

Abstract
CRISPR-Cas associated transposons (CASTs) are natural RNA-directed transposition systems. We demonstrate that transposon protein TniQ plays a central role in promoting R-loop formation by RNA-guided DNA-targeting modules. TniQ residues proximal to CRISPR RNA (crRNA) are required for recognizing different crRNA categories, revealing an unappreciated role of TniQ to direct transposition into different classes of crRNA targets. To investigate adaptations allowing CAST elements to utilize attachment sites inaccessible to CRISPR-Cas surveillance complexes, we compared and contrasted PAM sequence requirements in both I-F3b CAST and I-F1 CRISPR-Cas systems. We identify specific amino acids that enable a wider range of PAM sequences to be accommodated in I-F3b CAST elements compared to I-F1 CRISPR-Cas, enabling CAST elements to access attachment sites as sequences drift and evade host surveillance. Together, this evidence points to the central role of TniQ in facilitating the acquisition of CRISPR effector complexes for RNA-guided DNA transposition.
eTOC blurb
Park et al. investigate the mechanisms of the DNA-recognition module from I-F3 CRISPR-associated transposons, Cascade-TniQ. They demonstrate that TniQ functions in target engagement and regulation of target site choice with Cascade components. Comparing PAM recognition of I-F3 Cascade with canonical CRISPR-Cas reveals the structural basis of PAM ambiguity.
Graphical Abstract
Introduction
CRISPR-associated transposons (CASTs) have garnered substantial interest due to their capacity to direct a single cargo DNA insertion at a pre-programmed position.1 These transposition systems can be highly complex: in addition to 3–4 conserved transposition genes, they encode one or more CRISPR-Cas domain proteins from independent CRISPR effector subtypes.2 The largest group of sequenced CAST elements derived from type I-F1 CRISPR-Cas systems, referred to as type I-F3 CAST elements. These fall primarily into two large branches, I-F3a and I-F3b, that exhibit differences in both CRISPR array configuration and regulation.3
Type I-F3 CASTs have evolved mechanisms to target transposition in two classes of target sites, akin to prototypic Tn7 and other elements within the larger Tn7-like transposon family: 1) mobile elements and 2) a conserved position in the bacterial chromosome adjacent to an essential gene known as the attachment site.2,4 These two pathways facilitate the horizontal transfer of the transposon to new bacterial hosts by parasitizing the mobilization capabilities of targeted plasmids and bacteriophage. All I-F3 CASTs use CRISPR RNAs (crRNAs) encoded within a standard array to target transposition into mobile elements, but have evolved different adaptations to direct transposition into the attachment site in the two major I-F3 branches. The I-F3a elements have two CRISPR arrays that are differentially regulated. One array is expressed only when the element enters a naïve host.3 This array encodes a single crRNA that recognizes a conserved chromosomal attachment site. A second array encodes crRNAs that primarily recognize mobile plasmids. The I-F3b elements maintain a single array, but the final crRNA encoded in the array is functionally different, possessing a repeat sequence that is different from the other repeats in the array, called an atypical crRNA. The sequence differences between typical and atypical crRNA repeats have been described previously.3 The atypical crRNAs are found with spacers that recognize a conserved chromosomal attachment site, which in the I-F3b branch corresponds to the ffs gene encoding the RNA component of the signal recognition particle protein translocation system. The I-F3b effector complex recognizes both typical and atypical crRNAs, but an unknown mechanism allows a higher frequency of transposition with atypical crRNAs. This process therefore allows crRNAs to be categorized by favoring transposition into the ffs attachment site in the chromosome.
The mechanistic basis of this ability to discriminate between crRNAs is currently unknown, nor which CRISPR-Cas or transposition proteins might serve to discriminate between crRNA categories. To address this question, we focused our attention on components known to contact the crRNA, Cascade components, and TniQ. Previous cryo-EM studies of a I-F3a element revealed a direct physical association between the CRISPR effector (referred to throughout as the Cascade complex) and conserved transposon protein TniQ,5 which serves to recruit downstream transposition machinery. TniQ interacts with a AAA+ regulator, TnsC, to recruit the heteromeric transposase, TnsA and TnsB.4 All target-bound Cascade-TniQ complexes from I-F3 CAST systems examined to date exhibit incomplete R-loop formation,5–7 even when reconstituted with DNA substrates designed to promote R-loop formation.8 Therefore, it remained unclear if additional transposition components or unknown host factors were required for full R-loop formation and how they might engage the effector complex.
To investigate the molecular adaptations associated with CAST element evolution as well as mechanistically characterize the initial stages of CAST transposition, we examined the structure of DNA-bound type I-F3b CRISPR-effector complex, Cascade-TniQ. Our work reveals multiple previously unappreciated roles for TniQ in licensing transposition, acting to distinguish between typical and atypical crRNA categories, and as a platform for recruitment of downstream transposition components (akin to TnsD from prototypic Tn7, see discussion). Finally, we characterize mechanisms allowing I-F3b CAST elements to develop PAM ambiguity for host immune surveillance escape and to tolerate diversification of attachment sites recognized by the system.
Results
Cryo-EM reveals structural requirements for full R-loop engagement
We used cryo-electron microscopy (cryo-EM) to structurally characterize the I-F3b element (Tn6900) Cascade complex (including proteins Cas6, Cas7, and Cas8/5 in yellow, green, and purple shown in Figure 1A) associated with the TniQ transposition protein (pink) and target DNA (blue, Figure 1A&S1). The overall architecture of the I-F3b Cascade-TniQ complex closely matches previously published I-F3a cryo-EM structures.5 As a result of the conformational dynamics present within the complex, 3D reconstruction of the particle stack before classification (overall assessed to be 3.2 Å average resolution) contained comparatively weak TniQ density and variable local resolution throughout the structure (Figure S1). Nevertheless, we were able to capture two class averages, one with a full R-loop (Figure 1A-B) and one with partial R-loop, in which incomplete target DNA density is observed at the PAM-distal end of the complex (Figure 1C-D). The full R- loop structure showed cryo-EM density for the entire crRNA-hybridized 32-bp target strand (Figure 1B), as well as the density for the PAM-distal DNA (Figure 1A). On the other hand, the partial R-loop structure visualized only 28 bp of the target strand annealed with crRNA (Figure 1D). 3D variability analysis (3DVA)9 revealed that the dominant motion (i.e. separable using the first eigenvector) is the result of breathing motions in the complex separating partially-bound target DNA and fully engaged R-loop conformations (See Video S1). Full R-loop formation is accompanied by a 90 degrees rotation of the Cas8/5 fused helix bundle domain (red asterisk, Figure 1A, Video S1); a domain missing from prior I-F3a structures5 which more closely resembles the partial R-loop structure (Figure 1C). This rotation is correlated with a slight expansion of the Cascade complex (Video S1). The observed conformational change is reminiscent of a similar conformational change in type I-F1 Cascade, in which the helical-bundle of Cas8f rotates roughly 180 degrees upon target DNA binding.10 The conformational change we observe in the I-F3b particle population is tightly correlated with R-loop formation, which suggests that this is required prior to recruitment of downstream transposition factors (i.e., TnsC).
Figure 1. High-resolution cryo-EM reveals full R-loop and partial R-loop states of the DNA-bound Cascade-TniQ complex.
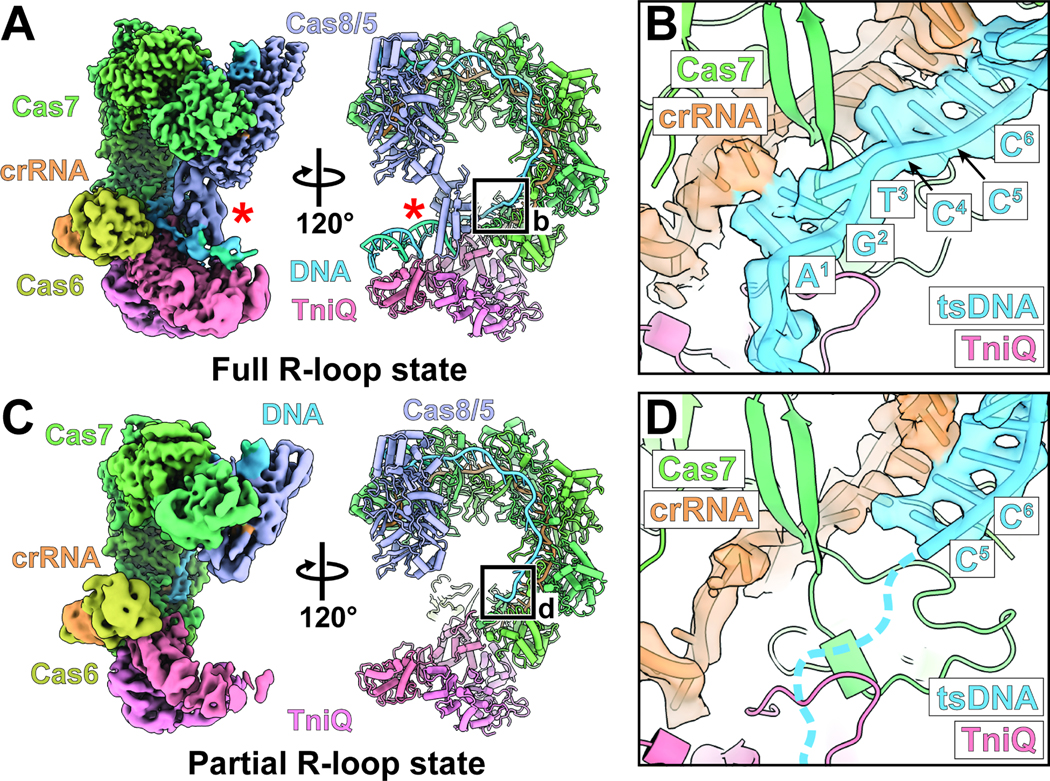
(A) Cryo-EM reconstructions and atomic model of the Cascade-TniQ complex in the full R-loop state (fully engaged RNA-DNA hybrid). The complex includes the following components: Cas8/5 (purple), Cas7 (green), Cas6 (olive), CRISPR RNA (crRNA, orange), TniQ (pink), and target DNA (blue). Red asterisks (*) indicate the Cas8/5 helix bundle domain. Black box defines the region for the close-up view in panel B. Local-resolution filtered maps from the focused refinements were combined for visualization (see Materials and Methods).
(B) Close-up view of the cryo-EM density and the atomic model of crRNA and target-strand DNA (tsDNA) from the full R-loop state conformation. The cryo-EM reconstruction visualizes the full engagement of tsDNA to crRNA.
(C) Cryo-EM structure of the Cascade-TniQ complex in the partial R-loop state. Color scheme is identical to the panel A. Black box indicates the region for detailed view in the panel D.
(D) Close-up view of PAM distal region of the R-loop from the cryo-EM structure of partial R-loop state. Base pair positions one through four are not resolved in the cryo-EM reconstruction of the partial R-loop conformation, indicated with dashed lines.
It was previously suggested that Cascade-TniQ might require other transposition components (i.e. TnsABC) for complete R-loop formation,5 Although we can’t rule out the possibility that another factor is required to stabilize association as shown in other Tn7-like transposons11 including the V-K CAST system,12 the structure presented implies that Cascade-TniQ itself is sufficient to form a full R-loop without additional host factor or transposition components. PAM-distal duplex DNA was not observed in previously determined I-F1 CRISPR-Cas structures,10 suggesting that, in this case, TniQ may serve as an additional platform in this system for stabilizing the distal end of the R-loop preparing the DNA substrate for transposition initiation. Although TniQ is associated with Cascade as a homodimer, only one monomer of TniQ (TniQ.1) associates with target DNA, which follows a path roughly parallel to the dimerization interface of TniQ (Figure 2A-B). Another TniQ subunit (TniQ.2) does not engage with the target DNA, potentially serving a structural role in the complex. The TniQ surface in contact with DNA is highly basic (Figure 2B) and has a footprint that spans 18 base pairs (Figure S2A). We also observed distortion in the PAM distal DNA corresponding to a slight lengthening and duplex DNA unwinding (Figure S2B). Though the local resolution of this region is slightly worse (5–6 Å) compared to the average resolution of the cryo-EM map (3.5 Å, Figure S1), the modeled substrate fits the cryo-EM map substantially better than an ideal B-form DNA model (cross-correlation is 0.71 vs 0.22 for distorted compared to ideal DNA). The DNA distortion is further accompanied by unwinding of approximately 3 base pairs past the expected protospacer region (colored red, Figure 2C), modeling which is supported by the model-map FSC (3.5 vs 3.7 Å for the gold-standard versus model-map FSC, respectively).
Figure 2. PAM distal end of target DNA in full R-loop state is further unwound, interacting with TniQ and Cas8/5 helix bundle domain.
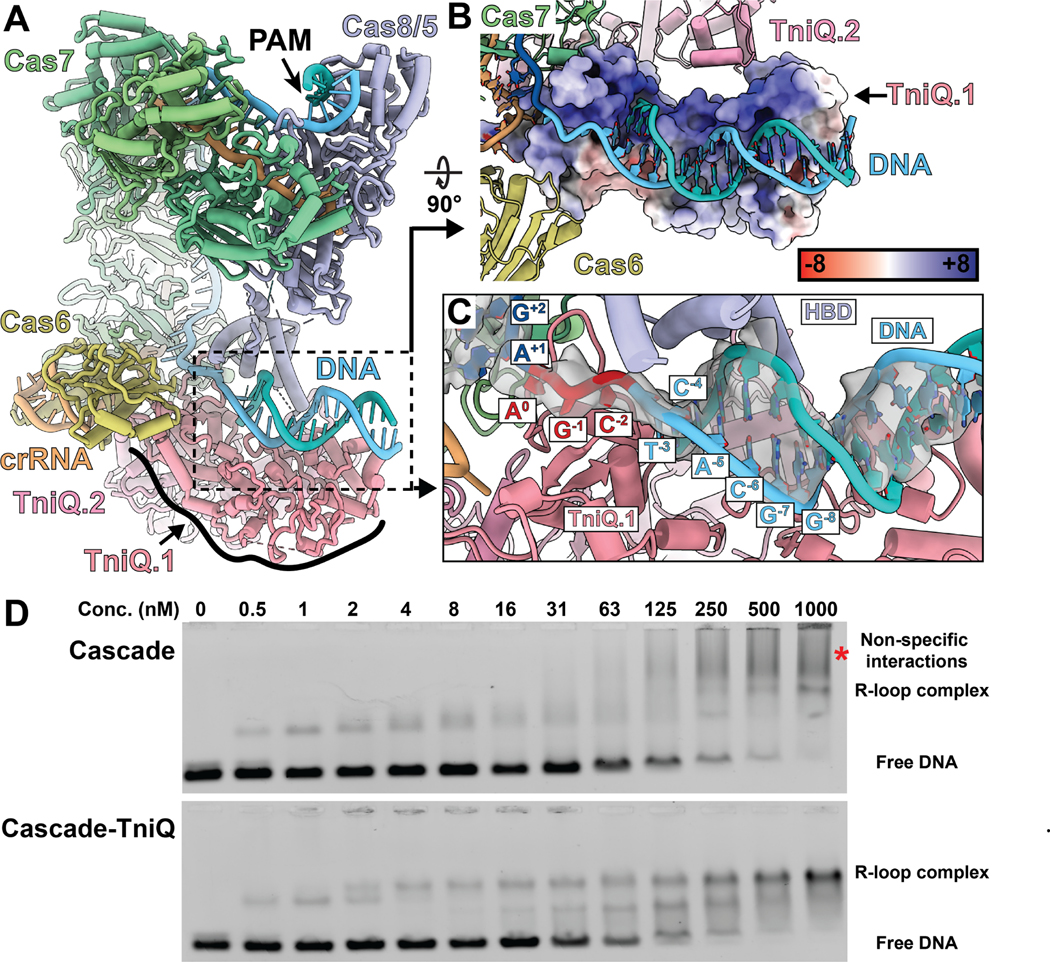
(A) Atomic model of a full R-loop formed Cascade-TniQ complex, shown for reference. The color scheme is consistent with Figure 1A. TniQ is distinguished by numbers (TniQ.1 or TniQ.2) to indicate which subunit interacts with the target DNA.
(B) One TniQ subunit (TniQ.1, surface representation) interacts with the PAM-distal DNA duplex (blue). DNA binding region of TniQ surface is positively charged, represented by the positive electrostatic potential. Legend indicates the color key for the dimensionless electrostatic potential calculated by APBS.
(C) The PAM-distal target DNA is unwound by additional three base pairs (red) following the protospacer (dark blue). Downstream double-stranded DNA interacts with the Cas8/5 helix-bundle domain (HBD, purple) and TniQ.1 subunit.
(D) Electrophoretic mobility shift assay (EMSA) reveals that TniQ promotes R-loop formation of Cascade with target DNA. At low concentrations., Cascade without TniQ shifts the DNA in a similar manner as Cascade-TniQ, but it forms a smeary band at high concentrations (≥ 250 nM, indicated with a red asterisk). On the other hand, Cascade associated with TniQ forms discrete bands in EMSA. The binding configurations that correspond to the band positions are indicated on the right of the gel image.
We propose a model in which the Cascade-TniQ complex presents target DNA for TnsC recruitment. Based on these structural observations (Figure 2A-C), we reasoned that TniQ may have a second, previously unappreciated role in stabilizing R-loop formation. To test this hypothesis, we probed the extent of R-loop formation by comparing the DNA binding of purified Cascade and Cascade-TniQ complexes using electrophoretic mobility shift assays (EMSA) (Figure 2D&S3A). We titrated fluorescently labeled target DNA (0.5 nM) with Cascade or Cascade-TniQ complex. We found similarly shifted products at low concentrations of Cascade or Cascade-TniQ (< 2 nM), suggesting that the initial DNA engagement with both complexes may be similar. However, at high concentrations of Cascade, most of the DNA substrate shifts to slower-migrating, smeared bands, suggesting non-specific binding (most prominent at 1000 nM, red asterisk on Figure 2D). Conversely, increasing concentrations of Cascade-TniQ complex exhibited substantially less non-specific binding than Cascade alone, instead resulting in two additional bands (Figure 2D). This suggests that different DNA-bound conformational states are present. To determine which band represents the full R-loop state, we conducted additional EMSA using pre-bubbled target DNA containing mismatched bases over the 32 bp protospacer sequence (Figure S3B). We reasoned that this pre-bubbled substrate would facilitate R-loop formation by reducing the energy required to melt the double-stranded DNA. We found that while the Cascade-TniQ resulted in both upper and lower bands with double-stranded target DNA, it formed only the upper band with pre-bubbled target DNA. This suggests that the upper band represents the full R-loop conformation. We speculate that the lower band may represent a partial R-loop state or another conformational state we have not captured in our cryo-EM analysis. Taken together, the EMSA data support the cryo-EM findings indicating that TniQ is required to allow the I-F3 Cascade to form a stable R-loop product.
TniQ is responsible for crRNA category discrimination
Type I-F Cascade engages with a 60 bp crRNA, including a 32 bp spacer-derived sequence (orange, Figure 3A-B) flanked by 8 bp 5’ handle (bound by Cas8/5, indicated with purple arrow, Figure 3A-B) and a 20 bp 3’ stem loop recognized by Cas6 (indicated with yellow star, Figure 3A-B). The sequences of typical and atypical crRNA of I-F3b CAST contain major differences in the 5 nucleotides adjacent to the 3’ stem loop (41GUGAA45 and 41AUUUU45 for typical and atypical crRNA, respectively) (Figure 3C). Closer inspection of this region of crRNA reveals differences between the I-F3a (PDB: 6PIJ, Figure 3C left in light blue)5 and I-F3b structures (this study, Figure 3C right in orange). In the I-F3a structure, the crRNA is too distant to make substantial interactions with TniQ (Figure 3C, left). However, the crRNA in the I-F3b structure reported here adopts a different path, with three bases (U42, U43, and U44) sufficiently close to interact with TniQ (2.9 Å, 3.7 Å, and 3.9 Å for U42, U43, and U44 respectively, Figure 3C right). This suggested that the molecular mechanisms for discriminating between different crRNA sequences (i.e., typical versus atypical, as reported previously3) may originate from TniQ.
Figure 3. Comparison of the crRNA bound structure of I-F3a and I-F3b point to a regulatory mechanism enacted by TniQ.
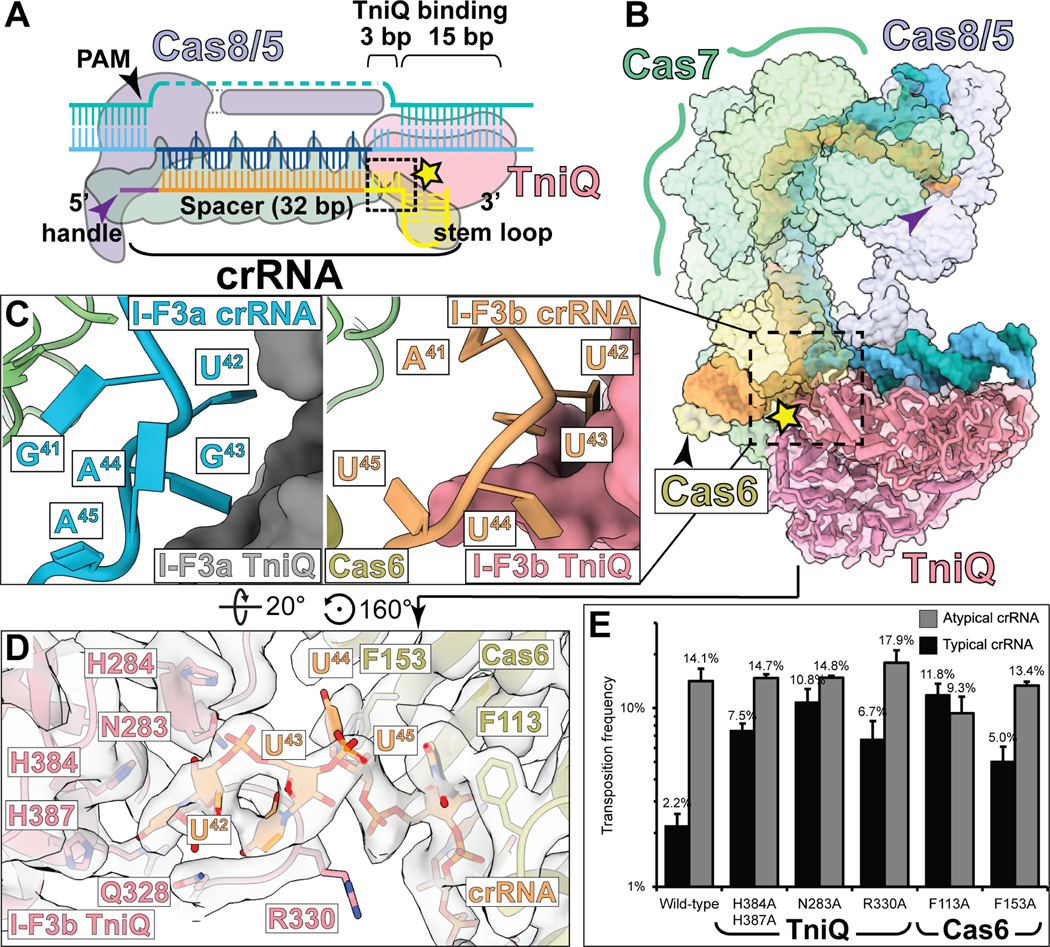
(A) Schematic of crRNA and target-DNA bound to Cascade-TniQ. Purple and black arrow indicate 5’ handle and PAM, respectively. Spacer and 3’ stem-loop of crRNA are colored orange and yellow, respectively. Black dashed box and yellow star indicate the interface between TniQ and crRNA.
(B) Structure of crRNA and bound DNA shown in opaque surface, protein shown in transparent surface. Dotted box indicates the region shown superimposed in panel C. Symbols are as indicated in A.
(C) Comparison of target-DNA bound structures of the I-F3a (PDB: 6PIJ, left) and I-F3b (this study, right) reveals striking differences in the structure of crRNA (blue for I-F3a, orange for I-F3b).
(D) The I-F3b atomic model built into the cryo-EM density (transparent) reveals potential residues which may form the basis of the regulatory function of TniQ.
(E) In vivo transposition frequency with TniQ and Cas6 mutants compared to wild-type with typical or atypical crRNA was monitored using the mate-out assay. Mutation of several crRNA-interacting residues abrogates the ability of I-F3b elements to regulate transposition as indicated by a restoration of typical crRNA transposition activity relative to atypical crRNA; data indicate mean +/− standard deviation (n=3 biological replicates).
From this observation, we identified multiple basic TniQ residues in the general vicinity of the crRNA.3 While we did not observe specific interactions with RNA, we nevertheless sought to ascertain a potential functional role of either TniQ or Cas6 in discriminating crRNAs. Surprisingly, alanine mutations of crRNA-proximal residues in either TniQ (N283, R330, H384, and H387) or Cas6 (F113 and F153) increase the transposition activity of typical crRNA close to the activity of atypical crRNA (Figure 3E), indicating defects in the mechanism allowing regulation of crRNA preference. These findings demonstrate that TniQ plays an important role in modulating crRNA preferences with I-F3b CAST elements.3
Comparative analysis of I-F3b PAM and I-F1 PAM requirements for transposition and interference.
Canonical I-F1 systems generally require a non-target strand CC PAM for interference activity.13 In contrast, previous work by multiple groups has highlighted expanded PAM flexibility with I-F3 CAST systems compared to the canonical I-F1 CRISPR-Cas defense systems.3,14–19 As part of our comparative analysis, we performed an unbiased PAM screen utilizing a target plasmid pool randomized at the PAM sequence (“−2” and “−1” positions, Figure 4A) associated with a plasmid-encoded ffs target sequence that is natively used as an attachment site for most of the I-F3b systems (Figure 4A). The target plasmid with the randomized PAM was then transformed into a cell population expressing the I-F3b Tn6900 CAST system or the 1-F1 P. aeruginosa PA14 CRISPR-Cas defense system (Figure 4A). The transposition potential of all possible PAM combinations (42) was assessed by looking for PAM sequence enrichment in the population of plasmids that were targeted for transposition (Figure 4B) or PAM sequence depletion for targets subject to interference (Figure 4C) when compared to input plasmid population.
Figure 4. PAM requirement for I-F3b CAST transposition is more promiscuous than I-F1 CRISPR-Cas interference.

(A) Schematic describing the workflow of the assay. Target plasmid with 2 base-pair degenerate sequence upstream protospacer (orange) was sequenced before and after selection for transposition with I-F3b Tn6900 CAST or interference with I-F1 P. aeruginosa PA14 CRISPR-Cas.
(B) Heatmap indicating PAM sequence enrichment/depletion score following selection for Tn6900 transposition of sixteen possible PAM sequences. Values >1 indicate preferred PAMs more abundant after selection for transposition; values <1 are less preferred than average.
(C) Sequence enrichment/depletion with PA14 interference is shown as in panel B. A lower score indicates higher interference activity.
(D-E). A subset of PAM sequences tested directly for transposition frequency in a mate-out assay (D) or interference activity by a transformation assay, with relative transformation efficiency compared to a non-target control plotted (E). Data indicate mean +/− standard deviation (n=3 biological replicates).
As expected, based on previous results,13 the I-F1 interference system had a strong preference for CC, which was completely depleted from the pool (i.e. “<0.005” in Figure 4C). More generally, I-F1 interference activity depleted plasmids with a C in the − 1 position (Figure 4C). By contrast, in the I-F3b transposition screen, we observe an overall loosening of PAM requirements (Figure 4B). While the CC PAM did show a 3-fold enrichment as a transposition target (Figure 4B), all of the PAM combinations in the pool were used as transposition targets, with at most a modest <10-fold reduction for the least favored AA PAM (Figure 4B). To further validate these results, a subset of PAM sequences were individually constructed and tested in the mate-out transposition assay for I-F3b CAST (Figure 4D) or interference assay for I-F1 CRISPR-Cas (Figure 4E). While the CC PAM was efficiently used as a target for transposition, many additional PAM combinations could be recognized as transposition targets, often only differing by a few percent with their transposition efficiency. This was markedly different than the result found with the interference assay with an almost two-order of magnitude difference being found between the appropriate CC PAM and any of the other PAM combinations (Figure 4D&E).
Molecular basis of I-F3 CAST PAM promiscuity and specificity
Close inspection of structures representing the I-F1 CRISPR-Cas associated Cascade complex (PDB: 6NE0)10 and the I-F3b CAST-associated Cascade-TniQ complex reveals candidate residues that could explain PAM sequence preferences (Figure 5A&S4A). Notably, serine residues S248 (3.6Å) and S130 (2.2Å) are within hydrogen-bonding distance of the −1 and −2 PAM bases, respectively, in the I-F3b CAST structure (Figure 5A). In comparison, the −1 and −2 PAM bases in the I-F1 CRISPR-Cas structure interact with N250 and N111, respectively (Figure 5B). Serine residues are generally thought to be less specific in recognizing base sequences compared to asparagine, due to its limited ability to form bidentate interactions.20 To explore the level of PAM specificity in the I-F3b system and validate the mechanisms suggested by our structural comparisons, we made changes in the S248 residue predicted to interact with the −1 position of the PAM (Figure 4A). We focused on this position as the I-F1 and I-F3b systems show major difference in the PAM preference at the −1 position (Figure 4B-C). Interestingly, neither the S248N nor the S248A mutants could re-establish any substantial level of PAM discrimination (Figure 5C). This indicates that PAM specificity may depend on other energetic effects besides the identified contacts, or it would require a more extensive change in the local environment than just the amino acid that directly interacts with the PAM to affect positioning of residues involved in these contacts.
Figure 5. Structure comparison and mutation analysis indicate key residues controlling PAM discrimination.

(A) Comparison of Cas8 (purple) interaction with PAM bases on the target-strand (TS, blue) in the target-DNA bound structures of I-F3b (this study, left) and I-F1 (6NE0, right). Distances are annotated for the sequence-specific hydrogen-bonding interactions between Cas8/5 residues and DNA bases. Non-target strand (NTS) and nucleotides other than G−1 and G−2 do not form sequence-specific interactions.
(B) Cartoon diagram describing the key interactions between DNA and Cas8/5 (I-F3b, left) or Cas8 (I-F1, right).
(C) PAM wheels indicating PAM preference of I-F3b Tn6900 CAST with Cas8/5 wild-type and mutants. Preference at the −1 position is represented by the inner ring; the −2 position is represented by the outer ring. While S248A, S248N, and A247T+S248N mutants showed modest change, A247Q+S248N mutation substantially increased −1 C preference.
To address if the local environment that positions the S248 residue was also important for PAM discrimination, we tested double mutations at A247 and S248 residues. We generated two mutants that mimic the local environment of Cas8/5 from I-F3a family (A247T+S248N, Figure S4B) or Cas8 from I-F1 Cascade (A247Q+S248N). Interestingly, the changes involving the residue S248N contacting the PAM and an adjacent residue increased sequence specificity at the −1 position to bias transposition to PAMs with a C at the −1 position, especially with the A247Q+S248N mutant resembling I-F1 Cas8 (Figure 5B&S5). Unlike wild-type Cas8/5 or the other mutants we examined, over half of the transposition events in the pool with the A247Q S248N change had the PAM with C-1 (Figure 5C).
Our results indicate that the PAM binding region of Cas8/5 in I-F3 CASTs has likely extensively evolved for PAM ambiguity since cooption from an ancestral canonical type I-F1 CRISPR-Cas system. Swapping the serine residue in I-F3b Cas8/5 that directly contacts the -PAM −1 position to the asparagine found in I-F1 Cas8 is not sufficient to reestablish PAM CC selectivity. However, more extensive changes involving multiple residues in the PAM binding pocket can start to reestablish the −1 C preferences suggesting rational design methods focused on the PAM binding pocket could reconfigure PAM selectivity. Interestingly, computational modeling of the I-F1, I-F3b, and I-F3b mutant (A247Q+S248N) generally captures the experimental trends reported here (Figure S6) and suggests that successful design strategies will incorporate both flexible backbone modeling as well as careful consideration of the full PAM-binding pocket. We suggest that two important factors have selected for PAM ambiguity in I-F3 transposition systems: capacity for privatization of chromosome-matching crRNAs from host CRISPR-Cas defense and diversification of attachment sites recognized by the system (see discussion).
Discussion
We find that co-option of type I-F Cascade for crRNA-directed transposition involved extensive reconfiguration of the control of the CRISPR-Cas effector. Our high-resolution structure reveals a series of new interactions between downstream DNA and both TniQ and as well as the Cas8/5 helix bundle domain (Figure 1 and 2). We propose that the new TniQ interactions we identify in the full R-loop complex reveal an important step for licensing a target for transposition in these systems (Figure 1 and 2). Additionally, these TniQ-DNA contacts result in a DNA distortion predicted to help accommodate TnsC-mediated transposase recruitment based on previous work with prototypic Tn7 (Figure 3).21,22 We show that TniQ acts to mediate crRNA selection, sorting typical and atypical crRNAs to regulate target choice in the system (Figure 3). Direct comparison between PAM usage in I-F3 transposition and I-F1 interference systems indicates the extent of PAM ambiguity in the transposition systems (Figure 4) and that extensive structural adaptations contribute to the process (Figure 5). These findings support a model where critical control features normally used by Cascade are now licensed by TniQ either directly or with the collaboration of the TnsA, TnsB, and TnsC transposition components.
TniQ regulates target-site preference
TniQ/TnsD proteins are known to have adapted to a variety of fixed attachment sites in bacteria and to programable attachment sites.3,23,24 Prototypic Tn7 distinguishes between target sites using either of two proteins that function in parallel targeting pathways: one allowing sequence-specific DNA-binding (TnsD, homologous to TniQ) or one recognizing specialized features of DNA replication found with mobile plasmids (TnsE).25–27 A sister group of the I-F3 CAST elements outside of the major I-F3a and I-F3b clades, type I-D CAST elements, and two independently coopted type I-B CAST elements use a hybrid approach half-way between prototypic Tn7 and I-F CAST elements, where pathway choice occurs via association with one of two separate TniQ proteins: either a sequence-specific TnsD/TniQ protein or a TniQ that functions with Cascade.3,18,28,29
Our work here indicates that in the I-F3 CAST systems, TniQ actively cooperates with Cascade to regulate transposition by sensing crRNA categories. This adds crucial insight into the function of TniQ, as previously it was unclear if TniQ simply acted as a physical connector between target-site and core transposition components.5 Not only do these findings provide a strong mechanistic basis for understanding previous studies highlighting complex regulatory behavior in I-F3b CAST3 and related elements,24,28 but they also reveal an evolutionarily conserved role for the TniQ/TnsD protein superfamily in facilitating decision making at attachment sites.
Cascade-TniQ forms a complete R-loop, distorting the target DNA to recruit transposition proteins
Previous studies on the I-F3a target-recognition module showed that Cascade, in the absence of TniQ, can also bind target DNA.19,30 Consistent with this, our EMSA result suggests that the initial PAM and seed recognition would be similar with and without TniQ. However, we found that the presence of TniQ promotes R-loop formation compared to Cascade alone, which forms non-specific interactions with DNA. Our cryo-EM reconstruction of Cascade-TniQ with a fully structured R-loop reveals the interaction between TniQ and the PAM-distal DNA, consistent with our ideas on how TniQ promotes R-loop formation. A recent study examining target site engagement with I-F3a CAST components in vivo using ChIP-seq found that Cascade-TniQ exhibited an enrichment at the target site greater than with Cascade alone.30 While the TniQ effect was largely discounted in the paper, in light of our results, an alternate explanation is that TniQ contributes to higher occupancy of the target site in vivo by stabilizing the full R-loop conformation. A unifying model holds that the ability of TniQ to stabilize a complete R-loop that we demonstrate is an important step for licensing TnsC recruitment as the complex progresses to a full transposition integration complex.
We show that the Cascade-TniQ complex unwinds and distorts downstream of the target DNA upon full R-loop formation. Extensive studies on prototypic Tn74 showed that DNA distortion, produced by the TniQ family protein TnsD, is a central feature in sequential recruitment of the TnsC regulator and, eventually, the transposase.21,25 The importance of this DNA distortion is highlighted by an artificial Tn7 targeting pathway in which a distortion induced by triplex-forming DNA is sufficient to recruit TnsABC for transposition.31 This model is further supported by a cryo-EM structure revealing that Tn7 TnsC specifically loads adjacent to a mismatched bubble in double-stranded DNA substrates.22 Thus, we find it compelling that a DNA distortion at the PAM-distal region, created by TniQ and the helix-bundle domain of Cas8/5 (Figure 3), may also serve as a signal for TnsC recruitment in I-F3 CASTs. We suggest our complete R-loop structure represents an active conformation for downstream factor recruitment with distorted DNA and TniQ in position to make simultaneous contacts with TnsC, as has been shown in the V-K CAST.32,33 However, we expect the recruitment of other transposition components may induce large reconfigurations as was also shown in a V-K CAST holo transposition complex.34 Therefore, it remains an intriguing question how the rest of the transposition components recognize the DNA distortion, and potentially rearrange the structure to drive transposition in a type I-F3 CAST system.
PAM ambiguity is a mechanistic adaptation of I-F3 systems, allowing attachment site drift tolerance and crRNA privatization
We speculate that PAM flexibility is important for two reasons in the I-F3 CAST transposition systems: 1. to maintain a fixed attachment site recognizable in diverse host chromosomes and 2. for privatizing attachment sites making them inaccessible to the host interference system. In fact, our experimental results extend generally to naturally occurring populations based on bioinformatics. By examining natural insertions of type I-F3b CAST elements into the native ffs attachment site we find that only a small percentage (3.4%, 8/235) have the canonical CC PAM (Table S1). The vast majority of these insertions (96.6%, 227/235) utilize a TC PAM (Table S1). This indicates that PAM flexibility is an essential feature allowing these elements to recognize their chromosomal attachment site. Overall, the CC PAM is only used 4.4 % (33/757) of the time across all of the I-F3 insertion sites identified in bacterial genomes suggesting this is a general attribute of this family of CAST elements (Table S1). Moreover, use of a TC or other non-CC PAM attachment site would also protect chromosomal attachment sites from canonical I-F1 CRISPR-Cas interference. Type I-F3 CAST elements can naturally reside in the same host as a canonical I-F1 interference system, and it has been shown that I-F1 systems are able to use typical I-F3 CAST-encoded crRNAs for interference.3 The TC PAM that is found 97% of the time in the ffs attachment site could therefore be hidden from the I-F1 system based on our interference assays while this same TC PAM would be readily used for transposition (Figure 4E).
For practical applications though, rational modification of PAM specificity would contribute to the development of orthogonally functioning, precise CAST elements. Our investigation takes one step further by establishing that extensive rearrangements must have been under selection to allow PAM ambiguity in the I-F3 systems (Figure 5). Our computational simulations suggest that accurate modeling must incorporate flexible backbone modeling of the entire PAM binding site, not just the positions directly interacting with the PAM motif (Figure S6).
Together, these results paint a picture in which multiple adaptive changes in both the CRISPR-effector (i.e., Cascade) and the associated TniQ work together to enhance the survival and distribution of CASTs. Our study demonstrates the crucial role of TniQ in the co-option of Cascade through its two important abilities: 1. it distinguishes different categories of insertion sites by sensing sequence differences in crRNAs, and 2. it licenses the substrate for transposition by completing R-loop formation at the target site. These adaptations provide another exciting example of the dynamic molecular mechanisms utilized to escalate the host-parasite arms race.
Limitations of the study
In this study, we showed that TniQ categorizes crRNAs to regulate transposition pathway choice. Although our structures of the Cascade-TniQ complex provide key insights into the residues responsible for regulation, it remains unclear how the crRNA sequence directly impacts transposition activity. A deeper understanding of this mechanism may require more than just snapshots of the complex in certain states and may involve the kinetics of Cascade-TniQ complex assembly or target DNA engagement. It is also possible that the crRNA sequence may affect downstream steps with recruitment of the TnsA and TnsB transposase components. Further biochemical and structural characterizations would be required to fully understand the molecular basis of the crRNA categorization.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Joseph E. Peters (joe.peters@cornell.edu).
Materials availability
Newly generated materials are available from the lead contact upon reasonable request.
Data and Code Availability
All data generated in this study, including atomic coordinates for Cascade-TniQ bound to target DNA for both full and partial R-loop states, have been deposited. Raw imaging data have been deposited at Mendeley data. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
All original code used for analysis have been deposited at Mendeley. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH5α | J. Peters Lab | N/A |
| Escherichia coli BW27783 attTn7::miniTn7(miniTn6900(kanR)) | J. Peters Lab, Pettasi et al., 2020 | MTP997 |
| Escherichia coli BL21-AI | Invitrogen | Cat# C607003 |
| Escherichia coli CW51 | J. Peters Lab | JP606 |
| T7 Express Competent Escherichia coli | NEB | Cat# C2566H |
| Critical commercial assays | ||
| Q5® High-Fidelity DNA polymerase | NEB | Cat# M0491L |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | Cat# E2621L |
| His-Pur™ Ni-NTA resin | ThermoFisher | Cat# 88221 |
| CloneJET PCR Cloning Kit | ThermoFisher | Cat# K1231 |
| GeneJET Gel Extraction Kit | ThermoFisher | Cat# K0691 |
| T4 DNA Ligase | ThermoFisher | Cat# EL0011 |
| Deposited data | ||
| Full R-loop Cascade-TniQ complex | This paper | PDB: 7U5D EMDB: 26348 |
| Partial R-loop Cascade-TniQ complex | This paper | PDB: 7U5E EMDB: 26349 |
| Custom code for Rosetta simulations and PAM analyses and plots | This paper | Mendeley Data DOI: 10.17632/gjgspgnxvy.1 |
| NGS data for PAM analysis | This paper, NCBI Sequence Read Archive | Bioproject: PRJNA935286 |
| Oligonucleotides | ||
| Oligo_cryo1, CCGCAAGAGGATGATTCGGGTGCTTACCTCCTGACCTTCTTTAGTAGGTTCAACCCCTGATCGAGTGCCGGGATGT | This paper | N/A |
| Oligo_cryo2, TGGCCCCATCAGCCACATCCCGGCACTCGAAGTCCCCAACTTGGATGATTTCTTCCAGTCCTGGTAAGCACCCGAATCATCCTCTTGCGG | This paper | N/A |
| Oligo_cryo3, GGCTGATGGGGCCACCACCTTGCGCTCGTTCGCCAGCCAG | This paper | N/A |
| Oligo_cryo4, CTGGCTGGCGAACGAGCGCAAGGTGG | This paper | N/A |
| F_EMSA, ACCTGCAGGCATGCAAG | This paper | N/A |
| R_EMSA, /Cy5/GCAGTTAAGTGCCTGCTGGCGAGGAAGCGGAAGAG | This paper | N/A |
| DNA substrate for EMSA, Top_ffs_bubble_EMSA Cy3/GCGGCACAGGTCTCACGCGTTCGTCACTACCAGGACTGGAAGAAATCATCCAAGTTGGGGACTTCGAGTGCCGGGATGTGGCTGATGGGGCCACCACCTGTTTCTGCACTTCAACCAGTTAGCCTCCAGAGCAACCGAGACCCGCTGCTG | This paper | N/A |
| DNA substrate for EMSA, Bottom_ffs_bubble_EMSA CAGCAGCGGGTCTCGGTTGCTCTGGAGGCTAACTGGTTGAAGTGCAGAAACAGGTGGTGGCCCCATCAGCCACATCCCGGCACTCGATTATTTTTTTAATTTATTAAATAATTTTATTAGGTAGTGACGAACGCGTGAGACCTGTGCCGC | This paper | N/A |
| Recombinant DNA | ||
| All plasmids used and constructed in this study | This paper | See Table S2 |
| Software and algorithms | ||
| Warp | Tegunov et al., 201935 | http://www.warpem.com/warp/ |
| cryoSPARC | Punjani et al., 201736 | https://cryosparc.com/ |
| Topaz | Bepler et al., 201937 | https://cb.csail.mit.edu/cb/topaz/ |
| RELION | Scheres, 201238 | https://relion.readthedocs.io/en/release-4.0/ |
| cryoSPARC 3DVA | Punjani et al., 202139 | https://cryosparc.com/ |
| UCSF Chimera | Pettersen et al., 200442 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 201843 | https://www.cgl.ucsf.edu/chimerax/ |
| I-TASSER | Yang et al., 201545 | https://zhanggroup.org/I-TASSER/ |
| Coot | Emsley et al., 201046 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| RosettaES | Frenz et al., 201747 | https://dimaiolab.ipd.uw.edu/software/ |
| MolProbity | Williams et al., 201848 | http://molprobity.biochem.duke.edu/ |
| Phenix | Afonine et al., 201849 | https://phenix-online.org/ |
| Biopython | Cock et al., 200962 | https://biopython.org/ |
| Matplotlib | Hunter, 200763 | https://matplotlib.org/ |
| Seaborn | Waskom, 202164 | https://seaborn.pydata.org/ |
| Krona | Ondov et al., 201165 | https://github.com/marbl/Krona/wiki |
| WebLogo 3 | Crooks et al., 200467 | http://weblogo.threeplusone.com/ |
| Cd-hit | Li et al., 200668 | https://github.com/weizhongli/cdhit |
| MUSCLE | Edgar, 200470 | http://www.drive5.com/muscle |
Experimental Model and Subject Details
Escherichia coli strains were grown at 30 or 37°C in lysogeny broth (LB) or on LB agar (unless stated otherwise in the method details) supplemented with the following concentrations of antibiotics when appropriate: 100 μg/mL carbenicillin, 10 μg/mL gentamicin, 30 μg/mL chloramphenicol, 8 μg/mL tetracycline, 50 μg/mL kanamycin, 100 μg/mL spectinomycin.
Method Details
Protein Purification
Expression plasmid sets were transformed into E. coli T7 Express cells (New England Biolabs). For Cascade-TniQ for cryo-EM imaging, plasmid set was pOPO066 (pETDuet-1-cas8/5-cas7), pOPO097 (pACYCDuet-1-cas6-cas7), pOPO127 (pCOLADuet-1-His6-tniQ), and pGS100 (pCDFDuet_crRNA-ffsx6). For Cascade-TniQ for EMSA, plasmid set was pOPO066 (pETDuet-1-cas8/5-cas7), pOPO097 (pACYCDuet-1-cas6-cas7), pOPO127 (pCOLADuet-1-His6-tniQ), and pMTP1277 (pCDFDuet_crRNA-ffsx6(alt)). For Cascade without TniQ for EMSA, plasmid set was pOPO065 (pETDuet-1-His6-cas8/5-cas7), pOPO097 (pACYCDuet-1-cas6-cas7), and pMTP1277 (pCDFDuet_crRNA-ffsx6(alt)). Cells were grown in LB with appropriate antibiotics at 37°C to O.D.600 0.8 and induced overnight at 16°C with 0.4mM IPTG. Cells were pelleted, resuspended in lysis buffer (500mM NaCl 25mM HEPES pH 7.5 10% glycerol 5mM DTT) plus 1mM PMSF, and lysed by sonication. After sonication cells were centrifuged at 12,000 rpm for 45 min and the supernatant was collected and imidazole was added to a final concentration of 20mM. The supernatant was loaded to 2mL Ni-NTA resin (ThermoFisher). The Ni-NTA resin was washed with 150mL of lysis buffer with graded increases of imidazole to 50mM. Complexes were eluted from the Ni-NTA resin with lysis buffer plus 300mM imidazole. Eluted cascade complexes were then purified with anion exchange chromatography (MonoQ 5/50GL cytiva). Peak fractions were collected and snap-frozen in liquid nitrogen for later use (Figure S3A).
DNA substrate preparation
Bubbled dsDNA substrate for cryo-EM sample preparation was created by heating 4 oligonucleotides (Oligo_cryo1, Oligo_cryo2, Oligo_cryo3, Oligo_cryo4, Key resources table) to 95°C for 10min in duplex buffer (30mM HEPES, pH 7.5; 100mM potassium acetate) followed by slow cooling. Annealed DNA was ligated with T4 ligase (ThemoFisher), ran on a 12% UREA-PAGE gel, and successfully ligated bands were cut and extracted from the gel. Purified ssDNA was then reannealed in duplex buffer, run on a 1% agarose gel to remove free ssDNA, and purified using GeneJet gel extraction kit (ThermoFisher).
Electrophoretic mobility shift assay
The ffs protospacer was cloned into pJET vectors (ThermoFisher). 214bp DNA target for EMSA was PCR amplified from the cloned pJET vectors using fluorescently labeled primers (F_EMSA and R_EMSA, Key resources table). Serial dilutions of Cascade or Cascade-TniQ were mixed with the DNA target (0.5nM) in EMSA buffer (100mM KCl, 5% glycerol, 5mM MgCl2, 2mM β-mercaptoethanol), and incubated at 37°C for 15 minutes. 20 μL of DNA binding reactions were run on a 1% agarose TBE gel for 1 hour and 15 minutes at 60 V at 4°C. The gel was imaged with an Amersham Typhoon Biomolecular Imager (GE Healthcare Life Sciences) and analyzed using ImageQuant 1D version 8.2 (GE Healthcare Life Sciences). The bubbled target was prepared using IDT ultramer oligos (Top_ffs_bubble_EMSA and Bottom_ffs_bubble_EMSA, Key resources table) which were annealed and gel purified. The 214bp dsDNA target from the previous EMSA was compared with the bubbled DNA target. 500nM of Cascade-TniQ was mixed with the dsDNA target or bubbled DNA target (0.5 nM) in EMSA buffer and incubated at 37°C for 15 minutes. 20 μL of DNA binding reactions were run on a 1% agarose TBE gel for 1 hour and 15 minutes at 60 V at 4°C. The gel was imaged with a ChemiDoc imaging system (Bio-Rad).
Cryo-EM sample preparation and imaging.
For the Cascade-TniQ with atypical crRNA, purified Cascade-TniQ sample was supplemented with 1.2-fold molar excess of target DNA with 32 base artificial bubble (see above). Then the Cascade-TniQ in the solution was diluted to 2 μM (~0.9 mg/mL) making the final buffer composition as follows: 25 mM HEPES pH 7.5, 150 mM NaCl. The sample was incubated for 30 minutes on ice before being vitrified using the Mark IV Vitrobot (ThermoFisher) set to 4°C and 100 % humidity. 4 μL of the reconstituted Cascade-TniQ-DNA sample is loaded on the QuantiFoil Cu 1.2/1.3 grids (QuantiFoil) that was freshly glow discharged using PELCO easiGlow (Ted Pella). Then the grids were immediately blotted for 6 seconds with blot force 6, followed by vitrification in the slurry of liquid ethane cooled with liquid nitrogen. The grids were first screened using Talos Arctica (ThermoFisher) operating at 200 kV, equipped with K3 direct electron detector (Gatan) and BioQuantum energy filter. Ice thickness, number of particles per image, and number of good squares are assessed to find the best grid for data collection. The chosen grid was imaged using Titan Krios G3 (ThermoFisher) operated at 300 kV, also equipped with K3 detector (Gatan) and BioQuantum energy filter (Gatan). The slit size of the energy filter was set to 20 eV. 13,800 micrographs were recorded at the 105,000X nominal magnification (corresponding to 0.873 Å per pixel) using 3 by 3 image shift, with the nominal defocus from −1.0 μm to −2.5 μm. Total 60 electrons were exposed per Å2 during 4.2 seconds, fractionated into 60 frames.
Image processing
Warp35 was used for beam-induced motion correction and CTF estimation of the total 13,800 movies. 12,024 Micrographs with 5 Å or higher CTF-fit resolution were imported to cryoSPARC36 for further processing. Initial particle picking was done using template-based picking in cryoSPARC, followed by 2D classification. Resulting 38,807 particles from 2D averages with high-resolution features were used to train topaz37 neural network. This trained network was applied to the filtered 12,024 micrographs to extract initial 1,338,135 particle picks. 2D classification was followed to remove “junk” particles, resulting in 1,075,078 particles from the selected 2D averages, which were then re-extracted using RELION38 with Fourier-cropping (420 pixels to 128 pixels, corresponding to 2.86 Å per pixel). This particle stack was subjected to 3D classification in RELION, which yielded 237,671 particles of intact Cascade-TniQ complex. This particle stack was then re-extracted without Fourier cropping (0.873 Å per pixel), followed by non-uniform refinement and heterogeneous refinement in cryoSPARC. One resulting class (53%, 126,320 particles) from the heterogeneous refinement showed significantly stronger TniQ density, thus selected for the downstream analysis. We noticed that even after the two rounds of classifications, resulting particle stack incorporated considerable level of conformational heterogeneity.
Following the non-uniform refinement of the selected class (126,320 particles), 3D variability analysis39 (3DVA) in cryoSPARC was used to analyze the conformational dynamics within the dataset. 3DVA visualization tool from cryoSPARC was used to cluster the input particle stack into three classes based on the identified eigenvectors and coordinates in the defined conformational space from the 3DVA. Each extreme of the space were identified as Cascade-TniQ complex with partial (28%, 34,800 particles) or full (20%, 26,306 particles) R-loop respectively, with 52% of particles in between showing disordered TniQ density. 34,800 particles that represent partial R-loop complex were then exported to RELION for Bayesian polishing,40 and CTF refinement41 of the complex, which resulted in 4.0 Å resolution of partial R-loop complex. In order to maximize the number of particles in the class of full R-loop complex, the earlier stack of 126,320 particles was subjected to two independent heterogeneous refinements. Each refinement job was set up using two volumes of the extreme clusters from the 3DVA eigenvector 1 and eigenvector 2, respectively. Each refinement resulted in 57% or 58% of Cascade-TniQ with full R-loop respectively, which were then merged and deduplicated. This stack of 91,051 particles was exported to RELION. Signals outside of the PAM-distal region is subtracted using the mask that includes Cas6, TniQ dimer, Cas8 helix bundle, and PAM-distal DNA (Figure S1B). Focused classification of the subtracted particles resulted in two major classes of PAM-distal DNA bound TniQ, but one class of high-resolution features (58%, 53,353 particles) was selected as the final particle stack for the Cascade-TniQ with full R-loop. CTF-refinement and Bayesian polishing of the particles resulted in 3.5 Å resolution of full R-loop complex. Focused refinement of the PAM-distal region resulted in 3.9 Å resolution. Final maps were sharpened using RELION postprocessing tool with automatically estimated B-factors. Local resolution estimation and filtering of the final reconstructions were done using cryoSPARC. For visualization, reconstructions from global and focused refinement were aligned and combined using UCSF Chimera42 command ‘fitmap’ and ‘vop maximum’ respectively. Figures describing cryo-EM reconstructions and atomic models were generated using UCSF ChimeraX.43
Model Building and Validation
The atomic models for Cas8/5, Cas6, Cas7, and TniQ from a homologous Cascade-TniQ complex from V. cholerae (PDB: 6PIF)44 were used as templates for homology models generated using the I-TASSER server45. These homology models were docked in the cryo-EM density map using UCSF Chimera42 and residue registers and backbone geometries were then corrected using Coot46. Extended loops that could not be modeled manually due to locally poorer EM density were rebuilt into the cryo-EM density maps using RosettaES47 and then manually refined in Coot in an iterative fashion. Refined protein models were subsequently relaxed into the EM density using Rosetta and were subjected to iterative rounds of relaxation in Rosetta and refinement in Coot to fix geometric and steric outliers identified by MolProbity48 during model validation. crRNA and DNA strands were manually built into the cryo-EM density map using Coot with resolution sufficient to distinguish purines and pyrimidines and, thus, confirm the register of each strand. Nucleic acid models were refined into an EM map ‘zoned’ (using UCSF Chimera) to remove protein density and using phenix49,50 real_space_refine subject to base-pair and stacking restraints generated by inspection. Details of the validation stats are summarized in Table 1.
Table 1.
Cryo-EM data collection, refinement, and validation statistics.
| Name | Cascade-TniQ with atypical crRNA, full R-loop | Cascade-TniQ with atypical crRNA, partial R-loop |
|---|---|---|
| PDB ID | 7U5D | 7U5E |
| EMDB ID | EMD-26348 | EMD-26349 |
| Data collection and Processing | ||
| Microscope | Titan Krios | Titan Krios |
| Voltage (keV) | 300 | 300 |
| Camera | K3 | K3 |
| Magnification | 105,000 | 105,000 |
| Pixel size at detector (Å/pixel) | 0.873 | 0.873 |
| Total electron exposure (e–/Å2) | 60 | 60 |
| Exposure rate (e-/pixel/sec) | 10.89 | 10.89 |
| Number of frames collected | 60 | 60 |
| Defocus range (μm) | −1.0 – −2.5 | −1.0 – −2.5 |
| Automation software | SerialEM | SerialEM |
| Energy filter slit width | 20 keV | 20 keV |
| Micrographs collected (no.) | 13,800 | 13,800 |
| Micrographs used (no.) | 12,024 | 12,024 |
| Total extracted particles (no.) | 1,338,135 | 1,338,135 |
| For each reconstruction: | ||
| Refined particles (no.) | 126,320 | 126,320 |
| Final particles (no.) | 53,353 | 34,800 |
| Symmetry | C1 | C1 |
| Resolution (global, Å) | ||
| FSC 0.5 (unmasked/masked) | 7.48/4.17 | 8.94/4.95 |
| FSC 0.143 (unmasked/masked) | 4.26/3.52 | 4.95/4.03 |
| Resolution range (local, Å) | 3.3 – 7.0 | 3.4 – 7.0 |
| Resolution range due to anisotropy (Å) | 3.30 – 3.86 | 3.78 – 4.64 |
| Map sharpening B factor (Å2) | −70 | −81 |
| Map sharpening methods | RELION | RELION |
| Model composition | ||
| Protein residues | 3,603 | 3,474 |
| Ligands | 0 | 0 |
| RNA/DNA | 141 | 100 |
| Model Refinement | ||
| Refinement package | Coot/Rosetta/Phenix | Coot/Rosetta/Phenix |
| - real or reciprocal space | Real | Real |
| - resolution cutoff (Å) | 3.5 | 4.0 |
| Model-Map scores | ||
| - CC (box) | 0.81 | 0.77 |
| - Average FSC (0.5 cutoff, Å) | 3.71 | 4.30 |
| R.m.s deviations from ideal values | ||
| Bond length (Å) | 0.012 | 0.012 |
| Bond angles (°) | 1.613 | 1.616 |
| Validation | ||
| MolProbity score | 1.74 | 1.78 |
| CaBLAM outliers (%) | 2.15 | 2.26 |
| Clashscore | 10.15 | 11.26 |
| Poor rotamers (%) | 0.20 | 0.14 |
| C-beta outliers (%) | 0.03 | 0.03 |
| EMRinger score | 2.90 | 2.06 |
| Ramachandran plot | ||
| Favored (%) | 96.66 | 96.66 |
| Outliers (%) | 0.17 | 0.12 |
Rosetta Refinement
Missing loops and initial protein models were paired with a ‘zoned’ and sharpened map that contained only the density corresponding to a single protein subunit of the broader Cascade-TniQ complex. Extended loops spanning 3–12 residues with locally poorer EM density were deleted from the protein model using UCSF Chimera and then rebuilt iteratively using RosettaES with the Rosetta xml script. The best scoring model generated by Rosetta was manually examined in Coot and the next missing model was rebuilt in another round of RosettaES modeling. This procedure was integrated with manual inspection and model-building. The RosettaES xml script is accessible at Mendeley Data, accession info in the key resources table.
Rosetta simulation of PAM specificity
PDB models for the DNA-bound I-F1 Cascade (PDB: 6NE0)10 and the full R-loop complex of the I-F3b system were used as inputs for specificity calculations. The model of I-F3b A247Q S248N mutant was generated from I-F3b full R-loop complex using fixbb application of the Rosetta modeling suite51–54. This application was used to fit an amino acid rotamer onto the fixed backbone of an input structure at specific positions. In order to generate 16 models of each system with 16 possible PAM combinations, −1 and −2 position nucleotides were substituted using the Simple Mutate tool from coot46, which replaces the base identity from the original model without altering other geometries. The RosettaScripts application55 was used to both optimize binding and calculate specificity values of each system with each possible PAM sequence. For I-F1 Cascade and I-F3b optimized binding, we allowed for rotamer packing of the residues at positions 247 and 248. This protocol also sampled backbone movement of this two-residue span and the four residues flanking either side of this region (total of 10 adjacent residues) to improve positioning of the two residues with user-defined target DNA bases. Rotamer packing was specifically optimized to improve binding between the residue pair and the PAM base pairs using a DNA-based energy function56–59. Total energy scores for each structure-PAM model were calculated after rotamer/backbone optimization. Total energy scores of each model were calculated using the same weight function, which considers both intra-protein interactions and protein-DNA interactions. These weights are composed of the standard Rosetta ref2015 energy function and additional optimized weights derived to optimize protein-DNA interactions60. The optimized weight function is included below (ST-DNA-10_11_21.wts). Ten independent optimization runs were performed for each Cas8/5-PAM combination to extensively sample conformational space. With each optimized model providing an energy score, ten energy scores were then averaged to be used as a Boltzmann energy term. The specificity of each design model was then calculated as a Boltzmann occupancy that compared the target structure against a partition function consisting of all competing PAM combinations. The command and the required files to run Rosetta scripts are accessible through Mendeley Data, see accession in key resources table, and can be generally used to determine the binding specificity of protein-DNA structural models. The ‘res_nums’ flag in Rosetta script file should be changed to represent the residue number to be sampled.
Plasmid construction
Plasmids for protein expression/purification, transposition and interference assays were constructed by standard methods including restriction/ligation, isothermal assembly, and golden gate cloning; sequences and source information (base vector sets for each system are available on Addgene) can be found in Table S2. F plasmid derivatives were made by recombineering as previously described3. The randomized 2-bp target plasmid for unbiased screening was constructed by amplification of plasmid backbone using primers with synthetic tails adding an ‘NN’-ffs target sequence and XhoI cut sites, digestion with XhoI and self-ligation.
Mate-out transposition assay
Mate-out assays were performed in strain MTP997 with F plasmid derivative as indicated in Table S2. Cells were made competent by growing in LB media to mid-log and washing/resuspending in ice cold CaCl2 solution61 and transformed with pMTP1293 (TnsABC), pMTP1261 (TniQ-Cascade) or mutant derivatives, and pMTP1379 (atypical crRNA targeting ffs) or pMTP1382 (typical crRNA targeting ffs) as indicated in Table S2 onto LB agar supplemented with 100 μg/mL carbenicillin, 30 μg/mL chloramphenicol, 8 μg/mL tetracycline, and 0.2% w/v glucose. After 16 hours incubation at 37°C, several hundred transformants were washed up in M9 minimal media supplemented with 0.2% w/v maltose and diluted to a calculated OD = 0.3 in M9 maltose supplemented with 100 μg/mL carbenicillin, 30 μg/mL chloramphenicol, 8 μg/mL tetracycline, 0.2% w/v arabinose, and 100 μM IPTG to induce transposition.
After 24 hours incubation with shaking at 30°C, a portion of induced cultures were washed once and resuspended in LB supplemented with 0.2% w/v glucose. After 1.5 hours incubation at 37°C induced pools were mixed with prepared mid-log CW51 recipient strain at a ratio of 1:5 donor:recipient and incubated with gentle agitation for 90 minutes at 37°C to allow mating. Cultures were then vortexed, placed on ice, serially diluted in LB 0.2% w/v glucose, and plated on LB supplemented with 20 μg/mL nalidixic acid, 100 μg/mL rifampicin, 100 μg/mL spectinomycin, 50 μg/mL X-gal, with or without 50 μg/mL kanamycin to sample the entire transconjugant population or select for transposition respectively. Plates were incubated at 37°C for 36 hours before colonies were counted.
P. aeruginosa CRISPR interference assay
Interference assays were performed in BL21-AI. BL21-AI was made competent by standard chemical methods61 and transformed with pOPO322 (cas1_cas2/3), pCsy_complex, and pOPO374 (PA14 crRNA-ffs) as indicated in Table S2 onto LB agar supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol, and 0.2% w/v glucose. Overnight cultures grown in LB agar supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol were diluted 1:50 in LB supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol, 100 μM IPTG and 1 mM arabinose. Cultures were grown to OD = 0.4 before electrocompetent cells were prepared by standard methods61 and transformed with 1 ng pOPO275 (CC-ffs target plasmid), alternate PAM derivative of pOPO275 or pOPO390 (non-target control). Cells were recovered in SOC at 37°C for one hour before being serially diluted and plated on LB supplemented with 100 μg/mL carbenicillin, 50 μg/mL kanamycin, 30 μg/mL chloramphenicol, and 100 μg/mL spectinomycin. Plates were incubated at 37°C for 16 hours before colonies were counted.
Randomized PAM transposition and interference assay
For Tn6900 unbiased PAM transposition screen, MTP997 (Escherichia coli BW27783 attTn7::miniTn7(miniTn6900(kanR))) transformed with pMTP1293 (TnsABC), pMTP1261 (TniQ-Cascade) or mutant derivatives, and pMTP1379 was made competent by standard chemical means61 and transformed with pMTP1412 (NN-ffs target plasmid). >50,000 colonies were washed up, washed and resuspended to a calculated OD = 0.3 in M9 maltose induction media (M9 maltose supplemented with 100 μg/mL carbenicillin, 30 μg/mL chloramphenicol, 8 μg/mL tetracycline, 10 μg/mL gentamycin, 0.2% w/v arabinose, and 100 μM IPTG) and induced for 24 hours at 30°C before plasmids were purified with Omega E.Z.N.A. Plasmid DNA Mini Kit.
To remove self-targeting transposition into the guide-RNA expression vector contaminating transformations, plasmid pools were digested with EcoRI and SalI before transformation into DH5alpha. Transformed cells were plated onto LB supplemented with 10 μg/mL gentamycin and 50 μg/mL kanamycin to select for target plasmid with mini-transposon. >10,000 colonies were washed up and combined for plasmid purification. Purified plasmid was subjected to 2×151 bp read Illumina total DNA sequencing by Seqcenter/MiGS (Microbial Genome Sequencing Center).
For I-F1 PA14 CRISPR-Cas unbiased PAM interference screen, BL21-AI transformed with pOPO322 (pACYCDuet-cas1_cas2/3), pCsy_complex, and pOPO374 (pCDFDuet-PA14-ffs) was grown overnight in LB agar supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol then diluted 1:50 in LB supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol, 100 μM IPTG and 1 mM arabinose. Cultures were grown to OD = 0.4 before electrocompetent cells were prepared by standard methods and transformed with 1 ng pMTP1412 (pBBR-genR-NNffs). Cells were recovered in SOC at 37°C for one hour before being plated on LB supplemented with 100 μg/mL carbenicillin, 50 μg/mL kanamycin, 30 μg/mL chloramphenicol, and 100 μg/mL spectinomycin. Plates were incubated at 37°C for 16 hours before >10,000 colonies were washed up, plasmids purified and 2×151 bp read Illumina total DNA sequencing was done by MiGS.
Reads were processed by custom python code62 to extract and count reads containing each sequence permutation at the 2-bp PAM region for the plasmid pool before and after transposition/interference assay. Data was plotted as heatmaps using matplotlib/seaborn63,64 and PAM wheels using Krona as previously described65,66. All code is available at Mendley Data, accession information in the key resources table.
Cas8/5 consensus sequence logo
The consensus sequence logos of Cas8/5 were made with WebLogo.67 The Cas8/5 sequences were collected with the information from a previous study.3 The protein sequences were deduplicated with CD-HIT68,69 using a sequence identity cut-off of 0.95, then aligned with MUSCLE v3.8.31.70 Sequences belonging to type I-F3a and I-F3b in the multiple alignments were then separated and used to make consensus sequence logos.
Quantification and Statistical Analysis
Statistical details are listed in the figure legends.
Supplementary Material
Table S1. Bioinformatics analysis of the PAM sequence used by I-F3 CAST elements, related to STAR Methods section.
Table S2. Plasmid constructs used in this study, related to STAR Methods section.
Video was generated using the first variability component (the most dominant component) from the 3D variability analysis. Each extreme cluster from the analysis corresponds to partial R-loop (blue) or full R-loop state (grey), respectively. Atomic models were docked into the cryo-EM density of each extreme cluster and colored in a same manner as the main figures.
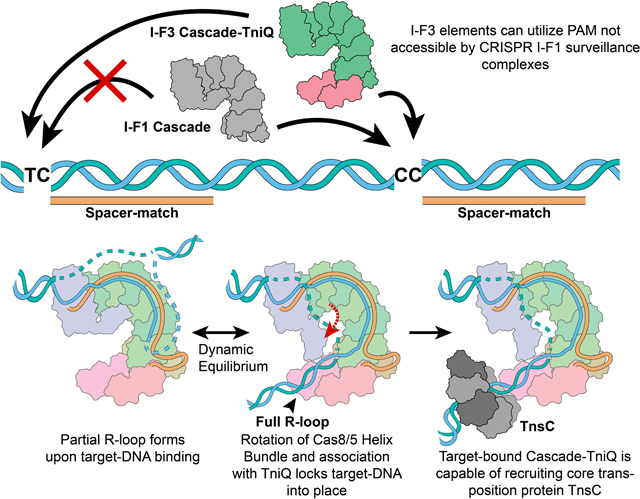
Figure 6. Schematic summarizing key mechanistic insights from this work.

I-F1 CRISPR surveillance complexes (grey) have stricter PAM sequence requirements. However, the I-F3 CAST family is able to make use of loosened PAM requirements. R-loop formation is accompanied by a conformational change in the Cascade complex and locks down on target-DNA via TniQ, revealing the mechanistic coupling between the CRISPR effector and core transposition protein, TniQ. This results in DNA distortions that most likely serve to recruit AAA+ regulator TnsC in order to direct DNA donor integration via the transposase, TnsA/B.
Highlights.
Structures of I-F3 CAST effector module reveal roles of transposon protein TniQ
TniQ participates in complete R-loop formation by engaging with target DNA
TniQ regulates target site choice through interactions with crRNA
Structural basis of PAM ambiguity in I-F3 CAST compared to canonical CRISPR-Cas
Acknowledgements
Supported by NIH grants R00-GM124463 (E.H.K.), R01GM129118 and R21AI148941 (J.E.P.), and GM118174 (A.K.). We thank the Cornell Center for Materials Research facility, as well as K. Spoth and M. Silvestry-Ramos, for maintenance of electron microscopes used for this research (NSF MRSEC program, DMR-1719875); XSEDE for computational resources used for image processing (MCB200090 to E.H.K.)
Footnotes
Declaration of Interests
Cornell University has filed patent applications related to this system with M.T.P., J.E.P., and E.H.K as inventors. The J.E.P. lab has corporate funding for research that is not directly related to the work in this publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 2.Peters JE (2019). Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol. Microbiol. 112, 1635–1644. 10.1111/mmi.14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petassi MT, Hsieh SC, and Peters JE (2020). Guide RNA Categorization Enables Target Site Choice in Tn7-CRISPR-Cas Transposons. Cell 183, 1757–1771 e1718. 10.1016/j.cell.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters JE (2015). Tn7. In Mobile DNA III L Nancy Craig, Rice P, Lambowitz A, Gellert M, and Sandmeyer SB, eds. (ASM Press; ), pp. In Press. [Google Scholar]
- 5.Halpin-Healy TS, Klompe SE, Sternberg SH, and Fernández IS (2020). Structural basis of DNA targeting by a transposon-encoded CRISPR-Cas system. Nature 577, 271–274. 10.1038/s41586-019-1849-0. [DOI] [PubMed] [Google Scholar]
- 6.Wang B, Xu W, and Yang H (2020). Structural basis of a Tn7-like transposase recruitment and DNA loading to CRISPR-Cas surveillance complex. Cell research 30, 185–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z, Zhang H, Xiao R, and Chang L (2020). Cryo-EM structure of a type IF CRISPR RNA guided surveillance complex bound to transposition protein TniQ. Cell research 30, 179–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia N, Xie W, de la Cruz MJ, Eng ET, and Patel DJ (2020). Structure-function insights into the initial step of DNA integration by a CRISPR-Cas-Transposon complex. Cell Res 30, 182–184. 10.1038/s41422-019-0272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Punjani A, and Fleet DJ (2020). 3D Variability Analysis: Directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images. Cold Spring Harbor Laboratory. 10.1101/2020.04.08.032466. [DOI] [PubMed] [Google Scholar]
- 10.Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, Santiago-Frangos A, Faith D, Lawrence CM, Lander GC, and Wiedenheft B (2019). Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell 74, 132–142 e135. 10.1016/j.molcel.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharpe PL, and Craig NL (1998). Host proteins can stimulate Tn7 transposition: a novel role for the ribosomal protein L29 and the acyl carrier protein. EMBO J 17, 5822–5831. 10.1093/emboj/17.19.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmitz M, Querques I, Oberli S, Chanez C, and Jinek M (2022). Structural basis for the assembly of the type V CRISPR-associated transposon complex. Cell 185, 4999–5010 e4917. 10.1016/j.cell.2022.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rollins MF, Schuman JT, Paulus K, Bukhari HS, and Wiedenheft B (2015). Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res 43, 2216–2222. 10.1093/nar/gkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rybarski JR, Hu K, Hill AM, Wilke CO, and Finkelstein IJ (2021). Metagenomic discovery of CRISPR-associated transposons. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2112279118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vo PLH, Ronda C, Klompe SE, Chen EE, Acree C, Wang HH, and Sternberg SH (2021). CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol 39, 480–489. 10.1038/s41587-020-00745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S, Zhang Y, Xu J, Zhang J, Zhang J, Yang J, Jiang Y, and Yang S (2021). Orthogonal CRISPR-associated transposases for parallel and multiplexed chromosomal integration. Nucleic Acids Res 49, 10192–10202. 10.1093/nar/gkab752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. 10.1038/s41586-019-1323-z. [DOI] [PubMed] [Google Scholar]
- 18.Klompe SE, Jaber N, Beh LY, Mohabir JT, Bernheim A, and Sternberg SH (2022). Evolutionary and mechanistic diversity of Type I-F CRISPR-associated transposons. Mol Cell. 10.1016/j.molcel.2021.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wimmer F, Mougiakos I, Englert F, and Beisel CL (2022). Rapid cell-free characterization of multi-subunit CRISPR effectors and transposons. Molecular Cell. 10.1016/j.molcel.2022.01.026. [DOI] [PubMed] [Google Scholar]
- 20.Luscombe NM, Laskowski RA, and Thornton JM (2001). Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res 29, 2860–2874. 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuduvalli P, Rao JE, and Craig NL (2001). Target DNA structure plays a critical role in Tn7 transposition. EMBO J. 20, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Y, Gomez-Blanco J, Petassi MT, Peters JE, Ortega J, and Guarné A (2022). Structural basis for DNA targeting by the Tn7 transposon. Nature Structural & Molecular Biology 29, 143–151. 10.1038/s41594-022-00724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc Natl Acad Sci U S A 114, E7358–E7366. 10.1073/pnas.1709035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh S-C, and Peters JE (2021). Tn7-CRISPR-Cas12K elements manage pathway choice using truncated repeat-spacer units to target tRNA attachment sites. bioRxiv. 10.1101/2021.02.06.429022. [DOI] [Google Scholar]
- 25.Mitra R, McKenzie GJ, Yi L, Lee CA, and Craig NL (2010). Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mob DNA 1, 18. 10.1186/1759-8753-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Q, Straus MR, Caron JJ, Wang H, Chung YS, Guarne A, and Peters JE (2015). Conformational toggling controls target site choice for the heteromeric transposase element Tn7. Nucleic Acids Res. 10.1093/nar/gkv913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parks AR, Li Z, Shi Q, Owens RM, Jin MM, and Peters JE (2009). Transposition into replicating DNA occurs through interaction with the processivity factor. Cell 138, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito M, Ladha A, Strecker J, Faure G, Neumann E, Altae-Tran H, Macrae RK, and Zhang F (2021). Dual modes of CRISPR-associated transposon homing. Cell. 10.1016/j.cell.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsieh S-C, and Peters JE (2022). Discovery and characterization of novel type I-D CRISPR-guided transposons identified among diverse Tn7-like elements in cyanobacteria. Nucleic Acids Research, 1–18. 10.1093/nar/gkac1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffmann FT, Kim M, Beh LY, Wang J, Vo PLH, Gelsinger DR, George JT, Acree C, Mohabir JT, Fernández IS, and Sternberg SH (2022). Selective TnsC recruitment enhances the fidelity of RNA-guided transposition. Nature. 10.1038/s41586-022-05059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao JE, Miller PS, and Craig NL (2000). Recognition of triple-helical DNA structures by transposon Tn7. Proc. Natl. Acad. Sci. USA 97, 3936–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JU, Tsai AW, Mehrotra E, Petassi MT, Hsieh SC, Ke A, Peters JE, and Kellogg EH (2021). Structural basis for target site selection in RNA-guided DNA transposition systems. Science 373, 768–774. 10.1126/science.abi8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Querques I, Schmitz M, Oberli S, Chanez C, and Jinek M (2021). Target site selection and remodelling by type V CRISPR-transposon systems. Nature. 10.1038/s41586-021-04030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J, Tsai A, Rizo A, Truong V, Wellner T, Schargel R, and Kellogg E (2022). Structures of the holo CRISPR RNA-guided transposon integration complex. Nature. 10.1038/s41586-022-05573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tegunov D, and Cramer P (2019). Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16, 1146–1152. 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 37.Bepler T, Morin A, Rapp M, Brasch J, Shapiro L, Noble AJ, and Berger B (2019). Positive- unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Methods 16, 1153–1160. 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Punjani A, and Fleet DJ (2021). 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J Struct Biol 213, 107702. 10.1016/j.jsb.2021.107702. [DOI] [PubMed] [Google Scholar]
- 40.Zivanov J, Nakane T, and Scheres SHW (2019). A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17. 10.1107/S205225251801463X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halpin-Healy TS, Klompe SE, Sternberg SH, and Fernandez IS (2020). Structural basis of DNA targeting by a transposon-encoded CRISPR-Cas system. Nature 577, 271–274. 10.1038/s41586-019-1849-0. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, and Zhang Y (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43, W174–181. 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frenz B, Walls AC, Egelman EH, Veesler D, and DiMaio F (2017). RosettaES: a sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat Methods 14, 797–800. 10.1038/nmeth.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Afonine PV, Klaholz BP, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, and Urzhumtsev A (2018). New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr D Struct Biol 74, 814–840. 10.1107/S2059798318009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Echols N, Grosse-Kunstleve RW, Afonine PV, Bunkoczi G, Chen VB, Headd JJ, McCoy AJ, Moriarty NW, Read RJ, Richardson DC, et al. (2012). Graphical tools for macromolecular crystallography in PHENIX. J Appl Crystallogr 45, 581–586. 10.1107/S0021889812017293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, and Baker D (2003). Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368. 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 52.Dantas G, Kuhlman B, Callender D, Wong M, and Baker D (2003). A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J Mol Biol 332, 449–460. 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 53.Hu X, Wang H, Ke H, and Kuhlman B (2007). High-resolution design of a protein loop. Proc Natl Acad Sci U S A 104, 17668–17673. 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leaver-Fay A, Kuhlman B, and Snoeyink J (2005). An adaptive dynamic programming algorithm for the side chain placement problem. Pac Symp Biocomput, 16–27. [PubMed] [Google Scholar]
- 55.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, et al. (2011). RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One 6, e20161. 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, and Baker D (2010). Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res 38, 5601–5608. 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, Stoddard BL, and Baker D (2009). Exploitation of binding energy for catalysis and design. Nature 461, 1300–1304. 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashworth J, and Baker D (2009). Assessment of the optimization of affinity and specificity at protein-DNA interfaces. Nucleic Acids Res 37, e73. 10.1093/nar/gkp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ Jr., Stoddard BL, and Baker D (2006). Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 441, 656–659. 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, et al. (2017). The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 13, 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters JE (2007). Gene Transfer in Gram-Negative Bacteria. In Methods for General and Molecular Microbiology, Reddy CA, Beveridge TJ, Breznak JA, Marzluf GA, Schmidt TM, and Snyder LR, eds. (ASM Press; ). 10.1128/9781555817497.ch31. [DOI] [Google Scholar]
- 62.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, and de Hoon MJ (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunter JD (2007). Matplotlib: A 2D graphics environment. Computing in science & engineering 9, 90–95. [Google Scholar]
- 64.Waskom ML (2021). Seaborn: statistical data visualization. Journal of Open Source Software 6, 3021. [Google Scholar]
- 65.Ondov BD, Bergman NH, and Phillippy AM (2011). Interactive metagenomic visualization in a Web browser. BMC bioinformatics 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, and Beisel CL (2016). Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Molecular cell 62, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W, and Godzik A (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 69.Fu L, Niu B, Zhu Z, Wu S, and Li W (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Bioinformatics analysis of the PAM sequence used by I-F3 CAST elements, related to STAR Methods section.
Table S2. Plasmid constructs used in this study, related to STAR Methods section.
Video was generated using the first variability component (the most dominant component) from the 3D variability analysis. Each extreme cluster from the analysis corresponds to partial R-loop (blue) or full R-loop state (grey), respectively. Atomic models were docked into the cryo-EM density of each extreme cluster and colored in a same manner as the main figures.
Data Availability Statement
All data generated in this study, including atomic coordinates for Cascade-TniQ bound to target DNA for both full and partial R-loop states, have been deposited. Raw imaging data have been deposited at Mendeley data. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
All original code used for analysis have been deposited at Mendeley. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH5α | J. Peters Lab | N/A |
| Escherichia coli BW27783 attTn7::miniTn7(miniTn6900(kanR)) | J. Peters Lab, Pettasi et al., 2020 | MTP997 |
| Escherichia coli BL21-AI | Invitrogen | Cat# C607003 |
| Escherichia coli CW51 | J. Peters Lab | JP606 |
| T7 Express Competent Escherichia coli | NEB | Cat# C2566H |
| Critical commercial assays | ||
| Q5® High-Fidelity DNA polymerase | NEB | Cat# M0491L |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | Cat# E2621L |
| His-Pur™ Ni-NTA resin | ThermoFisher | Cat# 88221 |
| CloneJET PCR Cloning Kit | ThermoFisher | Cat# K1231 |
| GeneJET Gel Extraction Kit | ThermoFisher | Cat# K0691 |
| T4 DNA Ligase | ThermoFisher | Cat# EL0011 |
| Deposited data | ||
| Full R-loop Cascade-TniQ complex | This paper | PDB: 7U5D EMDB: 26348 |
| Partial R-loop Cascade-TniQ complex | This paper | PDB: 7U5E EMDB: 26349 |
| Custom code for Rosetta simulations and PAM analyses and plots | This paper | Mendeley Data DOI: 10.17632/gjgspgnxvy.1 |
| NGS data for PAM analysis | This paper, NCBI Sequence Read Archive | Bioproject: PRJNA935286 |
| Oligonucleotides | ||
| Oligo_cryo1, CCGCAAGAGGATGATTCGGGTGCTTACCTCCTGACCTTCTTTAGTAGGTTCAACCCCTGATCGAGTGCCGGGATGT | This paper | N/A |
| Oligo_cryo2, TGGCCCCATCAGCCACATCCCGGCACTCGAAGTCCCCAACTTGGATGATTTCTTCCAGTCCTGGTAAGCACCCGAATCATCCTCTTGCGG | This paper | N/A |
| Oligo_cryo3, GGCTGATGGGGCCACCACCTTGCGCTCGTTCGCCAGCCAG | This paper | N/A |
| Oligo_cryo4, CTGGCTGGCGAACGAGCGCAAGGTGG | This paper | N/A |
| F_EMSA, ACCTGCAGGCATGCAAG | This paper | N/A |
| R_EMSA, /Cy5/GCAGTTAAGTGCCTGCTGGCGAGGAAGCGGAAGAG | This paper | N/A |
| DNA substrate for EMSA, Top_ffs_bubble_EMSA Cy3/GCGGCACAGGTCTCACGCGTTCGTCACTACCAGGACTGGAAGAAATCATCCAAGTTGGGGACTTCGAGTGCCGGGATGTGGCTGATGGGGCCACCACCTGTTTCTGCACTTCAACCAGTTAGCCTCCAGAGCAACCGAGACCCGCTGCTG | This paper | N/A |
| DNA substrate for EMSA, Bottom_ffs_bubble_EMSA CAGCAGCGGGTCTCGGTTGCTCTGGAGGCTAACTGGTTGAAGTGCAGAAACAGGTGGTGGCCCCATCAGCCACATCCCGGCACTCGATTATTTTTTTAATTTATTAAATAATTTTATTAGGTAGTGACGAACGCGTGAGACCTGTGCCGC | This paper | N/A |
| Recombinant DNA | ||
| All plasmids used and constructed in this study | This paper | See Table S2 |
| Software and algorithms | ||
| Warp | Tegunov et al., 201935 | http://www.warpem.com/warp/ |
| cryoSPARC | Punjani et al., 201736 | https://cryosparc.com/ |
| Topaz | Bepler et al., 201937 | https://cb.csail.mit.edu/cb/topaz/ |
| RELION | Scheres, 201238 | https://relion.readthedocs.io/en/release-4.0/ |
| cryoSPARC 3DVA | Punjani et al., 202139 | https://cryosparc.com/ |
| UCSF Chimera | Pettersen et al., 200442 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 201843 | https://www.cgl.ucsf.edu/chimerax/ |
| I-TASSER | Yang et al., 201545 | https://zhanggroup.org/I-TASSER/ |
| Coot | Emsley et al., 201046 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| RosettaES | Frenz et al., 201747 | https://dimaiolab.ipd.uw.edu/software/ |
| MolProbity | Williams et al., 201848 | http://molprobity.biochem.duke.edu/ |
| Phenix | Afonine et al., 201849 | https://phenix-online.org/ |
| Biopython | Cock et al., 200962 | https://biopython.org/ |
| Matplotlib | Hunter, 200763 | https://matplotlib.org/ |
| Seaborn | Waskom, 202164 | https://seaborn.pydata.org/ |
| Krona | Ondov et al., 201165 | https://github.com/marbl/Krona/wiki |
| WebLogo 3 | Crooks et al., 200467 | http://weblogo.threeplusone.com/ |
| Cd-hit | Li et al., 200668 | https://github.com/weizhongli/cdhit |
| MUSCLE | Edgar, 200470 | http://www.drive5.com/muscle |