

Summary
Pancreatic ductal adenocarcinoma (PDAC) is projected to become the second leading cause of cancer-related death. Hallmarks include desmoplasia with variable extracellular matrix (ECM) architecture and a complex microenvironment with spatially defined tumor, stromal, and immune populations. Nevertheless, the role of desmoplastic spatial organization in patient/tumor variability remains underexplored, which we elucidate using two technologies. First, we quantify ECM patterning in 437 patients, revealing architectures associated with disease-free and overall survival. Second, we spatially profile the cellular milieu of 78 specimens using codetection by indexing, identifying an axis of pro-inflammatory cell interactions predictive of poorer outcomes. We discover that clinical characteristics, including neoadjuvant chemotherapy status, tumor stage, and ECM architecture, correlate with differential stromal-immune organization, including fibroblast subtypes with distinct niches. Lastly, we define unified signatures that predict survival with areas under the receiver operating characteristic curve (AUCs) of 0.872–0.903, differentiating survivorship by 655 days. Overall, our findings establish matrix ultrastructural and cellular organizations of fibrosis linked to poorer outcomes.
Keywords: pancreatic ductal adenocarcinoma, desmoplasia, architecture, fibrosis, spatial biology, cancer, codex, cell interactions, cell networks, machine learning
Graphical abstract
Highlights
-
•
Matrix architecture of desmoplasia correlates with overall and disease-free survival
-
•
Pro-inflammatory cell spatial interactions further differentiate outcomes
-
•
Clinical characteristics correlate with distinct stromal-immune cell organization
-
•
A unified, machine-learning-based signature predicts PDAC patient survival
Mascharak and Guo et al. elucidate the role of desmoplastic spatial organization in PDAC patient outcomes. Their spatial analysis uncovers global matrix architectures associated with overall and disease-free survival, pro-inflammatory cell-cell interactions correlated with poorer outcomes, and a unified, machine-learning-based signature that predicts patient outcomes in an independent cohort.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains the only major cancer with a rising death rate in the United States and is projected to be the second leading cause of cancer-related deaths in the next decade.1 Recurrence of PDAC commonly occurs within 2 years after curative intent pancreatectomy and either preoperative and/or postoperative chemotherapy.2 Accordingly, the overall 5-year survival rate remains roughly 8%–10%.3 A deeper understanding of the factors driving PDAC progression, recurrence, and metastatic spread is necessary to develop more effective therapeutic strategies and prognostic tools.
PDAC and other solid tumors are defined by desmoplasia, an extensive fibrotic reaction that results from malignant cell crosstalk with stromal tissue.4,5 Interestingly, univariate morphological properties of the desmoplastic milieu, such as collagen density and fiber orientation, have been correlated with differential patient outcomes, though there is minimal consensus across findings.6,7,8,9 Critically, these approaches do not capture the full geometric complexity of matrix patterning observed in PDAC desmoplasia. Thus, the impact of complex desmoplastic architecture on PDAC patient outcomes remains unclear.
PDAC is further characterized by a heterogeneous tumor microenvironment (TME) with spatially defined tumor, stromal, and immune populations that drive overall disease progression and patient outcomes.4,10,11 Prior studies have focused on identification of a limited array of cell phenotypes that influence PDAC prognosis.12,13 For instance, immune subtypes, such as activated B lymphocytes and human leukocyte antigen, DR isotype (HLA-DR)+ macrophages, may influence PDAC outcomes via effects on immune regulation.8,14 Recent literature has also implicated cancer-associated fibroblasts (CAFs), classified into mechanoresponsive and inflammatory subtypes, in the deposition of fibrotic desmoplasia, pro-inflammatory signaling, and elaboration of tumor-modulating factors.6,15,16,17 However, these analyses have not fully evaluated the prognostic value of spatial cell organization, which includes a myriad of cell-cell interactions, spatially defined cell communities, and local extracellular matrix (ECM) architectures. Emerging literature suggests that spatially defined cell neighborhoods may shape and predict disease progression in other diseases such as colorectal and renal cancer,18,19,20 but this concept has not been thoroughly explored in PDAC. Thus, we hypothesized that cell- and matrix-based spatial organization may predict differential PDAC patient outcomes.
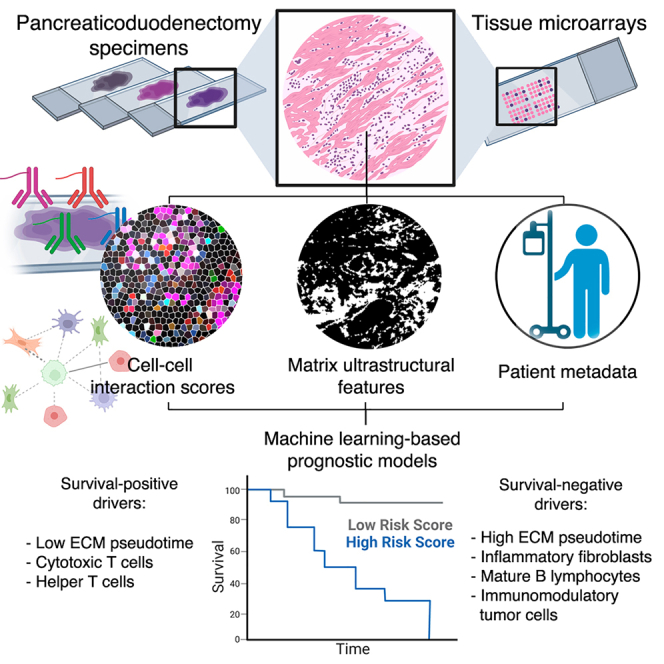
Here, we leveraged two complementary analyses to systematically profile the spatial heterogeneity of PDAC: (1) matrix analysis using an ultrastructural quantification algorithm and (2) cell spatial analysis using co-detection by indexing (CODEX). By integrating these analyses with clinical metadata using machine learning (ML), we uncovered holistic signatures that strongly differentiate patients’ overall survival (OS). Critically, we validated our prognostic signatures in an independent, blinded patient cohort. Our findings represent a desmoplastic-stromal signature that predicts patient outcomes in PDAC and suggests promising therapeutic targets.
Results
High-dimensional matrix architecture stratifies patient survival and disease recurrence
First, we analyzed the architecture of PDAC desmoplasia using an ultrastructural quantification algorithm developed in our laboratory for fibrotic tissue analysis.21 A total of 578 pancreaticoduodenectomy pathology specimens were collected across three separate cohorts—Stanford University, the University of Virginia (UVA), and commercial tissue microarrays (TMAs)—yielding 13,048 trichrome image tiles (Figure 1A; Table S1). Of these specimens, 437 PDAC specimens were included in downstream analysis, 129 were excluded because they did not have a diagnosis of PDAC, and 12 were excluded due to operative mortality without follow-up data, loss to follow-up, or metastasis at time of pancreaticoduodenectomy. Matrix ultrastructure in each Trichrome image was quantified using an automated analysis pipeline of 147 fiber features, which collectively capture architectural variation on local (individual fiber length, width, persistence, etc.) and global scales (alignment, packing, porosity, etc.) (Figure 1B).21 This high-dimensional feature matrix was reduced by uniform manifold approximation and projection (UMAP) to visualize differences in overall matrix architecture between images. We envisioned that ultrastructural states of the desmoplasia could be mathematically ordered based on stepwise deviations in the fiber feature matrix, similar to the relative ordering of transcriptional states based on quantitative shifts in a gene expression matrix.22 Thus, the DDRTree algorithm was utilized to learn a minimum-spanning-tree-based trajectory that connects datapoints based on similarity in ultrastructural parameters (Figure 1C), assigning pseudotime scores based on relative deviation from a root point near baseline histological architecture (healthy pancreas; Figure 1D).23,24
Figure 1.

Matrix ultrastructural analysis of PDAC desmoplasia
(A) CONSORT diagram of imaging and ultrastructural analysis for patients with PDAC.
(B) Ultrastructural quantification from histological images, including pseudotime modeling of ultrastructural states and integration with CODEX.
(C) Manifold of PDAC desmoplastic architecture with higher pseudotime representing increasingly disrupted, heterogeneous desmoplastic architecture. Boxed images show representative tiles along the pseudotime trajectory. Scale bars represent 100 μm.
(D) Visualization of healthy pancreas samples, which localize near the root point of the pseudotime trajectory.
(E and F) Integration of overall survival (E) and disease-free survival (F) with clinical metadata.
(G–I) Correlation of overall survival with patient-level pseudotime (G), stained quantity of fibrosis (H), and ultrastructural variance (I). Pearson coefficients and associated p values are shown.
(J–L) Correlation of disease-free survival with patient-level pseudotime (J), stained quantity of fibrosis (K), and ultrastructural variance (L). Pearson coefficients and associated p values are shown.
Pseudotime analysis revealed three general desmoplastic ECM patterns. The first, represented by images in the lower pseudotime (Figure 1C, left) region of the manifold (most similar to healthy pancreatic architecture), was characterized by intact glandular structures nested within regions of thinner matrix fibers (Figure 1C, bottom left). Increasing pseudotime involved gradual loss of glandular architecture and spindle-like fibrosis (Figure 1C, top left) until a split in the trajectory on the right region of the manifold into two branches: (1) a direct progression to high-pseudotime, terminal ECM patterning on the upper right and (2) progression through an intermediate state, followed by progression to terminal ECM patterning. The intermediate ECM pattern, represented by images in the bottom right quadrant (Figure 1C, bottom right), included confluent globules of matrix proteins and near-complete disruption of glandular architecture. The terminal ECM pattern, represented by images in the upper right quadrant (Figure 1C, top right), was characterized by thick, aligned cables of matrix and further disordered glandular architecture.
Initial visualization of clinical outcomes on the manifold suggested that architectural differences were associated with OS (death from time of pancreaticoduodenectomy; Figure 1E) and disease-free survival (DFS; recurrence or death from time of pancreaticoduodenectomy; Figure 1F). Thus, patient-specific ultrastructural data for PDAC specimens was averaged and integrated with clinical metadata (Figures 1G–1L), while healthy pancreas and non-PDAC neoplasms initially utilized for manifold training were censored and excluded from all downstream quantitative analyses. First, each patient’s average ultrastructural state was quantified by calculating the centroid (median) pseudotime of all patient-specific images. High median pseudotime values demonstrated a strong statistical correlation with decreased OS (Pearson coefficient [R] = −0.194, p = 0.006) and trended toward significance in predicting lower DFS (R = −0.093, p = 0.197) (Figures 1G and 1J). Furthermore, patient-level pseudotime demonstrated a stronger association with outcome than stained matrix area (R = −0.128/−0.092, p = 0.074/0.203 for OS and DFS, respectively), a parameter in the algorithm representing quantity of fibrosis (Figures 1H and 1K). Interestingly, intra-patient heterogeneity in matrix architecture, as quantified by sum of variances for all 147 ultrastructural parameters, also trended toward statistical significance in predicting lower OS and DFS (R = −0.089/−0.102, p = 0.212/0.153) (Figures 1I and 1L). Overall, these data suggested the strongest prognostic role for high-dimensional matrix architecture, particularly for predicting OS, rather than quantity of desmoplasia or intra-patient heterogeneity.
We next integrated patient-level pseudotime with clinical metadata to understand associations between matrix architectural progression and patient and tumor characteristics (Figures S1A and S1B). Interestingly, high pseudotime was correlated with more advanced patient age (R = 0.151, p = 0.002) but did not show a statistically significant association with patient gender (Figure S1B). Additionally, while pseudotime was statistically independent of tumor grade, it was correlated with more advanced American Joint Committee on Cancer (AJCC) staging (Spearman coefficient [Rs] = 0.183, p < 0.001). Further analysis indicated that this positive association was primarily driven by the N stage (Rs = 0.281, p < 0.001), representing involvement of regional lymph nodes, as opposed to the T stage, which characterizes tumor size/extent and exhibited a negative correlation (Rs = −0.105, p = 0.031). Higher matrix pseudotime was accordingly associated with a greater number of tumor-positive regional lymph nodes (R = 0.295, p < 0.001) but was agnostic to measured tumor size (R = 0.081, p = 0.259). Pseudotime was also statistically independent of CA19-9 levels or neoadjuvant chemotherapy status. As a whole, our analysis indicated that terminal ECM patterning was largely correlated with older patients and tumors with greater lymph node involvement. These findings suggested that complex stromal phenomena, particularly immune-stromal interactions, could be associated with differential matrix architecture and patient outcomes in PDAC.
Spatially defined cell-cell interactions among stromal, immune, and tumor cell populations predict differential patient outcomes
To analyze contributions of stromal cell organization to PDAC patient outcomes, we applied CODEX to spatially profile 78 pancreaticoduodenectomy specimens, consisting of >1,250,000 spatially indexed cells (Figure 2A; STAR Methods). Individual cell populations were identified across patient specimens based on protein expression (Figures 2A and S2A–S2D), including inflammatory (interleukin-6 [IL-6]+) and mechanoresponsive (alpha-smooth muscle actin [aSMA]+) CAF subpopulations, differentially activated (programmed death-ligand 1 [PDL1]/stromal cell-derived factor 1 [SDF1]/human leukocyte antigen [HLA1]+) tumor cells, immune cell subpopulations (B and T lymphocyte subtypes, macrophage subtypes), and vasculature-associated cells (endothelial cells, pericytes) (Table S2; Figures S3A–S3F). Interestingly, the annotated pericyte cluster (aSMAhigh/matrix gla protein [MGP]+; see Table S2) clustered directly adjacent to both endothelial cells and the "fibroblasts 3" cluster (CD26high/aSMAmed/IL-6low) and also expressed fibroblast-like markers (e.g., aSMA, collagen [COL] I). Thus, this population may contain an admixture of cell phenotypes such as pericytes and perivascular fibroblasts. First, cell phenotypic representation was correlated with OS and DFS at the patient level (Figures 2B and 2C), demonstrating that a high abundance of cytotoxic T lymphocytes (CD4low/CD8high) was the most strongly positive correlate of OS and DFS (R = 0.234/0.246, p = 0.028/0.021, respectively). In contrast, an abundance of a highly activated/immunomodulatory tumor cells 4 subtype (E-cadherinhigh/pan-cytokeratin [PANCK]med/PDL1high/SDF1high) was the most negative correlate of both OS and DFS (R = −0.223/−0.178, p = 0.036/0.096, respectively). To analyze the contributions of spatially defined cell-cell relationships, we computed 153 interaction scores (e.g., T lymphocyte-B lymphocyte, fibroblast-tumor subtype interactions) representing the frequency of specific cell-cell spatial colocalizations and integrated these scores with patient metadata (Figures 2D–2I). Our analysis revealed that poorer survival was strongly driven by two OS-negative interaction “nodes”: highly activated/immunomodulatory tumor cells 4 (E-cadherinhigh/PANCKmed/PDL1high/SDF1high) and mature/activated B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh), which both produced universally OS-negative cell interactions (Figures 2D, 3A, 3B, and S4A). Accordingly, the singular most survival-negative cell interaction was between tumor cells 4 and B lymphocytes 1 (R = −0.305, p = 0.004). Cytotoxic T lymphocytes and helper T lymphocytes, on the other hand, represented OS-positive nodes that drove predominantly survival-positive spatial interactions (Figures 2D, 3A, 3C, and S4A). The singular most survival-positive cell spatial interaction was between cytotoxic T lymphocytes and pericytes (R = 0.424, p < 0.001), implying T lymphocyte infiltration into the TME. Additionally, we examined associations of cell-cell spatial interactions with disease recurrence. Earlier disease recurrence was associated with interactions by tumor cells 4, B lymphocytes 1, and the relatively neutral fibroblasts 2 subtype (CD26med/aSMAlow/IL-6low) (Figures 2E, 3D, and S4B). Conversely, DFS-positive drivers included cytotoxic T lymphocytes and pericytes, whose interaction was, similar to OS, the most outcome-positive interaction (R = 0.474, p < 0.001) (Figures 2E, 3D, and S4B).
Figure 2.

Spatial phenotyping and cell spatial analysis of PDAC tumors
(A) CODEX spatial phenotyping and identification of cell phenotypes (e.g., more and less activated tumor cells, fibroblast subpopulations, macrophages, B and T lymphocytes, endothelial cells) by protein expression.
(B) Signatures of cell phenotypic representation for overall survival and disease-free survival.
(C) Representative patient specimens with spatially indexed cell phenotypes.
(D and E) Prognostic patient-level interactions for overall survival (D) and disease-free survival (E). Blue and red lines indicate outcome-positive and outcome-negative correlations, respectively. Width of line represents magnitude of the Pearson coefficient.
(F) Differential interactions in patients who did and did not receive neoadjuvant chemotherapy before pancreaticoduodenectomy. Blue and red lines represent interactions that are enriched and weakened, respectively, in patients who received neoadjuvant chemotherapy. Width of line represents the average difference in interaction score.
(G–I) Association of tumor grade (G), stage (H), and size (I) with cell interactions. Blue lines represent interactions that are associated with decreased (i.e., outcome-positive) clinical values, while red lines represent increased (i.e., outcome-negative) clinical values. Width of line represents magnitude of the Spearman coefficient for grade and stage and Pearson coefficient for size.
Figure 3.

Correlation between cell-cell spatial interactions and patient outcomes
(A) Pearson coefficients for overall survival (OS), ordered to highlight cell phenotypes driving OS-positive and OS-negative interactions.
(B and C) Representative patient specimens highlighting survival-negative (B) and survival-positive (C) interactions. Spatial plots are shown for both the overall cell phenotypic distribution (left) and areas of highest interaction score indicated in red (right). Boxes indicate an area of interest for each cell interaction, with raw CODEX staining shown, in addition to a magnified view (far right) of the area of interest.
(D) Pearson coefficients for disease-free survival (DFS).
Prognostic cell-cell interactions are primarily short-range associations
For the top outcome-positive and outcome-negative interactions, we used linear regression modeling to analyze the relationships between interaction distance and clinical correlation strength at length scales from 50 to 500 μm (Figure 4A), in order to infer mechanisms of paracrine vs. long-range interaction.25 Short-range cell-cell interactions were defined as weakening correlation strength (i.e., absolute value of Pearson coefficient) with increased distance, while long-range interactions were conversely defined as increasing strength with distance (Figure 4B). First, we analyzed range-dependent behavior of the most highly OS-positive interactions (Figure 4C). Pericyte interactions with cytotoxic T lymphocytes, helper T lymphocytes (CD4high/CD8low), and other pericytes were distinctly OS positive (R = 0.424/0.341/0.267, p < 0.001/p = 0.001/p = 0.012, respectively) and short range, suggesting that T lymphocyte infiltration into a vascularized TME serves as a positive clinical factor. Interestingly, interaction of cytotoxic T lymphocytes with inflammatory fibroblasts 5 (CD26low/aSMAmed/IL-6high) appeared to be long range and survival positive (R = 0.295, p = 0.005), implying potential interaction through secreted cytokines or intermediate actors rather than direct interaction.
Figure 4.

Distance-based analysis of top outcome-differentiating CODEX cell interactions
(A) Definition of short- and long-range correlations.
(B) Illustration of characteristic short-range (orange) and long-range (green) behavior for outcome-positive and outcome-negative interactions.
(C and D) Distance-based analysis of top survival-positive (C) and survival-negative (D) interactions. p values are indicated for linear regression models.
(E and F) Distance-based analysis of top recurrence-positive (E) and recurrence-negative (F) interactions. p values are indicated for linear regression models.
Next, we analyzed range dependence of the top OS-negative interactions (Figure 4D). B lymphocyte 1 interactions with highly activated/immunomodulatory tumor cells 4, relatively neutral fibroblasts 2 subtype (CD26med/aSMAlow/IL-6low), and helper T lymphocytes were short range and highly survival negative (R = −0.305/−0.259/−0.256, p = 0.004/0.015/0.016, respectively), indicating that mature B lymphocytes may correlate with poorer outcomes through an array of direct/paracrine interactions, particularly in a more activated TME. Accordingly, the activated tumor cells 4 phenotype drove its own subset of short-range, survival-negative (R = −0.232/−0.305, p = 0.030/0.004, respectively) interactions with immune cells such as helper T lymphocytes and B lymphocytes 1, as well as a survival-negative (R = −0.280, p = 0.008) but range-agnostic interaction with the mechanoresponsive fibroblasts 4 subtype (CD26high/aSMAhigh/IL-6med).
Further, we analyzed the range dependence of the most DFS-positive and DFS-negative interactions (Figures 4E and 4F). The top DFS-differentiating interactions largely mirrored the top OS-differentiating interactions in both identity and range-dependent behavior, with a few notable exceptions. For instance, interaction of endothelial cells with the less activated tumor cells 2 phenotype (E-cadherinmed/PANCKmed/PDL1med/SDF1med) was recurrence positive (R = 0.275, p = 0.010) and long range, indicating that co-occurrence of vasculature and less activated tumor cells within a single tumor, but not necessarily in close proximity, was correlated with longer duration until disease recurrence. Additionally, interaction between the neutral fibroblasts 2 and mechanoresponsive fibroblasts 4 subtypes was short range and recurrence negative (R = −0.215, p = 0.044), indicating that close colocalization of these fibroblast subtypes was associated with faster disease recurrence. Beyond this, short-range interactions involving cytotoxic T lymphocytes, most notably with pericytes in an inferred context of T lymphocyte infiltration, were positive factors for a longer DFS duration as previously observed for OS (Figures 4C and 4E). Similarly, a wealth of short-range, recurrence-negative interactions were driven by B lymphocytes—particularly the mature B lymphocyte 1 subtype—as well as the activated tumor cell 4 subtype (Figure 4F). Thus, outcome-negative cell spatial organization appeared to be largely driven by activated and immunomodulatory cell subpopulations in close proximity.
Neoadjuvant chemotherapy and tumor characteristics are associated with differences in cell spatial organization
We analyzed differences in the spatial TME associated with neoadjuvant chemotherapy, as well as clinically relevant metrics such as tumor grade, AJCC stage, and size. Patients who had received neoadjuvant chemotherapy exhibited highly enriched fibroblast-B lymphocyte interactions (e.g., fibroblasts 5-B lymphocytes 2, fibroblasts 5-B lymphocytes 1) compared with patients who did not receive neoadjuvant chemotherapy before pancreaticoduodenectomy (Figures 2F and 5A). In particular, the inflammatory fibroblast 5 subtype (CD26low/aSMAmed/IL-6high) appeared to drive the spatial TME associated with neoadjuvant chemotherapy, with this cell phenotype participating in 4 out of 5 (80%) of the top enriched interactions (Figure 5A). In contrast, neoadjuvant chemotherapy was associated with weakened interactions involving the relatively less activated tumor cell 1 (E-cadherinlow/PANCKmed/PDL1low/SDF1low) and tumor cell 2 (E-cadherinmed/PANCKmed/PDL1med/SDF1med) subtypes (e.g., tumor cells 1-endothelial cells, tumor cells 2-macrophages 1) (Figure 5A). These data suggest that neoadjuvant chemotherapy promotes a more inflammatory TME mediated by fibroblast-B lymphocyte interactions. Interestingly, higher tumor grade was largely associated with helper T lymphocyte-driven interactions, particularly with pericytes (Figures 2G, 5B, and S4C), while lower grade was associated with inter-fibroblast interactions. Higher AJCC stage, on the other hand, was associated with several fibroblast-driven interactions, particularly by fibroblasts 3 (CD26high/aSMAmed/IL-6low) and fibroblasts 4 (CD26high/aSMAhigh/IL-6med), while lower AJCC stage was associated with interactions involving endothelial cells and pericytes (Figures 2H, 5C, and S4D). Larger tumor size, which is incorporated in AJCC staging, was similarly correlated with fibroblast 4-mediated interactions, while smaller tumor size was correlated with inter-tumor cell interactions (Figures 2I, 5D, and S4E). Overall, these data suggested that clinical characteristics of PDAC were associated with highly differential CAF-immune cell spatial organization, such as enriched CAF-B lymphocyte interactions in tumors that received neoadjuvant chemotherapy.
Figure 5.

Differential cell spatial organization associated with neoadjuvant chemotherapy and clinically relevant tumor metrics
(A) Top 5 positively (top) and negatively (bottom) associated cell-cell interactions with neoadjuvant chemotherapy. p values are indicated for Student's t test.
(B and C) Spearman coefficients for ordinal categories of tumor grade (B) and AJCC stage (C).
(D) Pearson coefficients for continuous data on tumor size.
Inflammatory and mechanoresponsive fibroblasts exhibit distinct interactomes
While CAF heterogeneity has been investigated in the context of spatially agnostic tissue digests and/or in vitro culture,11,26 the in situ spatial niches of clinical CAFs have not been thoroughly explored. Thus, cell-cell spatial niches, i.e., interactomes, were examined for PDAC-associated CAFs via principal-component analysis (PCA) of all pairwise cell-cell interactions involving fibroblasts (Figure 6A). Overall, fibroblast subtypes appeared to exhibit distinct niches based on inflammatory vs. mechanically activated protein expression (Figures 6A and 6B). For instance, the inflammatory fibroblast 1 (CD26low/aSMAlow/IL-6med) and fibroblast 5 (CD26low/aSMAmed/IL-6high) subtypes clustered together by PCA centroids, suggesting similarity in interaction space (Figure 6B, right). Accordingly, both fibroblasts 1 and fibroblasts 5 were strongly defined by B lymphocyte 1 and 2 interactions (e.g., PC3, PC4, PC5). The mechanically activated fibroblast 3 (CD26high/aSMAmed/IL-6low) and fibroblast 4 (CD26high/aSMAhigh/IL-6med) subtypes also clustered together in PCA centroids (Figure 6B, right). These fibroblast subtypes were strongly defined by their interactions with the relatively less activated tumor cell 2 phenotype (E-cadherinmed/PANCKmed/PDL1med/SDF1med) (e.g., PC1, PC2). To a lesser extent, these two mechanically activated fibroblast subtypes were also defined by interactions with the least activated tumor cells 1 phenotype (E-cadherinlow/PANCKmed/PDL1low/SDF1low) (e.g., PC1, PC2) but did not appear to be strongly associated with more activated tumor cell phenotypes. Lastly, the neutral fibroblasts 2 subtype (CD26med/aSMAlow/IL-6low) was strongly defined by interactions with the most highly activated tumor cells 4 phenotype (E-cadherinhigh/PANCKmed/PDL1high/SDF1high) (e.g., PC3). Thus, mechanically activated and neutral fibroblast subtypes appeared to occupy a primarily tumor-adjacent spatial niche, while inflammatory fibroblasts resided in a predominantly B lymphocyte-adjacent niche. Critically, this juxtacrine analysis also revealed that mechanically activated fibroblast subtypes were associated with less activated tumor cell subpopulations while indicating that more neutral fibroblast subtypes were adjacent to more highly activated tumor cells, suggesting a relationship between fibroblast mechanical activation and tumor cell state.
Figure 6.

Interactomes of cancer-associated fibroblasts and B lymphocytes, as well as their variation with protein phenotype
(A) Principal-component analysis (PCA) of fibroblast interaction space up to 5 PCs, as determined by vertex of scree plot. Fibroblasts appear to exhibit distinct interaction patterns that are defined by differences in inflammatory vs. mechanically activated protein expression.
(B) Heatmaps of PCA coefficients and centroids for each fibroblast subtype.
(C) PCA of B lymphocyte interaction space up to 4 principal components, as determined by vertex of scree plot. B lymphocytes appear to exhibit distinct interaction patterns based on maturity of protein phenotype.
(D) Heatmaps of PCA coefficients and centroids for each B lymphocyte subtype.
Spatial interactomes were also examined for B lymphocytes, which consisted of the immature B lymphocytes 2 (CD20low/HLA-DRlow/IL-6low/Vimentinlow) and mature B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh) subtypes (Figure 6C). Overall, B lymphocytes exhibited a spectrum of spatial niches, starting from (1) immature phenotype and vasculature adjacent (e.g., endothelial cells and pericytes in PC3) to (2) mixed phenotype and neutral tumor/fibroblast adjacent (e.g., tumor cells 1 and fibroblasts 1 in PC2 and PC4), and finally, (3) mature phenotype and inflammatory fibroblast adjacent (e.g., fibroblasts 5 in PC4) (Figures 6C and 6D). B lymphocyte maturity thus appeared to be correlated with an increasingly fibroblast-adjacent and pro-inflammatory interaction space. Macrophages, on the other hand, exhibited weaker associations between a more mature phenotype (HLA-DRhigh) and helper T lymphocyte adjacency (Figures S5A and S5B), while tumor cells displayed highly heterogeneous interaction spaces (Figures S6A and S6B). Overall, across the single-cell interactomes (Figures 6, S5, and S6), more mature, antigen-presenting cell subtypes such as B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh) and macrophages 2 (HLA-DRhigh) were generally associated with increasingly inflammatory and/or immune-adjacent interaction spaces, which may include inflammatory CAFs.
Macrophage and fibroblast subpopulations colocalize with differential matrix architectures
In addition to assessing cell spatial niches, we sought to analyze associations between desmoplastic matrix architecture and cell spatial organization. To integrate matrix analysis with CODEX, we analyzed collagen I ultrastructure within 10 × 10 subsampled CODEX tiles (3,247 subsamples) and mapped the computed ultrastructural features to the original Trichrome manifold (Figure S7A), producing no significant difference in Trichrome vs. CODEX-defined pseudotime values at the patient level (p = 0.294). Interestingly, outcome-positive matrix architecture (low pseudotime) was strongly associated with macrophage-driven interactions, particularly by the less activated macrophage 1 (HLA-DRlow) subtype (Figure S7B, top). Outcome-negative CODEX architecture (high pseudotime), on the other hand, was associated with several interactions involving inflammatory fibroblasts 5 (CD26low/aSMAmed/IL-6high) and pericytes. Interestingly, interactions between fibroblasts 5 and mature/activated B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh), an outcome-negative cell interaction node, were also highly associated with high pseudotime. Given the associations of inflammatory, activated cell-cell interactions with both matrix pseudotime and worse patient outcomes, we examined how cell-type abundance varied in regions of differential matrix architecture, with a focus on immune composition (Figure S7B, bottom). Notably, high matrix pseudotime was associated with a high abundance of helper T lymphocytes, but not cytotoxic T lymphocytes, suggesting a relatively higher CD4/CD8 ratio in regions of outcome-negative architecture. While associations with B lymphocyte abundance were relatively weaker, it appeared that the macrophages 1 subtype (HLA-DRlow) was enriched in regions of outcome-positive architecture, in accordance with their involvement in multiple low-pseudotime cell interactions (Figure S7B, top). We additionally investigated associations between matrix architecture and tumor-endothelial cell interactions, which may represent metastatic spread, and found a potential trend between high pseudotime and endothelial cell interactions with more highly activated tumor cells (R = −0.053/−0.074/0.020/0.058 for tumor cells 1–4, respectively). Further, an examination of underlying ultrastructural features indicated that low pseudotime was strongly associated with individual fiber-level measures of dispersion (e.g., standard deviation of skeletonized fiber lengths and diameters), representing spindle-like fiber distributions observed in histology (Figure S7C). On the other hand, high pseudotime was associated with increasing topological porosity (Euler number) and texture heterogeneity (energy), consistent with its disrupted architecture of globular and sheet-like collagen fibers. To explore if other histological modalities such as hematoxylin and eosin (H&E) could be used to quantify matrix architecture and ECM-associated cellular features, we performed H&E staining on a pilot cohort of 48 sections taken consecutively to CODEX (Figure S7D). These samples were aligned one to one with the original CODEX data using a DAPI-based alignment mask, followed by subsampling of aligned H&E tiles (944 subsamples). H&E tiles were then color deconvoluted to isolate matrix components, quantified for 147 architectural features as previously described, and similarly mapped to the Trichrome manifold. High matrix pseudotime, based on H&E staining, revealed a similar set of cell spatial interactions that involved inflammatory fibroblasts 5 (CD26low/aSMAmed/IL-6high), mature/activated B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh), and T lymphocytes (Figure S7D). Low H&E pseudotime, on the other hand, did not correlate as strongly with macrophage-driven interactions but rather with inter-tumor cell interactions, including a cell-cell interaction between the less activated tumor cells 1 and tumor cells 2 subtypes that reflected their enrichment in regions of low pseudotime (Figure S7B, bottom). Thus, H&E staining may offer an alternative means of matrix architectural analysis to Trichrome and collagen I staining that warrants further exploration. Collectively, the integration of CODEX and matrix architecture indicated that the cellular niche of outcome-negative architecture may involve pro-inflammatory populations such as fibroblasts 5 and B lymphocytes 1, while the niche of outcome-positive matrix architecture may be driven by less mature/activated macrophages (HLA-DRlow) and/or inter-tumor cell interactions.
Unified spatial signatures predict patient outcomes using ML
Lastly, we unified cell spatial organization, matrix ultrastructure, and clinical metadata using ML modeling to produce holistic spatial signatures of PDAC survival (Figure 7). First, we trained six representative ML models—an artificial neural network (ANN), a generalized additive model (GAM), a k-nearest neighbors (KNN) model, a linear discriminant analysis (LDA), a random forest (RF), and a support vector machine (SVM)—using 3,247 subsampled CODEX tiles with computed cell interaction scores, ultrastructural parameters, and patient metadata. Categorical variables such as AJCC stage were ordinally encoded,27 and a pseudotime threshold of 24 was also applied to generate a binary “ECM architecture” variable representing tiles with terminal ECM patterning (Figure 1C, top right; STAR Methods). This dataset was processed by sequential feature selection (SFS) in MATLAB to prevent model overfitting,28 identifying a highly compact feature set of 9 parameters (5 cell interactions, 3 ultrastructural parameters, 1 clinical metadata feature; Figures S8A and S8B). These features were used to train the six ML models with automated Bayesian optimization of hyperparameters (10-fold cross-validation), producing training areas under the receiver operating characteristic curve (AUCs) of 0.884–0.953 (Figure 7A).
Figure 7.

Spatial signatures of PDAC patient prognosis using ML
(A) ML model training using cell interactions, matrix ultrastructure, and clinical metadata. Areas under the receiver operating characteristic curve (AUCs) ranged from 0.884–0.953 for six representative models: an artificial neural network (ANN), a generalized additive model (GAM), a k-nearest neighbors (KNN) model, a linear discriminant analysis (LDA), a random forest (RF), and a support vector machine (SVM).
(B) Blinded testing of prognostic ML models using an independent dataset of 40 patients with PDAC. AUCs ranged from 0.872–0.903, with the highest performance achieved by the ANN model.
(C and D) Kaplan-Meier analysis of ML spatial signature for blinded testing dataset (C) and entire set of patients (D). For the blinded testing dataset, the spatial signature successfully differentiated patient survival by a difference of 655 days (hazard ratio [HR] = 4.29; p = 0.00183). For the overall patient dataset, the survival curve for patients with spatial signature A did not cross 50%.
(E and F) Explanatory analysis of top-performing prognostic model using Shapley additive explanations (SHAP), including SHAP values (E) and average feature importance (F). Tumor grade and overall ECM architecture played the largest roles in explaining poor predicted survival, with additional support by individual ultrastructural parameters and cell interactions.
To validate prognostic efficacy, an independent, blinded cohort of 40 patients was processed by the established CODEX pipeline and classified by the previously trained ML models (Figure 7B). Briefly, new protein expression data were aligned by anchor-based transfer to the prior manifold (see Figure 2A, right-side manifold), generating predicted cell phenotypes. Cell interaction scores, ultrastructural parameters, and patient metadata were then generated for the new dataset of 1,714 tiles. Survival classification produced AUCs of 0.872–0.903, with the strongest performance (AUC = 0.903) generated by the ANN model with an associated sensitivity of 0.916, a specificity of 0.815, and a balanced accuracy of 0.866 (Table S3). To quantify discriminatory ability for OS duration, Kaplan-Meier survival analysis was performed on patient-averaged classes, termed spatial signatures A (survival positive) and B (survival negative; see STAR Methods for binary outcome classification scheme), for the blinded patient group and the overall patient dataset (Figures 7C and 7D). In the blinded testing group, spatial signature B predicted a worse median OS of 655 days (p = 0.00183), with an associated hazard ratio of 4.29 (Figure 7C). For the overall patient dataset, spatial signature B was associated with a hazard ratio of 3.39; the difference in median OS could not be predicted, as the Kaplan-Meier survival curve for patients with spatial signature A (survival positive) did not cross 50% (Figure 7D).
After establishing prognostic efficacy, we explained the contributions of individual parameters to the ML spatial signature using Shapley additive explanations (SHAP) analysis (Figures 7E and 7F).29 Tumor grade exhibited the highest feature importance (Figure 7F), with high feature values consistently contributing to worse predicted survival, as represented by the right side of the SHAP value distribution (Figure 7E). Interestingly, discretized ECM architecture (for which underlying pseudotime did not significantly covary with grade; Figure S1B) produced the second highest feature importance, with terminal ECM patterning similarly contributing to predictions of worse survival. Individual ultrastructural parameters and cell interactions also contributed prognostic value, albeit at a lesser magnitude compared with global metrics such as tumor grade and overall ECM architecture (Figure 7F). For instance, high values of collagen I grayscale homogeneity, representing increasingly sheet-like fiber organization as observed in terminal ECM patterning, contributed to predictions of poor survival (Figure 7E). Interestingly, interactions by the mature/activated macrophage 2 subtype (HLA-DRhigh) appeared to contribute primarily to survival-negative predictions (e.g., macrophages 2-endothelial cells, macrophages 2-cytotoxic T lymphocytes), while interactions driven by cytotoxic T lymphocytes (e.g., cytotoxic T lymphocytes-B lymphocytes 2, cytotoxic T lymphocytes-fibroblasts 5) appeared to contribute predominantly to survival-positive predictions (Figure 7E). Overall, our explanatory analysis indicated that poorer survival was predicted by a multifactorial signature that included tumor grade, desmoplastic architecture, and differential stromal-immune cell organization.
Discussion
The desmoplastic milieu of PDAC contains complex and highly variable mixtures of fibrotic tissue, for which the exact functional role is not fully understood.4 Furthermore, PDAC is associated with a heterogeneous mixture of cell phenotypes, including cancer cells, endothelial cells, pericytes, lymphocytes, macrophages, and CAFs.4,12 This study applies an advanced fibrosis grading algorithm to prognosticate PDAC in the postoperative setting, a scenario with increasing clinical relevance as more cancer centers take on locally advanced disease.1 Early recurrence after pancreaticoduodenectomy continues to trouble patients and physicians alike, as surgery represents a major trauma unlikely to be of benefit to this group.3 Similarly, histologic response grading to neoadjuvant therapies remains crude, with relatively few tumors demonstrating sufficient necrosis for scores to be prognostic.3 This study provides an additional histologic tool that may help meaningfully characterize responses to therapies, particularly emerging agents targeting mesenchymal cells, and assist physicians in prognostic discussions postoperatively. Spatially defined cell interactions provide further context to this algorithm in differentiating both patient outcomes and desmoplastic patterning.
In our first analysis, we uncovered global desmoplastic matrix architectures that predicted differences in patient outcome. The prognostic relevance of desmoplastic patterning was further underscored by the high feature importance values assigned to ECM architecture in our SHAP analysis. Recent literature has identified high collagen content as a positive suppressor of tumor growth in mouse models,6,7,8,17 and varying studies have also implicated collagen fiber alignment as either a negative or positive prognostic indicator in clinical PDAC.9,30 A study of stromal composition in clinical specimens of PDAC has, in contrast, implicated discrete tumor glands with higher stromal content as being negatively prognostic.31 Many studies have also applied qualitative evaluations of tumor and gland morphology to stratify patient outcomes.4,5,32 Nevertheless, these prior analyses evaluate either gross, univariate properties of collagen or subjective evaluations of tumor morphology, and they do not encapsulate the full spatial complexity of desmoplasia, which includes an array of quantifiable fiber-level properties such as averaged fiber dimensions, as well as higher-order features such as porosity and branching. Our architectural analysis quantifies complex histological patterning in PDAC desmoplasia using a multivariate signature of >147 local and global matrix features, which can be summarized using dimensional reduction techniques and applied to complementary analyses of cell spatial organization and clinical correlates (e.g., grade, stage). Our ultrastructural algorithm, like other emerging methods of graph representation learning in histopathology, consists of feature extraction from sampled histological images, followed by unsupervised learning of a trajectory that connects images based on feature similarity.33,34,35 We envision that future studies can apply this algorithm to study graph representations of matrix architecture in other solid tumors and fibrotic diseases.
Interestingly, several clinical metrics of tumor progression such as tumor grade and measured tumor size did not appear to be strongly correlated with position along the pseudotime trajectory, suggesting that desmoplastic architecture may be mediated by biological processes orthogonal to tumor cell dedifferentiation and other established correlates of PDAC progression. Future mechanistic studies will help elucidate the specific causal factors that drive these variations in desmoplastic architecture. Additionally, prior studies have implicated stromal heterogeneity in worse clinical outcomes,26 and our measure of architectural variability trended toward statistical significance in correlating with differential outcomes, suggesting that this concept may be a promising route for continued investigation. It is worth noting that survival-negative desmoplastic patterning was strongly associated with complex, emergent properties of global fiber organization such as increasing topological porosity and texture heterogeneity, features that have not been investigated in prior clinical literature and may serve as effective histopathological predictors of patient outcomes. These features and their associated global architectures may be directly quantified using our ultrastructural algorithm, which is designed as an automated image analysis pipeline. While our original ultrastructural analysis was performed with Trichrome due to its more specific coloration of matrix staining, our exploration of mapping H&E to Trichrome and one-to-one integration with CODEX suggested that H&E may be used analogously for the quantification of matrix architecture and identification of co-occurrent cell spatial features. We believe that further exploration of H&E-based image features may therefore be a promising route for future investigation that may be more readily adapted to existing clinical workflows. Clinical testing can further utilize this ultrastructural algorithm to investigate how treatment protocols, such as stroma-targeted antifibrotic drugs,36 influence the architecture of desmoplasia in PDAC and other solid tumors.
Furthermore, we investigated the contributions of cell-cell spatial interactions to PDAC prognosis. We observed an axis of activated, pro-inflammatory spatial interactions that were associated with worse outcomes, with mature B lymphocytes 1 (CD20high/HLA-DRhigh/IL-6high/Vimentinhigh) and highly activated/immunomodulatory tumor cells 4 (E-cadherinhigh/PANCKmed/PDL1high/SDF1high) acting as primary survival-negative drivers. Interestingly, the elevated expression of IL-6 and Vimentin that we observed in B lymphocytes 1 has been implicated in pro-inflammatory signaling and suppression of antitumor response by B lymphocytes and other immune cells in contexts outside of PDAC.37,38 Our data suggest that these effects may be mediated through a myriad of interactions with stromal cells such as CAFs. B lymphocytes, like CAFs, have been implicated in paradoxical roles of both tumor suppression and promotion. For instance, while B lymphocyte infiltration in the context of tertiary lymphoid structures (TLSs) is considered to be a positive prognostic factor, accumulation of mature, transforming growth factor β (TGF-β) signaling B lymphocytes has also been identified as negatively prognostic. The survival-negative associations of our IL-6high B lymphocyte subpopulation, relative to other B lymphocytes, suggest a significant role of B cell dysfunction, particularly within a pro-inflammatory context, in influencing PDAC outcomes. In contrast, survival-positive spatial interactions appeared to be largely associated with cytotoxic T lymphocytes, helper T lymphocytes, and vasculature-adjacent interactions, highlighting the positive influence of T lymphocyte infiltration. Our findings build upon the earlier work of Schürch et al. in establishing the importance of spatial context in tumor biology, particularly for immune cells and immune-interactive cell phenotypes.18 Similar to their findings of T cell-associated cellular communities promoting survival in colorectal cancer,18 we observed that T cell-associated spatial interactions were predictive of improved outcomes in PDAC.
Critically, a highly pro-inflammatory fibroblast 5 subtype (CD26low/aSMAmed/IL-6high) appeared to drive spatial interactions associated with outcome-negative desmoplastic architecture. This finding builds upon prior studies in CAF heterogeneity6,16,17,39 by establishing that inflammatory CAFs, rather than mechanically activated or neutral CAF subtypes, may mediate outcome-negative desmoplastic architecture via elaboration of a pro-inflammatory spatial niche. A prior study by Grünwald et al. demonstrated that CAFs, when cultured in vitro from PDAC specimens, exhibit distinct gene expression signatures depending on the histology of the originating tumor.26 Our data build upon this prior research by evaluating in situ stromal and cellular organization in PDAC, revealing direct co-occurrence of outcome-negative architecture with an inflammatory fibroblast niche. This niche-associated matrix architecture was specifically defined by a combination of local (e.g., standard deviation of fiber length, diameter) and global (e.g., porosity, texture) fiber features rather than by the quantity of fibrosis. Furthermore, our single-cell interactome analysis underscores the differential spatial interaction signatures of inflammatory and mechanically activated CAFs, which are distinctly immune cell adjacent or tumor cell adjacent, respectively. A prior study by Moncada et al. demonstrated colocalization of inflammatory CAFs with activated/inflammatory cancer cells in PDAC but did not directly evaluate non-inflammatory fibroblast populations.40 Our study builds upon this prior work by examining a spectrum of mechanoresponsive to inflammatory fibroblasts present in PDAC, revealing that more inflammatory fibroblast subpopulations are both more immune adjacent and spatially associated with outcome-negative stromal matrix architecture. Furthermore, our analysis of cellular niches supports prior findings that mechanically activated CAFs are proximal to tumor foci, while inflammatory CAFs are more distant to tumor cells.13,41 Subtype-specific analysis provides further granularity to these findings by suggesting that mechanically activated CAFs are in fact directly adjacent to less activated tumor cells and also by specifically implicating B lymphocytes as an immune component of the inflammatory CAF niche. The inflammatory fibroblast 5 subtype specifically appears to be associated by interactome analysis with B lymphocytes 1, a primary survival-negative driver in our patient-level analysis. For the reasons outlined above, inflammatory CAFs may represent a cell phenotype of interest for desmoplasia-targeted therapeutic development. This population could potentially be modulated by the targeting of IL-6 receptors or upstream signaling pathways to inflammatory CAF activation such as Janus kinase-signal transducer and activator of transcription (JAK-STAT).42 Additionally, our analysis indicated that macrophage-driven interactions were associated with outcome-positive matrix architecture. Recent studies in other tumor contexts such as colorectal cancer have suggested that the macrophage phenotype and the M1:M2 ratio within the stroma may predict differential patient outcomes,43 and future research may also investigate the role of more granular macrophage subpopulations in driving PDAC progression and desmoplastic composition.
Interestingly, patients who received neoadjuvant chemotherapy exhibited differential cell spatial organization but not significantly different desmoplastic architecture. The spatial TME of neoadjuvant-chemotherapy-treated patients was particularly enriched with pro-inflammatory fibroblast-B lymphocyte interactions. Prior studies have indicated that aSMA+ CAFs may be depleted after neoadjuvant chemotherapy, and our data suggest that inflammatory CAFs may dominate the stromal microenvironment in patients who received neoadjuvant chemotherapy, supporting a previously posited “push-pull” relationship between inflammatory and mechanoresponsive CAF subtypes.11,15,44 This association with CAFs may also help explain the shifts in collagen composition and volume previously observed for patients who have received neoadjuvant chemotherapy.45 Furthermore, while prior literature has demonstrated an increased B lymphocyte presence in PDAC following neoadjuvant chemotherapy, our findings elucidate that B lymphocyte infiltration may be primarily mediated by or associated with enhanced inflammatory fibroblast-B lymphocyte interactions. Future experiments may delineate whether these results are attributable to the presence of individual tissue-infiltrating B cells vs. the development of more organized TLSs. Other correlations of CODEX with clinical characteristics such as tumor size and AJCC stage confirmed previously established associations between aSMA+ CAFs and more progressed tumors, though knockout of these populations has paradoxically resulted in accelerated disease progression and reduced survival in the literature.13,17 Interestingly, higher tumor grade was associated with enriched interactions between CD4+ T lymphocytes and pericytes, which may relate to the elevation of CD4+ T lymphocyte infiltrate that has been associated with PDAC progression.46 Overall, our data established an association between neoadjuvant chemotherapy and an increasingly immunomodulated TME centered around inflammatory fibroblasts and B lymphocytes.
Lastly, we developed a holistic spatial signature of cell interactions, ultrastructural features, and clinical metadata to predict patient outcomes using ML. Our unified models produced AUCs of 0.872–0.903 on an independent, blinded testing cohort, representing superior performance compared with existing magnetic resonance imaging (MRI)- and lab-value-based ML models of PDAC classification (AUCs: ∼0.801–0.900).47,48 Furthermore, the ML spatial signature distinguished OS by a median of nearly 2 years—a substantial finding for a disease with a 5-year survival rate of 20%–25% in the postoperative setting.3 This approach could be used to stratify patients who may require more intensive therapy. For instance, patients classified at a higher risk may be recommended for more aggressive treatment regiments such as folinic acid-fluorouracil-irinotecan (FOLFIRI) chemotherapy ± radiation therapy.49 On the other hand, patients with lower risk may be recommended for less aggressive treatments such as gemcitabine + Abraxane without radiation.49 Importantly, our explanatory SHAP analysis revealed that ECM architecture played an outsize role in predicting differential patient outcomes. Furthermore, a subset of immune-stromal interactions—particularly with cytotoxic T lymphocytes—contributed prognostic value to the ML spatial signature, underscoring the relevance of cell spatial organization in predicting PDAC outcomes.
Ultimately, our findings establish the prognostic value of desmoplastic architecture and stromal-immune cellular organization. In addition to our prognostic findings, our data establish spatial biology with regards to CAFs and other desmoplasia-associated cells in the PDAC TME. These associations include inflammatory CAFs and outcome-negative desmoplastic architecture, links between neoadjuvant chemotherapy and enriched CAF-B lymphocyte interactions, and identification of highly activated B lymphocyte and tumor cell subtypes that drive outcome-negative spatial interactions. Overall, a more disordered and sheet-like desmoplastic architecture appears to coincide with a more activated and pro-inflammatory spatial TME, predicting worse patient outcomes in PDAC.
Limitations of the study
While the present study utilizes a pseudotime-based trajectory to model changes in high-dimensional matrix architecture, other graph learning and dimensional reduction techniques may be equally appropriate for the clustering and analysis of ultrastructural features. Furthermore, while this analysis was performed largely unsupervised, various input parameters such as minimum branch length, root point selection, etc., may be freely selected based on alternative biological criteria of interest to the field (e.g., proximity to PDAC or other carcinomas rather than healthy pancreas). Thus, we encourage readers to apply their own trajectories and analytic parameters to the raw histological image dataset (see STAR Methods). Additionally, while the present study defines cell phenotypes by protein expression, single-cell RNA sequencing (scRNA-seq)-based and/or chromatin-accessibility-based analyses may provide additional utility by assaying the underlying biological mechanisms involved. An inherent limitation of CODEX is its reliance on quantification of fluorescence from antibody-based staining, which may introduce artifacts related to non-specific antibody binding, sample-to-sample variations in imaging, etc. While these irregularities can be corrected to some degree through postprocessing methods such as stain normalization and batch correction, CODEX-defined cell populations may include non-specific markers or admixtures of multiple cell phenotypes. A notable limitation of this study is that sampling for each modality involved the capture of an isolated region of each tumor, which may not comprehensively represent the holistic biological features and/or intra-tumoral heterogeneity observed in PDAC and other cancers.12 This phenomenon may likely account for the relatively lower magnitude of Pearson correlation coefficients identified by the matrix architecture analysis, and future studies may therefore build upon this methodology by more comprehensively analyzing multiple regions within each tumor and directly exploring the clinical impact of intra-tumoral heterogeneity. In addition to its spatial complexity, the PDAC microenvironment evolves with time, and various transcriptionally defined stromal subtypes likely emerge during distinct stages of tumor growth, metastasis, etc., to drive disease progression.13 Thus, future studies should utilize mechanistic mouse models to evaluate the biological impact and kinetics of enriching or knocking out specific cell phenotypes or cell-cell interactions identified in the study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| aSMA, polyclonal | Abcam | Cat# ab5694; RRID: AB_2223021 |
| Vimentin, clone RV202 | BD Biosciences | Cat# 550513; RRID: AB_393716 |
| S100A4 (FSP1), clone S100A4 | Biolegend | Cat# 810101; RRID: AB_2564748 |
| PDGFRa, clone 16A1 | Abcam | Cat# ab96569; RRID: AB_10687154 |
| FAP, polyclonal | Millipore Sigma | Cat# SAB2900181; RRID: AB_10600445 |
| IL-6, clone 1.2-2B11-2G10 | Abcam | Cat# ab9324; RRID: AB_307175 |
| IL-1, polyclonal | Abcam | Cat# ab9722; RRID: AB_308765 |
| MGP, clone OTI8D6 | Abcam | Cat# ab273657; RRID: N/A |
| YAP, clone EPR19812 | Abcam | Cat# ab223126; RRID: N/A |
| CXCL-12 (SDF-1), polyclonal | Abcam | Cat# ab9797; RRID: AB_296627 |
| CD26, polyclonal | R&D AF1180 | Cat# AF1180; RRID: AB_354651 |
| CD56, clone MRQ-42 | Cell Marque | Custom; RRID: N/A |
| HLA Class I, clone EP1395Y | Abcam | Cat# ab216653; RRID: N/A |
| Collagen I, polyclonal | Abcam | Cat# ab34710; RRID: AB_731684 |
| Collagen IV, polyclonal | Abcam | Cat# ab6586; RRID: AB_305584 |
| Fibronectin, polyclonal | Abcam | Cat# ab23751; RRID: AB_447656 |
| PD-1, clone D4W2J | Cell Signaling Technologies | Custom; RRID: N/A |
| PD-L1, clone E1L3N | Cell Signaling Technologies | Custom; RRID: N/A |
| CTLA-4, clone CAL49 | Abcam | Cat# ab237712; RRID: AB_2905652 |
| Ki67, clone B56 | BD Biosciences | Cat# 556003; RRID: AB_396287 |
| CD4, clone EPR6855 | Abcam | Cat# ab181724; RRID: AB_2864377 |
| CD8, clone C8/144B | Biolegend | Cat# sc-53212; RRID: AB_1120718 |
| CD11c, clone EP1347Y | Abcam | Cat# ab216655; RRID: AB_2864379 |
| CD20, clone rIGEL/773 | Novus Biologicals | Cat# NBP2-54591; RRID: AB_2864380 |
| CD31, clone C31.3 + 31.7+31.10 | Novus Biologicals | Cat# NBP2-47785; RRID: AB_2864381 |
| CD68, clone KP-1 | Biolegend | Cat# 916104; RRID: AB_2616797 |
| HLA DR, clone EPR3692 | Abcam | Cat# ab215985; RRID: AB_2864390 |
| Pan Cytokeratin, clone AE-1+AE-3 | Biolegend | Cat# 914204; RRID: AB_2616960 |
| E-cadherin, clone 4A2C7 | Thermo Fisher | Cat# 33–4000; RRID: AB_2533118 |
| Biological samples | ||
| Archival tumor specimens | Stanford University, University of Virginia pathology repositories | Deidentified |
| Tissue microarrays | US Biomax, Inc. | Cat# PA2082a, PA806, PA1921a, PA501, PA803, PA961f |
| Critical commercial assays | ||
| CODEX Staining Kit | Akoya Biosciences | Cat# 7000008 |
| CODEX Antibody Conjugation Kit | Akoya Biosciences | Cat# 7000009 |
| Chemicals, peptides, and recombinant proteins | ||
| Assay Reagent for CODEX | Akoya Biosciences | Cat# 7000002 |
| Nuclear Stain for CODEX | Akoya Biosciences | Cat# 7000003 |
| CODEX Barcode/Reporter RX046 | Akoya Biosciences | Cat# 6150016 |
| CODEX Barcode/Reporter RX049 | Akoya Biosciences | Cat# 6150017 |
| CODEX Barcode/Reporter RX035 | Akoya Biosciences | Cat# 6250010 |
| CODEX Barcode/Reporter RX042 | Akoya Biosciences | Cat# 6550023 |
| CODEX Barcode/Reporter RX043 | Akoya Biosciences | Cat# 6150015 |
| CODEX Barcode/Reporter RX016 | Akoya Biosciences | Cat# 6150006 |
| CODEX Barcode/Reporter RX027 | Akoya Biosciences | Cat# 6550019 |
| CODEX Barcode/Reporter RX020 | Akoya Biosciences | Cat# 6250005 |
| CODEX Barcode/Reporter RX010 | Akoya Biosciences | Cat# 6150004 |
| CODEX Barcode/Reporter RX005 | Akoya Biosciences | Cat# 6250002 |
| CODEX Barcode/Reporter RX040 | Akoya Biosciences | Cat# 6150014 |
| CODEX Barcode/Reporter RX034 | Akoya Biosciences | Cat# 6150012 |
| CODEX Barcode/Reporter RX002 | Akoya Biosciences | Cat# 6250001 |
| CODEX Barcode/Reporter RX030 | Akoya Biosciences | Cat# 6550020 |
| CODEX Barcode/Reporter RX045 | Akoya Biosciences | Cat# 6550024 |
| CODEX Barcode/Reporter RX037 | Akoya Biosciences | Cat# 6150013 |
| CODEX Barcode/Reporter RX036 | Akoya Biosciences | Cat# 6550022 |
| CODEX Barcode/Reporter RX041 | Akoya Biosciences | Cat# 6250011 |
| CODEX Barcode/Reporter RX004 | Akoya Biosciences | Cat# 6150002 |
| CODEX Barcode/Reporter RX047 | Akoya Biosciences | Cat# 6250012 |
| CODEX Barcode/Reporter RX003 | Akoya Biosciences | Cat# 6550025 |
| CODEX Barcode/Reporter RX026 | Akoya Biosciences | Cat# 6250007 |
| CODEX Barcode/Reporter RX024 | Akoya Biosciences | Cat# 6550018 |
| CODEX Barcode/Reporter RX007 | Akoya Biosciences | Cat# 6150003 |
| CODEX Barcode/Reporter RX001 | Akoya Biosciences | Cat# 6150001 |
| CODEX Barcode/Reporter RX021 | Akoya Biosciences | Cat# 6550028 |
| CODEX Barcode/Reporter RX015 | Akoya Biosciences | Cat# 6550027 |
| CODEX Barcode/Reporter RX033 | Akoya Biosciences | Cat# 6550021 |
| CODEX Barcode/Reporter RX019 | Akoya Biosciences | Cat# 6150007 |
| CODEX Barcode/Reporter RX014 | Akoya Biosciences | Cat# 6250003 |
| Deposited data | ||
| Trichrome Histological Images | Github | Github: HageyLab/PDAC2023 |
| CODEX Phenotyping Data | Github | Github: HageyLab/PDAC2023 |
| Software and algorithms | ||
| R | R Foundation for Statistical Computing | 4.1 |
| RStudio | Posit PBC | 1.4.1106.0 |
| AdjacencyScore | Govek et al.50 | 0.1.0 |
| Batchelor | Haghverdi et al.51 | 1.6.3 |
| DDRTree | Mao et al.23 | 0.1.5 |
| Monocle3 | Trapnell et al.24 | 1.2.9 |
| RANN | Github | jefferislab/RANN (2.6.1) |
| Seurat | Hao et al.52 | 4.1.1 |
| STvEA | Govek et al.50 | 1.0 |
| MATLAB (Image Processing Toolbox, Statistics and Machine Learning Toolbox) | The MathWorks, Inc. | R2021a |
| Akoya Multiplex Analysis Viewer | Akoya Biosciences | 1.5 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Michael Longaker (longaker@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patients and samples
Whipple procedure paraffinized specimens were obtained from pathology archives at Stanford University Hospital and UVA Hospital under the respective institutions’ IRB approval. For the TMA cohort, de-identified core biopsy specimens were obtained through a commercial vendor (US Biomax, Inc.). Informed consent was exempted, given the retrospective coded nature of data collection. The study included only patients with pathology-confirmed pancreatic ductal adenocarcinoma without metastatic disease. Specimens containing normal pancreas or other pancreatic neoplasms (e.g., islet cell tumors, acinic cell carcinoma, duodenal adenocarcinoma, neuroendocrine carcinoma, solid pseudopapillary neoplasm) were excluded from downstream clinical analysis. Clinical metadata (age, gender, AJCC stage, grade, neoadjuvant chemotherapy, etc.) were either gathered from patient charts (Stanford, UVA cohorts) or provided in a de-identified fashion from the commercial third-party vendor (TMA cohort). For patient characteristics within each cohort, see Table S1.
Method details
Histological image acquisition
A total of 578 histologic specimens were gathered, sectioned, and stained with Masson’s Trichrome at the institution or vendor of origin. Slides were assigned random identifier numbers and imaged at 20× magnification on multiple microscopes (Leica DMI4000 B, DMI5000 B) in a blinded fashion by multiple participants not involved in data analysis. For Stanford and UVA specimens, slides were imaged in tiles, generating 20–100 images per specimen. For TMA specimens, 4 images were acquired per core biopsy. Images remained blinded throughout ultrastructural analysis and were subsequently categorized by cohort and patient of origin after integration with clinical metadata.
Matrix ultrastructural algorithm
A total of 13,048 trichrome histology image tiles were processed using a matrix ultrastructural algorithm previously utilized for fibrotic tissue analysis.21 Briefly, trichrome images were normalized by the red/green/blue (RGB) histogram method and color deconvoluted using the algorithm previously described by Ruifrok et al. wherein each pure stain is characterized by absorbances within the three RGB channels.53,54 Ortho-normal transformation of the histology images produced individual images corresponding to each color’s individual contribution to the image. This methodology produces deconvoluted blue images corresponding to ECM fibers alone,53,54 which were used for downstream analysis. Noise reduction of deconvoluted fibers was achieved using an adaptive Wiener filter, which tailors itself to local image variance within a pre-specified neighborhood (3-by-3 pixels). The filter preferentially smooths regions with low variance, thereby preserving sharp fiber edges. Smooth images were binarized using the im2bw command and processed through erosion filters with diamond-shaped structuring elements to select fiber-shaped objects. Finally, the fiber network was “skeletonized” using the bwmorph command and various parameters of the digitized map (fiber length, width, persistence, alignment, etc.) were measured.
Quantified matrix values were imported into RStudio and reduced to a two-dimensional UMAP manifold using uwot. The DDRTree algorithm, which has been utilized for graph representation learning in biological data, was used to fit all ultrastructural datapoints to a minimum spanning tree with a minimum branch length of 30, maximizing similarity in ultrastructural features between successive points along the trajectory and assigning pseudotime values based on geodesic distance to the root point.23 The trajectory’s root point, representing baseline histological architecture, was selected by identification of a branch terminus closely situated to normal pancreatic tissue (Figure 1D). Patient-level centroids along the pseudotime trajectory, representing average matrix architecture, were then calculated as the median value for all patient-specific images. Architectural heterogeneity was also quantified for each patient by taking the sum of variance in all 147 ultrastructural parameters for all patient-specific images.
CODEX spatial phenotyping
To spatially profile PDAC specimens, we used CODEX, an assay in which 30 or more protein markers are labeled with oligonucleotide-conjugated antibodies and iteratively imaged between cyclic additions and washouts of complementary dye-labeled oligonucleotides.18 A total of 118 unique patient specimens were assayed, of which 40 patients were held out for blinded testing. A custom 30-plex CODEX panel was designed for PDAC and validated on lymphoid tissues per manufacturer protocols for stromal cell populations, tumor cells, immune cells, and markers of fibroblast heterogeneity (PANCK, CD4, CD8, αSMA, YAP, etc.; see key resources table). Antibodies were individually barcoded and validated using the commercial supplier’s protocols (Akoya Biosciences). Custom microarrays containing circular tissue sections of 4 mm diameter and 8 μm thickness were deparaffinized, and antigens were retrieved by standard citrate-EDTA processing. Multiplexed CODEX images were acquired using the PhenoCycler platform and Keyence BZ-X800 epifluorescence microscope per standard protocols.55 The built-in CODEX processor was used for image processing, stitching, and cell segmentation using standard manufacturer settings and methodology previously established by Goltsev et al.56
To visualize and analyze CODEX data, we used the Multiplex Analysis Viewer (MAV) in ImageJ. Segmented cells were gated for marker expression and exported as.fcs files for concatenation in flowJo and further analysis in RStudio.50 Single-cell protein expression profiles were pre-processed in STvEA by filtering cell size to the 2.5–99% percentile range and normalizing individual protein channels to a Gaussian distribution.50 Following this, protein expression was batch corrected between specimens by imputing mutual nearest neighbors at k = 25 in Batchelor, then imported to Seurat for Louvain clustering and dimensional reduction of the entire protein marker set by principal component analysis (PCA) and UMAP, with a post-manifold threshold of >10,000 cells per cluster.51 Macrophages and T lymphocytes were additionally split into subtypes based on HLA-DR and CD8 expression, respectively (see Figures S3B and S3F).
Cell interaction analysis
For all possible cell type pairings, cell-cell spatial interactions were quantified first by extracting cell type vectors fa and fb representing the presence (1) or absence (0) of cell type a and b at each point within a CODEX specimen.50 Following this, an adjacency matrix, j, was extracted to identify neighboring points within k = 20 nearest neighbors of each other using AdjacencyScore. Single-cell interaction scores were then calculated using the dot product of paired cell type vectors with the adjacency matrix (i.e., fa·j·fb). The single-cell dataset of 153 cell interaction scores was reduced by PCA using prcomp to analyze the interactome of individual cell phenotypes. Scree plots were generated to identify the appropriate number of principal components (PCs) based on the plot vertex, and the resulting PCs were plotted and analyzed for the loadings assigned to each cell interaction (i.e., coefficients representing the contribution of each cell interaction to the PC). To assess correlations between cell phenotype and spatial niche, the PCA centroid (i.e., median per PC) was also calculated for each cell phenotype to determine how each cell phenotype clustered within PCA space.
To calculate normalized patient-level interaction scores, the single-cell interaction scores (i.e., fa·j·fb) for the patient were summed and divided by the number of patient-specific cells. To analyze the effects of interaction distance on prognostic strength, patient-level cell interactions were also quantified within radii of 50-500μm, with no constraint on k, followed by linear regression modeling of correlation strength (Pearson coef.) vs. distance (interaction radius, μm) using the lm function. Furthermore, differential interaction maps were generated in ggraph to illustrate prognostic correlations (Pearson coefficients for continuous comparisons, Spearman coefficients for ordinal comparisons, magnitude of difference for paired categorical comparisons) between patient-level cell interactions and clinical metrics.
Ultrastructure-CODEX integration
To integrate ultrastructural analysis with CODEX and generate paired parameters for matrix-cell interaction correlation, CODEX samples were first processed into 10 × 10 subsampled tiles with a threshold of ≥20 cells per tile, yielding a total sample set of 3,247 tiles. The subsampled CODEX tiles were converted to spatial plots of COL I expression, which were analyzed using the above matrix algorithm and mapped to the original Trichrome manifold using the model transformation pipeline in Monocle3.22 Pseudotime values were then assigned by nearest neighbors using RANN. Cell interactions and individual ultrastructural parameters were correlated with lower and higher ECM pseudotime using Pearson correlations.
Machine learning analysis of patient survival
Unified prognostic signatures were developed using six representative ML models (ANN, GAM, KNN, LDA, RF, SVM) using the Machine Learning and Parallel Computing toolboxes in MATLAB. For classification, patients were initially assigned to binary survival classes for ML prediction based on two-year overall survival (OS < or ≥730 days). To increase dimensionality of matrix ultrastructure, CODEX tiles were additionally converted to spatial plots of COL I and COL IV and analyzed using the matrix algorithm for 294 parameters in total. COL I ultrastructural parameters were mapped to pseudotime values as described above, and an additional binary “ECM Architecture” feature was generated to identify tiles with predominantly terminal ECM patterning (Figure 1C). The centroid of the right half of the ultrastructure manifold (24), which appeared to demarcate terminal ECM patterning (Figure 1C, top right) and approximated the upper 20th percentile of pseudotime scores, was utilized for this feature discretization. The unified feature set was composed of cell interaction scores (153 features), ultrastructural features (294 features), matrix pseudotime and architecture (2 features), and patient-level metadata (e.g., age, gender, AJCC stage, grade; 14 features). The feature matrix for all 3,247 tiles was then reduced by sequential feature selection (SFS) using sequentialfs to prevent model overfitting.28 This compact feature set was used to train all models with imputation of missing data using knnimpute and automated Bayesian optimization of hyperparameters with 10-fold cross validation using bayesopt.57 Average performance of the trained models was assessed using receiver operating characteristic (ROC) curves.
Blinded testing of prognostic models
To evaluate prognostic efficacy, an independent, blinded cohort of 40 patients was spatially sequenced using CODEX. Raw CODEX data was processed using the standardized pipeline described above and batch corrected to the original dataset at k = 25 in Batchelor. In Seurat, the new protein expression matrix was projected onto the PCA space of the original CODEX manifold by anchor-based transfer, predicting cell phenotypes for the new cells.58 After anchor-based transfer, cell interaction scores, ultrastructural parameters, and clinical metadata were generated for the new 10 × 10 subsampled dataset (1,714 tiles). The previously trained ML models were used to predict binary survival classes without a priori knowledge. Model performance on the blinded testing dataset was then assessed using ROC curves. To quantify discriminatory ability for OS, Kaplan-Meier survival analysis was performed on the patient-averaged classifications assigned by the highest performing ML model, determined by greatest AUC. SHAP analysis was performed in MATLAB to quantitatively explain the impact of individual features on model predictions.29 SHAP values were assigned on a per-sample basis to each parameter based on quantitative contribution to the sample/prediction’s deviation from average. SHAP values were visualized as a collective distribution using ggbeeswarm, and feature importance values were calculated as the mean absolute SHAP value across the dataset.
Quantification and statistical analysis
For Kaplan-Meier survival analysis and associated hazard ratios, log rank tests were performed at α = 0.05. Individual cell interactions and ultrastructural parameters were correlated with OS or DFS using Pearson correlations to determine prognostic relevance (α = 0.05). OS was defined as the duration between pancreaticoduodenectomy and time of death or alternatively, database lock (if patient still alive). DFS was defined as the duration between pancreaticoduodenectomy and clinically confirmed disease relapse/progression or alternatively, death (patients without recurrence and still alive at time of database lock were therefore censored). Correlations with clinical metadata were calculated as Pearson correlations for continuous metadata (e.g., tumor size) and Spearman correlations for ordinal metadata (e.g., grade, AJCC stage) (α = 0.05). To identify prognostically relevant parameters between discrete patient groups, one-way analysis of variance (ANOVA) or a two-tailed, unpaired Student’s t test was performed. Relevant p values were controlled for multiple hypothesis testing using the Benjamini-Hochberg procedure.
Acknowledgments
This work was supported by the Hagey Laboratory for Pediatric Regenerative Medicine (to J.A.N. and M.T.L.); the Taylor Family Cancer Research Fund (to J.A.N.); the Rantz Family Research Fund (to J.A.N.); NIH grant R01-GM136659 (to M.T.L.); NIH fellowship F32-HL167318 (to J.L.G.); the Wu Tsai Human Performance Alliance (to M.T.L.); and financial support from Judy and Warren Kaplan (to J.A.N.) and the Gunn/Olivier Fund (to M.T.L.).
Author contributions
S.M. and J.L.G. contributed equally to this work. Conceptualization, S.M., J.L.G., D.S.F., D.J.D., J.A.N., and M.T.L.; data curation, S.M., J.L.G., and D.S.F.; formal analysis, S.M. and J.L.G.; supervision, J.A.N. and M.T.L.; investigation, S.M., J.L.G., D.S.F., A.K., M.F.D., A.T.N., A.R.B., M.S.C., N.J.G., M.G., E.M., M.J., S.S.R., T.A.L., and D.J.D.; visualization, S.M., J.L.G., and A.K.; writing, review, and editing, all authors; resources, S.S.R., T.A.L., D.J.D., J.A.N., and M.T.L.; funding acquisition, J.A.N. and M.T.L.
Declaration of interests
The authors declare no competing interests.
Published: October 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101248.
Contributor Information
Jeffrey A. Norton, Email: janorton@stanford.edu.
Michael T. Longaker, Email: longaker@stanford.edu.
Supplemental information
Data and code availability
Data processing code, raw images, spatial phenotyping data, and deidentified patient data are all publicly available on Github (https://github.com/HageyLab/PDAC2023). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.da Costa W.L., Jr., Oluyomi A.O., Thrift A.P. Trends in the incidence of pancreatic adenocarcinoma in all 50 United States examined through an age-period-cohort analysis. JNCI Cancer Spectr. 2020;4:pkaa033. doi: 10.1093/jncics/pkaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takikawa T., Kikuta K., Hamada S., Kume K., Miura S., Yoshida N., Tanaka Y., Matsumoto R., Ikeda M., Kataoka F., et al. Clinical features and prognostic impact of asymptomatic pancreatic cancer. Sci. Rep. 2022;12:4262–4311. doi: 10.1038/s41598-022-08083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannon A., Thompson C., Hall B.R., Jain M., Kumar S., Batra S.K. Desmoplasia in pancreatic ductal adenocarcinoma: insight into pathological function and therapeutic potential. Genes Cancer. 2018;9:78–86. doi: 10.18632/genesandcancer.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whatcott C.J., Diep C.H., Jiang P., Watanabe A., LoBello J., Sima C., Hostetter G., Shepard H.M., Von Hoff D.D., Han H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015;21:3561–3568. doi: 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y., Kim J., Yang S., Wang H., Wu C.-J., Sugimoto H., LeBleu V.S., Kalluri R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–565.e6. doi: 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian C., Huang Y., Clauser K.R., Rickelt S., Lau A.N., Carr S.A., Vander Heiden M.G., Hynes R.O. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nat. Commun. 2021;12:2328–2416. doi: 10.1038/s41467-021-22490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knudsen E.S., Vail P., Balaji U., Ngo H., Botros I.W., Makarov V., Riaz N., Balachandran V., Leach S., Thompson D.M., et al. Stratification of pancreatic ductal adenocarcinoma: combinatorial genetic, stromal, and immunologic markers. Clin. Cancer Res. 2017;23:4429–4440. doi: 10.1158/1078-0432.CCR-17-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolm L., Zghurskyi P., Lapshyn H., Petrova E., Zemskov S., Vashist Y.K., Deichmann S., Honselmann K.C., Bronsert P., Keck T., Wellner U.F. Alignment of stroma fibers, microvessel density and immune cell populations determine overall survival in pancreatic cancer—An analysis of stromal morphology. PLoS One. 2020;15 doi: 10.1371/journal.pone.0234568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster D.S., Jones R.E., Ransom R.C., Longaker M.T., Norton J.A. The evolving relationship of wound healing and tumor stroma. JCI insight. 2018;3 doi: 10.1172/jci.insight.99911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster D.S., Januszyk M., Delitto D., Yost K.E., Griffin M., Guo J., Guardino N., Delitto A.E., Chinta M., Burcham A.R., et al. Multiomic analysis reveals conservation of cancer associated fibroblast phenotypes across species and tissue of origin. Cancer Cell. 2022;40:1392–1406.e7. doi: 10.1016/j.ccell.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougan S.K. The pancreatic cancer microenvironment. Cancer J. 2017;23:321–325. doi: 10.1097/PPO.0000000000000288. [DOI] [PubMed] [Google Scholar]
- 13.Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsuta E., Qi Q., Peng X., Hochwald S.N., Yan L., Takabe K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci. Rep. 2019;9:1310–1311. doi: 10.1038/s41598-018-37909-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biffi G., Tuveson D.A. Deciphering cancer fibroblasts. J. Exp. Med. 2018;215:2967–2968. doi: 10.1084/jem.20182069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhim A.D., Oberstein P.E., Thomas D.H., Mirek E.T., Palermo C.F., Sastra S.A., Dekleva E.N., Saunders T., Becerra C.P., Tattersall I.W., et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.-C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schürch C.M., Bhate S.S., Barlow G.L., Phillips D.J., Noti L., Zlobec I., Chu P., Black S., Demeter J., McIlwain D.R., et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. 2020;182:1341–1359.e19. doi: 10.1016/j.cell.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Errarte P., Larrinaga G., López J.I. The role of cancer-associated fibroblasts in renal cell carcinoma. An example of tumor modulation through tumor/non-tumor cell interactions. J. Adv. Res. 2020;21:103–108. doi: 10.1016/j.jare.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W., Li I., Reticker-Flynn N.E., Good Z., Chang S., Samusik N., Saumyaa S., Li Y., Zhou X., Liang R., et al. Identification of cell types in multiplexed in situ images by combining protein expression and spatial information using CELESTA. Nat. Methods. 2022;19:759–769. doi: 10.1038/s41592-022-01498-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mascharak S., desJardins-Park H.E., Davitt M.F., Griffin M., Borrelli M.R., Moore A.L., Chen K., Duoto B., Chinta M., Foster D.S., et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science. 2021;372 doi: 10.1126/science.aba2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Q., Wang L., Goodison S., Sun Y. Proceedings of the 21th ACM SIGKDD international conference on knowledge discovery and data mining. 2015. Dimensionality reduction via graph structure learning; pp. 765–774. [Google Scholar]
- 24.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. Pseudo-temporal ordering of individual cells reveals dynamics and regulators of cell fate decisions. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas S.K., Lee J., Beatty G.L. Paracrine and cell autonomous signalling in pancreatic cancer progression and metastasis. EBioMedicine. 2020;53 doi: 10.1016/j.ebiom.2020.102662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grünwald B.T., Devisme A., Andrieux G., Vyas F., Aliar K., McCloskey C.W., Macklin A., Jang G.H., Denroche R., Romero J.M., et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell. 2021;184:5577–5592.e18. doi: 10.1016/j.cell.2021.09.022. [DOI] [PubMed] [Google Scholar]
- 27.Potdar K., Pardawala T.S., Pai C.D. A comparative study of categorical variable encoding techniques for neural network classifiers. Int. J. Comput. Appl. 2017;175:7–9. [Google Scholar]
- 28.Aha D.W., Bankert R.L. Pre-proceedings of the Fifth International Workshop on Artificial Intelligence and Statistics. PMLR; 1995. A comparative evaluation of sequential feature selection algorithms; pp. 1–7. [Google Scholar]
- 29.Lundberg S.M., Lee S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017;30 [Google Scholar]
- 30.Drifka C.R., Loeffler A.G., Mathewson K., Keikhosravi A., Eickhoff J.C., Liu Y., Weber S.M., Kao W.J., Eliceiri K.W. Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget. 2016;7:76197–76213. doi: 10.18632/oncotarget.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ligorio M., Sil S., Malagon-Lopez J., Nieman L.T., Misale S., Di Pilato M., Ebright R.Y., Karabacak M.N., Kulkarni A.S., Liu A., et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell. 2019;178:160–175.e27. doi: 10.1016/j.cell.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.N Kalimuthu S., Wilson G.W., Grant R.C., Seto M., O’Kane G., Vajpeyi R., Notta F., Gallinger S., Chetty R. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut. 2020;69:317–328. doi: 10.1136/gutjnl-2019-318217. [DOI] [PubMed] [Google Scholar]
- 33.Pati P., Jaume G., Foncubierta-Rodríguez A., Feroce F., Anniciello A.M., Scognamiglio G., Brancati N., Fiche M., Dubruc E., Riccio D., et al. Hierarchical graph representations in digital pathology. Med. Image Anal. 2022;75 doi: 10.1016/j.media.2021.102264. [DOI] [PubMed] [Google Scholar]
- 34.Adnan M., Kalra S., Tizhoosh H.R. Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops. 2020. Representation learning of histopathology images using graph neural networks; pp. 988–989. [Google Scholar]
- 35.Choi H.-K., Bengtsson E., Jarkrans T., Vasko J., Wester K., Malmström P.-U., Busch C. Image Analysis and Processing: 8th International Conference, ICIAP’95 San Remo, Italy, September 13–15, 1995 Proceedings 8. Springer; 1995. Minimum spanning trees (MST) as a tool for describing tissue architecture when grading bladder carcinoma; pp. 615–620. [Google Scholar]
- 36.Hauge A., Rofstad E.K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 2020;18:207–211. doi: 10.1186/s12967-020-02376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bent E.H., Millán-Barea L.R., Zhuang I., Goulet D.R., Fröse J., Hemann M.T. Microenvironmental IL-6 inhibits anti-cancer immune responses generated by cytotoxic chemotherapy. Nat. Commun. 2021;12:1–13. doi: 10.1038/s41467-021-26407-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramos I., Stamatakis K., Oeste C.L., Pérez-Sala D. Vimentin as a multifaceted player and potential therapeutic target in viral infections. Int. J. Mol. Sci. 2020;21:4675. doi: 10.3390/ijms21134675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steele N.G., Biffi G., Kemp S.B., Zhang Y., Drouillard D., Syu L., Hao Y., Oni T.E., Brosnan E., Elyada E., et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021;27:2023–2037. doi: 10.1158/1078-0432.CCR-20-3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moncada R., Barkley D., Wagner F., Chiodin M., Devlin J.C., Baron M., Hajdu C.H., Simeone D.M., Yanai I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020;38:333–342. doi: 10.1038/s41587-019-0392-8. [DOI] [PubMed] [Google Scholar]
- 41.Yang D., Liu J., Qian H., Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 2023;55:1322–1332. doi: 10.1038/s12276-023-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H., Shi Y., Qian F. Opportunities and delusions regarding drug delivery targeting pancreatic cancer-associated fibroblasts. Adv. Drug Deliv. Rev. 2021;172:37–51. doi: 10.1016/j.addr.2021.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Väyrynen J.P., Haruki K., Lau M.C., Väyrynen S.A., Zhong R., Dias Costa A., Borowsky J., Zhao M., Fujiyoshi K., Arima K., et al. The Prognostic Role of Macrophage Polarization in the Colorectal Cancer Microenvironment. Cancer Immunol. Res. 2021;9:8–19. doi: 10.1158/2326-6066.CIR-20-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyashita T., Tajima H., Makino I., Okazaki M., Yamaguchi T., Ohbatake Y., Nakanuma S., Hayashi H., Takamura H., Ninomiya I., et al. Neoadjuvant chemotherapy with gemcitabine plus nab-paclitaxel reduces the number of cancer-associated fibroblasts through depletion of pancreatic stroma. Anticancer Res. 2018;38:337–343. doi: 10.21873/anticanres.12227. [DOI] [PubMed] [Google Scholar]
- 45.Nakajima K., Ino Y., Naito C., Nara S., Shimasaki M., Ishimoto U., Iwasaki T., Doi N., Esaki M., Kishi Y., et al. Neoadjuvant therapy alters the collagen architecture of pancreatic cancer tissue via Ephrin-A5. Br. J. Cancer. 2022;126:628–639. doi: 10.1038/s41416-021-01639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goulart M.R., Stasinos K., Fincham R.E.A., Delvecchio F.R., Kocher H.M. T cells in pancreatic cancer stroma. World J. Gastroenterol. 2021;27:7956–7968. doi: 10.3748/wjg.v27.i46.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaissis G., Ziegelmayer S., Lohöfer F., Algül H., Eiber M., Weichert W., Schmid R., Friess H., Rummeny E., Ankerst D. A machine learning model for the prediction of survival and tumor subtype in pancreatic ductal adenocarcinoma from preoperative diffusion-weighted imaging. Eur. Radiol. Exp. 2019;3:1–9. doi: 10.1186/s41747-019-0119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kapszewicz M., Małecka-Wojciesko E. Simple Serum Pancreatic Ductal Adenocarcinoma (PDAC) Protein Biomarkers—Is There Anything in Sight? J. Clin. Med. 2021;10:5463. doi: 10.3390/jcm10225463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.-L., Choné L., Francois E., Artru P., Biagi J.J., et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 50.Govek K.W., Troisi E.C., Miao Z., Aubin R.G., Woodhouse S., Camara P.G. Single-cell transcriptomic analysis of mIHC images via antigen mapping. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abc5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruifrok A.C., Johnston D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001;23:291–299. [PubMed] [Google Scholar]
- 54.Ruifrok A.C., Katz R.L., Johnston D.A. Comparison of quantification of histochemical staining by hue-saturation-intensity (HSI) transformation and color-deconvolution. Appl. Immunohistochem. Mol. Morphol. 2003;11:85–91. doi: 10.1097/00129039-200303000-00014. [DOI] [PubMed] [Google Scholar]
- 55.Black S., Phillips D., Hickey J.W., Kennedy-Darling J., Venkataraaman V.G., Samusik N., Goltsev Y., Schürch C.M., Nolan G.P. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 2021;16:3802–3835. doi: 10.1038/s41596-021-00556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goltsev Y., Samusik N., Kennedy-Darling J., Bhate S., Hale M., Vazquez G., Black S., Nolan G.P. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174:968–981.e15. doi: 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu D., Zhu H., Chu J., Wu J., Lei H., Deng S.-H. Hyperparameter optimization for machine learning models based on Bayesian optimization. J. Biotechnol. 2019;289:26–30. [Google Scholar]
- 58.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data processing code, raw images, spatial phenotyping data, and deidentified patient data are all publicly available on Github (https://github.com/HageyLab/PDAC2023). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.