexes with Smad4. These Smad complexes will translocate into nucleus and promote the transcription of TGF‐β pathway downstream genes. Sustained activation of TGF‐β signaling can lead to cancer drug resistance via inducing stable epithelial‐to‐mesenchymal transition (EMT) and regulating the expression of miR‐198 or miR423‐5p. Hence, agents, such as TGF‐β ligand traps, TGF‐β mAbs, and TGF‐β kinase inhibitors, have been developed to impair the activation of TGF‐β signaling pathway.
FGF/FGFR pathway is essential for early development of embryonic tissue or organ (such as the skeleton, lung, urinary system, and heart). 132 As one of the most diverse growth factor groups in vertebrates, the FGF family regulates lots of functions, comprising survival, proliferation, differentiation, and migration. 132 At present, 22 FGF ligands have been discovered in mice and humans that exert their pleiotropic effects through binding high‐affinity tyrosine kinase receptors, including FGFR1, FGFR2, FGFR3, FGFR4, and FGFRL1. 133 The binding of FGFs to FGFRs triggers conformational changes and the phosphorylation of tyrosine residues within the cytosolic tail of FGFRs, leading to dimerization and activation of cytosolic tyrosine kinases. 134 Then, the phosphorylated tyrosine residues provide docking sites for downstream signaling molecules and subsequently regulate their related pathways, including MAPK, PI3K/AKT, and STAT signaling. 134 In addition to embryonic development, accumulating evidence has revealed the significant functions of the FGF/FGFR pathway in the development of cancer. The overexpression of FGFs and FGFRs in cancer cells has been related to a poorer prognosis in a growing number of studies. 134 Moreover, FGF/FGFR axis‐dependent downstream signaling cascades play indispensable functions in this process. A recent research reveals the presence of key FGFR components in cervical cancer cell lines and their potential role in promoting invasive disease characteristics, highlighting the potential for therapeutic interventions targeting FGFR in cervical cancer treatment. 135 In HNSCC cells, FGFR3 overexpression activated MAPK signaling and upregulated the level of ERK, which in turn boosted FGF2 production and resistance to bevacizumab. 136 Similarly, the PI3K/AKT pathway was also identified as an important mediator in FGF/FGFR‐dependent cancer drug resistance. Overexpression of FGFR1 could increase AKT activation, leading to EMT and resistance to the first‐line EGFR inhibitor gefitinib in non‐small cell lung cancer (NSCLC). 137 Another downstream signaling pathway of the FGF/FGFR pathway involved in cancer is STAT signaling. In breast cancer cells, FGFR1 was shown to promote the synthesis of hyaluronan by activating STAT3 signaling. Blocking either hyaluronan synthesis or STAT3 activation reverses proliferation and doxorubicin resistance of breast cancer cells. 138
3. DORMANT CANCER CELL RESEMBLING EMBRYONIC DIAPAUSE
Insects utilize diapause in response to harsh environments (such as cold and nutrient deficiency). 139 , 140 , 141 Similarly, a large number of mammalian species, including mice, kangaroos, and deer, can also delay blastocyst implantation until they meet suitable conditions. 142 , 143 , 144 , 145 Before pregnancy, the blastocysts severely decrease their metabolic rate and block cell division for up to one year to prepare for future implantation. 146 , 147 Accordingly, dormant cancer cells also were found to enter an embryonic diapause‐like state following treatment with antitumor medications. 148 , 149 , 150
3.1. The definition of dormant cancer cell
Ever since Willis initially coined the term, “dormancy” has taken on varying interpretations among researchers, leading to potential misunderstandings, especially for individuals not directly engaged in this field. In clinical practice, the term “dormancy” is employed to describe the extended interval between primary tumor treatment and the recurrence of metastases in secondary locations. 151 Although dormant tumor cells and tumor stem cells share many similarities, such as drug resistance and their critical roles in recurrence, there are also significant differences between the two concepts. To begin with, there is no direct experimental evidence indicating that CSCs have experienced cell cycle arrest. Furthermore, not all dormant tumor cells can be detected with the same stemness markers as CSCs, such as SRY‐box 2 (SOX2) and Nanog Homeobox (NANOG). 152
Dormant cancer cells are a specific population that displays reversible cell cycle arrest and acquires the abilities to gain additional mutations, adapt to new environments and drive cancer drug resistance. 153 , 154 , 155 Conversely, extrinsic environmental signals (e.g., cell–matrix interface or chronic inflammation) and cellular regulatory mechanisms (e.g., the upregulation of Myc) are able to awaken quiescent tumor cells to re‐enter the proliferative cycle. 156 , 157 , 158 , 159 , 160 Presently, dormant cells have been found in several malignancies, including breast, colorectal, pancreatic, and ovarian cancers, acute myeloid leukemia, and GBM, and this rare subpopulation is thought to be responsible for lesions relapse. 152 , 161 , 162 , 163 , 164 , 165 , 166 However, key mechanisms, such as how dormant cells utilize their specific state to adapt to new ecological niches, resist initial drug assaults, and transit between dormancy and activation states, have been poorly identified. 151
3.2. Dormant cancer cells and cancer progression
Most theories agree that dormant cancer cells are caused by genetic variants rather than nongenetic variants. 167 , 168 , 169 Moreover, whole‐exome and whole‐genome sequencing studies showed little difference in the mutational landscape between primary and metastatic cancer cells (arising from dormant cells). 170 , 171 , 172 , 173 , 174 Is it possible that niches can be reprogrammed to induce alterations within dormant cells? In support of this, a recent study indicated that stromal changes in aged lungs result in the occurrence of melanoma dormancy. 175 The role of lung aged fibroblasts in inducing the transition from dormant phenotype to outgrowth was revealed by intradermally injecting melanoma cells into young or old C57BL6 mice, implying the niche dependence of dormant cells. Meanwhile, dormant cancer cells within BRAFV600E‐mutated mice and human melanoma are tightly linked with the activation of CAFs. BRAF inhibition‐induced activation of CAFs stimulates the remodeling of the fibronectin‐rich matrix, consequently contributing to melanoma cell persistence by the reactivation of ERK. 176 Moreover, CAFs are internalized and degraded by breast cancer cell cannibalism. 177 The modulation of cell cannibalism is determined by Jun N‐terminal kinase, EMT, and stem cell‐like markers, which lead to the activation of deceleration programs and drug resistance in vitro. 178 , 179 , 180 In addition, CAF‐derived secretory proteins, such as hepatocyte growth factor (HGF), the ligand of MET, trigger the activation of the PI3K–AKT signaling pathway. 181 , 182 , 183 This pathway endows resistance to BRAF inhibition in melanoma, colorectal, and glial tumor cells. 184 , 185 , 186 , 187 In basal‐like HER2+ breast cancer, CAF‐secreted HGF also promotes resistance to HER2 inhibitor. 188 A recent study proposed that the TME could drive cell state transition and drug response in pancreatic cancer. 189 , 190 Combining systematic profiling of metastatic pancreatic cancer biopsies and matched organoid models, the functions of TME in modulating the cell state to impact drug response were illustrated. Transcriptional state representation of pancreatic cancer shows strong culture‐specific biases under stimulation with different conditions, highlighting the crucial functions of the niche in regulating cell state (Figure 7).
FIGURE 7.
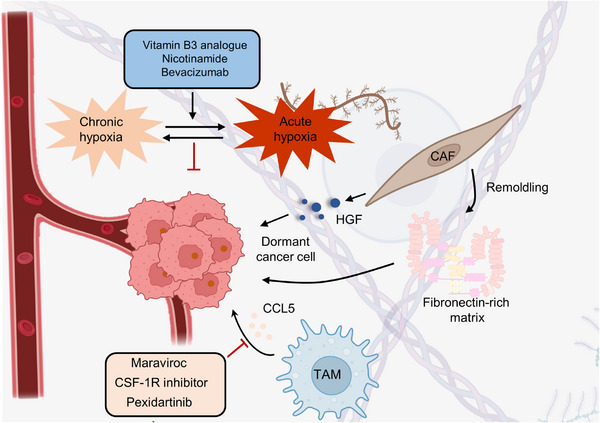
The important roles of the niche in dormant cancer cells. Tumor recurrence is closely related to dormant cancer cells, and the maintenance of dormant cells is significantly associated with the TME (such as the hypoxic microenvironment, CAFs, and TAMs). CAFs favor dormant cancer cells by secreting HGF or stimulating the remodeling of the fibronectin‐rich matrix. By altering the TME, the dormancy of tumor cells can be activated, and the recurrence of lesions could be inhibited. Vitamin B3 analogs, nicotinamide, and bevacizumab could augment radiotherapy by changing the oxygen content in the TME. Meanwhile, by applying maraviroc, CSF‐1R, and pexidartinib, the recruitment of TAMs could be abolished, enhancing therapeutic outcomes in various cancers.
The above evidence supports the opinion that dormant cancer cells need complex dynamic interactions with different cell types in the niche. 178 , 191 , 192 Therefore, we wondered whether environmental factors could also cause the cell cycle arrest of dormant cells. Physical factors in the niche, such as hypoxia, may inhibit cell proliferation and thus maintain a state of dormancy. 193 , 194 , 195 Indeed, breast cancer cells cultured in chronic intermittent conditions in vitro have been shown to become dormant, characterized by G0‐G1 cell cycle arrest, and hypoxia also results in dormancy in vivo. 196 , 197 Moreover, in prostate and breast cancers, crosstalk between cells and their secreted molecules leads to the inhibition of proliferation. For instance, endothelial cell‐secreted thrombospondin 1 has been proven to induce breast cancer cells to exit the cell cycle. 198 , 199 In addition to the endothelial niche, the endosteal niche has been shown to regulate the proliferation of multiple myeloma cells and acute lymphoblastic leukemia (ALL). 200 Coculturing with MC3T3 osteoblast precursor cells and primary osteoblasts conditioned medium can suppress the proliferation of 5TGM1 multiple myeloma cells in vitro. 201 In this regard, extracellular osteopontin in the endosteal niche can also promote ALL cell cycle arrest and dormant transit. 202
It is clear that the transition from cancer cells to dormant cancer cells is a major challenge in clinical practice. 203 , 204 This transition results in functional alteration of tumor cells to escape damage from chemotherapy drugs as well as immunosurveillance. 205 , 206 , 207 Most antitumor drugs are designed to target cancer cells with high proliferation features and neglect slow‐cycling cells that cause lesion relapse. 208 , 209 Several core driver genes and pathways have been reported in this process, including Myc, mTOR, GPX4, Mex3a, oxidative phosphorylation and LINE‐1 repression in 2D or 3D cell models. 210 , 211 , 212 , 213 Once antitumor drugs are removed, these cells exit dormancy by controlling notable vital factors, such as reactivation of Myc, and grow into a population. 214 , 215 , 216 Finally, these clones provide an opportunity to gain resistance mutations and induce drug resistance. 217 It is well explained why ALL patients benefit from long‐term oral low‐dose chemotherapy from the end of intensive chemotherapy, even in chemosensitive ALL subtypes. 218
Regarding how dormant cells evade the surveillance of the immune system, a body of data indicates that this process could be achieved by repressing endogenous antigen presentation, such as major histocompatibility complex class I (MHC I). 219 , 220 Indeed, the expression of tumor antigens and MHC I is frequently deficient in individual metastatic cancer cells. 221 , 222 , 223 Moreover, another mechanism by which dormant cancer cells avoid T‐cell recognition and elimination was unveiled in PDAC, whereby tumor cells in the liver that lacked expression of tumor antigen cytokeratin 19 and MHC I selectively responded to endoplasmic reticulum stress. 224 In contrast to MHC I, MHC II expression may contribute to immunosuppression in patients with melanoma and lung cancer. 225 , 226 , 227 Dormant cancer cells exposed to interferon in the niche could drive the expression of MHC II and other cell surface molecules linked to myeloid‐lineage cells. As a result, myeloma cells are mistaken for niche‐specific local immune cells and shield themselves from immune clearance. Dormant myeloma cells are recognized as myeloid cells, such as osteal macrophages and CD169+ bone marrow macrophages, and evade immune surveillance by this mechanism. 228 , 229 , 230
These findings show that the entire life cycle of dormant cancer cells, from quiescence to reactivation, results from interaction with the local niches. Furthermore, the life cycle of dormant cancer cells, accompanied by cell‐intrinsic and cell‐extrinsic control, can be divided into five stages: niche occupancy, niche interaction and engagement, cellular reprogramming for adaption to the niche, long‐term dormancy, and relapse. In this regard, further advances are needed to comprehensively understand the recognition of cell‐extrinsic control of dormant state via the niche.
4. ONCOFETAL REPROGRAMMING OF TME
The ability of tumor cells to avoid immunotherapy by altering the microenvironment is similar to embryo implantation, which can maintain an active state of maternal immune tolerance through CD4+ regulatory T cells (Tregs). 231 For example, to extensively characterize the cellular landscape of the human liver from development to disease, single‐cell RNA (scRNA) sequencing was employed, which revealed remarkable fetal‐like reprogramming of the TME. 232 Specifically, the results showed that the hepatocellular carcinoma (HCC) ecosystem displayed characteristics reminiscent of fetal development, including re‐emergence of fetal‐associated endothelial cells (PLVAP/VEGFR2) and fetal‐like (FOLR2) TAMs. In addition, the distinct roles of NK cells in the initiation and resolution of inflammation in different phases of pregnancy also indicate the plasticity of the fetal immune microenvironment. 233 More importantly, this fetal‐like feature has been observed in cancer cells, emphasizing the link between embryogenesis and cancer development and progression. This section focuses primarily on TAMs and MDSCs and their roles in regulating TME.
4.1. Tumor‐associated macrophages
TAMs have long been recognized to exert crucial roles in immunosuppression, and the increased abundance of TAMs is associated with a worse prognosis for cancer patients. 234 In the primary tumor environment, TAMs enhancing tumor cell invasion, intravasation, and the viability of tumor stem cells. 235 Numerous experiments have provided detailed insights into the mechanisms through which TAMs facilitate tumor cell migration and invasion. For instance, in the RIP tag model of pancreatic islet cancer, tumor cells capable of synthesizing IL‐4 can induce TAMs to produce cathepsin proteases B and S. These proteases play a role in degrading and remodeling the extracellular matrix, thereby facilitating the detachment of tumor cells from the tumor. 236 Meanwhile, in mouse models of breast cancer, TAMs play a significant role in sustaining the survival of CSCs through juxtacrine signaling, 237 and in HCC mouse models, they achieve this through signaling mediated by TGF β‐1. 238 At metastatic sites, macrophages associated with metastasis support processes such as extravasation, ensuring tumor cell survival and sustained growth. 235 Interestingly, in certain contexts, they can also be involved in maintaining tumor cell dormancy. 239 In both primary and metastatic locations, TAMs exert suppressive effects on the activities of cytotoxic T cells and natural killer cells, which possess the potential to eliminate tumors.
TAMs also play essential roles in many types of ICB‐resistant cancer cells. In lung cancer, P2X7, a crucial sensor of extracellular ATP, is highly expressed in immunosuppressive cells such as TAMs. 240 TAMs that highly express P2X7 promote “M2‐like” polarization by downregulating STAT6 and IRF4 phosphorylation in vivo and in vitro. Meanwhile, the P2X7‐expressing TAMs in lung cancer are associated with anti‐PD‐1 antibody resistance, which can be overcome by P2X7 inhibitors O‐ATP, A‐740003, and A‐438079 hydrochloride. TAMs were also reported to confer anti‐PD‐1 resistance by expressing c‐Maf. 241 The inhibition of c‐Maf partly overcomes resistance to anti‐PD‐1 therapy in a subcutaneous LLC tumor model. Furthermore, in an experimental model of melanoma, CD163‐expressing TAMs specifically maintain immune suppression to resist anti‐PD‐1 checkpoint therapy. 242 These findings highlight the heterogeneity of the TME and the numerous roles of TAMs in regulating tumor progression.
4.2. Myeloid‐derived suppressor cells
Based on their density, morphology, and phenotype, MDSCs primarily fall into two subsets: polymorphonuclear (PMN)‐MDSCs and monocytic (M)‐MDSCs. Initially, PMN‐MDSCs were referred to as granulocytic (G)‐MDSCs, but gradually, the term PMN‐MDSCs became more widely adopted due to distinguishing features in morphology and phenotype compared with steady‐state neutrophils. These features include altered buoyancy, reduced granules, decreased expression of CD16 and CD62L, and upregulated CD11b and CD66b. 243 Furthermore, a unique population of fibrocystic MDSCs (F‐MDSCs) has been identified and characterized in humans. 244 MDSCs are a heterogeneous population of immature myeloid cells that can inhibit T‐cell and NK‐cell activity and govern tumor growth, premetastatic niche development, and immunotherapy resistance. 245 In mouse tumor models, tumor‐infiltrating M‐MDSCs were shown to promote an EMT/CSC phenotype, aiding the dissemination of tumor cells from primary sites. Conversely, PMN‐MDSCs infiltrating the lungs supported metastatic tumor growth by reversing the EMT/CSC phenotype and promoting tumor cell proliferation. 246 In several mouse tumor models, the inhibition of S100A8/A9, the regulatory factors of MDSCs, has been shown to restrict tumor growth by diminishing the accumulation of MDSCs. 247 A recent study revealed that the number of MDSCs is related to the antitumor immune response induced by a PD‐1 antibody in mouse models of gastric cancer. 248 5‐FU can increase the effects of anti‐PD‐1 by reducing the number of MDSCs. In brief, PD‐L1 expressed by gastric epithelial cells increases the accumulation of MDSCs, which promotes tumor growth and worsens the immune response to PD‐1. Furthermore, MDSCs are involved in KRAS‐interferon regulatory factor 2 (IRF2) axis‐induced immunotherapy resistance in CRC. 249 Oncogenic KRASG12D can suppress the expression of IRF2, which leads to direct inhibition of CXCL3 expression. High expression of CXCL3 binds to CXCR2 on MDSCs and promotes the development of immune therapy resistance. Another component within cancer cells that mediates reciprocal communication between tumor cells and MDSCs is cell cycle‐related kinase (CCRK). 250 Simultaneous overexpression of CCRK and MDSC markers (CD11b/CD33) in HCC significantly reduces the efficacy of immunotherapy. Mechanistically, hepatic CCRK activates the nuclear factor‐κB (NF‐κB)/IL‐6 axis, resulting in the accumulation of (PMN)‐MDSCs resistant to PD‐L1 blockade. Apart from the above conditions, MDSCs have been proven to decrease the efficacy of PD‐L1 blockade in many kinds of cancers, including lung cancer, pancreatic cancer, and melanoma, highlighting the potential of targeting MDSCs for reversing resistance to immunotherapy. 251 , 252 , 253
5. OVEREXPRESSION OF EMBRYO/FETAL TRANSPORTERS HAMPERS CANCER THERAPY
The human placenta is generally regarded as the functional barrier between fetal blood circulation and the mother, which protects the fetus from heterologous substances such as therapeutic agents, drugs of abuse, and other xenobiotics circulating in the mother's metabolic system. 254 , 255 However, this concept was reconsidered after the thalidomide disaster. 256 Subsequent research has shown that most xenobiotics and metabolites can cross the placenta and even be transported by the placenta. 257 , 258 , 259 Furthermore, genes involved in the delivery of drugs and metabolites have been identified and termed ABC transporter genes. 260 In the placenta and the fetal blood–brain barrier (BBB), these transporters can function as efflux pumps of xenobiotics from the maternal circulation. 261 , 262 To date, 48 human membrane transporters involved in distinct biological processes have been identified and classified into seven subfamilies. These include the ABC subfamilies A‐G, which regulate the levels of hormones, amino acids, xenobiotics, and other macromolecules by transferring them across cell membranes. 263 , 264 , 265 , 266 It should be noted that only a small proportion of these transporters have low substrate specificity, with the majority having a much broader spectrum. 267 This characteristic therefore provides opportunities for cancer cells to develop multidrug resistance. Ample reports suggest that overexpression of ABC transporters enhances the capacity to transport antitumor drugs and is closely associated with drug resistance. 268 , 269 , 270 Among the more than 15 drug resistance‐related ABC transporters identified, multidrug resistance protein 1 (MDR1), also termed P‐glycoprotein or P‐gp, encoded by ABCB1, multidrug resistance‐associated protein 1 (MRP1), encoded by ABCC1, and breast cancer‐resistant protein (BCRP), encoded by ABCG2 are thought to be the dominant drug efflux transporters. 271 Here, we focus on recent studies about these three canonical drug resistance proteins and highlight their dominant functions in resistant cancer cells (Figure 8).
FIGURE 8.
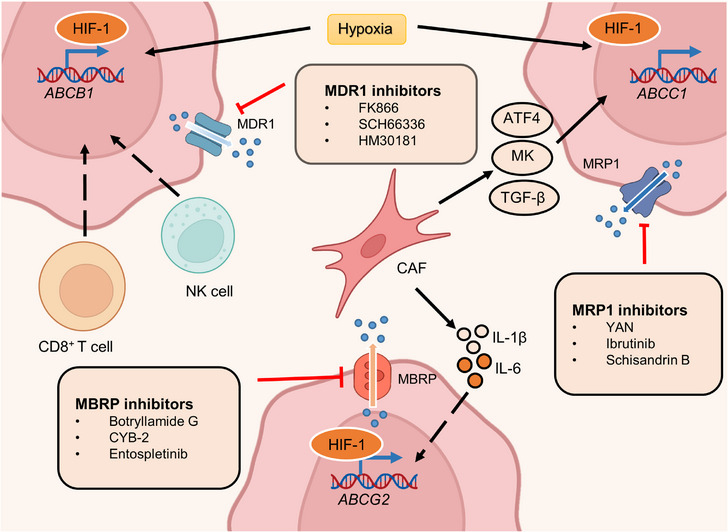
ATP‐binding cassette (ABC) transporter inhibitors and factors that regulate its expression. The transcription factor HIF‐1 is significantly upregulated in the hypoxic niche and subsequently promotes the transcription of ABCB1, ABCC1, and ABCG2. CAFs, one of the most plentiful stromal components in the TME, induce ABC gene overexpression by secreting growth factors and interleukins. CAF‐derived growth factors, including ATF4, MK, and TGF‐β, enhanced MRP1 expression. Meanwhile, IL‐1β and IL‐6 derived from CAFs transcriptionally activate BCRP expression. Immune cells within the TME, such as NK cells and CD8+ T cells, also contribute to ABCB1 activation, but the underlying mechanism is unknown. The activity of MDR1 could be blocked by many targeting agents, such as FK866, SCH66336, and HM30181. Several inhibitors, such as botryllamide G, CYB‐2, and entospletinib, have been developed to decrease MBRP function. YAN, ibrutinib, Schisandrin B, and other drugs also inhibit MRP1 activation.
5.1. Multidrug resistance protein 1
MDR1 overexpression has been observed in most drug‐resistant cancer cells and is frequently associated with hypoxia. 272 , 273 HIF‐1, a hypoxia marker, is significantly associated with MDR1 expression in cancer and normal tissue. 274 , 275 , 276 For example, both physiological and chemical hypoxia can increase the expression of HIF‐1 and MDR1, resulting in doxorubicin resistance in prostate multicellular tumor spheroids. 277 Conversely, the inhibition of HIF‐1 caused by antisense oligonucleotides (ASOs) significantly decreases MDR1 expression and enhances sensitivity to doxorubicin. The pro‐oxidants H2O2 and buthionine sulfoximine reduce HIF‐1α and MDR1 expression, indicating the importance of reactive oxygen species (ROS) in drug resistance. 278 , 279 In addition to ROS, a high calcium concentration also reduces MDR1 levels by downregulating HIF‐1α expression. 280 , 281 Interestingly, in NSCLC hypoxia can reverse doxorubicin resistance by reducing MDR1 expression. 282 Hence, HIF‐1 may play distinct roles in regulating MDR1 expression under different conditions besides hypoxia.
Ample evidence suggests that MDR1 is also regulated by noncoding RNAs in breast, prostate, lung, pancreatic, and ovarian cancer. 283 , 284 , 285 , 286 , 287 , 288 For instance, CRC patients with high lncRNA CCAL expression have a worse response to adjuvant chemotherapy and shorter overall survival. 289 The overexpression of CCAL dramatically inhibits activator protein 2α, a suppressor of Wnt signaling, and in turn upregulates MDR1 levels. Additionally, in breast cancer, CCAL can enhance the expression of MDR1 via epigenetic regulation. 290 Mechanistically, methyltransferase‐like 3 increases the expression of miR‐221‐3p by promoting pri‐miR‐221‐3p m6A mRNA methylation, which further triggers the transcription of MDR1 and BCRP.
5.2. Multidrug resistance‐associated protein 1
MRP1, the second transporter identified in the ABC transporter family, also plays a crucial role in cancer multidrug resistance. 291 , 292 , 293 MRP1 was found in a small cell lung cancer cell line that showed multidrug resistance. 294 In 1992, the regulatory gene involved in mediating resistance was first reported in the same cell line. 295 Since then, the mechanisms by which MRP1 induces drug resistance have been widely revealed. Most of the drug resistance mediated by MRP1 is associated with its aberrant expression. 296 , 297 , 298 Specifically, changes in the extracellular environment, such as oxygen content, can lead to the upregulation of several TFs, which enhances the expression of MRP1. 299 , 300 , 301 , 302 Hypoxia, one of the most common features of cancer, stimulates the expression of TFs and further augments MRP1 levels in various cancer cells. 303 , 304 , 305 For example, the coexpression of HIF‐1α and MRP1 was observed using immunohistochemical techniques in most of the 50 chordoma specimens, which implied decreased sensitivity to chemotherapy. 306 However, the mechanism by which HIF‐1α regulates the expression of MRP1 is unclear. A subsequent study of chemoresistant HepG2 cells revealed that ERK/MAPK signaling mediates the activity of HIF‐1α by altering phosphorylation levels. 307 Active HIF‐1α induces the expression of MDR‐related genes, such as MRP1, and drives chemoresistance. In addition to hypoxia, some CAF‐secreted growth factors can also promote the expression of MRP1. 308 A recent study indicated that CAF‐derived activating transcription factor 4 (ATF4) positively correlated with malignancy and gemcitabine resistance in PDAC. Further investigation found that CAFs secreted TGF‐β1 to activate the SMAD2/3 pathway, which enhanced the expression of ATF4. Consequently, ATF4 directly bound to the ABCC1 promoter region and upregulated the expression of MRP1.
Midkine (MK), a heparin‐binding growth factor associated with both carcinogenesis and chemoresistance, is another CAF‐derived growth factor that modulates MRP1 levels. 309 CAFs enhance the expression of lncRNA ANRIL by secreting MK and in turn augment MRP1 expression. Additionally, several metabolites also govern the occurrence of multidrug resistance by upregulating MRP1. In NSCLC, increased glycolysis and lactate production within the TME harmonize the TGF‐β1/Snail and TAZ/AP‐1 pathways to form the Snail/TAZ/AP‐1 complex at the ABCC1 promoter, thus enhancing MRP1 transcription. 310 The addition of NaHCO3 reversed lactate‐induced MRP1 overexpression and overcame etoposide (VP‐16) resistance.
5.3. Breast cancer resistant protein
The third identified ABC transporter is BCRP. Similar to the above two transporters, BCRP was detected in numerous types of cancers and contributed to resistance to various antitumor drugs, such as tamoxifen. 311 , 312 , 313 As the most prescribed hormonal agent for the treatment of estrogen receptor α (ERα)‐positive breast cancer, tamoxifen rarely prolongs the overall survival of patients due to drug resistance. 314 Microarray analysis revealed a higher level of Dicer, an RNase III‐containing enzyme, in tamoxifen‐resistant metastatic breast cancers. Dicer overexpression significantly elevated the level of BCRP and governed resistance to tamoxifen in vivo and in vitro. 311 Moreover, hypoxia was reported to enhance the stability of HIF, which targeted the promoter of ABCG2 and thus upregulated BCRP expression. 315 The HIF/BCRP regulatory axis has been discovered in several types of tumors, including breast, ovarian, renal cell, and anaplastic thyroid cancer. 316 , 317 , 318 , 319 , 320 In the context of hypoxia and oxidative stress, SP PC populations function to increase the expression of HIF‐2α, which transactivates ABCG2 and promotes cytoprotection. 321 Recent comprehensive reports have shown crosstalk between N6‐methyladenosine (m6A) modification and drug resistance. 322 , 323 , 324 Alteration of the m6A modification contributes to the expression of BCRP, which regulates drug efficacy. In addition, CAF‐derived cytokines comprising interleukin 1β (IL‑1β) and interleukin 6 (IL‑6) were also found to promote BCRP expression. 325 , 326 These two CAF‐secreted cytokines were detected in breast cancer cells and associated with BCRP‐dependent drug resistance. Glucose can also enhance the expression of BCRP by activating the AKT pathway and subsequently conferring resistance to a small fraction of cancer cells with stem‐like properties, termed side population (SP) cells, within tumors. 327 However, 3‐BrOP, an inhibitor of glycolysis, could significantly reverse the tumorigenesis ability induced by SP cells, which may provide a potential drug for drug‐resistant SP cells.
It is well known that many types of ABC transporters could be present in a solitary cancer type. 328 An analysis of the expression of ABC transporters in AML samples showed that coexpression of transporter genes significantly reduced overall survival in patients. 329 Additionally, quantities of factors enhance the expression of ABC transporters, including gene mutation, epigenetic regulation, and metabolic reprogramming. 330 , 331 , 332 , 333 Therefore, accurately determining the key ABC transporters involved in drug resistance might provide an approach to benefit clinical outcomes.
6. TARGETING ONCOFETAL REPROGRAMMING IN CANCER THERAPY
Understanding the underlying mechanisms of how cancer cells enter the embryonic‐like state is essential to enhance the outcome of patients with drug resistance. In this section, we introduce the inhibitors or potential strategies that target the core factors of embryonic development‐related signaling pathways, transporter proteins, the drivers of dormant cancer cells, and immune cell subpopulations implicated in immunotherapeutic resistance.
6.1. Targeting developmental signaling pathways
Dysregulated developmental pathways are commonly detected in cancer cells and proven to mediate drug resistance and recurrence. Given its critical functions, many targeting agents have been developed to aid cancer treatment.
Wnt signaling pathway inhibitors can be grouped into four main categories: ligand or receptor inhibitory agents involved in Wnt signaling; porcupine antagonists that target the processing or secretion of Wnt ligands; agents that maintain the function of the destruction complex by activating caspases or inhibiting tankyrase, thereby enhancing the deregulation of β‐catenin; and blockades of β‐catenin–TCF axis‐dependent transcription 334 , 335 , 336 (Figure 2). To date, each class of agents has achieved good results in preclinical studies, and several drugs have entered clinical testing (Table 1). In the canonical Wnt pathway, inhibition of FZD receptors is linked to β‐catenin deregulation and suppression of Wnt signaling, which indicates a potential strategy for oncotherapy. 337 , 338 To achieve this goal, several target agents, such as ipafricept, 339 OMP‐18R5 (vantictumab), 340 , 341 , 342 OTSA‐101, 343 F2. A, 344 and Fz7‐21, 345 have been utilized to competitively bind to the FZD family, thereby inhibiting Wnt‐dependent oncogenesis. In addition, DKN‐01, a mAb to DKK1 that blocks Wnt signaling by enhancing the internalization of low‐density lipoprotein (LDL)‐related protein 5/6 (LRP5/6) coreceptors, has entered clinical trials. 14 , 346 , 347 The development of porcupine antagonists is another approach to suppress the Wnt pathway by affecting the key factor for the secretion of Wnt ligands. 348 , 349 For instance, WNT974 (LGK974) has been proven to abolish Wnt secretion and exert effective antitumor activity in epithelial ovarian cancer. 350 , 351 WNT974 monotherapy for patients with cervical squamous cell carcinoma, pancreatic cancer, and triple‐negative breast cancer (TNBC) has entered a phase I clinical trial (NCT01351103). 352 In preclinical or clinical trials, β‐catenin is one of the most common targets of inhibitors due to its pivotal role in Wnt signaling. CWP232291, a first‐in‐class peptidomimetic drug, decreases β‐catenin by activating caspases and shows anticancer activity against relapsed or refractory myeloma. 353 Moreover, several compounds, such as LF3 and KYA1797K/KY1220, also effectively decrease the activation of Wnt pathway by targeting the β‐catenin/TCF complex. 354 , 355 , 356 In preclinical studies, both LF3 and KYA1797K/KY1220 effectively suppress the growth of colon cancer by disrupting the critical interaction between β‐catenin and TCF4. 357 , 358
TABLE 1.
Small molecules targeting Wnt signaling and their clinical trials.
| Compound | Tumor type | Phase (Clinicaltrials.gov identifier) | Efficacy outcomes | Status | References |
|---|---|---|---|---|---|
| PRI‐724 with gemcitabine | Advanced metastatic pancreatic cancer | Phase Ib (NCT01764477) | 8 patients had SD (40.0%) | Clinical studies ongoing | 359 |
| DKN‐01 | Advanced‐stage DKK1‐positive esophageal cancer or gastroesophageal junction tumors | Phase Ib study (NCT02013154) | Encouraging early efficacy signals (no further details reported) | Clinical studies ongoing | 360 |
| Ipafricept | Advanced‐stage solid tumors | Phase I (NCT01608867) | 3 patients had SD for more than 6 months (1 with germ cell cancer and 2 with desmoid tumor) | Clinical studies ongoing | 361 |
| Vantictumab | Advanced solid tumors | Phase I | SD in 3 patients with NET | Clinical studies ongoing | 362 |
| Vantictumab | Stage IV pancreatic ductal adenocarcinoma | Phase I (NCT02005315) | 53% evaluable patients (8 out of 15) had a PR and 27% had SD (4); Median PFS: 7.2 months | Clinical studies ongoing | 363 |
| Vantictumab | Metastatic HER2‐negative breast cancer | Phase Ib (NCT01973309) | 33% evaluable patients (7 of 21) had a PR and 29% (6) had SD | Updated results pending | 364 |
| Cirmtuzumab | Chronic lymphocytic leukemia | Phase I (NCT02222688) | 17 of 22 evaluable patients had SD | Clinical studies ongoing | 365 |
| OTSA101 | Synovial sarcoma | Phase I (NCT01469975) | 77% evaluable patients (3 of 8) had SD | Recruiting | 343 |
| CWP232291 alone or with lenalidomide and dexamethasone | Myeloma (relapsed or refractory) | Phase Ia/b (NCT02426723) | 27% patients (3 of 11) had SD | Clinical studies ongoing | 366 |
| CWP232291 (CWP291) | Relapsed and/or refractory AML or myelodysplastic syndrome | Phase I (NCT01398462) | 1 CR in a patient with AML | Clinical studies ongoing | 353 |
Abbreviations: AML, acute myeloid leukemia; CR, complete response; DKK1, Dickkopf‐related protein 1; Fzd, Frizzled; n, number of patients; NET, neuroendocrine tumor; PFS, progression‐free survival; PR, partial response; SD, stable disease.
The development of Notch signaling inhibitors necessitates a thorough understanding of its diverse and complex role in various cancers 367 , 368 , 369 (Figure 3). Given the importance of Notch signaling pathways in cancer progression, such as angiogenesis and stemness maintenance, several classes of agents have been developed that target the Notch pathway in distinct ways 370 , 371 (Table 2). The γ‐secretase inhibitors (GSIs) are the first and largest class of small molecule antagonists that block the second proteolytic cleavage of Notch receptors and thus inhibit the activation of Notch downstream. 372 GSIs exert strong antitumor activities in many preclinical trials. For instance, the combination of MRK‐003 and trastuzumab fully eliminated HER2‐positive breast cancer cells in a mouse model. 373 Moreover, in an NSCLC model, another GSI, BMS‐906024 also displayed synergistic therapy potential when combined with cisplatin, crizotinib, PTX, and docetaxel. 374 , 375 Notch ligands are also targets for antitumor drug development. A phase I first‐in‐human study of enoticumab, a human anti‐DLL4 IgG1 mAb, showed potential therapeutic effect in patients with advanced solid tumors. 376 In addition to enoticumab, many other humanized antibodies that target DLL‐3, such as rovalpituzumab tesirine, have also been tested in clinical trials and have shown signs of efficacy in patients with recurrent small cell lung cancer. 377 Monoclonal antibodies targeting Notch receptors (such as brontictuzumab and tarextumab) have been well demonstrated to show moderate antitumor activities in different phases of studies. 378 , 379 , 380 CB‐103 is a novel small molecule inhibitor that targets Notch signaling by disrupting the interaction of the Notch transcription complex. 381 Preclinical studies showed that CB‐103 significantly inhibited the growth of tumors in a GSI‐resistant TNBC model. Moreover, another inhibitor targeting the Notch coactivator protein, IMR‐1, inhibits the growth of Notch‐dependent cell lines and patient‐derived tumor xenografts. 380
TABLE 2.
Small molecules targeting Notch signaling and their clinical trials.
| Compound | Tumor type | Phase (Clinicaltrials.gov identifier) | Efficacy outcomes | Status | References |
|---|---|---|---|---|---|
| MK‐0752 | T‐ALL and AML | Phase I (NCT00100152) | 1 patient with T‐ALL had a 45% reduction in a mediastinal mass | Discontinued | 382 |
| MK‐0752 with gemcitabine | Unresectable PDAC | Phase I (NCT01098344) | 47% patients had SD (9 of 19); PRs: 5% patients (1 of 19) | Discontinued | 383 |
| MK‐0752 followed by docetaxel | Advanced‐stage breast cancer | Phase I/II (NCT00645333) | Not reported | Discontinued | 384 |
| MK‐0752 with ridaforolimus | Advanced‐stage solid tumors | Phase I (NCT01295632) | 11% patients (2 of 18) with HNSCC had responses; 1 patient had SD lasting ≥6 months | Discontinued | 385 |
| PF‐03084014 | Advanced‐stage solid tumors | Phase I (NCT00878189) | CR: 2% evaluable patients (1 of 46; thyroid cancer); PRs: 11% patients (5 of 46; all desmoid tumors); 30% patients had SD (14 of 46) | Clinical studies ongoing | 386 |
| PF‐03084014 | Advanced‐stage TNBC | Phase II (NCT02299635) | Not reported | Clinical studies ongoing | 387 |
| PF‐03084014 | T‐ALL or T‐LBL | Phase I (NCT00878189) | CR: 12.5% patients (1 of 8) | Clinical studies ongoing | 388 |
| PF‐03084014 | Desmoid tumors | Phase II (NCT01981551) | PR: 29% patients (5 of 16); 29% patients had SD (5 of 16) | Clinical studies ongoing | 389 |
| PF‐03084014 with gemcitabine and nab‐paclitaxel | Metastatic PDAC | Phase Ib/II (NCT02109445) | ORR: 0% | Clinical studies ongoing | |
| RO4929097 | Metastatic CRC | Phase II (NCT01116687) | 18.2% evaluable patients had SD (6 of 33); Median OS: 6.0 months; Median PFS: 1.8 months | Discontinued | 390 |
| RO4929097 with gemcitabine | Advanced‐stage solid tumors | Phase I | PR: 5.6% patient with nasopharyngeal carcinoma; 3 patients had SD | Discontinued | 391 |
| RO4929097 with temsirolimus | Advanced‐stage solid tumors | Phase Ib (NCT01198184) | 73% patients had SD | Discontinued | 392 |
| BMS‐906024 | T‐ALL | Phase I | (32%) patients (8) had ≥50% reduction in bone marrow blasts | Clinical studies ongoing | 393 |
| Brontictuzumab | Hematological malignancies | Phase I | PR in 4.3% patient with TMF (1); 8.7% patients had SD in (2) | No phase II or III trials ongoing; discontinued in hematological malignancies | 394 |
| Tarextumab or placebo with gemcitabine and nab‐paclitaxel | Metastatic PDAC | Phase Ib/II (NCT01647828) | Placebo vs. tarextumab arms: Median OS: 7.9 months vs. 6.4 months Median PFS: 5.5 months vs. 3.7 months; ORR: 31.8 vs. 20.2% | Discontinued | 395 |
| Tarextumab or placebo with etoposide and cisplatin | Extensive‐stage SCLC | Phase Ib/II (NCT01859741) | ORR in 84% patients of phase Ib part; placebo vs. tarextumab arms in phase II part: Median PFS: 10.3 months vs. 9.3 months; ORR: 70.8 vs. 68.5%; | Discontinued | 396 |
| Demcizumab or placebo with carboplatin and pemetrexed | Metastatic nonsquamous NSCLC | Phase I (NCT01189968) | 3% patients had CR patients (1 of 40); 48% patients had PR (19 of 40); 38% patients had SD (15 of 40) | Progressed to the randomized phase II study (NCT02259582) | 397 |
|
Demcizumab with gemcitabine, with or without nab‐paclitaxel |
Advanced‐stage PDAC | Phase Ib (NCT01189929) | 50% evaluable patients had PR (14 of 28); 39.3% patients had SD (11 of 28); Median OS: 10.1 months; Median PFS: 9.0 months | Progressed to the randomized phase II study (NCT02289898) | 398 |
| Rovalpituzumab tesirine | SCLC | Phase I (NCT01901653) | 17% patients had PR or CR (11 of 65 patients); 54% patients had SD (35 of 65); Median OS: 4.6 months; Median PFS: 3.1 months | Progressed to the phase II study (NCT02674568) | 377 |
Abbreviations: AML, acute myeloid leukemia; CR, complete response; CRC, colorectal cancer; DoR, duration of response; HNSCC, head and neck squamous cell carcinoma; n, number of patients; NSCLC, non‐small cell lung cancer; ORR, objective response rate; OS, overall survival; PDAC, pancreatic adenocarcinoma; PFS, progression‐free survival; PR, partial response; SCLC, small‐cell lung cancer; SD, stable disease; T‐ALL, T‐cell acute lymphocytic leukemia; T‐LBL, T‐cell lymphoblastic lymphoma; TMF, transformed mycosis fungoides; TNBC, triple‐negative breast cancer.
Among the components involved in the Hedgehog pathway, its ligands, Smo and Gli transcription factors, are the most common targets in Hedgehog pathway‐directed oncotherapy 399 , 400 , 401 (Figure 4). Agents that target Smo and Gli are currently in clinical trials 402 (Table 3). Vismodegib and sonidegib are two United States Food and Drug Administration‐approved antagonists of SMO for treating patients with metastatic BCC and/or recurrent locally advanced BCC. 403 These two Hedgehog pathway inhibitors yielded overall objective response rates (ORRs) from 44 to 68% in different stages of clinical trials. 403 , 404 , 405 , 406 , 407 Glasdegib, combined with low‐dose cytarabine to treat patients with newly diagnosed AML aged over 75 years old and ineligible for intensive induction chemotherapy, was an important advance in the development of SMO inhibitors in 2018. 408 Meanwhile, retarding transcriptional activity of Hedgehog signaling is an attractive therapeutic option, and agents targeting Gli‐mediated transcription, such as GANT58 and GANT61a, are utilized to overcome cancer resistance to SMO inhibitors. 409 , 410 GANT61 exhibited significant antitumor activity in several preclinical models, including breast, ovarian, pancreatic, lung, and liver cancer. 411 , 412 , 413 , 414 In addition, GANT58 decreased the growth of prostate cancer cells by suppressing the expression of PTCH1 and Gli1 in vivo. 415
TABLE 3.
Small molecules targeting Hedgehog signaling and their clinical trials.
| Compound | Tumor type | Phase (Clinicaltrials.gov identifier) | Efficacy outcomes | Status | References |
|---|---|---|---|---|---|
| Vismodegib vs. placebo | Recurrent epithelial ovarian, primary peritoneal cancer or fallopian tube in second or third CR | Phase II (NCT00739661) | Median PFS: 7.5 months vs. 5.8 months with placebo | Magnitude of sought increase in PFS not achieved | 416 |
| Vismodegib or placebo in combination with FOLFOX or FOLFIRI plus bevacizumab | mCRC | Phase II | PFS: HR 1.25, 90% CI 0.89−1.76, p = 0.28; 1‐year OS: 81.4 vs. 80.1%; ORR: 51% in vismodegib arm vs. 46% in placebo arm | Development for CRC terminated | 417 |
| Vismodegib with gemcitabine | Advanced‐stage PDAC (25 with elevated SHH on pretreatment biopsy) | Phase II (NCT01195415) | Fibrosis decreased in 45.4% of 22 patients and Ki67 levels decreased in 52.9% of 17 patients; Gli1 and PTCH1 expression decreased in 95.6 and 82.6%, respectively, of 23 evaluable patients; Median OS: 5.3 months; DCR: 65.2%; Median PFS: 2.8 months; ORR: 21.7% | Development for PDAC terminated | 418 |
| Vismodegib or placebo with gemcitabine | Advanced‐stage PDAC | Phase I/II (NCT01064622) | Median OS: 6.9 months vs. 6.1 months (HR 1.04, 95% CI 0.69−1.58); PR rate: 8 vs. 11%; SD rate: 51 vs. 38%; Median PFS: 4.0 months vs. 2.5 months (HR 0.81, 95% CI 0.54−1.21); CR rate: 0% in vismodegib arm vs. 2% in placebo arm | Development for PDAC terminated | 419 |
| Vismodegib preoperatively and/or postoperatively | Recurrent resectable glioblastoma | Phase II (NCT00980343) | Median PFS and OS were 1.8 months and 8.3 months, respectively | Development for glioblastoma terminated | 420 |
| Vismodegib or placebo with FOLFOX | Advanced stage gastric or gastroesophageal junction adenocarcinoma | Phase II (NCT00982592) | Median PFS: 7.3 months vs. 8.0 months in the placebo group; ORR: 35% in both arms; Median OS: 11.5 months vs. 14.9 months with placebo | Development terminated for these diseases | 421 |
| Taladegib | Advanced‐stage cancer | Phase I (NCT01226485) | ORR: 26.2%; SD rate: 28.6%; | Clinical studies ongoing | 407 |
| Saridegib | Advanced‐stage solid tumors | Phase I | 6 PRs observed in 22 patients with HH inhibitor‐naïve BCC (27%) | Clinical studies ongoing | 422 |
| Saridegib (with cetuximab) |
Recurrent metastatic head and neck squamous cell carcinoma |
Phase I (NCT01255800) | Median PFS: 77 days; 12.5% evaluable patients had PR (1 of 8) and 50% had SD (4 of 8) | Clinical studies ongoing | 423 |
| Itraconazole | Biochemically relapsed prostate cancer | Phase II (NCT01787331) | 47% evaluable patients (9 of 19) had PSA declines by week 12 | Development for prostate cancer terminated | 424 |
| Itraconazole and pemetrexed | Metastatic nonsquamous non‐small cell lung cancer | Phase II | 15 patients received pemetrexed and itraconazole and 8 received pemetrexed alone); OS: 32 months vs. 8 months; Median PFS: 5.5 months vs. 2.8 months | Discontinued | 425 |
| Itraconazole with arsenic trioxide | Refractory metastatic BCC | Phase I | SD in 3 patients | Study ongoing, further results pending | 426 |
Abbreviations: ALT, alanine transaminase; AML, acute myeloid leukemia; AST, aspartate transaminase; BCC, basal cell carcinoma; CR, complete response; CRC, colorectal cancer; DCR, disease control rate; DoR, duration of response; FOLFIRI, folinic acid, 5‐fluorouracil and irinotecan; FOLFOX, folinic acid, 5‐fluorouracil and oxaliplatin; HH, Hedgehog; HR, hazard ratio; mCRC, metastatic colorectal cancer; ORR, objective response rate; OS, overall survival; PDAC, pancreatic ductal adenocarcinoma; PFS, progression‐free survival; PR, partial response; PSA, prostate‐specific antigen; SD, stable disease; SHH, Sonic hedgehog.
LATS1/2 and MST1/2 activators are currently the most used agents in Hippo‐targeted therapeutics 427 , 428 (Figure 5). Due to the distinctive characteristics of the Hippo pathway, in which loss‐of‐function mutations of LATS1/2 and MST1/2 are always linked to oncogenesis, the development of targeted agents presents more challenges than other antagonists. 429 , 430 Indeed, to date, only a few agents specifically targeting Hippo signaling have been brought into clinical testing. For example, pevonedistat, a first‐in‐class NEDD8‐activating enzyme inhibitor, blocks CRL4DCAF1‐mediated degradation of LATS1/2 and thereby attenuates YAP/TAZ activity. 431 , 432 Pevonedistat has been tested in combination with azacitidine in a phase I trial involving patients with AML or MDS, which resulted in 83% attenuation of ORRs in patients who received more than 6 cycles of treatment. 156
A variety of receptor kinase inhibitors, monoclonal antibodies, ligand traps, and ASOs have been developed to block TGF‐β signaling, and most agents have been or are being tested in clinical trials to provide more effective treatments (Figure 6). The small molecule inhibitors of TGF‑β receptor kinase were mainly designed to bind to the ATP‐binding domain of TGF‐β R kinase, therefore inhibiting ATP kinase activity and abolishing the downstream signaling cascade. 102 For example, vactosertib (MedPacto), an oral inhibitor that targets TGF‐βRI/ALK‐5 (IC50 = 12.9 nM), acts pleiotropically on diverse cancer types, including CRC, gastric cancer, and NSCLC, through intrinsic and extrinsic mechanisms. 433 , 434 Galunisertib is another kinase inhibitor that has shown antitumor effects in lung and breast cancer cell lines, and its safety was proven in phase I studies. 435 , 436 Moreover, many other agents targeting TGF‐βRI, such as LY3200882, LY573636, and A83‐01, have also been reported to display certain antitumor activity in various tumors by weakening the activity of TGF‐β signaling. 437 , 438 , 439 Another potential method for reversing the tumorigenic and immune suppressive responses induced by the TGF‐β pathway is the administration of antibodies that can obstruct the binding of TGF‐β ligand to its receptor. Indeed, the highly selective inhibitor of the TGF‐β1 isoform SRK181‐mIgG1 was shown to overcome primary resistance to checkpoint inhibitor therapy, such as anti‐PD‐(L) 1 Abs. 440 Meanwhile, in a phase I trial of 28 patients with malignant melanoma, Fresolimumab, a type of human mAb that neutralizes TGF‐β1 and TGF‐β2, showed a partial response in 25% of patients. 441 The development of TGF‑β ligand traps is an approach to handle the exogenous‐dependent hyperactivation of TGF‑β signaling. 442 In this context, cancer patients might gain more precise treatment. Currently, there is an increasing number of ways to inhibit the TGF‑β pathway, such as ASOs and vaccine‑based approaches to modulate TGF‑β signaling, which again demonstrates their irreplaceable role in cancer. 443 , 444
Most FGF/FGFR signaling inhibitors fall into three groups: small‐molecule FGFR TKIs, anti‐FGFR antibodies, and FGF ligand traps 445 (Figure 7). The most widely used therapeutic approach are FGFR TKIs, which include pan‐FGFR inhibitors (such as FIIN‐2, JNJ‐42756493, LY2874455, and ponatinib) and FGFR‐specific TKIs (such as the selective FGFR4 inhibitor BLU9931). 446 Furthermore, according to the interaction pattern between the inhibitor and the kinase domain, FGFR TKIs can be classified into irreversible or reversible inhibitors. Irreversible inhibitors (such as erdafitinib and pemigatinib) are thought to have a better binding affinity and selectivity, but the early phases of clinical trials showed limited efficacy or demonstrated minimal clinical benefit. 447 , 448 Other molecules that have been developed as investigational agents targeting FGF/FGFR signaling include the FGF traps FP‐1039 (HGS1036), msFGFR2c, sFGFR3 sm27, and NSC12 449 , 450 , 451 ; the anti‐FGF2 mAbs 3F12E7 452 and H3L3 453 ; the anti‐FGFR2 mAb hFR2‐14 454 ; the anti‐FGFR4 mAb U3‐1784 455 ; and the anti‐FGFR1 antibody–drug conjugate (ADC) LY3076226. 456 The benefits of FGFR inhibitors have been proven in clinical trials in subsets of patients, including those with lung, breast, and gastric cancer. However, the low response rates among patients with FGFR alterations and the existence of responders without detectable FGFR alterations still hamper treatment outcomes.
6.2. Eliminating dormant cancer cells
Previous comprehensive reviews have summarized the therapeutic strategies to change the TME and improve therapy outcomes. 457 , 458 , 459 , 460 , 461 Here, we focus on advances in the field of dormant cancer cells‐related treatment by altering the TME. Forcing dormant cancer cells to enter distinct oxygen conditions, such as the switch between chronic hypoxia and acute hypoxia, has been proposed to improve the efficacy of antitumor drugs. 462 , 463 The combined treatment showed that the vitamin B3 analog nicotinamide, which prevents temporary fluctuations in tumor blood flow, increases the oxygen level and shrinks the extent of tumor metastasis. 464 In addition, mild temperature hyperthermia, another approach to elevate tumor blood flow, was reported to relieve acute hypoxia and enhance drug susceptibility. 465 Altering hypoxia at various degrees of irradiation increased radiosensitivity, particularly to X‐rays. 466 For example, nicotinamide or mild temperature hyperthermia enhances the radiosensitivity of total and dormant cancer cells when given X‐rays in combination with high‐dose‐rate irradiation. 462 , 467 , 468 Therefore, irradiation combined with nicotinamide or mild temperature hyperthermia could significantly improve the patient's prognosis. Moreover, bevacizumab, a drug targeting vascular endothelial growth factor (VEGF), impairs oxygen supply and shows antitumoral effects in various drug‐tolerant persister cells. 469 , 470 , 471 Given the roles of VEGF in the formation of blood vessels, bevacizumab could dramatically augment the hypoxic niche by influencing oxygen transport and exerting greater antineoplastic activity when combined with chemotherapy. 472 , 473 However, in patients with refractory breast cancer, the addition of bevacizumab to PTX treatment seems to be ineffective under hypoxic conditions and may induce hypoxia and increase cytokine secretion related to cancer progression. 474 These findings suggest that targeting hypoxia might be a promising way to eradicate tumor cells and dormant cancer cells, although many unknown mechanisms need further study.
A substantial body of evidence suggests the crucial roles of TAMs in inducing chronic inflammation and contributing to cancer development. 475 , 476 , 477 The inflammatory microenvironment is now recognized as one of the hallmarks of cancer 478 and tightly correlated with dormant malignant cells due to its multifaceted functions in retaining tumor dormancy and reawakening dormant tumor cells. 479 , 480 , 481 Hence, macrophage‐targeting therapeutic strategies might be a promising way to change the dormant state of cancer cells and thus inhibit tumor relapse. A number of potential strategies targeting macrophages have been reported. 482 , 483 , 484 In general, TAM‐focused therapeutic approaches are classified into either restraining the interplay by inhibiting the localization of these cells at tumor sites or reactivating their antineoplastic activities. As previously discussed, chemokines (such as CCL5) are responsible for the recruitment of macrophages to tumors. 485 In research on residual breast cancer cells, Her2 downregulation‐driven CCL5 has been reported to promote breast cancer recurrence via macrophage recruitment. 486 CCL5, a product of cancer cells and macrophages, is usually linked to a worse outcome in diverse types of breast cancer. 487 , 488 Blocking CCL5 with maraviroc, the cognate receptor for CCL5, is associated with biological and clinical responses in advanced stage CRC. 489 Thus, drugs targeting the CCL5–CCR5 axis are worthy of further study, considering that the functions of this signaling pathway are well established in the pathogenesis of breast cancer, GBM, and gastric cancer. 490 , 491 , 492 CSF‐1R, the key modulator of monocyte‐macrophage lineage growth and differentiation, is abundantly expressed in numerous tumor types. 493 , 494 , 495 Given the direct interference with TAMs, CSF1R has become a promising therapeutic target, and several related inhibitors (such as small molecules and antibody antagonists) have been tested in different preclinical models. 496 , 497 , 498 For instance, in glioma macrophage populations after radiotherapy, a CSF‐1R inhibitor combined with radiotherapy enhanced the outcome in preclinical models, accompanied by decreased recruitment of monocyte‐derived macrophages in GBM. 499 Moreover, pexidartinib, a drug used for recurrent GBM, can reduce the number of circulating CD14dim/CD16+ monocytes. Although the primary 6‐month progression‐free survival (8.6%) was not satisfactory in the 37 patients after treatment, rational combination therapy approaches may significantly augment the outcome. 500 In general, macrophage‐targeting therapeutic strategies have the potential to complement and synergize with both chemotherapy and immunotherapy.
6.3. Regulation of tumor immune microenvironment
Reciprocal communication between cancer cells and different immune cell subpopulations enables tumor cells to escape immune responses and develop resistance to ICBs. 501 , 502 , 503 Hence, the heterogeneity of the immune microenvironment is tightly linked to the outcomes of ICB in immunotherapy. Combined treatment was used to address this issue, considerably enhancing the treatment effect.
In a phase III GBM trial, regulatory Tregs were shown to play crucial role in resistance to ICBs. 504 Therefore, targeting glucocorticoid‐induced TNFR‐related receptor (GITR) in Treg cells using an agonistic antibody (αGITR) significantly reversed the suppression of antitumor immune response. The functionality of intratumoral macrophages can reflect the response to ICBs in melanoma. Mechanistically, the activation of ER promotes melanoma growth in murine models by skewing macrophage polarization, which leads to ICB resistance. 505 Targeting ER by using fulvestrant, a selective ER inhibitor greatly decreased tumor growth and promoted the antitumor efficacy of ICBs. Nanobased combinational treatment is also a potent way to target immunotherapeutic resistant cancer cells by reprogramming the immune environment. To overcome tumor immunological tolerance against ICBs, a versatile nanomodulator was designed that could point‐to‐point counteract immune suppressors and promote the infiltration of tumor T cells. 506 Small interfering RNAs targeting indoleamine 2, 3‐dioxygenase‐1 and gemcitabine delivered by biocompatible nanocages account for targeting Tregs and MDSCs. Meanwhile, O2‐producible mineralization tattooed on the surface of the nanocarriers accounts for suppressing the immune inhibition of M2 macrophages. A multifunctional nanomodulator was used to reverse the immunosuppressive state and overcome tumor immunological tolerance after decorating the therapeutic ICB antibodies on the mineralized shell. In many situations, targeting the tumor immune environment may be feasible to decrease resistance to ICBs.
6.4. Inhibition of ABC transporters
Targeting ABC transporters has been considered a promising approach to suppressing or eliminating drug resistance in oncotherapy since discovering the vital role of ABC proteins in tumor drug resistance. 507 , 508 As early as 1981, Tsuruo's group found that verapamil could weaken drug resistance in leukemia cells. 509 However, high dosages of verapamil trigger cardiovascular toxicity, limiting its further applications. 510 The second generation of P‐gp inhibitors, such as dexverapamil, 511 valspodar (PSC 833), 512 and biricodar citrate (VX‐710), 513 failed to cause drug–drug interactions with other antineoplastic agents, as did the first generation. 514 , 515 , 516 Continuing failures with antidrug resistance agents have driven the development of third‐generation inhibitors. These novel drugs are not only more than 200‐fold more potent at reversing drug resistance than before but also block the interaction with other chemotherapeutic drugs. 517 , 518 For example, tariquidar (XY9576), a drug currently in clinical trials, binds to P‐gp with high affinity and inhibits its ATPase activity at very low concentrations. 519 Clinical trials have demonstrated tariquidar's potential as a candidate, even if additional studies indicated that it might work in vivo either as a substrate or an inhibitor of P‐gp depending on the dosage. 520
With the development of nanotechnology approaches, novel strategies for targeting ABC transporters have been established, paving the way for more effective treatment of drug‐resistant cancer. 521 , 522 , 523 The diameter of nanoparticles (NPs) varies from one to several hundred nm. NPs can load a range of antitumor molecules, such as antineoplastic agents, P‐gp inhibitors, and RNAi fragments. 524 , 525 Mesoporous silica material (MSNPS)‐based drug delivery is one example, as it has a high surface area, large pore volume, biocompatibility, and tunable pore size. MSNPS loaded with siRNA and anticancer drugs showed significant antitumor activity by inhibiting P‐gp expression and increasing intracellular drug concentrations. 526
7. CONCLUSIONS
During the development of mammals, fetal stem cells can differentiate into diverse types of cells to ensure normal body function. 527 , 528 These observations imply that cells can switch from one lineage to another, greatly expanding our understanding of cell identity and fate determination. Recently, this hallmark of cancer cells has been comprehensively reviewed by Hanahan and termed unlocking phenotypic plasticity. 529 In his opinion, cellular plasticity could be selectively regulated by three mechanisms: dedifferentiation, blocked differentiation, and transdifferentiation, which are tightly linked to carcinogenesis. Indeed, to encounter diverse pressures from diverse niches, cancer cells can alter their state to gain stronger adaptability. 530 , 531 Embryonic development is a special phase in the progression from a fertilized egg to a mature individual. 532 On the one hand, embryos need to activate developmental signaling pathways to ensure their rapid proliferation and accurate differentiation. 84 , 533 , 534 , 535 On the other hand, deficiency in embryonic detoxification systems requires more effective efflux of toxic substances, which is mainly performed by ABC transporters. 536 , 537 In some harsh circumstances, the embryo will even stop developing until favorable conditions are restored. 538 , 539 Meanwhile, fetuses can regulate the immune microenvironment to evade attacks from the maternal immune system. 540 , 541 , 542 , 543 , 544
Cancer therapy is still a major challenge because each patient with cancer has their underlying mechanisms of tumorigenesis, metastasis as well as resistance, and there is a defining set of characteristics that dictate tumor deterioration, which may eventually result in death. 545 , 546 , 547 With the continuous advancement in cancer research, many new drugs and treatment approaches have emerged in cancer therapy, including multiple drug delivery‐based nanocarriers, CAR‐T or ICB immunotherapy, CRISPR‐based gene therapy, and so on. Though these approaches have shown significant potential in clinical studies, not all cancer patients can benefit from the advances. For example, CAR‐T therapy has demonstrated promising results in the treatment of hematological malignancies such as ALL and lymphoma, but its efficacy is significantly reduced in solid tumors. 548 Therefore, there is a growing awareness of the critical role of tumor heterogeneity in cancer therapy and the importance of precision medicine. Precision medicine in cancer care offers numerous advantages, including personalized treatment tailored to an individual's genetic and biological characteristics, minimizing side effects, increasing treatment success rates, avoiding trial‐and‐error approaches, early risk prediction and prevention measures, real‐time treatment monitoring, and contributing to future cancer research through data collection. However, the development of precision medicine must be built upon the foundation of preclinical research and clinical translation. Future studies need to focus on the mechanistic links between cellular stress sensors and embryo‐like transitions in different molecular contexts and tumor types, which enabling better understanding of the complexity and heterogeneity of tumors and facilitates the development and application of novel treatment approaches.
It is well understood that abnormal genetic or nongenetic alterations in normal cells can drive cancer onset and progression. However, the described changes can be viewed as a process of empowering tumor cells, enabling them to possess a range of malignant functions, including resistance to immune clearance, sustenance of their own growth, and the capability to metastasize within the body. To date, researchers have made significant strides in understanding that cancer cells exhibit the remarkable capacity to mimic various specialized cell types. They can, for example, emulate neutrophils by entering the circulatory system and infiltrating different organs, engage in asymmetric differentiation akin to stem cells, and even establish communication with the nervous system. 540 , 541 , 542 For these reasons, many scientists now believe that the genesis of cancer cells is, in fact, a form of atavism. 543 Meanwhile, the concept of oncofetal reprogramming has been proposed due to the striking resemblance between cancer cells and the high pluripotency potential exhibited by cells during early embryonic development in various higher animals. 544 Here, we have discussed representative embryonic development hallmarks contributing to tumor development and progression via genetic, epigenetic, or TME alterations. In addition, investigating the intricate mechanisms by which tumor cells respond to embryonic developmental genes and pathways can lead to new therapeutic approaches for cancer treatment in patients, increasing the possibility of a cure. Although methods for solving this problem appear challenging, we systematically revisit cancer development from an embryonic development perspective, which provides new insight into this intricate issue. Despite the disadvantages, it does seem feasible, at least conceptually, to draw a roadmap for confronting the problem of cancer therapy.
AUTHOR CONTRIBUTION
J. C., Z. Z., and L. Z. searched for original articles and wrote the manuscript. M. L., L. L., and B. L. prepared the figures. E. C. N. revised the manuscript. Z. S., W. H., and C. H. conceived and critically discussed the clinical unmet problems, underlying mechanisms and wrote the manuscript. All authors have read and approved the final version of the manuscript.
CONFLICT OF INTERESTS STATEMENT
Author Canhua Huang is an Editorial board member of Medcomm. Author Canhua Huang was not involved in the journal's review of or decisions related to this manuscript. The other authors declared no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
All images are created in whole or in part using BioRender (www.biorender.com), and they have all obtained licenses.
Cao J, Zhang Z, Zhou L, et al. Oncofetal reprogramming in tumor development and progression: novel insights into cancer therapy. MedComm. 2023;4:e427. 10.1002/mco2.427
Contributor Information
Weifeng He, Email: whe761211@hotmail.com.
Shaojiang Zheng, Email: zhengshaojiang@hainmc.edu.cn.
Canhua Huang, Email: hcanhua@scu.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Zhai J, Xiao Z, Wang Y, Wang H. Human embryonic development: From peri‐implantation to gastrulation. Trends Cell Biol. 2022;32(1):18‐29. [DOI] [PubMed] [Google Scholar]
- 2. Sidrat T, Rehman ZU, Joo MD, Lee KL, Kong IK. Wnt/β‐catenin pathway‐mediated PPARδ expression during embryonic development differentiation and disease. Int J Mol Sci. 2021;22(4):1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Telfer EE, Grosbois J, Odey YL, Rosario R, Anderson RA. Making a good egg: Human oocyte health, aging, and in vitro development. Physiol Rev. 2023;103(4):2623‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Souilhol C, Serbanovic‐Canic J, Fragiadaki M, et al. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat Rev Cardiol. 2020;17(1):52‐63. [DOI] [PubMed] [Google Scholar]
- 5. Hall ET, Cleverdon ER, Ogden SK. Dispatching sonic Hedgehog: Molecular mechanisms controlling deployment. Trends Cell Biol. 2019;29(5):385‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neto F, Klaus‐Bergmann A, Ong YT, et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife. 2018;7:e31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu MY, Hill CS. Tgf‐beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16(3):329‐343. [DOI] [PubMed] [Google Scholar]
- 8. Renfree MB, Fenelon JC. The enigma of embryonic diapause. Development. 2017;144(18):3199‐3210. [DOI] [PubMed] [Google Scholar]
- 9. Hussein AM, Wang Y, Mathieu J, et al. Metabolic control over mTOR‐dependent diapause‐like state. Dev Cell. 2020;52(2):236‐250. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu X, Zhou Y, Wei H. Roles of HLA‐G in the maternal‐fetal immune microenvironment. Front Immunol. 2020;11:592010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Al‐Enazy S, Ali S, Albekairi N, El‐Tawil M, Rytting E. Placental control of drug delivery. Adv Drug Deliv Rev. 2017;116:63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gardner DK, Pool TB, Lane M. Embryo nutrition and energy metabolism and its relationship to embryo growth, differentiation, and viability. Semin Reprod Med. 2000;18(2):205‐218. [DOI] [PubMed] [Google Scholar]
- 13. Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015;14(1):29‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—a clinical update. Nat Rev Clin Oncol. 2020;17(4):204‐232. [DOI] [PubMed] [Google Scholar]
- 15. Konieczna A, Lichnovka R, Erdosova B, Ehrmann J. The Role of PPARs in MDR—a lesson from embryonic development. Neoplasma. 2009;56(4):279‐283. [DOI] [PubMed] [Google Scholar]
- 16. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Das PK, Islam F, Lam AK. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells. 2020;9(6):1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho YH, Ro EJ, Yoon JS, et al. 5‐FU promotes stemness of colorectal cancer via p53‐mediated WNT/β‐catenin pathway activation. Nat Commun. 2020;11(1):5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang N, Zou C, Zhu Y, et al. HIF‐1ɑ‐regulated miR‐1275 maintains stem cell‐like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/β‐catenin and Notch signaling. Theranostics. 2020;10(6):2553‐2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cordenonsi M, Zanconato F, Azzolin L, et al. The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell. 2011;147(4):759‐772. [DOI] [PubMed] [Google Scholar]
- 21. Vidal AC, Howard LE, Moreira DM, Castro‐Santamaria R, Andriole GL, Freedland SJ. Aspirin, NSAIDs, and risk of prostate cancer: Results from the REDUCE study. Clin Cancer Res. 2015;21(4):756‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan J, To KKW, Chen ZS, Fu L. ABC transporters affects tumor immune microenvironment to regulate cancer immunotherapy and multidrug resistance. Drug Resist Updat. 2023;66:100905. [DOI] [PubMed] [Google Scholar]
- 23. Giddings EL, Champagne DP, Wu MH, et al. Mitochondrial ATP fuels ABC transporter‐mediated drug efflux in cancer chemoresistance. Nat Commun. 2021;12(1):2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L. Overcoming ABC transporter‐mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur J Med Chem. 2017;142:271‐289. [DOI] [PubMed] [Google Scholar]
- 25. Matei D, Filiaci V, Randall ME, et al. Adjuvant chemotherapy plus radiation for locally advanced endometrial cancer. N Engl J Med. 2019;380(24):2317‐2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rehman SK, Haynes J, Collignon E, et al. Colorectal cancer cells enter a diapause‐like DTP state to survive chemotherapy. Cell. 2021;184(1):226‐242. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qin S, Li B, Ming H, Nice EC, Zou B, Huang C. Harnessing redox signaling to overcome therapeutic‐resistant cancer dormancy. Biochim Biophys Acta Rev Cancer. 2022;1877(4):188749. [DOI] [PubMed] [Google Scholar]
- 28. Mikubo M, Inoue Y, Liu G, Tsao MS. Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J Thorac Oncol. 2021;16(11):1798‐1809. [DOI] [PubMed] [Google Scholar]
- 29. Chan T, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann Oncol. 2019;30(1):44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: From hype to hope. Cell Stem Cell. 2019;24(1):25‐40. [DOI] [PubMed] [Google Scholar]
- 31. Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer. 2021;21(1):5‐21. [DOI] [PubMed] [Google Scholar]
- 32. Errico A. Targeted therapies: Hippo effector YAP1 inhibition—towards a new therapeutic option to overcome drug resistance. Nat Rev Clin Oncol. 2015;12(4):190. [DOI] [PubMed] [Google Scholar]
- 33. Eberl M, Mangelberger D, Swanson JB, et al. Tumor architecture and notch signaling modulate drug response in basal cell carcinoma. Cancer Cell. 2018;33(2):229‐243. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99‐109. [DOI] [PubMed] [Google Scholar]
- 35. Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem‐cell compartments in the small intestine of mice lacking Tcf‐4. Nat Genet. 1998;19(4):379‐383. [DOI] [PubMed] [Google Scholar]
- 36. Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta‐Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105(4):533‐545. [DOI] [PubMed] [Google Scholar]
- 37. Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17(14):1709‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843‐850. [DOI] [PubMed] [Google Scholar]
- 39. Nusse R, Clevers H. Wnt/β‐catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985‐999. [DOI] [PubMed] [Google Scholar]
- 40. Wu Q, Ma J, Wei J, Meng W, Wang Y, Shi M. lncRNA SNHG11 promotes gastric cancer progression by activating the Wnt/β‐catenin pathway and oncogenic autophagy. Mol Ther. 2021;29(3):1258‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Asad M, Wong MK, Tan TZ, et al. FZD7 drives in vitro aggressiveness in Stem‐A subtype of ovarian cancer via regulation of non‐canonical Wnt/PCP pathway. Cell Death Dis. 2014;5(7):e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song M, Yeku OO, Rafiq S, et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat Commun. 2020;11(1):6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Y, Ye YC, Chen Y, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β‐catenin signaling promotes M2‐like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9(8):793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang H, Jiang R, Zhou J, et al. CTL attenuation regulated by PS1 in cancer‐associated fibroblast. Front Immunol. 2020;11:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kukcinaviciute E, Jonusiene V, Sasnauskiene A, Dabkeviciene D, Eidenaite E, Laurinavicius A. Significance of Notch and Wnt signaling for chemoresistance of colorectal cancer cells HCT116. J Cell Biochem. 2018;119(7):5913‐5920. [DOI] [PubMed] [Google Scholar]
- 46. Han P, Li JW, Zhang BM, et al. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR‐181a‐5p‐mediated regulation of Wnt/β‐catenin signaling. Mol Cancer. 2017;16(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang M, Zhang D, Wu JY, et al. Wnt‐mediated endothelial transformation into mesenchymal stem cell‐like cells induces chemoresistance in glioblastoma. Sci Transl Med. 2020;12(532):eaay7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan Y, Liu F, Han L, et al. HIF‐2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. 2018;37(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yue X, Lan F, Xia T. Hypoxic glioma cell‐secreted exosomal miR‐301a activates Wnt/β‐catenin signaling and promotes radiation resistance by targeting TCEAL7. Mol Ther. 2019;27(11):1939‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patel S, Alam A, Pant R, Chattopadhyay S. Wnt signaling and its significance within the tumor microenvironment: Novel therapeutic insights. Front Immunol. 2019;10:2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. DeVito NC, Sturdivant M, Thievanthiran B, et al. Pharmacological Wnt ligand inhibition overcomes key tumor‐mediated resistance pathways to anti‐PD‐1 immunotherapy. Cell Rep. 2021;35(5):109071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pinyol R, Sia D, Llovet JM. Immune exclusion‐Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res. 2019;25(7):2021‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruiz de Galarreta M, Bresnahan E, Molina‐Sánchez P, et al. β‐Catenin activation promotes immune escape and resistance to anti‐PD‐1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9(8):1124‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature. 2015;523(7559):231‐235. [DOI] [PubMed] [Google Scholar]
- 55. Lu L, Ling W, Ruan Z. TAM‐derived extracellular vesicles containing microRNA‐29a‐3p explain the deterioration of ovarian cancer. Mol Ther Nucleic Acids. 2021;25:468‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Du L, Lee JH, Jiang H, et al. β‐Catenin induces transcriptional expression of PD‐L1 to promote glioblastoma immune evasion. J Exp Med. 2020;217(11):e20191115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guo X, Qiu W, Wang J, et al. Glioma exosomes mediate the expansion and function of myeloid‐derived suppressor cells through microRNA‐29a/Hbp1 and microRNA‐92a/Prkar1a pathways. Int J Cancer. 2019;144(12):3111‐3126. [DOI] [PubMed] [Google Scholar]
- 58. Hu B, Tang WG, Fan J, Xu Y, Sun HX. Differentially expressed miRNAs in hepatocellular carcinoma cells under hypoxic conditions are associated with transcription and phosphorylation. Oncol Lett. 2018;15(1):467‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Collu GM, Hidalgo‐Sastre A, Brennan K. Wnt‐Notch signalling crosstalk in development and disease. Cell Mol Life Sci. 2014;71(18):3553‐3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cai X, Gong P, Huang Y, Lin Y. Notch signalling pathway in tooth development and adult dental cells. Cell Prolif. 2011;44(6):495‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9(1):49‐61. [DOI] [PubMed] [Google Scholar]
- 62. Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97(4):1235‐1294. [DOI] [PubMed] [Google Scholar]
- 63. Pourquié O. Skin development: Delta laid bare. Curr Biol. 2000;10(11):R425‐R428. [DOI] [PubMed] [Google Scholar]
- 64. Formosa‐Jordan P, Ibañes M, Ares S, Frade JM. Lateral inhibition and neurogenesis: Novel aspects in motion. Int J Dev Biol. 2013;57(5):341‐350. [DOI] [PubMed] [Google Scholar]
- 65. Bigas A, Espinosa L. The multiple usages of Notch signaling in development, cell differentiation and cancer. Curr Opin Cell Biol. 2018;55:1‐7. [DOI] [PubMed] [Google Scholar]
- 66. Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4(10):a011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. 2018;34(4):536‐548. [DOI] [PubMed] [Google Scholar]
- 68. Cao Z, Ding BS, Guo P, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell. 2014;25(3):350‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ayyanan A, Civenni G, Ciarloni L, et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch‐dependent mechanism. Proc Natl Acad Sci USA. 2006;103(10):3799‐3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rodilla V, Villanueva A, Obrador‐Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci USA. 2009;106(15):6315‐6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim W, Khan SK, Gvozdenovic‐Jeremic J, et al. Hippo signaling interactions with Wnt/β‐catenin and Notch signaling repress liver tumorigenesis. J Clin Invest. 2017;127(1):137‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kopan R, Ilagan MX. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell. 2009;137(2):216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bray SJ. Notch signalling: A simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7(9):678‐689. [DOI] [PubMed] [Google Scholar]
- 74. Boelens MC, Wu TJ, Nabet BY, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159(3):499‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. De Francesco EM, Maggiolini M, Musti AM. Crosstalk between Notch, HIF‐1α and GPER in breast cancer EMT. Int J Mol Sci. 2018;19(7):2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsuyada A, Chow A, Wu J, et al. CCL2 mediates cross‐talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768‐2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Huang TX, Guan XY, Fu L. Therapeutic targeting of the crosstalk between cancer‐associated fibroblasts and cancer stem cells. Am J Cancer Res. 2019;9(9):1889‐1904. [PMC free article] [PubMed] [Google Scholar]
- 78. De Luca A, Cerrato V, Fucà E, Parmigiani E, Buffo A, Leto K. Sonic hedgehog patterning during cerebellar development. Cell Mol Life Sci. 2016;73(2):291‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Laufer E, Kesper D, Vortkamp A, King P. Sonic hedgehog signaling during adrenal development. Mol Cell Endocrinol. 2012;351(1):19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ericson J, Briscoe J, Rashbass P, van Heyningen V, Jessell TM. Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb Symp Quant Biol. 1997;62:451‐466. [PubMed] [Google Scholar]
- 81. Ingham PW, McMahon AP. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001;15(23):3059‐3087. [DOI] [PubMed] [Google Scholar]
- 82. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445‐3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416‐429. [DOI] [PubMed] [Google Scholar]
- 85. Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2010;16(8):337‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tang YA, Chen YF, Bao Y, et al. Hypoxic tumor microenvironment activates GLI2 via HIF‐1α and TGF‐β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci USA. 2018;115(26):E5990‐E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Whitson RJ, Lee A, Urman NM, et al. Noncanonical hedgehog pathway activation through SRF‐MKL1 promotes drug resistance in basal cell carcinomas. Nat Med. 2018;24(3):271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jiang J. Hedgehog signaling mechanism and role in cancer. Semin Cancer Biol. 2022;85:107‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Youssef KK, Van Keymeulen A, Lapouge G, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010;12(3):299‐305. [DOI] [PubMed] [Google Scholar]
- 90. Sánchez‐Danés A, Hannezo E, Larsimont JC, et al. Defining the clonal dynamics leading to mouse skin tumour initiation. Nature. 2016;536(7616):298‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yao Y, Zhou D, Shi D, et al. GLI1 overexpression promotes gastric cancer cell proliferation and migration and induces drug resistance by combining with the AKT‐mTOR pathway. Biomed Pharmacother. 2019;111:993‐1004. [DOI] [PubMed] [Google Scholar]
- 92. Tang CT, Zeng CY, Chen YX. Letter to editor regarding “GLI1 overexpression promotes gastric cancer cell proliferation and migration and induces drug resistance by combining with the AKT‐mTOR pathway”. Biomed Pharmacother. 2020;122:109792. [DOI] [PubMed] [Google Scholar]
- 93. Alonso S, Hernandez D, Chang YT, et al. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J Clin Invest. 2016;126(12):4460‐4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yan GN, Yang L, Lv YF, et al. Endothelial cells promote stem‐like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol. 2014;234(1):11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Madison BB, Braunstein K, Kuizon E, Portman K, Qiao XT, Gumucio DL. Epithelial hedgehog signals pattern the intestinal crypt‐villus axis. Development. 2005;132(2):279‐289. [DOI] [PubMed] [Google Scholar]
- 96. Regan JL, Schumacher D, Staudte S, et al. Non‐canonical Hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 2017;21(10):2813‐2828. [DOI] [PubMed] [Google Scholar]
- 97. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19(7):480‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886‐897. [DOI] [PubMed] [Google Scholar]
- 99. Wang Y, Yu A, Yu FX. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell. 2017;8(5):349‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang Y, Zhang H, Zhao B. Hippo signaling in the immune system. Trends Biochem Sci. 2018;43(2):77‐80. [DOI] [PubMed] [Google Scholar]
- 101. Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9(5):534‐546. [DOI] [PubMed] [Google Scholar]
- 102. Zheng Y, Pan D. The Hippo signaling pathway in development and disease. Dev Cell. 2019;50(3):264‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the Cancer Genome Atlas. Cell. 2018;173(2):321‐337. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee CK, Jeong SH, Jang C, et al. Tumor metastasis to lymph nodes requires YAP‐dependent metabolic adaptation. Science. 2019;363(6427):644‐649. [DOI] [PubMed] [Google Scholar]
- 105. Janse van Rensburg HJ, Azad T, Ling M, et al. The Hippo pathway component TAZ promotes immune evasion in human cancer through PD‐L1. Cancer Res. 2018;78(6):1457‐1470. [DOI] [PubMed] [Google Scholar]
- 106. Feng J, Yang H, Zhang Y, et al. Tumor cell‐derived lactate induces TAZ‐dependent upregulation of PD‐L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36(42):5829‐5839. [DOI] [PubMed] [Google Scholar]
- 107. Li Z, Razavi P, Li Q, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell. 2018;34(6):893‐905. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lin L, Sabnis AJ, Chan E, et al. The Hippo effector YAP promotes resistance to RAF‐ and MEK‐targeted cancer therapies. Nat Genet. 2015;47(3):250‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Garcia‐Rendueles ME, Ricarte‐Filho JC, Untch BR, et al. NF2 loss promotes oncogenic RAS‐induced thyroid cancers via YAP‐dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov. 2015;5(11):1178‐1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Uchihara T, Miyake K, Yonemura A, et al. Extracellular vesicles from cancer‐associated fibroblasts containing annexin A6 induces FAK‐YAP activation by stabilizing β1 integrin, enhancing drug resistance. Cancer Res. 2020;80(16):3222‐3235. [DOI] [PubMed] [Google Scholar]
- 111. Yang Y, Ma Y, Yan S, et al. CAF promotes chemoresistance through NRP2 in gastric cancer. Gastric Cancer. 2022;25(3):503‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ghiso E, Migliore C, Ciciriello V, et al. YAP‐Dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia. 2017;19(12):1012‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Xu MZ, Chan SW, Liu AM, et al. AXL receptor kinase is a mediator of YAP‐dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30(10):1229‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zanconato F, Battilana G, Forcato M, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24(10):1599‐1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zuo Q, Liu J, Huang L, et al. AXL/AKT axis mediated‐resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene. 2018;37(24):3275‐3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti‐cancer approach: Functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wu G, Ma Z, Cheng Y, et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kariolis MS, Miao YR, Diep A, et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J Clin Invest. 2017;127(1):183‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang C, Jin H, Wang N, et al. Gas6/Axl axis contributes to chemoresistance and metastasis in breast cancer through Akt/GSK‐3β/β‐catenin signaling. Theranostics. 2016;6(8):1205‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Antony J, Tan TZ, Kelly Z, et al. The GAS6‐AXL signaling network is a mesenchymal (Mes) molecular subtype‐specific therapeutic target for ovarian cancer. Sci Signal. 2016;9(448):ra97. [DOI] [PubMed] [Google Scholar]
- 121. Rankin EB, Fuh KC, Castellini L, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci USA. 2014;111(37):13373‐13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chiu KC, Lee CH, Liu SY, et al. Polarization of tumor‐associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol. 2015;51(7):683‐689. [DOI] [PubMed] [Google Scholar]
- 123. Orlova A, Neubauer HA, Moriggl R. The stromal microenvironment provides an escape route from FLT3 inhibitors through the GAS6‐AXL‐STAT5 axis. Haematologica. 2019;104(10):1907‐1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF‐β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Li Y, Yan J, Chang HM, Chen ZJ, Leung PCK. Roles of TGF‐β superfamily proteins in extravillous trophoblast invasion. Trends Endocrinol Metab. 2021;32(3):170‐189. [DOI] [PubMed] [Google Scholar]
- 126. Mueller S, Engleitner T, Maresch R, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554(7690):62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nie E, Jin X, Miao F, et al. TGF‐β1 modulates temozolomide resistance in glioblastoma via altered microRNA processing and elevated MGMT. Neuro Oncol. 2021;23(3):435‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shan G, Gu J, Zhou D, et al. Cancer‐associated fibroblast‐secreted exosomal miR‐423‐5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF‐β signaling pathway. Exp Mol Med. 2020;52(11):1809‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yi M, Zhang J, Li A, et al. The construction, expression, and enhanced anti‐tumor activity of YM101: a bispecific antibody simultaneously targeting TGF‐β and PD‐L1. J Hematol Oncol. 2021;14(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Katsuno Y, Meyer DS, Zhang Z, et al. Chronic TGF‐β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. 2019;12(570):eaau854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: Learning from mouse models. Bone Res. 2014;2:14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics. 2000;69(2):275‐279. [DOI] [PubMed] [Google Scholar]
- 134. Xie Y, Su N, Yang J, et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther. 2020;5(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mahmood HA, Tomas Bort E, Walker AJ, Grose RP, Chioni AM. FGF signalling facilitates cervical cancer progression. Febs J. 2022;289(12):3440‐3456. [DOI] [PubMed] [Google Scholar]
- 136. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A proangiogenic signature is revealed in FGF‐mediated bevacizumab‐resistant head and neck squamous cell carcinoma. Mol Cancer Res. 2013;11(12):1585‐1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kurimoto R, Iwasawa S, Ebata T, et al. Drug resistance originating from a TGF‐β/FGF‐2‐driven epithelial‐to‐mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int J Oncol. 2016;48(5):1825‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bohrer LR, Chuntova P, Bade LK, et al. Activation of the FGFR‐STAT3 pathway in breast cancer cells induces a hyaluronan‐rich microenvironment that licenses tumor formation. Cancer Res. 2014;74(1):374‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Meuti ME, Denlinger DL. Evolutionary links between circadian clocks and photoperiodic diapause in insects. Integr Comp Biol. 2013;53(1):131‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Denlinger DL, Armbruster PA. Mosquito diapause. Annu Rev Entomol. 2014;59:73‐93. [DOI] [PubMed] [Google Scholar]
- 141. Tougeron K, Devogel M, van Baaren J, Le Lann C, Hance T. Trans‐generational effects on diapause and life‐history‐traits of an aphid parasitoid. J Insect Physiol. 2020;121:104001. [DOI] [PubMed] [Google Scholar]
- 142. Fenelon JC, Banerjee A, Murphy BD. Embryonic diapause: Development on hold. Int J Dev Biol. 2014;58(2‐4):163‐174. [DOI] [PubMed] [Google Scholar]
- 143. Liu WM, Cheng RR, Niu ZR, et al. Let‐7 derived from endometrial extracellular vesicles is an important inducer of embryonic diapause in mice. Sci Adv. 2020;6(37):eaaz7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Renfree MB. Embryonic diapause in marsupials. J Reprod Fertil Suppl. 1981;29:67‐78. [PubMed] [Google Scholar]
- 145. van der Weijden VA, Ulbrich SE. Embryonic diapause in roe deer: A model to unravel embryo‐maternal communication during pre‐implantation development in wildlife and livestock species. Theriogenology. 2020;158:105‐111. [DOI] [PubMed] [Google Scholar]
- 146. Deng L, Li C, Chen L, Liu Y, Hou R, Zhou X. Research advances on embryonic diapause in mammals. Anim Reprod Sci. 2018;198:1‐10. [DOI] [PubMed] [Google Scholar]
- 147. Fenelon JC, Renfree MB. The history of the discovery of embryonic diapause in mammals. Biol Reprod. 2018;99(1):242‐251. [DOI] [PubMed] [Google Scholar]
- 148. Basu S, Dong Y, Kumar R, Jeter C, Tang DG. Slow‐cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin Cancer Biol. 2022;78:90‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chang CA, Jen J, Jiang S, et al. Ontogeny and vulnerabilities of drug‐tolerant persisters in HER2+ breast cancer. Cancer Discov. 2022;12(4):1022‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Dhanyamraju PK, Schell TD, Amin S, Robertson GP. Drug‐tolerant persister cells in cancer therapy resistance. Cancer Res. 2022;82(14):2503‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. 2020;20(7):398‐411. [DOI] [PubMed] [Google Scholar]
- 152. Khoo WH, Ledergor G, Weiner A, et al. A niche‐dependent myeloid transcriptome signature defines dormant myeloma cells. Blood. 2019;134(1):30‐43. [DOI] [PubMed] [Google Scholar]
- 153. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell. 2013;155(4):750‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Roesch A, Fukunaga‐Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow‐cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Aberger F, Hutterer E, Sternberg C, Del Burgo PJ, Hartmann TN. Acute myeloid leukemia—strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun Signal. 2017;15(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zhang J, Si J, Gan L, et al. Research progress on therapeutic targeting of quiescent cancer cells. Artif Cells Nanomed Biotechnol. 2019;47(1):2810‐2820. [DOI] [PubMed] [Google Scholar]
- 158. Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence entry, maintenance, and exit in adult stem cells. Int J Mol Sci. 2019;20(9):2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dhimolea E, de Matos Simoes R, Kansara D, et al. An embryonic diapause‐like adaptation with suppressed Myc activity enables tumor treatment persistence. Cancer Cell. 2021;39(2):240‐256. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ohta Y, Fujii M, Takahashi S, et al. Cell‐matrix interface regulates dormancy in human colon cancer stem cells. Nature. 2022;608(7924):784‐794. [DOI] [PubMed] [Google Scholar]
- 161. Vera‐Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9(1):1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Lah TT, Novak M, Breznik B. Brain malignancies: Glioblastoma and brain metastases. Semin Cancer Biol. 2020;60:262‐273. [DOI] [PubMed] [Google Scholar]
- 163. Tsakiris N, Fauvet F, Ruby S, et al. Combined nanomedicines targeting colorectal cancer stem cells and cancer cells. J Control Release. 2020;326:387‐395. [DOI] [PubMed] [Google Scholar]
- 164. Fox DB, Garcia NMG, McKinney BJ, et al. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat Metab. 2020;2(4):318‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation‐resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bast RC Jr. Molecular approaches to personalizing management of ovarian cancer. Ann Oncol. 2011;22:viii5‐viii15. Suppl 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Balaban NQ, Helaine S, Lewis K, et al. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol. 2019;17(7):441‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Marine JC, Dawson SJ, Dawson MA. Non‐genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743‐756. [DOI] [PubMed] [Google Scholar]
- 169. Brock A, Chang H, Huang S. Non‐genetic heterogeneity–a mutation‐independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10(5):336‐342. [DOI] [PubMed] [Google Scholar]
- 170. Hu Z, Ding J, Ma Z, et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat Genet. 2019;51(7):1113‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hedberg ML, Goh G, Chiosea SI, et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest. 2016;126(4):1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Dobson SM, García‐Prat L, Vanner RJ, et al. Relapse‐fated latent diagnosis subclones in acute b lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020;10(4):568‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Fane ME, Chhabra Y, Alicea GM, et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature. 2022;606(7913):396‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hirata E, Girotti MR, Viros A, et al. Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27(4):574‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Plava J, Cihova M, Burikova M, Matuskova M, Kucerova L, Miklikova S. Recent advances in understanding tumor stroma‐mediated chemoresistance in breast cancer. Mol Cancer. 2019;18(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bartosh TJ, Ullah M, Zeitouni S, Beaver J, Prockop DJ. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc Natl Acad Sci USA. 2016;113(42):E6447‐E6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Martins I, Raza SQ, Voisin L, et al. Anticancer chemotherapy and radiotherapy trigger both non‐cell‐autonomous and cell‐autonomous death. Cell Death Dis. 2018;9(7):716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Overholtzer M, Mailleux AA, Mouneimne G, et al. A nonapoptotic cell death process, entosis, that occurs by cell‐in‐cell invasion. Cell. 2007;131(5):966‐979. [DOI] [PubMed] [Google Scholar]
- 181. Gesualdi L, Leonetti E, Cucina A, et al. The PI3K/AKT pathway is activated by HGF in NT2D1 non‐seminoma cells and has a role in the modulation of their malignant behavior. Int J Mol Sci. 2020;21(22):8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Xu J, Liu S, Yang X, Cao S, Zhou Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c‐Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020;263:118523. [DOI] [PubMed] [Google Scholar]
- 183. Zhang KL, Zhu WW, Wang SH, et al. Organ‐specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. 2021;11(13):6560‐6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Straussman R, Morikawa T, Shee K, et al. Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Sánchez‐Hernández I, Baquero P, Calleros L, Chiloeches A. Dual inhibition of (V600E)BRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK‐independent mechanism. Cancer Lett. 2012;314(2):244‐255. [DOI] [PubMed] [Google Scholar]
- 186. Byeon HK, Na HJ, Yang YJ, et al. c‐Met‐mediated reactivation of PI3K/AKT signaling contributes to insensitivity of BRAF(V600E) mutant thyroid cancer to BRAF inhibition. Mol Carcinog. 2016;55(11):1678‐1687. [DOI] [PubMed] [Google Scholar]
- 187. Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF‐mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Watson SS, Dane M, Chin K, et al. Microenvironment‐mediated mechanisms of resistance to HER2 inhibitors differ between HER2+ breast cancer subtypes. Cell Syst. 2018;6(3):329‐342. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Raghavan S, Winter PS, Navia AW, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell. 2021;184(25):6119‐6137. e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Biffi G, Tuveson DA. Diversity and biology of cancer‐associated fibroblasts. Physiol Rev. 2021;101(1):147‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Plaks V, Kong N, Werb Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Zhang X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun (Lond). 2019;39(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Gooding S, Olechnowicz SWZ, Morris EV, et al. Transcriptomic profiling of the myeloma bone‐lining niche reveals BMP signalling inhibition to improve bone disease. Nat Commun. 2019;10(1):4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kopecka J, Salaroglio IC, Perez‐Ruiz E, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updat. 2021;59:100787. [DOI] [PubMed] [Google Scholar]
- 196. Carcereri de Prati A, Butturini E, Rigo A, et al. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J Cell Biochem. 2017;118(10):3237‐3248. [DOI] [PubMed] [Google Scholar]
- 197. Fluegen G, Avivar‐Valderas A, Wang Y, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19(2):120‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15(7):807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Tao WY, Liang XS, Liu Y, Wang CY, Pang D. Decrease of let‐7f in low‐dose metronomic Paclitaxel chemotherapy contributed to upregulation of thrombospondin‐1 in breast cancer. Int J Biol Sci. 2015;11(1):48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Braham MVJ, Ahlfeld T, Akkineni AR, et al. Endosteal and perivascular subniches in a 3D bone marrow model for multiple myeloma. Tissue Eng Part C Methods. 2018;24(5):300‐312. [DOI] [PubMed] [Google Scholar]
- 201. Lawson MA, McDonald MM, Kovacic N, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015;6:8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood. 2013;121(24):4821‐4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Risson E, Nobre AR, Maguer‐Satta V, Aguirre‐Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer. 2020;1(7):672‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol. 2013;10(1):41‐51. [DOI] [PubMed] [Google Scholar]
- 205. Ebinger S, Özdemir EZ, Ziegenhain C, et al. Characterization of rare, dormant, and therapy‐resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30(6):849‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Shimizu T, Sugihara E, Yamaguchi‐Iwai S, et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014;74(22):6531‐6541. [DOI] [PubMed] [Google Scholar]
- 207. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells—mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. 2017;14(3):155‐167. [DOI] [PubMed] [Google Scholar]
- 208. Ding D, Allman BL, Salvi R. Review: Ototoxic characteristics of platinum antitumor drugs. Anat Rec (Hoboken). 2012;295(11):1851‐1867. [DOI] [PubMed] [Google Scholar]
- 209. Dembic Z. Antitumor drugs and their targets. Molecules. 2020;25(23):5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Echeverria GV, Ge Z, Seth S, et al. Resistance to neoadjuvant chemotherapy in triple‐negative breast cancer mediated by a reversible drug‐tolerant state. Sci Transl Med. 2019;11(488):eaav0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Álvarez‐Varela A, Novellasdemunt L, Barriga FM, et al. Mex3a marks drug‐tolerant persister colorectal cancer cells that mediate relapse after chemotherapy. Nat Cancer. 2022;3(9):1052‐1070. [DOI] [PubMed] [Google Scholar]
- 212. Guler GD, Tindell CA, Pitti R, et al. Repression of stress‐induced LINE‐1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell. 2017;32(2):221‐237. e13. [DOI] [PubMed] [Google Scholar]
- 213. Sharma SV, Lee DY, Li B, et al. A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16(4):313‐317. [DOI] [PubMed] [Google Scholar]
- 215. Lin WC, Rajbhandari N, Liu C, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73(6):1821‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hu X, Liu R, Hou J, et al. SMARCE1 promotes neuroblastoma tumorigenesis through assisting MYCN‐mediated transcriptional activation. Oncogene. 2022;41(37):4295‐4306. [DOI] [PubMed] [Google Scholar]
- 217. Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. 2019;40(2):128‐141. [DOI] [PubMed] [Google Scholar]
- 218. Rehe K, Wilson K, Bomken S, et al. Acute B lymphoblastic leukaemia‐propagating cells are present at high frequency in diverse lymphoblast populations. EMBO Mol Med. 2013;5(1):38‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185‐S198. Suppl. [DOI] [PubMed] [Google Scholar]
- 220. Balasubramanian A, John T, Asselin‐Labat ML. Regulation of the antigen presentation machinery in cancer and its implication for immune surveillance. Biochem Soc Trans. 2022;50(2):825‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Pantel K, Schlimok G, Kutter D, et al. Frequent down‐regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991;51(17):4712‐4715. [PubMed] [Google Scholar]
- 222. Erdogdu IH. MHC class 1 and PDL‐1 status of primary tumor and lymph node metastatic tumor tissue in gastric cancers. Gastroenterol Res Pract. 2019;2019:4785098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Garrido F, Aptsiauri N. Cancer immune escape: mHC expression in primary tumours versus metastases. Immunology. 2019;158(4):255‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Pommier A, Anaparthy N, Memos N, et al. Unresolved endoplasmic reticulum stress engenders immune‐resistant, latent pancreatic cancer metastases. Science. 2018;360(6394):eaao4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Johnson DB, Estrada MV, Salgado R, et al. Melanoma‐specific MHC‐II expression represents a tumour‐autonomous phenotype and predicts response to anti‐PD‐1/PD‐L1 therapy. Nat Commun. 2016;7:10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Rodig SJ, Gusenleitner D, Jackson DG, et al. MHC proteins confer differential sensitivity to CTLA‐4 and PD‐1 blockade in untreated metastatic melanoma. Sci Transl Med. 2018;10(450):eaar3342. [DOI] [PubMed] [Google Scholar]
- 227. Johnson DB, Nixon MJ, Wang Y, et al. Tumor‐specific MHC‐II expression drives a unique pattern of resistance to immunotherapy via LAG‐3/FCRL6 engagement. JCI Insight. 2018;3(24):e120360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Winkler IG, Sims NA, Pettit AR, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116(23):4815‐4828. [DOI] [PubMed] [Google Scholar]
- 229. Chow A, Lucas D, Hidalgo A, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208(2):261‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208(3):421‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Robertson SA, Care AS, Moldenhauer LM. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. 2018;128(10):4224‐4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Sharma A, Seow JJW, Dutertre CA, et al. Onco‐fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183(2):377‐394. e21. [DOI] [PubMed] [Google Scholar]
- 233. Zhang X, Wei H. Role of decidual natural killer cells in human pregnancy and related pregnancy complications. Front Immunol. 2021;12:728291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Cassetta L, Fragkogianni S, Sims AH, et al. Human tumor‐associated macrophage and monocyte transcriptional landscapes reveal cancer‐specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588‐602. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Cassetta L, Pollard JW. A timeline of tumour‐associated macrophage biology. Nat Rev Cancer. 2023;23(4):238‐257. [DOI] [PubMed] [Google Scholar]
- 236. Gocheva V, Wang HW, Gadea BB, et al. IL‐4 induces cathepsin protease activity in tumor‐associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Lu H, Clauser KR, Tam WL, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16(11):1105‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Fan QM, Jing YY, Yu GF, et al. Tumor‐associated macrophages promote cancer stem cell‐like properties via transforming growth factor‐beta1‐induced epithelial‐mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352(2):160‐168. [DOI] [PubMed] [Google Scholar]
- 239. Borriello L, Coste A, Traub B, et al. Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nat Commun. 2022;13(1):626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Qin J, Zhang X, Tan B, et al. Blocking P2×7‐mediated macrophage polarization overcomes treatment resistance in lung cancer. Cancer Immunol Res. 2020;8(11):1426‐1439. [DOI] [PubMed] [Google Scholar]
- 241. Liu M, Tong Z, Ding C, et al. Transcription factor c‐Maf is a checkpoint that programs macrophages in lung cancer. J Clin Invest. 2020;130(4):2081‐2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Etzerodt A, Tsalkitzi K, Maniecki M, et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell‐mediated tumor regression. J Exp Med. 2019;216(10):2394‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid‐derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zhang H, Maric I, DiPrima MJ, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013;122(7):1105‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Law AMK, Valdes‐Mora F, Gallego‐Ortega D. Myeloid‐derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9(3):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Ouzounova M, Lee E, Piranlioglu R, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8:14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Kinoshita R, Sato H, Yamauchi A, et al. Newly developed anti‐S100A8/A9 monoclonal antibody efficiently prevents lung tropic cancer metastasis. Int J Cancer. 2019;145(2):569‐575. [DOI] [PubMed] [Google Scholar]
- 248. Kim W, Chu TH, Nienhüser H, et al. PD‐1 signaling promotes tumor‐infiltrating myeloid‐derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology. 2021;160(3):781‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Liao W, Overman MJ, Boutin AT, et al. KRAS‐IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(4):559‐572. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Zhou J, Liu M, Sun H, et al. Hepatoma‐intrinsic CCRK inhibition diminishes myeloid‐derived suppressor cell immunosuppression and enhances immune‐checkpoint blockade efficacy. Gut. 2018;67(5):931‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Bockorny B, Semenisty V, Macarulla T, et al. BL‐8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat Med. 2020;26(6):878‐885. [DOI] [PubMed] [Google Scholar]
- 252. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol. 2020;65:99‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Huang F, Gonçalves C, Bartish M, et al. Inhibiting the MNK1/2‐eIF4E axis impairs melanoma phenotype switching and potentiates antitumor immune responses. J Clin Invest. 2021;131(8):e140752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Koren G, Ornoy A. The role of the placenta in drug transport and fetal drug exposure. Expert Rev Clin Pharmacol. 2018;11(4):373‐385. [DOI] [PubMed] [Google Scholar]
- 255. Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res. 2004;114(5‐6):397‐407. [DOI] [PubMed] [Google Scholar]
- 256. Sulamaa M, Ryoeppy S. Early treatment of congenital bone defects of the extremities: Aftermath of thalidomide disaster. Lancet. 1964;1(7325):130‐132. [DOI] [PubMed] [Google Scholar]
- 257. Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med. 1998;338(16):1128‐1137. [DOI] [PubMed] [Google Scholar]
- 258. Myllynen P, Pasanen M, Vähäkangas K. The fate and effects of xenobiotics in human placenta. Expert Opin Drug Metab Toxicol. 2007;3(3):331‐346. [DOI] [PubMed] [Google Scholar]
- 259. Cerrillo I, Granada A, López‐Espinosa MJ, et al. Endosulfan and its metabolites in fertile women, placenta, cord blood, and human milk. Environ Res. 2005;98(2):233‐239. [DOI] [PubMed] [Google Scholar]
- 260. Gros P, Ben Neriah YB, Croop JM, Housman DE. Isolation and expression of a complementary DNA that confers multidrug resistance. Nature. 1986;323(6090):728‐731. [DOI] [PubMed] [Google Scholar]
- 261. Baello S, Iqbal M, Bloise E, Javam M, Gibb W, Matthews SG. TGF‐β1 regulation of multidrug resistance P‐glycoprotein in the developing male blood‐brain barrier. Endocrinology. 2014;155(2):475‐484. [DOI] [PubMed] [Google Scholar]
- 262. Baardman ME, Kerstjens‐Frederikse WS, Berger RM, Bakker MK, Hofstra RM, Plösch T. The role of maternal‐fetal cholesterol transport in early fetal life: Current insights. Biol Reprod. 2013;88(1):24. [DOI] [PubMed] [Google Scholar]
- 263. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug‐resistant cancer. Nat Rev Cancer. 2018;18(7):452‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Szakács G, Paterson JK, Ludwig JA, Booth‐Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219‐234. [DOI] [PubMed] [Google Scholar]
- 265. Ambudkar SV, Kimchi‐Sarfaty C, Sauna ZE, Gottesman MM. P‐glycoprotein: From genomics to mechanism. Oncogene. 2003;22(47):7468‐7485. [DOI] [PubMed] [Google Scholar]
- 266. Trowitzsch S, Tampé R. ABC transporters in dynamic macromolecular assemblies. J Mol Biol. 2018;430(22):4481‐4495. [DOI] [PubMed] [Google Scholar]
- 267. Maqbool A, Horler RS, Muller A, Wilkinson AJ, Wilson KS, Thomas GH. The substrate‐binding protein in bacterial ABC transporters: Dissecting roles in the evolution of substrate specificity. Biochem Soc Trans. 2015;43(5):1011‐1017. [DOI] [PubMed] [Google Scholar]
- 268. Mlejnek P, Kosztyu P, Dolezel P, Bates SE, Ruzickova E. Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem Biol Interact. 2017;273:171‐179. [DOI] [PubMed] [Google Scholar]
- 269. Yuan T, Sun J, Tian J, Hu J, Yin H, Yin J. Involvement of ABC transporters in the detoxification of non‐substrate nanoparticles in lung and cervical cancer cells. Toxicology. 2021;455:152762. [DOI] [PubMed] [Google Scholar]
- 270. Gao Q, Li XX, Xu YM, et al. IRE1α‐targeting downregulates ABC transporters and overcomes drug resistance of colon cancer cells. Cancer Lett. 2020;476:67‐74. [DOI] [PubMed] [Google Scholar]
- 271. Bar‐Zeev M, Livney YD, Assaraf YG. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist Updat. 2017;31:15‐30. [DOI] [PubMed] [Google Scholar]
- 272. Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter‐mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8(6):1996‐2011. [DOI] [PubMed] [Google Scholar]
- 273. Cheng X, He L, Xu J, et al. Oxygen‐producing catalase‐based prodrug nanoparticles overcoming resistance in hypoxia‐mediated chemo‐photodynamic therapy. Acta Biomater. 2020;112:234‐249. [DOI] [PubMed] [Google Scholar]
- 274. Ding Z, Yang L, Xie X, et al. Expression and significance of hypoxia‐inducible factor‐1 alpha and MDR1/P‐glycoprotein in human colon carcinoma tissue and cells. J Cancer Res Clin Oncol. 2010;136(11):1697‐1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Liang Y, Zheng T, Song R, et al. Hypoxia‐mediated sorafenib resistance can be overcome by EF24 through Von Hippel‐Lindau tumor suppressor‐dependent HIF‐1α inhibition in hepatocellular carcinoma. Hepatology. 2013;57(5):1847‐1857. [DOI] [PubMed] [Google Scholar]
- 276. Xie J, Li DW, Chen XW, Wang F, Dong P. Expression and significance of hypoxia‐inducible factor‐1α and MDR1/P‐glycoprotein in laryngeal carcinoma tissue and hypoxic Hep‐2 cells. Oncol Lett. 2013;6(1):232‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Wartenberg M, Ling FC, Müschen M, et al. Regulation of the multidrug resistance transporter P‐glycoprotein in multicellular tumor spheroids by hypoxia‐inducible factor (HIF‐1) and reactive oxygen species. Faseb J. 2003;17(3):503‐505. [DOI] [PubMed] [Google Scholar]
- 278. Cui Q, Wang JQ, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1‐25. [DOI] [PubMed] [Google Scholar]
- 279. Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol Res. 2015;100:170‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Liu J, Zhu C, Xu L, et al. Nanoenabled intracellular calcium bursting for safe and efficient reversal of drug resistance in tumor cells. Nano Lett. 2020;20(11):8102‐8111. [DOI] [PubMed] [Google Scholar]
- 281. Riganti C, Doublier S, Viarisio D, et al. Artemisinin induces doxorubicin resistance in human colon cancer cells via calcium‐dependent activation of HIF‐1alpha and P‐glycoprotein overexpression. Br J Pharmacol. 2009;156(7):1054‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Chen YL, Yang TY, Chen KC, Wu CL, Hsu SL, Hsueh CM. Hypoxia can impair doxorubicin resistance of non‐small cell lung cancer cells by inhibiting MRP1 and P‐gp expression and boosting the chemosensitizing effects of MRP1 and P‐gp blockers. Cell Oncol (Dordr). 2016;39(5):411‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Zhou D, Gu J, Wang Y, et al. Long non‐coding RNA NEAT1 transported by extracellular vesicles contributes to breast cancer development by sponging microRNA‐141‐3p and regulating KLF12. Cell Biosci. 2021;11(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Cao C, Sun G, Liu C. Long non‐coding RNA SNHG6 regulates the sensitivity of prostate cancer cells to paclitaxel by sponging miR‐186. Cancer Cell Int. 2020;20:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Gong F, Dong D, Zhang T, Xu W. Long non‐coding RNA FENDRR attenuates the stemness of non‐small cell lung cancer cells via decreasing multidrug resistance gene 1 (MDR1) expression through competitively binding with RNA binding protein HuR. Eur J Pharmacol. 2019;853:345‐352. [DOI] [PubMed] [Google Scholar]
- 286. Gao ZQ, Wang JF, Chen DH, et al. Long non‐coding RNA GAS5 antagonizes the chemoresistance of pancreatic cancer cells through down‐regulation of miR‐181c‐5p. Biomed Pharmacother. 2018;97:809‐817. [DOI] [PubMed] [Google Scholar]
- 287. Zou H, Li H. Knockdown of long non‐coding RNA LINC00152 increases cisplatin sensitivity in ovarian cancer cells. Exp Ther Med. 2019;18(6):4510‐4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Wang Y, Wang Y, Qin Z, et al. The role of non‐coding RNAs in ABC transporters regulation and their clinical implications of multidrug resistance in cancer. Expert Opin Drug Metab Toxicol. 2021;17(3):291‐306. [DOI] [PubMed] [Google Scholar]
- 289. Ma Y, Yang Y, Wang F, et al. Long non‐coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β‐catenin signalling pathway via suppression of activator protein 2α. Gut. 2016;65(9):1494‐1504. [DOI] [PubMed] [Google Scholar]
- 290. Pan X, Hong X, Li S, Meng P, Xiao F. METTL3 promotes adriamycin resistance in MCF‐7 breast cancer cells by accelerating pri‐microRNA‐221‐3p maturation in a m6A‐dependent manner. Exp Mol Med. 2021;53(1):91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol. 2014;54:95‐117. [DOI] [PubMed] [Google Scholar]
- 292. Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat. 2016;26:1‐9. [DOI] [PubMed] [Google Scholar]
- 293. Boumendjel A, Baubichon‐Cortay H, Trompier D, Perrotton T, Di Pietro A. Anticancer multidrug resistance mediated by MRP1: recent advances in the discovery of reversal agents. Med Res Rev. 2005;25(4):453‐472. [DOI] [PubMed] [Google Scholar]
- 294. Mirski SE, Gerlach JH, Cole SP. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res. 1987;47(10):2594‐2598. [PubMed] [Google Scholar]
- 295. Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug‐resistant human lung cancer cell line. Science. 1992;258(5088):1650‐1654. [DOI] [PubMed] [Google Scholar]
- 296. Chen XY, Yang Y, Wang JQ, Wu ZX, Li J, Chen ZS. Overexpression of ABCC1 confers drug resistance to Betulin. Front Oncol. 2021;11:640656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Martin‐Broto J, Gutierrez AM, Ramos RF, et al. MRP1 overexpression determines poor prognosis in prospectively treated patients with localized high‐risk soft tissue sarcoma of limbs and trunk wall: An ISG/GEIS study. Mol Cancer Ther. 2014;13(1):249‐259. [DOI] [PubMed] [Google Scholar]
- 298. Poulain S, Lepelley P, Preudhomme C, et al. Expression of the multidrug resistance‐associated protein in myelodysplastic syndromes. Br J Haematol. 2000;110(3):591‐598. [DOI] [PubMed] [Google Scholar]
- 299. Lv Y, Zhao S, Han J, Zheng L, Yang Z, Zhao L. Hypoxia‐inducible factor‐1α induces multidrug resistance protein in colon cancer. Onco Targets Ther. 2015;8:1941‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Hyun JY, Chun YS, Kim TY, Kim HL, Kim MS, Park JW. Hypoxia‐inducible factor 1alpha‐ mediated resistance to phenolic anticancer. Chemotherapy. 2004;50(3):119‐126. [DOI] [PubMed] [Google Scholar]
- 301. Liu L, Ning X, Sun L, et al. Hypoxia‐inducible factor‐1 alpha contributes to hypoxia‐induced chemoresistance in gastric cancer. Cancer Sci. 2008;99(1):121‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Kolenda J, Jensen SS, Aaberg‐Jessen C, et al. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma‐derived spheroids. J Neurooncol. 2011;103(1):43‐58. [DOI] [PubMed] [Google Scholar]
- 303. Guo D, Xu S, Huang Y, et al. Platinum(IV) complex‐based two‐in‐one polyprodrug for a combinatorial chemo‐photodynamic therapy. Biomaterials. 2018;177:67‐77. [DOI] [PubMed] [Google Scholar]
- 304. Li C, Guo D, Tang B, Zhang Y, Zhang K, Nie L. Notch1 is associated with the multidrug resistance of hypoxic osteosarcoma by regulating MRP1 gene expression. Neoplasma. 2016;63(5):734‐742. [DOI] [PubMed] [Google Scholar]
- 305. Zhu H, Chen XP, Luo SF, Guan J, Zhang WG, Zhang BX. Involvement of hypoxia‐inducible factor‐1‐alpha in multidrug resistance induced by hypoxia in HepG2 cells. J Exp Clin Cancer Res. 2005;24(4):565‐574. [PubMed] [Google Scholar]
- 306. Ji Z, Long H, Hu Y, et al. Expression of MDR1, HIF‐1α and MRP1 in sacral chordoma and chordoma cell line CM‐319. J Exp Clin Cancer Res. 2010;29(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Zhu H, Luo SF, Wang J, et al. Effect of environmental factors on chemoresistance of HepG2 cells by regulating hypoxia‐inducible factor‐1α. Chin Med J (Engl). 2012;125(6):1095‐1103. [PubMed] [Google Scholar]
- 308. Wei L, Lin Q, Lu Y, et al. Cancer‐associated fibroblasts‐mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF‐β1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021;12(4):334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Zhang D, Ding L, Li Y, et al. Midkine derived from cancer‐associated fibroblasts promotes cisplatin‐resistance via up‐regulation of the expression of lncRNA ANRIL in tumour cells. Sci Rep. 2017;7(1):16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Dong Q, Zhou C, Ren H, et al. Lactate‐induced MRP1 expression contributes to metabolism‐based etoposide resistance in non‐small cell lung cancer cells. Cell Commun Signal. 2020;18(1):167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Selever J, Gu G, Lewis MT, et al. Dicer‐mediated upregulation of BCRP confers tamoxifen resistance in human breast cancer cells. Clin Cancer Res. 2011;17(20):6510‐6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Peña‐Solórzano D, Stark SA, König B, Sierra CA, Ochoa‐Puentes C. ABCG2/BCRP: Specific and nonspecific modulators. Med Res Rev. 2017;37(5):987‐1050. [DOI] [PubMed] [Google Scholar]
- 313. Lee J, Kang J, Kwon NY, et al. Dual inhibition of P‐gp and BCRP improves oral topotecan bioavailability in rodents. Pharmaceutics. 2021;13(4):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Shi Q, Li Y, Li S, et al. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat Commun. 2020;11(1):5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia‐induced transcription. Proc Natl Acad Sci USA. 1998;95(20):11715‐11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Xiang L, Liu ZH, Huan Q, et al. Hypoxia‐inducible factor‐2a is associated with ABCG2 expression, histology‐grade and Ki67 expression in breast invasive ductal carcinoma. Diagn Pathol. 2012;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Ming H, Li B, Tian H, et al. A minimalist and robust chemo‐photothermal nanoplatform capable of augmenting autophagy‐modulated immune response against breast cancer. Mater Today Bio. 2022;15:100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Wang WJ, Sui H, Qi C, et al. Ursolic acid inhibits proliferation and reverses drug resistance of ovarian cancer stem cells by downregulating ABCG2 through suppressing the expression of hypoxia‐inducible factor‐1α in vitro. Oncol Rep. 2016;36(1):428‐440. [DOI] [PubMed] [Google Scholar]
- 319. Li F, Aljahdali IAM, Zhang R, Nastiuk KL, Krolewski JJ, Ling X. Kidney cancer biomarkers and targets for therapeutics: Survivin (BIRC5), XIAP, MCL‐1, HIF1α, HIF2α, NRF2, MDM2, MDM4, p53, KRAS and AKT in renal cell carcinoma. J Exp Clin Cancer Res. 2021;40(1):254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Di Desidero T, Orlandi P, Gentile D, Bocci G. Effects of pazopanib monotherapy vs. pazopanib and topotecan combination on anaplastic thyroid cancer cells. Front Oncol. 2019;9:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Martin CM, Ferdous A, Gallardo T, et al. Hypoxia‐inducible factor‐2alpha transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res. 2008;102(9):1075‐1081. [DOI] [PubMed] [Google Scholar]
- 322. Li B, Jiang J, Assaraf YG, Xiao H, Chen ZS, Huang C. Surmounting cancer drug resistance: New insights from the perspective of N(6)‐methyladenosine RNA modification. Drug Resist Updat. 2020;53:100720. [DOI] [PubMed] [Google Scholar]
- 323. Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation‐treated cardiomyocytes. Autophagy. 2019;15(8):1419‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m⁶A‐demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113(14):E2047‐E2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. De Marco P, Lappano R, De Francesco EM, et al. GPER signalling in both cancer‐associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci Rep. 2016;6:24354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Dittmer A, Lange T, Leyh B, Dittmer J. Protein‑ and growth‑modulatory effects of carcinoma‑associated fibroblasts on breast cancer cells: Role of interleukin‑6. Int J Oncol. 2020;56(1):258‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Liu PP, Liao J, Tang ZJ, et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014;21(1):124‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Li W, Zhang H, Assaraf YG, et al. Overcoming ABC transporter‐mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. 2016;27:14‐29. [DOI] [PubMed] [Google Scholar]
- 329. Marzac C, Garrido E, Tang R, et al. ATP Binding Cassette transporters associated with chemoresistance: Transcriptional profiling in extreme cohorts and their prognostic impact in a cohort of 281 acute myeloid leukemia patients. Haematologica. 2011;96(9):1293‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Shen DW, Fojo A, Chin JE, et al. Human multidrug‐resistant cell lines: Increased mdr1 expression can precede gene amplification. Science. 1986;232(4750):643‐645. [DOI] [PubMed] [Google Scholar]
- 331. Modlich U, Kustikova OS, Schmidt M, et al. Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis. Blood. 2005;105(11):4235‐4246. [DOI] [PubMed] [Google Scholar]
- 332. Mazerska Z, Mróz A, Pawłowska M, Augustin E. The role of glucuronidation in drug resistance. Pharmacol Ther. 2016;159:35‐55. [DOI] [PubMed] [Google Scholar]
- 333. Maeda K, Sugiyama Y. Transporter biology in drug approval: Regulatory aspects. Mol Aspects Med. 2013;34(2‐3):711‐718. [DOI] [PubMed] [Google Scholar]
- 334. Harb J, Lin PJ, Hao J. Recent development of wnt signaling pathway inhibitors for cancer therapeutics. Curr Oncol Rep. 2019;21(2):12. [DOI] [PubMed] [Google Scholar]
- 335. Fu WB, Wang WE, Zeng CY. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharmacol Sin. 2019;40(1):9‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Kongkham PN, Northcott PA, Croul SE, Smith CA, Taylor MD, Rutka JT. The SFRP family of WNT inhibitors function as novel tumor suppressor genes epigenetically silenced in medulloblastoma. Oncogene. 2010;29(20):3017‐3024. [DOI] [PubMed] [Google Scholar]
- 337. King TD, Zhang W, Suto MJ, Li Y. Frizzled7 as an emerging target for cancer therapy. Cell Signal. 2012;24(4):846‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Sun Y, Wang W, Zhao C. Frizzled receptors in tumors, focusing on signaling, roles, modulation mechanisms, and targeted therapies. Oncol Res. 2021;28(6):661‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP‐54F28. Pharmacol Ther. 2015;146:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA. 2012;109(29):11717‐11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Pavlovic Z, Adams JJ, Blazer LL, et al. A synthetic anti‐Frizzled antibody engineered for broadened specificity exhibits enhanced anti‐tumor properties. MAbs. 2018;10(8):1157‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Flanagan DJ, Barker N, Costanzo NSD, et al. Frizzled‐7 is required for wnt signaling in gastric tumors with and without Apc mutations. Cancer Res. 2019;79(5):970‐981. [DOI] [PubMed] [Google Scholar]
- 343. Giraudet AL, Cassier PA, Iwao‐Fukukawa C, et al. A first‐in‐human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer. 2018;18(1):646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Zhao Y, Ren J, Hillier J, Lu W, Jones EY. Antiepileptic drug carbamazepine binds to a novel pocket on the Wnt receptor Frizzled‐8. J Med Chem. 2020;63(6):3252‐3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Nile AH, de Sousa EMF, Mukund S, et al. A selective peptide inhibitor of Frizzled 7 receptors disrupts intestinal stem cells. Nat Chem Biol. 2018;14(6):582‐590. [DOI] [PubMed] [Google Scholar]
- 346. Wall JA, Klempner SJ, Arend RC. The anti‐DKK1 antibody DKN‐01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin Investig Drugs. 2020;29(7):639‐644. [DOI] [PubMed] [Google Scholar]
- 347. Goyal L, Sirard C, Schrag M, et al. Phase I and biomarker study of the Wnt pathway modulator DKN‐01 in combination with gemcitabine/cisplatin in advanced biliary tract cancer. Clin Cancer Res. 2020;26(23):6158‐6167. [DOI] [PubMed] [Google Scholar]
- 348. Torres VI, Godoy JA, Inestrosa NC. Modulating Wnt signaling at the root: Porcupine and Wnt acylation. Pharmacol Ther. 2019;198:34‐45. [DOI] [PubMed] [Google Scholar]
- 349. Kabiri Z, Numata A, Kawasaki A, Edison, Tenen DG, Virshup DM. Wnts are dispensable for differentiation and self‐renewal of adult murine hematopoietic stem cells. Blood. 2015;126(9):1086‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta‐catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Agarwal P, Zhang B, Ho Y, et al. Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood. 2017;129(8):1008‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Rodon J, Argilés G, Connolly RM, et al. Phase 1 study of single‐agent WNT974, a first‐in‐class Porcupine inhibitor, in patients with advanced solid tumours. Br J Cancer. 2021;125(1):28‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Lee JH, Faderl S, Pagel JM, et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020;4(9):2032‐2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Lepourcelet M, Chen YN, France DS, et al. Small‐molecule antagonists of the oncogenic Tcf/beta‐catenin protein complex. Cancer Cell. 2004;5(1):91‐102. [DOI] [PubMed] [Google Scholar]
- 355. Lei Y, Yang Q, Nie Y, Wan J, Deng M. Small‐molecule inhibitor LF3 restrains the development of pulmonary hypertension through the Wnt/β‐catenin pathway. Acta Biochim Biophys Sin (Shanghai). 2021;53(10):1277‐1289. [DOI] [PubMed] [Google Scholar]
- 356. Ruan Z, Liang M, Lai M, Shang L, Deng X, Su X. KYA1797K down‐regulates PD‐L1 in colon cancer stem cells to block immune evasion by suppressing the β‐catenin/STT3 signaling pathway. Int Immunopharmacol. 2020;78:106003. [DOI] [PubMed] [Google Scholar]
- 357. Fang L, Zhu Q, Neuenschwander M, et al. A small‐molecule antagonist of the β‐catenin/TCF4 interaction blocks the self‐renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016;76(4):891‐901. [DOI] [PubMed] [Google Scholar]
- 358. Cha PH, Cho YH, Lee SK, et al. Small‐molecule binding of the axin RGS domain promotes β‐catenin and Ras degradation. Nat Chem Biol. 2016;12(8):593‐600. [DOI] [PubMed] [Google Scholar]
- 359. Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC‐0449. N Engl J Med. 2009;361(12):1173‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Klempner SJ, Bendell JC, Villaflor VM, et al. Safety, efficacy, and biomarker results from a phase Ib study of the anti‐DKK1 antibody DKN‐01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther. 2021;20(11):2240‐2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Jimeno A, Gordon M, Chugh R, et al. A first‐in‐human phase I study of the anticancer stem cell agent ipafricept (OMP‐54F28), a decoy receptor for wnt ligands, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(24):7490‐7497. [DOI] [PubMed] [Google Scholar]
- 362. Diamond JR, Becerra C, Richards D, et al. Phase Ib clinical trial of the anti‐frizzled antibody vantictumab (OMP‐18R5) plus paclitaxel in patients with locally advanced or metastatic HER2‐negative breast cancer. Breast Cancer Res Treat. 2020;184(1):53‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Davis SL, Cardin DB, Shahda S, et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab‐paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest New Drugs. 2020;38(3):821‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Mita MM, Becerra C, Richards DA, et al. Phase 1b study of WNT inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel (P) in patients (pts) with 1st‐to 3rd‐line metastatic HER2‐negative breast cancer (BC). J Clin Oncol. 2016;34:2516. 15_suppl.27269942 [Google Scholar]
- 365. Choi MY, Widhopf GF II, Ghia EM, et al. Phase I trial: Cirmtuzumab inhibits ROR1 signaling and stemness signatures in patients with chronic lymphocytic leukemia. Cell Stem Cell. 2018;22(6):951‐959. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Cortes J, Faderl S, Pagel J, et al. Phase 1 study of CWP232291 in relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). J Clin Oncol. 2015;33(15):7044‐7044. [Google Scholar]
- 367. Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66(3):1517‐1525. [DOI] [PubMed] [Google Scholar]
- 368. Carvalho FL, Simons BW, Eberhart CG, Berman DM. Notch signaling in prostate cancer: A moving target. Prostate. 2014;74(9):933‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Büchler P, Gazdhar A, Schubert M, et al. The Notch signaling pathway is related to neurovascular progression of pancreatic cancer. Ann Surg. 2005;242(6):791‐800. discussion 800–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem‐like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. 2017;36(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27(38):5132‐5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Wang K, Zhang Q, Li D, et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple‐negative breast cancer sensitive to a γ‐secretase inhibitor. Clin Cancer Res. 2015;21(6):1487‐1496. [DOI] [PubMed] [Google Scholar]
- 373. Pandya K, Meeke K, Clementz AG, et al. Targeting both Notch and ErbB‐2 signalling pathways is required for prevention of ErbB‐2‐positive breast tumour recurrence. Br J Cancer. 2011;105(6):796‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Sosa Iglesias V, Theys J, Groot AJ, et al. Synergistic effects of NOTCH/γ‐secretase inhibition and standard of care treatment modalities in non‐small cell lung cancer cells. Front Oncol. 2018;8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Morgan KM, Fischer BS, Lee FY, et al. Gamma secretase inhibition by BMS‐906024 enhances efficacy of paclitaxel in lung adenocarcinoma. Mol Cancer Ther. 2017;16(12):2759‐2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Chiorean EG, LoRusso P, Strother RM, et al. A phase I first‐in‐human study of enoticumab (REGN421), a fully human delta‐like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res. 2015;21(12):2695‐2703. [DOI] [PubMed] [Google Scholar]
- 377. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3‐targeted antibody‐drug conjugate, in recurrent small‐cell lung cancer: A first‐in‐human, first‐in‐class, open‐label, phase 1 study. Lancet Oncol. 2017;18(1):42‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Ferrarotto R, Eckhardt G, Patnaik A, et al. A phase I dose‐escalation and dose‐expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. 2018;29(7):1561‐1568. [DOI] [PubMed] [Google Scholar]
- 379. Schultheis B, Reuter D, Ebert MP, et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first‐line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann Oncol. 2017;28(10):2429‐2435. [DOI] [PubMed] [Google Scholar]
- 380. Astudillo L, Da Silva TG, Wang Z, et al. The small molecule IMR‐1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016;76(12):3593‐3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Lehal R, Zaric J, Vigolo M, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci USA. 2020;117(28):16292‐16301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Deangelo D. A phase I clinical trial of the notch inhibitor MK‐0752 in patients with T‐cell acute lymphoblastic leukemia/lymphoma T‐ALL and other leukemias. J Clin Oncol. 2006;24(18):658S. [Google Scholar]
- 383. Cook N, Basu B, Smith DM, et al. A phase I trial of the γ‐secretase inhibitor MK‐0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer. 2018;118(6):793‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Schott A, Chang J, Krop I, et al. Abstract P6‐15‐03: phase Ib trial of the gamma secretase inhibitor (GSI), MK‐0752 followed by docetaxel in locally advanced or metastatic breast cancer. Cancer Res. 2010;70(24_Supplement):P6‐15‐03. [Google Scholar]
- 385. Piha‐Paul SA, Munster PN, Hollebecque A, et al. Results of a phase 1 trial combining ridaforolimus and MK‐0752 in patients with advanced solid tumours. Eur J Cancer. 2015;51(14):1865‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Messersmith WA, Shapiro GI, Cleary JM, et al. A Phase I, dose‐finding study in patients with advanced solid malignancies of the oral γ‐secretase inhibitor PF‐03084014. Clin Cancer Res. 2015;21(1):60‐67. [DOI] [PubMed] [Google Scholar]
- 387. Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose‐finding study in patients with advanced solid malignancies of the oral γ‐secretase inhibitor PF‐03084014γ‐secretase inhibitor PF‐03084014 evaluation in solid tumors. Clin Cancer Res. 2015;21(1):60‐67. [DOI] [PubMed] [Google Scholar]
- 388. Papayannidis C, DeAngelo DJ, Stock W, et al. A Phase 1 study of the novel gamma‐secretase inhibitor PF‐03084014 in patients with T‐cell acute lymphoblastic leukemia and T‐cell lymphoblastic lymphoma. Blood Cancer J. 2015;5(9):e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Kummar S, O'Sullivan Coyne G, Do KT, et al. Clinical activity of the γ‐secretase inhibitor PF‐03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol. 2017;35(14):1561‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. De Jesus‐Acosta A, Laheru D, Maitra A, et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs. 2014;32(4):739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Richter S, Bedard PL, Chen EX, et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL‐078/CTEP 8575). Invest New Drugs. 2014;32(2):243‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Diaz‐Padilla I, Hirte H, Oza AM, et al. A phase Ib combination study of RO4929097, a gamma‐secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2013;31(5):1182‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Zweidler‐McKay PA, DeAngelo DJ, Douer D, et al. The safety and activity of BMS‐906024, a Gamma Secretase Inhibitor (GSI) with anti‐notch activity, in patients with relapsed T‐cell acute lymphoblastic leukemia (T‐ALL): initial results of a phase 1 trial. Blood. 2014;124(21):968. [Google Scholar]
- 394. Casulo C, Ruan J, Dang NH, et al. Safety and preliminary efficacy results of a phase I first‐in‐human study of the novel Notch‐1 targeting antibody brontictuzumab (OMP‐52M51) administered intravenously to patients with hematologic malignancies. Blood. 2016;128(22):5108. [Google Scholar]
- 395. Hu ZI, Bendell JC, Bullock A, et al. A randomized phase II trial of nab‐paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019;8(11):5148‐5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Gracian AC, Dean A, Muñoz A, et al. YOSEMITE: A 3 arm double‐blind randomized phase 2 study of gemcitabine, paclitaxel protein‐bound particles for injectable suspension, and placebo (GAP) versus gemcitabine, paclitaxel protein‐bound particles for injectable suspension and either 1 or 2 truncated courses of demcizumab (GAD). Ann Oncol. 2017;28:v211. [Google Scholar]
- 397. Hughes B, Dean A, Markman B, et al. Abstract A084: dENALI: A 3‐arm double‐blind randomized phase 2 study of carboplatin, pemetrexed, and placebo (CPP) versus carboplatin, pemetrexed, and either 1 or 2 truncated courses of demcizumab (CPD) in patients with non‐squamous non‐small cell lung cancer (NSCLC). Mol Cancer Ther. 2018;17(1_Supplement):A084‐A084. [Google Scholar]
- 398. Hidalgo M, Cooray P, Carrato A, et al. A phase 1b study of the anti‐cancer stem cell agent demcizumab (DEM) and gemcitabine (GEM) +/‐ nab‐paclitaxel in patients with pancreatic cancer. J Clin Oncol. 2016;34:341‐341. 4_suppl. [Google Scholar]
- 399. Wang D, Nagle PW, Wang HH, et al. Hedgehog pathway as a potential intervention target in esophageal cancer. Cancers (Basel). 2019;11(6):821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Kubo M, Nakamura M, Tasaki A, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64(17):6071‐6074. [DOI] [PubMed] [Google Scholar]
- 401. Bhateja P, Cherian M, Majumder S, Ramaswamy B. The Hedgehog signaling pathway: A viable target in breast cancer? Cancers (Basel). 2019;11(8):1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog‐signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29(4):469‐481. [DOI] [PubMed] [Google Scholar]
- 403. Basset‐Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open‐label trial. Eur J Cancer. 2017;86:334‐348. [DOI] [PubMed] [Google Scholar]
- 404. Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal‐cell carcinoma. N Engl J Med. 2012;366(23):2171‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Lear JT, Migden MR, Lewis KD, et al. Long‐term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30‐month analysis of the randomized phase 2 BOLT study. J Eur Acad Dermatol Venereol. 2018;32(3):372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Xie P, Lefrançois P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta‐analysis. J Am Acad Dermatol. 2018;79(6):1089‐1100. e17. [DOI] [PubMed] [Google Scholar]
- 407. Bendell J, Andre V, Ho A, et al. Phase I study of LY2940680, a Smo antagonist, in patients with advanced cancer including treatment‐naïve and previously treated basal cell carcinoma. Clin Cancer Res. 2018;24(9):2082‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high‐risk myelodysplastic syndrome. Leukemia. 2019;33(2):379‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Vanderburgh JP, Kwakwa KA, Werfel TA, et al. Systemic delivery of a Gli inhibitor via polymeric nanocarriers inhibits tumor‐induced bone disease. J Control Release. 2019;311‐312:257‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Li J, Cai J, Zhao S, et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J Exp Clin Cancer Res. 2016;35(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Huang L, Walter V, Hayes DN, Onaitis M. Hedgehog‐GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014;20(6):1566‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Didiasova M, Singh R, Wilhelm J, et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. Faseb J. 2017;31(5):1916‐1928. [DOI] [PubMed] [Google Scholar]
- 413. Zou WJ, Huang Z, Jiang TP, et al. Pirfenidone inhibits proliferation and promotes apoptosis of hepatocellular carcinoma cells by inhibiting the Wnt/β‐catenin signaling pathway. Med Sci Monit. 2017;23:6107‐6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Chen Q, Xu R, Zeng C, et al. Down‐regulation of Gli transcription factor leads to the inhibition of migration and invasion of ovarian cancer cells via integrin β4‐mediated FAK signaling. PLoS One. 2014;9(2):e88386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI‐mediated transcription and tumor cell growth by small‐molecule antagonists. Proc Natl Acad Sci USA. 2007;104(20):8455‐8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Kaye SB, Fehrenbacher L, Holloway R, et al. A phase II, randomized, placebo‐controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012;18(23):6509‐6518. [DOI] [PubMed] [Google Scholar]
- 417. Berlin J, Bendell JC, Hart LL, et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19(1):258‐267. [DOI] [PubMed] [Google Scholar]
- 418. Kim EJ, Sahai V, Abel EV, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC‐0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. 2014;20(23):5937‐5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33(36):4284‐4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Sloan AE, Nock CJ, Ye X, et al. Targeting glioma‐initiating cells in GBM: aBTC‐0904, a randomized phase 0/II study targeting the Sonic Hedgehog‐signaling pathway. J Clin Oncol. 2014;32(15_suppl):2026‐2026. [Google Scholar]
- 421. Cohen DJ, Christos PJ, Kindler HL, et al. Vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX for first‐line therapy of patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: A New York Cancer Consortium led phase II randomized study. J Clin Oncol. 2013;31:4011‐4011. 15_suppl. [Google Scholar]
- 422. Rudin CM, Jimeno A, Miller WH, et al. A phase I study of IPI‐926, a novel hedgehog pathway inhibitor, in patients (pts) with advanced or metastatic solid tumors. J Clin Oncol. 2011;29:3014‐3014. 15_suppl. [Google Scholar]
- 423. Bowles DW, Keysar SB, Eagles JR, et al. A pilot study of cetuximab and the hedgehog inhibitor IPI‐926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016;53:74‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Lee M, Hong H, Kim W, et al. Itraconazole as a noncastrating treatment for biochemically recurrent prostate cancer: A phase 2 study. Clin Genitourin Cancer. 2019;17(1):e92‐e96. [DOI] [PubMed] [Google Scholar]
- 425. Rudin CM, Brahmer JR, Juergens RA, et al. Phase 2 study of pemetrexed and itraconazole as second‐line therapy for metastatic nonsquamous non‐small‐cell lung cancer. J Thorac Oncol. 2013;8(5):619‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Ally MS, Ransohoff K, Sarin K, et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016;152(4):452‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Qi S, Zhu Y, Liu X, et al. WWC proteins mediate LATS1/2 activation by Hippo kinases and imply a tumor suppression strategy. Mol Cell. 2022;82(10):1850‐1864. e7. [DOI] [PubMed] [Google Scholar]
- 428. Park J, Kim JS, Nahm JH, Kim SK, Lee DH, Lim DS. WWC1 and NF2 prevent the development of intrahepatic cholangiocarcinoma by regulating YAP/TAZ activity through LATS in mice. Mol Cells. 2020;43(5):491‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Blume‐Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355‐365. [DOI] [PubMed] [Google Scholar]
- 430. Guo L, Teng L. YAP/TAZ for cancer therapy: Opportunities and challenges (review). Int J Oncol. 2015;46(4):1444‐1452. [DOI] [PubMed] [Google Scholar]
- 431. Sekido Y. Targeting the Hippo pathway is a new potential therapeutic modality for malignant mesothelioma. Cancers (Basel). 2018;10(4):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Lan C, Ni B, Zhao T, et al. An integrative pan‐cancer analysis revealing MLN4924 (Pevonedistat) as a potential therapeutic agent targeting Skp2 in YAP‐driven cancers. Front Genet. 2022;13:866702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Jin CH, Krishnaiah M, Sreenu D, et al. Discovery of N‐((4‐([1,2,4]triazolo[1,5‐a]pyridin‐6‐yl)‐5‐(6‐methylpyridin‐2‐yl)‐1H‐imidazol‐2‐yl)methyl)‐2‐fluoroaniline (EW‐7197): a highly potent, selective, and orally bioavailable inhibitor of TGF‐β type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem. 2014;57(10):4213‐4238. [DOI] [PubMed] [Google Scholar]
- 434. Kim BG, Malek E, Choi SH, Ignatz‐Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF‐β pathway. J Hematol Oncol. 2021;14(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor‐beta signaling pathway. Drug Des Devel Ther. 2015;9:4479‐4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Rodón J, Carducci M, Sepulveda‐Sánchez JM, et al. Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor‐β receptor I kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Invest New Drugs. 2015;33(2):357‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Yap TA, Vieito M, Baldini C, et al. First‐in‐human phase I study of a next‐generation, oral, TGFβ receptor 1 inhibitor, LY3200882, in patients with advanced cancer. Clin Cancer Res. 2021;27(24):6666‐6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Gordon MS, Ilaria R Jr, de Alwis DP, et al. A phase I study of tasisulam sodium (LY573636 sodium), a novel anticancer compound, administered as a 24‐h continuous infusion in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(1):21‐27. [DOI] [PubMed] [Google Scholar]
- 439. Tojo M, Hamashima Y, Hanyu A, et al. The ALK‐5 inhibitor A‐83‐01 inhibits Smad signaling and epithelial‐to‐mesenchymal transition by transforming growth factor‐beta. Cancer Sci. 2005;96(11):791‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Martin CJ, Datta A, Littlefield C, et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. 2020;12(536):eaay8456. [DOI] [PubMed] [Google Scholar]
- 441. Morris JC, Tan AR, Olencki TE, et al. Phase I study of GC1008 (fresolimumab): a human anti‐transforming growth factor‐beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Komrokji RS. Activin receptor II ligand traps: New treatment paradigm for low‐risk MDS. Curr Hematol Malig Rep. 2019;14(4):346‐351. [DOI] [PubMed] [Google Scholar]
- 443. Gleave ME, Monia BP. Antisense therapy for cancer. Nat Rev Cancer. 2005;5(6):468‐479. [DOI] [PubMed] [Google Scholar]
- 444. Oh J, Barve M, Matthews CM, et al. Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol. 2016;143(3):504‐510. [DOI] [PubMed] [Google Scholar]
- 445. Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318‐332. [DOI] [PubMed] [Google Scholar]
- 446. Katoh M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole‐body homeostasis (Review). Int J Mol Med. 2016;38(1):3‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Porta R, Borea R, Coelho A, et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256‐267. [DOI] [PubMed] [Google Scholar]
- 448. Liu FT, Li NG, Zhang YM, et al. Recent advance in the development of novel, selective and potent FGFR inhibitors. Eur J Med Chem. 2020;186:111884. [DOI] [PubMed] [Google Scholar]
- 449. Presta M, Chiodelli P, Giacomini A, Rusnati M, Ronca R. Fibroblast growth factors (FGFs) in cancer: fGF traps as a new therapeutic approach. Pharmacol Ther. 2017;179:171‐187. [DOI] [PubMed] [Google Scholar]
- 450. Tolcher AW, Papadopoulos KP, Patnaik A, et al. A phase I, first in human study of FP‐1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Ann Oncol. 2016;27(3):526‐532. [DOI] [PubMed] [Google Scholar]
- 451. Garcia S, Dirat B, Tognacci T, et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med. 2013;5(203):203ra124. [DOI] [PubMed] [Google Scholar]
- 452. de Aguiar RB, Parise CB, Souza CR, et al. Blocking FGF2 with a new specific monoclonal antibody impairs angiogenesis and experimental metastatic melanoma, suggesting a potential role in adjuvant settings. Cancer Lett. 2016;371(2):151‐160. [DOI] [PubMed] [Google Scholar]
- 453. Wang S, Qin Y, Wang Z, et al. Construction of a human monoclonal antibody against bFGF for suppression of NSCLC. J Cancer. 2018;9(11):2003‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105‐122. [DOI] [PubMed] [Google Scholar]
- 455. Bartz R, Fukuchi K, Ohtsuka T, et al. Preclinical development of U3‐1784, a novel FGFR4 antibody against cancer, and avoidance of its on‐target toxicity. Mol Cancer Ther. 2019;18(10):1832‐1843. [DOI] [PubMed] [Google Scholar]
- 456. Kollmannsberger C, Britten CD, Olszanski AJ, et al. A phase 1 study of LY3076226, a fibroblast growth factor receptor 3 (FGFR3) antibody‐drug conjugate, in patients with advanced or metastatic cancer. Invest New Drugs. 2021;39(6):1613‐1623. [DOI] [PubMed] [Google Scholar]
- 457. Li Y, Liu J, Gao L, et al. Targeting the tumor microenvironment to overcome immune checkpoint blockade therapy resistance. Immunol Lett. 2020;220:88‐96. [DOI] [PubMed] [Google Scholar]
- 458. McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev. 2010;15(3):264‐272. [PubMed] [Google Scholar]
- 459. Roma‐Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4):840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721‐732. [DOI] [PubMed] [Google Scholar]
- 461. Semenza GL. Pharmacologic targeting of hypoxia‐inducible factors. Annu Rev Pharmacol Toxicol. 2019;59:379‐403. [DOI] [PubMed] [Google Scholar]
- 462. Masunaga S, Matsumoto Y, Kashino G, et al. Significance of manipulating tumour hypoxia and radiation dose rate in terms of local tumour response and lung metastatic potential, referring to the response of quiescent cell populations. Br J Radiol. 2010;83(993):776‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Zhang Z, Qin S, Chen Y, et al. Inhibition of NPC1L1 disrupts adaptive responses of drug‐tolerant persister cells to chemotherapy. EMBO Mol Med. 2022;14(2):e14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Masunaga SI, Sakurai Y, Tano K, et al. Effect of bevacizumab combined with boron neutron capture therapy on local tumor response and lung metastasis. Exp Ther Med. 2014;8(1):291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Masunaga SI, Sanada Y, Moriwaki T, et al. Significance of fractionated administration of thalidomide combined with γ‐ray irradiation in terms of local tumor response and lung metastasis. World J Oncol. 2014;5(4):155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Masunaga S, Hirayama R, Uzawa A, et al. Influence of manipulating hypoxia in solid tumors on the radiation dose‐rate effect in vivo, with reference to that in the quiescent cell population. Jpn J Radiol. 2010;28(2):132‐142. [DOI] [PubMed] [Google Scholar]
- 467. Masunaga S, Liu Y, Tanaka H, et al. Reducing intratumour acute hypoxia through bevacizumab treatment, referring to the response of quiescent tumour cells and metastatic potential. Br J Radiol. 2011;84(1008):1131‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Masunaga S, Matsumoto Y, Hirayama R, et al. Significance of manipulating intratumor hypoxia in the effect on lung metastases in radiotherapy, with reference to its effect on the sensitivity of intratumor quiescent cells. Clin Exp Metastasis. 2009;26(7):693‐700. [DOI] [PubMed] [Google Scholar]
- 469. Lee EQ, Duda DG, Muzikansky A, et al. Phase I and biomarker study of plerixafor and bevacizumab in recurrent high‐grade glioma. Clin Cancer Res. 2018;24(19):4643‐4649. [DOI] [PubMed] [Google Scholar]
- 470. Hattingen E, Jurcoane A, Bähr O, et al. Bevacizumab impairs oxidative energy metabolism and shows antitumoral effects in recurrent glioblastomas: A 31P/1H MRSI and quantitative magnetic resonance imaging study. Neuro Oncol. 2011;13(12):1349‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Monk BJ, Sill MW, Burger RA, Gray HJ, Buekers TE, Roman LD. Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: A gynecologic oncology group study. J Clin Oncol. 2009;27(7):1069‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Bagri A, Berry L, Gunter B, et al. Effects of anti‐VEGF treatment duration on tumor growth, tumor regrowth, and treatment efficacy. Clin Cancer Res. 2010;16(15):3887‐3900. [DOI] [PubMed] [Google Scholar]
- 473. Brenner A, Zuniga R, Sun JD, et al. Hypoxia‐activated evofosfamide for treatment of recurrent bevacizumab‐refractory glioblastoma: A phase I surgical study. Neuro Oncol. 2018;20(9):1231‐1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Ueda S, Saeki T, Osaki A, Yamane T, Kuji I. Bevacizumab induces acute hypoxia and cancer progression in patients with refractory breast cancer: Multimodal functional imaging and multiplex cytokine analysis. Clin Cancer Res. 2017;23(19):5769‐5778. [DOI] [PubMed] [Google Scholar]
- 475. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature. 2008;454(7203):436‐444. [DOI] [PubMed] [Google Scholar]
- 476. Galdiero MR, Marone G, Mantovani A. Cancer inflammation and cytokines. Cold Spring Harb Perspect Biol. 2018;10(8):a028662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829‐845. e20. [DOI] [PubMed] [Google Scholar]
- 478. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 479. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211‐217. [DOI] [PubMed] [Google Scholar]
- 480. Manjili MH. Tumor dormancy and relapse: From a natural byproduct of evolution to a disease state. Cancer Res. 2017;77(10):2564‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Demkow U. Neutrophil extracellular traps (NETs) in cancer invasion, evasion and metastasis. Cancers (Basel). 2021;13(17):4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Pathria P, Louis TL, Varner JA. Targeting tumor‐associated macrophages in cancer. Trends Immunol. 2019;40(4):310‐327. [DOI] [PubMed] [Google Scholar]
- 483. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor‐associated macrophage (TAM)‐targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Cassetta L, Pollard JW. Targeting macrophages: Therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887‐904. [DOI] [PubMed] [Google Scholar]
- 485. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. 2019;8:e43653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Frankenberger C, Rabe D, Bainer R, et al. Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor‐associated macrophages. Cancer Res. 2015;75(19):4063‐4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Svensson S, Abrahamsson A, Rodriguez GV, et al. CCL2 and CCL5 are novel therapeutic targets for estrogen‐dependent breast cancer. Clin Cancer Res. 2015;21(16):3794‐3805. [DOI] [PubMed] [Google Scholar]
- 489. Halama N, Zoernig I, Berthel A, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti‐CCR5 therapy in cancer patients. Cancer Cell. 2016;29(4):587‐601. [DOI] [PubMed] [Google Scholar]
- 490. Zhang XN, Yang KD, Chen C, et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5‐CCR5 paracrine signaling. Cell Res. 2021;31(10):1072‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Nie Y, Huang H, Guo M, et al. Breast phyllodes tumors recruit and repolarize tumor‐associated macrophages via secreting CCL5 to promote malignant progression, which can be inhibited by CCR5 inhibition therapy. Clin Cancer Res. 2019;25(13):3873‐3886. [DOI] [PubMed] [Google Scholar]
- 492. Aldinucci D, Casagrande N. Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int J Mol Sci. 2018;19(5):1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Noy R, Pollard JW. Tumor‐associated macrophages: From mechanisms to therapy. Immunity. 2014;41(1):49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF‐1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Hume DA, MacDonald KP. Therapeutic applications of macrophage colony‐stimulating factor‐1 (CSF‐1) and antagonists of CSF‐1 receptor (CSF‐1R) signaling. Blood. 2012;119(8):1810‐1820. [DOI] [PubMed] [Google Scholar]
- 496. Goswami S, Sahai E, Wyckoff JB, et al. Macrophages promote the invasion of breast carcinoma cells via a colony‐stimulating factor‐1/epidermal growth factor paracrine loop. Cancer Res. 2005;65(12):5278‐5283. [DOI] [PubMed] [Google Scholar]
- 497. Manthey CL, Johnson DL, Illig CR, et al. JNJ‐28312141, a novel orally active colony‐stimulating factor‐1 receptor/FMS‐related receptor tyrosine kinase‐3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther. 2009;8(11):3151‐3161. [DOI] [PubMed] [Google Scholar]
- 498. Ries CH, Cannarile MA, Hoves S, et al. Targeting tumor‐associated macrophages with anti‐CSF‐1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846‐859. [DOI] [PubMed] [Google Scholar]
- 499. Akkari L, Bowman RL, Tessier J, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF‐1R inhibition as a strategy to overcome resistance. Sci Transl Med. 2020;12(552):eaaw7843. [DOI] [PubMed] [Google Scholar]
- 500. Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18(4):557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Mao X, Xu J, Wang W, et al. Crosstalk between cancer‐associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol Cancer. 2021;20(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74‐80. [DOI] [PubMed] [Google Scholar]
- 503. Tian H, Cao J, Li B, et al. Managing the immune microenvironment of osteosarcoma: The outlook for osteosarcoma treatment. Bone Res. 2023;11(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Amoozgar Z, Kloepper J, Ren J, et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun. 2021;12(1):2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Chakraborty B, Byemerwa J, Shepherd J, et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J Clin Invest. 2021;131(23):e151347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Chen C, Li A, Sun P, et al. Efficiently restoring the tumoricidal immunity against resistant malignancies via an immune nanomodulator. J Control Release. 2020;324:574‐585. [DOI] [PubMed] [Google Scholar]
- 507. Bugde P, Biswas R, Merien F, et al. The therapeutic potential of targeting ABC transporters to combat multi‐drug resistance. Expert Opin Ther Targets. 2017;21(5):511‐530. [DOI] [PubMed] [Google Scholar]
- 508. Lou H, Dean M. Targeted therapy for cancer stem cells: The patched pathway and ABC transporters. Oncogene. 2007;26(9):1357‐1360. [DOI] [PubMed] [Google Scholar]
- 509. Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41(5):1967‐1972. [PubMed] [Google Scholar]
- 510. Pennock GD, Dalton WS, Roeske WR, et al. Systemic toxic effects associated with high‐dose verapamil infusion and chemotherapy administration. J Natl Cancer Inst. 1991;83(2):105‐110. [DOI] [PubMed] [Google Scholar]
- 511. Saville MW, Lietzau J, Pluda JM, et al. Treatment of HIV‐associated Kaposi's sarcoma with paclitaxel. Lancet. 1995;346(8966):26‐28. [DOI] [PubMed] [Google Scholar]
- 512. Tidefelt U, Liliemark J, Gruber A, et al. P‐Glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P‐glycoprotein‐positive acute myeloid leukemia. J Clin Oncol. 2000;18(9):1837‐1844. [DOI] [PubMed] [Google Scholar]
- 513. Minderman H, O'Loughlin KL, Pendyala L, Baer MR. VX‐710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P‐glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res. 2004;10(5):1826‐1834. [DOI] [PubMed] [Google Scholar]
- 514. Gottesman MM, Ludwig J, Xia D, Szakács G. Defeating drug resistance in cancer. Discov Med. 2006;6(31):18‐23. [PubMed] [Google Scholar]
- 515. Kathawala RJ, Gupta P, Ashby CR Jr, Chen ZS. The modulation of ABC transporter‐mediated multidrug resistance in cancer: A review of the past decade. Drug Resist Updat. 2015;18:1‐17. [DOI] [PubMed] [Google Scholar]
- 516. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8(5):411‐424. [DOI] [PubMed] [Google Scholar]
- 517. Raza A, Kopp SR, Jabbar A, Kotze AC. Effects of third generation P‐glycoprotein inhibitors on the sensitivity of drug‐resistant and ‐susceptible isolates of Haemonchus contortus to anthelmintics in vitro. Vet Parasitol. 2015;211(1‐2):80‐88. [DOI] [PubMed] [Google Scholar]
- 518. Falasca M, Linton KJ. Investigational ABC transporter inhibitors. Expert Opin Investig Drugs. 2012;21(5):657‐666. [DOI] [PubMed] [Google Scholar]
- 519. Lubelski J, van Merkerk R, Konings WN, Driessen AJ. Nucleotide‐binding sites of the heterodimeric LmrCD ABC‐multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry. 2006;45(2):648‐656. [DOI] [PubMed] [Google Scholar]
- 520. Kannan P, Telu S, Shukla S, et al. The “specific” P‐glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci. 2011;2(2):82‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Shapira A, Davidson I, Avni N, Assaraf YG, Livney YD. β‐Casein nanoparticle‐based oral drug delivery system for potential treatment of gastric carcinoma: Stability, target‐activated release and cytotoxicity. Eur J Pharm Biopharm. 2012;80(2):298‐305. [DOI] [PubMed] [Google Scholar]
- 522. Tiram G, Segal E, Krivitsky A, et al. Identification of dormancy‐associated microRNAs for the design of osteosarcoma‐targeted dendritic polyglycerol nanopolyplexes. ACS Nano. 2016;10(2):2028‐2045. [DOI] [PubMed] [Google Scholar]
- 523. Tian H, Zhang T, Qin S, et al. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J Hematol Oncol. 2022;15(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Kapse‐Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Wen Y, Kolonich HR, Kruszewski KM, Giannoukakis N, Gawalt ES, Meng WS. Retaining antibodies in tumors with a self‐assembling injectable system. Mol Pharm. 2013;10(3):1035‐1044. [DOI] [PubMed] [Google Scholar]
- 526. Chen AM, Zhang M, Wei D, et al. Co‐delivery of doxorubicin and Bcl‐2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug‐resistant cancer cells. Small. 2009;5(23):2673‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Wang H, Yang Y, Liu J, Qian L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat Rev Mol Cell Biol. 2021;22(6):410‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z. Stem cells: Past, present, and future. Stem Cell Res Ther. 2019;10(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022;12(1):31‐46. [DOI] [PubMed] [Google Scholar]
- 530. Martínez‐Reyes I, Chandel NS. Cancer metabolism: Looking forward. Nat Rev Cancer. 2021;21(10):669‐680. [DOI] [PubMed] [Google Scholar]
- 531. Hara T, Chanoch‐Myers R, Mathewson ND, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal‐like states in glioblastoma. Cancer Cell. 2021;39(6):779‐792. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Shahbazi MN, Zernicka‐Goetz M. Deconstructing and reconstructing the mouse and human early embryo. Nat Cell Biol. 2018;20(8):878‐887. [DOI] [PubMed] [Google Scholar]
- 533. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell. 2006;127(3):469‐480. [DOI] [PubMed] [Google Scholar]
- 534. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. MacGrogan D, Münch J, de la Pompa JL. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat Rev Cardiol. 2018;15(11):685‐704. [DOI] [PubMed] [Google Scholar]
- 536. Ganal‐Vonarburg SC, Hornef MW, Macpherson AJ. Microbial‐host molecular exchange and its functional consequences in early mammalian life. Science. 2020;368(6491):604‐607. [DOI] [PubMed] [Google Scholar]
- 537. Thomas C, Tampé R. Structural and mechanistic principles of ABC transporters. Annu Rev Biochem. 2020;89:605‐636. [DOI] [PubMed] [Google Scholar]
- 538. Lopes FL, Desmarais JA, Murphy BD. Embryonic diapause and its regulation. Reproduction. 2004;128(6):669‐678. [DOI] [PubMed] [Google Scholar]
- 539. Renfree MB, Shaw G. Diapause. Annu Rev Physiol. 2000;62:353‐375. [DOI] [PubMed] [Google Scholar]
- 540. Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell. 2023;186(8):1564‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer. 2021;148(12):2884‐2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Magnon C, Hondermarck H. The neural addiction of cancer. Nat Rev Cancer. 2023;23(5):317‐334. [DOI] [PubMed] [Google Scholar]
- 543. Erenpreisa J, Salmina K, Anatskaya O, Cragg MS. Paradoxes of cancer: Survival at the brink. Semin Cancer Biol. 2022;81:119‐131. [DOI] [PubMed] [Google Scholar]
- 544. Sharma A, Blériot C, Currenti J, Ginhoux F. Oncofetal reprogramming in tumour development and progression. Nat Rev Cancer. 2022;22(10):593‐602. [DOI] [PubMed] [Google Scholar]
- 545. Dagogo‐Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81‐94. [DOI] [PubMed] [Google Scholar]
- 546. Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. 2019;12(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell. 2020;37(4):471‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Sterner RC, Sterner RM. CAR‐T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.