

Summary
The role of tumor mutational burden (TMB) in shaping tumor immunity is a key question that has not been addressable using genetically engineered mouse models (GEMM) of lung cancer. To induce TMB in lung GEMMs, we expressed an ultra-mutator variant of DNA polymerase-E (POLE) P286R in lung epithelial cells. Introduction of PoleP286R allele into KrasG12D and KrasG12D; p53L/L (KP) models significantly increase their TMB. Immunogenicity and sensitivity to immune checkpoint blockade (ICB) induced by Pole is partially dependent on p53. Corroborating these observations, survival of NSCLC patients whose tumors have TP53 nonsense mutations is shorter than those with TP53WT with immunotherapy. Immune resistance is in part through reduced antigen presentation and in part due to mutational heterogeneity. Total STING protein levels are elevated in Pole mutated KP tumors creating a vulnerability. A stable polyvalent STING agonist or p53 induction increases sensitivity to immunotherapy offering therapeutic options in these polyclonal tumors.
Graphical Abstract
Background:
Lung cancer is the leading cause of cancer-related deaths for both men and women 1. Programmed death 1/Programmed death ligand 1 (PD-1/PD-L1) or combination of anti PD-1/PD-L1 and anti- Cytotoxic T-Lymphocyte Associated Protein-4 (CTLA)4 antibodies-immune checkpoint blockade (ICB) treatments were approved for the treatment of non-small cell lung cancer (NSCLC) and are the standard of care 2–5. However, only a subset of patients derives durable benefit from these treatments. 5-year survival of NSCLC patients whose tumors have >50% PD-L1 expression is 21–31%, indicating that novel combination treatments are needed to improve durable ICB response 6,7. Currently, there are three FDA-approved predictive biomarkers for ICB: PD-L1 expression, microsatellite instability (MSI)/deficient mismatch repair(dMMR), and high tumor mutational burden (TMB) 8. MSI/dMMR is common in endometrial carcinoma, colon adenocarcinoma and gastric adenocarcinoma, but less common in lung cancer 9. Since MMR safeguards genome fidelity during DNA replication and in response to external perturbation, dMMR results in a high rate of base substitutions/missense mutations and microsatellite instability. dMMR triggers anti-tumor immunity through neoantigens and activation of Type 1 interferon (IFN) signaling through second messenger cyclic GMP–AMP (cGAS) and stimulator of interferon genes cGAS/STING pathway 10,11.
Polymerase ε encoded by the POLE gene is the housekeeping leading strand DNA polymerase. A small number of highly specific and recurring amino acid substitutions identified in human cancers (most commonly P286R) render it highly error-prone, with an error rate much higher than deletion of the proofreading domain of POLE. Concordantly, such tumor-driving POLE mutations are gain-of-function and behave in a genetically dominant manner 12. POLE mutations are frequently detected in the brain, colon, rectum, and uterine tumors. POLE mutations are also detected in 2.8–6% of NSCLCs and associated with favorable prognosis 13,14. POLE-driven cancers fall in the high TMB category and respond well to immunotherapy 15.
TMB refers to the number of somatic mutations in the tumor exome or per megabase (MB) of DNA. Average TMB in NSCLC patients is around 8 mutations/Mb, ranking one of the highest among all cancer types 16. NSCLCs with high TMB respond better to anti-PD-1 and anti-CTLA-4 combination therapy 17. However, TMB does not entirely correlate with ICB response. In a retrospective study correlating TMB with response to ICB found that TMB only correlated with ICB response in certain subsets of cancers18. In some cases, there is no correlation with tumor neo-antigen load and CD8T cell infiltration in a subset of the tumors18. Small cell lung cancer (SCLC) is one of the cancer types associated with high TMB. However, they are relatively resistant to ICB19. These observations highlight the need to better understand the significance of TMB in assessing response to ICB. Tumor onco-genotype also influences the response to ICB. Inactivating mutations in Serine/threonine kinase 11(STK11/LKB1), Kelch like ECH associated protein 1(KEAP1) and Phosphatase and tensin homolog (PTEN) are associated with poor overall survival in NSCLC patients receiving ICB therapy 20,21.
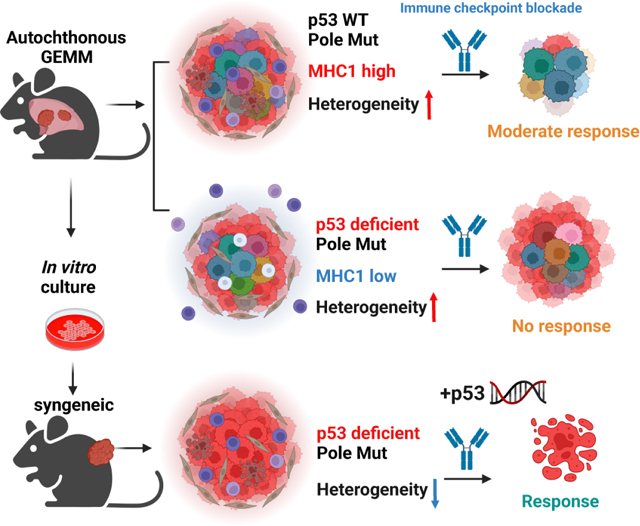
Murine NSCLC models are standard tools to study lung cancer biology. Autochthonous lung cancer mouse models are unique because they recapitulate many features of human tumor initiation and progression: spontaneity and growth in orthotopic environment allowing studies of tumor initiation and interactions with the microenvironment. However, previous studies showed that GEMM models carry far fewer TMB as compared to human lung tumors of either smoker or non-smoker patients 22. Kras p53 mutant GEMMs were shown to respond to PD-1 blockade only in the setting of combination treatments with epigenetic modifiers or chemotherapy 23,24. This prompted us to investigate whether increasing TMB can promote development of more immunogenic lung tumors and better model human lung cancer. We induced expression of PoleP86R in lung epithelial cells with a conditional PoleLSL-P86R mouse model. These mice in monoallelic condition developed lung tumors with a low frequency indicating that PoleP286R is not sufficient to cause initiation of lung cancer. In the setting of KrasG12D(K), KrasG12D; p53L/L (KP) most mutated oncogene and tumor suppressor respectively in lung adenocarcinoma, introduction of PoleP86R caused significantly different outcomes. Pole only caused increased immunogenicity in the setting of K tumors. Loss of p53 caused an immunosuppressive tumor microenvironment and ICB resistance in the PoleP86R mutant GEMM model. However, syngeneic models derived from these GEMMs were moderately sensitive to ICB. Therapeutic sensitivity of the syngeneic model positively correlated with the percentage of shared mutations within the tumor population and heterogeneity drove resistance. Genetic or pharmacological p53 restoration induced MHC1 expression and sensitivity to ICB.
RESULTS:
PoleP286R is not a driver of lung tumor initiation by itself but addition of PoleP286R to KrasG12D/+ Increases TMB
To dissect the role of high TMB in the development and progression of NSCLC, we first induced PoleP286R expression in lung of PoleLSL-P286R/+ GEMM by intranasal Adeno-Cre instillation (Figure 1A). We observed adenoma or carcinoma in only 4 of the 29 Adeno-Cre induced and aged PoleP286R/+ animals and 1 out of 10 of the uninduced PoleP286R/+ mice, a difference that is not statistically significant (p=0.7) Figure 1A). Overall survival between the two groups was also similar (Figure 1B). Only 1 of the induced mice died from a non-cancer related cause from the induced cohort. This is quite different than the significant tumor incidence including lung cancers and reduced in survival of mice expressing of PoleP286R in the whole body 26. These results indicate that PoleP286R alone is not a strong driver of lung cancer initiation in the context of a functional immune system.
Figure 1. PoleP286R is not a driver of lung tumor initiation by itself but addition of PoleP286R to KrasG12D/+ increases their TMB.

(A) Schema for GEMM tumor induction by intranasal Adeno-Cre. Pie chart showing incidence of tumors in the lung of uninduced PoleLSL-P286R/+ control and induced PoleP286R/+ mice. Representative H&E staining of tumor free lung and a tumor in the lung of uninduced PoleLSL-P286R/+ control and induced PoleP286R/+ mice. Control n=10, induced n=29.
(B) Survival curves for PoleLSL-P286R/+ control and induced PoleP286R/+ animals analyzed by Log-rank test. All animals except for the one that died were euthanatized at ~70 weeks for observation of tumors in the lungs. Control n=10, induced n=24.
(C) Tumor progression of K and KO GEMMs was measured by lung weight (left) and MRI (middle) at 12 (n=22 for K, n=19 for KO) and 20 weeks (n=7 for K, n=14 for KO) respectively (Unpaired t-test, Error bar: SD). 12-week tumors were not always detectable my MRI. Survival curves for K/KO mice are on the right (n=20 for K, n=23 for KO, Log-rank test). Mice were recorded as dead when they either died or reached euthanasia point.
(D) Representative H&E-stained lung sections of K and KO GEMMs (12-week: n=21 for K, n=18 for KO. Unpaired t-test was utilized. Error bar: standard deviation (SD)). Tumor area is quantified on the right.
(E) Ki67 immunohistochemistry (IHC) on K and KO GEMM lung tissues (12-week: n=21 for K, n=18 for KO. 20-week: n=7 for K, n=6 for KO, Unpaired t-test. Error bar: SD). Representative images (left) and quantification of staining (right) shown.
(F) Combination of PD-L1 and CTLA-4 antibodies immune checkpoint blockade (ICB) treatment of K and KO GEMMs. Representative MRI images (left) and quantification of lung tumors at 4 weeks after treatment initiation (right). Yellow arrows point to tumors. H: heart. N=8 for K-isotype, n=12 for K-ICB, n=7 for KO-isotype, n=14 for KO-ICB. Unpaired t-test. Error bar: SD.
(G) Overall survival (OS) and progression-free survival (PFS) of K/KO mice under isotype or ICB treatment analyzed by Log-rank test. Progression is defined by the timepoint when tumor volume increases more than 20% of baseline tumor volume. N=7 for K-isotype, n=9 for K-ICB, n=12 for KO-isotype, n=9 for KO-ICB. Log-rank test. For all figures, *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001.
Also see Figure S1.
Next, we crossed PoleLSL-P286R/+ (O) to KrasLSL-G12D/+ (K) GEMM to induce tumorigenesis in the setting of a driver oncogene. We induced tumors with the same dose of Adeno-Cre (10x=2.5×10^8) as KrasWt mice to reduce latency as 1X resulted in long latency with some of the mice living beyond a year in B6 background (Figure 1SA). KrasG12D/+; PoleP286R/+(KO) showed delayed tumor growth as compared to KrasG12D/+(K) GEMM shown by lung weights 12 weeks after tumor induction and by MRI at 20 weeks after tumor induction. KO mice also lived significantly longer as compared to K mice (mean survival 146 days for K mice group, mean survival 196 days for KO mice group, p=0.029, Figure 1C). Tumor volumes were quantified from MRI images as in Fig S1B. H&E-stained tissue sections of K and KO mice indicate significantly reduced tumor area in the lung tissues of KO mice quantified from the H&E sections (Figure 1D, ,Figure S1C) consistent with the survival and MRI data. This is surprising and potentially resulted from tumor extrinsic factors because Ki67 IHC on tumor tissue identified higher proliferation rate of KrasG12D/+;PoleP286R/+ tumor at early stage (12 weeks after induction) but similar proliferation at later stage (20 weeks, Figure 1E). To determine whether T cells are critical in restricting the growth of KO tumors, we depleted CD8T cells in K/KO mice after tumor confirmation by MRI. Depletion of CD8 T cells from KO mice but not K mice resulted in significantly increased tumor growth and decreased survival (Figure 1SD & 1SE). Whole exome sequencing (WES) was completed to assess the TMB of tumors. As predicted, KO tumors had a five-fold increase in TMB as compared to K tumors (1.14 vs 0.22mut/MB, Figure 1SF).
To test whether increase in the TMB causes increased sensitivity to ICB, we treated tumor-bearing K and KO GEMMs with combination of PD-L1 and CTLA-4 antibodies (ICB) after tumor confirmation by MRI. While some of the mice from both groups showed some response to ICB, KO GEMMs overall responded significantly better to ICB than K. This resulted in a significant difference in the tumor volume change at 4-weeks (Figure 1F) and significantly increased survival of ICB treated KO mice as compared to isotype treated KO mice (Figure 1G, p=0.02). While survival of K mice was also increased with ICB treatment, difference in survival did not reach significance (p=0.57). ICB treatment also increased the progression free survival (PFS) of KO mice (Figure 1H).
Addition of PoleP286R accelerated the progression of KrasG12D/+;p53−/− tumors.
To study the impact of high TMB in the context of concurrent Kras and Trp53 mutation we incorporated PoleLSL-P286R (O) into the KrasLSL-G12D/+;p53fl/fl (KP, KPO) GEMM, which develops lung adenocarcinomas with a 100% penetrance. The role of p53 in tumor progression is well established while its role in immune evasion is less clear. We induced expression of Kras G12D and deletion of Trp53 with 1x dose of intranasal Adeno-Cre (Figure 2A) as it was previously shown that Trp53 inactivation significantly reduces survival of Kras mice38. As expected, all mice developed lung tumors. Tumors in both KP and KPO cohorts were high-grade lung adenocarcinomas (Figure S1G). Histology of tumor-bearing GEMMs indicated that KPO mice presented significantly higher tumor area calculated from the H&E-stained histology sections p=0.02, Figure 2B). There was a small but significant difference between the survival of KP and KPO mice Mean survival was 122 days for KPO mice and 135 days for KP mice KP (p=0.049, Figure 2C). Per Ki67 immunohistochemistry (IHC), the mitotic index was higher in KPO than KP (Figure 2D). PoleP286R/+ tumor cells exhibited significantly higher number of DNA double strand breaks as indicated by γH2A.X IHC (Figure 2E).
Figure 2. Addition of PoleP286R accelerated the progression of KrasG12D/+;p53−/− tumors.

(A) Schema for GEMM tumor induction by intranasal Adeno-Cre.
(B) Representative H&E stains of lungs from KP and KPO GEMMs with tumor area quantification as % of normal area quantified on the right. N=6 for KP, n=10 for KPO, Unpaired t-test, Error bar: SD.
(C) Survival of KP and KPO GEMMs after tumor induction. N=9 for KP, n=11 for KPO, Log-rank test.
(D) Ki67 IHC on KP and KPO GEMM lung tissues. Representative image is shown on the left and stained area is quantified on the right. N=6 for KP, n=10 for KPO. Statistical test: Unpaired t-test, Error bar: SD.
(E) γH2AX IHC on KP and KPO GEMM lung tissues. Representative images are shown on the left and quantification of stained area is shown on the right. N=6 for KP, n=10 for KPO. Statistical test: Unpaired t-test, Error bar: SD.
(F) Total mutations/MB determined by whole exome sequencing of KP vs KPO tumors. N=3 for KP, n=5 for KPO, Mann Whitney test, Error bar: standard error of the mean (SEM).
(G) Distribution of the type of mutations in KP vs KPO tumors.
(H) TMB (Tumor Mutational Burden) in NSCLC (Non-Small Cell Lung Carcinoma) patients classified based on TP53 status from MSKCC35 and DFCI cohort. Sample n: sample number.
(I) Lung tumor volume quantification of KP and KPO mice treated with ICB after tumor confirmation by MRI. Changes in tumor volumes at indicated timepoints after treatment initiation is shown. N=10 for KP-isotype, n=14 for KP-ICB, n=14 for KPO-isotype, n=15 for KPO-ICB, Error bar: SD.
(J) Representative MRI images from the mice in H, 2 weeks after treatment initiation. Arrows point to tumors. H: heart.
(K) Survival of treated mice in H.
Also see Figure S1.
KPO tumors exhibited about a 10-fold increase in TMBs, as compared to KP tumors (0.59mut/MB to 5.9mut/MB, p=0.03) (Figure 2 F and G) by WES. One potential reason for detection of higher TMB counts in KPO tumors could be technical. It was previously shown that tumor multiplicity depends on Adeno-Cre titer 39. We observed significantly increased (p<0.0001) number of lesions in K/KO than KP/KPO lung tissues from which sequencing samples were prepared (Figure S1H). High number of lesions in p53wt tumors may potentially reduce the power of mutation detection 40. Another and more likely reason for high TMB in Trp53 mutant tumors could be due to the roles p53 plays in DNA repair. In response to DNA damage, proteins such as ataxia-telangiectasia mutated (ATM) are activated, they phosphorylate MDM2, the negative regulator of p53, to promote accumulation and activation of p53. p53 upregulates several genes involved in various DNA repair mechanisms including Ku86, Mlh1, Msh2, Polk and etc. Thus, Trp53 deficiency can influence the ability to maintain genomic stability and DNA repair efficiency 41,42. Consistent with the mouse tumors, patient tumors with TP53 mutations have significantly higher TMB as compared to patients whose tumors are wild-type for TP53 in DFCI and MSKCC35 datasets. In MSKCC dataset mean TMB is 5.69 vs 12.40 vs 13.37 in TP53 wild-type, missense, and truncating mutant tumors respectively (Figure 2H). In DFCI dataset mean TMB was 8.85 vs 11.97 vs 12.91 in TP53 wild-type, missense, and truncating mutant tumors respectively (Figure 2H).
To determine whether higher TMB would increase sensitivity to ICB of KP models, we treated tumor-bearing KP and KPO GEMMs with ICB antibodies after lung tumors were confirmed by MRI. KP or KPO GEMMs were similarly resistant to ICB (Figure 2I and J). Changes in tumor volumes were similar in the two cohorts and unlike K/KO models, survival curves for control and ICB treated mice overlapped. Mean survival was 36 days for KP-isotype group and 44 for KP-ICB group (p=0.14 between these 2 groups, Figure 2K). Mean survival was 38.5 days for KPO-isotype group and 40 days for KPO-ICB group (p=0.23 between these 2 groups, Figure 2K).
To determine the clinical translation of our findings, we utilized a large publicly available dataset composed of a large number of NSCLC (both Non-Squamous and Squamous Carcinoma) clinical samples from patients treated with ICB treatments35. TP53 truncating mutations (nonsense and frameshift mutations) which would be functionally equivalent to p53 genetic deletion in our KP/KPO GEMMs correlated with significantly lower survival in NSCLC patients (Figure 3A, p=0.00046). Median survival of 61 patients whose tumors had nonsense TP53 mutations was 8 months as compared to the 19 months of survival of 133 patients whose tumors carried wild-type TP53. Of note, due to its known role in resistance to ICB, we excluded STK11/KEAP1 mutant samples from the analysis for all the clinical samples. We additionally analyzed 3 other datasets: Pembrolizumab treated patients in AACR-Genie dataset, Stand Up to Cancer (SU2C) Dataset36 and our (Dana-Farber Cancer Institute) dataset. In all of the three cohorts, p53 truncating mutations were associated with shorter survival. Median survival was 21 months for patients whose tumors had wild-type TP53 vs 9 months for truncating TP53 mutations in (Figure 3B, p=0.01) in AARC-Genie set. SU2C dataset showed a similar pattern of median survival of 26 months for wild-type TP53 and 14 months for truncating TP53, p=0.07 by Log-rank test and p=0.0048 with Wilcoxon test, Fig 3C). Truncating TP53 mutations were associated with lower survival in the DFCI dataset too with a median survival of 15 months with wild-type TP53 and 10 months for truncating TP53, although this difference was not significant (p=0.22 and 0.15 by Log-rank vs Wilcoxon tests, Fig 3D). These data confirmed our observations with mice regarding p53 loss maybe associated with poor prognosis with ICB in lung cancer patients. In patient tumors missense mutations are observed more commonly and the types of TP53 mutations differentially influenced survival in NSCLC patients treated with immunotherapy. Although there was a trend towards decreased survival with TP53 missense mutations in MSKCC and Genie datasets, this difference was not significant in any of the four datasets analyzed (Figure 3A–D).
Figure 3. TP53 truncating mutations decrease overall survival of NSCLC patients receiving ICB.

Overall survival of NSCLC patients carrying TP53 wild-type, TP53 truncating mutations (nonsense and frameshift mutations combined) and TP53 missense mutations after receiving ICB. STK11/KEAP1 WT cases were included in the analysis. Source of the data are as follows.
(A) Samstein et al35, Nature Genetics, 2019. Log-rank test.
(B) AACR Genie, patients treated with pembrolizumab. Log-rank test.
(C) Ravi et al 36, Nature Genetics, 2023.
(D) NSCLC patients from DFCI cohort.
P53 inactivation modulates tumor immune microenvironment of KrasG12D/+ and KrasG12D/+; PoleP28R/+ GEMM lung tumors
Despite the high number of detectable mutations, KPO tumors were resistant to ICB treatment. Differences in the tumor microenvironment or potential defects in presentation of these mutations as antigens could account for the differences in response to ICB between p53+/+ and p53−/− PoleP286R lung tumors. We profiled the tumor microenvironment of tumors collected at the same time point after tumor induction (16 weeks) by flow cytometry. At this time point, KP/KPO lung weights were larger than K/KO. However, this difference was not significant (Figure 4A). p53-deficient tumors regardless of Pole status had also had lower levels of CD45+ infiltration into the tumors (Figure 4B) even though this was not significant. Total T lymphocyte and cytotoxic lymphocyte percentages (CD3+ out of CD45+ and CD8+ out of CD45+) were significantly lower in Trp53−/− tumors as compared to p Trp53+/+ tumors (Figure 3C, CD3+: p<0.05 for K vs KP, p<0.05 for KO vs KPO. CD8+: p<0.01 for K vs KP, p<0.0001 for KO vs KPO. Percentage of CD4+ T cells or NK cells did not display significant differences (Fig S2A). Proportion of regulatory T cells (Treg) were significantly higher in KPO as compared to KP (Fig S2B). CD8/Treg ratios were significantly lower in p53 deficient tumors (Figure 4D). Macrophage percentage (CD11c+CD11b−CD103−) was significantly higher in Trp53−/− tumors as compared to Trp53+/+ tumors but neutrophil (Cd11b+ Ly6G+) percentage was similar (Fig S2C). We next determined cytokine production in these tumors. Both KP and KPO tumors produced significantly higher levels of CCL2 potentially accounting for increased macrophages (Figure 4E) 43.
Figure 4. Immune profile of Pole+/+ or PoleP286R/+ GEMMs.

Tumor microenvironment of K/KO, KP/KPO mouse lungs were analyzed by flow cytometry and immunohistochemistry.
(A) Lung weights of analyzed of the mice included in analysis. Mice were euthanatized at 16 weeks after tumor induction. N=4 for K, n=5 for KO, n=12 for KP, n=6 for KPO. Error bar: SD.
(B) Total CD45+ cell percentage out of all live cells in K, KO, KP, and KPO tumors determined by flow cytometry. N=4 for K, n=5 for KO, n=12 for KP, n=6 for KPO. Error bar: SEM.
(C) Total T cell (CD3+) and cytotoxic T cell (CD3+CD8+) percentages out of all CD45+ cells in K, KO, KP, KPO tumors. N=4 for K, n=5 for KO, n=12 for KP, n=6 for KPO. Error bar: SEM. Unpaired t-test.
(D) Quantification of CD8/Treg ratios in K, KO, KP, and KPO tumors. N=4 for K, n=5 for KO, n=7 for KP, n=6 for KPO. Error bar: SD. Unpaired t-test.
(E) CCL2 levels in lung tumors were determined by qPCR of RNA extracted from lung tumor tissues. N=14 for K, n=7 for KO, n=11 for KP, n=4 for KPO. Error bar: SEM. Unpaired t-test.
(F) Expression of MHC1 on GEMM tumor cells (Epcam+) determined by flow cytometry of K, KO, KP, and KPO lung tissue. N=3 for K, n=5 for KO, n=11 for KP, n=5 for KPO. Error bar: SEM. Unpaired t-test.
(G) Quantification of CD8 T cells from the IHC staining of K, KO, KP and KPO GEMM lung tissues. N=20 for K, n=19 for KO, n=6 for KP, n=4 for KPO. Error bar: SD. Statistical test: Unpaired t-test.
(H) Representative IHC images of CD8 IHC in K, KO, KP and KPO GEMMs.
Also see Figure S2.
As for presentation of antigens resulting from PoleP286R in these models, KP/KPO tumors expressed significantly lower levels of MHC1 as compared to K/KO potentially affecting the presentation of the antigens (Figure 4F, (p=0.03)). mRNA levels for the two of the key genes in antigen processing, Transporter associated with antigen processing (TAP1) and endoplasmic reticulum aminopeptidase-1 (ERAP1) were significantly lower in Trp53−/− tumors tumors as compared to Trp53+/+ tumors (Figure S2D). Among the immune cells, CD8+ T cells were confirmed by IHC in situ. CD8+ T cells were lower in KP/KPO as compared to K/KO. KPO also displayed reduced CD8 T cell counts as compared to KP (Figure 4G, H).
To determine what may account for differences in survival of K/KO mice, we comparatively analyzed checkpoint receptor expression and T cell activation K/KO tumors. CD3+ T cells with expression of checkpoint receptors PD-1 or CTLA-4 expression were lower at initial stages of disease in KO mice as compared to K (12-week or 16-week after induction (Figure S2E). Tregs were significantly lower and proliferating cytotoxic T cells (CD3+CD8+Ki67+) were significantly higher in KO tumors compared to K. Activated (IFNγ+) NK cells were significantly higher in KO mice as compared to K at 12 weeks (Fig S2E).
PoleP286R subcutaneous syngeneic models are moderately sensitive to immune checkpoint blockade
To further explore the potential mechanisms of immune escape in Trp53−/− PoleP286R tumors. We established syngeneic cell lines from KrasG12D/+;p53−/− (KP) and KrasG12D/+;p53−/−;PoleP286R/+ (KPO) GEMM tumors. Cell lines were cultured for 20 passages to accumulate mutations before in vivo studies. These pooled cells were implanted subcutaneously in wild type B6 mice for therapeutic studies. PoleP286R/+ lines KPO24 and KPO105 exhibited significantly improved therapeutic response to ICB as compared to Pole+/+ lines KP67–1 and KP9–1 (Figure 5A and Figure S3A–C). WES of Pole+/+ line KP67–1 and PoleP286R/+ line KPO24 at passage 1 revealed that Pole P286R/+ cells have significantly higher number of mutations at baseline (1mut/MB vs 40mut/MB). This was significantly higher than the TMB observed in KPO tumors. TMB in KPO line increased to 60 mut/MB by passage 21 (Figure 5B). In conclusion, we generated a syngeneic lung cancer model with high TMB.
Figure 5. Tumor heterogeneity contributes to immune escape in KPO syngeneic model.

(A) Growth curve for subcutaneously implanted KP67–1 (n=8 for isotype, n=8 for ICB, Error bar: SEM. Unpaired t-test.) derived from KP tumors and KPO24 (n=4 for isotype, n=6 for ICB, Error bar: SEM. Unpaired t-test.) derived from KrasG12D/+;p53−/;PoleP286R/+ syngeneic models treated with isotype control or ICB. ns: not significant.
(B) Left: Tumor mutational burden (TMB) of KP cell line (passage 1) and KPO cell line clones KPO24–1, KPO24–2, and KPO24–3 at passage 1 and passage 21. Right: Distribution of mutation types in KP vs KPO syngeneic cell lines. N=3 per group. Error bar: SEM. Unpaired t-test.
(C) Schema for generating single cell clones from KPO24. In vivo tumor growth of single clones 1–3 (n=6 for isotype, n=6 for ICB, Error bar: SEM in curve, SD in scatterplot. Unpaired t-test.) derived from syngeneic cell line KPO24 implanted subcutaneously and treated with isotype or ICB. Weights of dissected tumors shown on the right.
(D) Lung tumor incidence for KP67–1 and KPO24 pool or single cell clones derived from KPO24 cells injected intravenously (IV) into mice. Any tumor presence was recorded as an incidence. Log-rank test.
(E) Re-challenge experiment with pool vs single clone models. Growth of KPO24 single clone on either tumor/treatment-naïve mice or mice which were previously implanted with corresponding single clone KPO tumors that regressed upon ICB (left). Growth of KPO24 pool syngeneic line on either tumor/treatment naïve mice or mice which previously were implanted with single clone KPO tumors that regressed with ICB (left). Error bar: SEM.
(F) Quantification of MATH (Mutant-Allele Tumor Heterogeneity) score of KPO syngeneic lines between passage 1 and passage 21 (left). Venn diagram showing the number of shared mutations between KPO single clone and corresponding pool in passage 1 vs 21 (right). N=3 for p1, n=5 for >p20. Error bar: SEM. Unpaired t-test.)
Also see Figures S3, S4, and S5.
Differences in the TMB and the microenvironment may contribute to increased sensitivity to ICB in the lung vs subcutaneous tissue. We implanted KP67–1 and KPO24 subcutaneously into mice, harvested tumors and performed flow cytometry analysis. The subcutaneous environment highlighted increased immunogenicity in Pole P286R/+ tumors when tumors were established from cell lines. Syngeneic KPO tumors had higher overall CD45+ infiltration as compared to KP tumors (p<0.0001, Figure S3D). There were also more neutrophils and dendritic cells in KPO syngeneic model as compared to KP (p<0.0001, Figure S3D). KPO model also showed decreased expression of immune checkpoint receptors (PD-1, LAG-3 and CTLA-4) and increased NK cell activation (CD107a, Granzyme B) as compared to KP (Figure S3D).
Tumor heterogeneity further contributes to immune escape in KPO syngeneic model
We did not observe complete tumor rejection in KPO syngeneic models indicating presence of potential mechanisms of immune resistance. To further dissect the potential mechanisms of immune escape in these hypermutated tumors, we used the clones generated for WES (KPO24–1,2,3) and compared their response to ICB with that of KPO24 pool. Two of the three clones (KPO24–1 and 3) showed delayed tumor growth as compared to parental cells in vivo (Figure 5B & 5C). All three single clones of KPO24 showed superior sensitivity to ICB as compared to the parental pool population. Some of the mice completely cleared their tumors (Figure 5C). To determine the contribution of CD8+ T cells in immunogenicity of these tumors, we depleted CD8+ T cells with anti-CD8a antibody in mice subcutaneously implanted with these tumors. Depletion of CD8+ T cells significantly promoted growth of KPO single clones (p=0.003) but did not change the growth of KP clones (Figure S3E and S3F). To further evaluate the role of heterogeneity even in the context of immunogenic tumors, we expanded 4 clones (KPO24 1,2,3, and 4 (Polewt) and mixed them at equal ratios for in vitro and in vivo experiments right before experiment setup (Figure S4A). Mutant Pole allele was deleted in clone 4 by Crispr-Cas9 resulting in Polewt status. Only clone-4 grew faster in vitro than other lines; however, the difference was not statistically significant (Figure S4B). Clone 4 is also responsive to ICB although not as sensitive as clones 1 or 3 (Figure S3C). Interestingly, mixture tumors even though implanted at the same total number as single clones (2×106) grew significantly faster in vivo with tumors reaching mean volume of ~500mm3 in about 12-days as compared to single clones reaching this volume at around 30 days or later (Figure S4D). This mixture was resistant to ICB under these conditions (Figure S4D).
To develop syngeneic tumors in mouse lungs we implanted KP/KPO cells by tail vein injection. There was a significant delay in growth of KPO line as compared to KP line similar to the subcutaneous model (Figure 5D). For the therapy experiment, tumor cells were implanted by IV and mice received ICB treatment for ~4 weeks. Mice were euthanatized around 50 days after tumor implantation to collect lungs. KPO24 line was sensitive to ICB in the context of lung with lower tumor areas in the lungs of treated mice as compared to controls while KP67–1 remained insensitive (Figure S4E and Figure S4F). Single clones grew even less efficiently with some of the mice not developing any tumors (evaluated microscopically) by 200 days confirming the increased immunogenicity of the single cell derived clones even in the lung tissue. (Figure 5D).
Since some of the mice carrying single KPO clones exhibited durable complete therapeutic response to ICB, we further investigated development of immune memory. We implanted KPO24 pool or single cell clones on mice implanted with single clones cured by ICB at least 12 weeks prior to re-challenge. All mice rejected the corresponding single cell clones suggesting that these GEMM derived syngeneic models carry immunogenic antigens (Figure 5E). There was also strong immunity against the KPO24 pool as most of the mice rejected tumors (n=6 out of 7). Pool tumors developing in one of the mice escaped immune surveillance. This argues that although shared antigens between the pool and clones provide protective immunity, heterogeneity within the pool may drive the immune escape (Figure 5E). We further analyzed sequencing data to determine whether shared antigens could play a role in immune protection. There were shared mutations between pool and single cell clones at both passages 1 and 21. Interestingly, cells passaged to P21 showed significantly higher number of shared mutations between pool and single cell clones both in numbers and percentage of total mutations (from 9.6% at passage 1 (236 mutations) to 71% at passage 21 (1622), Figure 5F). Quantification of Mutant-Allele Tumor Heterogeneity (MATH) score of KPO syngeneic lines between passage 1 and passage 21 indicates decreased heterogeneity in passage 21.
Pole P286R induced accumulation of C>A/C>T substitutions
KP tumor samples have a random distribution of substitutions while KPO tumor samples have more frequent C>A/C>T substitutions and fewer C>G/ T>A substitutions similar to the reported PoleP286R mutational signature 44–46 (Figure S5A) and this was significant (p=0.0002, Figure S5B). C>G are also observed more commonly in tumors from smoker patients than non-smoker lung cancer patients 47. Mutational signatures in KP and KPO syngeneic cell lines were also examined by WES. KP line KP67–1 presented a random distribution of different substitutions while KPO line KPO24 acquired higher numbers of C>A, C>T and T>G substitutions as compared to C>G and C>T (Figure S5C and Figure S5D). POLE mutations (P286R and V411L) in human tumors are associated with COSMIC signature 10. According to this signature C>A and C>T substitutions are more frequent compared to others. Of note, C>A is biased to TCT context and T>G substitution is biased to TTT context 46. Mutations in KPO tumors were consistent with this signature. We further performed hierarchical clustering analysis based on variants. Among the syngeneic cell lines, while KP lines cluster together, KPO lines cluster separately (Figure S5E). KPO pools cluster with corresponding single clones at each passage, demonstrating clonal relationship (Figure S5E). GEMM tumors exhibit similar separation of mutations detected in KP and KPOs (Figure S5F).
p53 induction further induces T cell killing and immunogenicity of KPO tumors
As we observed that p53 status was partially responsible for mediating resistance to ICB in GEMMs, we set up a study to functionally confirm the role of p53 in immune resistance using the polyclonal syngeneic model we characterized. We induced p53 expression in the KP and KPO models with a doxycycline inducible system (Fig 6A and Figure S6A). P53 inducible Tap1 expression was lower in p53 deficient tumors consistent with the literature (Figure S2D) 48. Induction of p53 in p53 deficient KP/KPO lines resulted in decreased survival of cells in vitro, as would be expected (Figure S6B). p53 expression also induced Tap1 mRNA levels in both KP and KPO lines (Figure 6B). Increase in MHC1 levels were confirmed in two KPO models by flow cytometry (Figure 6 C–E). We wondered whether missense mutations of Trp53 regulate MHC-1 as a previous study showed TP53R273C retained weak binding to ERAP1 promoter in a human line 49. We expressed missense mutant Trp53 R172H and R270H- equivalent to human hotspot mutations in codons R175 and R273 in KPO24 (Figure S6C). Missense mutants forms p53 did not increase baseline MHC1 levels. However, they increased inducible MHC1 in response to IFNγ (p<0.05, Figure S6D and E).
Figure 6. p53 induction increases immunogenicity of tumor cells and induces T cell cytotoxicity.

(A) Western blot for p53 and vinculin in KP, KPO lines stably transduced with pLVX-Tet-ON Advanced and pLVX-Tight puro-Trp53 and treated with doxycycline for 48 hours.
(B) qPCR for Tap1 in KP and KPO-TetOp-Trp53 lines treated with doxycycline for the indicated times. N=3 per group. Error bar: SD.
(C) Representative contour plot showing MHC1 expression in KPO105-TetOp-Trp53 model treated with doxycycline for 48 hours as determined by flow cytometry.
(D) Quantification of MHC1 expression determined by flow cytometry in KPO105-TetOp-Trp53 model with and without doxycycline. N=3 for control, n=3 for +dox. Error bar: SEM. Unpaired t-test.
(E) Quantification of MHC1 expression in KPO24-TetOp-Trp53 model treated with doxycycline for 48 hours as determined by flow cytometry. N=3 for control, n=3 for +dox. Error bar: SEM. Unpaired t-test.
(F) T cell specific killing measured by LDH release of co-cultures of p53 inducible KPO lines (KPO105 and KPO24) expressing OVA with OT-1 T cells isolated from OT-1 transgenic mouse spleens. Apoptosis of tumor cells was evaluated by Annexin V staining and analyzed by flow cytometry. LDH lysis: n=5 for control, n=5 for +dox. Error bar: SEM. Unpaired t-test. Annexin V staining: n=3 for control, n=3 for +dox. Error bar: SEM. Unpaired t-test.
(G) MHC1 expression in human NSCLC lines A549 and H23 treated with 20μM Nutlin-3a for 48h determined by flow cytometry. N=4 for vehicle, n=4 for Nutlin-3a. Error bar: SEM. Unpaired t-test.
(H) MHC1 expression in MC38 cells treated with varying Nutlin-3a doses for 96h as determined by flow cytometry. N=3 per group. Error bar: SEM. Unpaired t-test.
(I) T cell specific killing measured by LDH release in co-cultures of nutlin-3a pre-treated OVA expressing MC38 cells with OT-1 T cells. Apoptosis of tumor cells was measured by Annexin V staining and analyzed by flow cytometry. LDH lysis: n=5 for control, n=5 for +dox. Error bar: SEM. Unpaired t-test. Annexin V staining: n=3 for control, n=3 for +dox. Error bar: SEM. Unpaired t-test.
(J) TP53 and TAP1 mRNA level in NSCLC lines with wild-type, missense or nonsense TP53 mutations from Cancer Cell Line Encyclopedia (CCLE).
(K) p53 and TAP1 protein levels in NSCLC lines with WT, missense or nonsense TP53 mutations (data from CCLE).
Also see Figure S6.
To determine whether induction of p53 expression can induce T cell killing of cancer cells, we expressed model antigen chicken ovalbumin (OVA) in syngeneic lines and utilized an OVA-OT1 system. OT-1 T cell receptor (TCR) recognizes the OVA-derived peptide OVA257–264 (SIINFEKL) bound to mouse MHC-1 H-2Kb. Induction of p53 by doxycycline in OVA expressing KPO lines TetOp KPO24, KPO24–4 and KPO105 resulted in significantly increased tumor cell lysis and apoptosis by the OT-1 cells isolated from OT-1 transgenic mice (Figure 6F and Fig S6F). Increase in MHC1 levels by p53 induction was also confirmed with an inhibitor of E3 ubiquitin ligase MDM2-nutlin-3a. Treatment of human NSCLC lines A549 (TP53 wild-type) and H23 (TP53 missense mutant) resulted in increased MHC1 by flow (Figure 6G). Nutlin-3a also induced expression of MHC1 in mouse syngeneic line expressing p53: MC38 (Trp53 missense mutant) (Figure 6H). Treatment with nutlin-3a induced cytotoxicity of OT-1 T cells against MC38 cells expressing OVA (Figure 6I).
To confirm correlations between TP53 mutation status and TAP1 expression in human NSCLC lines, we analyzed the Cancer Cell Line Encyclopedia (CCLE) 50. TP53 missense mutant tumors had the highest mRNA for TP53 followed by wild-type TP53. TP53 nonsense mutant tumors had very low levels of TP53 mRNA as expected (Figure 6J). TAP1 mRNA was significantly lower in TP53 nonsense mutant lines as compared to both TP53 wild-type and missense mutant lines (Figure 6J). Protein levels through reverse phase protein array (RPPA) were available for a subset of NSCLC lines50. As expected TP53 missense mutant lines had the highest level of p53 protein. TAP1 levels were significantly lower in nonsense and missense mutant lines as compared to TP53 wild-type lines (Figure 6K).
We evaluated the in vivo impact of p53 restoration in the polyclonal tumors using the stable cell KPO105-Tetop-Trp53 line in vivo. Tumors only grew in 2 out of 15 B6 mice likely due to rtTA immunogenicity 51 or the two-vector system (Figure S6G) because tumors were not rejected in NSG mice (not shown). We isolated tumors grown in B6 mice pooled and re-implanted into B6 mice. All of the mice grew tumors and tumors expressed p53 upon doxycycline treatment (Figure S6G). We performed combination treatment experiments with doxycycline and ICB. Tumors were responsive to ICB, dox, and combination treatments with starting baseline volumes around 100mm3 as in the other experiments, (Figure 7A, B). Some of the combination treated mice had complete regression of tumors (Figure 7C). Since KPO105-Tetop-Trp53 tumors were very sensitive to ICB, to be able to observe synergy between p53 expression and ICB we repeated this experiment with higher baseline tumor volumes (~250mm3). We also reduced frequency/dose of doxycycline and ICB (Figure 7A, C). Induction of p53 expression did not reduce the growth of these large tumors but caused significantly increased sensitivity to ICB (Figure 7C). Tumor tissue had p53 expression by western blots (Fig S6H) although we do not know whether Trp53 is possibly mutated at later timepoints in these tumors. p53 induction in these tumors increased tumor MHC1 expression, proliferation of T cells and NK cells, and additionally reduced expression of checkpoint receptors PD-1 and LAG-3 (Figure 7D). Dox and ICB combination treated tumors exhibited significantly decreased proliferation (Ki67+) and increased apoptosis as shown by cleaved caspase-3 (Figure 7E & 7F). To concurrently address heterogeneity and p53 expression in tumor growth and response to ICB, we isolated single cell clones of KPO24 and KPO105-TetOp-Trp53 and expanded in vitro. Consistent with single clones without TetOp system, these lines also exhibited increased immunogenicity and either did not grow or grew at a slower rate than the pool population in B6 mice not allowing in vivo experiments (Figure S6I).
Figure 7. p53 induction increases sensitivity to ICB in vivo.

(A) Schedules for combination treatments of doxycycline and ICB.
(B) Growth of subcutaneous KPO105-TetOp-Trp53 tumors treated with doxycycline, ICB, or combination of doxycycline and ICB with schedule 1 at baseline volume of 100mm3. Final measurements of tumor volumes are shown on the right. n=8 for control. n=8 for Control+ICB. n=8 for +dox control. n=8 for +dox+ICB. Error bar: SEM. Unpaired t-test. ***: comparison of control and +dox groups.
(C) Growth of subcutaneous KPO105-TetOp-Trp53 tumors treated with doxycycline (dox), ICB, or combination of doxycycline and ICB with schedule 2 at baseline volume of 250mm3 (left) and weights of final dissected tumors are shown on the right. n=8 for control. n=6 for Control+ICB. n=8 for +dox control. n=8 for +dox+ICB. Error bar: SEM. Unpaired t-test.
(D) Flow cytometry analysis of KPO105-TetOp-Trp53 tumors from mice treated with vehicle or doxycycline showing expression on MHC1 on tumor cells (Epcam+) (n=5 for control, n=6 for +dox), and other markers (Ki67, PD-1, LAG-3+) (n=6 for control, n=6 for +dox) on CD3+ T cells, CD8+ T cells or NK cells. Error bar: SD. Unpaired t-test.
(E) IHC for Ki67 on KPO105-TetOp-Trp53 tumors treated with doxycycline, ICB, or combination of doxycycline and ICB (schedule 1). Representative images are shown on the left and quantification of stained area are shown on the right. n=8 for control. n=6 for Control+ICB. n=8 for +dox control. n=8 for +dox+ICB. Error bar: SD. Unpaired t-test.
(F) IHC for cleaved caspase-3 on KPO105-TetOp-Trp53 tumors treated with doxycycline, ICB, or combination of doxycycline and ICB (schedule 1). Representative images are shown on the left and quantification of stained area on the right. n=8 for control. n=6 for Control+ICB. n=8 for +dox control. n=8 for +dox+ICB. Error bar: SD. Unpaired t-test.
Also see Figure S6.
Elevated STING is a therapeutic target in heterogenous p53 mutant tumors with high TMB
We noted increased DNA damage indicated by higher number of γH2A.X positive tumor cells in KrasG12D/+;Trp53−/−;PoleP286R/+ lung tumor tissues compared to KrasG12D/+;Trp53−/− (Figure 2E). This was further validated in KP and KPO lines (Figure S7A). PoleP286R/+ KPO24 line showed a higher percentage of γH2A.X positive cells compared to the Pole+/+ lines KP9–1 and KP9–3 by both immunofluorescence and western blotting (Figure S7A and Figure S7B). As a result of genomic instability, higher number of micronuclei were detected in KPO line as compared to KP lines (Figure S7C, p<0.0001). Based on these observations, we further investigated the cGAS-STING pathway, the responder to cytosolic DNA as micronuclei in syngeneic cell lines. Total STING expression was higher in KPO lines as compared to KP. However, cGAS and phosphorylated STING (p-STING) remained unchanged (Figure S7B). Attenuating cGAS-STING pathway in hypermutated tumors may be a potential escape mechanism 52. The presence of elevated level of total STING but lack of activated STING presented as a potential therapeutic target in this model.
We further investigated cGAS-STING pathway in GEMM tumors. KrasG12D/+;Trp53−/−;PoleP286R/+ tumor nodules dissected from GEMMs showed increases STING expression overall, but expression levels were variable between tumor nodules from different mice as shown by western blot and immunohistochemistry (Figures 8A, Figure 8B). We did not observe this heterogeneity in p53 wt K and KO mouse tumors (Figure S8A, B). STING can be degraded through TBK1 induced p62/SQSTM1-dependent autophagy following ubiquitination53,54. We investigated p62 expression in KP, KPO, K and KO tumors. PoleP286R/+ tumors (KPO and KO) also express higher level p62, which may counteract elevated STING levels in KPO tumor to impede anti-tumor immunity stimulated by cGAS-STING pathway (Figure 8C and Figure S8C). To harness increased total STING and increase potential bystander killing by the T cells in PoleP286R/+ polyclonal tumors where less of the antigens are shared, we utilized a nanoparticle-loaded STING agonist (polySTING) we recently developed 37. PolySTING significantly suppressed primary tumor growth of KPO tumors through intra-tumoral administration (Figure 8D). PolySTING also increased efficacy of ICB, and significantly inhibited tumor growth as compared to ICB alone as shown in tumor growth curves and final weights of dissected tumors (Figure 8D). PolySTING triggered antitumor immunity by increasing activation of cytotoxic CD8+ T cells and NK cells as determined by flow cytometry of treated tumor tissues (IFNγ+) (Figure 8E). Sting agonist treated tumors increased phosphorylated TBK1 (pTBK1) indicating activation of cGAS-STING pathway and cleaved caspase-3 (Figure 8F).
Figure 8. Sting is expressed heterogeneously in PoleP286R lung tumors and is a therapeutic target.

(A) Western blot for STING, pSTING, and vinculin on tumors from KP and KPO GEMMs (top left).
(B) Representative IHC for STING on lung tumors from KP and KPO GEMMs (left). Positive and negative stained tumor nodules are shown from the same mouse tissue scan and quantification of STING IHC in tumor nodules (right). N=41 for KP, N=91 for KPO. Error bar: SEM. Unpaired t-test.
(C) Western blot for p62 and vinculin on tumors from KP and KPO GEMMs. Relative protein amount was quantified by image densitometry. N= 4 for KP, n=9 for KPO. Error bar: SEM. Unpaired t-test.
(D) In vivo tumor growth of KrasG12D/+;Trp53−/;PoleP286R/+ syngeneic cell line KPO105 subcutaneous tumors treated with isotype, Sting agonist (PolySTING), ICB or combination of ICB and Sting agonist. The final weight of dissected tumors at experiment termination was graphed. n=6 for control, n=6 for ICB, n=6 for Sting agonist, n=6 for Sting agonist+ICB. Error bar: SEM. Unpaired t-test.
(E) Flow cytometry analysis of KPO syngeneic tumors treated with control or Sting agonist. Quantification of Intracellular staining for IFNγ+ on CD8+ T cells or NK cells. n=5 for control, n=5 for Sting agonist. Error bar: SD. Unpaired t-test.
(F) Western blot for pTBK1 and cleaved caspase-3 on KPO105 subcutaneous tumors treated with vehicle or STING agonist. Relative protein amount was quantified by image densitometry. n=3 for control, n=3 for Sting agonist. Error bar: SD. Unpaired t-test.
Also see Figures S7 and S8.
Discussion:
Mice expressing only Pole P286R in lung epithelial cells developed lung tumors at a low frequency similar to aged-matched controls. Turnover rate of the cell of origin may play a role in the lack of lung tumors in these mice. Pole P286R in endometrium 55 and MSH2 inactivation in colon without a driver oncogene can initiate endometrial and colon carcinoma, respectively 56. Turnover rate for endometrium (~a month in humans and 4–5 days in mice) and colonic epithelial cells (<10 days in humans), is much higher than for bronchial epithelial cells (>100 days) 57,58. The presence of activating Kras resulted in an increase in TMB but not complete ICB sensitivity unlike what we observed in endometrium 55. Pole P286R driven endometrial tumors have longer latency allowing for higher TMB accumulation and are likely more clonal due to Pole P286R being the driver potentially resulting in sensitivity to ICB.
PoleP286R in the context of the KP model induced development of autochthonous lung tumors with TMBs similar to that of some of the human lung adenocarcinomas. Increase in TMB with Pole P286R is higher than that of Msh2 inactivation in the context of KP model 59. This is not surprising as PoleP286R mutations in human tumors result in higher TMB than tumors with mismatch repair deficiencies 60. We highlight several immune suppressive mechanisms such as heterogeneity, reduced antigen presentation and lymphocyte infiltration partially accounting for lack of any clinical response to ICB in KPO model. A functional screen identified preferential loss of p53 in immune-competent but not in immune compromised mice supporting the notion that loss of p53 may be required for immune escape of certain tumors 61. In PoleP286R driven endometrial cancers, p53 loss was observed as a late event again suggesting a cooperation between Pole P286R and p53 to promote tumor growth 26.
Our mouse studies were limited to complete genetic loss of Trp53 while missense mutations are more common in human tumors and distinct roles of these mutations in tumor development are being discovered. Missense mutations of TP53 but not nonsense was shown to suppress innate immune signaling through inhibiting TBK1 62. TP53 mutations were least observed in the immune favorable Immune subtype of lung adenocarcinoma as determined by gene expression profiling 63. A previous study associated TP53 nonsense mutations with poor prognosis in ICB in NSCLC 64. Some of the studies evaluating prognosis with TP53 mutations under ICB showed a positive correlation between TP53 mutations and response to ICB- TP53 mutations in this study were determined by a positive staining by immunohistochemistry which favors detection of missense mutations 65,66. Other studies also associated TP53 mutations with concurrent KRAS mutations with better response to ICB than wild-type TP5321. TP53 may also be potentially generally prognostic in NSCLC. In a prospective study with stage 1 lung cancer patients whose tumors have mutant TP53 lived significantly shorter than patients whose tumors have wild-type TP53 67. Further research into the types of TP53 mutations in tumors of patients receiving immunotherapy is warranted.
We focused on mutations in TP53 in regulating TAP1 expression and antigen presentation. Alternative mechanisms of loss of p53 function include amplification of MDM genes coding for E3 ubiquitin ligases that regulate p53 protein levels in the cells 68. For example, gain of chromosome 1q carrying MDM4 caused decreased p53 signaling in the context of wild-type TP53 69. Gain of 1q was associated with wild-type TP53 in 789 human cell lines and MDM4 was essential for 216 of those lines70. Future studies that integrate both mutation and copy number analysis of TP53 and other genes involved in regulation of p53 such as MDM2 and MDM4 may potentially resolve the differences between different patient datasets and mouse/human tumors. Genes involved in antigen processing were recently implicated in predicting response to ICB 36. Expression of these may also be considered in conjunction with mutations for better selection for combination treatments.
Limitations of the Study:
A limitation of our study is that some of the experiments were performed in the flank tumors due to robust growth in the flank. In this model p53 loss did not cause complete resistance to ICB and heterogeneity was a more dominant immune suppressor. Heterogeneity as an immune suppressive mechanism is consistent with recent melanoma studies that found that intratumoral heterogeneity drives immune escape in an hypermutated syngeneic melanoma model 71. Another limitation of our study is that we utilized experimental systems to test the impact of p53 restoration and targeting STING intratumorally to sensitize polyclonal tumors to ICB. These strategies can be improved for clinical testing. Our data suggests MDM2 inhibitors such as nutlin-3a can induce MHC1 expression. Other drugs such as APR-246 which can restore wild-type p53 function with mutant p53 can also be tested in future studies to determine their effect on antigen presentation. STING agonists that allow systemic delivery will be investigated in future studies.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Esra A. Akbay, Ph.D. Esra.Akbay@utsouthwestern.edu.
Materials availability
Plasmids and cell lines generated in this study are available upon request with a valid material transfer agreement (MTA).
Data and code availability
Whole exome sequencing (WES) data of mouse tissues have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 488 anti-mouse CD4 | Biolegend | 100423 |
| Alexa Fluor® 488 anti-mouse CD45 | Biolegend | 103122 |
| Alexa Fluor® 488 anti-mouse CD8a | Biolegend | 100723 |
| APC anti-mouse CD4 | Biolegend | 100516 |
| APC anti-mouse IL-2 | Biolegend | 503810 |
| APC Rat IgG2b, κ Isotype Ctrl | Biolegend | 400612 |
| APC/Cy7 anti-mouse CD45 | Biolegend | 103116 |
| APC/Cy7 anti-mouse CD8a | Biolegend | 100714 |
| Brilliant Violet 421™ anti-mouse CD19 | Biolegend | 115538 |
| Brilliant Violet 421™ anti-mouse Ki-67 | Biolegend | 652411 |
| PE/Cy7 anti-mouse CD326 (Ep-CAM) | Biolegend | 118216 |
| PE/Cy7 anti-mouse CD3ε | Biolegend | 100320 |
| PerCP/Cy5.5 anti-mouse CD335 (NKp46) | Biolegend | 137610 |
| Alexa Fluor® 488 Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400233 |
| PE Rat IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400636 |
| APC Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400220 |
| APC anti-human HLA-A, B, C Antibody | Biolegend | 311410 |
| APC anti-mouse CD335 (NKp46) Antibody | Biolegend | 137607 |
| PE anti-mouse H-2K b/H-2D b Antibody | Biolegend | 114607 |
| PE Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400212 |
| Brilliant Violet 421™ Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400341 |
| PE anti-mouse RAE-1δ Antibody | Biolegend | 133203 |
| PE Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400111 |
| APC Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400319 |
| APC Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400511 |
| APC Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400119 |
| PerCP/Cy5.5 anti-mouse CD8a Antibody | Biolegend | 100733 |
| APC anti-mouse IgG2a Antibody | Biolegend | 407110 |
| APC Rat IgG1, κ Isotype Ctrl Antibody | Biolegend | 400411 |
| Brilliant Violet 421™ Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400157 |
| PE/Cy7 Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400125 |
| PerCP/Cy5.5 anti-mouse CD3ε Antibody | Biolegend | 100328 |
| Alexa Fluor® 488 anti-mouse CD3ε Antibody | Biolegend | 100321 |
| PE anti-mouse CD4 Antibody | Biolegend | 100512 |
| APC anti-mouse CD107a (LAMP-1) Antibody | Biolegend | 121614 |
| PerCP/Cy5.5 anti-mouse CD107a (LAMP-1) Antibody | Biolegend | 121626 |
| Brilliant Violet 421™ Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400549 |
| PE anti-mouse IL-2 Antibody | Biolegend | 503808 |
| TruStain fcX™ (anti-mouse CD16/32) Antibody | Biolegend | 101320 |
| Brilliant Violet 421™ anti-mouse CD3ε Antibody | Biolegend | 100341 |
| PE Rat IgG1, κ Isotype Ctrl Antibody | Biolegend | 400408 |
| PE/Cy7 Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400326 |
| PE/Cy7 anti-mouse CD3ε Antibody | Biolegend | 100320 |
| PE Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400508 |
| PE anti-human HLA-A,B,C Antibody | Biolegend | 311406 |
| PE Mouse IgG2a, κ Isotype Ctrl (FC) Antibody | Biolegend | 400214 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | 114608 |
| Brilliant Violet 421™ anti-mouse IL-2 Antibody | Biolegend | 503826 |
| FITC anti-human/mouse Granzyme B Recombinant Antibody | Biolegend | 372206 |
| PerCP/Cyanine5.5 anti-mouse CD8a Antibody | Biolegend | 100734 |
| FITC Mouse IgG1, κ Isotype Ctrl (ICFC) Antibody | Biolegend | 400138 |
| PE anti-mouse NK-1.1 Antibody | Biolegend | 108708 |
| Alexa Fluor® 488 anti-mouse CD49b (pan-NK cells) Antibody | Biolegend | 108913 |
| APC anti-mouse CD49b (pan-NK cells) Antibody | Biolegend | 108910 |
| PE/Cy7 Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400522 |
| PE/Cy7 Rat IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400617 |
| PE anti-mouse IFN-γ Antibody | Biolegend | 505808 |
| APC/Cyanine7 anti-mouse CD45 Antibody | Biolegend | 103116 |
| InVivoMAb rat IgG2b isotype control | BioXcell | BE0090 |
| InVivoMAb anti-mouse CD8α | BioXcell | BE0117 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXcell | BP0164 |
| p53 (1C12) Mouse mAb | Cell signaling Technology | 2524 |
| Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb | Cell signaling Technology | 9718 |
| Ki-67 Recombinant Rabbit Monoclonal Antibody (SP6) | Thermofisher | MA5–14520 |
| CD8α (C8/144B) Mouse mAb | Cell signaling Technology | 70306 |
| STING (D2P2F) Rabbit mAb | Cell signaling Technology | 13647 |
| Phospho-STING (Ser365) (D8F4W) Rabbit mAb | Cell signaling Technology | 72971 |
| Vinculin (E1E9V) XP® Rabbit mAb | Cell signaling Technology | 13901 |
| GAPDH (D16H11) XP® Rabbit mAb | Cell signaling Technology | 5174 |
| Phospho-TBK1/NAK (Ser172) (D52C2) XP® Rabbit mAb | Cell signaling Technology | 5483 |
| Caspase-3 Antibody | Cell signaling Technology | 9662 |
| DAPI | Cell signaling Technology | 4083 |
| Bacterial and virus strains | ||
| NEB Stable Competent E. coli | NEB | C3040H |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Nutlin-3a | MedChemExpress | HY-10029 |
| Recombinant Murine IL-2 | PeproTech | 212–12 |
| OVA Peptide (323–339) | Genscript | RP10610 |
| Collagenase, Type IV, powder | Themofisher | 17104019 |
| DNase I recombinant, RNase-free | Millipore | 4716728001 |
| Cell Activation Cocktail (with Brefeldin A) | Biolegend | 423304 |
| Critical commercial assays | ||
| CyQUANT™ LDH Cytotoxicity Assay | Invtrogen | C20300 |
| MojoSort™ Mouse CD8 T Cell Isolation Kit | Biolegend | 480007 |
| DNeasy Blood & Tissue Kits | Qiagen | 69504 |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 |
| RNeasy Mini Kit | Qiagen | 74104 |
| PowerUp™ SYBR™ Green Master Mix | Thermofisher | A25742 |
| Deposited data | ||
| Tumor mutational burden in genetically engineered-mouse model | SRA | PRJNA933600 |
| Experimental models: Cell lines | ||
| A549 | UTSW | Hamon Center for Therapeutic Oncology Research |
| H23 | UTSW | Hamon Center for Therapeutic Oncology Research |
| MC38 | Kerafast | ENH204-FP |
| KP9–1 | Akbay et al, 2017, JTO25 | |
| KP9–3 | Akbay et al, 2017, JTO25 | |
| KP67–1 | Generated in this study | |
| KPO24 | Generated in this study | |
| KPO24–1 | Generated in this study | |
| KPO24–2 | Generated in this study | |
| KPO24–3 | Generated in this study | |
| KPO24–4 | Generated in this study | |
| KPO105 | Generated in this study | |
| KPO24-TetOp-p53 | Generated in this study | |
| KPO105-TetOp-p53 | Generated in this study | |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| B6.129S6-Poletm1Dcas/J | Jackson Laboratory | 037051 |
| B6(Cg)-Krastm5Tyj/J | Jackson Laboratory | 023590 |
| B6.129S2-Trp53tm1Tyj/J | Jackson Laboratory | 002101 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | 003831 |
| Oligonucleotides | ||
| mouse Actb Forward primer: GCCCTGAGGCTCTTTTCCAG | sigma | |
| mouse Actb Reverse primer: TGCCACAGGATTCCATACCC |
sigma | |
| mouse Tap1 Forward primer: GCTGTTCAGGTCCTGCTCTC | sigma | |
| mouse Tap1 reverse primer: CACTGAGTGGAGAGCAAGGAG | sigma | |
| mouse Erap1 Forward primer: CGAGGACCTGTGGAATAGCATG | sigma | |
| mouse Erap1 reverse primer: CATCTACAACCTCCTGACGCCA | sigma | |
| Mouse Ccl2 Forward primer: CATCCACGTGTTGGCTCA |
||
| Mouse Ccl2 reverse primer: GATCATCTTGCTGGTGAATGAGT |
||
| Recombinant DNA | ||
| cDNA clone for Mus musculus transformation related protein 53 (Trp53), transcript variant 2, mRNA. | Genscript | NM_001127233.1 |
| Software and algorithms | ||
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| BD FACSAria™ III System | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-sorters/facsaria-iii |
| GraphPad Prism software 9.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| Image J | NIH | imagej.nih.gov/ij/download/ |
| 3D slicer | NIH | https://www.slicer.org |
| Incucyte Base Analysis Software | Sartorius | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-base-software |
| NDP.view2 | Hamamatsu | https://www.hamamatsu.com/eu/en/product/life-science-and-medical-systems/digital-slide-scanner/U12388–01.html |
| Trim Galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| Burrows-Wheeler Aligner (BWA, v0.7.17) | https://academic.oup.com/bioinformatics/article/25/14/1754/225615 | |
| Picard | https://broadinstitute.github.io/picard | |
| Genome Analysis Toolkit (GATK, 4.1.4.0) | https://www.nature.com/articles/ng.806 | |
| Minimap2 (v2.24-r1122) | https://academic.oup.com/bioinformatics/article/34/18/3094/4994778?login=true | |
| SAMtools (v1.9) | https://academic.oup.com/bioinformatics/article/25/16/2078/204688?login=true | |
| R | R foundation | https://www.r-project.org |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mus musculus of 6–10-week-old from both sexes were included in the study. PoleP286R mice were of mixed C57BL/6J and 129S6/SvEvTac and backcrossed at least 8 times to C57BL/6J mice generate syngeneic models in C57BL/6J background. All animal work described in this manuscript was approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
Human cell lines were obtained from the Hamon Center for Therapeutic Oncology at UTSW. Cells were maintained by routine fingerprinting and under mycoplasma free conditions. Human cell lines were maintained in RPMI-1640(Sigma Cat# R8758) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin (10000U/m) at 37°C with 5% CO2.
Sex of mouse syngeneic lines used in the manuscript: KP9–1 25 (male), KPO24 (male), KPO105 (female), KP67–1 (Male). Mouse cell lines were maintained in RPMI-1640(Sigma Cat# R8758) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin (10000U/m) at 37°C with 5% CO2.
METHOD DETAILS
Co-culture experiments
OT-1 T cells were collected from splenocytes of OT-1 transgenic mice (JAX # 003831). T cells were cultured with IL-2 (10ng/ml) and OVA peptides (5μg/ml) for 72h. Inducible p53 lines were treated with doxycycline (1μg/ml) or vehicle for 48h. Other lines were pre-treated with 5μM or other indicated concentration of Nutlin-3a or vehicle for 48h. Then, CD8+ T cells were enriched by MojoSort™ Mouse CD8 T Cell Isolation Kit (Biolgend Cat# 480007). Equal numbers of tumor cells were seeded and co-cultured with isolated OT-1 CD8+ T cells at effector: tumor 1:1 ratio overnight. 50μl of cell culture medium was harvested for LDH cytotoxicity assay. Cytotoxicity was evaluated by CyQUANT™ LDH Cytotoxicity Assay Kit (Thermofisher, Catalog # 20300) following manufacturer’s instructions. Final percentage of lysis was calculated by the formula %lysis=100*(Release-Tumor spontaneous release-CD8 spontaneous release)/ (Tumor maximum release- Tumor spontaneous release). Remaining tumor cells were collected and stained with Annexin V antibody (Biolgend Cat# 640907) and analyzed by flow cytometry.
Genetically Engineered Mouse Models (GEMMs)
All mice were housed in a barrier facility and maintained on standard chow. p53fl/fl (Cat# 008462) and KrasLSL-G12D/+(Cat# 008179) mice were purchased from Jackson laboratory. PoleLSL-P286R/+ mice were previously generated 26. PoleLSL-P286R/+, KrasLSL-G12D/+(K), KrasLSL-G12D; PoleLSL-P286R/+(KO) were induced intranasally with 2.5×108 Adeno-CMV-Cre (purchased from University of Iowa Viral Vector Core) resuspended in Eagle’s minimal essential medium (Sigma Cat# M4526) with 9.6mM CaCl2. KrasLSL-G12D/+; p53fl/fl(KP), KrasLSL-G12D/+;p53fl/fl;PoleLSL-P286R/+(KPO) mice were induced with 2.5×107 Adeno-CMV-Cre resuspended in Eagle’s minimal essential medium with 9.6mM CaCl2. All animal work described in this manuscript was approved by the University of Texas Southwestern Institutional Animal Care and Use Committee. Mice were randomized for therapeutic studies. Unless otherwise indicated on the figure, survival curves were analyzed by Log-rank test.
Syngeneic models
Unless otherwise indicated 2×106 KP or KPO cells (in 50% matrigel (Corning, Cat# 354230)) were implanted subcutaneously into 4–8-week-old C57B6/J (The Jackson Laboratory, Cat# 000644) mice to induce tumors in the flanks. For experiments with mixed tumor clones: 5×105 individual clones were mixed to obtain 2×106 total cancer cells. Both male and female mice were included in all studies with similar distribution and mice were randomized for the therapeutic experiments. Experiments were repeated at least twice. Width (shorter dimension) and length (longer dimension) of tumors were measured by digital caliper and volume was calculated using the following formula. Volume (mm3) = width (mm) x width (mm) x length (mm)/2. For intravenous (IV) model, 1×106 KP/KPO cells were injected by tail vein. 200μg of each of the anti-PD-L1 (Tecentriq) and anti-CTLA-4 (Bioxcell Cat# BE0164) antibodies and the isotype controls were administered by intraperitoneal (I.P.) injection 3 times a week where indicated. 50μg STING agonist was administrated by intra-tumoral injection on days 10, 14 and 17 after tumor inoculation.
RNA Extraction & qRT-PCR
RNA was extracted by Zymo Direct-zol RNA Miniprep (Cat# R2051). 1μg of total RNA was reverse transcribed to cDNA by Applied Biosystems TaqMan High-Capacity RNA-to-cDNA Kit (fisher, Cat# 43–874-06) in 20μl system. 1μl product was used for qPCR in 10μl system by Taqman mastermix (Thermofisher Cat# 4369016). Amplification was assessed by QuantStudio 3 Real-Time PCR System.
Western Blotting
Homogenized mouse tissues and pelleted cells were lysed by RIPA buffer supplemented with proteinase/phosphatase inhibitor cocktail (Cell Signaling Technology Cat# 5872S). Protein concentration was quantified by Thermo Scientific™ Pierce™ BCA™ Protein Assay (Fisher Scientific Cat# PI23225). Then 30μg protein was loaded to precast gel (Thermo Fisher Cat# NP0326BOX) and ran at 120V for 2h. Later protein was transferred to PVDF membrane and blocked with 5% skimmed milk in TBST for 1h. Primary antibodies were diluted at 1:1000 ratio in skimmed milk and incubated with the membrane at 4°C overnight. Membranes were washed 3 times,10 minutes with TBST. Then incubated with appropriate secondary antibodies at 1:10000 ratio in skimmed milk or 2% BSA. The membranes were washed again 3 times for 10 minutes with TBST. ECL reagents were added to the membrane and blot was developed by ChemiDoc Imaging System.
Immunofluorescence
Cells were grown on chamber slides (ThermoFisher #177380), fixed with 4% PFA for 20min at RT. Then, permeabilized with PBS+0.1% TritonX-100. Cells were washed with PBS once and blocked with 0.1% BSA in PBS for 1h at RT. Cells were incubated with primary antibody diluted in PBS with 0.1% BSA at 1:500) for 1h at RT. Cells were washed with PBS once and incubated with secondary antibody (1:1000 diluted in PBS with 0.1% BSA) for 1h in the dark. Cells were washed with PBS once and incubated with DAPI at 1μg/ml for 5min at RT. After staining, cells were washed with PBS once and mounted. Stained slides were visualized under fluorescent microscope.
Hematoxylin and Eosin (H&E) Staining
Mouse tissues were fixed with 10% formalin for 48h and submitted to tissue management core for paraffin embedding. Paraffin blocks were sectioned at 5μm thickness by microtome. Sections were transferred from the following solutions with indicated times. Xylene(5min)-Xylene(5min) -Xylene(5min)-100% Ethanol(30s) −100% Ethanol(30s)-100% Ethanol(30s)-95%Ethanol(30s)-Water(1min)-Hematoxylin(7min)-Water(1min)-Water(1min)-HCl Water(0.1% HCl, 7s)-Water(1min)-Bluing solution(15s)-Water(1min) −95% Ethanol(1min)-Eosin Y(10s)-95% Ethanol(1min)-95% Ethanol(30s) −100% Ethanol(30s)-100% Ethanol(30s)-100% Ethanol(30s)-Xylene(1min)-Xylene(1min). After staining, the slides were air dried and covered.
Immunohistochemistry
Slides were hydrated by soaking in xylene for 10min twice, hydrated in 100% ethanol for 3min twice, then in 95% ethanol for 3min twice, and rinsed in water for 3min once. Tissues were incubated in gently boiling 35mM sodium citrate solution for 15min for antigen retrieval. Slides were cooled down to RT and blocked with 3% H2O2 for 30min. Slides were blocked with 1% BSA in PBS for 1min at RT. Primary antibody (Cell signaling Technology, γH2A.X, Cat# 9718S, STING, Cat# 13647, Thermo Fisher, Ki67, Cat# MA5–14520) were diluted in PBS with 2% BSA at 1:500 ratio and incubated with the slides at RT for 1h. Slides were washed with TBST for 5 minutes 4 times. Slides were incubated with secondary antibodies ( ImmPRESS HRP Goat Anti-Rabbit IgG Polymer Detection Kit (Vector Lab, Cat# MP-7451) 1hr at RTs. Slides were then washed with TBST for 5 minutes 4 times. DAB Substrate, Peroxidase (Vector Lab, Cat# SK-4105) was applied and color development was observed. Slides were counterstained with hematoxylin, washed under tap water, air dried, and cover slipped for visualization.
Flow cytometry analysis
Human NSCLC lines were treated with 20μM Nutlin-3a for 48h. Cell lines were first stained with fixable live/dead cell stain (Fisher Cat# 50–112-1528) RT for 8min to gate the live cells. Then they were stained with fluorophore conjugated antibodies (in FACS buffer) for 20min on ice. Samples were washed with FACS buffer (2% FBS in PBS). Stained samples were analyzed using BDFACS Canto. Data was analyzed using FlowJo. 10ng/ml IFNγ was incubated with cells to induce MHC1 expression where indicated.
Mouse tissues were minced and digested at 37°C for 1h (100 units/ml collagenase Fisher Cat# 17104019, 10μg/ml DNase I Sigma Cat# DN25, 10% heat-inactivated FBS in RPMI) to dissociate cells. Red blood cells were lysed in the dissociated tissue with ACK lysis buffer (Fisher Cat# A1049201). Following lysis, tissues were passed through a 70μm cell strainer to generate single cell suspension. Cells were stained with fixable live/dead cell stain (Molecular probes #L34959) at room temperature for 8 min. Cells were incubated with CD16/32 antibody (Biolegend, Cat# 101320) for 20min on ice. Next, they were incubated with fluorophore conjugated antibodies diluted in FACS buffer for 20min on ice to stain for surface markers. Samples were then washed with FACS buffer after fixatives or antibodies. Intracellular staining protocol used for intracellular markers.
Intracellular staining was performed using eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermofisher, Cat# 00–5523-00). Samples were permeabilized with fixation/permeabilization buffer at 4°C overnight. Then they were incubated with fluorophore conjugated antibodies (IFNγ, Ki67, granzyme) diluted in FACS buffer for 20min on ice. Samples were washed with permeabilization buffer after every incubation with antibody. All antibodies were diluted in permeabilization buffer.
To determine T cell activation lymphocytes were enriched using Ficoll-Paque (GE healthcare) following the protocol. Enriched samples were incubated with PMA/ionomycin/Golgi plug for ex vivo stimulation for 6 hours. Cells were then first stained with surface markers (for lineage markers) then intracellular markers (for IFNγ, Ki67 and Granzyme b) as detailed above. Samples were run on BDFACS Canto, and flow data was analyzed using FlowJo.
Whole Exome Sequencing (WES)
Genomic DNA was extracted from tumor tissues or cells using Qiagen DNA isolation kit (69504) following the kit instructions. DNA concentrations were quantified and then shipped to BGI Americas for sequencing on dry ice. 1μg genomic DNA was fragmented by Covaris. Fragment with average size between 150–250bp was purified by magnetic beads. The fragments were subjected to end-repair and then was 3’ adenylated. Adapters were ligated to the ends of these 3’ adenylated fragments. Then fragments with adaptors were amplified by PCR and purified by Agencourt AMPure XP-Medium kit (Bechman, Cat# A63882). PCR products were used for hybridization to capture exomes. Agilent SureSelect XT Mouse All Exon Kit 50M was used for target enrichment. The captured fragments were amplified by PCR and recovered by the Agencourt AMPure XP-Medium kit. The double stranded PCR products were heat denatured and circularized by the splint oligo sequence. The single strand circle DNA (ssCir DNA) were formatted as the final library. The library was qualified by Qubit ssDNA kit. The library was amplified to make DNA nanoball (DNB) which have more than 300 copies of one molecular. The DNBs were loaded into the patterned nanoarray, and pair-end 100 bases reads were generated in the way of sequenced by combinatorial Probe-Anchor Synthesis (cPAS).
Whole exome sequencing (WES) data analysis
Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used for quality and adapter trimming. The mouse reference genome sequence and gene annotation data, mm10, were downloaded from Illumina iGenomes (https://support.illumina.com/sequencing/sequencing_software/igenome.html). The sequencing reads were aligned to the reference genome sequence using Burrows-Wheeler Aligner (BWA, v0.7.17)27. Picard (2.21.3) (https://broadinstitute.github.io/picard) was used to remove PCR duplicates and Genome Analysis Toolkit (GATK, 4.1.4.0)28,29 was used to recalibrate base qualities. Calling variants and genotyping were performed using GATK HaplotypeCaller and low-quality variant calls were excluded by the following filtering thresholds: QD (Variant Confidence/Quality by Depth) < 2, FS (Phred-scaled p-value using Fisher’s exact test to detect strand bias) > 60, MQ (RMS Mapping Quality) < 40, DP (Approximate read depth) < 3, GQ (Genotype Quality) < 7. Custom Perl scripts (https://github.com/jiwoongbio/Annomen) were used to annotate variants with RefSeq30 mouse transcripts and proteins, mitochondrial genes (NC_012920.1), dbSNP (build 151)31. The RefSeq and GenBank genome sequences of mouse strains were downloaded from NCBI FTP and were aligned to the reference genome sequence using Minimap2 (v2.24-r1122)32. SAMtools (v1.9)33 was employed to sort the genome alignments and convert them to pileup format. A custom Perl script was used to extract mouse strain variants from the pileup format. Somatic mutations were identified by GATK Mutect2 using multiple mouse tail samples as multiple normal control samples. The variants and somatic mutations outside the exome-capture regions were excluded. The dbSNP and mouse strain variants were excluded from the somatic mutations. The mutational burdens were calculated by the numbers of filtered non- synonymous somatic mutations divided by the length of the exome-capture regions. Jaccard distances between samples were calculated by variants on target regions and used to perform hierarchical clustering of samples using R. MATH (Mutant-Allele Tumor Heterogeneity) score was calculated according to pervious publication 34
Analysis of patient datasets:
Data for MSKCC35 and AACR-Genie were downloaded from cBioPortal, Stand Up to Cancer Dataset was from the article 36. Samples carrying frameshift, nonsense, and frameshift-nonsense or nonsense-missense mutations were included in the truncating mutation category. Missense mutations included samples carrying missense mutations or missense mutations and all other mutations. Only samples with WT STK11/KEAP1 status were included in the analysis. Unless otherwise indicated on the figure, survival curves were analyzed by Log-rank test. There are overlapping samples in between the DFCI-SU2C-Genie and MSKCC-SU2C-Genie datasets that we could not exclude due to not being able to identify them.
Magnetic resonance imaging (MRI)
Mice were under anesthesia with 2.5% isoflurane, 5% oxygen air flow. Breathing and heart rate were monitored during the procedure. Images were acquired by the 3 Tesla MRS 3017 system. Mice were transferred to 38°C heating pad for recovery after the procedure. Tumor volume was quantified by the 3D slicer software by drawing the area covered by the tumor in subsequent slices. Volume obtained from the software was used in the graphs.
Tumor area quantification from the H&E sections
Total lung areas in H&E images were calculated through image J by adjusting threshold function to cover the whole lung areas. Tumor areas in H&E images were calculated through image J by adjusting threshold function to cover the tumor areas. Tumor area percentage was calculated by normalizing tumor area with whole lung area. For immunohistochemistry, positive staining was quantified using image J IHC toolbox plugin in.
Stable expression cell lines:
Mouse Trp53 cDNA was purchased from Genscript (Cat# OMu22847). Trp53 fragment was cloned from plasmids and cloned into pLVX-Tight-puro from Lenti-X Tet-On Advanced inducible expression system (Clontech, Cat#632162). P53 mutant (R172H, R270H) plasmids were generated through QuikChange Lightning Site-Directed Mutagenesis Kit (Aligent Cat#210518). Ligated plasmid (pLVX-Tight-puro-Trp53) was selected on Luria-Bertani (LB) agar plate and then validated by sanger sequencing. pLVX-Tet-On Advanced and pLVX-Tight-puro-p53 were individually co-transfected with pMD2.G (Addgene Cat# 12259) and psPAX (Addgene Cat# 12260) to HEK293FT to package lentivirus. Then collected virus medium was combined supplemented with 8μg/ml polybrene to infect KP or KPO cell lines. Later infected cells were selected with 4μg/ml puromycin and 200μg/ml geneticin for 7d. Established doxycycline-inducible stable cell lines were cultured with 1μg/ml doxycycline for 48h to induce p53 expression for downstream analysis. p53 expression was confirmed by western blotting. For in vivo study, 2mg doxycycline or PD-L1+CTLA-4 antibody cocktail (100μg or 200μg of each as indicated in the figure) were i.p. administrated into mice 3 times per week. The last dose of doxycycline was given 24 hours before euthanasia.
STING agonist nanoparticle formulation
PEG-b-PC7A block copolymer was synthesized and purified in accordance with previously reported method 37. 2’3’-cGAMP was purchased commercially (Invivogen Cat# tlrl-nacga23). For polySTING formulation, the polymer was dissolved in methanol at 4 mg/mL. A 2 mg/mL stock of 2’3’-cGAMP in H2O was added to the polymer solution at a ten-fold dilution. The input loading capacity is 5% (wt. polymer/wt. cGAMP). The solution was added dropwise to an eight-fold excess of H2O under sonication to induce micelle self-assembly. Methanol and free 2’3’-cGAMP were removed with consecutive water washes under centrifugation in a 100 kDa centrifugal filter unit (Millipore Cat# UFC910024) according to the manufacturer’s specifications. The supernatant was diluted to 10 mg/mL in filtered H2O and centrifuged at 10 krcf to remove residual aggregates. The resulting nanoparticle was characterized with dynamic light scattering for size (number mean diameter = 27.4 ± 1.9 nm; n = 3) and HPLC for encapsulation efficiency of cGAMP loading (efficiency = 61.5 ± 0.9%; n =2).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 9 and R (4.3.1) were used for statistical analysis. Specific statistical details for graphs can be found in figure legends. All centers in this graph in this manuscript represent mean of the data. Unpaired t tests and log-rank tests were used to identify statistical differences unless otherwise specified.
Supplementary Material
Video S1.
p53 induces expression of MHC1 and T cell cytotoxicity against cancer cells
Significance of Findings:
Zhu et al, generated an autochthonous lung GEMM with high TMB using a hypermutator Pole allele. Data from these models and patient samples suggest that p53 status and mutational heterogeneity may influence clinical outcome. STING or p53 can be activated in these polyclonal tumors to promote anti-tumor immunity.
Highlights:
Oncogenotype influences TMB induced immunogenicity of tumors.
Genetic or pharmacological p53 induction stimulates antigen presentation.
Single cell clones derived from high TMB GEMM tumors are highly immunogenic.
Shared mutations are protective and provide protective immunity.
Acknowledgements:
We would like to thank Drs David Gerber, Shohei Koyama, Ralf Kittler, and James Malter for helpful discussions; Dr Nan Yan for sharing OT-1 mice; Dr James Chen for sharing Sting knockout mice; and Drs Yang-Xin Fu and Jian Qiao for the OVA plasmid. We thank Daniella Yang for help with mouse colonies and Jade Lee for administrative assistance. We thank UTSW Tissue Management Shared resource for tumor tissue embedding and sectioning (P30CA142543).
Grant support:
EAA is a Cancer Prevention and Research Institute of Texas (CPRIT) Scholar in Cancer Research. This work was supported by CPRIT Scholar Award RR160080, Department of Defense W81XWH-21-1-0856, Welch Foundation grant (1975-20190330), A Breath of Hope Lung Foundation Fellowship Award (ABOHLF 2020), NCCN Foundation Young Investigator Award (NCCN 2021), American Cancer Society Research Scholar Award RSG-22-051-01-IBCD, and Mary Kay Ash Foundation grants to EAA, NIH 5P50CA070907 to JDM and EAA, and R01CA237405 and R01CA265884 to DHC. Mouse Preclinical MRI Core facility was supported by CPRIT (RP210099), and Preclinical Radiation Core Facility where some of the MRI was performed was supported by CPRIT (RP180770).
Declaration of Conflict of Interests:
JG has patents related to this work and is scientific co-founder and advisor of OncoNano Medicine, Inc. JDM receives licensing fees from the NCI and UT Southwestern to distribute cell lines. JVA receives Advisory Board fee from BMS. BR served as consultant/advisory board for Regeneron, AMGEN, and AstraZeneca and received honoraria from Targeted Oncology. MMA reports serving as a consultant to Merck, Bristol-Myers Squibb, Genentech, AstraZeneca, Nektar, Maverick, Blueprint Medicine, Syndax, AbbVie, Gritstone, ArcherDX, Mirati, NextCure, and EMD Serono; receiving research funding from Bristol-Myers Squibb, Eli Lilly, Genentech, and AstraZeneca.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Siegel RL, Miller KD, Fuchs HE, and Jemal A. (2022). Cancer statistics, 2022. CA Cancer J Clin 72, 7–33. 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Chae Y., Arya A., Iams W., Cruz M., Chandra S., Choi J., and Giles F. (2018). Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). Journal For Immunotherapy of Cancer 6, ARTN 39. 10.1186/s40425-018-0349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378, 2078–2092. 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 4.Lee S, Shim HS, Ahn BC, Lim SM, Kim HR, Cho BC, and Hong MH (2021). Efficacy and safety of atezolizumab, in combination with etoposide and carboplatin regimen, in the first-line treatment of extensive-stage small-cell lung cancer: a single-center experience. Cancer Immunol Immunother. 10.1007/s00262-021-03052-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puri S, and Shafique M. (2020). Combination checkpoint inhibitors for treatment of non-small-cell lung cancer: an update on dual anti-CTLA-4 and anti-PD-1/PD-L1 therapies. Drugs Context 9. 10.7573/dic.2019-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. (2021). Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer WithPD-L1 Tumor Proportion Score >/= 50. J Clin Oncol 39, 2339–2349. 10.1200/JCO.21.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Castro G Jr., Kudaba I, Wu YL, Lopes G, Kowalski DM, Turna HZ, Caglevic C, Zhang L, Karaszewska B, Laktionov KK, et al. (2023). Five-Year Outcomes With Pembrolizumab Versus Chemotherapy as First-Line Therapy in Patients With Non-Small-Cell Lung Cancer and Programmed Death Ligand-1 Tumor Proportion Score >/= 1% in the KEYNOTE-042 Study. J Clin Oncol 41, 1986–1991. 10.1200/JCO.21.02885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Tong Z, Zhang W, Zhang W, Buzdin A, Mu X, Yan Q, Zhao X, Chang H, Duhon M, et al. (2021). FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Frontiers in Oncology 11, ARTN 683419. 10.3389/fonc.2021.683419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, Reeser JW, Yu L, and Roychowdhury S. (2017). Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis Oncol 2017. 10.1200/PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Shih D, and Lin S. (2020). Role of DNA repair defects in predicting immunotherapy response. Biomarker Research 8, ARTN 23. 10.1186/s40364-020-00202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, Stephens D, Zhang H, Zhu J, Yang M, et al. (2021). DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 39, 96-+. 10.1016/j.ccell.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kane DP., and Shcherbakova PV. (2014). A common cancer-associated DNA polymerase epsilon mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res 74, 1895–1901. 10.1158/0008-5472.CAN-13-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y, Zheng Y, Wang PP, Chen YY, and Ding ZY (2022). Immunotherapy for a POLE Mutation Advanced Non-Small-Cell Lung Cancer Patient. Front Pharmacol 13, 817265. 10.3389/fphar.2022.817265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L, Ruiz J, O’Neill SS, Grant SC, Petty WJ, Yang M, Chen K, Topaloglu U, Pasche B, and Zhang W. (2018). Favorable outcome of patients with lung adenocarcinoma harboring POLE mutations and expressing high PD-L1. Mol Cancer 17, 81. 10.1186/s12943-018-0832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X, Dong L, Liu X, Ou K, and Yang L. (2022). POLE/POLD1 mutation and tumor immunotherapy. J Exp Clin Cancer Res 41, 216. 10.1186/s13046-022-02422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 17.Hellmann M, Nathanson T, Rizvi H, Creelan B, Sanchez-Vega F, Ahuja A, Ni A, Novik J, Mangarin L, Abu-Akeel M, et al. (2018). Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 33, 843-+. 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGrail DJ, Pilie PG, Rashid NU, Voorwerk L, Slagter M, Kok M, Jonasch E, Khasraw M, Heimberger AB, Lim B, et al. (2021). High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol 32, 661–672. 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal C, Thompson JC, Chien AL, Quinn KJ, Hwang WT, Black TA, Yee SS, Christensen TE, LaRiviere MJ, Silva BA, et al. (2020). Baseline Plasma Tumor Mutation Burden Predicts Response to Pembrolizumab-based Therapy in Patients with Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res 26, 2354–2361. 10.1158/1078-0432.CCR-19-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. (2018). STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 8, 822–835. 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. (2016). Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A 113, E6409-E6417. 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, Vlahos R, Grondine M, Dolgalev I, Ivanova EV, et al. (2018). BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol Res 6, 1234–1245. 10.1158/2326-6066.CIR-18-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akbay EA., Koyama S., Liu Y., Dries R., Bufe LE., Silkes M., Alam MM., Magee DM., Jones R., Jinushi M., et al. (2017). Interleukin-17A Promotes Lung Tumor Progression Through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J Thorac Oncol. 10.1016/j.jtho.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li HD, Cuevas I, Zhang M, Lu C, Alam MM, Fu YX, You MJ, Akbay EA, Zhang H, and Castrillon DH (2018). Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J Clin Invest 128, 4179–4191. 10.1172/JCI122095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo M. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, Philippakis A, del Angel G, Rivas M, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics 43, 491−+. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Leary N, Wright M, Brister J, Ciufo S, McVeigh D, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research 44, D733–D745. 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherry S, Ward M, and Sirotkin K. (1999). dbSNP - Database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Research 9, 677–679. [PubMed] [Google Scholar]
- 32.Li H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD, and Proc GPD (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mroz EA, Tward AD, Hammon RJ, Ren Y, and Rocco JW (2015). Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med 12, e1001786. 10.1371/journal.pmed.1001786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51, 202–206. 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravi A., Hellmann MD., Arniella MB., Holton M., Freeman SS., Naranbhai V., Stewart C., Leshchiner I., Kim J., Akiyama Y., et al. (2023). Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Nat Genet 55, 807–819. 10.1038/s41588-023-01355-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li S, Luo M, Wang Z, Feng Q, Wilhelm J, Wang X, Li W, Wang J, Cholka A, Fu YX, et al. (2021). Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng 5, 455–466. 10.1038/s41551-020-00675-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DuPage M, Dooley AL, and Jacks T. (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248. 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt D. (2020). Best practices for variant calling in clinical sequencing. Genome Medicine 12, ARTN 91. 10.1186/s13073-020-00791-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eischen CM (2016). Genome Stability Requires p53. Cold Spring Harb Perspect Med 6. 10.1101/cshperspect.a026096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams AB, and Schumacher B. (2016). p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 6. 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fei L, Ren X, Yu H, and Zhan Y. (2021). Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front Immunol 12, 771210. 10.3389/fimmu.2021.771210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shinbrot E, Henninger E, Weinhold N, Covington K, Goksenin A, Schultz N, Chao H, Doddapaneni H, Muzny D, Gibbs R, et al. (2014). Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Research 24, 1740–1750. 10.1101/gr.174789.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haradhvala N, Kim J, Maruvka Y, Polak P, Rosebrock D, Livitz D, Hess J, Leshchiner I, Kamburov A, Mouw K, et al. (2018). Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nature Communications 9, ARTN 1746. 10.1038/s41467-018-04002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexandrov L, Nik-Zainal S, Wedge D, Aparicio S, Behjati S, Biankin A, Bignell G, Bolli N, Borg A, Borresen-Dale A, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415−+. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. (2016). Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622. 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu K, Wang J, Zhu J, Jiang J, Shou J, and Chen X. (1999). p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 18, 7740–7747. 10.1038/sj.onc.1203235. [DOI] [PubMed] [Google Scholar]
- 49.Wang B, Niu D, Lai L, and Ren EC (2013). p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun 4, 2359. 10.1038/ncomms3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghandi M., Huang FW., Jane-Valbuena J., Kryukov GV., Lo CC., McDonald ER 3rd, Barretina J., Gelfand ET., Bielski CM., Li H., et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stieger K, Belbellaa B, Le Guiner C, Moullier P, and Rolling F. (2009). In vivo gene regulation using tetracycline-regulatable systems. Adv Drug Deliv Rev 61, 527–541. 10.1016/j.addr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, Stephens D, Zhang H, Zhu J, Yang M, et al. (2021). DNA Sensing in Mismatch Repair-Deficient Tumor Cells IsEssential for Anti-tumor Immunity. Cancer Cell 39, 96–108 e106. 10.1016/j.ccell.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang R, and Cui J. (2016). TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Molecular Cell 64, 105–119. 10.1016/j.molcel.2016.08.025. [DOI] [PubMed] [Google Scholar]
- 54.Prabakaran T, Bodda C, Krapp C, Zhang B, Christensen M, Sun C, Reinert L, Cai Y, Jensen S, Skouboe M, et al. (2018). Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. Embo Journal 37, ARTN e97858. 10.15252/embj.201797858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li HD, Lu C, Zhang H, Hu Q, Zhang J, Cuevas IC, Sahoo SS, Aguilar M, Maurais EG, Zhang S, et al. (2020). A PoleP286R mouse model of endometrial cancer recapitulates high mutational burden and immunotherapy response. JCI Insight 5. 10.1172/jci.insight.138829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kucherlapati MH, Lee K, Nguyen AA, Clark AB, Hou H Jr., Rosulek A, Li H, Yang K, Fan K, Lipkin M, et al. (2010). An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology 138, 993–1002 e1001. 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sender R, and Milo R. (2021). The distribution of cellular turnover in the human body. Nat Med 27, 45–48. 10.1038/s41591-020-01182-9. [DOI] [PubMed] [Google Scholar]
- 58.Cousins FL, Pandoy R, Jin S, and Gargett CE (2021). The Elusive Endometrial Epithelial Stem/Progenitor Cells. Front Cell Dev Biol 9, 640319. 10.3389/fcell.2021.640319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Usatorre A, Kadioglu E, Boivin G, Cianciaruso C, Guichard A, Torchia B, Zangger N, Nassiri S, Keklikoglou I, Schmittnaegel M, et al. (2021). Overcoming microenvironmental resistance to PD-1 blockade in genetically engineered lung cancer models. Sci Transl Med 13. 10.1126/scitranslmed.abd1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma X, Riaz N, Samstein RM, Lee M, Makarov V, Valero C, Chowell D, Kuo F, Hoen D, Fitzgerald CWR, et al. (2022). Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity. Nat Genet 54, 996–1012. 10.1038/s41588-022-01108-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin TD., Patel RS., Cook DR., Choi MY., Patil A., Liang AC., Li MZ., Haigis KM., and Elledge SJ. (2021). The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 373, 1327–1335. 10.1126/science.abg5784. [DOI] [PubMed] [Google Scholar]
- 62.Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, van der Velden AWM, and Martinez LA (2021). Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 39, 494–508 e495. 10.1016/j.ccell.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang F, Gao X, Wang P, He H, Chen P, Liu Z, Chen Y, Zhou H, Chen W, Yi X, et al. (2022). Immune Subtypes in LUAD Identify Novel Tumor Microenvironment Profiles With Prognostic and Therapeutic Implications. Front Immunol 13, 877896. 10.3389/fimmu.2022.877896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun H, Liu S, Zhou J, Xu J, Zhang H, Yan H, Huan J, Dai P, Xu C, Su J, et al. (2020). Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. Ebiomedicine 60, ARTN 102990. 10.1016/j.ebiom.2020.102990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Assoun S, Theou-Anton N, Nguenang M, Cazes A, Danel C, Abbar B, Pluvy J, Gounant V, Khalil A, Namour C, et al. (2019). Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer 132, 65–71. 10.1016/j.lungcan.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 66.Murnyak B, and Hortobagyi T. (2016). Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget 7, 64910–64920. 10.18632/oncotarget.11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahrendt SA, Hu Y, Buta M, McDermott MP, Benoit N, Yang SC, Wu L, and Sidransky D. (2003). p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J Natl Cancer Inst 95, 961–970. 10.1093/jnci/95.13.961. [DOI] [PubMed] [Google Scholar]
- 68.Chinnam M, Xu C, Lama R, Zhang X, Cedeno CD, Wang Y, Stablewski AB, Goodrich DW, and Wang X. (2022). MDM2 E3 ligase activity is essential for p53 regulation and cell cycle integrity. PLoS Genet 18, e1010171. 10.1371/journal.pgen.1010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Girish V, Lakhani AA, Scaduto CM, Thompson SL, Brown LM, Hagenson RA, Sausville EL, Mendelson BE, Lukow DA, Yuan ML, et al. (2023). Oncogene-like addiction to aneuploidy in human cancers. bioRxiv. 10.1101/2023.01.09.523344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hullein J, Slabicki M, Rosolowski M, Jethwa A, Habringer S, Tomska K, Kurilov R, Lu J, Scheinost S, Wagener R, et al. (2019). MDM4 Is Targeted by 1q Gain and Drives Disease in Burkitt Lymphoma. Cancer Res 79, 3125–3138. 10.1158/0008-5472.CAN-18-3438. [DOI] [PubMed] [Google Scholar]
- 71.Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, Levy R, Jiménez-Sánchez A, Trabish S, Lee JS, et al. (2019). UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 179, 219–235.e221. 10.1016/j.cell.2019.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1.
p53 induces expression of MHC1 and T cell cytotoxicity against cancer cells
Data Availability Statement
Whole exome sequencing (WES) data of mouse tissues have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 488 anti-mouse CD4 | Biolegend | 100423 |
| Alexa Fluor® 488 anti-mouse CD45 | Biolegend | 103122 |
| Alexa Fluor® 488 anti-mouse CD8a | Biolegend | 100723 |
| APC anti-mouse CD4 | Biolegend | 100516 |
| APC anti-mouse IL-2 | Biolegend | 503810 |
| APC Rat IgG2b, κ Isotype Ctrl | Biolegend | 400612 |
| APC/Cy7 anti-mouse CD45 | Biolegend | 103116 |
| APC/Cy7 anti-mouse CD8a | Biolegend | 100714 |
| Brilliant Violet 421™ anti-mouse CD19 | Biolegend | 115538 |
| Brilliant Violet 421™ anti-mouse Ki-67 | Biolegend | 652411 |
| PE/Cy7 anti-mouse CD326 (Ep-CAM) | Biolegend | 118216 |
| PE/Cy7 anti-mouse CD3ε | Biolegend | 100320 |
| PerCP/Cy5.5 anti-mouse CD335 (NKp46) | Biolegend | 137610 |
| Alexa Fluor® 488 Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400233 |
| PE Rat IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400636 |
| APC Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400220 |
| APC anti-human HLA-A, B, C Antibody | Biolegend | 311410 |
| APC anti-mouse CD335 (NKp46) Antibody | Biolegend | 137607 |
| PE anti-mouse H-2K b/H-2D b Antibody | Biolegend | 114607 |
| PE Mouse IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400212 |
| Brilliant Violet 421™ Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400341 |
| PE anti-mouse RAE-1δ Antibody | Biolegend | 133203 |
| PE Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400111 |
| APC Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400319 |
| APC Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400511 |
| APC Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400119 |
| PerCP/Cy5.5 anti-mouse CD8a Antibody | Biolegend | 100733 |
| APC anti-mouse IgG2a Antibody | Biolegend | 407110 |
| APC Rat IgG1, κ Isotype Ctrl Antibody | Biolegend | 400411 |
| Brilliant Violet 421™ Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400157 |
| PE/Cy7 Mouse IgG1, κ Isotype Ctrl Antibody | Biolegend | 400125 |
| PerCP/Cy5.5 anti-mouse CD3ε Antibody | Biolegend | 100328 |
| Alexa Fluor® 488 anti-mouse CD3ε Antibody | Biolegend | 100321 |
| PE anti-mouse CD4 Antibody | Biolegend | 100512 |
| APC anti-mouse CD107a (LAMP-1) Antibody | Biolegend | 121614 |
| PerCP/Cy5.5 anti-mouse CD107a (LAMP-1) Antibody | Biolegend | 121626 |
| Brilliant Violet 421™ Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400549 |
| PE anti-mouse IL-2 Antibody | Biolegend | 503808 |
| TruStain fcX™ (anti-mouse CD16/32) Antibody | Biolegend | 101320 |
| Brilliant Violet 421™ anti-mouse CD3ε Antibody | Biolegend | 100341 |
| PE Rat IgG1, κ Isotype Ctrl Antibody | Biolegend | 400408 |
| PE/Cy7 Mouse IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400326 |
| PE/Cy7 anti-mouse CD3ε Antibody | Biolegend | 100320 |
| PE Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400508 |
| PE anti-human HLA-A,B,C Antibody | Biolegend | 311406 |
| PE Mouse IgG2a, κ Isotype Ctrl (FC) Antibody | Biolegend | 400214 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | 114608 |
| Brilliant Violet 421™ anti-mouse IL-2 Antibody | Biolegend | 503826 |
| FITC anti-human/mouse Granzyme B Recombinant Antibody | Biolegend | 372206 |
| PerCP/Cyanine5.5 anti-mouse CD8a Antibody | Biolegend | 100734 |
| FITC Mouse IgG1, κ Isotype Ctrl (ICFC) Antibody | Biolegend | 400138 |
| PE anti-mouse NK-1.1 Antibody | Biolegend | 108708 |
| Alexa Fluor® 488 anti-mouse CD49b (pan-NK cells) Antibody | Biolegend | 108913 |
| APC anti-mouse CD49b (pan-NK cells) Antibody | Biolegend | 108910 |
| PE/Cy7 Rat IgG2a, κ Isotype Ctrl Antibody | Biolegend | 400522 |
| PE/Cy7 Rat IgG2b, κ Isotype Ctrl Antibody | Biolegend | 400617 |
| PE anti-mouse IFN-γ Antibody | Biolegend | 505808 |
| APC/Cyanine7 anti-mouse CD45 Antibody | Biolegend | 103116 |
| InVivoMAb rat IgG2b isotype control | BioXcell | BE0090 |
| InVivoMAb anti-mouse CD8α | BioXcell | BE0117 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXcell | BP0164 |
| p53 (1C12) Mouse mAb | Cell signaling Technology | 2524 |
| Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb | Cell signaling Technology | 9718 |
| Ki-67 Recombinant Rabbit Monoclonal Antibody (SP6) | Thermofisher | MA5–14520 |
| CD8α (C8/144B) Mouse mAb | Cell signaling Technology | 70306 |
| STING (D2P2F) Rabbit mAb | Cell signaling Technology | 13647 |
| Phospho-STING (Ser365) (D8F4W) Rabbit mAb | Cell signaling Technology | 72971 |
| Vinculin (E1E9V) XP® Rabbit mAb | Cell signaling Technology | 13901 |
| GAPDH (D16H11) XP® Rabbit mAb | Cell signaling Technology | 5174 |
| Phospho-TBK1/NAK (Ser172) (D52C2) XP® Rabbit mAb | Cell signaling Technology | 5483 |
| Caspase-3 Antibody | Cell signaling Technology | 9662 |
| DAPI | Cell signaling Technology | 4083 |
| Bacterial and virus strains | ||
| NEB Stable Competent E. coli | NEB | C3040H |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Nutlin-3a | MedChemExpress | HY-10029 |
| Recombinant Murine IL-2 | PeproTech | 212–12 |
| OVA Peptide (323–339) | Genscript | RP10610 |
| Collagenase, Type IV, powder | Themofisher | 17104019 |
| DNase I recombinant, RNase-free | Millipore | 4716728001 |
| Cell Activation Cocktail (with Brefeldin A) | Biolegend | 423304 |
| Critical commercial assays | ||
| CyQUANT™ LDH Cytotoxicity Assay | Invtrogen | C20300 |
| MojoSort™ Mouse CD8 T Cell Isolation Kit | Biolegend | 480007 |
| DNeasy Blood & Tissue Kits | Qiagen | 69504 |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 |
| RNeasy Mini Kit | Qiagen | 74104 |
| PowerUp™ SYBR™ Green Master Mix | Thermofisher | A25742 |
| Deposited data | ||
| Tumor mutational burden in genetically engineered-mouse model | SRA | PRJNA933600 |
| Experimental models: Cell lines | ||
| A549 | UTSW | Hamon Center for Therapeutic Oncology Research |
| H23 | UTSW | Hamon Center for Therapeutic Oncology Research |
| MC38 | Kerafast | ENH204-FP |
| KP9–1 | Akbay et al, 2017, JTO25 | |
| KP9–3 | Akbay et al, 2017, JTO25 | |
| KP67–1 | Generated in this study | |
| KPO24 | Generated in this study | |
| KPO24–1 | Generated in this study | |
| KPO24–2 | Generated in this study | |
| KPO24–3 | Generated in this study | |
| KPO24–4 | Generated in this study | |
| KPO105 | Generated in this study | |
| KPO24-TetOp-p53 | Generated in this study | |
| KPO105-TetOp-p53 | Generated in this study | |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| B6.129S6-Poletm1Dcas/J | Jackson Laboratory | 037051 |
| B6(Cg)-Krastm5Tyj/J | Jackson Laboratory | 023590 |
| B6.129S2-Trp53tm1Tyj/J | Jackson Laboratory | 002101 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | 003831 |
| Oligonucleotides | ||
| mouse Actb Forward primer: GCCCTGAGGCTCTTTTCCAG | sigma | |
| mouse Actb Reverse primer: TGCCACAGGATTCCATACCC |
sigma | |
| mouse Tap1 Forward primer: GCTGTTCAGGTCCTGCTCTC | sigma | |
| mouse Tap1 reverse primer: CACTGAGTGGAGAGCAAGGAG | sigma | |
| mouse Erap1 Forward primer: CGAGGACCTGTGGAATAGCATG | sigma | |
| mouse Erap1 reverse primer: CATCTACAACCTCCTGACGCCA | sigma | |
| Mouse Ccl2 Forward primer: CATCCACGTGTTGGCTCA |
||
| Mouse Ccl2 reverse primer: GATCATCTTGCTGGTGAATGAGT |
||
| Recombinant DNA | ||
| cDNA clone for Mus musculus transformation related protein 53 (Trp53), transcript variant 2, mRNA. | Genscript | NM_001127233.1 |
| Software and algorithms | ||
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| BD FACSAria™ III System | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-sorters/facsaria-iii |
| GraphPad Prism software 9.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| Image J | NIH | imagej.nih.gov/ij/download/ |
| 3D slicer | NIH | https://www.slicer.org |
| Incucyte Base Analysis Software | Sartorius | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-base-software |
| NDP.view2 | Hamamatsu | https://www.hamamatsu.com/eu/en/product/life-science-and-medical-systems/digital-slide-scanner/U12388–01.html |
| Trim Galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| Burrows-Wheeler Aligner (BWA, v0.7.17) | https://academic.oup.com/bioinformatics/article/25/14/1754/225615 | |
| Picard | https://broadinstitute.github.io/picard | |
| Genome Analysis Toolkit (GATK, 4.1.4.0) | https://www.nature.com/articles/ng.806 | |
| Minimap2 (v2.24-r1122) | https://academic.oup.com/bioinformatics/article/34/18/3094/4994778?login=true | |
| SAMtools (v1.9) | https://academic.oup.com/bioinformatics/article/25/16/2078/204688?login=true | |
| R | R foundation | https://www.r-project.org |