

Abstract
Acute myeloid leukemia (AML) is a heterogeneous hematopoietic neoplasm which results in clonal proliferation of abnormally differentiated hematopoietic cells. In this review, mechanisms contributing to myeloid leukemogenesis are summarized, highlighting aberrations of epigenetics, transcription factors, signal transduction, cell cycling, and the bone marrow microenvironment. The mechanisms contributing to AML are detailed to spotlight recent findings that convey clinical impact. The applications of current and prospective therapeutic targets are accentuated in addition to reviews of treatment paradigms stratified for each characteristic molecular lesion – with a focus on exploring novel treatment approaches and combinations to improve outcomes in AML.
Keywords: Acute myeloid leukemia, Leukemogenesis, Epigenetics, Transcription factor, Oncoprotein, Signal transduction, Cell cycle, Novel strategies, Targeted therapy
1. Introduction
Acute myeloid leukemia is a clonal bone marrow disorder reflecting an expansion of a progenitor cell arrested in development, often resulting in hyperproliferation of hematopoietic precursor cells. The SEER-reported age-adjusted incidence of AML has gradually increased over the last few decades, currently at 4 per 100,000 per year [1]. It is primarily a disease of older adults, with the median age at diagnosis of 68 years [2]. Despite recent advances in refinement of predictive and prognostic markers and the assessment of a multitude of novel agents, both FDA-approved and in clinical trials, the mortality rate of AML has remained stable over the past two decades, currently the fifth worst 5-year overall survival when stratified by cancer type at 24.0%, with an approximate median overall survival of 8.5 months [1].
Complicating the analysis of predictive markers for therapeutic regimens in AML is the existence of clonal hematopoiesis of indeterminate potential (CHIP), in which otherwise healthy patients devoid of overt disease harbor mutations in genes commonly mutated in myeloid neoplasms, such as DNMT3A, TET2, ASXL1, and TP53 [3]. While most patients with CHIP do not exhibit AML or myelodysplastic syndrome (MDS), clonal hematopoiesis was found to be associated with decreased overall survival, with age being the largest contributor to the risk of mutation emergence [3]. Consequently, there is a need to identify the precise mechanisms of mutations implicated in leukemogenesis to tailor rational approaches to therapy and improve outcomes.
Recent years have witnessed dramatic developments in molecular biology, genomics, the understanding of the role of the bone marrow microenvironment, as well as their corresponding effects on leukemogenesis. It is widely recognized that the evolution of AML is unlikely to represent the result of a single biological aberration – but instead the consequence of multiple and synergistic aberrations in epigenetic events, cell cycling, proliferation, signal transduction, and apoptosis. The diverse mechanisms culminating in AML support the notion of a distinctly heterogeneous disease that extends beyond prior and current classifications. In this article, we review and examine the major contributors leading to the development of AML with an emphasis on recent insights and future directions in targeted therapies designed to rationally exploit implicated pathways.
2. Epigenetics
2.1. Overview of epigenetics in AML
Understanding leukemogenesis requires an appreciation of two concepts – the function of proteins of commonly mutated genes and how the relevant genes are expressed. Gene expression is a tightly regulated process with derangements observed frequently in AML. Nucleosomes represent a length of DNA coiled around packing proteins known as histones, which function as transcriptional regulators. Common histone alterations include methylation, acetylation, and ubiquitination – and each histone modification can lead to differential effects on gene transcription or gene repression. In addition to histone alternations, DNA methylation and noncoding RNA species also modify transcriptional activity, and collectively constitute an overarching regulatory process of gene expression known as epigenetic modifications, summarized in Table 1.
Table 1.
Major regulators of epigenetics in AML.
| Role in epigenetics | Implicated proteins or molecules |
|---|---|
| DNA methylation | • DNMT family |
| DNA demethylation | • TET family • IDH1/2 (via supporting catalytic function of TET proteins) |
| Histone methylation | • EZH2 • KMT2A (MLL) • DOT1L |
| Histone demethylation | • LSD1 (reviewed elsewhere) |
| Histone acetylation | • Histone acetyltransferases |
| Histone deacetylation | • Histone deacetylases (HDACs) |
| Histone deubiquitiniation | • ASXL1 • BAP1 |
| MicroRNA | • miR-9 (reviewed elsewhere) |
DNA methylation is the most well-studied epigenetic modification in AML. Cytosine-guanine dinucleotides, also known as CpGs, control gene expression in clusters that overlap or are adjacent to promoter regions. Methylated CpGs are found in repressed chromatin characterized by transcriptionally silenced activity and are generally associated with tumor suppressor genes. Conversely, unmethylated CpGs promote active transcription and are often found in association with housekeeping genes [4]. While methylated promoters are always repressed, absence of methylation does not always lead to enhanced promoter activity [5]. In neoplastic development, the degree of CpG-island hypermethylation (as observed in leukemia and lymphoma) or global genomic hypomethylation (as observed in colon, lung, or breast cancer) increases with progression from a benign to a malignant state [6].
DNA hypomethylating agents (HMAs) used in treatment of AML, such as decitabine or azacitidine, reduce methylation on a genome-wide scale by forming a covalent bond with DNA methyltransferases (DNMTs) [4], but the effects of these agents are not permanent [7,8]. In the absence of these agents, re-methylation is commonly observed and is thought to culminate in disease relapse. This provides a theoretical basis for the indefinite continuation of hypomethylating agents after an induction period until disease progression. However, the precise mechanisms of azacitidine and decitabine in the treatment of AML have not been definitively resolved. While both agents do result in hypomethylation through inhibition of DNMTs [9], additional potential therapeutic mechanisms include induction of tumor suppressor genes and incorporation into DNA with decitabine or RNA with azacitidine [10,11]. In clinical practice, hypomethylating agents, with or without additional therapies, are used in the treatment of older patients with AML and unfavorable risk cytogenetics, for those who are not candidates for intensive induction as monotherapy, as well as in the post-remission setting.
MicroRNA (miRNA) represent noncoding RNA that targets and down-regulates mRNA [12]. Following nuclear processing, mature miRNA binds to the RNA-induced silencing complex (RISC) and pairs with complementary mRNAs, which results in decreased translation and protein expression. Dysregulated miRNAs can be associated with oncogenesis (oncomiR) or they can down-regulate mRNA encoding for tumor suppressors and influence a diverse range of leukemic processes [13,14]. MiRNAs implicated in leukemia are frequently disrupted by epigenetic silencing via RUNX1-RUNX1T1 while cooperating with the PI3K pathway, resulting in increased signal transduction [15] and dysregulation of transcription factors [16]. Reversing aberrant epigenetic changes may lead to restoration of appropriate miRNA expression, with either hypomethylating agents, or with targeted therapy to disrupt overactive downstream pathways such as PI3K. Despite pharmacokinetic challenges, administration of tumor suppressor miRNA mimics or locked nucleic acid oligonucleotide inhibitors, which feature a fixed conformation of the ribose ring of RNA, provide additional avenues of miRNA modulation [17].
2.2. DNA methyltransferases
The DNMTs regulate epigenetic modifications via methylation of cytosine primarily at CpG dinucleotides to create 5-methylcytosine (Fig. 1A). There are five primary DNMTs: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L – and of these, DNMT1, 3A, and 3B regulate most of the core processes of DNA methylation in humans. DNMT1 is the primary housekeeper and maintains existing DNA methylation patterns. In contrast, DNMT3A and 3B establish new methylation patterns and are known as de novo DNA methyltransferases. Loss of DNMT activity results in altered methylation patterns, culminating in aberrations of critical regulators of HSC differentiation, such as PU.1, IKAROS, and RUNX1 – impairing terminal differentiation and leading to AML [18].
Fig. 1.
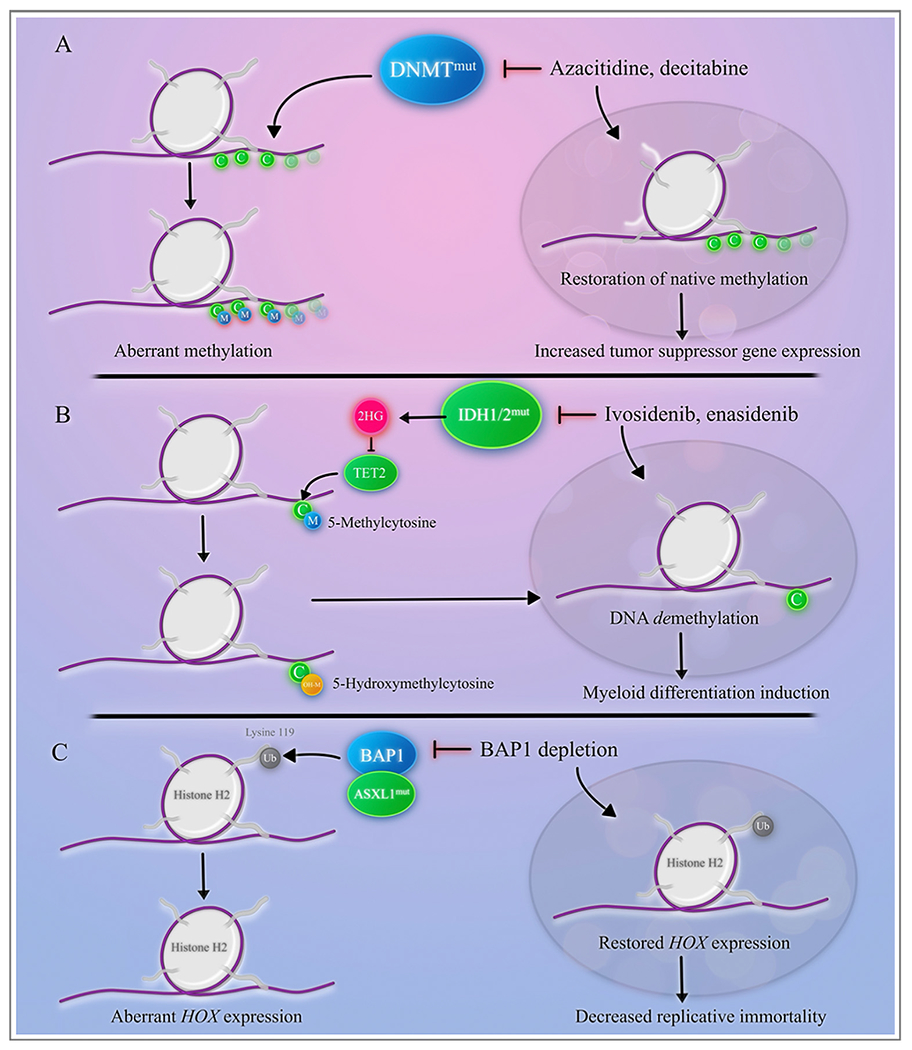
Epigenetic regulators in acute myeloid leukemia.
A. The DNMT family converts cytosine residues to 5-methylcytosine and are responsible for somatic methylation patterns. DNMTmut creates aberrant methylation patterns and disruption of gene expression. Hypo-methylating agents, including azacitidine and decitabine, restore methylation through inhibition of DNMTmut, allowing for increased tumor suppressor gene expression.
B. TET2 modifies 5-methylcytosine to 5-hydroxymethylcytosine, resulting in DNA demethylation. TET2 is inhibited by 2-hydroxyglutarate (2HG), produced by mutated IDH1 or IDH2. IDH1 and IDH2 inhibitors indirectly restore methylation patterns and result in myeloid differentiation C. ASXL1 cooperates with BAP1 to deubiquitinate lysine 119 on histone H2, contributing to a gain-of-function effect and resulting in a differentiation block. Depletion of BAP1 restores HOX gene expression by decreasing HOX-mediated replicative immortality.
Perturbed transcription of the DNMT genes, particularly DNMT3A/B, results in a truncated, non-catalytic isoform with a higher frequency of locus-specific irregularities that accelerates tumorigenesis [19–22]. DNMT aberrations result in dysregulation of methylation patterns and alteration of gene expression, and a clear link has been demonstrated between tumorigenesis and methylation of tumor suppressor genes [23]. Mutations in both DNMT3A (commonly in codon R882) and DNMT3B have been associated with decreased overall survival, increased disease relapse, and a poorer prognosis in patients with AML [24–26]. Smaller studies have suggested that hypomethylating agents may have utility in treating patients with AML and DNMT3A mutations, given their superior overall response rate and median overall survival compared to DNMT3A-wild type patients, although these results did not reach statistical significance [27].
2.3. TET2 and IDH1/2
Aberrant TET2 is present in up to 24% of myeloid neoplasms, including primary myelofibrosis, chronic myelomonocytic leukemia, MDS, and AML [28–30]. Similar to the DNMT family, TET2 and IDH1/2 mutations also regulate epigenetic expression, although the TET and IDH family proteins are responsible for DNA demethylation. Ten-eleven translocation methylcytosine deoxygenase 2 (TET2) converts 5-methylcytosine to 5-hydroxymethylcytosine ( Fig. 1B) [31,32]. Following conversion of 5-methylcytosine by the TET proteins, DNA demethylation subsequently occurs through base excision and opposes the action of DNA methyltransferases [33]. The leukemogenic mechanism appears to be enhancement of HSC self-renewal through aberrant methylation associated with loss of functional TET2 [34]. Consequently, wild-type TET2 acts as a tumor suppressor to maintain hematopoiesis through maintenance of appropriate demethylation, with TET2mut leading to hypermutagenicitiy and a tendency to develop mutations in FLT3 and NOTCH1 [35]. Loss-of-function of TET2 is associated with a poorer prognosis in AML patients with intermediate cytogenetics, particularly when combined with additional negative prognostic markers [36]. In a recent study of cell lines with hypermethylated TET2, decitabine reversed TET2 methylation, resulting in induced expression of TET2 and decreased chromosomal instability [37]. Consequently, hypomethylating agents appear to hold promise in the treatment for TET2mut or TET2 silenced AML.
Cytosolic isocitrate dehydrogenase (IDH1) and its mitochondrial homolog (IDH2) are enzymes involved in the citric acid cycle that convert isocitrate to α-ketoglutarate. TET2 mutations appear to be mutually exclusive with IDH1/2 mutations and are associated with similar loss-of-function epigenetic defects, as mutations in IDH1/2 impair TET2 catalytic function [38]. The leukemogenic mechanism of IDH1/2mut is due to the production of an aberrant metabolite, 2-hydroxyglutarate (2HG), a structural analog of α-ketoglutarate. Thus, 2HG inhibits TET2 and induces DNA hypermethylation – the dominant feature of IDH1/2mut AML [38–40].
Enasidenib is a small molecule inhibitor of mutated IDH2 and has been evaluated as monotherapy and in combination with hypomethylating agents. Single-agent enasidenib demonstrated an overall response rate (ORR) of 40% in the relapsed or refractory setting and a median overall survival of 8.0–12.4 months [41]. Similarly, ivosidenib is a small molecule inhibitor of mutated IDH1, demonstrating an ORR of 41.6% in the relapsed or refractory setting [42]. Both ivosidenib and enasidenib are FDA-approved for relapsed or refractory AML harboring IDH1mut or IDH2mut, respectively. These agents are approved as monotherapy and feature a relatively delayed median time to response compared to more intensive strategies. The median time to response of enasidenib as salvage therapy was 1.9 months, with nearly half of the cohort achieving maximum response by cycle 4, and 80% achieving maximum response by cycle 6 [41]. Similar findings were observed with ivosidenib, with the median time to response at 1.9 months and the median time to complete remission at 2.8 months [42]. In addition to use in the relapsed or refractory settings, ivosidenib monotherapy is approved in the first-line setting for IDH1mut AML ineligible for intensive induction. IDH differentiation syndrome is a significant toxicity associated with IDH inhibitors, with rates of grade 3 or higher toxicity reported at 6.4% [41,42].
Combining hypomethylating agents with IDH inhibitors appears to produce augmented responses, with enasidenib and azacitidine demonstrating a composite response rate (CRR; CR + CRi) of 100% in the front-line setting and the median overall survival was not reached at a median follow-up time of 13.1 months [43]. Similar survival benefits were seen with ivosidenib and azacitidine [44]. On the basis of these findings, ivosidenib has been approved in combination with azacitidine for treatment of IDH1mut AML. Multiple clinical trials are underway investigating other combinations of IDH inhibitors with hypomethylating agents [45], the BCL-2 inhibitor venetoclax [46], and intensive induction strategies [47], summarized in Table 2.
Table 2.
Clinical trials in IDH1mat or IDH2mat AML.
| Mutation | Study regimen | Trial identifier |
|---|---|---|
| IDH1mut or IDH2mut | AML | |
| IDH1 mut | Ivosidenib or placebo + azacitidine | NCT03173248 |
| IDH1 mut | Ivosidenib and CPX-351 | NCT04493164 |
| IDH1 mut | Olutasidenib with azacitidine or cytarabine | NCT02719574 |
| IDH2 mut | Enasidenib and venetoclax | NCT04092179 |
| IDH1mut or IDH2mut | Ivosidenib or enasidenib with 7 + 3 followed by cytarabine or mitoxantrone/etoposide consolidation | NCT02632708 |
2.4. Bromodomain-containing proteins
The bromodomain (BRD)-containing proteins are epigenetic modifiers that bind to acetylated lysine on histones [48]. Because the BRD-containing proteins regulate a multitude of cellular processes, they have been implicated in several stages of leukemogenesis, including potentiation of signal transduction events [49]. This class of proteins represents a diverse family including chromatin remodeling proteins, transcriptional coactivators, and the bromodomain and extraterminal domain-containing (BET) proteins [50]. The BET proteins (BRDT, BRD2, BRD3, and BRD4) are responsible for the regulation of RNA transcription and activation of RNA polymerase II, which modulates cell cycle progression through cyclin T1 and CDK9 [51]. Therefore, up-regulation of the BET proteins, particularly BRD4, results in remodeling of chromatin and activates gene transcription through targeting of aurora B kinase (AURKB), FOS, and MYC [52]. Maintenance of AML was found to operate through BRD4-mediated MYC activation, creating persistent self-renewal, while inhibition of BRD4 resulted in cell cycle arrest and induction of apoptosis [53].
BET inhibitors, such as mivebresib (ABBV-075) and birabresib (MK-8628/OTX015), interfere with the binding of BRD-containing proteins to acetylated histones. In turn, BET inhibitors reverse the effects of BRD up-regulation by promoting cell cycle arrest and MYC suppression [54]. Additionally, BET inhibitors were found to block the transcription of BCL2 [55] and MCL1 [56]. As MCL1 is up-regulated in cells exhibiting resistance to venetoclax, such findings provide a rationale for treatment with a BET inhibitor and a BCL-2 inhibitor – either in combination or in sequence. Furthermore, mutated nucleophosmin impairs transport of BRD4 between the nucleus and the cytoplasm, leading to a loss of repression of BRD4 and an increase in the expression of MYC and BCL2 [57]. This suggests patients with NPM1 mutations may be sensitive to BET inhibitors, with or without venetoclax-based combination strategies. Clinical trials are underway evaluating BET inhibitors as monotherapy or in combination approaches, such as with venetoclax [58,59] or azacitidine [60].
2.5. Histone acetyltransferases, deacetylases, and methyltransferases
Unlike the BRD-containing proteins, which function primarily as the “readers” of histone acetylation, histone acetyltransferases are responsible for the addition of acetyl groups to lysine residues. Consequently, histone acetyltransferases can be thought of as the “writers” of histone modifications and histone deacetylases as the “erasers”. Following histone acetylation, recruitment of BET proteins and transcription factors occurs, resulting in assembly of transcription machinery and subsequent gene activation [61]. Malignant cells were associated with relative histone hypoacetylation compared to normal tissues [62], and dysregulated histone deacetylase (HDAC) activity in leukemia has been a subject of intense study. In particular, chimeric proteins frequently seen in AML, such as RUNX1-RUNX1T1 and CBFB-MYH11, recruit HDACs to corepressor complexes and subsequently block transcription of target genes that are ultimately responsible for myeloid differentiation [63].
Inhibitors of histone deacetylation (HDACIs) induce expression of inappropriately silenced genes through induction of a more transcriptionally permissible chromatin structure, resulting in a reversal of a malignant acetylation signature [64]. Following treatment with HDACIs, restoration of differentiation, apoptosis, autophagy, and control of cell-cycle progression occurs [65]. HDACIs as monotherapy have shown limited responses and clinical benefit in AML, with a phase II study of belinostat in relapsed or refractory AML demonstrating no patients achieving a complete or partial response and 25% of patients with stable disease for at least five cycles [64,66]. Combinations of HDACIs with hypomethylating agents, venetoclax, and targeted therapies have been explored to augment clinical response. Despite initial reports of synergism between sequential administration of HDACIs and hypomethylating agents [67,68], this strategy has not resulted in clinical benefit [69]. Phase I trials investigating belinostat, an HDACI, in combination with pevonedistat, a selective NEDD8-activating enzyme inhibitor, are underway following demonstration of reduced tumor burden and improved survival in AML xenograft models [70].
Similarly, methylation of histones plays a critical role in regulation of gene expression responsible for hematopoietic development. Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase that trimethylates lysine 27 of histone 3 (H3K27me3), creating a repressive transcriptional signature [71]. EZH2 is a member of the polycomb group proteins and composes part of the polycomb repressive complex 2 (PRC2), which functions as a direct inhibitor of gene transcription through chromatin compaction. EZH2 overexpression in myeloid malignancies has been correlated with increased methylation of the tumor suppressor p15INK4B, encoded by CDKN2B, and results in poor clinical outcomes [72]. In the context of gain-of-function mutations, EZH2 was shown to function as an oncogene, particularly during disease maintenance [73]. In contrast, in loss-of-function mutations, EZH2 demonstrated tumor suppressor characteristics during AML induction [71,73], and recent studies have demonstrated that loss of EZH2 correlated with dysregulation of HOX gene expression and resistance to chemotherapy in AML [74]. In sum, either gain- or loss-of-function of EZH2 may contribute to leukemic progression.
Inhibitors of EZH2 have been developed, and the most well-studied has been 3-deazaneplanocin (DZNep) [75]. DZNep was shown to induce differentiation and apoptosis in AML and demonstrated synergism with HDACIs, resulting in reduction of HOXA9 expression [76]. The combination of DZNep and decitabine has also been explored in the preclinical setting, with combination therapy demonstrating reactivation of previously silenced target genes, leading to reduced proliferation in leukemic cells [77]. A dual inhibitor of EZH2 and EZH1, UNC1999, was shown to inhibit growth of MLL-rearranged leukemia cells and prolonged survival in murine models [78]. More recently, a second dual inhibitor of EZH1/2, DS-3201 (valemetostat), was discovered to recruit LSCs into the cell cycle, with EZH1/2 inhibition and G-CSF potentiating apoptosis following exposure to venetoclax and a hypomethylating agent [79]. Further utility of EZH2 inhibition in AML remains to be explored in clinical trials.
2.6. Mixed-lineage leukemia
The mixed-lineage leukemia gene (MLL, also known as KMT2A) encodes for a histone methyltransferase that acts as a master regulator of gene expression during hematopoiesis, and whose protein product controls the HOX gene family [80,81]. MLL rearrangements are found in approximately 10% of myeloid leukemias [82,83] and are more common in secondary or therapy-related leukemias, particularly after agents that target topoisomerase II. MLL rearrangements are also seen in adult and pediatric acute lymphoblastic leukemia, as well as mixed-phenotype acute leukemia, and are associated with poor clinical outcomes [84]. The native function of MLL is to maintain the activity of target genes through what was historically thought to be the histone 3 lysine 4 (H3K4) methyltransferase activity of the MLL protein [85]. However, it has recently been discovered that it is the increase in recruitment and activity of the males-absent on the first (MOF) histone 4 lysine 16 (H4K16) acetyltransferases rather than the MLL-intrinsic histone methyltransferase activity that is more directly implicated in maintenance of MLL target genes [86]. Rearrangements of MLL are created by the addition of a fusion partner to produce the MLL fusion oncoprotein, similar to the leukemogenesis patterns exhibited by RUNX1-RUNX1T1, although the variety of fusion partners for MLL-associated rearrangements is vast with over 70 reported fusion proteins described [87].
Understanding leukemogenesis driven by MLL requires understanding the function of HOXA9 and MEIS1. HOXA9 is part of the highly conserved homeobox family responsible for tightly controlling cell differentiation and proliferation, and constitutive expression is critical in maintaining hematopoietic stem cell populations [88]. In leukemias harboring MLL rearrangements, HOXA9 is constitutively expressed during development, leading to persistent replicative immortality of leukemic stem cells [89]. Increased levels of HOXA9 expression due to the presence of the MLL rearrangement are associated with increased histone 3 lysine 79 (H3K79) dimethylation – regulated by the histone methyltransferase disruptor of telomeric silencing 1-like protein, DOT1L [90]. Collectively, these findings have led to the development of clinical trials investigating small molecule inhibitors that target DOTIL or other modulators of chromatin regulation [91].
MEIS1 is a protein dimerization partner of HOXA9, which enhances and regulates expression of the HOX proteins. Consequently, in the context of MLL-rearranged leukemia, it is partially responsible for the differentiation block, leukemic progression, and self-renewal capacity of the leukemic stem cells [92]. Concomitant expression of HOXA9 and MEIS1 or a second cofactor, PBX3, promotes AML development [93,94]. Although the interactions between HOXA9, MEIS1, and PBX3 have been the subject of intense investigation over the last three decades, a unifying explanation for the development of leukemogenesis across all possible MLL fusion partners has not yet been fully elucidated.
Additional therapeutic targets in MLL-rearranged AML include BRD4 [95], the RAS pathway, and menin, which stabilizes the binding of MLL to chromatin and is critical for MLL fusion-driven gene expression [96,97]. Small molecule inhibitors targeting DOTIL, BRD4, MEK (downstream of RAS), and menin are attractive targets either alone or in combination [98] with additional targeted therapy or cytotoxic chemotherapy to improve outcomes in MLL-rearranged leukemia, summarized in Table 3. For example, menin-MLL inhibition has been shown to synergize with FLT3 inhibitors in preclinical models [99] as well as with venetoclax in NPM1mut and MLL-rearranged AML harboring a FLT3 mutation [100]. Additionally, as cyclin-dependent kinase 9 (CDK9) comprises a component of the positive transcription elongation factor b (P-TEFb), a target of MLL, combined CDK9 and BET inhibition has shown preclinical activity in MLL-rearranged AML [101]. Given the complexify and interactions of the members within the transcription complex and the diverse fusion partners in the oncoprotein, MLL-rearranged leukemias represent a highly heterogeneous category, and further studies are needed to facilitate the development of targeted therapies.
Table 3.
Clinical trials in MLL-rearranged AML.
| Mutation | Study regimen | Trial identifier |
|---|---|---|
| MLL-rearranged AML | ||
| MLL-rearranged or NPM1mut | DSP-5336 (menin-MLL inhibitor) | NCT04988555 |
| MLL-rearranged or NPM1mut | SNDX-5613 (menin-MLL inhibitor) | NCT04065399 |
| None specified; relapsed or refractory AML | KO-539 (menin-MLL inhibitor) | NCT04067336 |
| MLL-rearranged AML | EPZ-5676 (DOT1L inhibitor) | NCT01684150 |
2.7. ASXL1
The role of additional sex combs like 1 (ASXL1) in hematopoiesis and leukemogenesis has yet to be fully elucidated. Recent work has revealed that ASXL1 regulates histone modifications through the interaction of ASXL1 and a histone deubiquitinase, BRCA1 associated protein 1 (BAP1). BAP1 is an essential component of the polycomb repressive deubiquitinase (PR-DUB) complex, a member of the polycomb group proteins. PR-DUB opposes the ubiquitination of histones catalyzed by the polycomb repressive complex 1 (PRC1) [102], which down-regulates the transcription of genes controlling stem cell pluripotency and regulation of cell death [102]. More specifically, the PR-DUB complex, composed of BAP1 activated by ASXL1 [103], is recruited to DNA and forms a DNA-protein complex which regulates genes responsible for hematopoietic development, including the HOX gene family [104] (Fig. 1C). ASXL1 is therefore thought to modulate the balance between polycomb and HOX gene expression [105].
ASXL1 mutations promote HSC aberrations while maintaining survival, creating a predisposition to leukemic transformation by cooperating with the acquisition of mutant RUNX1 [106], MLL [106], NRAS [107], or loss-of-function of TET2 [108]. These mutations in ASXL1 frequently occur as frameshift or nonsense mutations that characteristically create a C-terminally truncated protein which increases the function of BAP1 in the modification of histones – including enhancement of deubiquitination of histone H2A [102] and down-regulation of methylated histone H3 [108,109]. Ultimately, the complex of mutated ASXL1 and BAP1 leads to gain-of-function synergistic interactions between the two proteins and is thought to confer a myeloid differentiation block by deubiquitinating H2AK119 located at HOX gene regions [110].
ASXL1 mutations confer adverse-risk disease with lower complete remission rates and inferior survival [111]. Depletion of BAP1 using CRISPR/Cas9 created a profound reduction in BAP1 – and when ASXL1mut was transduced in BAP1-depleted cells, the differentiation block was reversed [110]. This finding prompted the identification of BAP1 small-molecule inhibitors through biochemical screening that inhibit ASXL1-driven leukemogenesis [112]. These studies set the stage for future pre-clinical work involving small-molecule BAP1 inhibitors for ASXL1mut AML in combination with anthracycline-based or venetoclax-based therapeutic approaches.
3. Transcription factors
3.1. Biallelic CEBPA
The CCAAT-enhancer binding protein alpha (CEBPα) is a transcription factor that regulates differentiation and proliferation in myeloid progenitors. Functional CEBPα directly interacts with coactivators and repressors dependent on cell type and allows progression from the myeloblast to granulocytemacrophage progenitors [113]. CEBPA is translated into a 30 kDa isoform and a 42 kDa isoform by initiating translation at different start sites. In mutated CEBPA, which occurs in up to 14% of AML, the synthesis of the 42 kDa protein is impaired and the expression of the 30 kDa protein is preserved, conferring a differentiation block during myeloid maturation [114–117].
There are two main types of CEBPA mutations: an N-terminal frameshift resulting in production of the 30 kDa protein and a C-terminal mutation in the leucine zipper (bZIP), which affects DNA binding, homodimerization, and heterodimerization [118]. These mutations disrupt the activation of transcription of the granulocyte colony-stimulating factor (G-CSF) receptor and the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor, resulting in the differentiation block [119,120]. Additionally, loss of functional CEBPα results in activation of genes responsible for maintaining leukemic cells in their dedifferentiated states [121–123]. Biallelic CEBPA mutations produce two mutated 30 kDa mutations (p30/p30 homodimers): one with the N-terminal mutation and the other with the C-terminal mutation, creating a distinct clinical entity with a favorable prognosis when occurring with normal cytogenetics and without additional molecular aberrations [124]. Since biallelic CEBPAmut AML confers a favorable prognosis, current recommendations involve treatment with intensive cytotoxic chemotherapy with cytarabine and daunorubicin, with or without the anti-CD33 antibody-drug conjugate, gemtuzumab ozogamicin. As additional druggable targets are discovered, their implementation into clinical trials will evolve.
3.2. Nucleophosmin
Nucleophosmin (NPM1) is a multifunctional nucleocytoplasmic shuttling phosphoprotein that is found primarily in the nucleolus [125]. NPM1 regulates the assembly of primordial ribosomal constituents and facilitates their transport through the nuclear membrane to the cytoplasm [125,126]. It is also responsible for maintenance of the cell cycle (targeting CDK2-cyclin E during the initiation of centrosome duplication) [127] and promotion of p53-mediated tumor-suppressor activity [128] . NPM1 is involved in several chromosomal rearrangements that promote leukemogenesis by activating the oncogenic effects of a fused partner, particularly with the retinoic acid receptor α (NPM1-RARα) [129], anaplastic lymphoma kinase (NPM1-ALK) [130], and myeloid leukemia factor 1 (NPM1-MLF1) [131].
In the absence of a partnered gene fusion, it is thought that NPM1mut exerts its leukemogenic effect through aberrant shuttling function, since mutations in NPM1 appear to increase the concentration of the aberrant protein in the cytoplasm and decrease its concentration in the nucleus. This results in rapid up-regulation of HOX gene expression, which is coupled to a gain-of-function interaction between NPM1mut and exportin 1 (XPO1), another protein that facilitates nuclear export [132]. This potentially provides a molecular rationale for induction chemotherapy coupled with an XPO1 inhibitor (such as selinexor) in patients with NPM1mut AML. Uniting nuclear transport and transcriptional machinery appears to promote HOX expression and resultant AML, partially explaining the contribution of NPM1mut to leukemogenesis. New drugs are in development to either restore appropriate nuclear localization of NPM1, suppress HOX expression [133], disrupt oligomerization [134], or induce proteasomal degradation to facilitate apoptosis [135].
In patients with AML and normal cytogenetics, NPM1mut in isolation is associated with a favorable prognosis and good response to front-line induction chemotherapy [126]. In patients with NPM1mut and FLT3-ITD mutations with a high allelic ratio (defined as greater than 0.5), the favorable prognosis is abrogated and the risk is reclassified to the intermediate category [111]. NPM1mut AML was shown to benefit from high-dose daunorubicin (90 mg/m2) compared to lower-dose daunorubicin (45 mg/m2) during induction for patients younger than 60 years, regardless of the presence of a FLT3-ITD mutation [136]. However, when stratified by cytogenetic risk, daunorubicin 90 mg/m2 failed to demonstrate clear benefit in any category in randomized trials compared to daunorubicin 60 mg/m2, although the study included a large fraction of patients older than 60 years and did not specifically investigate the impact of dose escalation on NPM1mut AML [137]. Finally, NPM1mut AML strongly expresses CD33 [138], which establishes a molecular rationale for the use of gemtuzumab ozogamicin (GO) in combination with cytarabine and daunorubicin in this subset of favorable risk disease. A recent randomized trial investigating this notion demonstrated significantly fewer relapses in the GO arm, although at the expense of an increased risk of early death [139].
3.3. Core binding factor
Similar to the prognostic significance of NPM1mut and biallelic CEBPAmut, core-binding factor (CBF) AML is also associated with a favorable prognosis. Runt-related transcription factor 1 (RUNX1, also known as AML1) encodes the α subunit of CBF while core-binding factor β (CBFB) encodes the β subunit. Both CBFα and CBFβ combine to form the CBF complex that is involved in normal hematopoietic progression [140,141]. A loss of function of the CBF complex results in a differentiation block and culminates in CBF-AML [142]. Specific chromosomal rearrangements are seen in this subset of AML: translocation of chromosomes 8 and 21 or inversion of chromosome 16, resulting in the gene fusion products RUNX1-RUNX1T1 (also known as AML1-ETO) and CBFB-MYH11, respectively. Notably, translocations involving CBFs do not induce AML on their own and are postulated to promote leukemogenesis by partnering with additional mutations – particularly those implicated in cell signal transduction, such as NBAS, KRAS, FLT3, and KIT [143–146].
RUNX1-RUNX1T1 exerts its effects through recruitment of gene repression complexes, which alter the chromosomal architecture and epigenetic landscape of target genes. In the fusion protein, RUNX1 contains a DNA binding domain that is fused to a transcriptional corepressor, RUNX1T1 [147]. Therefore, RUNX1-RUNX1T1 negatively interacts with transcription factors that regulate hematopoietic progression by binding to DNA with RUNX1 binding sites and repressing genes responsible for hematopoietic differentiation [148–150]. Thus, RUNX1-RUNX1T1 outcompetes RUNX1, resulting in inhibition of RUNX1-mediated gene expression and maintenance of the HSC-like state. Compounds that inhibit altered gene expression due to RUNX1-RUNX1T1 were identified through a bioinformatics search followed by biological activity testing, subsequently revealing that budesonide and dexamethasone demonstrated potent antileukemic activity and synergy with conventional chemotherapy in murine models via inhibition of RUNX1-RUNX1T1 mediated gene expression and restoration of targets of RUNX1 [151]. A phase II clinical trial of dexamethasone in combination with intensive therapy in older adults with AML is ongoing (NCT03609060).
The DNA-binding domain of RUNX1 forms a heterodimer with CBFß, which stabilizes the binding of RUNX1 to DNA [152]. As these proteins unite to form the CBF complex, it stands to reason that disruptions of either RUNX1 or CBFB will have similar effects in patients with AML. Indeed, the fusion between CBFB-MYH11 represents a disruption of CBFß that stems from the translocation of the smooth muscle myosin heavy chain gene (SMMHC; encoded by MYH11) and is associated with a clinical outcome similar to that observed with RUNX1-RUNX1T1 due to the formation of an aberrant core-binding factor complex. The CBFß-SMMHC fusion protein restricts RUNX1 to the cytoplasm, acting as a dominant repressor [153–155], and mediating leukemogenesis through the RUNX1-dependent mechanisms mentioned above. Collectively, CBF-AML is often treated with intensive induction with cytarabine and daunorubicin, with or without gemtuzumab ozogamicin [156].
Direct inhibition of CBFß-SMMHC-RUNX1 has been investigated using a FRET assay to screen for compounds that inhibited oncoprotein interactions with RUNX1. This led to the identification and modification of a series of small-molecule inhibitors against the oncoprotein, one which eventually came to be designated as AI-10-49. When administered to mice transplanted with inv(16) leukemic cells, AI-10-49 significantly improved survival with increased apoptosis of leukemic cells and repression of MYC [157]. Combination approaches such as synergistic BET inhibition (counteracting BRD4-mediated MYC activation) in conjunction with anti-transcription factor small molecule inhibitors represent a promising area of study for this subset of AML [157].
3.4. Mutated TP53
There are few tumor suppressor genes as well-studied as TP53, which encodes for a transcription factor that surveils cellular stress and activates numerous pathways that truncate cell proliferation. The function of intact p53 in hematopoietic precursor cells is to oppose a leukemic phenotype, with either loss- or gain-of-function mutations favoring leukemogenesis and susceptibility to mutational accumulation. Mutations in TP53 occur in up to 10% of patients with AML [158], with missense mutations among the most common – particularly in R282, which represents a structural mutation. In contrast, truncating or contact mutations, such as R248 or R273, impair p53 binding to DNA [159]. Nevertheless, disruptions of TP53 encompass an AML subtype with an adverse prognosis due to a higher rate of relapse and poorer survival [160],
Murine models suggest that the loss of a single TP53 allele may be sufficient for the activation of a multistep leukemogenic pathway [161]. Additionally, down-regulation of DNA repair, apoptosis, and cell cycle regulation due to TP53mut may also play a role in the promotion of leukemogenesis. In these circumstances, leukemogenesis is likely not due to a single mutation in TP53, but rather the consequence of additional acquired mutations stemming from the dysfunctional p53 protein, resulting in accumulation of cytogenetic abnormalities as well as mutations in epigenetic regulators (IDH1, IDH2, TET2, DNMT), transcription factors (CEBPA, RUNX1, NPM1), or activating signaling pathways (FLT3, RAS, and KIT), arising through clonal evolution [162–164].
Patients with TP53mut treated with front-line anthracycline-based cytotoxic chemotherapy show poor responses and inferior survival compared to cytogenetically normal, TP53 wild-type AML [165]. Decitabine has emerged as a preferred therapy backbone in this setting, with studies showing relatively high response rates, but not durable durations of remission [166,167]. Initial findings with decitabine led to alternative strategies to augment responses and improve survival in TP53mut AML, culminating in the addition of venetoclax, which appears to exert its effects independently of p53 [168]. Following treatment with azacitidine and venetoclax, suppression of oxidative phosphorylation through disruption of the tricarboxylic acid cycle selectively targeted and eradicated LSCs [169]. The addition of venetoclax demonstrated an overall response rate of 47% in de novo TP53mut AML and 24% in relapsed or refractory disease, with the duration of remission still disappointingly short (6.4 months and 3.6 months, respectively) [170]. Further work is needed to delineate prognostic markers for venetoclax-based therapy, particularly in TP53mut AML. A recent analysis of venetoclax-based therapy in relapsed or refractory AML demonstrated durable remissions in NPM1mut and IDH2mut, but resistance to venetoclax was seen in FLT3-ITDmut and loss of TP53 [171]. The impact of cooperating mutations in TP53mut AML undergoing venetoclax-based treatment is less clear.
Targeted strategies have emerged in the treatment of TP53mut AML, with one encouraging innovation being the development of eprenetapopt (APR-246), which binds p53mut and promotes the regulation of transcription factors involved in apoptosis [172], allowing for reacquisition of cell cycle arrest and apoptosis. Eprenetapopt and azacitidine demonstrated an overall response rate of 64% and a complete response rate of 36% in 11 patients [173]. Resistance to BCL-2 inhibition in AML can be overcome through p53-mediated suppression of the RAS pathway, resulting in MCL-1 phosphorylation and degradation, shown in Fig. 2 [174]. Thus, the addition of venetoclax to eprenetapopt with or without a hypomethylating agent represents a theoretical synergistic regimen, with a phase I trial ongoing (NCT04214860).
Fig. 2.
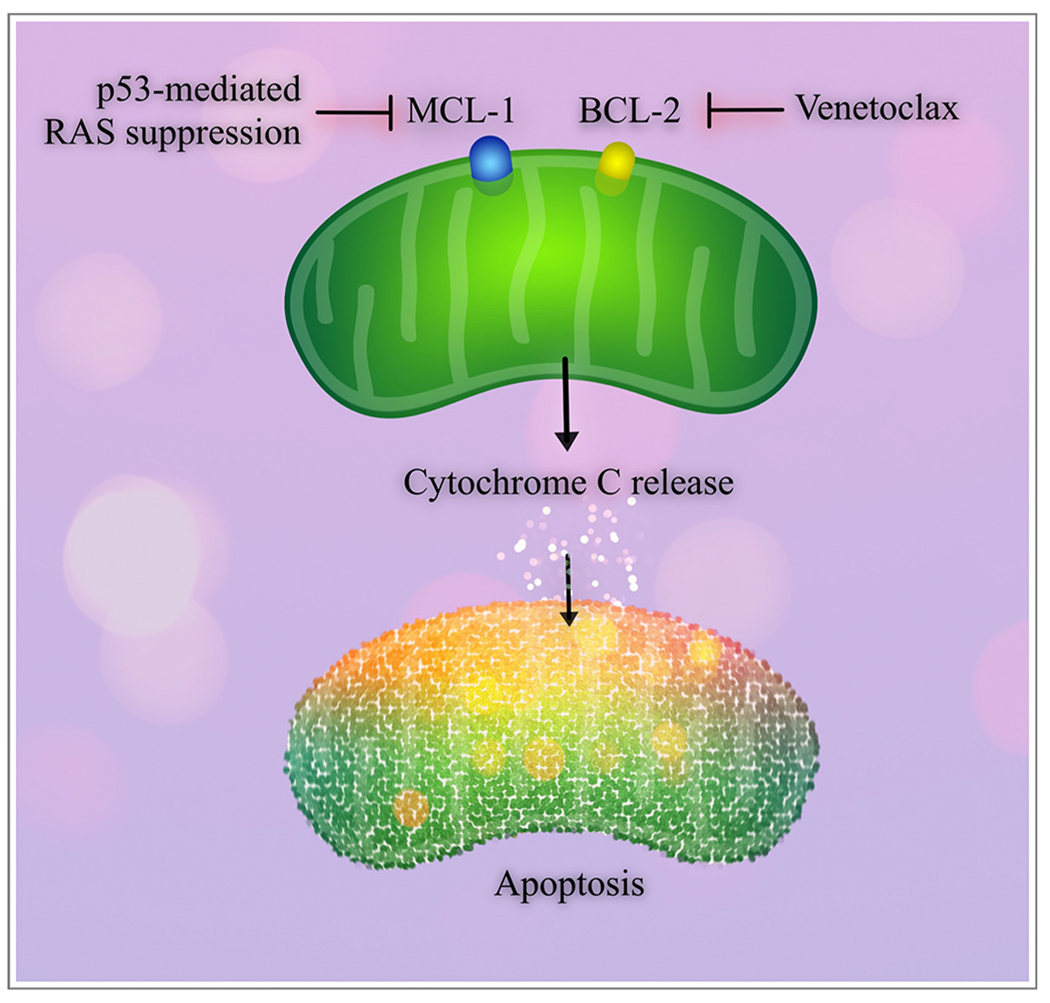
Pro-survival proteins and apoptosis in acute myeloid leukemia.
MCL-1 and BCL-2 are pro-survival proteins that are frequently targeted or dysregulated in AML. Venetoclax is a BCL-2 inhibitor that results in apoptosis through mitochondrial cytochrome C release. BCL-2 resistance can occur through up-regulation of MCL-1, which can be overcome through suppression of the RAS pathway by p53.
MDM2 is a negative regulator of TP53, which inhibits transcription of TP53 and targets p53 for proteasomal degradation [175,176]. Inhibitors of MDM2, e.g., idasanutlin, result in stabilization of p53, induction of apoptosis, and resultant cell-cycle arrest [177]. The addition of idasanutlin to cytarabine in the relapsed or refractory setting was found to improve the CRR at 38.8% compared to 22.0% with cytarabine alone, although there was no difference in the overall survival [178]. However, idasanutlin was found to overcome acquired resistance to venetoclax, with the combination of venetoclax and idasanutlin demonstrating a CRR of 29% and the median survival was 5.7 months [179]. Attempts to dampen proliferation while promoting apoptosis have led to a trial of MEK inhibition with cobimetinib alongside venetoclax and idasanutlin, which is currently underway (NCT02670044).
While not entirely specific to TP53 mutants, immunotherapy has shown promise in the treatment of TP53mut AML. CD47 is a cell-surface molecule that interacts with SIRPα to inhibit phagocytosis of healthy cells by macrophages, and is constitutively up-regulated in myeloid leukemia cells to evade phagocytosis [180]. Magrolimab, a monoclonal antibody to CD47, results in leukemic cell phagocytosis, and was assessed in combination with azacitidine in treatment-naive AML and MDS, 28% of which had TP53mut. In the TP53mut cohort, the composite response rate (CR + CRi) was 88%, and the median OS was not reached at a median follow-up time of 4.9 months [181]. Additionally, LSCs were eliminated in 63% of responding patients in the overall cohort. Importantly, as CD47 expression is lost during red blood cell senescence, a notable event is that magrolimab may potentiate clearance of aging erythrocytes and potentially leads to severe or prolonged anemia [182]. Ongoing clinical trials in TP53mut AML are summarized in Table 4.
Table 4.
Clinical trials in TP53mut AML.
| Mutation | Study regimen | Trial identifier |
|---|---|---|
| TP53mut AML | ||
| TP53mut AML | Eprenetapopt + azacitidine | NCT03931291 |
| TP53mut AML | Decitabine monotherapy | NCT03063203 |
| TP53mut AML | Magrolimab + azacitidine versus venetoclax + azacitidine or intensive chemotherapy | NCT04778397 |
| None specified; relapsed or refractory AML | Idasanutlin, venetoclax, and cobimetinib | NCT02670044 |
| TP53mut/TP53wt and FLT3mut AML | Siremadlin (TP53/MDM2 inhibitor) + midostaurin | NCT04496999 |
4. Signaling pathways
4.1. Mutated RAS
The rat sarcoma (RAS) proteins are signal transducers that relay to downstream targets to regulate cell proliferation [183]. They are commonly implicated in leukemogenesis as cooperating mutations – partners to additional molecular lesions that accelerate tumor formation and subclonal evolution [184]. Activation of the RAS pathway in this context is generally through one of two mechanisms: autonomous signaling of a protein upstream of RAS (such as FLT3 or KIT) [185] or inactivation of a RAS GTPase-activating protein that serves to hydrolyze GTP, resulting in a persistently active GTP-bound form [186].
The ultimate consequence of persistent RAS signaling is an increased sensitivity to GM-CSF and a skewing of the development of the hematopoietic stem cell toward the common myeloid and granulocyte-macrophage progenitor, producing a strong proliferative advantage [187,188]. However, inhibition of MEK, which targets the pathway downstream of RAS by inactivating MAPK/ERK, has had disappointing results as monotherapy [189]. A potential explanation for these findings is that additional cooperative mutations commonly occur in such patients, leading to expansions of subclonal lines that produce disparate cellular phenotypes. A notable exception is that the addition of a TET2 disruption to a RAS mutation potentiates sensitivity of cells to MEK inhibitors and identifies a subset of AML that may preferentially respond to combined MEK inhibition in combination with other known active agents [190]. The addition of venetoclax to cobimetinib, a MEK inhibitor, modulates sensitivity to apoptosis by increasing the action of the pro-apoptotic pathway through suppression of MCL-1 following inhibition of MEK [191].
Further illustrating the importance of cooperative mutations involving the RAS pathway in myeloid leukemogenesis, it was found that up-regulated MAPK signaling conveyed resistance to enasidenib in IDH2 mutants [192], providing a molecular rationale for combined use of enasidenib and cobimetinib – with the potential for additional agents (e.g., venetoclax, hypomethylating therapy, or antibody-drug conjugates). Together, such findings raise the possibility that RAS mutations, which cooperate with additional drivers of leukemogenesis, may promote resistance to therapy – supporting the notion that combined targeting of appropriate aberrant pathways may lead to more effective therapeutic strategies.
4.2. Mutated FLT3
Among the most common activating signaling mutations in AML are the mutations of FLT3. FLT3 encodes FMS-like tyrosine kinase 3, a transmembrane receptor tyrosine kinase that recognizes an extracellular FLT3 ligand. Following binding of the FLT3 ligand, the FLT3 receptor dimerizes and activates the intrinsic tyrosine kinase through phosphorylation, promoting a cascade of downstream parallel signaling pathways including the RAS and phosphatidylinositol 3-kinase (PI3K) pathways, which lead to cell survival, proliferation, and differentiation [193]. PI3K generates a second messenger, PIP3, which binds and activates the AKT family of serine-threonine kinases, subsequently triggering cell growth through up-regulation of the mammalian target of rapamycin (mTOR) and cell survival through negative regulation of FOXO and other pro-apoptotic proteins [194]. Signaling through the RAS pathway, as discussed above, supports the other arm of leukemogenesis downstream from FLT3 – promotion of cell proliferation (Fig. 3).
Fig. 3.
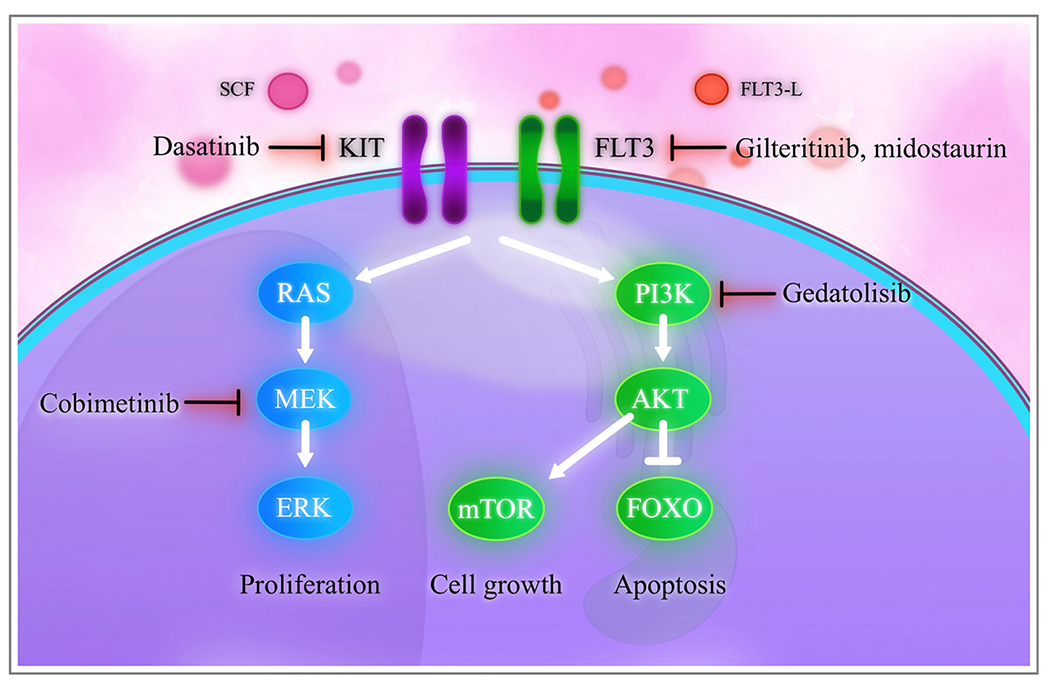
Signaling pathways in acute myeloid leukemia.
Stem cell factor (SCF) binds to the KIT tyrosine kinase receptor, triggering activation of downstream RAS and PI3K. Similarly, the FLT3 ligand (FLT3-L) binds to the FLT3 tyrosine kinase receptor and activates RAS and PI3K. RAS cascades into MEK and ERK, resulting in cell proliferation. PI3K activates AKT, which leads to mTOR-driven cell growth. AKT also inhibits FOXO and other pro-apoptotic proteins, leading to cell survival.
In AML, there are two common constitutively activating FLT3 mutations: an internal tandem duplication (ITD) and a tyrosine kinase domain (TKD) mutation. FLT3-ITD reflects a duplication of a region in the juxtamembrane domain of FLT3, leading to ligand-independent constitutive autophosphorylation, while FLT3-TKD mutations reflect aberrations of the inhibitory property of the activation loop, leading to constitutive tyrosine kinase activation [193]. Patients with FLT3-ITDmut were shown to have inferior survival and increased risk of relapse when compared to FLT3 wild-type [195]. In contrast, the prognostic impact of FLT3-TKDmut is less well defined [196]. Investigation of FLT3 inhibition with tyrosine kinase inhibitors has led to the FDA approval of midostaurin for newly diagnosed AML in combination with cytotoxic chemotherapy [197] and second-generation gilteritinib in the relapsed or refractory setting [198].
With the advent of more selective TKIs targeting FLT3, the role of FLT3 inhibition during induction has become better defined. However, the use of FLT3 inhibitors during consolidation or maintenance remains to be established and represents an area of active study [199], although the use of FLT3 inhibition following allogeneic stem cell transplant has clearly shown benefit [200]. Nevertheless, resistance to FLT3 inhibition has been problematic. For example, while FLT3 inhibition can be achieved at the level of the tyrosine kinase, constitutive activation of downstream pathways still occurs, such as in PI3K or RAS [201]. This raises the possibility of employing multiple TKIs, either in combination or in sequence, to suppress the constitutive activation of parallel signaling pathways. Indeed, the observation that activated PI3K signaling occurred during sorafenib-mediated FLT3 inhibition in FLT3mut AML led to the discovery of FLT3 resistance mediated through the parallel PI3K signaling pathway. Notably, PI3K-mediated resistance was overcome by the addition of the PI3K inhibitor gedatolisib, which induced apoptosis and blocked cell proliferation, demonstrated in Fig. 3 [202]. While monotherapy with PI3K inhibition has not produced clinical benefit [203], combination therapy has not yet been fully explored.
To add to the complexity, recent findings demonstrated that relapsed or refractory AML is associated with a larger FLT3 allelic burden than newly diagnosed disease [204]. The optimal FLT3 inhibitor and sequence of FLT3 inhibition after disease progression remain unknown in the era of newly discovered targeted agents. For example, BCL-2 inhibition may trigger apoptosis and circumvent resistance to FLT3 inhibitors, with phase I trials demonstrating impressive blast clearance in FLT3 mutants [205]. Analogous to the downstream action of MEK inhibition with cobimetinib, gilteritinib down-regulates the pro-survival protein MCL-1. This raises the possibility that the combination of gilteritinib and venetoclax may synergistically circumvent MCL-1-mediated venetoclax resistance [206]. These findings provide a rationale for multi-targeted therapy in FLT3mut disease. A comprehensive review of FLT3 inhibitors in AML has recently been published which covers these concepts in further detail [207].
With respect to cooperative mutations, a potential therapeutic avenue can be envisioned in NPM1mut AML that is concurrently FLT3 mutated. With inhibition of XPO1, e.g., by nuclear transport inhibitors such as selinexor, NPM1mut remains in the nucleus and promotes nuclear localization of p53 and other tumor suppressor proteins [208]. Therefore, mutations in NPM1 may confer a vulnerability to selinexor. In support of this concept, simultaneous targeting of XPO1 and FLT3 with selinexor and either gilteritinib or midostaurin demonstrated enhanced antileukemic activity in mouse models [209]. As NPM1mut AML has been shown to strongly express CD33, the combination of XPO1 and FLT3 inhibition with gemtuzumab ozogamicin remains to be explored. As noted in studies involving RAS mutations, the effect of multiple cooperating mutations requires further investigation to determine the optimal combination of targeted therapies. A summary of ongoing clinical trials in FLT3mut AML is provided in Table 5.
Table 5.
Clinical trials in FLT3mut AML.
| Mutation | Study regimen | Trial identifier |
|---|---|---|
| FLT3mut AML | ||
| FLT3-ITDmut | Gilteritinib + gemtuzumab ozogamicin | NCT05199051 |
| FLT3-ITDmut | Quizartinib and 7 + 3 | NCT04676243 |
| FLT3-ITDmut | Quizartinib + venetoclax | NCT03735875 |
| FLT3mut AML | Gilteritinib + azacitidine | NCT02752035 |
| FLT3mut AML | Gilteritinib + azacitidine and venetoclax | NCT04140487 |
| FLT3mut AML | Gilteritinib + decitabine, cedazuridine, and venetoclax | NCT05010122 |
| FLT3mut AML | Gilteritinib + CPX-351 | NCT05024552 |
| FLT3mut AML | Gilteritinib + atezolizumab | NCT03730012 |
| FLT3mut AML | Gilteritinib versus midostaurin | NCT03836209 |
| FLT3mut AML | Crenolanib versus midostaurin | NCT03258931 |
| FLT3mut AML | CLN-049 (FLT3-CD3 bispecific antibody) | NCT05143996 |
4.3. Mutated kit
The KIT (CD117) protein is a receptor tyrosine kinase that is structurally related to FLT3 and binds to stem cell factor (SCF). Following the binding of SCF to KIT at the surface of the cell membrane, autophosphorylation of the intrinsic tyrosine residues ensues and allows for signal transduction through the RAS, PI3K, and JAK/STAT pathways. In KITmut AML, constitutive activation of the tyrosine kinase leads to persistent signal transduction, creating cytokine-independent growth and reduced apoptosis [210].
Similar to FLT3mut and RASmut, mutations in KIT often cooperate to promote leukemogenesis, although they generally appear later in the process [158]. Constitutively active KIT mutations are commonly found in t(8;21) AML, which features the previously discussed fusion oncoprotein RUNX1-RUNX1T1. Cooperative mutations of KIT in t(8;21) AML correspond to a poorer prognosis than t(8;21) alone, resulting in a higher relapse risk and shorter overall survival in adult AML [211], although the same does not appear to be true in pediatric AML [212].
Nevertheless, exposure of KITmut cells to KIT inhibitors such as dasatinib resulted in endocytosis of the KIT receptor tyrosine kinase and activation of the intrinsic apoptotic pathway [213]. These findings suggest that dasatinib added to intensive cytotoxic chemotherapy with or without gemtuzumab ozogamicin may enhance responses in this subset of CBF-AML [214].
4.4. Hedgehog pathway
There are three extracellular proteins that activate the hedgehog pathway in mammals: sonic hedgehog (SHH), desert hedgehog, and Indian hedgehog. These three proteins are ligands for the patched-1 transmembrane receptor (PTCH1), which acts to inhibit a transmembrane protein known as smoothed (SMO) when the hedgehog ligands are unbound to PTCH1 [215]. Therefore, following binding of the hedgehog ligands, PTCH1 will inhibit SMO, whose function is to trigger the activation of the glioma-associated zinc finger (GLI) family of transcription factors [216]. The GLI transcription factors then accumulate inside the nucleus, where they regulate the hedgehog target genes that control cell cycle progression, differentiation, and survival and are crucial in the self-renewal of hematopoietic stem cells [217,218]. In contrast, in the absence of hedgehog ligand binding, the GLI transcription factors remain inactive in the cytoplasm due to a protein complex known as suppressor of the fused (SUFU).
In AML, there exists increased expression of the sonic hedgehog ligand and downstream GLI transcription factors [219]. One of the first advances in modulation of hedgehog signaling showed that treatment of AML cells with decitabine reduced the levels of hedgehog-interacting proteins and partially reduced SMO activity in leukemic cells by reducing cell proliferation [220]. Further studies prompted the development of SMO inhibitors. Treatment with inhibitors of SMO, such as glasdegib, allows leukemic cells to overcome cytarabine resistance [221]. Sensitivity of the hedgehog pathway to azacitidine was identified through an RNA interference assay, which demonstrated azacitidine-sensitizing hits to SHH, SMO, and GLI with the administration of the SMO inhibitor, erismodegib, in combination with azacitidine. This combination demonstrated inhibition of long-term repopulation capacity in AML and MDS [222]. These findings paved the way for the clinical rationale behind co-administration of glasdegib and low-dose cytarabine, and similarly, with glasdegib and azacitidine [223].
Constitutive activation of the hedgehog pathway may cooperate with persistently aberrant FLT3 signal transduction to promote leukemogenesis [224]. Treatment with the combination of FLT3 and SMO inhibitors resulted in a synergistic reduction of malignant hematopoietic stem cell growth [224]. The importance of these findings is that they support the strategy of targeting parallel pathways in AML. In addition, hyperactivation of the hedgehog pathway maintains chemoresistant cells [225]. While inhibition of the hedgehog pathway represents a prospective therapeutic avenue to overcome drug resistance, a caveat is that eradication of leukemic stem cells is significantly more challenging than eliminating the more differentiated leukemic clones.
5. The bone marrow microenvironment
One of the prominent contributors to AML is the environment in which hematopoietic stem cells (HSCs) reside – the bone marrow. The bone marrow architecture is characterized by specialized stem cell niches at the perivascular and endosteal sites of the intramedullary space, where the bulk of HSCs are found [226]. In addition to de novo leukemia, alterations occurring in the bone marrow niche are also thought to contribute to the pathogenesis of MDS and myeloproliferative neoplasms that predispose to leukemia [226,227]. For example, in the perivascular distribution of the bone marrow, granulocytes and megakaryocytes regulate the release of mature progenitors to the peripheral vasculature. However, in primary myelofibrosis, the marrow microenvironment becomes fibrotic and subsequently dysregulated, leading to the release of leukemic blasts [227]. This exemplifies the importance of the bone marrow microenvironment in the pre-leukemic state, potentially culminating in AML.
In contrast to the vascular niche, which regulates the release of matured bone marrow precursors, the endosteal niche represents the site where most HSCs are located and thereby functions as a concentrated stem cell reserve. In MDS, marrow microenvironment alterations are well documented, including intensified vascularization resulting in increased microvessel density often observed in AML [228,229]. However, data are still in conflict regarding whether or not angiogenesis represents a mechanism causally responsible for progression of MDS to AML [230–233]. For example, microvessel density, expression of associated vascular endothelial growth factor receptor 2 (VEGFR2), angio-poietins, and basic fibroblast growth factor (FGF) were lower in AML arising from MDS (secondary AML) than from de novo AML [231,232]. This suggests neoplastic independence from angiogenesis, a phenomenon that could have clinical implications in the treatment of this distinct subset of AML arising from multilineage dysplasia.
In a more general sense, it is recognized that residual leukemic stem cells (LSCs) residing in the bone marrow represent a major determinant of treatment failure and lead to early relapse [234]. The CD34+ CD123+ LSCs usually reside in the G0 phase of the cell cycle and exhibit multiple mechanisms of drug resistance, including synthesis of proteins such as BCL-2 as well as P-glycoprotein, a broad-spectrum efflux pump responsible for extrusion of xenobiotics and multiple anti-neoplastic agents [235,236]. These residual leukemic cells alter the bone marrow microenvironment, fueling a self-sustaining cycle by generating leukemic clones that have lost the ability to respond appropriately to antiproliferative signals from the surrounding niche [237,238]. The significance of this point can be illustrated through the CXCL12-CXCR4 pathway: CXC motif chemokine ligand 12 (CXCL12) binds to CXC motif chemokine receptor 4 (CXCR4) and serves as a pivotal mediator for stem cell homing, migration, quiescence, and survival within the bone marrow microenvironment [239]. Blocking CXCL12 or its receptor, CXCR4, with either small molecule inhibitors (such as BPRCX807) or monoclonal antibodies has been of recent interest, particularly in combination with targeted therapy, hypomethylating agents, or intensive cytotoxic chemotherapy [238,240]. Similar pharmacological applications can be used for alternative interactions between leukemic and stromal cells, including blockade of VLA-4/VCAM1 [241] and CD44 [242], both of which regulate homing and retention of HSCs in the osteogenic niche. Particularly, CD44, a cell adhesion molecule, appears to promote CXCL12-mediated resistance to venetoclax-mediated apoptosis and preclinical studies have demonstrated that CD44 knockout sensitized AML cells to venetoclax [243].
Additionally, the up-regulation of cytokines in the leukemic bone marrow niche promotes clonal proliferation and inhibits apoptosis through a paracrine mechanism [244] as well as by modulation of mesenchymal stromal cell transcriptosomes by leukemic blasts to suppress normal cell cycle progression [226]. Another relevant phenomenon is the up-regulation of BCL-2 by stroma-supported leukemic blasts, which antagonizes apoptosis and promotes cell survival. This event has been directly targeted by BH3-mimetics such as venetoclax in the treatment of both the acute and chronic leukemias [245]. In sum, the vascular and endosteal leukemic cell niches, in cooperation with contributions from nervous system signaling, aberrant signal transduction, neoangiogenesis, marrow remodeling [226], and metabolic dysregulation [246] may all play roles in facilitating leukemogenesis in the bone marrow [229,247]. Consequently, each of these processes may provide opportunities for therapeutic interventions [229].
Finally, distinct from the above processes, down-regulation of immune detection in the marrow microenvironment has been hypothesized to contribute to a phenomenon of immune escape, protecting leukemia cells from immune destruction [248]. The interaction between programmed cell death-1 (PD-1) and its ligand (PD-L1) functions as an immune checkpoint to promote self-tolerance through suppression of T-cell inflammatory activity. Lower PD-L1 expression in CD34+ cells was found to be associated with prolonged survival in untreated AML patients [249]. However, blockade of PD-1 and PD-L1 via monoclonal antibodies has not shown impressive results as monotherapy in patients with relapsed AML [250]. Hypomethylating agents were shown to induce PD-L1 expression in AML and up-regulation of PD-L1 is postulated to be a mechanism of resistance to these therapies [249]. Concurrent treatment with anti-PD-L1 nivolumab and azacitidine produced a composite response rate of 22% and a median survival of 6.3 months in relapsed or refractory AML, which compared favorably to a cohort of historical controls that received HMA-based salvage [251].
6. Other novel approaches and future directions
In addition to the previously described mechanisms and corresponding strategies in AML, several new avenues are beginning to be explored. For example, the ubiquitin-proteasome system, which controls timely destruction of intracellular proteins, has emerged as an attractive therapeutic target. The cullin protein family are scaffold proteins that provide support for ubiquitin ligases, which control the ubiquitination and destruction of mediators of cell cycle progression, signal transduction, or DNA damage response [252]. Neddylation regulates the activity of these cullins through the NEDD8 conjugation pathway, controlled by the NEDD8-activating enzyme (NAE) [253]. NAE inhibitors were shown to activate the intrinsic and extrinsic apoptotic pathways [254], and NAE inhibitors such as pevonedistat have been evaluated in combination with venetoclax and azacitidine in clinical trials [255]. A clinical trial of pevonedistat and belinostat is ongoing (NCT03772925).
Additional novel strategies include inhibition of the cyclin-dependent kinases (CDKs) – particularly CDK7 and CDK9. CDK7 functions as a catalytic subunit of the CDK-activating kinase (CAK), which functions to activate multiple other CDKs and also activates RNA polymerase II [256]. A multi-target CDK 7/12/13 inhibitor, THZ1, was evaluated in AML cell lines with RUNX1-RUNX1T1 and demonstrated loss of proximal (5′) pausing of RNA polymerase and concomitant suppression of RNA polymerase at the distal (3′) end of genes [257]. Similarly, CDK9 is a transcriptional regulator and comprises the super elongation complex which controls RNA polymerase II. Fusion partners of MLL also constitute the super elongation complex and therefore inappropriately unite CDK9 to HOX promoters, contributing to aberrant CDK9 function. Inhibition of CDK9 with AZD3573 revealed significant MCL-1-mediated apoptotic induction, which was potentiated by venetoclax [258]. Additional preclinical work in this setting demonstrated that concurrent treatment with venetoclax and A-1467729, another small-molecule inhibitor of CDK9, exhibited synergistic cell death [259]. More recently, CDK9 inhibitors were found to result in a reduction of secondary AML burden in xenograft models, with improvement of the overall survival after three weeks of treatment [260]. Additionally, CDK9 inhibitors were found to demonstrate synergistic lethality with anti-BCL-2 strategies in the context of secondary AML [260]. As novel targets are discovered that contribute to or maintain a leukemic state, their application to specifically defined genetic sub-types of AML should present opportunities for newer precision medicine-based treatment strategies. A summary of the mechanisms of leukemogenesis and potential targets is available in Table 6.
Table 6.
Mechanisms of leukemogenesis and potential targets.
| Class | Target | Mechanism | Potential drugs or combinations |
|---|---|---|---|
| Transcription | Factors RUNX1-RUNX1T1 or CBFB-MYH11 | Alteration of epigenetic landscape, negative regulation of transcription factors that regulate hematopoietic progression, cell cycle arrest, repression of tumor suppressors | • AI-10-49 • Anthracycline-based induction with • Anthracycline-based induction with gemtuzumab ozogamicin if favorable risk |
| TP53mut (or TP53 deleted) | Down-regulation of DNA repair, apoptosis, cell cycle regulation; accrual of sequential mutations | • Eprenetapopt (APR-246) • Magrolimab Idasanutlin or other MDM2 inhibition (siremadlin) Hypomethylating agent + venetoclax |
|
| Signal Transduction | RAS mut | Increased sensitivity to GM-CSF, skewed development of the hematopoietic stem cell to the myeloid line | • MEK inhibition (cobimetinib) • RAS inhibition • With or without cytotoxic chemotherapy or venetoclax-based therapy |
| FLT3 mut | Activation of RAS and PI3K pathways | • Midostaurin • Gilteritinib • Quizartinib • Crenolanib • Momelotinib (FLT3/JAK inhibition) • With or without cytotoxic chemotherapy or venetoclax-based therapy |
|
| KIT mut | Activation of RAS, PI3K, and JAK/STAT pathways | Dasatinib with or without cytotoxic chemotherapy or venetoclax-based therapy | |
| Hedgehog pathway | Increased expression of the sonic hedgehog ligand and GLI transcription factors | Glasdegib with hypomethylating agents or low-dose cytarabine | |
| Other Novel Approaches | NEDD8-activating enyzme | Ubiquitination of proteins controlling cell cycle progression, signal transduction, and DNA damage response | Pevonedistat with or without venetoclax-based strategies |
| CDK7/9 | Activation of RNA polymerase II, HOX dysregulation | • THZ1 for CDK7 • AZD3573 for CDK9 • With or without cytotoxic chemotherapy or venetoclax-based therapy |
|
| Bone Marrow Microenvironment | CXCL12 or CXCR4 | Up-regulation of cytokines, promotion of clonal proliferation, inhibition of apoptosis, BCL-2 up-regulation | • BPRCX807 • AMD3100 • With or without venetoclax-based therapy |
| PD-1 or PD-L1 | Immune escape; protecting leukemia cells from immune destruction | Anti-PD-L1/PD-1 with or without HMAs | |
| Epigenetics | CARM1 dysregulated | Histone methylation silencing target genes | • HDAC inhibition (belinostat, givinostat, vorinostat) • With or without cytotoxic chemotherapy or venetoclax-based therapy |
| miR-9 | HOXA9-mediated transformation, hypermethylation, reduction of FOXO | • Locked nucleic acid oligonucleotide inhibitors/tumor suppressor miRNA mimetics • Hypomethylating agents • PI3K inhibition |
|
| DNMT mut | Aberrant methylation and alteration of gene expression | Hypomethylating agents with venetoclax | |
| TET2mut or silenced | Loss of conversion of 5-methylcytosine to 5-hydroxymethylcytosine, resulting in dysregulated methylation | Hypomethylating agents for hypermethylated TET2 with venetoclax | |
| IDH1mut and IDH2mut | Formation of 2-hydroxyglutarate, inhibition of TET2, DNA hypermethylation | • Ivosidenib or olutasidenib for IDH1 • Enasidenib for IDH2 • With or without hypomethylating agent backbones, venetoclax under investigation |
|
| ASXL1 mut | Increases the function of BAP1 in the modification of histones, HOX dysregulation | • Small-molecule inhibitors of BAP1 • With or without cytotoxic chemotherapy or venetoclax-based therapy |
|
| BET proteins | Dysregulated transcriptional activation through RNA polymerase II, DNA repair, and MYC signal transduction | • Mivebresib • Birabresib • Both with or without venetoclax-based therapies |
|
| Histone deacetylases | Blocking transcription of genes responsible for differentiation due to aberrant histone acetylation | • Belinostat • Vorinostat • Pracinostat • In combination with additional targeted therapy, hypomethylating agents, or venetoclax |
|
| EZH2 mut | Methylation of p15INK4B, loss of CEBPα myeloid differentiation, dysregulated HOX gene expression | • DZNep with or without HDACI or hypomethylating agents • UNC1999 • GSK126 • DS-3201 with or without venetoclax-based therapies |
|
| KMT2Amut (or MLL rearranged) | HOXA9 constitutive expression due to DOT1L dysregulation, expression of MEIS1 with PBX3 | • DOT1L inhibitor • MEK inhibitor • BET inhibition (BRD4) • Menin inhibition • With cytotoxic chemotherapy or venetoclax-based therapy |
|
| Transcription Factors | Biallelic CEBPAmut | Disruption of G-CSF and GM-CSF receptors, activation of genes responsible for maintaining leukemic cells in dedifferentiated states | Anthracycline-based induction with gemtuzumab ozogamicin if favorable risk |
| NPM1 mut | Aberrant shuttling between nucleus and cytoplasm, up-regulation of HOX expression | • Selinexor • Menin inhibition • Anthracy cline-based induction with gemtuzumab ozogamicin if favorable risk |
7. Conclusions
Future trials investigating novel combinations involving the aforementioned pathways will be necessary to develop innovative treatment regimens and improve survival in AML. Building upon the backbones of either anthracycline- or venetoclax-based regimens (e.g., venetoclax and azacitidine) appear to be the most promising avenue to improve patient outcomes, with careful selection of novel drugs that are either synergistic or act to delay or mitigate the development of drug resistance or disease relapse. In addition, if tolerable and justified by underlying leukemogenic aberrations, one can envision future rational three-agent regimens involving targeted agents – for example, with venetoclax, CDK9 inhibitors, and FLT3 antagonists for the treatment of FLT3mut MLL-rearranged AML. Notably, as investigations continue to expand the armamentarium of novel agents to be combined with established therapeutic backbones, care must be taken to assess the risk and benefits of such approaches in the light of potential additional toxicities. Further preclinical work coupled with careful conduct of clinical trials will be necessary to develop such innovative treatment strategies for AML.
The numerous paths that lead to the genesis of leukemia are frequently intertwined, and cooperating mutations involving epigenetic regulators, oncoproteins, transcription factors, tumor suppressors, and activating signaling mutations contribute to the enormous complexity of leukemogenesis. A better understanding of these mechanisms will be essential for the implementation of developing new and more effective treatment strategies for this highly heterogeneous disease. It is likely that the development of rational combinations of such inhibitors offers the best chance for overcoming resistance and improving survival in AML. Consequently, the results of novel combination therapies and the discovery of future targets are eagerly anticipated.
Practice points.
There are numerous mechanisms which cooperate to promote leukemogenesis. These include aberrations in epigenetics, transcription factors, signal transduction, DNA damage control, and cell cycling.
As understanding of these mechanisms improves, discovery of potential targets will facilitate the development of novel therapies.
Given the relatively modest activity of many targeted therapies and the frequency of multiple cytogenetic or molecular lesions in AML, combination strategies involving targeted agents should be vigorously explored.
In addition to the use of targeted therapies during induction or treatment of relapsed disease, consideration should be given to evaluating novel agents during consolidation or maintenance to delay relapse.
The use of novel combination regimens in the setting of specific genetically defined AML sub-types is likely necessary for optimal activity.
Care must be taken to select combinations of novel therapies with established approaches to minimize toxicity and maximize efficacy.
Research agenda.
Improvement in the understanding of the molecular mechanisms of leukemogenesis.
Identification of new mechanisms or signaling pathways that can be therapeutically targeted.
Identification and/or development of novel small molecule inhibitors that disrupt or truncate the aforementioned mechanisms.
Demonstration of preclinical activity of novel agents as monotherapy and in synergy with existing conventional or targeted strategies.
Identification of subsets of AML that optimally respond to newer agents.
Demonstration of superiority of novel combination strategies to current standard of care strategies.
Acknowledgements
R01CA205607, P30CA16059, UM1CA186644, Leukemia and Lymphoma Society of America #R6508.
References
- [1].Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev 2019;36:70–87. [DOI] [PubMed] [Google Scholar]
- [2].Song X, Peng Y, Wang X, Chen Y, Jin L, Yang T, et al. Incidence, survival, and risk factors for adults with acute myeloid Leukemia not otherwise specified and acute myeloid Leukemia with recurrent genetic abnormalities: analysis of the surveillance, epidemiology, and end results (SEER) database, 2001-2013. Acta Haematol 2018;139(2):115–27. [DOI] [PubMed] [Google Scholar]
- [3].Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jasielec J, Saloura V, Godley LA. The mechanistic role of DNA methylation in myeloid leukemogenesis. Leukemia. 2014;28(9):1765–73. [DOI] [PubMed] [Google Scholar]
- [5].Estecio MR, Issa JP. Dissecting DNA hypermethylation in cancer. FEBS Lett 2011;585(13):2078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Esteller M. Epigenetics in cancer. N Engl J Med 2008;358(11):1148–59. [DOI] [PubMed] [Google Scholar]
- [7].Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol 2005;23(17):3948–56. [DOI] [PubMed] [Google Scholar]
- [8].Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, et al. DNA methylation changes after 5-aza-2′ -deoxycytidine therapy in patients with leukemia. Cancer Res 2006;66(10):5495–503. [DOI] [PubMed] [Google Scholar]
- [9].Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–28. [DOI] [PubMed] [Google Scholar]
- [10].Lavelle D, DeSimone J, Hankewych M, Kousnetzova T, Chen YH. Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk Res 2003;27(11):999–1007. [DOI] [PubMed] [Google Scholar]
- [11].Oz S, Raddatz G, Rius M, Blagitko-Dorfs N, Lubbert M, Maercker C, et al. Quantitative determination of decitabine incorporation into DNA and its effect on mutation rates in human cancer cells. Nucleic Acids Res 2014;42(19):e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2): 281–97. [DOI] [PubMed] [Google Scholar]
- [13].Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A 2008;105(40):15535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Starczynowski DT, Morin R, McPherson A, Lam J, Chari R, Wegrzyn J, et al. Genome-wide identification of human microRNAs located in leukemia-associated genomic alterations. Blood. 2011;117(2):595–607. [DOI] [PubMed] [Google Scholar]
- [15].Li Y, Gao L, Luo X, Wang L, Gao X, Wang W, et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood. 2013;121(3):499–509. [DOI] [PubMed] [Google Scholar]
- [16].Senyuk V, Zhang Y, Liu Y, Ming M, Premanand K, Zhou L, et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc Natl Acad Sci U S A 2013;110(14):5594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Y, Gao DY, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev 2015;81:128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2011;44(1):23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shah MY, Vasanthakumar A, Barnes NY, Figueroa ME, Kamp A, Hendrick C, et al. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res 2010;70 (14):5840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shannon K, Armstrong SA. Genetics, epigenetics, and leukemia. N Engl J Med 2010;363(25):2460–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ostler KR, Yang Q, Looney TJ, Zhang L, Vasanthakumar A, Tian Y, et al. Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Res 2012;72(18):4714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang J, Walsh G, Liu DD, Lee JJ, Mao L. Expression of Delta DNMT3B variants and its association with promoter methylation of p16 and RASSF1A in primary non-small cell lung cancer. Cancer Res 2006;66(17):8361–6. [DOI] [PubMed] [Google Scholar]
- [24].Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363(25):2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hayette S, Thomas X, Jallades L, Chabane K, Charlot C, Tigaud I, et al. High DNA methyltransferase DNMT3B levels: a poor prognostic marker in acute myeloid leukemia. PLoS One 2012;7(12):e51527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 2012;12(9):599–612. [DOI] [PubMed] [Google Scholar]
- [27].Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009;23(7): 1343–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tefferi A, Lim KH, Levine R. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;361(11):1117. author reply -8. [DOI] [PubMed] [Google Scholar]
- [31].Saint-Martin C, Leroy G, Delhommeau F, Panelatti G, Dupont S, James C, et al. Analysis of the ten-eleven translocation 2 (TET2) gene in familial myeloproliferative neoplasms. Blood. 2009;114(8):1628–32. [DOI] [PubMed] [Google Scholar]
- [32].Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333 (6047):1303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat Commun 2017;8:15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–10. [DOI] [PubMed] [Google Scholar]
- [37].Wang J, He N, Wang R, Tian T, Han F, Zhong C, et al. Analysis of TET2 and EZH2 gene functions in chromosome instability in acute myeloid leukemia. Sci Rep 2020;10(1):2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18(6):553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366(12):1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in preleukemic stem cells in acute myeloid leukemia. Int J Hematol 2013;98(6):648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stein EM, DiNardo CD, Fathi AT, Pollyea DA, Stone RM, Altman JK, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018;378(25):2386–98. [DOI] [PubMed] [Google Scholar]
- [43].Venugopal S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J 2022;12(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med 2022;386(16):1519–31. [DOI] [PubMed] [Google Scholar]
- [45].DiNardo CD, Stein AS, Stein EM, Fathi AT, Frankfurt O, Schuh AC, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol 2021;39(1):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lachowiez CA, Borthakur G, Loghavi S, Zeng Z, Kadia TM, Masarova L, et al. A phase Ib/II study of ivosidenib with venetoclax +/− azacitidine in IDH1-mutated myeloid malignancies. J Clin Oncol 2021;39(15_suppl):7012. [Google Scholar]
- [47].Stein EM, DiNardo CD, Fathi AT, Mims AS, Pratz KW, Savona MR, et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: a phase 1 study. Blood. 2021;137(13):1792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735): 491–6. [DOI] [PubMed] [Google Scholar]
- [49].Reyes-Garau D, Ribeiro ML, Roue G. Pharmacological targeting of BET bromodomain proteins in acute myeloid leukemia and malignant lymphomas: from molecular characterization to clinical applications. Cancers (Basel) 2019;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 2005;19(4):523–34. [DOI] [PubMed] [Google Scholar]
- [52].Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol 2016;23(6):540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011; 478(7370):524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6(19):17698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hogg SJ, Newbold A, Vervoort SJ, Cluse LA, Martin BP, Gregory GP, et al. BET inhibition induces apoptosis in aggressive B-cell lymphoma via epigenetic regulation of BCL-2 family members. Mol Cancer Ther 2016;15(9):2030–41. [DOI] [PubMed] [Google Scholar]
- [56].Mill CP, Cai T, Fiskus W, Borthakur G, Kornblau SM, Kadia TM, et al. Mechanisms underlying superior efficacy of co-targeting BET proteins and anti-apoptotic BCL2 or MCL1 protein against AML blast progenitor cells. Blood. 2018;132(Supplement 1):1351. [Google Scholar]
- [57].Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28(2):311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Borthakur G, Odenike O, Aldoss I, Rizzieri DA, Prebet T, Chen C, et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer. 2021,127(16):2943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ramsey HE, Greenwood D, Zhang S, Childress M, Arrate MP, Gorska AE, et al. BET inhibition enhances the antileukemic activity of low-dose venetoclax in acute myeloid leukemia. Clin Cancer Res 2021;27(2):598–607. [DOI] [PubMed] [Google Scholar]
- [60].Pemmaraju N, Bhalla KN, Daver N, Wilson NR, Fiskus WC, Ravandi F, et al. Phase 1 results of novel combination therapy: BET inhibitor PLX51107 with azacitidine in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Blood 2021;138(Supplement1):3421 [Google Scholar]
- [61].Gaub A, Sheikh BN, Basilicata MF, Vincent M, Nizon M, Colson C, et al. Evolutionary conserved NSL complex/BRD4 axis controls transcription activation via histone acetylation. Nat Commun 2020;11(1):2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 2007;18(4):769–74. [PubMed] [Google Scholar]
- [63].Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol 1998;18(12):7185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25(2):226–35. [DOI] [PubMed] [Google Scholar]
- [65].Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–52. [DOI] [PubMed] [Google Scholar]
- [66].Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM, et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California cancer consortium study. Leuk Lymphoma 2014;55(10):2301–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 2006;66(12):6361–9. [DOI] [PubMed] [Google Scholar]
- [68].Marchi E, Zullo KM, Amengual JE, Kalac M, Bongero D, McIntosh CM, et al. The combination of hypomethylating agents and histone deacetylase inhibitors produce marked synergy in preclinical models of T-cell lymphoma. Br J Haematol 2015,171(2):215–26. [DOI] [PubMed] [Google Scholar]
- [69].Pan T, Qi J, You T, Yang L, Wu D, Han Y, et al. Addition of histone deacetylase inhibitors does not improve prognosis in patients with myelodysplastic syndrome and acute myeloid leukemia compared with hypomethylating agents alone: a systematic review and meta-analysis of seven prospective cohort studies. Leuk Res 2018;71:13–24. [DOI] [PubMed] [Google Scholar]
- [70].Zhou L, Chen S, Zhang Y, Kmieciak M, Leng Y, Li L, et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood. 2016;127(18):2219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014;28(1):44–9. [DOI] [PubMed] [Google Scholar]
- [72].Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, et al. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol 2011;90(6):643–53. [DOI] [PubMed] [Google Scholar]
- [73].Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med 2019;216(4):966–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gollner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med 2017;23(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, et al. 3-deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun 1986;135(2):688–94. [DOI] [PubMed] [Google Scholar]
- [76].Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin a and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Momparler RL, Idaghdour Y, Marquez VE, Momparler LF. Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res 2012;36(8):1049–54. [DOI] [PubMed] [Google Scholar]
- [78].Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125(2):346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chang KH, Alaniz Z, Nishida Y, Dos Santos CE, Slosberg E, Daver N, et al. Inhibition of EZH1 and EZH2 restores chemosensitivity of leukemia stem cells in acute myeloid leukemia by recruitment of quiescent AML stem/progenitor cells. Blood. 2020;136(Supplement 1):27–8. [Google Scholar]
- [80].Giampaolo A, Pelosi E, Valtieri M, Montesoro E, Sterpetti P, Samoggia P, et al. HOXB gene expression and function in differentiating purified hematopoietic progenitors. Stem Cells 1995;13(Suppl. 1):90–105. [PubMed] [Google Scholar]
- [81].Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll mutant mice. Nature. 1995;378(6556):505–8. [DOI] [PubMed] [Google Scholar]
- [82].Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, et al. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res 1998;58(1):55–9. [PubMed] [Google Scholar]
- [83].Munoz L, Nomdedeu JF, Villamor N, Guardia R, Colomer D, Ribera JM, et al. Acute myeloid leukemia with MLL rearrangements: clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia. 2003;17(1):76–82. [DOI] [PubMed] [Google Scholar]
- [84].Chen CS, Sorensen PH, Domer PH, Reaman GH, Korsmeyer SJ, Heerema NA, et al. Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood. 1993;81(9):2386–93. [PubMed] [Google Scholar]
- [85].Ballabio E, Milne TA. Epigenetic control of gene expression in leukemogenesis: cooperation between wild type MLL and MLL fusion proteins. Mol Cell Oncol 2014;1(2):e955330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep 2014;7(4):1239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 1998;17(13):3714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Imamura T, Morimoto A, Takanashi M, Hibi S, Sugimoto T, Ishii E, et al. Frequent co-expression of HoxA9 and Meis1 genes in infant acute lymphoblastic leukaemia with MLL rearrangement. Br J Haematol 2002;119(1):119–21. [DOI] [PubMed] [Google Scholar]
- [90].Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev 2008;22(24):3403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011;20(1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev 2007;21(21):2762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol 2004;24(2):617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Li Z, Zhang Z, Li Y, Arnovitz S, Chen P, Huang H, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121(8):1422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008;14(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A Menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell 2019;36(6):660–73 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Dafflon C, Craig VJ, Mereau H, Grasel J, Schacher Engstler B, Hoffman G, et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia. 2017;31(6):1269–77. [DOI] [PubMed] [Google Scholar]
- [99].Dzama MM, Steiner M, Rausch J, Sasca D, Schonfeld J, Kunz K, et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood. 2020;136(21):2442–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Carter BZ, Tao W, Mak PY, Ostermann LB, Mak D, McGeehan G, et al. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood. 2021;138(17):1637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].McCalmont H, Li KL, Jones L, Toubia J, Bray SC, Casolari DA, et al. Efficacy of combined CDK9/BET inhibition in preclinical models of MLL-rearranged acute leukemia. Blood Adv 2020;4(2):296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun 2016;7:10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306(5701):1574–7. [DOI] [PubMed] [Google Scholar]
- [105].Milne TA, Sinclair DA, Brock HW. The additional sex combs gene of drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol Gen Genet 1999;261(4–5):753–61. [DOI] [PubMed] [Google Scholar]
- [106].Nagase R, Inoue D, Pastore A, Fujino T, Hou HA, Yamasaki N, et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med 2018;215(6):1729–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest 2013;123(11):4627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun 2015;6:7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012;22(2):180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T, et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun 2018;9(1):2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wang L, Birch NW, Zhao Z, Nestler CM, Kazmer A, Shilati A, et al. Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat Cancer 2021;2(5):515–26. [DOI] [PubMed] [Google Scholar]
- [113].Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007;7(2):105–17. [DOI] [PubMed] [Google Scholar]
- [114].Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 2001;27(3): 263–70. [DOI] [PubMed] [Google Scholar]
- [115].Snaddon J, Smith ML, Neat M, Cambal-Parrales M, Dixon-McIver A, Arch R, et al. Mutations of CEBPA in acute myeloid leukemia FAB types M1 and M2. Genes Chromosomes Cancer 2003;37(1):72–8. [DOI] [PubMed] [Google Scholar]
- [116].Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113(13): 3088–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011;117(8):2469–75. [DOI] [PubMed] [Google Scholar]
- [118].Nerlov C C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 2004;4(5):394–400. [DOI] [PubMed] [Google Scholar]
- [119].Hohaus S, Petrovick MS, Voso MT, Sun Z, Zhang DE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol 1995;15(10):5830–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH, et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99(4):1332–40. [DOI] [PubMed] [Google Scholar]
- [121].Alberich-Jorda M, Wouters B, Balastik M, Shapiro-Koss C, Zhang H, Di Ruscio A, et al. C/EBPgamma deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest 2012;122(12):4490–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A, et al. Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell 2013;24(5):575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Jakobsen JS, Laursen LG, Schuster MB, Pundhir S, Schoof E, Ge Y, et al. Mutant CEBPA directly drives the expression of the targetable tumor-promoting factor CD73 in AML. Sci Adv 2019;5(7). eaaw4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ohlsson E, Schuster MB, Hasemann M, Porse BT. The multifaceted functions of C/EBPalpha in normal and malignant haematopoiesis. Leukemia. 2016;30(4):767–75. [DOI] [PubMed] [Google Scholar]
- [125].Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56(3):379–90. [DOI] [PubMed] [Google Scholar]
- [126].Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005;352(3):254–66. [DOI] [PubMed] [Google Scholar]
- [127].Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103(1):127–40. [DOI] [PubMed] [Google Scholar]
- [128].Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol 2002;4(7):529–33. [DOI] [PubMed] [Google Scholar]
- [129].Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. 1996;87(3):882–6. [PubMed] [Google Scholar]
- [130].Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. [DOI] [PubMed] [Google Scholar]
- [131].Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12(2):265–75. [PubMed] [Google Scholar]
- [132].Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang YH, Ramabadran R, et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018;34(3):499–512 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov 2016;6(10):1166–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Balusu R, Fiskus W, Rao R, Chong DG, Nalluri S, Mudunuru U, et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood. 2011;118(11):3096–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].AI Nabbouh, Hleihel RS, Saliba JL, Karam MM, Hamie MH, Wu HJM, et al. Imidazoquinoxaline derivative EAPB0503: a promising drug targeting mutant nucleophosmin 1 in acute myeloid leukemia. Cancer. 2017;123(9):1662–73. [DOI] [PubMed] [Google Scholar]
- [136].Luskin MR, Lee JW, Fernandez HF, Abdel-Wahab O, Bennett JM, Ketterling RP, et al. Benefit of high-dose daunorubicin in AML induction extends across cytogenetic and molecular groups. Blood. 2016;127(12):15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Burnett AK, Russell NH, Hills RK, Kell J, Cavenagh J, Kjeldsen L, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood. 2015;125(25):3878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].De Propris MS, Raponi S, Diverio D, Milani ML, Meloni G, Falini B, et al. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica. 2011;96(10): 1548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Schlenk RF, Paschka P, Krzykalla J, Weber D, Kapp-Schwoerer S, Gaidzik VI, et al. Gemtuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: early results from the prospective randomized AMLSG 09-09 phase III study. J Clin Oncol 2020;38(6):623–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wang SW, Speck NA. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol 1992;12(1):89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wang S, Wang Q, Crate BE, Melnikova IN, Keller SR, Speck NA. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol Cell Biol 1993;13(6):3324–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Dombret H Gene mutation and AML pathogenesis. Blood. 2011;118(20):5366–7. [DOI] [PubMed] [Google Scholar]
- [143].Rhoades KL, Hetherington CJ, Harakawa N, Yergeau DA, Zhou L, Liu LQ, et al. Analysis of the role of AML1-ETO in leukemogenesis, using an inducible transgenic mouse model. Blood. 2000;96(6):2108–15. [PubMed] [Google Scholar]
- [144].Downing JR. The core-binding factor leukemias: lessons learned from murine models. Curr Opin Genet Dev 2003;13(1):48–54. [DOI] [PubMed] [Google Scholar]
- [145].Jourdan E, Boissel N, Chevret S, Delabesse E, Renneville A, Cornillet P, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121(12):2213–23. [DOI] [PubMed] [Google Scholar]
- [146].Duployez N, Marceau-Renaut A, Boissel N, Petit A, Bucci M, Geffroy S, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016;127(20):2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Thiel VN, Giaimo BD, Schwarz P, Soller K, Vas V, Bartkuhn M, et al. Heterodimerization of AML1/ETO with CBFbeta is required for leukemogenesis but not for myeloproliferation. Leukemia. 2017;31(11):2491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Friedman AD. Leukemogenesis by CBF oncoproteins. Leukemia. 1999;13(12):1932–42. [DOI] [PubMed] [Google Scholar]
- [149].Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255–62. [DOI] [PubMed] [Google Scholar]
- [150].Al-Harbi S, Aljurf M, Mohty M, Almohareb F, Ahmed SOA. An update on the molecular pathogenesis and potential therapeutic targeting of AML with t(8;21) (q22;q22.1);RUNX1-RUNX1T1. Blood Adv 2020;4(1):229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lu L, Wen Y, Yao Y, Chen F, Wang G, Wu F, et al. Glucocorticoids inhibit oncogenic RUNX1-ETO in acute myeloid leukemia with chromosome translocation t(8;21). Theranostics. 2018,8(8):2189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hyde RK, Liu PP. RUNX1 repression-independent mechanisms of leukemogenesis by fusion genes CBFB-MYH11 and AML1-ETO (RUNX1-RUNX1T1). J Cell Biochem 2010;110(5):1039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Adya N, Stacy T, Speck NA, Liu PP. The leukemic protein core binding factor beta (CBFbeta)-smooth-muscle myosin heavy chain sequesters CBFalpha2 into cytoskeletal filaments and aggregates. Mol Cell Biol 1998;18(12):7432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lutterbach B, Hou Y, Durst KL, Hiebert SW. The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A 1999;96(22):12822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Tang YY, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, et al. Energetic and functional contribution of residues in the core binding factor beta (CBFbeta) subunit to heterodimerization with CBFalpha. J Biol Chem 2000;275(50):39579–88. [DOI] [PubMed] [Google Scholar]
- [156].Borthakur G, Kantarjian H. Core binding factor acute myelogenous leukemia-2021 treatment algorithm. Blood Cancer J 2021;11(6):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Pulikkan JA, Hegde M, Ahmad HM, Belaghzal H, Illendula A, Yu J, et al. CBFbeta-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell. 2018;174(5):1325. [DOI] [PubMed] [Google Scholar]
- [158].Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374(23):2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Zhao D, Tahaney WM, Mazumdar A, Savage MI, Brown PH. Molecularly targeted therapies for p53-mutant cancers. Cell Mol Life Sci 2017;74(22):4171–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Seifert H, Mohr B, Thiede C, Oelschlagel U, Schakel U, Illmer T, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009;23(4):656–63. [DOI] [PubMed] [Google Scholar]
- [161].Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531(7595):471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–21. [DOI] [PubMed] [Google Scholar]
- [163].Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–98. [DOI] [PubMed] [Google Scholar]
- [164].Fontana MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, Luserna Ghelli, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia. 2018;32(7):1609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012;120(15):2963–72. [DOI] [PubMed] [Google Scholar]
- [166].Yi L, Sun Y, Levine A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget. 2014;5(19):8924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Welch JS, Petti AA, Ley TJ. Decitabine in TP53-mutated AML. N Engl J Med 2017;376(8):797–8. [DOI] [PubMed] [Google Scholar]
- [168].Schiffer CA. Promoting apoptosis with venetoclax - a benefit for older patients with AML. N Engl J Med 2020;383(7):677–9. [DOI] [PubMed] [Google Scholar]
- [169].Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018;24(12):1859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Shoukier M, Konopleva M, Dinardo CD, Ravandi F, Andreeff M, Garcia-Manero G, et al. Activity of venetoclax-based therapy in TP53-mutated acute myeloid leukemia. J Clin Oncol 2019;37(15_suppl):7034. [Google Scholar]
- [171].DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lehmann BD, Pietenpol JA. Targeting mutant p53 in human tumors. J Clin Oncol 2012;30(29):3648–50. [DOI] [PubMed] [Google Scholar]
- [173].Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol 2021;39(14):1584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell 2017;32(6):748–60 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999;55(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett 2013;4(5):466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Pan R, Kojima K, Zheng Z, Ruvolo VR, Nichols G, Leverson JD, et al. Activation of p53 by novel MDM2 antagonist RG7388 overcomes AML inherent and acquired resistance to Bcl-2 inhibitor ABT-199 (GDC-0199). Blood. 2014;124(21):2162.25278563 [Google Scholar]
- [178].Konopleva MY, Röllig C, Cavenagh J, Deeren D, Girshova L, Krauter J, et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv 2022;6(14):4147–56. 10.1182/bloodadvances.2021006303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Daver NG, Garcia JS, Jonas BA, Kelly KR, Assouline S, Brandwein JM, et al. Updated results from the venetoclax (Ven) in combination with idasanutlin (Idasa) arm of a phase 1b trial in elderly patients (pts) with relapsed or refractory (R/R) AML ineligible for cytotoxic chemotherapy. Blood. 2019;134(Supplement_1):229. [Google Scholar]
- [180].Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Sallman DA, Asch AS, Al Malki MM, Lee DJ, Donnellan WB, Marcucci G, et al. The first-in-class anti-CD47 antibody Magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. Blood. 2019;134(Supplement_1):569. [Google Scholar]
- [182].Safety concerns prompt pause of magrolimab trials. Cancer Discov 2022,12(4):877–8. [DOI] [PubMed] [Google Scholar]
- [183].Ulku AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat Res 2003;115:189–208. [PubMed] [Google Scholar]
- [184].Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H, et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther 2020;5(1):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Zhao Z, Chen CC, Rillahan CD, Shen R, Kitzing T, McNerney ME, et al. Cooperative loss of RAS feedback regulation drives myeloid leukemogenesis. Nat Genet 2015;47(5):539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Bos JL. ras oncogenes in human cancer: a review. Cancer Res 1989;49(17):4682–9. [PubMed] [Google Scholar]
- [187].Van Meter ME, Diaz-Flores E, Archard JA, Passegue E, Irish JM, Kotecha N, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109(9):3945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Fatrai S, van Gosliga D, Han L, Daenen SM, Vellenga E, Schuringa JJ. KRAS (G12V) enhances proliferation and initiates myelomonocytic differentiation in human stem/progenitor cells via intrinsic and extrinsic pathways. J Biol Chem 2011;286(8):6061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Jain N, Curran E, Iyengar NM, Diaz-Flores E, Kunnavakkam R, Popplewell L, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res 2014;20(2):490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kunimoto H, Meydan C, Nazir A, Whitfield J, Shank K, Rapaport F, et al. Cooperative epigenetic remodeling by TET2 loss and NRAS mutation drives myeloid transformation and MEK inhibitor sensitivity. Cancer Cell 2018;33(1):44–59 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Han L, Zhang Q, Dail M, Shi C, Cavazos A, Ruvolo VR, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020;105(3):697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev 2012;6(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Goncalves MD, Hopkins BD, Cantley LC. Phosphatidylinositol 3-kinase, growth disorders, and cancer. N Engl J Med 2018;379(21):2052–62. [DOI] [PubMed] [Google Scholar]
- [195].Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–9. [DOI] [PubMed] [Google Scholar]
- [196].Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters–an analysis of 3082 patients. Blood. 2008;111(5):2527–37. [DOI] [PubMed] [Google Scholar]
- [197].Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377(5):454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med 2019;381(18):1728–40. [DOI] [PubMed] [Google Scholar]
- [199].Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Burchert A, Bug G, Fritz LV, Finke J, Stelljes M, Rollig C, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol 2020;38(26):2993–3002. [DOI] [PubMed] [Google Scholar]
- [201].Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109(4):1643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir NN, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016;35(39):5119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Vargaftig J, Farhat H, Ades L, Briaux A, Benoist C, Turbiez I, et al. Phase 2 trial of single agent gedatolisib (PF-05212384), a dual PI3K/mTOR inhibitor, for adverse prognosis and relapse/refractory AML: clinical and transcriptomic results. Blood 2018;132(Supplement 1):5233. [Google Scholar]
- [204].Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Perl AE, Daver NG, Pratz KW, Maly J, Hong W-J, Bahceci E, et al. Venetoclax in combination with gilteritinib in patients with relapsed/refractory acute myeloid leukemia: a phase 1b study. Blood. 2019;134(Supplement_1):3910. [Google Scholar]
- [206].Ma J, Zhao S, Qiao X, Knight T, Edwards H, Polin L, et al. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer Res 2019;25(22):6815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev 2021;100905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Talati C, Sweet KL. Nuclear transport inhibition in acute myeloid leukemia: recent advances and future perspectives. Int. J Hematol Oncol 2018;7(3). IJH04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Brinton LT, Sher S, Williams K, Canfield D, Orwick S, Wasmuth R, et al. Cotargeting of XPO1 enhances the antileukemic activity of midostaurin and gilteritinib in acute myeloid leukemia. Cancers (Basel) 2020;12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Malaise M, Steinbach D, Corbacioglu S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr Hematol Malig Rep 2009;4(2):77–82. [DOI] [PubMed] [Google Scholar]
- [211].Paschka P, Marcucci G, Ruppert AS, Mrozek K, Chen H, Kittles RA, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv (16) and t(8;21): a cancer and leukemia group B study. J Clin Oncol 2006;24(24):3904–11. [DOI] [PubMed] [Google Scholar]
- [212].Pollard JA, Alonzo TA, Gerbing RB, Ho PA, Zeng R, Ravindranath Y, et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood. 2010;115(12):2372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Heo SK, Noh EK, Kim JY, Jeong YK, Jo JC, Choi Y, et al. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci Rep 2017;7(1):15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Tarlock K, Alonzo TA, Wang YC, Gerbing RB, Ries R, Loken MR, et al. Functional properties of KIT mutations are associated with differential clinical outcomes and response to targeted therapeutics in CBF acute myeloid leukemia. Clin Cancer Res 2019;25(16):5038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6. [DOI] [PubMed] [Google Scholar]
- [216].Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126(17):3915–24. [DOI] [PubMed] [Google Scholar]
- [217].Bhardwaj G, Murdoch B, Wu D, Baker DP, Williams KP, Chadwick K, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol 2001;2(2):172–80. [DOI] [PubMed] [Google Scholar]
- [218].Chahal KK, Parle M, Abagyan R. Hedgehog pathway and smoothened inhibitors in cancer therapies. Anticancer Drugs 2018;29(5):387–401. [DOI] [PubMed] [Google Scholar]
- [219].Bai LY, Chiu CF, Lin CW, Hsu NY, Lin CL, Lo WJ, et al. Differential expression of Sonic hedgehog and Gli1 in hematological malignancies. Leukemia. 2008;22(1):226–8. [DOI] [PubMed] [Google Scholar]
- [220].Kobune M, Iyama S, Kikuchi S, Horiguchi H, Sato T, Murase K, et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J 2012;2:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, et al. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci 2016;107(10):1422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Tibes R, Al-Kali A, Oliver GR, Delman DH, Hansen N, Bhagavatula K, et al. The Hedgehog pathway as targetable vulnerability with 5-azacytidine in myelodysplastic syndrome and acute myeloid leukemia. J Hematol Oncol 2015;8:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Zeidan AM, Schuster MW, Krauter J, Maertens JA, Gyan E, Joris M, et al. Clinical benefit of Glasdegib in combination with azacitidine or low-dose cytarabine in patients with acute myeloid leukemia. Blood. 2019;134(Supplement_1:3916 [Google Scholar]
- [224].Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Transl Med 2015;7(291).291ra96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. 2010;29(48):6314–22. [DOI] [PubMed] [Google Scholar]
- [226].Kim JA, Shim JS, Lee GY, Yim HW, Kim TM, Kim M, et al. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res 2015;75(11):2222–31. [DOI] [PubMed] [Google Scholar]
- [227].Lataillade JJ, Pierre-Louis O, Hasselbalch HC, Uzan G, Jasmin C, Martyre MC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008;112(8):3026–35. [DOI] [PubMed] [Google Scholar]
- [228].Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000;95(1):309–13. [PubMed] [Google Scholar]
- [229].Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Korkolopoulou P, Apostolidou E, Pavlopoulos PM, Kavantzas N, Vyniou N, Thymara I, et al. Prognostic evaluation of the microvascular network in myelodysplastic syndromes. Leukemia. 2001,15(9):1369–76. [DOI] [PubMed] [Google Scholar]
- [231].Campioni D, Punturieri M, Bardi A, Moretti S, Tammiso E, Lanza F, et al. “In vitro” evaluation of bone marrow angiogenesis in myelodysplastic syndromes: a morphological and functional approach. Leuk Res 2004;28(1):9–17. [DOI] [PubMed] [Google Scholar]
- [232].Lundberg LG, Hellstrom-Lindberg E, Kanter-Lewensohn L, Lerner R, Palmblad J. Angiogenesis in relation to clinical stage, apoptosis and prognostic score in myelodysplastic syndromes. Leuk Res 2006;30(3):247–53. [DOI] [PubMed] [Google Scholar]
- [233].Wimazal F, Krauth MT, Vales A, Bohm A, Agis H, Sonneck K, et al. Immunohistochemical detection of vascular endothelial growth factor (VEGF) in the bone marrow in patients with myelodysplastic syndromes: correlation between VEGF expression and the FAB category. Leuk Lymphoma 2006;47(3):451–60. [DOI] [PubMed] [Google Scholar]
- [234].Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28(12):2276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].de Figueiredo-Pontes LL, Pintao MC, Oliveira LC, Dalmazzo LF, Jacomo RH, Garcia AB, et al. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin Cytom 2008;74(3):163–8. [DOI] [PubMed] [Google Scholar]
- [236].Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12(3):329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Le PM, Andreeff M, Battula VL. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica. 2018;103(12):1945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Wang A, Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. 2018;23(10):729–39. [DOI] [PubMed] [Google Scholar]
- [239].Cho BS, Kim HJ, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med 2017;32(2):248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Zhang Y, Saavedra E, Tang R, Gu Y, Lappin P, Trajkovic D, et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci Rep 2017;7(1):7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Hsieh Y-T, Jiang E, Pham J, Kim H-N, Abdel-Azim H, Khazal S, et al. VLA4 blockade in acute myeloid leukemia. Blood. 2013;122(21):3944. [Google Scholar]
- [242].Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006;12(10): 1167–74. [DOI] [PubMed] [Google Scholar]
- [243].Yu X, Munoz-Sagredo L, Streule K, Muschong P, Bayer E, Walter RJ, et al. CD44 loss of function sensitizes AML cells to the BCL-2 inhibitor venetoclax by decreasing CXCL12-driven survival cues. Blood. 2021;138(12):1067–80. [DOI] [PubMed] [Google Scholar]
- [244].Hatfield K, Ryningen A, Corbascio M, Bruserud O. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int J Cancer 2006;119(10):2313–21. [DOI] [PubMed] [Google Scholar]
- [245].Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia. 2002;16(9):1713–24. [DOI] [PubMed] [Google Scholar]
- [246].Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014; 158(6):1309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Seshadri M, Qu CK. Microenvironmental regulation of hematopoietic stem cells and its implications in leukemogenesis. Curr Opin Hematol 2016;23(4):339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Hamdi W, Ogawara H, Handa H, Tsukamoto N, Nojima Y, Murakami H. Clinical significance of regulatory T cells in patients with myelodysplastic syndrome. Eur J Haematol 2009;82(3):201–7. [DOI] [PubMed] [Google Scholar]
- [249].Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res 2008;14(10):3044–51. [DOI] [PubMed] [Google Scholar]
- [251].Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov 2019;9(3):370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 2005;6(1):9–20. [DOI] [PubMed] [Google Scholar]
- [253].Ohh M, Kim WY, Moslehi JJ, Chen Y, Chau V, Read MA, et al. An intact NEDD8 pathway is required for Cullin-dependent ubiquitylation in mammalian cells. EMBO Rep 2002;3(2): 177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Ochiiwa H, Ailiken G, Yokoyama M, Yamagata K, Nagano H, Yoshimura C, et al. TAS4464, a NEDD8-activating enzyme inhibitor, activates both intrinsic and extrinsic apoptotic pathways via c-Myc-mediated regulation in acute myeloid leukemia. Oncogene. 2021;40(7):1217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Short NJ, Sedarati F, Zhao D, Tsukurov O, Friedlander S, Faller DV. A randomized phase 2 study of pevonedistat, venetoclax, and azacitidine versus venetoclax plus azacitidine in adults with newly diagnosed acute myeloid leukemia (AML) who are unfit for intensive chemotherapy. Blood. 2020;136(Supplement_1):34–5. [Google Scholar]
- [256].Sun B, Mason S, Wilson RC, Hazard SE, Wang Y, Fang R, et al. Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers. Oncogene. 2020;39(1):50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Sampathi S, Acharya P, Zhao Y, Wang J, Stengel KR, Liu Q, et al. The CDK7 inhibitor THZ1 alters RNA polymerase dynamics at the 5′ and 3′ ends of genes. Nucleic Acids Res 2019;47(8):3921–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res 2020;26(4):922–34. [DOI] [PubMed] [Google Scholar]
- [259].Phillips DC, Jin S, Gregory GP, Zhang Q, Xue J, Zhao X, et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia. 2020;34(6):1646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Fiskus W, Manshouri T, Birdwell C, Mill CP, Masarova L, Bose P, et al. Efficacy of CDK9 inhibition in therapy of post-myeloproliferative neoplasm (MPN) secondary (s) AML cells. Blood Cancer J 2022;12(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]