

Summary
Association studies describe genetic associations between noncoding variants and disease susceptibility; however, they do not provide functional insight into the underlying molecular mechanisms of these variants. We present a protocol to assay the regulatory potential of thousands of noncoding variants using massively parallel reporter assays. We describe steps for oligo design, generating a plasmid pool, and extracting tag-seq libraries from cells to quantify the tested sequences.
For complete details on the use and execution of this protocol, please refer to Oliveros and Delfosse et al.1
Subject areas: Genetics, Genomics, High Throughput Screening, Gene Expression, Systems biology
Graphical abstract
Highlights
-
•
Key steps for performing massively parallel reporter assays (MPRAs)
-
•
Protocol for high-throughput cloning, transfection, and MPRA library preparation
-
•
MPRA design to assay thousands of regulatory regions and/or genetic variants
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Association studies describe genetic associations between noncoding variants and disease susceptibility; however, they do not provide functional insight into the underlying molecular mechanisms of these variants. We present a protocol to assay the regulatory potential of thousands of noncoding variants using massively parallel reporter assays. We describe steps for oligo design, generating a plasmid pool, and extracting tag-seq libraries from cells to quantify the tested sequences.
Before you begin
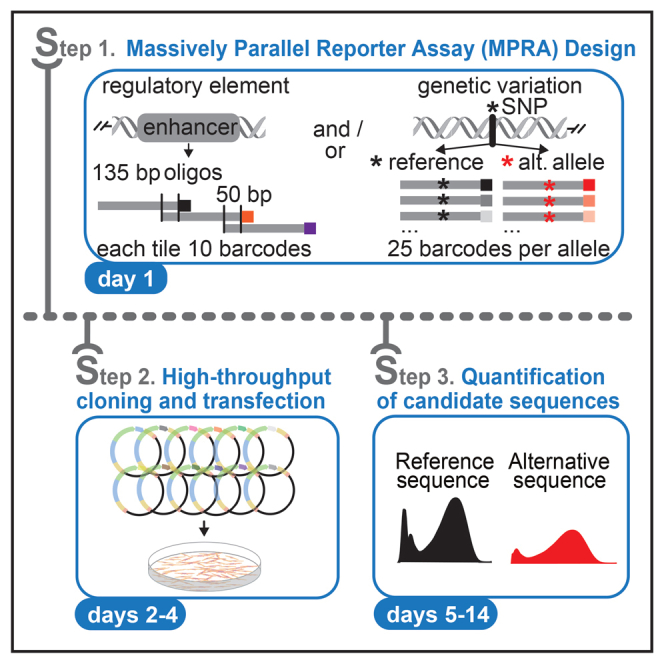
Design and order your oligonucleotide pool in advance. The type and number of sequences you plan to assay will influence the lengths of oligos and contribute to the complexity of the plasmid pool. For example, this protocol can be adapted to identify and to measure regulatory activity of candidate genomic regions by tiling oligonucleotides to determine regulatory sequences2,3 (Figure 1A). Genetic variants identified by Genome Wide Association Studies (GWAS) and variants in high linkage disequilibrium can be tested by MPRA technology to identify causal variants for a trait1 (Figure 1A).
Figure 1.

MPRA design
(A) Scheme of MPRA design. Candidate regions can be tested for regulatory elements (i.e., enhancer) by a tiling oligonucleotide design (left, each tile 10 barcodes), and/or alleles of genetic variants are tested by 25 barcodes each (right).
(B) Schemes of oligonucleotide design with candidate region, KpnI and XbaI sites and barcode (BC, top), ePCR amplification to add SfiI sites (middle), and final tag-seq library product for next generation sequencing (NGS, bottom). Asterisk highlights position of Illumina index in NGS adapter.
Design oligo pool
Timing: 3 days
Design an oligo pool of genetic elements to be tested, positive, and negative controls. For example, a previous study1 had 4608 variants × 50 barcodes each (4608 reference alleles × 25 barcodes each + 4608 alternative alleles × 25 barcodes) + 400 negative controls × 5 barcodes + 10 positive controls × 100 barcodes = total 233 k oligos.
-
1.Procure a list of variants or sequences that are to be assayed (previously shown for 4,608 genetic variants derived from blood pressure GWAS).
-
a.Extract the sequence of interest with appropriate genomic context.
-
b.If testing SNPs, ensure that both the reference allele and alternative allele are included in the pool.
-
a.
-
2.Design control elements.
-
a.Design negative controls by generating scrambled sequences of the same length (recommendation: 300-400 negative control sequences with five barcodes each).
-
b.Select positive controls (recommendation: 5–10 positive control sequences with 100 barcodes each → see Critical).
-
a.
-
3.Computational assembly of all oligo components.
-
a.Add restriction sites. Addition of KpnI and XbaI restriction sites is required for downstream cloning of promoter:reporter cassette in between the candidate element and barcode in cloning step 2.
-
i.KpnI RE site: GGTACC.
-
ii.XbaI RE site: TCTAGA.
-
i.
-
b.Add barcodes. The barcodes serve as unique molecular identifiers for the quantification by next generation sequencing (NGS).Note: When assaying genetic variants 25 barcodes are recommended per allele to provide adequate statistical power. When determining regulatory activity of candidate genomic regions (i.e. enhancer characterization), we recommend tiling oligos with 10 barcodes each. (Figure 1A). Recommended barcode length is 11 bp.
-
c.Add universal priming sites. These sites are used for amplification of the oligo pool by emulsion PCR (ePCR), as well as for NGS library preparation (Figure 1B).
-
i.universal primer site 1: ACTGGCCGCTTCACTG.
-
ii.universal primer site 2: AGATCGGAAGAGCGTCG.Example oligo (Figure 1B):5’ – universal primer site 1 – candidate region with genetic variant (G) [centered] – KpnI site – XbaI site – unique barcode (11 bp) – universal primer site 2 - 3’ACTGGCCGCTTCACTG –ACTAATGGCTGGAAAATGTTTCTACTTCTAAAAATAGTGAAAGAATGAGATCTGTATGTAAACTGATGAGCCTCATGCAGACGCCAAACAAAATTCTAAAATGGATGATTTGTAGGCAAGATACAGTAAACAAAT – GGTACC – TCTAGA – NNNNNNNNNNN – AGATCGGAAGAGCGTCG
 CRITICAL: It is important to include both context-specific and context-agnostic positive controls. Recently, we used four context-agnostic variants as positive controls that showed regulatory activity in different MPRAs across cell types (rs117104239, rs60002611, rs6445040, rs92708981,3). Moreover, adding variants that have been identified as context and cell-type specific regulatory variants supports the quality control steps and the functional readout.1 However, each MPRA requires the identification of known functional loci in the area of study to use as specific positive controls. In different MPRA studies,1,2,3 we experienced that 25 barcodes per allele are required to successfully test the regulatory effects of genetic variants and 10 barcodes for tiling designs. Too few barcodes per candidate element can lead to unusable data.
CRITICAL: It is important to include both context-specific and context-agnostic positive controls. Recently, we used four context-agnostic variants as positive controls that showed regulatory activity in different MPRAs across cell types (rs117104239, rs60002611, rs6445040, rs92708981,3). Moreover, adding variants that have been identified as context and cell-type specific regulatory variants supports the quality control steps and the functional readout.1 However, each MPRA requires the identification of known functional loci in the area of study to use as specific positive controls. In different MPRA studies,1,2,3 we experienced that 25 barcodes per allele are required to successfully test the regulatory effects of genetic variants and 10 barcodes for tiling designs. Too few barcodes per candidate element can lead to unusable data.
-
i.
-
a.
-
4.
Order as double stranded oligos.
Order primers
-
5.Order ePCR primers for amplification of the oligo pool and addition of SfiI sites for downstream cloning.CRITICAL: These primers anneal to the universal priming sites to amplify the oligo pool by ePCRs, which supports evenly distributed amplifications of oligos and prevent PCR-induced amplification artifacts (‘jackpotting of oligos’). Exact sequences are available in the key resources table.
-
a.ePCR Forward Primer/ePCR Reverse Primer composition (Figure 1B): 5’ – extra bases (6) – SfiI cut site compatible with cloning vector –universal priming site – 3′.
-
a.
-
6.Design library prep primers for ePCR amplification product and first cloning verification (Library Prep Primers A).Note: These primers will 1) amplify the oligo both after the ePCR and from the MPRA vector after the first cloning step and 2) append Illumina adapters and indices (enables multiplexing). This will facilitate downstream sequencing of oligos and allow for assessment of barcode representation.
-
a.Library Prep Reverse Primer A composition: 5’ – Illumina adapter – universal priming site – 3′.
-
b.Library Prep Forward Primer A composition: 5’ – Illumina adapter – TruSeq Index X – Illumina adapter – universal priming site – 3′.
-
c.Example sequences for Illumina indices are below:
Index number Sequence 1 CGTGAT 2 ACATCG 3 GCCTAA 4 TGGTCA 5 CACTGT 6 ATTGGC 7–12 etc. CRITICAL: Some Illumina indices are not compatible with one another. Please refer to instructions provided by Illumina to ensure optimal combinations.
-
a.
-
7.Design library prep primers for second and third cloning verification and experimental libraries (Library Prep Primers B). These primers will amplify oligo and barcode, as well as 90 bp of the adjacent GFP reporter. Assessing barcode distribution is again important at these steps.
-
a.Library Prep Reverse Primer B composition: same as above.
-
b.Library Prep Forward Primer B composition: 5’ – Illumina adapter – TruSeq Index X – Illumina adapter – GFP priming site – 3′.
-
c.Incorporate various Illumina adapters as above.
-
a.
-
8.
Order GFP and GAPDH qPCR primers to measure DNA (plasmid) contamination in RNA from cells transfected with plasmid library using qPCR.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| NEB 5-alpha competent E. coli (high efficiency) | NEB | Cat#C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| Q5 high-fidelity DNA polymerase | NEB | Cat#M0491L |
| Acetylated BSA | Invitrogen | Cat#AM2614 |
| 2-Butanol | Sigma-Aldrich | Cat#294810-1L |
| PhiX control v3 | Illumina | Cat#FC-110-3001 |
| SfiI | NEB | Cat#R0123L |
| Alkaline phosphatase, calf intestinal | NEB | Cat#M0525 |
| T4 DNA ligase | NEB | Cat#M0202M |
| T4 DNA ligase reaction buffer | NEB | Cat#B0202S |
| XbaI | NEB | Cat#R0145L |
| KpnI-HF | NEB | Cat#R3142L |
| dNTP solution mix - 40 μmol of each | NEB | Cat#N0447L |
| Formamide (deionized) | Invitrogen | Cat#AM9342 |
| Agencourt AMPure XP | Beckman Coulter | Cat#A63881 |
| TRIzol reagent | Life Technologies | Cat#15596018 |
| RNase-free DNase set | QIAGEN | Cat#79254 |
| DNaseI recombinant | Worthington Biochemical | Cat#LS006355 |
| PowerUp SYBR Green master mix | ABI | A25742 |
| Critical commercial assays | ||
| Micellula DNA Emulsion & Purification Kit | Chimerx | Cat#E3600 |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | Cat#12965 |
| Qubit dsDNA quantitation, high sensitivity | Thermo Fisher Scientific | Cat#Q32851 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| SuperScript III first strand synthesis | Invitrogen | Cat#18080051 |
| Monarch PCR & DNA Cleanup Kit | NEB | Cat#T1030L |
| Monarch DNA Gel Extraction Kit | NEB | Cat#T1020L |
| TruSeq Stranded Total RNA Ribo-Zero Gold | Illumina | Cat#RS-122-2301 |
| Oligonucleotides | ||
| ePCR forward primer: GCTAAGGGCCTAACTGGCCGCTTCACTG | Mattioli et al.3 | N/A |
| ePCR reverse primer: GTTTAAGGCCTCCGAGGCCGACGCTCTTC | Mattioli et al.3 | N/A |
| Library prep forward primer A: AATGATACGGCGACCACCGAGA TCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
Mattioli et al.3 | N/A |
| Library prep reverse primer A: caagcagaagacggcatacgagatCGTG ATgtgactggagttcagacgtgtgctcttccgatctACTGGCCGCTTCACTG |
Mattioli et al.3 | N/A |
| Library prep forward primer B: AATGATACGGCGACCACCGAGA TCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
Mattioli et al.3 | N/A |
| Library prep reverse primer B: caagcagaagacggcatacgagatCGTG ATgtgactggagttcagacgtgtgctcttccgatctCGCCGCGTGGAGGAGGA |
Mattioli et al.3 | N/A |
| GFP qPCR forward: TGATGGGCTACGGCTTCTAC | Mattioli et al.3 | N/A |
| GFP qPCR reverse: GTGCCCATCACCTTGAAGTC | Mattioli et al.3 | N/A |
| GAPDH qPCR forward: AAGGTGAAGGTCGGAGTCAAC | N/A | N/A |
| GAPDH qPCR reverse: CACTCACTCCTGGAAGATGGT | N/A | N/A |
| Recombinant DNA | ||
| MPRA empty vector | Mattioli et al.3 | Addgene 204865 |
| MPRA_minCMV+GFP | Mattioli et al.3 | Addgene 204866 |
| Software and algorithms | ||
| MPRAnalyze | Ashuach et al.4 | https://bioconductor.org/packages/release/bioc/html/MPRAnalyze.html |
| Other | ||
| 2100 Bioanalyzer instrument | Agilent | Cat#G2939BA |
| QIAxcel advanced instrument | QIAGEN | Cat#9001941 |
| Qubit fluorometer 2, 3, or 4 | Invitrogen | Cat#Q33216 |
| Illumina NovaSeq 6000 or equivalent | Illumina | N/A |
| DynaMag-2 magnet | Invitrogen | 12321D |
| LoBind protein or genomic microcentrifuge tubes – 1.5 mL | Eppendorf | CA80077-230 |
| LoBind protein or genomic microcentrifuge tubes – 2 mL | Eppendorf | CA80077-234 |
Step-by-step method details
Amplification of oligo pool
In the following steps, the oligo pool is amplified by ePCRs. Subsequently, ePCR amplicons are purified, size-selected, and ultimately sequenced. Recommended input and scaling are provided but should be further optimized by the user.
-
1.Dilute and quantify the oligo pool.
-
a.Serially dilute oligo pool in two steps to ∼10 ng/μL in TE buffer.
-
b.Quantify DNA concentration using Qubit High Sensitivity kit.
-
c.Calculate the volume required as input for ePCR (108–109 molecules per reaction).
-
a.
Note: Example calculation is provided below.
Oligo pool is 13.1 ng/μL. Calculate number of molecules in 1 μL
-
2.Amplify oligo pool by ePCR as per manufacturer’s instruction:
-
a.Mix the emulsion components, in the order indicated below, for the number of reactions needed plus 2, at 20°C–25°C. This is the oil phase.
Reagent Amount Emulsion component 1 220 μL Emulsion component 2 20 μL Emulsion component 3 60 μL Note: component 2 is very viscous, therefore it is recommended to use either low retention wide-bore tips or to cut tip ends slightly to increase end diameter.-
i.Mix thoroughly by vortexing.
-
ii.Keep on ice until ready to use.
-
i.
-
b.Create the PCR water phase by mixing all components for the number of reactions needed plus 1 reaction for the non-emulsion PCR control.Alternatives: The kit recommends EURx Taq but Q5 polymerase from NEB has been used successfully.Note: Using the recommended 108–109 molecules per reaction × 9 reactions has been sufficient for downstream steps in previous amplifications.
Reagent Amount 5× Q5 reaction buffer 10 μL 10 μM dNTPs 1 μL ePCR Forward Primer (10 μM) 2.5 μL ePCR Reverse Primer (10 μM) 2.5 μL 100 μg/mL Acetylated BSA 5 μL Diluted oligo pool variable Q5 polymerase 0.5 μL ddH2O To 50 μL CRITICAL: Acetylated BSA is required to coat the hydrophobic/hydrophilic interface at the micelle border. The amount of BSA required depends on total micelle border surface area and therefore overall micelle count and size. Excess BSA is also an inhibitor of PCR amplifications. Therefore, the amount of BSA must be optimized. The amounts shown here are offered as a starting point for such optimization.-
i.Mix by inverting several times.
-
ii.Keep on ice.
-
i.
-
c.Combine the oil and water phases and run the PCR.
-
i.Add 300 μL of emulsion into 1.5 mL LoBind tubes (as many as needed).
-
ii.Add 50 μL of the PCR water phase into each tube (keep what is left for the PCR control reaction) and mix by inverting a few times.
-
iii.Vortex for 5 min at 4°C.CRITICAL: pre-cool on ice and perform the next steps on ice.
-
iv.Split each reaction into 3 tubes of a PCR strip.
-
v.Transfer the PCR control reaction to a tube of another PCR strip.
-
vi.Run reaction according to the cycling conditions below.
Steps Temperature Time Cycles Initial denaturation 98°C 30 s 1 Denaturation 98°C 10 s ×30 Annealing 55°C 10 s Extension 72°C 10 s Final Extension 72°C 2 min 1 Hold 10°C forever
-
i.
-
a.
-
3.DNA recovery step 1: emulsion breaking and phase separation.
-
a.Break the emulsion by adding 1 mL of 2-butanol into one 2 mL LoBind tube for each reaction.
-
b.Transfer all of the 3 PCRs for each reaction into one of the butanol-containing tubes.
-
c.Transfer 150 μL of butanol from the 2 mL tube back into the first PCR tube and then to the second and third, mixing by pipetting each time to dissolve all remaining emulsion. Then transfer from the third PCR tube back into the 2 mL tube.
-
d.Repeat the above steps for all reactions.
-
e.Vortex all tubes until the solution becomes transparent.
-
f.Add 400 μL of Orange-DX buffer and mix by gentle agitation (by hand or on a rotator for 2 min).
-
g.Centrifuge for 2 min at maximum speed (e.g., 16 000 × g).
-
h.Remove the upper organic phase (it should be approximately 1.2–1.3 mL) leaving a small volume on top of the interphase. Transfer upper phase to a clean tube and store at 4°C until completion of the purification.
-
i.Continue immediately with purification of lower aqueous phase.
-
a.
-
4.DNA recovery step 2: column purification.
-
a.Add 40 μL of Activation Buffer DX onto the spin-column provided with the ePCR kit (do not spin) and keep it at 20°C–25°C.
-
b.Transfer all of the lower aqueous phase (former step: ‘bottom’ aqueous phase) + interphase (max. 600 μL) into a spin-column/receiver tube assembly pre-loaded with Activation Buffer DX and centrifuge at 11,000 × g for 1 min.
-
c.Remove spin column, discard flow-through and replace the spin-column in the collection tube.Note: If the total volume of aqueous phase + interphase exceeds 600 μL repeat steps b-c using the same spin column.
-
d.Add 500 μL of Wash-DX1 buffer and centrifuge at 11,000 × g for 1 min.
-
e.Remove spin column, discard flow-through and replace the spin-column in the collection tube.
-
f.Add 650 μL of Wash-DX2 buffer and centrifuge at 11,000 × g for 1 min.
-
g.Remove spin column, discard flow-through and replace the spin-column in the collection tube.
-
h.Centrifuge again at 11,000 × g for 2 min to remove traces of Wash-DX buffer.
-
i.Place spin-column into new 1.5 mL LoBind collection tube and add 50 μL of Elution-DX buffer.Note: It is possible to reduce the volume of eluting buffer below 50 μL (no less than 20 μL). However, recovery of DNA will gradually decrease.
-
j.Incubate for 2 min at 20°C–25°C and centrifuge at 11,000 × g for 1 min.
-
k.Remove spin columns and pool all eluted DNA into one single LoBind tube. Store at −20°C.Note: the pool can be concentrated by vacuum centrifugation (optional heat [30°C]) to decrease the volume. Extended concentration may decrease oligo pool integrity.
 Pause point: Purified DNA can be stored at −20°C until the next step.
Pause point: Purified DNA can be stored at −20°C until the next step.
-
a.
-
5.Purify and quantify amplified oligo pool.
-
a.Bring the AMPure (SPRI) beads to 20–25°C and prepare fresh 80% ethanol.
-
b.Add 1.6× volume of vortexed AMPure beads to ePCR product (640 μL–400 μL pool DNA from above) and mix by pipetting– gently to minimize foaming. SPRI beads in 1.6× excess will remove everything smaller than 100 bp.
-
c.Incubate 10 min at 20°C–25°C.
-
d.Place mixture on magnetic separator for 2 min or until supernatant is clear.
-
e.With the tube still on the magnetic separator, remove supernatant.
-
f.To wash, add 200 μL 80% ethanol incubate 30 s and discard ethanol.
-
g.Repeat step e and discard all ethanol.
-
h.Dry beads at 20°C–25°C for 5 min.
-
i.Elute from beads by removing the tube from the magnetic separator and adding 32 μL ddH2O. Mix by pipetting – gently to minimize foaming.
-
j.Incubate 2 min at 20°C–25°C.
-
k.Place mixture on magnetic separator for 2 min or until supernatant is clear.
-
l.Transfer 30 μL of the supernatant to a new prelabeled LoBind tube.
-
m.Repeat steps i to l to recover all of the cleaned pool.
-
n.Transfer 30 μL of the supernatant to the prelabeled tube from step l.
-
o.Measure concentration by Qubit HS-DNA (use 4 μL) and check size on QIAXcel or Bioanalyzer (Figure 3A).
-
p.Store at −20°C.
-
a.
-
6.Prepare library for assessment of ePCR product by NGS.
-
a.Prepare sequencing library.
Reagent Amount 5× Q5 reaction buffer 10 μL 10 μM dNTPs 1 μL Library Prep Forward Primer A (2 μM) 2.5 μL Library Prep Reverse Primer A (2 μM) 2.5 μL ePCR product 1 ng Q5 polymerase 0.5 μL ddH2O To 50 μL -
b.Mix gently by pipetting to avoid foaming. Run according to the cycling conditions below.
Step Temperature Time Cycles Initial denaturation 98°C 30 s 1 Denaturation 98°C 10 s ×12 Annealing 55°C 10 s Extension 72°C 10 s Final Extension 72°C 2 min 1 Hold 10°C forever -
c.Keep 2 μL for QIAxcel or Bioanalyzer and use the remainder for SPRI cleanup. This is important to ensure proper size and concentration of sequencing libraries before and after cleanup.
-
a.
-
7.Purify library with triple SPRI bead cleanup.
-
a.Bring the AMPure beads to 20°C–25°C and prepare fresh 70% ethanol.
-
b.Clean up with 0.8× SPRI to keep fragments ∼<300 bp.
-
i.Transfer 46 μL of each library to a LoBind microcentrifuge tube.
-
ii.Add 36.8 μL AMPure beads and mix by pipetting – gently to minimize foaming.
-
iii.Incubate for 5 min at 20°C–25°C.
-
iv.Place mixture on magnetic separator for 5 min.
-
v.Transfer the supernatant to a new tube. It is usually possible to get 70 μL without disturbing the beads. If less or more, record the volume of the supernatant.
-
i.
-
c.Clean up with 1.6× SPRI to remove fragments <100 bp.
-
i.Add AMPure beads to 1.6× (112 μL for 70 μL supernatant from above) and mix by pipetting – gently to minimize foaming.
-
ii.Incubate for 5 min at 20°C–25°C.
-
iii.Place mixture on magnetic separator for 5 min.
-
iv.Discard supernatant with the tube still on the magnetic separator.
-
v.Wash with 200 μL 70% ethanol, incubate 30 s, and discard ethanol.
-
vi.Repeat wash step and discard all ethanol.
-
vii.Briefly spin tube.
-
viii.Place back on magnetic separator for 1 min and discard all remaining ethanol.
-
ix.Dry beads for 2 min at 20°C–25°C.
-
x.Resuspend with 42 μL water, mix by pipetting – gently to minimize foaming.
-
xi.Incubate 2 min at 20°C–25°C.
-
xii.Place mixture on magnetic separator for 5 min.
-
xiii.Transfer 40 μL of the supernatant to a new tube.
-
i.
-
d.Clean up with 1× SPRI to keep fragments ∼100–300 bp
-
i.Add 40 μL AMPure beads to 1.0× and mix by pipetting – gently to minimize foaming.
-
ii.Incubate 5 min at 20°C–25°C.
-
iii.Place mixture on magnetic separator for 5 min.
-
iv.Discard supernatant with the tube still on the magnetic separator.
-
v.Wash with 200 μL 70% ethanol, incubate for 30 s, and discard ethanol.
-
vi.Repeat wash step and discard all ethanol.
-
vii.Briefly spin tube.
-
viii.Place back on magnetic separator for 1 min and discard all remaining ethanol.
-
ix.Dry beads for 3–4 min at 20°C–25°C.
-
x.Resuspend with 16 μL water, mix by pipetting – gently to minimize foaming.
-
xi.Incubate for 2 min at 20°C–25°C.
-
xii.Place tube on magnetic separator for 5 min.
-
xiii.Transfer 15 μL of supernatant to a prelabeled PCR strip.
-
xiv.Use 1 μL for concentration (Qubit HS-DNA) and 1 μL for Bioanalyzer/QIAxcel
-
i.
-
e.Run library preparation on Bioanalyzer or QIAxcel to verify amplicon length and concentration (Figures 3A and 3B).
-
a.
-
8.
Discuss together with NGS sequencing facility if adding 20% PhiX is required (→ low complexity library sequencing), then load libraries in equimolar ratios to lanes of flow cell.
Note: Single-end sequencing with 50 bp reads. The sequencing will inform how uniform the ePCR amplification was as it will allow the user to assess barcode representation and unimodal distribution (Figure 2A). The sequencing results will represent your ‘DNA input’ sample to analyze your MPRA and to quantify the barcodes in transfected cells.
Note: Aim for ∼400–600× coverage, however, do not sequence more than one order of magnitude deeper than the final MPRA replicates, i.e. 300× coverage for replicate = 600× coverage for DNA input.
-
9.
If the ePCR was successful (Figure 3B), up to 99.7% of barcodes can be found, and up to 92.4% of reads can align to barcodes.
Figure 3.

MPRA library preparation and test transfection
(A) Example of non-specific amplification from QIAxcel measurement. In addition to expected ePCR library, non-specific peaks (∼140 bp, ∼220 bp) preclude this sample from further experimental steps.
(B) Successful amplification and library purification of ePCR products (QIAxcel).
(C) Successful library amplification and library purification of tag-seq library from transfected cells (Bioanalyzer).
(D) Agarose gel showing an example of KpnI-digested oligopool after cloning step 2 where cloning step 3 is not required (left lane). Kpn I and XbaI digest to gel-extract fragment for insertion into oligopool from cloning step 1 (right lane).
(E) An example of transfection efficiency (∼>40%) that is required to generate a tag-seq library for a successful and meaningful MPRA. White cells (VSMCs1) depict GFP test transfection.
Figure 2.

Unimodal oligo distribution and cloning
(A) Example of unimodally distributed oligos in density plot after NGS.
(B) Example of 1 × 10 cm Petri dish with >4000 bacterial colonies.
(C) Schemes of cloning steps 10–21. In step 1, candidate region with KpnI and XbaI sites and barcode (BC) is cloned into ‘empty MPRA vector’ via SfiI sites, whilst in cloning step 2, KpnI and XbaI sites are used to insert CMV - GFP insert into the ordered oligo.
High-throughput cloning of plasmid pool
This step integrates the oligos (candidate region + barcode), and subsequently a minimal promoter and a reporter gene, into the MPRA empty vector (Figures 2B and 2C). The reporter gene that we used was a destabilized turboGFP-dest1 (Evrogen) with a shorter half-life to prevent cell stress.
-
10.Digest backbone and insert.
-
a.Activate SfiI restriction sites on both the MPRA empty vector1,3 and the ePCR-amplified oligo pool by digesting with SfiI in separate reactions (Figure 2C). Combine the following in LoBind tubes:
Reagent Amount MPRA backbone 6 μg 10× CutSmart Buffer 5 μL SfiI 50 U ddH2O To 50 μL Reagent Amount ePCR-amplified oligo pool 50 ng 10× CutSmart Buffer 5 μL SfiI 50 U ddH2O To 50 μL -
b.Incubate at 50°C for 90 min.
-
c.Dephosphorylate backbone only by adding 1.5 μL of calf intestinal phosphatase (CIP) and incubating at 37°C for 30 min.
-
d.Column purify the digested oligo pool using a Monarch PCR & DNA Cleanup Kit (NEB) or equivalent.
-
e.Gel extract the linearized backbone (2503 bp) after running on a 1% agarose gel using a Monarch DNA Gel Extraction Kit (NEB) or equivalent.
-
f.Measure concentration of both using Qubit HS-DNA kit.
-
a.
-
11.Ligate components vector and inserts.
-
a.Combine the following reagents in each of two PCR tubes. Insert: vector should be a 4:1 M ratio. Calculate the required ng of insert based on the length of the oligos in the digested pool.
Reagent Amount T4 DNA ligase buffer (10×) 2 μL Digested MPRA backbone 100 ng Digested oligo pool 34.67 ng T4 DNA ligase 1 μL ddH2O To 20 μL Note: An example calculation is provided below. -
b.Gently mix and incubate for 16–20 h at 16°C.
-
a.
-
12.Transform into competent cells.
-
a.Split both ligation reactions evenly across 32× 50 μL tubes of DH5α chemically competent cells.
-
b.Transform according to standard heat shock procedures.
-
c.Spread transformations across 80 10 cm LB ampicillin plates and incubate 16–20 h at 37°C. Alternative plating materials with equivalent total surface area, such as bioassay plates or larger petri dishes (15 cm), can also be used. The photo in Figure 2B depicts a 10 cm plate with more than 4000 bacterial colonies. Consider adjusting the number of transformation reactions and the number of plates to generate a number of bacterial colonies comparable to the number of designed oligos.
-
a.
-
13.Purify plasmid DNA.
-
a.Pool bacterial colonies from all plates by scraping them with cell scraper into liquid LB media.
-
i.Example: add 5 mL LB amp to each of 4 plates.
-
ii.Scrape each plate and prop it up on its lid so the liquid pools.
-
iii.Collect liquid and add to a 50 mL tube.
-
iv.Use 10 mL LB to wash all 4 plates, scraping any leftover colonies that were missed.
-
v.Repeat for all plates.
-
i.
-
b.Pellet bacterial by spinning at 2,500 × g for 15 min at 4°C and remove supernatant. Combine all pellets and redistribute across 12 tubes (scale up, if necessary, based on size of pellet as per QIAGEN Plasmid Plus Maxi Kit recommendation).
-
c.Purify DNA using 12 endotoxin-free QIAGEN Plasmid Plus Maxi prep columns.
-
a.
-
14.Check barcode representation.
-
a.Prepare the following reaction:
Reagent Amount 5× Q5 reaction buffer 10 μL Formamide (5%) 2.5 μL 10 μM dNTPs 1 μL Library Prep Forward Primer A (2 μM) 2.5 μL Library Prep Reverse Primer A (2 μM) 2.5 μL Cloning step 1 plasmid maxi prep 50 ng Q5 polymerase 0.5 μL ddH2O To 50 μL -
b.Run according to the cycling conditions below.
Step Temperature Time Cycles Initial denaturation 98°C 30 s 1 Denaturation 98°C 10 s ×18 Annealing 55°C 10 s Extension 72°C 10 s Final Extension 72°C 2 min 1 Hold 10°C forever -
c.Check concentration after 18 cycles while keeping the tube on ice. Run 1 μL on QIAxcel or Bioanalyzer DNA-HS.
-
i.If peak is too small or non-specific amplification occurred (Figure 3A), run 2–4 more cycles; if the molarity is sufficient for NGS, do the final extension (72°C for 10 min).
-
i.
-
d.Perform triple SPRI bead cleanup as in step 7.
-
a.
- 15.
-
16.
Sequence single read, 50 bp reads. This will inform how uniformly the ePCR amplicon pool was integrated into the plasmid.
-
17.Sequentially digest the plasmid pool with KpnI and XbaI restriction enzymes. The digestion is done sequentially because KpnI requires intact flanking sequence for effective enzyme activity (Figure 2C).
-
a.Digest with KpnI.
Reagent Amount Plasmid pool (cloning step 1) 6 μg 10× CutSmart Buffer 5 μL KpnI-HF 50 U ddH2O To 50 μL -
b.Incubate at 37°C for 60 min.
-
c.Run digestion on a 1% agarose gel and extract linearized product (2642 bp).
-
d.Digest linear plasmid pool with XbaI.
Reagent Amount Plasmid pool (cloning step 1) 6 μg 10× CutSmart Buffer 5 μL XbaI 50 U ddH2O To 50 μL -
e.Incubate at 37°C for 60 min.
-
f.Add 1.5 μL CIP and incubate at 37°C for 30 min.
-
g.Column purify digestion using a PCR clean-up kit.
-
a.
-
18.Digest CMV promoter and GFP ORF from MPRA_minCMV+GFP.
Reagent Amount MPRA_minCMV+GFP 6 μg 10× CutSmart Buffer 5 μL XbaI 50 U KpnI-HF 50 U ddH2O To 50 μL -
a.Incubate at 37°C for 90 min.
-
b.Run digestion on a 1% agarose gel and extract digestion product (833 bp). Measure concentration on a NanoDrop or Qubit fluorometer.
-
a.
-
19.Ligate CMV-GFP into plasmid pool.
-
a.Set up the following reaction in 2–3 tubes, each with a 4:1 insert to vector ratio.
Reagent Amount T4 DNA ligase buffer (10×) 2 μL Digested MPRA backbone 100 ng Digested CMV-GFP fragment variable T4 DNA ligase 1 μL ddH2O To 20 μL -
b.Gently mix and incubate for 16–20 h at 16°C.
-
c.Transform and maxi prep as in step 12–13.Pause point: Maxi prep can be stored at −20°C until the next step.
-
a.
-
20.Remove unsuccessfully ligated plasmids that do not contain CMV-GFP (Figure 3D).
-
a.Digest plasmid pool with KpnI.
Reagent Amount Plasmid pool 6 μL 10× CutSmart Buffer 5 μL KpnI 50 U ddH2O To 50 μL -
b.Incubate at 37°C for 90 min.
-
c.Run digestion on a 1% agarose gel to size separate the plasmids. Plasmids containing the CMV-GFP will be 3.3 kb and those not containing CMV-GFP will be 2.5 kb.Note: If the digestion shows one clear band at the ∼3.3 kb mark, this indicates a highly efficient ligation. In this case, plasmids from cloning step 2 can be used for transfection and subsequent cloning steps are not necessary (Figure 3D).
-
d.Gel extract upper band.
-
e.Re-ligate and transform as in steps 11–12.
-
f.Maxi prep and sequence as in step 13.
-
a.
-
21.Prepare library and sequence according to step 14–16
-
a.These libraries should be prepared with Library Prep Primers B (forward/reverse).
-
a.
Transfection of cells
This protocol will describe how MPRA plasmid pools can be transfected into mammalian cells. Specific details for different cells can be found here.2,3 The following steps provide a reference and approximation of the required number of cells and DNA content and how to determine a successful tag-seq library for NGS that addresses the regulatory effects of the studied candidate elements. The user should adapt transfection guidelines for their cell types of interest (Figure 3E).
-
22.Transfect the cells in bulk with the plasmid pool.
-
a.Seed at least one 10 cm dish to be 80% confluent the following day. Use > 1 × 106 cells when investigating thousands of candidate elements.
-
b.24 h later, transfect the cells with 10–12 μg of MPRA plasmid pool.
-
c.Harvest total RNA after 48 h post-transfection using 1 mL TRIzol and store at −80°C. Alternatively, commercial RNA isolation kits such as the QIAGEN RNeasy Mini Kit can be used.
-
a.
-
23.Perform double DNase I digest to remove all genomic and plasmid DNA.Note: Due to the large quantity of plasmid DNA being transfected into the cells, one DNase I digestion is often insufficient. For this reason, two rounds of DNase I digestion are recommended, followed by a qPCR quantification of the remaining DNA (plasmid) contamination. This is important to prevent plasmid DNA from being amplified during the subsequent library preparation step.
-
a.Perform an in-solution digestion using QIAGEN RNase-free DNase set as per the manufacturer’s instructions.
-
b.Purify this digestion using the RNeasy Mini Kit as per the manufacturer’s instructions. Elute in 30 μL.
-
c.Digest entire volume again using DNase I from Worthington Biochemical. Do not heat-inactivate.
-
a.
-
24.Measure the plasmid DNA contamination of RNA sample.
-
a.Prepare cDNA from each RNA sample with (+RT) and without (-RT) reverse transcriptase using SuperScript III 1st Strand Synthesis Kit (Invitrogen) or equivalent.
-
b.Perform a qRT-PCR using PowerUp SYBR Green Master Mix or equivalent. Use these samples with GFP and GAPDH primers (GAPDH only for +RT). Save the remainder of the cDNA for downstream sequencing.
-
c.Calculate the ΔCT by subtracting GAPDH CT from GFP CT (+RT samples).
-
d.Calculate ΔΔCT by subtracting GFP CT from -RT sample from ΔCT.
-
e.Calculate GFP relative expression = 2-ΔΔCT (base should be 1+primer efficiency. For example, if primers are 95% efficient, relative expression is 1.95-ΔΔCT).
-
f.Calculate percent DNA contamination = GFP relative expression × 100.
-
a.
-
25.
Use the rest of the cDNA in the subsequent steps.
Library preparation and sequencing
These steps describe the library preparation and next-generation sequencing of a MPRA. The libraries created here will be sequenced and the barcodes, and therefore corresponding oligos, will be quantified (Figure 1B).
-
26.Amplify cDNA samples to prepare libraries for NGS as in step 21.
-
a.Run library preparation on Bioanalyzer or QIAxcel to verify amplicon length and concentration.
-
a.
-
27.
Discuss sequencing conditions with NGS core facility. We added 20% PhiX and loaded libraries in equimolar ratios to lanes of flow cell.1
-
28.
Sequence single read, 50 bp reads. Approximately 200–300× coverage, which corresponds to 40–60 million reads when having an oligopool of ∼200,000 barcodes.
Expected outcomes
Include information about the anticipated outcomes of the protocol, e.g., estimated yield of DNA extraction, images of protein expression pattern. We encourage authors to provide figures to illustrate the expected outcomes (see examples here) and describe the expected results in paragraphs.
Completion of the above steps will result in an evenly distributed oligo pool and plasmid pool of high complexity, efficiently transfected cells (>40%), and cDNA libraries with sufficient barcode recovery (>50%). The user will now have the materials needed to complete their MPRA analysis. In previous studies that have employed this protocol, these materials have facilitated the identification of functional variants implicated in blood pressure gene regulation,1 measurement of cis and trans effects of regulatory elements between mouse and human,2 and dissecting the relationship between lncRNA core promoter sequence composition and activity.3
Quantification and statistical analysis
DNA and RNA reads can be trimmed and quality filtered using CutAdapt.5 Next, reads should be filtered based on a minimum barcode recovery and a background model must be built based on positive and negative controls.1 Upon completing these initial computational analysis steps, the user can perform desired statistical analysis using various methods that best suit their study. MPRA activity can be quantified using the publicly available R package MPRAnalyze.4
Limitations
This approach is limited by the cell types the assay is performed in. Many results will be cell type- or tissue type-specific. It is important to select relevant cell lines to assay and it is recommended to assay more than one. Low transfection efficiencies in a given cell type will also negatively impact the success of the MPRA as this will cause some (or many) candidate elements to be lost from the experiment.
Troubleshooting
Problem 1
Poor representation of barcode diversity after ePCR or during cloning.
Potential solution
-
•
The number of molecules used as input in the ePCR was not appropriate for the number of micelles.
-
•
ePCR cycling conditions based on amplicon length may require optimization.
-
•
Transform more bacterial cells and ensure scraping and washing of plates are thorough.
Problem 2
Low barcode recovery in MPRA after transfections.
Potential solution
-
•
Ensure your barcode recovery in previous cloning steps was sufficient.
-
•
Optimize transfection conditions by altering transfection reagent, reagent amount, or DNA amount, change number of cells.
-
•
Test whether the selected in vitro model expresses the CMV-GFP construct.
Problem 3
Poor amplification of cDNA libraries.
Potential solution
-
•
Adjust PCR conditions and alter cycle numbers.
-
•
Check library before and after SPRI cleanup to validate successful cleanup.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Philipp Maass (philipp.maass@sickkids.ca).
Materials availability
Plasmids generated in this study have been deposited to Addgene, [MPRA Empty Vector #204865; MPRA_minCMV+GFP #204866].
Data and code availability
This study did not generate code.
Acknowledgments
We thank The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada for assistance with high-throughput sequencing. This project was supported by the Canadian Institutes of Health Research (CIHR PJT 173542 [P.G.M.]) and the Canada Research Chairs Program.
Author contributions
Conceptualization and funding acquisition, P.G.M.; methodology, K.D. and C.G.; resources, J.L.R.; writing – review and editing, K.D., C.G., J.L.R., and P.G.M.
Declaration of interests
The authors declare no competing interests.
References
- 1.Oliveros W., Delfosse K., Lato D.F., Kiriakopulos K., Mokhtaridoost M., Said A., McMurray B.J., Browning J.W.L., Mattioli K., Meng G., et al. Systematic characterization of regulatory variants of blood pressure genes. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mattioli K., Oliveros W., Gerhardinger C., Andergassen D., Maass P.G., Rinn J.L., Melé M. Cis and trans effects differentially contribute to the evolution of promoters and enhancers. Genome Biol. 2020;21:210. doi: 10.1186/s13059-020-02110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mattioli K., Volders P.J., Gerhardinger C., Lee J.C., Maass P.G., Melé M., Rinn J.L. High-throughput functional analysis of lncRNA core promoters elucidates rules governing tissue specificity. Genome Res. 2019;29:344–355. doi: 10.1101/gr.242222.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashuach T., Fischer D.S., Kreimer A., Ahituv N., Theis F.J., Yosef N. MPRAnalyze: statistical framework for massively parallel reporter assays. Genome Biol. 2019;20:183. doi: 10.1186/s13059-019-1787-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate code.