

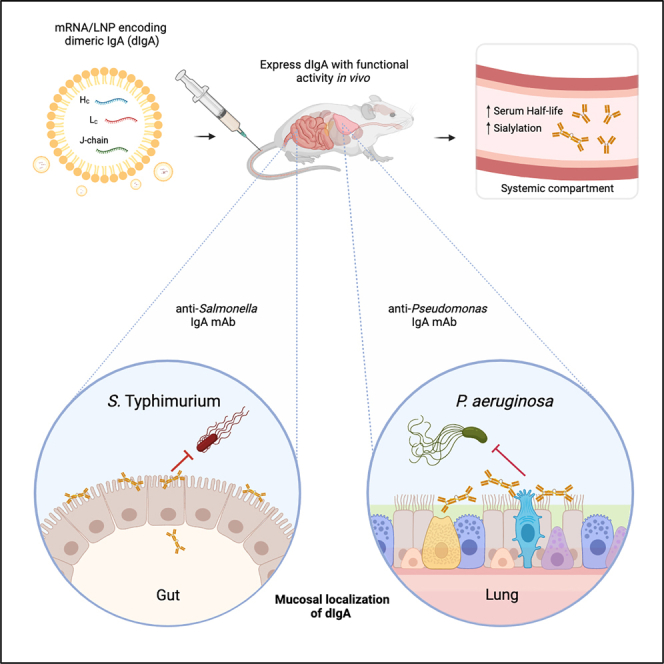
Summary
Colonization of the gut and airways by pathogenic bacteria can lead to local tissue destruction and life-threatening systemic infections, especially in immunologically compromised individuals. Here, we describe an mRNA-based platform enabling delivery of pathogen-specific immunoglobulin A (IgA) monoclonal antibodies into mucosal secretions. The platform consists of synthetic mRNA encoding IgA heavy, light, and joining (J) chains, packaged in lipid nanoparticles (LNPs) that express glycosylated, dimeric IgA with functional activity in vitro and in vivo. Importantly, mRNA-derived IgA had a significantly greater serum half-life and a more native glycosylation profile in mice than did a recombinantly produced IgA. Expression of an mRNA encoded Salmonella-specific IgA in mice resulted in intestinal localization and limited Peyer’s patch invasion. The same mRNA-LNP technology was used to express a Pseudomonas-specific IgA that protected from a lung challenge. Leveraging the mRNA antibody technology as a means to intercept bacterial pathogens at mucosal surfaces opens up avenues for prophylactic and therapeutic interventions.
Keywords: mRNA, mucosal, IgA, antibody, infection, therapy, dimeric, Salmonella, Pseudomonas
Graphical abstract
Highlights
-
•
Synthetic mRNAs encoding IgA heavy (HC), light (LC), and J chains express dimeric IgA
-
•
In vivo mucosal localization of IgA delivered by synthetic mRNA/LNP
-
•
Higher sialylation and serum half-life with IgA from mRNA/LNP than recombinant protein
-
•
Anti-Salmonella IgA mAb from mRNA/LNPs reduced invasion during in vivo challenge
Deal et al. report that mRNA/LNPs encoding IgA heavy, light, and J chains express glycosylated dimeric IgA that localizes to mucosal secretions and has a greater serum half-life over recombinant IgA. mRNA-expressed pathogen-specific IgA mAbs limited Salmonella Peyer’s patch invasion and reduced mortality from Pseudomonas acute lung pneumonia in mouse models.
Introduction
The COVID-19 pandemic revealed the potential of synthetic mRNA-based vaccine technology to combat infectious diseases.1 An inherent advantage of mRNA as a platform technology over more conventional vaccines is the ability to bypass the need for large-scale protein manufacturing and purification.2 Rather, difficult-to-manufacture proteins and protein complexes, like the trimeric spike proteins of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), are expressed in situ. While most attention to date on mRNA technology has focused on vaccines, mRNA-based platforms are amenable to the expression of a wide variety of human immune-associated macromolecules. For example, August and colleagues recently completed a phase I clinical trial to evaluate the safety and pharmacology of a lipid nanoparticle (LNP)-encapsulated mRNA encoding the heavy and light chains of a Chikungunya virus (ChikV)-specific neutralizing monoclonal antibody (mAb).3 The potential of the mRNA/LNP platform for in vivo co-expression of a tripartite cocktail of broadly neutralizing antibodies directed against the HIV-1 envelope protein has also been demonstrated.4
Immunoglobulin A (IgA) is the predominant antibody isotype in mucosal secretions of the human gastrointestinal (GI) tract and upper airways, where it functions as a first line of defense against pathogenic and opportunistic bacterial pathogens.5,6 Humans have two IgA subclasses, IgA1 and IgA2, that differ in hinge length, degrees of O-linked glycosylation,5 and mucosal localization. IgA-secreting B cells are generated in mucosa-associated lymphoid tissues (MALTs) such as Peyer’s patches in the small intestine and adenoids in the upper airways in response to environmental antigens and mucosal pathogens.7 B cells derived from the MALTs take up residence in mucosal tissues as resident plasma cells that secrete IgA as a dimer (dIgA) due to co-expression of joining (J) chains.8 dIgA is actively transported across certain epithelial cell barriers by the polymeric Ig receptor (pIgR) and released into mucosal secretions as secretory IgA (SIgA).9 Neither IgG nor monomeric IgA (mIgA), which is the predominant form of IgA in circulation, are actively transported into mucosal secretions by the pIgR.10 As such, there exists a distinct compartmentalization between systemic and mucosal antibody pools.
The mucosal surfaces that line the GI tract and the upper airways are constantly exposed to a myriad of resident microbiota, as well as opportunistic and pathogenic bacteria. Foodborne pathogens like Salmonella enterica serovar Typhimurium (STm) readily invade intestinal tissues, and while antibiotic treatment can clear STm infections, subpopulations of antibiotic-resistant bacteria may persist.11,12 Similarly, nosocomial infections like Pseudomonas aeruginosa (PA) are notoriously difficult to eradicate once entrenched in the lung, especially considering the degree of antibiotic resistance that exists in clinical isolates.13 With that in mind, there is a pressing need for both prophylactic and therapeutic interventions aimed at preventing bacterial pathogens from colonizing vulnerable tissues in the first place, as well as clearing infections once established. IgA is ideally suited to accomplish these tasks, especially if pathogen-specific antibodies can be delivered into mucosal secretions at sufficient concentrations to interfere with the earliest steps in the infection process.
Passive immunization with IgA is not a new concept but one that has proven challenging to implement because of the technical obstacles in producing recombinant IgA (IgAR) and delivering sufficient quantities to be effective for sustained periods.14 Moreover, human IgA is heavily glycosylated, which poses challenges from a biopharmaceutical standpoint, as N-glycosylation can influence conformation, thermal stability, folding efficiency, solubility, and susceptibility to proteolytic degradation.15,16,17,18 In addition, IgAR is intractable for most clinical purposes, as it exhibits a shorter serum half-life than IgG due to its inability to recycle through the neonatal Fc receptor,19 and displays faster clearance from circulation as compared to endogenous human IgA as a result of incomplete sialylation.20 Efforts to generate IgG/IgA chimeras with desired Fc receptor interactions, serum half-life, and mucosal delivery are ongoing21,22,23 but have not reached clinical-stage readiness.
In this report, we investigated the use of synthetic mRNA for in vivo production of structurally and functionally intact human IgA. We employed two monoclonal antibodies (mAbs) that have been extensively characterized as recombinant proteins in mouse models of mucosal challenge. Sal4 is an IgA mAb that recognizes the O5− antigen of lipopolysaccharide from STm24 and has been shown to reduce invasion of STm into intestinal Peyer’s patches when administered passively.24,25 CAM003 is an IgG1 mAb that binds to PA biofilm component Psl and has demonstrated protection in multiple different PA animal models, including acute lung pneumonia.26,27 Here, Sal4 and CAM003 heavy chains (HCs), light chains (LCs), and J chains (JCs) were encoded as mRNA as human IgA2 (IgA2mRNA) or IgA1 (IgA1mRNA), respectively, and were characterized in vitro and in vivo as compared to analogous IgAR to assess the potential of the mRNA platform to generate potent and protective mucosal mAbs.
Results
Biophysical, functional, and pharmacokinetic properties of mRNA encoded IgA2
To explore the potential of synthetic mRNA to encode functional monoclonal IgA, mRNA encoding the STm-specific Sal4 HCs and LCs were packaged in LNPs, with or without mRNA encoding the JC.24,25 Transfection of EXPI293 cells resulted in IgA2mRNA antibody that recognized STm by enzyme-linked immunosorbent assay (ELISA) to a similar level as IgA2R (Figure 1A). Sal4 IgA2mRNA transfected with the JC was bound by immobilized pIgR similar to dIgA2R, confirming the formation of dIgA from mRNA (Figure 1B). The mIgA2R product bound pIgR 10-fold less than dIgA2R, whereas mIgA2mRNA had no demonstrable interaction with pIgR. Using size-exclusion chromatography (SEC), we observed that dIgA2 predominated over mIgA2 upon JC co-expression in vitro and was distinct from mIgA2 and IgA2 aggregates assembled in the absence of the JC (Figure 1C).28 Differences in aggregate assembly were especially evident following gel electrophoresis of SEC-separated peaks; whereas dIgA2 exhibited bands at the size of JCs (10–15 kDa), these bands were largely absent from the aggregate peak (Figure 1D). Notably, SEC retention times of the dIgA2 and mIgA2 peaks were nearly identical between IgA2mRNA and IgA2R, further demonstrating the expression of similar products (Figure S1). Negative-stain electron microscopy (EM) images of the dIgA2mRNA preparations revealed Igs in a tail-to-tail oligomerization configuration identical to that of dIgA2R, demonstrating the ability of mRNA to encode for oligomeric mAbs (Figures 1E and S1). Finally, the functionality of IgA2mRNA was assessed in an in vitro assay of STm HeLa cell invasion. mRNA transfections (with JCs) of EXPI293 resulted in functional IgA2mRNA that reduced STm invasion of HeLa cells to a level similar to IgA2R (Figure 1F),25 confirming that mRNA can encode for functional oligomeric IgA proteins.
Figure 1.

Characterization of Sal4 IgA2mRNA from in vitro transfection
(A) Binding of mRNA transfection supernatant and recombinant protein of Sal4 IgA2 to STm.
(B) Binding of SEC-separated Sal4 IgA2R and supernatant from mRNA-transfected Sal4 IgA2 to immobilized human pIgR.
(C) Analytical SEC of affinity-purified IgA2 from mRNA transient transfection. Dimer (d) and monomer (mon) peaks are denoted. Transfections with JC are in blue and without JC are in orange, with aggregates (ag) labeled.
(D) SDS-PAGE of designated SEC peaks from mRNA transient transfection under reducing (R) and non-reducing (NR) conditions. dimer and monomer for NR are denoted along with HCs, LCs, and JCs in R conditions.
(E) Representative image of negative-stain EM 2D class averages of the dimeric peak isolated from mRNA transfection with J chain.
(F) Effect of IgA2R or IgA2mRNA on STm invasion of HeLa cells. Each symbol represents a replicate, and data is graphed mean ± SD of 3 replicates, ∗∗p < 0.05.
IgA2mRNA exhibits an increased serum half-life and N-linked sialylation of all asparagine sites as compared to IgA2R
While the mucosal targeting and oligomeric properties of IgA are clinically appealing, these proteins have traditionally suffered from poor pharmacodynamic properties and, in particular, a short serum half-life.20,29 To assess how in vivo production of IgA from formulated mRNA (containing HCs, LCs, and JCs) impacts pharmacodynamics, BALB/c mice were injected with 5 mg/kg IgA2R (containing a mixture of monomeric and dimeric IgA2), 4.5 mg/kg polyclonal human IgA isolated from serum (mostly monomeric), and mRNA/LNPs encoding IgA2mRNA. Analogous to previous reports,29 IgA2R exhibited a terminal half-life of 0.64 days and was detected for 2 days, while polyclonal human serum IgA was detected for 12 days with a terminal half-life of 0.93 days (Figure 2A). In comparison, IgA2mRNA exhibited a terminal half-life of 1.63 days, demonstrating a fundamental divergence from that of IgA2R (Figures 2A and 2B).
Figure 2.

Characterization of IgA2mRNA serum half-life and site-specific glycosylation patterns in vivo as compared to IgA2R
BALB/c mice were injected intravenously with 1 mg/kg formulated IgA2mRNA, 5 mg/kg Sal4 IgA2R, or 4.5 mg/kg IgA isolated from human serum. Concentrations of antibody were measured in serum over time by isotype-specific ELISA and modeled using a flexible linear mixed-effects model.
(A and B) Observed animal-level expression by ELISA and (A) model-based estimates of the mean and 95% credible interval and (B) model-based half-life estimates with 95% credible intervals are shown.
(C) Examples of three main types of N-glycan structure: complex, hybrid, and high mannose.
(D) IgA2 N-glycan compositions observed at 4 asparagine residues (N190, N274, N348, and N470).
(E) Sialylation of IgA2 asparagine residues, expressed as a percentage of complex and hybrid glycans that are sialylated.
(F) Fucosylation of IgA2 asparagine residues, expressed as a percentage of complex, hybrid, and high-mannose glycans that are fucosylated. Consistent detection of glycopeptides was observed across three technical executions in a serum pool from 50 animals.
IgA2R produced in HEK293 cells has been reported to be under-sialylated compared with IgA isolated from human serum,29 resulting in rapid clearance from circulation by the asialoglycoprotein receptor (ASGPR) and other mechanisms.20,30 To determine if we observe glycosylation differences, we quantified the levels of sialylation and overall glycosylation patterns of Sal4 IgA2mRNA purified from mouse serum (Figures 2C–2F and S2). As serum IgA is mostly monomeric, glycosylation patterns of IgA2mRNA were compared to isolated mIgA2R or IgA2R containing monomers and dimers. IgA2mRNA exhibited greater N-glycosylation of the complex type (90%) compared with IgA2R (30%) and mIgA2R (19%) (Figure S2). In contrast, high-mannose-type branched structures were predominant in IgA2R (66%) and mIgA2R (76%), as compared with the low levels (5%) found in IgA2mRNA. Levels of high-mannose structures correlated with a low level of sialylation (13%–14%) in mIgA2R and IgA2R, as compared with the high levels of sialylation (91% of glycan structures in all asparagine sites) of IgA2mRNA (Figures 2C–2F and S2). In addition, IgA2mRNA and mIgA2R have minimal fucosylation (13%–14% of glycan structures at all asparagine sites; Figure S2) compared with IgA2R (24%). High-mannose-type branched structures of JCs were found to predominate in IgA2R, while IgA2mRNA and mIgA2R were undetectable (Figure S2), further indicating the mostly monomeric nature of IgA2mRNA in circulation.
When N-glycosylation was examined at the individual residue level, similar trends in glycosylation patterns were observed (Figures 2C–2F). Residues N190 and N348 of IgA2mRNA were found to have nearly 100% complex-type glycans. In contrast, N190 and N348 of IgA2R had 31%–45% complex-type glycans. However, compared to N190 and N348, lower levels of complex-type glycans were observed on N274 and N470 in all the IgA2 preparations tested. In addition, N470 was glycosylated at low occupancy (55% for IgA2mRNA and 58% for mIgA2R) relative to the nearly complete occupancy observed at most other sites (Figures 2C–2F).
Distribution profiles of mRNA encoded IgG1 and dIgA
As dIgA is actively transported into mucosal secretions, we investigated whether administration of mRNA/LNP-formulated Sal4 IgA2mRNA results in accumulation of dIgAmRNA in mucosal tissues. We compared Sal4 dIgA2R, IgA2mRNA, and IgG serum kinetics and mucosal localization over time. Intravenous administration of IgG1R (2.5 mg/kg) to BALB/c mice resulted in the immediate appearance of Sal4 IgG in circulation (Figure 3A). At no time point was Sal4 IgG1R detectable in fecal pellets, despite the high circulating concentrations in serum (Figure 3B). Intravenous administration of an equivalent amount of dIgA2R (2.5 mg/kg), on the other hand, was undetectable in the serum, potentially due to rapid transport into mucosal tissues (Figure 3A). Indeed, intravenous injection of Sal4 dIgA2R resulted in detectable Sal4 IgA in fecal pellets at 6 h (Figure 3B).
Figure 3.

IgA2mRNA expresses in serum, traffics to mucosa, and can block STm invasion into mouse Peyer’s patches
(A–C) Adult BALB/c mice were injected intravenously with 1 mg/kg formulated antibody encoded by mRNA modified with 3′idT, 2.5 mg/kg IgG1R, or 2.5 mg/kg dIgA2R. Concentrations of antibody were measured in (A) serum and (B) feces over time by isotype-specific ELISA.
(C) Comparison of serum and feces levels at 24 h following administration in mice receiving different test articles.
(D) STm challenge outlined for (E)–(H) where adult BALB/c mice were injected intravenously with 1 mg/kg formulated antibody encoded by mRNA modified with 3′idT, 5 mg/kg recombinant antibody protein, or saline. Mice were challenged 24 h later with an oral gavage of a one-to-one mixture of O5+ and O5− strains (4 × 107 colony-forming units [CFUs] total).
(E and F) Concentrations of antibody were measured in (E) serum and (F) feces prior to challenge by isotype-specific ELISA.
(G) Comparison of serum and fecal antibody levels immediately prior to challenge.
(H) Competitive indices of O5+ and O5− STm. Dashed line represents the limit of detection of the assay.
Each group in (A)–(C) represents the mean ± SD of 4–8 mice per group. Each symbol in (D)–(G) represents an individual mouse with mean ± SD depicted. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
In contrast to the recombinant proteins, mRNA-LNP (1 mg/kg) encoding IgG1mRNA and IgA2mRNA reached peak systemic titers of 69 and 27 μg/mL, respectively, between 24 and 48 h following injection (Figure 3A). Undetectable levels of dIgA2R in circulation suggests that levels of IgA2mRNA in the serum at later time points are mostly monomeric, but this finding was not formally investigated. However, IgG1mRNA was not detectable in fecal pellets collected at 24 h, suggesting that Sal4 IgG1mRNA was transported into the gut at undetectable levels and/or that its half-life in intestinal secretions was extremely short (Figure 3B). In contrast, mice injected with mRNA/LNP encoding IgA2mRNA had Sal4 IgA in fecal pellets at 24 h (67 ng/mL) and remained detectable for 96 h (Figures 3B, 3C, S3A, and S3B).
IgA2mRNA administration results in reduced intestinal invasion by STm
We employed an established mouse model of STm intestinal invasion to determine if expression of Sal4 IgA2mRNA resulted in reduced infection of intestinal mucosa (Figure 3C).25 Mice were administered 1 mg/kg mRNA/LNP encoding for Sal4 IgA2mRNA or IgGmRNA or 5 mg/kg analogous recombinant antibody protein. Prior to challenge, serum IgA2mRNA levels were an average of 11 μg/mL, while IgA2R was undetectable (Figure 3D). In contrast, IgG1mRNA and IgG1R levels were 291 and 84 μg/mL, respectively (Figure 3D). However, in the fecal pellets, IgA2mRNA was found at the highest concentrations (average: 125 ng/mL), followed by Sal4 IgG1mRNA (average: 52 ng/mL) and IgG1R (average: 10 ng/mL) (Figure 3E). Detection of Sal4 IgG in the fecal pellet extracts in this experiment compared with the previous experiment may be a consequence of using mice sourced from different vendors, which can change the microbiome composition and, consequently, the intestinal barrier function.31 The elevated levels of fecal IgA2mRNA relative to serum antibody levels are consistent with active transport of Sal4 IgA2mRNA into intestinal secretions, while the observed fecal IgG levels are consistent with transudation (Figure 3F). Importantly, IgA2mRNA significantly reduced invasion of intestinal tissues by STm more effectively than either IgA2R or IgG1 (Figure 3G). Protection, as defined by a competitive index (CI) of <0.5, was observed in mice with fecal Sal4 IgA2mRNA levels that were >100 ng/mL, suggesting a threshold of protection associated with mRNA-based delivery (Figure S3).
IgA1mRNA and IgG1mRNA protected mice against lethal challenge in a PA acute pneumonia model
We next investigated the capacity of IgA1mRNA to protect from PA in an acute lethal pneumonia model. CAM003, a well-characterized IgG antibody that binds Psl of PA, was class-switched to IgA1, the most common IgA isotype in the respiratory tract.32 CAM003 IgA1mRNA retained its affinity for PA, bound to immobilized pIgR, and expressed at high levels in mouse serum (Figure S4). To determine baseline efficacy of this antibody, administration of both CAM003 IgG1R and IgG1mRNA demonstrated dose-dependent expression protection in a mouse model of PA acute pneumonia (Figure S4). Interestingly, IgG1mRNA administration resulted in higher levels of protection with lower systemic expression levels compared with IgG1R, suggesting better localization of IgG1mRNA to lung tissue. This infection model was repeated to compare the efficacy between IgG and IgA. IgG1mRNA and IgA1mRNA administration resulted in systemic averages of 69 and 8 μg/mL, respectively, before intranasal challenge (Figure 4A). Despite significant differences in circulating antibody titers, both IgA1mRNA and IgG1mRNA protected mice from lethal challenge, consistent with the ability of IgA1mRNA to migrate to mucosal sites and demonstrating the ability of mRNA encoded IgA to protect in this pneumonia challenge model (Figure 4B).
Figure 4.

CAM003 IgA1mRNA and IgG1mRNA protect mice against lethal challenge in a P. aeruginosa acute pneumonia model
Adult BALB/c mice were injected intravenously with 1 mg/kg formulated mRNA encoding CAM003 as an IgG1 or an IgA1 and were intranasally challenged 24 h later with 6.75 log10 CFU P. aeruginosa strain PA01. Mice were monitored for survival for 6 days following challenge.
(A) Circulating antibody concentration in serum prior to challenge by isotype-specific ELISA (n = 10/group). Each symbol represents an individual mouse, and data is graphed mean ± SD, ∗∗∗∗p < 0.0001
(B) Kaplan-Meier survival curves.
Discussion
As the predominant antibody transported into mucosal secretions, IgA serves as a first line of defense in immunity to a range of respiratory and enteric pathogens. This contrasts with IgG, which collects in secretions primarily by transudation, often following inflammation and/or physical breaks in barrier integrity.33 The half-life of IgA, once in external fluids, also exceeds that of IgG, due to the presence of the secretory component and glycans that shield IgA from resident proteases.34 These attributes, along with the demonstrated ability of certain IgAs to intercept and incapacitate pathogens prior to accessing mucosal tissues, have raised the prospect of deploying IgA mAbs as interventions to combat infectious diseases of the gut and upper airways. However, advances on this front have been stymied by the cost of generating uniform human dIgAR mAbs at scale.
In this study, we report on the use of synthetic mRNA to encode human IgA mAbs that resemble native IgA in their biochemical, biophysical, and functional properties. Importantly, IgAmRNA displayed pharmacodynamics in vivo more like endogenous human IgA than IgAR. We hypothesize that the observed dichotomy in serum half-life between IgA2mRNA and IgA2R could be due to differences in glycosylation patterns as a result of different cell-type expression.20,29,35 The higher level of sialylation observed for IgA2mRNA is reminiscent of IgA2 isolated from human serum, and this could contribute to increased serum half-life via protection from receptor-mediated clearance. Alternatively, the high mannose glycosylation of IgA2R may contribute to faster clearance, which has been observed for IgG.36 Lastly, observed differences in fucosylation levels between IgA2mRNA and IgA2R could contribute to differences in half-life37; however, the roles of the IgA2 core fucose in transcytosis and receptor engagement are currently unknown.38 Importantly, glycosylation was only determined for an IgA2m1 isotype; it is currently unknown what level of sialylation is occurring on IgA1. As abnormally O-linked glycosylation on IgA1 has been associated with IgA nephropathy,39 we are investigating both glycosylation and the pathology of repeated IgAmRNA dosing to fully characterize and understand the risks associated with this isotype.
Better pharmacodynamic properties in vivo translated to enhanced protection from mucosal bacterial challenge. mRNA/LNPs delivered intravenously resulted in accumulation of levels of antigen-specific IgA in the gut and respiratory tract to attenuate STm and PA infections. In a stringent model of STm invasion, Sal4 IgA2mRNA resulted in significantly decreased bacterial burden as compared to IgAR and IgG due to its accumulation at the site of infection. The degree of protection obtained from Sal4 IgA2mRNA was similar to that achieved with purified, recombinant Sal4 SIgA2 administered orally.40 Our results are consistent with Sal4 IgA2mRNA intercepting STm in the intestinal lumen and blocking uptake into Peyer’s patch tissues. The exact mechanism of IgA2 superiority compared with IgG in these studies is unknown; however, there are several plausible mechanisms. It has previously been demonstrated that bacteria agglutination is more efficient via Sal4 SIgA than IgG in vitro, potentially preventing bacterial invasion in vivo.40 Alternatively, SIgA has been shown to prevent STm bacterial division, which results in “enchained growth,” further accelerating pathogen clearance.41 Lastly, the privileged access of IgA to the GI lumen cannot be discounted as a mechanism that results in superior protection from challenge compared with IgG.
We were also able to demonstrate protection from a lethal PA challenge in the lung with intravenously administered CAM003 IgA1mRNA. Despite significant differences in circulating antibody titers, IgA1mRNA protected mice more efficiently from a lethal PA challenge than the analogous IgGmRNA. Previously, greater protection in the lung over a native IgG was achieved by fusing a pIgR binding peptide to the Fc of CAM003 IgG1 to achieve better mucosal trafficking.27 We hypothesize that class-switching CAM003 to IgA1 resulted in a similar phenomenon, with higher titers of antibody localized to the lung, which resulted in increased protection. Unfortunately, levels of antibody were not measured in the lung, but despite lower IgA titers in circulation, we still observed similar levels of protection from mortality between IgA and IgG groups, possibly due to IgA transport to the mucosa. While these results may not directly translate to humans, as the mucosal localization of IgA may differ significantly between humans and mice due to pIgR localization,42,43 monoclonal IgA produced from mRNA can still serve as an important modality in the battle against infectious diseases.
Limitations of the study
While these studies represent a significant advance in technology to enable delivery of IgA mAbs, there are several limitations. All investigative studies described herein represent proof of concept for synthetic mRNA delivery of IgA mAbs. In humans, administration of an mRNA/LNP encoded for a ChikV mAb induced elevated transient cytokine responses at doses as low as 0.3 mg/kg3. Doses utilized in these mouse models (1 mg/kg) exceeded those tested in humans (0.1–0.6 mg/kg); additional pathology would be required to evaluate dose and safety prior to clinical use.
The mRNA/LNP platform was investigated only in the context of prophylactic delivery. This was somewhat by design, as our focus first and foremost was on establishing the platform technology using well-established mouse models in which IgA has been shown to be protective. Indeed, prophylactic IgA delivery could serve as means to interrupt outbreaks the likes of norovirus in confined situations such as on a cruise ship44 or even to serve as a firewall in nosocomial outbreaks of Klebsiella pneumoniae, which currently does not have a licensed vaccine.45 The potential of IgA delivery for mucosal infections is vast, and we are now actively focused on therapeutic applications of this technology. The data presented here and in previous studies3,4 establish mRNA as a central platform for evaluating basic biology and therapeutics in a manner previously unattainable with protein-based approaches.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CAM003 IgG | GeneArt | N/A |
| Sal4 IgG | Genscript | N/A |
| Sal4 IgA2m1 | GeneArt | N/A |
| CAM003 IgA1 | GeneArt | N/A |
| Goat anti human IgG Fc fragment | Bethyl | A80-104A; RRID:AB_67060 |
| Goat anti human IgA | Bethyl | A80-102A; RRID:AB_67044 |
| Goat anti human IgG HRP | Southern Biotech | 2040–05; RRID:AB_2795644 |
| Goat anti human lambda HRP | Bethyl | A80-116P; RRID:AB_67591 |
| Goat anti human IgA HRP | Southern Biotech | 2050–05; RRID:AB_2687526 |
| Bacterial and virus strains | ||
| Salmonella enterica serovar Typhimurium (STm) | ATCC | 14028s |
| S. Typhimurium strain GGW445 (sj8101::kan oafA126::TN10d-Tc fkpA-lacZ) | This study | GGW445 |
| S. Typhimurium strain GGW444 (zj8101::kan) | This study | GGW444 |
| Pseudomonas aeruginosa PA01 | ATCC | 15692 |
| Biological samples | ||
| Normal goat serum | Gibco | 16210072 |
| Fetal bovine serum | Gibco | 16140089 |
| Mouse serum, feces, and intestines | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Opti-MEM I | Gibco | 31895–070 |
| Dulbeco’s Modified Media (DMEM) | Gibco | 11965–092 |
| Expi 293 expression medium | Gibco | A14351-01 |
| CaptureSelect™ IgA affinity matrix | ThermoFisher | 194288010 |
| MabSelect SuRe™ | Cytiva | 17547402 |
| carbonate-bicarbonate buffer capsules | Sigma | C3041-100CAP |
| Recombinant human pIgR | R&D Biosystems | 2717-PG-05 |
| 10x PBS-T (0.5% Tween 20 pH 7.4) | Boston Bioproducts Inc | IBB-171 |
| Superblock PBST | ThermoFisher | 37515 |
| Sureblue TMB 1-C substrate | SeraCare | 5120–0074 |
| TMB Stop solution | SeraCare | 5150–0021 |
| 1X Phosphate buffered saline (PBS) | Cytiva | SH30256.02 |
| Hanks’ Balanced Salt Solution (HBSS) | Sigma-Aldrich | H9269-500ML |
| Kanamycin | Sigma-Aldrich | K4000 |
| X-gal | RPI | B71800 |
| Gentamicin | Sigma-Aldrich | G1264 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| Hydrochloric Acid | Fisher Scientific | A144S-212 |
| glycine | Sigma-Aldrich | 50046-50G |
| 1M Tris-HCl, pH8.0 | ThermoFisher | 15568025 |
| 4x Laemmli Sample buffer | Bio-Rad | 1610747 |
| Precision Plus Protein™ standard, strep-tagged recombinant | Bio-Rad | 1610363 |
| Novex™ Tris-Glycine SDS Running Buffer (10X) | ThermoFisher | LC2675 |
| 2-mercaptoethanol (>99%) | Millipore Sigma | M6250-100ML |
| Protease inhibitor mini tablets | ThermoFisher | A32953 |
| Peptide M agarose | Invivogen | Gel-pdm-5 |
| Urea | Sigma-Aldrich | U5378-500G |
| Tris-HCl buffer, pH 7.8 | Invitrogen | 15567–027 |
| Dithiothreitol | Sigma-Aldrich | D9779-5G |
| Iodoacetamide | Sigma-Aldrich | I1149-5G |
| Glu-Glu | Sigma-Aldrich | G3640 |
| Endoproteinase GluC | New England Biolabs | P8100S |
| Trypsin | Promega | V5111 |
| Gibco | 25300–054 | |
| Formic acid | Fisher Scientific | A11710X1-AMP |
| Ammonium bicarbonate | Sigma-Aldrich | A6141-25G |
| PNGase F | Sigma-Aldrich | F8435-50UN |
| ZIC-HILIC particles | Nest Group Inc | 200-001-0050 |
| LC-MS grade water | Fisher Scientific | W64 |
| LC-MS grade acetonitrile | Fisher Scientific | A955-4 |
| LC-MS grade water with 0.1% Trifluoroacetic Acid | Fisher Scientific | LS119-1 |
| LC-MS grade acetonitrile with 0.1% Trifluoroacetic Acid | Fisher Scientific | LS121-1 |
| Critical commercial assays | ||
| trans IT mRNA transfection kit | Mirus Bio LLC | MIR 2256 |
| Amicon Ultra-15 Centrifugal Filter 50kDa MWCO | Millipore | UFC905024 |
| Amicon Ultra-15 Centrifugal Filter 100kDa MWCO | Millipore | UFC910024 |
| 96 well NUNC Maxisorp plates | ThermoScientific | 12565135S23 |
| Immunlon 4HBX plates | Thermo Fisher Scientific | 3855 |
| 4-20% Mini-PROTEAN® TGX Stain-Free™ protein gels, 10 well, 30uL | Bio-Rad | 4568093 |
| Carbon-coated copper grid | Electron Microscopy | CF300-Cu |
| HiTrap Protein An HP | Cytiva | 17040301 |
| Superdex 200 Increase 10/300 GL gel filtration column | Cytiva | 28990944 |
| 2.8mm ceramic beads | Thermo Fisher | 15-340-154 |
| 100kDa Pierce Protein Concentrators PES | Thermo Fisher | 88533 |
| Hypersep C18 Cartridges | Thermo Scientific | 03-251-259 |
| C4 HyperSep tips | Thermo Fisher | 60109–211 |
| Easy-Spray PepMap Neo C18 75umx500mm column | Thermo Fisher | ES75500PN |
| 20mm nanoEase C18 Trap Column | Water Corporation | 186008821 |
| Experimental models: Cell lines | ||
| EXPI293F | ThermoFisher | A14527 |
| HeLa | ATCC | CCL-2 |
| Experimental models: Organisms/strains | ||
| Balb/c mice (female) | Charles River Laboratories | BALB/cAnNCrl |
| Balb/c mice (female) | Taconic Biosciences | BALB-F |
| Balb/c mice (female) | Envigo Laboratories | BALB/cAnNHsd |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad | 9.3.1 |
| Relion 4.0 | N/A | |
| R package brms (version 2.17.3) | N/A | |
| Proteome Discoverer | Thermo Fisher | 2.5.0.400 |
| Byonic | Protein Metrics Inc | V3.2.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents can be directed to the lead contact, Cailin Deal (cailin.deal@modernatx.com)
Materials availability
All reagents are commercially available and this study did not result in any new reagents.
Experimental model and subject details
Cell lines
Expi293F cells were obtained from ThermoFisher (A14527) and maintained in Expi293 expression medium (ThermoFisher) in an Infors incubator with 5% CO2 shaking at 120 rpm. HeLa cells were obtained from ATCC (Cat. # CCL-2) and maintained in Dulbecco’s Modified Media (DMEM) with 10% fetal bovine serum at 37°C and 5% CO2.
Bacterial strains and growth conditions
Salmonella enterica serovar Typhimurium (STm) was purchased from ATCC (ATCC 14028s). S. Typhimurium strains GGW445 (sj8101::kan oafA126::TN10d-Tc fkpA-lacZ) and GGW444 (zj8101::kan) with the O4 and O5 O-Ag, respectively, are derivatives of ATCC 14028s25,46,47 Pseudomonas aeruginosa PA01 strain was purchased from ATCC (ATCC 15692).
Mice
Female BALB/c mice, 6 to 10 week old, were obtained from Charles River Laboratory and maintained at Moderna in Cambridge, MA. All mouse studies were approved by the Animal Care and Use Committee at Moderna. For studies performed at the Wadsworth Center, female Balb/c mice, aged 8–12 weeks, were obtained from Taconic Biosciences and were cared for by the Wadsworth Center Animal Core Facility. All experiments were performed in accordance with protocols approved by the Wadsworth Center’s IACUC. Finally, all mouse studies performed at the University of North Texas Health Science Center at Fort Worth were approved by their Animal Care and Use Committee and conducted according to IACUC protocol 2019-0030. These studies utilized female Balb/c mice, aged 6–8 weeks old, that were obtained from Envigo Laboratories.
Method details
Recombinant antibody proteins
Recombinant protein versions of all monoclonal antibodies were purchased from Genscript or GeneArt (ThermoFisher). CAM003 IgG (GeneArt) was purified from transient transfection of a 2:1 ratio of HC to LC by weight in EXPI293 with HiTrap Protein An HP and polished using HiLoad Superdex 200 26/600 prep. Sal4 IgG (Genscript) was purified from transient transfection of HD CHO-S cells with MabSelect SuRe LX. Sal4 IgA2m1 (GeneArt) was purified from transient transfection of a 4:4:1 ratio of HC to LC to JC (by weight) in EXPI293 with CaptureSelect IgA Affinity matrix. All protein concentrations were determined by absorption at 280nm using NanoDrop.
Generation of modified mRNA and LNPs
Sequence-optimized mRNA encoding functional IgA monoclonal antibodies were synthesized in vitro using an optimized T7 RNA polymerase-mediated transcription reaction with complete replacement of uridine by N1-methyl-pseudouridine.48 The reactions included a DNA template containing an open reading frame flanked by 5′ untranslated region (UTR) and 3′ UTR sequences with a terminal encoded polyA tail. Where indicated, inverted deoxythymidine (idT) were appended to the 3′ terminus of fully synthesized mRNA by phosphodiester linkage of a modifying oligo with 3′ idT. Free and end-stabilized mRNA were purified, buffer exchanged and sterile filtered.
Lipid nanoparticle-formulated mRNA was produced through a modified ethanol-drop nanoprecipitation process as described previously.35 Briefly, ionizable, structural, helper, and polyethylene glycol lipids were mixed with mRNA (2:1 HC to LC for IgG; 4:4:1 HC to LC to JC for IgA) in acetate buffer at a pH of 5.0 and at a ratio of 3:1 (lipids:mRNA). The mixture was neutralized with Tris-Cl at a pH 7.5, sucrose was added as a cryoprotectant, and the final solution was sterile filtered. Vials were filled with formulated LNP and stored frozen at −70°C until further use. The drug product underwent analytical characterization, which included the determination of particle size and polydispersity, encapsulation, mRNA purity, osmolality, pH, endotoxin and bioburden, and the material was deemed acceptable for in vivo study.
mRNA transfection for mAb expression
EXPI293 cells were transiently transfected with the corresponding mRNA encoding for Sal4 IgG, Sal4 IgA2m1, CAM003 IgG or CAM003 IgA1 HC, LC and/or JC using trans IT-mRNA Transfection Kit (Mirus Bio LLC) per the manufacturer’s recommendations. For all IgG isotypes, this required co-transfection of HC and LC at a 2:1 ratio by weight, whereas all IgA isotypes consisted of HC, LC, and JC transfections at a 4:4:1 ratio by weight, unless otherwise stated that JC was not included. EXPI293 cells were diluted to 1x106 cells/mL with EXPI293 Expression Medium (ThermoFisher). Using a ratio of 1 μg mRNA per 1 mL of culture, mRNA was first added to Opti-MEM I SFM (Gibco), followed by addition of TransIT-mRNA transfection reagent (Mirus Bio) at a 1:2 ratio of mRNA to TransIT. The complex was allowed to form for 3 min before addition to the suspension culture. For DNA transfections, EXPI293 cells were transfected at 3x106 cells/mL using 1 μg plasmid DNA per mL of culture. DNA complexes were formed by separately diluting DNA and Turbo293 Transfection reagent (Speed Biosystems) in Opti-MEM I, then combined and allowed to incubate at RT for 20 min before adding to suspension culture. At 48 h following transfection, cell cultures were centrifuged, and supernatant was collected. Unless being purified, majority of supernatant was further concentrated in 50–100 kdA Amicon filters for 40 min at 3000 x g. Transfection supernatants and concentrated supernatants were further used to quantitate and characterize by enzyme-linked immunosorbent assay (ELISA).
Enzyme-linked immunosorbent assay (ELISA)
To quantitate total human IgG (hIgG) or IgA (hIgA), 96-well NUNC Maxisorp plates (ThermoScientific) or Immulon 4HBX plates (Thermo Fisher Scientific) were coated with 0.1 mL per well of goat anti-human IgG Fc fragment (Bethyl A80-104A) or goat anti-human IgA (Bethyl A80-102A) at 1:100 dilution in 0.05 M carbonate-bicarbonate (Sigma C30411) overnight at 4°C. To determine binding to human pIgR, Nunc maxisorp plates were coated with 2 μg/mL of recombinant human pIgR (R&D Biosystems 2717-PG-05) at 4°C overnight. For both isotype-specific and pIgR ELISA, plates were washed with an automated plate washer (Biotek) or 4x with 0.05% PBS-T and were subsequently blocked with 0.2 mL per well of Superblock PBST (ThermoFisher) or 2% goat serum in PBS-T for 2 h at room temperature. Using purified antibodies as a standard, mRNA transfection supernatant or mouse serum was serially diluted in PBST in a dilution plate and 0.1mL per well was transferred to the coated plates and incubated for 1 to 2 h at room temperature. Following incubation, plates were subsequently washed and incubated with 0.1 mL per well of goat anti-human IgG horseradish peroxidase (HRP; Southern Biotech; 1:5000), goat anti-human lambda HRP (Bethyl; 1:10000), goat anti-human kappa HRP (Bethyl; 1:10000) or goat anti-human IgA HRP (Southern Biotech; 1:5000) for 1 h at room temperature. Plates were subsequently washed and incubated with 0.1 mL per well of Sureblue TMB 1-C substrate (Fisher Scientific) for 5 min. The reaction was stopped with 0.1 mL per well of TMB Stop solution (SeraCare) and read at an absorbance of 450nm on a SpectraMax ABS Microplate Reader or a SpectraMax iD3 Microplate Reader (Molecular Devices). Absolute quantities of human antibody in transfection supernatant or mouse serum were extrapolated in GraphPad Prism 9 using a standard curve that was generated with the appropriate purified isotype version of Sal4 or CAM003.
Bacterial whole cell ELISA
For STm whole cell ELISA (WCE), an overnight culture of STm (ATCC 14028s) was inoculated with one colony from a freshly streaked plate into 5mL of LB broth. The following day, the overnight STm culture was subcultured 1:50 in LB broth and grown to OD600 of 0.7–0.8, with a final adjusted concentration of OD600 0.7. Cells were washed 2x with sterile PBS and resuspended in the prewash volume. NUNC MaxiSorp or Immulon 4HBX plates (Thermo Fisher Scientific) were coated with 0.1 mL per well of the STm bacterial solution, covered and incubated at 4°C overnight. For P. aeruginosa PA01 WCE, 10 to 50 mL of LB broth was inoculated with one colony from a freshly streaked plate. Bacteria were grown to an OD650 of ∼2.0 and subsequently diluted with LB broth to an OD650 of 0.5, followed by an additional 50% dilution with sterile PBS. NUNC MaxiSorp plates were coated with 0.1 mL per well of the PA01 bacterial solution, covered and incubated at 4°C overnight. For STm WCE, wells were washed 4x with 0.5% PBS-T and blocked in either 0.2 mL of Superblock PBST (Thermo Fisher Scientific) or 2% goat serum in PBS-T for 2 h at room temperature. For PA01 WCE, wells were washed 4x with 0.5% PBST in an automated plate washer (Biotek) and subsequently blocked with 0.2mL Superblock PBST (ThermoFisher) for 2 h at room temperature. Subsequent ELISA steps are identical to what has been described previously.
HeLa cell invasion assay
HeLa cells were obtained from the ATCC and maintained in Dulbecco’s Modified Eagle Media (DMEM) with 10% fetal bovine serum at 37°C and 5% CO2. The HeLa cell invasion assay was performed as described.25 Cells were seeded at 5x105 cells/mL in 96-well plates and grown for 24 h until 70%–90% confluent. Prior to invasion assays, cells were washed 3x with serum-free DMEM. Overnight cultures of GGW444 (O5+) and GGW445 (O5-) were diluted 1:50 into LB at 37°C and 220 rpm and adjusted to an OD600 of 0.7. Strains were mixed 1:1 and washed 2x by centrifugation (6,000 x g for 4 min) and resuspended in PBS with a pH of 7.4. Bacteria were diluted 1:10 in Hanks’ Balanced Salt Solution (HBSS, Sigma-Aldrich); an aliquot was plated on LB agar supplemented with kanamycin (100 μg/mL) and X-gal (40 μg/mL) for subsequent determination of CFUs associated with bacterial input.
For the invasion assay, bacterial mixtures were incubated with 15 μg/mL of Sal4 IgA for 15 min at 37°C. The source of Sal4 IgA used in these studies was either recombinant purified protein (GeneArt) or concentrated supernatant 2 days following transfection of EXPI-293 with Sal4 IgAmRNA. Treated bacteria were applied to HeLa cell monolayers and the culture plate was centrifuged at 1,000 x g for 10 min. The microtiter plates were then incubated for 60 min at 37°C and the cells washed 3x with HBSS and treated with gentamicin (100 μg/mL) to eliminate extracellular bacteria. Cells were then washed 3 times with HBSS and lysed with 1% Triton X-100 (in Ca2+ and Mg2+-free PBS)., The cells were then serially diluted and plated on LB agar containing kanamycin (100 μg/mL) and X-gal (40 μg/mL), and then incubated overnight at 37°C. The competitive index (CI) [(%strain A recovered/% B strain recovered)/(%strain A inoculated/% strain B inoculated)] was calculated for each treatment group.
Purification of Sal4 IgA
Purification proceeded identically for both mRNA and DNA transfections. To harvest, suspension cultures were centrifuged at 4,000 rcf for 30 min and pellets were discarded. Cell supernatant was filtered through a 0.22 μm vacuum filter followed by gravity chromatography with columns packed with CaptureSelect IgA Affinity Matrix (ThermoFisher). Columns were washed with PBS before eluting in 0.1 M Glycine pH 3 and were then immediately neutralized with 1 M Tris with a pH of 8. Affinity purified IgA was then injected onto a Superdex 200 Increase 10/300 GL gel filtration column (Cytiva) using an AKTA Pure FPLC.
SDS-PAGE
For SDS-PAGE analysis, 20 μL of SEC fractions were mixed with 4X Laemmli Sample Buffer (Bio-Rad). Reducing conditions were achieved with 5% (v/v) 2-mercaptoethanol in sample buffer. Samples were heated to 95°C for 5 min prior to loading onto 4–20% Mini-Protean TGX Stain-Free gel (Bio-Rad), with Precision Plus Protein Unstained Protein Standards included as marker (Bio-Rad). Gels were run for 30 min at 200 V in Tris-Glycine-SDS running buffer (Invitrogen), the stain-free dye was developed via UV activation and then imaged on ChemiDoc MP gel imager (Bio-Rad).
Transmission electron microscopy (TEM) imaging
For each sample, a ∼3.5 μL aliquot was applied to a freshly glow-discharged, carbon-coated copper grid (Electron Microscopy Sciences, CF300-Cu) and allowed to absorb for 30 s. After blotting the excess solution, grids were stained with 1% uranyl acetate solution (Electron Microscopy Sciences) for 1 min and then air-dried. A Thermo Fisher Scientific Talos L120C electron microscope equipped with a 4k x 4k Ceta CMOS Camera was used for data collection. Images were captured at a nominal magnification of 57,000× and 73,000x (pixel size 2.60 Å/pixel and 2.04 Å/pixel, respectively) with a defocus range of ∼ −1.5 to −2 μm.
Expression of mAbs in mice
Six-to 8-week-old female BALB/c mice (Charles River Laboratories), in groups of 5–28 mice each, were injected intravenously with the indicated recombinant protein, mRNA/LNP, or PBS, at the indicated dose in 100 μL. All in vivo studies with mRNA/LNP were coformulations of HC and LC for IgG and HC, LC, and JC for IgA. For IgA, this results in a mixture of monomeric, dimeric, and polymeric IgA. Unless otherwise stated, all IgAR injected into mice is a mixture of monomeric, dimeric, and polymeric. At the doses used major adverse reactions, pathology or mortality was not observed or linked to mRNA/LNP treatment in animals. Mice were bled via submandibular vein at the indicated time points and serum was isolated for antibody quantification by ELISA. At the final indicated time point, mice were euthanized via CO2 asphyxiation and a terminal bleed was collected via cardiac puncture.
Mouse tissue processing for mAb quantification
For fecal samples, three to four fresh fecal pellets were collected at each of the indicated time points following intravenous administration of test articles. Total weight of the fecal pellets per mouse were recorded and fecal extract buffer (PBS containing 10% normal goat serum [Gibco] and 1 protease inhibitor per 50mL solution [Pierce A32953]) was added to achieve a final concentration of 200 mg/mL. Pellets were vortexed and manually broken until the entire pellet was disrupted. Following centrifugation at 13,000 rpm for 10 min at 4°C, supernatants were collected and mAb concentrations were quantified via ELISA.
Where indicated, intestines were harvested at 168 h after injection following CO2 asphyxiation and washed in PBS to remove any mucous or fecal debris. Each mouse intestine (comprising both the small and large intestine) was weighed and homogenized (2x at 5 m/s for 45 s, with a 10 s break between sets) at 2 g/mL in homogenization buffer (PBS +10% normal goat serum) in 2.0 mL homogenizing tubes containing 2.8 mm ceramic beads (Fisherbrand) using the BeadMill homogenizer (Fisherbrand). Eppendorf tubes containing homogenized intestinal tissue were spun at 13,000 rpm for 10 h at 4°C to obtain debris-free supernatant which was stored for further analysis by ELISA.
Purification of IgA2 from mouse serum
Fifty Balb/c mice per group (8–10 weeks old; Charles River Laboratory) were intravenously administered either 1 mg/kg of the indicated mRNA/LNP formulations or PBS and terminal bleed serum was collected 24 h following administration (Figure S2). At this dose, major adverse reactions, pathology or mortality was not observed or linked to mRNA/LNP treatment in animals. Sal4 IgA2 was purified from pooled mouse sera using peptide M agarose (Invivogen) following the manufacturer’s instructions. Briefly, the sera were concentrated by 100kDa Pierce Protein Concentrators PES (Thermo Fisher), then buffer exchanged with PBS buffer. The sera whereas then incubated with peptide M agarose resin overnight at 4°C, and the resin was washed using PBS buffer. After another wash step with 0.1 M glycine buffer (pH 5.0), IgA2 was eluted with 0.2 M glycine buffer (pH 2.5) and incubated for 5 min at room temperature. The elution step was repeated 3x, and the eluted IgA2 concentration of the administered group and control group were measured by ELISA.
Isolation of recombinant IgA2 monomer
Sal4 IgA2 expressed by EXPI293 cell line as described above was further using the AKTA pure protein purification system. The monomeric IgA2 (mIgA2R) was isolated for glycosylation comparison with mRNA expressed IgA2 in vivo (Figure S2). Briefly, 1mg of the total IgA2 was injected to Superdex 200 Increase 10/300 GL column (Cytiva) and then IgA2 was eluted by PBS buffer at a constant flow rate of 0.25 mL/min. The eluent was collected by AKTA fraction collector F9-C (0.5 mL per fraction). The fractions of monomer were determined using Western blot and combined for the next step of the experiment.
Protein digestion
Sal4 IgA2 proteins from different sources were concentrated to 20 μL by 100 kDa Pierce Protein Concentrators PES, then buffer exchanged with UA buffer (8 M urea in 50 mM Tris-HCl buffer pH 7.8). Dithiothreitol (Sigma-Aldrich) was added to the solution to a concentration of 10 mM; the solution was then heated at 95°C for 10 min. Once cooled to room temperature, iodoacetamide (Sigma-Aldrich) was added to the solution to a concentration of 20 mM. The solution was then incubated in darkness at room temperature for 40 min and another 10 mM dithiothreitol was added to quench the reaction. The solution was then diluted with 200 μL of 0.5 mM Glu-Glu (Sigma-Aldrich) in 50 mM Tris-HCl buffer at a pH of 7.8. Endoproteinase GluC (New England Biolabs) was added to make GluC and IgA2 at a 1:10 ratio (w/w), and the solution was incubated at 37°C overnight. The sample was further digested with Trypsin to a final protease:protein ratio of 1:50 (w/w) for 4 h at 37°C. The reaction was quenched by adding 0.1% Formic Acid (Fisher Scientific) and the digested peptides were desalted using HyperSep C18 Cartridges (Fisher Scientific). The desalted peptides were then dried by Savant SpeedVac Concentrator (Thermo Fisher Scientific) and stored at −80°C.
N-glycosylation site analysis
N-linked glycosylation sites of Sal4 IgA2 were de-glycosylated and labeled with 18O using a previously reported method (Figure S2).49 Sal4 IgA2 proteins from different sources were dissolved in 30 μL of 20 mM ammonium bicarbonate (Sigma-Aldrich) in H218O. One unit of PNGase F (Sigma-Aldrich) was then added, and the solution was incubated for 1 h at 37°C to label the N-glycosylation sites. The solution was stored at −80°C for mass spectrometry analysis (Figure S2).
Glycopeptide enrichment
N-linked intact glycopeptides of Sal4 IgA2 were enriched by HILIC using a previously reported method.50 ZIC-HILIC particles (The Nest Group, Inc.) were packed in C4 HyperSep tips (Thermo Fisher Scientific). The HILIC tip was then conditioned 3x in LC-MS grade water (Fisher Scientific) followed by conditioning (3x) in 1 mL of 80% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA). Sal4 IgA2 proteins from different sources were dissolved 3x in 80% ACN and 0.1%TFA and loaded onto HILIC tips. The HILIC tips were then washed 3x with 80% ACN and 0.1%TFA, and the glycopeptides were eluted 3x using 100% water, 0.1%TFA. The eluent was dried by lyophilizer (Labconco) and store at −80°C for mass spectrometry analysis (Figure S2).
Mass spectrometry analysis
Peptides or glycopeptides were dissolved in different volumes of 0.1% formic acid (FA) and separated (in three technical executions) through a Waters ACQUITY UPLC M-Class liquid chromatography system equipped with an Easy-Spray PepMap Neo C18 75 μm × 500 mm column (Thermo Fisher Scientific) coupled to a 20-mm nanoEase C18 Trap Column trap column (Water Corporation). The mobile phase flow rate was 0.3 μL/min with 0.1% FA in water (A) and 0.1% FA 100% acetonitrile (B). The gradient profile was set as the following: 1%–35% B for 80 min, 35%–60% B for 10 min, 60%–95% B for 10 min, 95% B for 5 min, and equilibrated in 1% B for 10 min. MS analysis was performed using a Thermo Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). The spray voltage was set at 1.9 kV. Spectra (maximum IT of 100 ms) were collected from 400 to 2000 m/z at a resolution of 120 K followed by data-dependent HCD MS/MS (at a resolution of 6K, stepped NCE 20,30,40, intensity threshold of 1 × 105 and maximum IT of 100 ms) of the 20 most abundant ions using an isolation window of 2 m/z. Charge-state screening was set to only include ions with more than one and less than eight charges.
Data analysis
N-glycosylation sites data from the 18O label experiment was processed with Proteome Discoverer 2.5 (Thermo Fisher Scientific). Glycopeptides were identified by Byonic software (Protein Metrics Inc.). The LC-MS/MS spectra of combined GluC/tryptic digests of glycoproteins were searched against the FASTA sequence of Sal4 IgA2 by choosing RKDE as cleavage sites with a maximum of two missed cleavage sites. MS/MS spectra were matched with a tolerance of 10 ppm on precursor mass and 50 ppm on a fragment mass. All protein identification hits had an FDR rate ≤1%. Carbamidomethylation was set as fixed modification, oxidation of methionine, and deamidation of asparagine and glutamine were used as variable modifications. In Proteome Discoverer, 18O labeling of Asn (Δm = 2.9848) was set as variable modification to determine N-glycosylation sites and N-glycan occupancy (Figure S2). Intact-glycopeptide data were searched against the common mammalian N-glycans database in Byonic. Glycans were manually categorized according to their composition using the following criteria: HexNAc(2)Hex(9−4) was classified as high-mannose. HexNAc(3)Hex(5−6)X was classified as hybrid (X can be fucose or sialic acid). Other compositions were classified as complex-type glycans. If any of the compositions had a fucose, it was assigned as a fucosylated glycan. Any glycan containing at least one sialic acid was counted as sialylated.51 Glycopeptides with more than five MS/MS spectral identified (PSM#) were used for quantifying the percentage of glycans belonging to any of the three categories, which include complex, hybrid, or high-mannose (Figure S2).52 The same method was used to determine the percentage of fucosylation and sialylation among all glycans. IgA2 was undetectable in both HILIC-enriched and 18O-labeled aliquots of the serum pool from PBS-administered animals.
STm intragastric challenge
Four mice per group of female Balb/c mice aged 8 to 12 weeks (Taconic Biosciences) were used for each challenge and the challenge was repeated four separate times (16 mice in total per group). The challenge experiments were performed as previously described with some modifications.25 This model utilizes a competitive infection assay to normalize natural variations in challenge inoculum using two STm strains premixed prior to oral delivery: one expressing the Sal4 epitope, the O5 antigen (O5+), and an O5-null strain (O5-) that expresses β-galactosidase to easily distinguish the two strains. A reduction in competitive index (CI) only occurs when Sal4 reduces the number of O5+ in mouse Peyer’s patches.
Briefly, mice were injected intravenously with saline, 1 mg/kg of formulated mRNA modified with 3′idT, or 5 mg/kg of recombinant protein, 24 h prior to bacterial challenge. At the doses used major adverse reactions, pathology or mortality was not observed or linked to mRNA-LNP treatment in the animals. Overnight cultures of GGW444 (O5+) and GGW445 (O5-) were subcultured to an OD600 of 0.7, combined 1:1 (v/v) and resuspended in sterile PBS pH 7.4. Following this, an aliquot was plated on LB agar containing kanamycin (100 μg/mL) and X-gal (40 μg/mL) at the start of the experiment. Mice were given an STm gavage (∼4x107 CFUs in 200 μLs) and sacrificed (CO2 asphyxiation followed by cervical dislocation) 24 h after challenge, following which the small intestine was removed above the cecum for each mouse. The Peyer’s Patches from each mouse were then pooled in 1 mL sterile PBS and placed on ice. Samples were then homogenized 3x for 30 s each using a Bead Mill 4 Homogenizer (Fisher Scientific) and the homogenates serially diluted and plated on LB agar (containing 100 μg/mL kanamycin and 40 μg/mL X-gal) and incubated overnight at 37°C. The number of blue and white colonies were determined and the competitive index (CI) was calculated as [(%strain A recovered/% B strain recovered)/(%strain A inoculated/% strain B inoculated)]. Whole plate dilutions at 100 μL per plate were required in order to observe the required number of colonies to calculate CI. Samples containing fewer than 30 CFUs/100 μL were removed and considered “too few to count”.
P. aeruginosa lethal acute pneumonia mouse model
Female Balb/c mice (22 ± 2g) (Envigo Laboratories), 6–8 weeks old, were administered the indicated dose of mRNA/LNP formulations or recombinant protein intravenously (n = 8–10/group) and serum was collected 24 h later to quantitate circulating antibody titers prior to challenge. At the doses used major adverse reactions, pathology or mortality was not observed or linked to mRNA-LNP treatment in the animals. Mice were subsequently challenged intranasally with P. aeruginosa PAO1 and monitored for mortality for 6 days. Results were graphed in a Kaplan-Meier survival curve.
Quantification and statistical analysis
Data was processed using GraphPad Prism 9 and unless otherwise stated, all statistical analysis was performed in this software. Mean and standard deviation are presented as averages and error bars. In vitro tests were performed in triplicate and either averaged or a single representative experiment was shown. Where applicable, Student’s T test was used to analyze the statistical differences between independent groups in in vitro experiments. Unless otherwise stated, all in vivo experiments comprised of 5–28 mice per group. For STm intragastric challenge, four mice per group were used and the challenge was repeated four separate times. Statistical significance was determined by a Kruskal-Wallis test and Dunn’s post hoc test. Kaplan-Meier survival curves were utilized for the P. aeruginosa acute pneumonia challenge model and differences in survival were calculated by the log rank test for IgA1 versus IgG1. All differences were considered significant when p values were less than 0.05 and significance is denoted by asterisks (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
For EM images, reference-free 2D classification was performed using Relion 4.0 software. Glycosylation data analysis was performed using Proteome Discoverer 2.5 (Thermo Fisher Scientific) to process the N-glycosylation sites data from the 18O label experiment. Additionally, glycopeptides were identified by the Byonic Software (Protein Metrics Inc)
Statistical model for IgA half-life modeling
Since each analyte was measured at different time points, we fit the following model to each analyte separately. We estimated the population-level half-life of each analyte using a flexible linear mixed effects model that incorporates a first-order kinetic component for terminal measurements. To do so, we modeled the terminal decay phase of the data using a first-order kinetic model but employed a flexible spline basis to model the earlier phases. Let be the measured analyte concentration for animal at time , where and . To treat the early and terminal phase of the time course differently, we introduce variables and , defined as
| (Equation 1) |
where is some fixed threshold after which the terminal decay phase has begun. Then,
| (Equation 2) |
where is the concentration of the analyte for animal at time prior to censoring by the lower limit of quantification (LLOQ), is the LLOQ of the assay, is a threshold after which the decay process has entered the terminal phase, and is a -dimensional thin plate regression spline basis.53
We allow for animal-specific random intercepts and slopes, where the random slopes imply animal-specific deviations about the population-level terminal IgA half-life. Therefore, we have
| (Equation 3) |
We allow residual variance to change with time following a 7-dimensional thin plate spline regression basis, giving .
In order to choose the threshold t, for each analyte, we selected the latest t to allow for ≥3 time points where the majority of measurements were above the LLOQ. Since all IgA2R measurements were above the LLOQ, we selected the t such that the last 3 time points were used to estimate the terminal half-life. For IgA2mRNA and human serum IgA, the last 4 time points were used to estimate terminal half-life. The dimension of the thin plate regression spline basis f was chosen to be as large as possible, given the threshold t.
All modeling was implemented using the R package brms (version 2.17.3)54 using default, non- or weakly-informative prior specifications.
Acknowledgments
We thank Dr. Gregory Hendricks and Dr. Kyoung Hwan Lee from the Electron Microscopy Facility at UMass Chan Medical School for their support of the use of Talos L120C. At the Wadsworth Center, we thank Dr. Graham Willsey for STm strain construction, Dr. Jennifer Yates for assistance with mouse studies, and the Media and Cell Culture core facility for bacterial growth media. Medical writing and editorial assistance, under the direction of the authors, was provided by Jared Mackenzie, PhD, of MEDiSTRAVA in accordance with Good Publication Practice (GPP3) guidelines and was funded by Moderna, Inc. The graphical abstract was made using a BioRender license. All experiments and studies were funded by Moderna.
Author contributions
Conceptualization, C.E.D., A.F.R., T.Y., D.L., R.K., N.J.M., A.C., and O.J.P.; methodology, C.E.D., A.F.R., M.J.M., Z.W., Y.-T.L., B.R.F., S.H., E.N., D.L., R.K., S.L., H.K., M.K., M.E.P., W.J.W., J.E.D., and S.K.L.; formal analysis, C.J.H., H.K., M.K., and I.L.R.; investigation, C.E.D., A.F.R., T.Y., M.J.M., Z.W., Y.-T.L., D.L., M.E.P., W.J.W., J.E.D., and S.K.L.; resources, B.R.F., S.H., E.N., S.L., M.E.P., W.J.W., N.J.M., A.C., and O.J.P.; writing – original draft, C.E.D. and D.L.; writing – review & editing, C.E.D., A.F.R., Y.-T.L., D.L., R.K., S.L., W.J.W., N.J.M., A.C., and O.J.P.; validation, A.F.R., T.Y., J.E.D., and S.K.L.; supervision, Y.-T.L., R.K., S.L., N.J.M., A.C., and O.J.P.; project administration, N.J.M., W.J.W., A.C., and O.J.P.
Declaration of interests
C.E.D., A.F.R., T.Y., M.J.M., Z.W., Y.-T.L., B.R.F., S.H., D.L., R.K., S.L., C.J.H., I.L.R., H.K., M.K., A.C., and O.J.P. are employees of and shareholders in Moderna, Inc. C.E.D. and O.J.P. are co-inventors on international patent WO 2022/212191 A1. E.N. was an employee of and shareholder in Moderna Inc. at the time of the study.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 1, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101253.
Contributor Information
Cailin E. Deal, Email: cailin.deal@modernatx.com.
Andrea Carfi, Email: andrea.carfi@modernatx.com.
Obadiah J. Plante, Email: obadiah.plante@modernatx.com.
Supplemental information
Data and code availability
All data supporting the findings of this study are found within the paper and its supplemental information. They are also available from the lead contact upon request.
References
- 1.Kyriakidis N.C., López-Cortés A., González E.V., Grimaldos A.B., Prado E.O. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines. 2021;6:28. doi: 10.1038/s41541-021-00292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaudhary N., Weissman D., Whitehead K.A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021;20:817–838. doi: 10.1038/s41573-021-00283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.August A., Attarwala H.Z., Himansu S., Kalidindi S., Lu S., Pajon R., Han S., Lecerf J.M., Tomassini J.E., Hard M., et al. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat. Med. 2021;27:2224–2233. doi: 10.1038/s41591-021-01573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayanan E., Falcone S., Elbashir S.M., Attarwala H., Hassett K., Seaman M.S., Carfi A., Himansu S. Rational design and in vivo characterization of mRNA-encoded broadly neutralizing antibody combinations against HIV-1. Antibodies. 2022;11:67. doi: 10.3390/antib11040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steffen U., Koeleman C.A., Sokolova M.V., Bang H., Kleyer A., Rech J., Unterweger H., Schicht M., Garreis F., Hahn J., et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020;11:120. doi: 10.1038/s41467-019-13992-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y., Jin L., Chen T. The effects of secretory IgA in the mucosal immune system. BioMed Res. Int. 2020;2020:2032057. doi: 10.1155/2020/2032057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertrand Y., Sánchez-Montalvo A., Hox V., Froidure A., Pilette C. IgA-producing B cells in lung homeostasis and disease. Front. Immunol. 2023;14:1117749. doi: 10.3389/fimmu.2023.1117749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandtzaeg P., Prydz H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature. 1984;311:71–73. doi: 10.1038/311071a0. [DOI] [PubMed] [Google Scholar]
- 9.Macpherson A.J., Geuking M.B., McCoy K.D. Homeland security: IgA immunity at the frontiers of the body. Trends Immunol. 2012;33:160–167. doi: 10.1016/j.it.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto M.L. Molecular mechanisms of multimeric assembly of IgM and IgA. Annu. Rev. Immunol. 2022;40:221–247. doi: 10.1146/annurev-immunol-101320-123742. [DOI] [PubMed] [Google Scholar]
- 11.Hausmann A., Böck D., Geiser P., Berthold D.L., Fattinger S.A., Furter M., Bouman J.A., Barthel-Scherrer M., Lang C.M., Bakkeren E., et al. Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site-specific bacterial PAMP expression. Mucosal Immunol. 2020;13:530–544. doi: 10.1038/s41385-019-0247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakkeren E., Huisman J.S., Fattinger S.A., Hausmann A., Furter M., Egli A., Slack E., Sellin M.E., Bonhoeffer S., Regoes R.R., et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature. 2019;573:276–280. doi: 10.1038/s41586-019-1521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings L.K., Dreifus J.E., Reichhardt C., Storek K.M., Secor P.R., Wozniak D.J., Hisert K.B., Parsek M.R. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep. 2021;34:108782. doi: 10.1016/j.celrep.2021.108782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards A., Baranova D., Mantis N.J. The prospect of orally administered monoclonal secretory IgA (SIgA) antibodies to prevent enteric bacterial infections. Hum. Vaccines Immunother. 2022;18:1964317. doi: 10.1080/21645515.2021.1964317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamane-Ohnuki N., Satoh M. Production of therapeutic antibodies with controlled fucosylation. mAbs. 2009;1:230–236. doi: 10.4161/mabs.1.3.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solá R.J., Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharmaceut. Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flintegaard T.V., Thygesen P., Rahbek-Nielsen H., Levery S.B., Kristensen C., Clausen H., Bolt G. N-glycosylation increases the circulatory half-life of human growth hormone. Endocrinology. 2010;151:5326–5336. doi: 10.1210/en.2010-0574. [DOI] [PubMed] [Google Scholar]
- 18.Li C.W., Lim S.O., Xia W., Lee H.H., Chan L.C., Kuo C.W., Khoo K.H., Chang S.S., Cha J.H., Kim T., et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer S., Nederend M., Jansen J.H.M., Reiding K.R., Jacobino S.R., Meeldijk J., Bovenschen N., Wuhrer M., Valerius T., Ubink R., et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. mAbs. 2016;8:87–98. doi: 10.1080/19420862.2015.1106658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rouwendal G.J., van der Lee M.M., Meyer S., Reiding K.R., Schouten J., de Roo G., Egging D.F., Leusen J.H., Boross P., Wuhrer M., et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. mAbs. 2016;8:74–86. doi: 10.1080/19420862.2015.1102812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Y., Zhang Y., Wang Z., Zhao W., Xu X., Chen X., Tan F., Sun Z., Huang B., Tian K. A therapeutic chimeric IgG/IgA expressed by CHO cells for oral treatment of PED in piglets. Front. Microbiol. 2022;13:1018748. doi: 10.3389/fmicb.2022.1018748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albaugh G.P., Dutta S.K., Iyengar V., Shami S., Lohani A., Sainz E., Kessie G., Nair P., Lagerholm S., Kamra A., et al. Identification of a native novel oncolytic immunoglobulin on exfoliated colon epithelial cells: A bispecific heterodimeric chimera of IgA/IgG Open. Open J. Prev. Med. 2020;10:126–150. [Google Scholar]
- 23.Li C., An X., Butt A.M., Zhang B., Zhang Z., Wang X., Huang Y., Zhang W., Zhang B., Mi Z., Tong Y. Construction of a chimeric secretory IgA and its neutralization activity against avian influenza virus H5N1. J. Immunol. Res. 2014;2014:394127. doi: 10.1155/2014/394127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michetti P., Mahan M.J., Slauch J.M., Mekalanos J.J., Neutra M.R. Monoclonal secretory immunoglobulin A protects mice against oral challenge with the invasive pathogen Salmonella typhimurium. Infect. Immun. 1992;60:1786–1792. doi: 10.1128/iai.60.5.1786-1792.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards A.F., Doering J.E., Lozito S.A., Varrone J.J., Willsey G.G., Pauly M., Whaley K., Zeitlin L., Mantis N.J. Inhibition of invasive salmonella by orally administered IgA and IgG monoclonal antibodies. PLoS Neglected Trop. Dis. 2020;14:e0007803. doi: 10.1371/journal.pntd.0007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiGiandomenico A., Warrener P., Hamilton M., Guillard S., Ravn P., Minter R., Camara M.M., Venkatraman V., Macgill R.S., Lin J., et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J. Exp. Med. 2012;209:1273–1287. doi: 10.1084/jem.20120033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borrok M.J., DiGiandomenico A., Beyaz N., Marchetti G.M., Barnes A.S., Lekstrom K.J., Phipps S.S., McCarthy M.P., Wu H., Dall'Acqua W.F., et al. Enhancing IgG distribution to lung mucosal tissue improves protective effect of anti-Pseudomonas aeruginosa antibodies. JCI Insight. 2018;3:e97844. doi: 10.1172/jci.insight.97844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y., He L., Lugano R., Zhang Y., Cao H., He Q., Chao M., Liu B., Cao Q., Wang J., et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight. 2021;6:e150861. doi: 10.1172/jci.insight.150861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lombana T.N., Rajan S., Zorn J.A., Mandikian D., Chen E.C., Estevez A., Yip V., Bravo D.D., Phung W., Farahi F., et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. mAbs. 2019;11:1122–1138. doi: 10.1080/19420862.2019.1622940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rifai A., Fadden K., Morrison S.L., Chintalacharuvu K.R. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J. Exp. Med. 2000;191:2171–2182. doi: 10.1084/jem.191.12.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov I.I., Atarashi K., Manel N., Brodie E.L., Shima T., Karaoz U., Wei D., Goldfarb K.C., Santee C.A., Lynch S.V., et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janeway C A., Travers P., M W. 5th edition Edition. Garland Science; 2001. Immunobiology: The Immune System in Health and Disease. [Google Scholar]
- 33.Castro-Dopico T., Clatworthy M.R. IgG and Fcgamma receptors in intestinal immunity and inflammation. Front. Immunol. 2019;10:805. doi: 10.3389/fimmu.2019.00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chintalacharuvu K.R., Chuang P.D., Dragoman A., Fernandez C.Z., Qiu J., Plaut A.G., Trinh K.R., Gala F.A., Morrison S.L. Cleavage of the human immunoglobulin A1 (IgA1) hinge region by IgA1 proteases requires structures in the Fc region of IgA. Infect. Immun. 2003;71:2563–2570. doi: 10.1128/IAI.71.5.2563-2570.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J., et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goetze A.M., Liu Y.D., Zhang Z., Shah B., Lee E., Bondarenko P.V., Flynn G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21:949–959. doi: 10.1093/glycob/cwr027. [DOI] [PubMed] [Google Scholar]
- 37.Ferrara C., Grau S., Jäger C., Sondermann P., Brünker P., Waldhauer I., Hennig M., Ruf A., Rufer A.C., Stihle M., et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider M., Al-Shareffi E., Haltiwanger R.S. Biological functions of fucose in mammals. Glycobiology. 2017;27:601–618. doi: 10.1093/glycob/cwx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H., Moldoveanu Z., Hall S., Brown R., Vu H.L., Novak L., Julian B.A., Tomana M., Wyatt R.J., Edberg J.C., et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Invest. 2008;118:629–639. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards A.F., Baranova D.E., Pizzuto M.S., Jaconi S., Willsey G.G., Torres-Velez F.J., Doering J.E., Benigni F., Corti D., Mantis N.J. Recombinant human secretory IgA induces salmonella typhimurium agglutination and limits bacterial Invasion into gut-associated lymphoid tissues. ACS Infect. Dis. 2021;7:1221–1235. doi: 10.1021/acsinfecdis.0c00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moor K., Diard M., Sellin M.E., Felmy B., Wotzka S.Y., Toska A., Bakkeren E., Arnoldini M., Bansept F., Co A.D., et al. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature. 2017;544:498–502. doi: 10.1038/nature22058. [DOI] [PubMed] [Google Scholar]
- 42.P Vaerman J., Lemaître-Coelho I.M., Limet J.N., et al. In: Hepatic transfer of polymeric IgA from plasma to bile in rats and other mammals: a survey. Strober W., Hanson L.A., Sell K., editors. Raven Press; 1982. [Google Scholar]
- 43.Snoeck V., Peters I.R., Cox E. The IgA system: a comparison of structure and function in different species. Vet. Res. 2006;37:455–467. doi: 10.1051/vetres:2006010. [DOI] [PubMed] [Google Scholar]
- 44.Alvarado G., Ettayebi K., Atmar R.L., Bombardi R.G., Kose N., Estes M.K., Crowe J.E., Jr. Human Monoclonal Antibodies That Neutralize Pandemic GII.4 Noroviruses. Gastroenterology. 2018;155:1898–1907. doi: 10.1053/j.gastro.2018.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen T.S., Pelletier M., Cheng L., Pennini M.E., Bonnell J., Cvitkovic R., Chang C.S., Xiao X., Cameroni E., Corti D., et al. Anti-LPS antibodies protect against Klebsiella pneumoniae by empowering neutrophil-mediated clearance without neutralizing TLR4. JCI Insight. 2017;2:e92774. doi: 10.1172/jci.insight.92774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forbes S.J., Eschmann M., Mantis N.J. Inhibition of Salmonella enterica serovar typhimurium motility and entry into epithelial cells by a protective antilipopolysaccharide monoclonal immunoglobulin A antibody. Infect. Immun. 2008;76:4137–4144. doi: 10.1128/iai.00416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forbes S.J., Martinelli D., Hsieh C., Ault J.G., Marko M., Mannella C.A., Mantis N.J. Association of a protective monoclonal IgA with the O antigen of Salmonella enterica serovar Typhimurium impacts type 3 secretion and outer membrane integrity. Infect. Immun. 2012;80:2454–2463. doi: 10.1128/iai.00018-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson J., Sorensen E.W., Mintri S., Rabideau A.E., Zheng W., Besin G., Khatwani N., Su S.V., Miracco E.J., Issa W.J., et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020;6:eaaz6893. doi: 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao L., Diedrich J.K., Ma Y., Wang N., Pauthner M., Park S.K.R., Delahunty C.M., McLellan J.S., Burton D.R., Yates J.R., Paulson J.C. Global site-specific analysis of glycoprotein N-glycan processing. Nat. Protoc. 2018;13:1196–1212. doi: 10.1038/nprot.2018.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu D., Wang S., Zhang J., Xiao W., Miao C.H., Konkle B.A., Wan X.F., Li L. Site-specific N- and O-glycosylation analysis of human plasma fibronectin. Front. Chem. 2021;9:691217. doi: 10.3389/fchem.2021.691217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varki J., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Mohnen D., Kinoshita T., Packer N.H., Prestegard J.H., et al. Cold Spring Harbor (NY): Laboratory Press; 2015. Essentials of Glycobiology. [PubMed] [Google Scholar]
- 53.Wood S.N. Thin plate regression splines. J. Roy. Stat. Soc. B. 2003;65:95–114. doi: 10.1111/1467-9868.00374. [DOI] [Google Scholar]
- 54.Bürkner P.-C. brms: An R Package for Bayesian Multilevel Models Using Stan. J. Stat. Software. 2017;80:1–28. doi: 10.18637/jss.v080.i01. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are found within the paper and its supplemental information. They are also available from the lead contact upon request.