

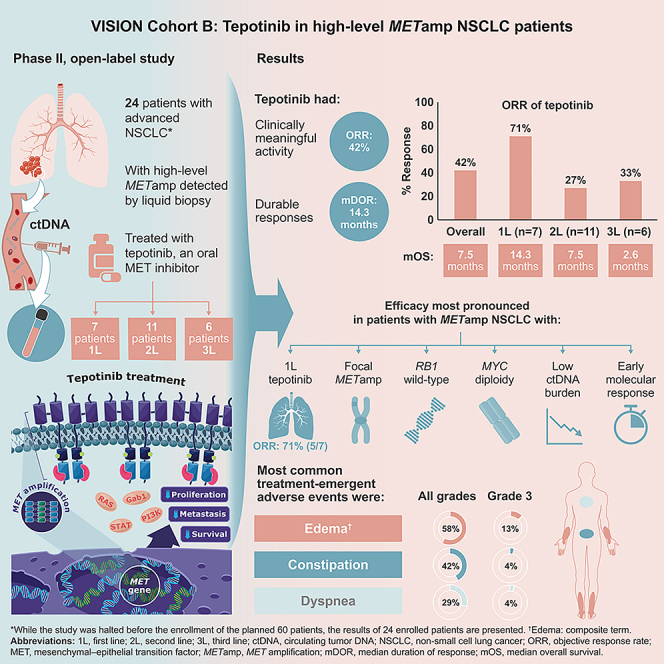
Summary
High-level MET amplification (METamp) is a primary driver in ∼1%–2% of non-small cell lung cancers (NSCLCs). Cohort B of the phase 2 VISION trial evaluates tepotinib, an oral MET inhibitor, in patients with advanced NSCLC with high-level METamp who were enrolled by liquid biopsy. While the study was halted before the enrollment of the planned 60 patients, the results of 24 enrolled patients are presented here. The objective response rate (ORR) is 41.7% (95% confidence interval [CI], 22.1–63.4), and the median duration of response is 14.3 months (95% CI, 2.8–not estimable). In exploratory biomarker analyses, focal METamp, RB1 wild-type, MYC diploidy, low circulating tumor DNA (ctDNA) burden at baseline, and early molecular response are associated with better outcomes. Adverse events include edema (composite term; any grade: 58.3%; grade 3: 12.5%) and constipation (any grade: 41.7%; grade 3: 4.2%). Tepotinib provides antitumor activity in high-level METamp NSCLC (ClinicalTrials.gov: NCT02864992).
Keywords: biomarkers, MET amplification, MET inhibitor, non-small cell lung cancer, tepotinib
Graphical abstract
Highlights
-
•
High-level MET amplification (METamp) is a primary driver in ∼1%–2% of NSCLCs
-
•
VISION Cohort B evaluates tepotinib in 24 patients with high-level METamp NSCLC
-
•
Tepotinib shows antitumor activity: ORR is 41.7%, and median DOR is 14.3 months
-
•
Safety is manageable, with mostly mild/moderate AEs and no new safety signals
High-level MET amplification (METamp) is a primary driver in ∼1%–2% of non-small cell lung cancers (NSCLCs). Le et al. report that tepotinib provides antitumor activity in patients with high-level METamp NSCLC in VISION trial Cohort B. Tepotinib is a promising option for these patients, who urgently require new treatments.
Introduction
In non-small cell lung cancers (NSCLCs), up to 5% of the tumors harbor MET amplification.1,2,3 Depending on the methods and cutoff values used, high-level MET amplification can be defined as a MET:CEP7 ratio ≥2.0 or ≥2.2, or as a MET gene copy number (GCN) ≥6 or ≥10, as identified by fluorescence in situ hybridization (FISH) or next-generation sequencing (NGS) on tissue biopsies.4,5,6,7,8 Studies have shown that the stringent criterion of MET GCN ≥10 on tissue biopsy selects ∼1%–2% of NSCLCs, which rarely harbor other oncogenic drivers.6,7,8,9 Furthermore, treatment with anti-MET therapies in those patients with high-level MET amplification NSCLC induced clinical response,10 indicating that high-level MET amplification is a primary oncogenic driver for these NSCLCs.1,2,3
MET amplification is an independent poor prognostic factor,6,11,12,13,14 which defines an aggressive, treatment-resistant malignancy with a very short median overall survival (OS) of 4 months6,9 Despite expression (≥1%) of programmed death-ligand 1 (PD-L1) in 85% of lung adenocarcinomas with MET amplification,15 outcomes with immunotherapies are poor.9,16,17 Therefore, patients with high-level MET amplification NSCLC have an unmet need for better treatment options.
Although no targeted therapy is approved specifically for metastatic NSCLC with high-level MET amplification, MET tyrosine kinase inhibitors (TKIs) have demonstrated promising efficacy3,10,18 and are recommended in the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines).19 Tepotinib, a highly selective and potent MET TKI,20 is approved in multiple countries for treatment of MET exon 14 (METex14) skipping NSCLC21 based on the clinical activity demonstrated in the phase 2 VISION trial.22,23 In preclinical models of NSCLC with MET amplification, tepotinib induced complete regression of cell-line- and patient-derived xenografts, including after orthotopic implantation in the brain.24,25 In addition, antitumor activity has also been observed with tepotinib plus gefitinib or osimertinib in patients with epidermal growth factor receptor (EGFR)-mutant NSCLC and MET amplification.26,27,28
Cohort B of the phase 2 VISION trial evaluated tepotinib in patients with advanced NSCLC with high-level MET amplification as detected by a liquid biopsy assay. The MET GCN cutoff in liquid biopsy was chosen to be ≥2.5, which selects ∼1.5%–2% of NSCLCs, corresponding to the same fraction of patients with high-level MET amplification identified using a MET GCN cutoff of ≥10 in tissue biopsies.8,9,29 Tumors with EGFR, ALK, or METex14 skipping oncogenic alterations were excluded, further ensuring the enrollment of a population with MET amplification as the primary driver. Clinical efficacy, safety, and exploratory biomarker analyses were performed.
Results
Patients
Among all patients prescreened using the Guardant360 liquid biopsy assay (Guardant Health, Redwood City, CA, USA) for molecular eligibility, 70/3,205 (2.2%) (with evaluable test results) were positive for high-level MET amplification and negative for METex14 skipping (Figure S1). Baseline tissue samples were not mandatory and were only available in six patients, of which four indicated the absence of METex14 skipping alteration and two were not evaluable. Thirty-two patients were further screened for enrollment, and 24 were treated between September 2018 and January 2020.
The median age was 63.4 years (Table 1). Most patients were male (87.5%), current/former smokers (87.5%), and had Eastern Cooperative Oncology Group performance status (ECOG PS) 1 (87.5%). Tepotinib was administered as first-, second-, and third-line treatment in seven (29.2%), 11 (45.8%), and six (25.0%) patients, respectively. Ten patients (41.7%) had prior immunotherapy, with a best response of partial response (PR) in only one patient (10.0%).
Table 1.
Baseline characteristics
| Characteristic | Overall (n = 24) | By line of therapy |
||
|---|---|---|---|---|
| First line (n = 7) | Second line (n = 11) | Third line (n = 6) | ||
| Male, n (%) | 21 (87.5) | 7 (100.0) | 10 (90.9) | 4 (66.7) |
| Median age, years (range) | 63.4 (38–73) | 66.8 (59–71) | 60.5 (38–73) | 64.2 (61–70) |
| Race, n (%) | ||||
| White | 17 (70.8) | 5 (71.4) | 7 (63.6) | 5 (83.3) |
| Asian | 7 (29.2) | 2 (28.6) | 4 (36.4) | 1 (16.7) |
| Current/former smoker, n (%) | 21 (87.5) | 6 (85.7) | 10 (90.9) | 5 (83.3) |
| ECOG PS, n (%) | ||||
| 0 | 3 (12.5) | 1 (14.3) | 2 (18.2) | 0 (0) |
| 1 | 21 (87.5) | 6 (85.7) | 9 (81.8) | 6 (100) |
| Median tumor load of target lesionsa (IRC), mm (range) | 95.6 (26.9–231.9) | 55.0 (26.9–168.8) | 99.6 (66.5–231.9) | 102.1 (31.4–160.3) |
| Histology, n (%) | ||||
| Adenocarcinoma | 16 (66.7) | 6 (85.7) | 7 (63.6) | 3 (50.0) |
| NOSb | 4 (16.7) | 1 (14.3) | 3 (27.3) | 0 (0) |
| Neuroendocrine carcinomac | 3 (12.5) | 0 (0) | 1 (9.1) | 2 (33.3) |
| Squamous cell carcinoma | 1 (4.2) | 0 (0) | 0 (0) | 1 (16.7) |
| Median time since initial diagnosis, months (range) | 5.5 (0.1–62.6) | 0.8 (0.1–7.1) | 6.2 (0.2–62.6) | 8.3 (1.0–29.4) |
| Brain metastases at baseline, n (%)d | 2 (8.3) | 0 (0) | 2 (18.2) | 0 (0) |
| MET GCN, median (range) | 2.9 (2.5–26.9) | 3.6 (2.5–10.2) | 2.8 (2.5–26.9) | 2.9 (2.5–4.0) |
ECOG PS, Eastern Cooperative Oncology Group performance status; GCN, gene copy number; IRC, independent review committee; NOS, not otherwise specified; NSCLC, non-small cell lung cancer.
Sum of longest diameters for non-nodal lesions and short axes for nodal lesions.
Comprising NOS (n = 2), NSCLC (n = 1), and non-squamous NSCLC (n = 1).
Comprising large-cell neuroendocrine carcinoma (n = 2) and carcinoma with neuroendocrine morphology (n = 1).
Brain metastases were non-target lesions.
The study was halted early before the enrollment of the planned number of 60 patients because of the high rate of early progression (during the first 3 months of tepotinib treatment) in eight out of the 24 enrolled patients. These early progressions likely reflected the patients’ poor prognosis and the aggressive nature of the disease. However, the halting of the study was to allow for the full analysis of the 24 patients to best identify those patients who were potentially most likely to benefit from tepotinib and to minimize risks.
Efficacy in the overall population
Objective response rate (ORR) by independent review committee (IRC) was 41.7% (95% confidence interval [CI], 22.1–63.4), and the clinical benefit rate (CBR; defined as complete response [CR] + PR + stable disease [SD]) was 45.8% (95% CI, 25.6–67.2) (Table 2). The best overall response by IRC was CR in one patient (4.2%), PR in nine patients (37.5%), SD in one patient (4.2%), and progressive disease (PD) in five patients (20.8%). Of eight patients (33.3%) with a best response of not evaluable (NE), four discontinued before the response was confirmed due to investigator-assessed PD, three discontinued due to unrelated adverse events (AEs), and one discontinued due to consent withdrawal. Tumor shrinkage was attained in 16 patients (66.7%; Figures 1A and 1B). Responses were rapid: median time to response was 1.4 months (range, 1.3–11.1), and 7/10 responses occurred by the first assessment.
Table 2.
Efficacy outcomes in the overall population and according to line of therapy
| Outcomea | Overall (n = 24) | By line of therapy |
||
|---|---|---|---|---|
| First line (n = 7) | Second line (n = 11) | Third line (n = 6) | ||
| Best overall response rate, n (%) | – | – | – | – |
| CR | 1 (4.2) | 1 (14.3) | 0 (0) | 0 (0) |
| PR |
9 (37.5) | 4 (57.1) | 3 (27.3) | 2 (33.3) |
| SD |
1 (4.2) | 0 (0) | 1 (9.1) | 0 (0) |
| PD |
5 (20.8) | 1 (14.3) | 3 (27.3) | 1 (16.7) |
| NE |
8 (33.3) | 1 (14.3) | 4 (36.4) | 3 (50.0) |
| ORR, n (%) [95% CI] | 10 (41.7) [22.1–63.4] | 5 (71.4) [29.0–96.3] | 3 (27.3) [6.0–61.0] | 2 (33.3) [4.3–77.7] |
| CBR, n (%) [95% CI] | 11 (45.8) [25.6–67.2] | 5 (71.4) [29.0–96.3] | 4 (36.4) [10.9–69.2] | 2 (33.3) [4.3–77.7] |
| DOR, median (95% CI), months | 14.3 (2.8–ne) | 14.3 (2.8–ne) | ne (ne–ne) | ne (3.2–ne) |
| OS, median (95% CI), months | 7.5 (4.0–15.6) | 14.3 (4.0–ne) | 7.5 (1.9–24.0) | 2.6 (0.6–ne) |
CBR, clinical benefit rate; CI, confidence interval; CR, complete response; DOR, duration of response; IRC, independent review committee; ne, not estimable; NE, not evaluable; ORR, objective response rate; OS, overall survival; PD, progressive disease; PR, partial response; SD, stable disease.
Best overall response, ORR, CBR, and DOR are per IRC assessment.
Figure 1.

Objective response and OS by IRC
(A) Waterfall plot showing percent change in sum of longest diameters between baseline and best post-baseline assessment in the overall population. Labels indicate BOR. Three patients were excluded due to lack of post-baseline assessments, and five patients had a BOR of NE due to treatment discontinuation before response was confirmed.
(B) Spider plot showing percentage of change in sum of longest diameters at each assessment in the overall population. Solid lines connect on-treatment assessments; dotted lines connect the last on-treatment assessment, with the cross indicating treatment discontinuation as well as any post-treatment assessments. Three patients were excluded due to lack of post-baseline assessments.
(C) Kaplan-Meier plot of OS according to line of therapy.
BOR, best overall response; CI, confidence interval; CR, complete response; IRC, independent review committee; ne, not estimable; NE, not evaluable; OS, overall survival; PD, progressive disease; PR, partial response; SD, stable disease.
Median follow-up was 26.8 months (95% CI, 20.4–not estimable [ne]). Median duration of response (DOR) was 14.3 months (95% CI, 2.8–ne) (Figure 2A), and median progression-free survival (PFS) was 4.2 months (95% CI, 1.4–15.6). PFS events were recorded for 14 patients (58.3%), of whom nine (37.5%) had early progression/death during the first 3 months. At the data cutoff, 18 patients (75.0%) had died, and median OS was 7.5 months (95% CI, 4.0–15.6) (Figure S2A).
Figure 2.

DOR by independent review committee
(A and B) Kaplan-Meier plots showing DOR in the overall population (A) and DOR according to line of therapy (B). CI, confidence interval; DOR, duration of response; ne, not estimable.
Median duration of tepotinib treatment was 3.6 months (range, 0.1–26.8). Treatment duration was ≥12 months in five patients (20.8%) and ≥24 months in two patients (8.3%), both of whom had treatment ongoing at the data cutoff (August 20, 2021; Figure S3). One of these patients is still receiving tepotinib as of June 2023. The other patient discontinued tepotinib due to edema, after which the edema resolved, and the patient's tumor continues to respond, without additional treatment. Six patients (25.0%) received post-study anticancer therapy, including chemotherapy (n = 6; 25.0%) and immunotherapy (n = 3; 12.5%), specifically chemotherapy (carboplatin, cisplatin, docetaxel, paclitaxel, pemetrexed, or tegafur), immunotherapy (atezolizumab, nivolumab, pembrolizumab), and antiangiogenic therapy (ramucirumab, bevacizumab).
Efficacy according to therapy line
Patients treated with tepotinib in the first-line setting attained an ORR by IRC of 71.4% (5/7 patients; 95% CI, 29.0–96.3; Table 2), a median (95% CI) DOR of 14.3 months (2.8–ne; Figure 2B), and a median (95% CI) OS of 14.3 months (4.0–ne; Figure 1C). In second and third lines, respectively, the ORRs were 27.3% (3/11 patients; 95% CI, 6.0–61.0) and 33.3% (2/6 patients; 95% CI, 4.3–77.7), the median DOR was not estimable due to the low number of patients (events recorded in 0/3 and 1/2 patients), and the median OSs (95% CI) were 7.5 (1.9–24.0) and 2.6 months (0.6–ne; Figure 1C).
Safety
Treatment-emergent AEs (TEAEs; Table 3) were reported at any grade in 23 patients (95.8%), with grade ≥3 in 16 (66.7%). Treatment-related AEs (TRAEs) were reported in 17 (70.8%) patients, with grade ≥3 in seven (29.2%). TEAEs led to dose reduction in five patients (20.8%), treatment interruption in 12 patients (50.0%), and permanent discontinuation in five patients (20.8%; none were TR; Table S1). Serious TEAEs were reported in 13 patients (54.2%; TR, n = 2 [8.3%]) (Table S1). Seven patients had fatal TEAEs, including disease progression recorded as an AE (n = 3, 12.5%) and respiratory failure (n = 2; 8.3%), none of which were TR.
Table 3.
TEAEs reported at any grade in ≥10% of patients, irrespective of causality
| TEAE | Patients, n (%) (n = 24) |
|
|---|---|---|
| All grades | Grade 3a | |
| Edema (composite term) | 14 (58.3) | 3 (12.5) |
| Peripheral edema | 12 (50.0) | 2 (8.3) |
| Generalized edema | 5 (20.8) | 2 (8.3) |
| Edema (preferred term) | 5 (20.8) | 1 (4.2) |
| Constipation | 10 (41.7) | 1 (4.2) |
| Dyspnea | 7 (29.2) | 1 (4.2) |
| Asthenia | 5 (20.8) | 1 (4.2) |
| Blood creatinine increased | 5 (20.8) | 0 (0) |
| Diarrhea | 5 (20.8) | 0 (0) |
| Hypoalbuminemia | 5 (20.8) | 2 (8.3) |
| Nausea | 4 (16.7) | 1 (4.2) |
| Abdominal pain | 3 (12.5) | 1 (4.2) |
| Alanine aminotransferase increased | 3 (12.5) | 1 (4.2) |
| Anemia | 3 (12.5) | 1 (4.2) |
| Aspartate aminotransferase increased | 3 (12.5) | 0 (0) |
| Cough | 3 (12.5) | 0 (0) |
| Disease progression | 3 (12.5) | 0 (0) |
| Hypoproteinemia | 3 (12.5) | 0 (0) |
| Pneumonia | 3 (12.5) | 1 (4.2) |
| Productive cough | 3 (12.5) | 0 (0) |
| Pyrexia | 3 (12.5) | 0 (0) |
| Vomiting | 3 (12.5) | 0 (0) |
TEAE, treatment-emergent adverse event.
For the events shown, there were no grade 4 TEAEs, and the only grade 5 TEAEs were disease progression (n = 3; 12.5%) and pneumonia (n = 1; 4.2%), which were unrelated to treatment.
HRQoL
Health-related quality of life (HRQoL) was evaluated using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core-30 and Lung Cancer-13 (EORTC QLQ-C30 and QLQ-LC13) and EuroQol 5-dimension 5-level (EQ-5D-5L) questionnaires, which had high completion rates (Table S2). EQ-5D-5L visual analog scale and EORTC QLQ-C30 global health scores showed stability of overall HRQoL (Figures S4A and S4B; Table S3). EORTC QLQ-LC13 symptom scores indicated early improvement in chest pain and stability of dyspnea and cough (Figure S4C).
Exploratory analysis of clinical characteristics associated with clinical benefit
Exploratory analyses were conducted to identify baseline characteristics (Figure S5) and biomarkers (Figure 3; Table 4) associated with clinical benefit.
Figure 3.

Association between response to treatment and clinical characteristics and biomarkers
Co-occurring mutations were most commonly detected in TP53, NF1, ARID1A, MET, PDGFRA, and RB1. The genes most frequently co-amplified with MET were CDK6, EGFR, BRAF, CCNE1, and PDGFRA. ADC, adenocarcinoma; BOR, best overall response; CR, complete response; ctDNA, circulating tumor DNA; DOR, duration of response; IRC, independent review committee; NE, not evaluable; NOS, not otherwise specified; PD, progressive disease; PR, partial response; Q3, SCC, squamous cell carcinoma; SD, stable disease; SOLD, sum of target lesion diameters.
Table 4.
ORR by IRC, DOR, and OS according to MET amplification focality, RB1 mutation, MYC amplification, and ctDNA burden at baseline, and early molecular response at 6–8 weeks
 |
CI, confidence interval; ctDNA, circulating tumor DNA; DOR, duration of response; IRC, independent review committee; ne, not estimable; ORR, overall response rate, OS, overall survival; Q3, third quartile.
aA total of 18 patients were evaluable for early molecular response, defined as disappearance of MET amplification in ctDNA at 6–8 weeks.
bFive patients (adenocarcinoma, n = 3; not otherwise specified histology, n = 2) had a total of six RB1 mutations (nonsense, n = 4; splice site, n = 2), all expected to cause loss of function.
Eleven patients had clinical benefit, as defined by best overall response by IRC of SD or better (i.e., CR + PR + SD). These patients attained a median OS of 24.0 months (95% CI, 8.3–ne) (Figure S2B) and clinically meaningful chest pain improvement (Table S3). Clinical benefit was reported in 52.4% (11/21) of male patients, 50% (7/14) of younger patients (<65 years), 57.1% (4/7) of Asian patients, and 62.5% (10/16) of patients with adenocarcinoma (Figure S5). The median tumor load (defined as the sum of lesion diameters by IRC) in the overall population was 95.6 mm (Table 1) but was numerically lower in patients with clinical benefit than patients without clinical benefit (91.6 versus 103.7 mm) (Figure 3).
Exploratory biomarker analyses
In this trial, circulating tumor DNA (ctDNA) was collected at baseline, week 6, and end of treatment and analyzed using the Guardant360 assay. We evaluated associations between clinical outcomes and baseline biomarkers and on-treatment early molecular response, along with potential resistance mechanisms. Baseline biomarker profiles, including co-occurring mutations and co-amplified genes, were available for all patients (Figure 3). Five patients had other MET mutations (G1144A, G1280R, Q1067fs, D414_R417delinsG, and N680H), none of which were known to cause oncogenic MET activation or resistance to MET inhibitors. Focal MET amplification was defined by a co-amplification ≤1 of three other chromosome 7 genes (EGFR, BRAF, and CDK6). A total of 14 patients (58.3%) had focal MET amplification, which was potentially associated with better ORR and OS than non-focal MET amplification (Figure 3; Table 4). Analysis comparing the frequency of baseline biomarker alterations between patients with or without benefit from tepotinib also identified RB1 and MYC as biomarkers, although patient numbers are small (Figure S6). Outcomes were better in patients with RB1 wild-type (n = 19) versus mutant (n = 5) status and in patients with MYC diploidy (n = 18) versus amplification (n = 6) (Table 4).
Median baseline ctDNA burden, defined by the maximum baseline variant allele fraction of any cancer-specific alteration, was 10.7% (interquartile range [IQR], 7.5–26.0). Low ctDNA burden (whether defined as ≤median or ≤third quartile [Q3]) was associated with greater efficacy (Table 4). Due to the sample size, statistical significance was not assessed for ORR or OS for the biomarker subset analysis.
Eighteen patients had matched baseline and on-treatment samples, of whom 14 (77.8%) attained an early molecular response, as defined by undetectable MET amplification 6–8 weeks after tepotinib first dose. Patients with early molecular response had a high clinical response rate (ORR, 71.4% [5/7]), whereas those with MET amplification persistence in ctDNA at 6–8 weeks showed a lack of clinical response. Of nine patients with available end-of-treatment biomarker profiles, two (22.2%) showed emergence of MET kinase domain mutations (D1228H/N/Y, Y1230C/H, and D1231N in one patient, and D1213N, D1228N/H, and Y1230H in the other). Both patients attained PR, with PFS >4 months, and showed re-emergence of MET amplification at the end of treatment.
Discussion
In this study, tepotinib provided antitumor activity in patients with NSCLC with high-level MET amplification detected by liquid biopsy: ORR was 41.7%, CBR was 45.8%, and median DOR was 14.3 months. The Cohort B data provided further evidence to support that high-level MET amplification is an actionable driver in NSCLC. Tepotinib safety was manageable, with mostly mild/moderate AEs and no discontinuations due to TRAEs, and consistent with that seen in patients with METex14 skipping,22,23,30 with no new safety signals.
Eight patients with high-level MET amplification NSCLC had rapid progression, underlying that it is an independent poor prognostic factor. The planned sample size for Cohort B of the VISION trial was 60 patients. However, Cohort B was halted early due to the high rate of early progression in these eight patients, leading to the early stopping of enrollment at 24 patients. In Cohort B, 13 molecularly eligible patients had clinical deterioration that prevented enrollment (Figure S1), and 8 of the 24 enrolled patients discontinued treatment due to PD during the first 3 months of treatment. This high rate of early progression led to the decision of halting enrollment at N = 24 for full analysis to identify patients who can potentially derive the most benefit from tepotinib. This early progression observation is most likely reflecting the underlying aggressive nature of MET amplification NSCLC, as similar results were reported in crizotinib and capmatinib studies.3,10 Our liquid biopsy ctDNA and tumor load analyses confirmed that VISION Cohort B patients (MET amplification) had poorer baseline prognostic factors than VISION Cohort A and C patients (METex14 skipping, ctDNA cohort only), with higher median tumor load (95.6 versus 68.0 mm) and greater prevalence of ECOG PS 1 (88% versus 76%).31 Tumor load and ctDNA burden were also higher relative to other advanced lung cancer studies.32,33,34 Lower tumor load and ctDNA burden were associated with better outcomes. Due to the poor prognosis of patients with this disease, it is important that an effective treatment is given in the first-line setting. In VISION Cohort B, efficacy appeared most pronounced in the first-line setting, with a notably high ORR of 71.4% (5/7) and a long median DOR (14.3 months). The present analysis further supports the National Comprehensive Cancer Network (NCCN) recommendation of tepotinib as a treatment option for patients with high-level MET amplification metastatic NSCLC,19 which was based on the analysis of this cohort.35
The VISION Cohort B was enrolled solely based on liquid biopsy for detecting MET amplification. As the copy number gain of MET gene is a continuous variable, the choice of cutoff is particularly important to identify the appropriate patient population most likely to respond to a MET inhibitor. Using liquid biopsies with a MET GCN ≥2.5, high-level MET amplification was detected in 2.2% (70/3,205) of the patients with NSCLC who were prescreened for VISION Cohort B. This finding corresponds to the reported high-level MET amplification occurrence of ∼1%–2% of NSCLCs using tissue biopsies with a MET GCN ≥10.7,9 In VISION Cohort B, tepotinib was an effective treatment, which further supports our current knowledge that high-level MET amplification is an actionable driver in NSCLC and that those tumors respond to a MET inhibitor.
It has been widely accepted that liquid biopsy is more convenient and less invasive compared with tissue biopsy and that it enables molecular testing even when tumor tissue is unavailable.36 Considering quick laboratory turnaround (median of 10 days in the VISION trial)22 alongside simple operational requirements for sample collection, liquid biopsy enables timely initiation of targeted therapy for this aggressive subtype. Furthermore, liquid biopsy also allows longitudinal monitoring of molecular response. We observed association between early molecular response and clinical response, which adds to the growing evidence supporting a role for liquid biopsy in serial monitoring of response and resistance, with a view toward refining the therapeutic approach to improve outcomes.37
While liquid biopsies have many merits for clinical practice, they also present several challenges. Different thresholds were applied in tissue- as well as liquid-biopsy-based assays for claiming the presence of MET amplification. With the Guardant360 assay, MET plasma GCNs as low as 2.2 were applied to define MET amplification.38 In VISION Cohort B, a MET GCN cutoff of ≥2.5 was used to be stringent and to select patients with NSCLC with a high likelihood of deriving benefit from MET inhibition. These differences in defining MET amplification need to be considered when interpreting data from different studies and applying the findings to clinical practice. Second, the detection of cancer-specific alterations in liquid biopsies is less sensitive compared with tissue-based testing.39 This is also true for MET amplification detection rate by ctDNA versus tissue samples, screened in the TATTON, SAVANNAH, ORCHARD, and INSIGHT 2 studies.40,41,42,43 The positive percentage agreement (PPA) of MET amplification detection between tissue and liquid biopsy can vary between 23% and 67% depending on factors such as methods used and the quality of the sample,41,42,43,44,45 and tissue biopsy should be considered after a negative liquid biopsy result for detecting missed alterations.43,44,45,46 Third, liquid biopsy positivity requires adequate ctDNA shedding, which is usually associated with larger tumor burden.47 In particular, detection of gene amplification is dependent on a high ctDNA fraction in circulation.48,49 Therefore, ctDNA-based analysis may select a poorer prognostic group of patients compared with tissue-based screening. This is supported by the associations of higher ctDNA burden with poorer outcomes and/or tumor load in our trial as well as studies in other oncogene-driven subtypes.31,50,51,52 Nonetheless, the use of liquid biopsies offers advantages over tissue biopsies in terms of convenience, accessibility, and being less invasive.53,54 VISION Cohort B confirmed that liquid biopsy can identify NSCLC with high-level MET amplification and that those patients could benefit from MET-targeted therapy.
The ORR and DOR with tepotinib compare favorably with data from crizotinib and capmatinib trials in NSCLC with high-level MET amplification by FISH.3,10 In PROFILE-1001, crizotinib provided an ORR of 38.1% and a median DOR of 5.2 months in 21 patients with a MET:CEP7 ratio ≥4.0, of whom three were treatment naive.3 In patients with MET GCN ≥10 in the GEOMETRY mono-1 trial of capmatinib, ORR was 40% in first line (n = 15) with a median DOR of 7.5 months, and in second or later lines (n = 69), the ORR was 29% with a median DOR of 8.3 months10 Tepotinib, crizotinib, and capmatinib have all consistently demonstrated benefit for this population and are recommended treatment options for high-level MET amplification metastatic NSCLC in NCCN Guidelines.19
With the observation that some patients progressed early and rapidly, but some other patients sustained benefit from tepotinib, we performed exploratory analyses integrating both a tumor’s clinical characteristics and biomarkers. While patient numbers are small, we observed that baseline MYC diploidy and RB1 wild-type status were associated with better outcomes with tepotinib, which is consistent with the function of MYC and RB1 as signal transducers downstream of MET.55 Prior studies have implicated MYC alterations in primary or acquired resistance to other MET inhibitors.56,57,58,59 Interestingly, RB1 loss and MYC copy number gain were also negative clinical predictors for EGFR-mutant NSCLC, both in the adjuvant setting59 and in the metastatic resistant setting with an association of transformation to small cell lung cancer.60,61,62 Acquired MET kinase domain mutations identified in two patients at the end of treatment are known type 1 MET-inhibitor-resistance mechanisms63 and are reported here for the first time as resistance mechanisms in MET amplification NSCLC with MET TKI treatment.
In conclusion, tepotinib demonstrated antitumor activity in NSCLC with high-level MET amplification. Tepotinib is a promising option for patients with high-level MET amplification as a primary driver who have exceptionally poor outcomes with current standard-of-care therapies6 and urgently require new treatments.
Limitations of the study
Study limitations include the halt of enrollment to investigate predictors of tepotinib benefit (which limited the sample size) and lack of histology selection. Furthermore, exploratory biomarker analyses were limited to ctDNA and did not include tumor tissue assessments. Nonetheless, the analyses presented herein provide valuable insights that can inform the development of effective treatment strategies for this population.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Blood samples | Participating study centers | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Tepotinib | the healthcare business of Merck KGaA, Darmstadt, Germany | N/A |
| Critical commercial assays | ||
| Guardant360® | Guardant Health, Redwood City, CA, USA | N/A |
| Software and algorithms | ||
| Statistical Analysis System, windows version 9.2 or higher | SAS Institute, Cary, NC, USA | RRID: SCR_008567 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andreas Johne (andreas.johne@emdgroup.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Subject to the healthcare business of Merck KGaA, Darmstadt, Germany, Data Sharing Policy, data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental models and study participant details
VISION (ClinicalTrials.gov, NCT02864992) evaluated tepotinib for treatment of advanced non-small cell lung cancer (NSCLC) with MET alterations. We report results from Cohort B, which enrolled 24 patients with high-level MET amplification. Cohorts A and C enrolled patients with MET exon 14 (METex14) skipping, as reported elsewhere.22,23 Patients were aged ≥18 years and had Eastern Cooperative Oncology Group performance status (ECOG PS) 0–1, histologically/cytologically confirmed, measurable, locally advanced/metastatic NSCLC with MET amplification, and 0–2 prior treatment lines. Exclusion criteria were: symptomatic brain metastases with neurologic instability; EGFR, ALK, or METex14 skipping alterations (other MET mutation types were allowed); unresolved Grade ≥2 toxicity; prior hepatocyte growth factor- or MET-targeted therapy; and inadequate organ function.
Cohort B was introduced in protocol v5 (May 10, 2018) and used the same liquid biopsy assay and prescreening procedures as Cohort A.22 MET amplification was centrally evaluated in circulating tumor DNA (ctDNA) from freshly collected plasma samples using a 73-gene NGS-based assay (Guardant360; Guardant Health, Redwood City, CA, USA). Guardant360 is a liquid biopsy (ctDNA) method allowing for comprehensive molecular analysis. A list of the 73 genes that Guardant360 analyses is shown in Table S4, which includes analyses of point mutations, indels, amplifications and fusions. The Guardant360 lower limit of MET gene copy number (GCN) gain was defined as ≥2.2. In the VISION Cohort B, criteria of MET GCN ≥2.5 was used for molecular selection, which represents a highly stringent selection criterion identifying the top 1.5%–2% of MET-amplified NSCLCs.29
The study complied with the Declaration of Helsinki, International Council on Harmonisation Good Clinical Practice, local laws and regulatory requirements. Independent Ethics Committees or Institutional Review Boards approved the protocol. Patients provided written informed consent.
Method details
Study procedures and endpoints
VISION is a multicohort, single-arm, phase 2 trial. Patients received tepotinib 500 mg (450 mg active moiety), orally, once daily, until disease progression (PD), intolerable toxicity or consent withdrawal. Tumor assessments were conducted by computed tomography or magnetic resonance imaging at baseline, every 6 weeks during the first 9 months, and every 12 weeks thereafter. Response was evaluated by an independent review committee (IRC) and investigators according to Response Evaluation Criteria in Solid Tumors v1.1. Objective responses were confirmed ≥4 weeks after response was first observed.
Health-related quality of life (HRQoL) was evaluated using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core-30 and Lung Cancer-13 (EORTC QLQ-C30 and QLQ-LC13) and EuroQol 5-dimension 5-level (EQ-5D-5L) questionnaires. Adverse events (AEs) were assessed for severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03.
The primary endpoint was confirmed objective response by IRC. Secondary endpoints included objective disease control, duration of response (DOR), progression-free survival (PFS), overall survival (OS), HRQoL, and safety.
Biomarker assessments
Exploratory biomarker analyses were conducted in blood samples using the Guardant360 assay. Focal MET amplification was defined by co-amplification of ≤1 of three other chromosome 7 genes (EGFR, BRAF and CDK6). ctDNA burden was defined as the maximum baseline variant allele fraction of any cancer-specific alteration among all analyzed genes in each patient and was dichotomized at the median or third quartile (Q3) in separate analyses. Early molecular response was defined as undetectable MET amplification after 6–8 weeks after the first dose of tepotinib (i.e., in Week 6 or, if the patient discontinued after ≤8 weeks, end-of-treatment samples).
Quantification and statistical analysis
The trial targeted an objective response rate (ORR) by IRC of 40%–50%, with a lower limit of the corresponding 95% confidence interval (CI) of >20% across therapy lines. Enrollment of 60 patients would provide a maximum 95% CI width of 26.4% across the target ORR range. The protocol defined an early futility analysis requiring an ORR of ≥25% for continuation. While this target was reached and the trial continued, early discontinuation in a subset of patients prompted a halt of enrollment at 24 patients and longer follow-up to investigate predictors of tepotinib benefit.
The data cutoff was August 20, 2021. Efficacy and safety were analyzed descriptively in patients who received ≥1 tepotinib dose. Objective response and disease control were summarized as rates with two-sided exact Clopper–Pearson 95% CIs. Time-dependent endpoints were analyzed using Kaplan–Meier methods. Changes from baseline in HRQoL scores were summarized as empirical means and, in analyses based on an earlier data cutoff (February 1, 2021), using linear mixed models including a covariate for IRC response. Prespecified subgroup analyses were performed by therapy line. Tumor load was defined as the sum of longest diameters for non-nodal target lesions and short axes for target nodal lesions by IRC. Exploratory analyses evaluated characteristics and outcomes according to clinical benefit (i.e., best overall response by IRC of stable disease or better).
Acknowledgments
The authors would like to thank patients and their families, investigators, co-investigators, and the study teams at all participating centers, as well as the healthcare business of Merck KGaA, Darmstadt, Germany. The trial was sponsored by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945). These results were presented in part at ASCO Annual Meetings in 2021 (June 4–8, 2021, abstract no. 9021) and 2022 (June 3–7, 2022, Chicago, IL; abstract no. 9121).
Author contributions
Conceptualization, C.S., D.J., R.B., A.J., P.K.P., and X.L.; data curation: C.S., D.J., H.V., R.B., G.O., and A.J.; formal analysis, R.B.; investigation, X.L., L.G.P.-A., J.V.M., S.V., C.C.G., E.F.S., M.G., R.V., D.V.B., J.F.P., M.S., T.K., Y.-C.K., S.S.Y., J.-Y.H., J.-H.K., C.-H.S., Y.J.C., and P.K.P.; methodology, G.O., A.J., and H.V.; resources, G.O. and A.J.; validation: C.S., D.J., H.V., R.B., G.O., and A.J.; writing – review & editing, all authors.
Declaration of interests
X.L. reported personal/consulting fees from EMD Serono during the conduct of the study; personal or consulting fees from AstraZeneca, Spectrum Pharmaceuticals, Novartis, Eli Lilly, Boehringer Ingelheim, Janssen, Blueprint Medicines, Bayer, and Albion; grants from ArriVent, Eli Lilly, Boehringer Ingelheim, and Regeneron; and personal fees from AbbVie outside the submitted work. L.G.P.-A. reported consulting roles with AstraZeneca, Lilly, EMD Serono, Spectrum Pharmaceuticals, and Daiichi Sankyo/Eli Lilly; research funding from Lilly and Boehringer Ingelheim; leadership roles from Genomica and ALTUM Sequencing; speakers bureau from Merck & Co., Kenilworth, NJ, Bristol-Myers Squibb, Roche, Pfizer, Lilly, AstraZeneca, and the healthcare business of Merck KGaA, Darmstadt, Germany; travel/accommodations/expenses from Roche, AstraZeneca, Merck & Co., Kenilworth, NJ, Bristol-Myers Squibb, Pfizer, and Takeda; and honoraria from Roche, Lilly, Pfizer, Bristol-Myers Squibb, Merck & Co., Kenilworth, NJ, AstraZeneca, the healthcare business of Merck KGaA, Darmstadt, Germany, PharmaMar, Novartis, Celgene, Amgen, Sanofi, Ipsen, Servier, Bayer, Blueprint Medicines, Mirati Therapeutics, and Takeda outside the submitted work. J.V.M. reported an advisory role with Amgen outside the submitted work. S.V. reported consulting or advisory role from the healthcare business of Merck KGaA, Darmstadt, Germany, AbbVie, Bristol-Myers Squibb, AstraZeneca, Merck & Co., Kenilworth, NJ, and Roche; non-financial support from OSE Immunotherapeutics; and personal fees from Janssen and Puma Biotechnology outside the submitted work. C.C.G. reported a consulting/advisory role with Boehringer Ingelheim and travel/accommodations/expenses from Roche and Merck & Co., Kenilworth, NJ, outside the submitted work. E.F.S. reported an advisory/consultancy role (institution) with Eli Lilly, AstraZeneca, Boehringer Ingelheim, Roche/Genentech, Bristol-Myers Squibb, the healthcare business of Merck KGaA, Darmstadt, Germany, Merck & Co., Kenilworth, NJ, Takeda, Bayer, Regeneron, Novartis, Daiichi Sankyo, and Seattle Genetics; and research funding (institution) from Boehringer Ingelheim, Bayer, Roche/Genentech, AstraZeneca, and Bristol-Myers Squibb outside the submitted work. M.G. reported personal fees from the healthcare business of Merck KGaA, Darmstadt, Germany, during the conduct of the study; grants and personal fees from AstraZeneca; and personal fees from the healthcare business of Merck KGaA, Darmstadt, Germany, Bayer, Bristol-Myers Squibb, AbbVie, Takeda, Janssen, Roche, Sanofi, Boehringer Ingelheim, Daiichi Sankyo, Eli Lilly, Novartis, and Blueprint outside the submitted work. R.V. reported research funding from the healthcare business of Merck KGaA, Darmstadt, Germany, during the conduct of the study; personal consulting fees from Janssen; personal speaker fees from Bristol-Myers Squibb and Takeda; personal speaker bureau fees from Amgen, Sanofi, Roche, and AstraZeneca; and travel fees from Pfizer Travel and Janssen outside the submitted work. D.V.B. reported advisory/consultancy honoraria from Roche, the healthcare business of Merck KGaA, Darmstadt, Germany, Bristol-Myers Squibb, AstraZeneca, Pfizer, Boehringer Ingelheim, and Takeda; and speaker honoraria from Roche, the healthcare business of Merck, KGaA, Darmstadt, Germany, Bristol-Myers Squibb, AstraZeneca, Pfizer, and Boehringer Ingelheim outside the submitted work. J.F.P. reported consulting/advisory roles with Roche, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Merck & Co., Kenilworth, NJ, and GlaxoSmithKline outside the submitted work. M.S. reported an advisory/consulting role with Roche, AstraZeneca, Bristol-Myers Squibb, and Merck & Co., Kenilworth, NJ, outside the submitted work. T.K. reported grants and personal fees from Chugai Pharmaceutical Co., AstraZeneca, Eli Lilly Japan, Taiho Pharmaceutical Co., Bristol-Myers Squibb, Merck & Co., Kenilworth, NJ, and Kyowa Hakko Kirin; personal fees from Ono Pharmaceutical, Pfizer Japan, Nippon Boehringer Ingelheim, Nippon Kayaku, Novartis, and Daiichi-Sankyo; and grants from the healthcare business of Merck KGaA, Darmstadt, Germany, outside the submitted work. Y.-C.K. reported honoraria from AstraZeneca, Roche, Boehringer Ingelheim, Merck & Co., Kenilworth, NJ, Pfizer, Ono, Bristol-Myers Squibb, Daiichi Sankyo, and Yuhan; and research funding from AstraZeneca, Roche, and Boehringer Ingelheim outside the submitted work. S.S.Y. reported honoraria from AstraZeneca, Roche, Boehringer Ingelheim, Merck & Co., Kenilworth, NJ, Pfizer, Ono Pharmaceutical, Bristol-Myers Squibb, Daiichi Sankyo, and Yuhan; and research funding from AstraZeneca, Roche, and Boehringer Ingelheim outside the submitted work.
J.-Y.H. reported research funding from Hoffmann-La Roche, Ltd., Ono Pharmaceutical, Pfizer, and Takeda outside the submitted work. J.-H.K. reported honoraria from Roche, Boehringer Ingelheim, Merck & Co., Kenilworth, NJ, and Bristol-Myers Squibb; consulting roles with Roche, Boehringer Ingelheim, Merck & Co., Kenilworth, NJ, AstraZeneca, and Yuhan; speakers bureau for Pfizer, Merck & Co., Kenilworth, NJ, and Roche; and research funding from Boehringer Ingelheim, AstraZeneca, Daiichi Sankyo, and Yuhan outside the submitted work. Y.J.C. reported consulting/advisory role with Astella, Yuhan, Merck & Co., Kenilworth, NJ, Roche, and Chong Kun Dang outside the submitted work. C.S. is an employee of the healthcare business of Merck KGaA, Darmstadt, Germany. D.J. is an employee of the healthcare business of Merck KGaA, Darmstadt, Germany, and holds stock in Merck KGaA, Darmstadt, Germany. H.V. is an employee of the healthcare business of Merck KGaA, Darmstadt, Germany. R.B. is an employee of the healthcare business of Merck KGaA, Darmstadt, Germany, and holds stock in Merck KGaA, Darmstadt, Germany. G.O. was an employee of the healthcare business of Merck KGaA, Darmstadt, Germany, at the time of the study and holds stock in Novartis. A.J. is an employee of the healthcare business of Merck KGaA, Darmstadt, Germany, and holds stock in Merck KGaA, Darmstadt, Germany. P.K.P. reported an advisory/consulting role from Takeda, Xencor, Janssen, CrownBio, Bicara, Mirati, and EMD Serono; and research funding (institution) from Bicara, Boehringer Ingelheim, and EMD Serono outside the submitted work.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101280.
Supplemental information
References
- 1.Drilon A., Cappuzzo F., Ou S.-H.I., Camidge D.R. Targeting MET in lung cancer: Will expectations finally be MET? J. Thorac. Oncol. 2017;12:15–26. doi: 10.1016/j.jtho.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang H., Wang M. MET oncogene in non-small cell lung cancer: Mechanism of MET dysregulation and agents targeting the HGF/c-MET axis. OncoTargets Ther. 2020;13:2491–2510. doi: 10.2147/OTT.S231257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camidge D.R., Otterson G.A., Clark J.W., Ignatius Ou S.H., Weiss J., Ades S., Shapiro G.I., Socinski M.A., Murphy D.A., Conte U., et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021;16:1017–1029. doi: 10.1016/J.JTHO.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Califano R., Morgillo F., De Mello R.A., Mountzios G. Role of mesenchymal-epithelial transition amplification in resistance to anti-epidermal growth factor receptor agents. Ann. Transl. Med. 2015;3:81. doi: 10.3978/J.ISSN.2305-5839.2015.03.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo R., Luo J., Chang J., Rekhtman N., Arcila M., Drilon A. MET-dependent solid tumours — molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020;17:569–587. doi: 10.1038/s41571-020-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kron A., Scheffler M., Heydt C., Ruge L., Schaepers C., Eisert A.-K., Merkelbach-Bruse S., Riedel R., Nogova L., Fischer R.N., et al. Genetic Heterogeneity of MET-Aberrant NSCLC and Its Impact on the Outcome of Immunotherapy. J. Thorac. Oncol. 2021;16:572–582. doi: 10.1016/j.jtho.2020.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Schildhaus H.U., Schultheis A.M., Rüschoff J., Binot E., Merkelbach-Bruse S., Fassunke J., Schulte W., Ko Y.D., Schlesinger A., Bos M., et al. MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. 2015;21:907–915. doi: 10.1158/1078-0432.CCR-14-0450. [DOI] [PubMed] [Google Scholar]
- 8.Le X. Heterogeneity in MET-Aberrant NSCLC. J. Thorac. Oncol. 2021;16:504–506. doi: 10.1016/j.jtho.2021.01.1609. [DOI] [PubMed] [Google Scholar]
- 9.Overbeck T.R., Cron D.A., Schmitz K., Rittmeyer A., Körber W., Hugo S., Schnalke J., Lukat L., Hugo T., Hinterthaner M., et al. Top-level MET gene copy number gain defines a subtype of poorly differentiated pulmonary adenocarcinomas with poor prognosis. Transl. Lung Cancer Res. 2020;9:603–616. doi: 10.21037/tlcr-19-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf J., Seto T., Han J.Y., Reguart N., Garon E.B., Groen H.J.M., Tan D.S.W., Hida T., de Jonge M., Orlov S.V., et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 2020;383:944–957. doi: 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- 11.Tong J.H., Yeung S.F., Chan A.W.H., Chung L.Y., Chau S.L., Lung R.W.M., Tong C.Y., Chow C., Tin E.K.Y., Yu Y.H., et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016;22:3048–3056. doi: 10.1158/1078-0432.CCR-15-2061. [DOI] [PubMed] [Google Scholar]
- 12.Cappuzzo F., Marchetti A., Skokan M., Rossi E., Gajapathy S., Felicioni L., Del Grammastro M., Sciarrotta M.G., Buttitta F., Incarbone M., et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo B., Cen H., Tan X., Liu W., Ke Q. Prognostic value of MET gene copy number and protein expression in patients with surgically resected non-small cell lung cancer: a meta-analysis of published literatures. PLoS One. 2014;9 doi: 10.1371/JOURNAL.PONE.0099399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin W., Cheng J., Tang Z., Toruner G., Hu S., Guo M., Robinson M., Medeiros L.J., Tang G. MET Amplification (MET/CEP7 Ratio ≥ 1.8) Is an Independent Poor Prognostic Marker in Patients With Treatment-naive Non-Small-cell Lung Cancer. Clin. Lung Cancer. 2021;22:e512–e518. doi: 10.1016/J.CLLC.2020.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Feng Z., Pignon J.-C., Sharaf R., Romanov N., Doudement J., Albacker L.A., Kurata N., Smith N.R., Matsushita N., Scheuenpflug J. Abstract 5737: Precision oncology-driven real world clinical genomics data mining of MET alterations, TMB, and PD-L1 to empower indication agnostic patient enrollment and combination strategies. Cancer Res. 2022;82:5737. doi: 10.1158/1538-7445.AM2022-5737. [DOI] [Google Scholar]
- 16.Zhang Y., Yang Q., Zeng X., Wang M., Dong S., Yang B., Tu X., Wei T., Xie W., Zhang C., et al. MET Amplification Attenuates Lung Tumor Response to Immunotherapy by Inhibiting STING. Cancer Discov. 2021;11:2726–2737. doi: 10.1158/2159-8290.CD-20-1500. [DOI] [PubMed] [Google Scholar]
- 17.Mazieres J., Drilon A., Lusque A., Mhanna L., Cortot A.B., Mezquita L., Thai A.A., Mascaux C., Couraud S., Veillon R., et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019;30:1321–1328. doi: 10.1093/annonc/mdz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuler M., Berardi R., Lim W.T., de Jonge M., Bauer T.M., Azaro A., Gottfried M., Han J.Y., Lee D.H., Wollner M., et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: clinical and biomarker results from a phase I trial. Ann. Oncol. 2020;31:789–797. doi: 10.1016/j.annonc.2020.03.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.National Comprehensive Cancer Network . 2022. Non-Small Cell Lung Cancer Version 3.2022. [Google Scholar]
- 20.Falchook G.S., Kurzrock R., Amin H.M., Xiong W., Fu S., Piha-Paul S.A., Janku F., Eskandari G., Catenacci D.V., Klevesath M., et al. First-in-man Phase I trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin. Cancer Res. 2020;26:1237–1246. doi: 10.1158/1078-0432.CCR-19-2860. [DOI] [PubMed] [Google Scholar]
- 21.Markham A. Tepotinib: First Approval. Drugs. 2020;80:829–833. doi: 10.1007/s40265-020-01317-9. [DOI] [PubMed] [Google Scholar]
- 22.Paik P.K., Felip E., Veillon R., Sakai H., Cortot A.B., Garassino M.C., Mazieres J., Viteri S., Senellart H., Van Meerbeeck J., et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020;383:931–943. doi: 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le X., Sakai H., Felip E., Veillon R., Garassino M.C., Raskin J., Cortot A.B., Viteri S., Mazieres J., Smit E.F., et al. Tepotinib efficacy and safety in patients with MET exon 14 skipping NSCLC: Outcomes in patient subgroups from the VISION study with relevance for clinical practice. Clin. Cancer Res. 2022;28:1117–1126. doi: 10.1158/1078-0432.CCR-21-2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bladt F., Faden B., Friese-Hamim M., Knuehl C., Wilm C., Fittschen C., Grädler U., Meyring M., Dorsch D., Jaehrling F., et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin. Cancer Res. 2013;19:2941–2951. doi: 10.1158/1078-0432.ccr-12-3247. [DOI] [PubMed] [Google Scholar]
- 25.Friese-Hamim M., Clark A., Perrin D., Crowley L., Reusch C., Bogatyrova O., Zhang H., Crandall T., Lin J., Ma J., et al. Brain penetration and efficacy of tepotinib in orthotopic patient-derived xenograft models of MET-driven non-small cell lung cancer brain metastases. Lung Cancer. 2022;163:77–86. doi: 10.1016/J.LUNGCAN.2021.11.020. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y.L., Cheng Y., Zhou J., Lu S., Zhang Y., Zhao J., Kim D.W., Soo R.A., Kim S.W., Pan H., et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020;8:1132–1143. doi: 10.1016/S2213-2600(20)30154-5. [DOI] [PubMed] [Google Scholar]
- 27.Liam C.K., Ahmad A.R., Hsia T.C., Zhou J., Kim D.W., Soo R.A., Cheng Y., Lu S., Shin S.W., Yang J.C.H., et al. Randomized Trial of Tepotinib Plus Gefitinib versus Chemotherapy in EGFR-Mutant NSCLC with EGFR Inhibitor Resistance Due to MET Amplification: INSIGHT Final Analysis. Clin. Cancer Res. 2023;29:1879–1886. doi: 10.1158/1078-0432.CCR-22-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazieres J., Kim T.M., Lim B.K., Wislez M., Dooms C., Finocchiaro G., Hayashi H., Liam C.K., Raskin J., Tho L.M., et al. LBA52 - Tepotinib + osimertinib for EGFRm NSCLC with MET amplification (METamp) after progression on first-line (1L) osimertinib: Initial results from the INSIGHT 2 study. Ann. Oncol. 2022;33:S1419–S1420. doi: 10.1016/annonc/annonc1089. [DOI] [Google Scholar]
- 29.Le X., Kowalski D., Cho B.C., Conte P., Felip E., Garassino M.C., Viteri S., Chang G.-C., Richart J., Paz-Ares L., et al. Abstract 3385: Liquid biopsy to detect MET exon 14 skipping (METex14) and MET amplification in patients with advanced NSCLC: Biomarker analysis from VISION study. Cancer Res. 2020;80:3385. doi: 10.1158/1538-7445.AM2020-3385. [DOI] [Google Scholar]
- 30.Veillon R., Sakai H., Le X., Felip E., Cortot A.B., Smit E.F., Park K., Griesinger F., Britschgi C., Wu Y.-L., et al. Safety of Tepotinib in Patients with MET Exon 14 Skipping NSCLC and Recommendations for Management. Clin. Lung Cancer. 2022;23:320–332. doi: 10.1016/j.cllc.2022.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felip E., Garassino M.C., Sakai H., Le X., Veillon R., Smit E., Mazieres J., Cortot A., Raskin J., Thomas M., et al. P45.03 - Tepotinib in Patients with MET exon 14 (METex14) Skipping NSCLC as Identified by Liquid (LBx) or Tissue (TBx) biopsy. J. Thorac. Oncol. 2021;16:S1085. [Poster P45.03] [Google Scholar]
- 32.Pairawan S., Hess K.R., Janku F., Sanchez N.S., Mills Shaw K.R., Eng C., Damodaran S., Javle M., Kaseb A.O., Hong D.S., et al. Cell-free Circulating Tumor DNA Variant Allele Frequency Associates with Survival in Metastatic Cancer. Clin. Cancer Res. 2020;26:1924–1931. doi: 10.1158/1078-0432.CCR-19-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandara D.R., Paul S.M., Kowanetz M., Schleifman E., Zou W., Li Y., Rittmeyer A., Fehrenbacher L., Otto G., Malboeuf C., et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018;24:1441–1448. doi: 10.1038/S41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 34.Hopkins A.M., Kichenadasse G., McKinnon R.A., Rowland A., Sorich M.J. Baseline tumor size and survival outcomes in lung cancer patients treated with immune checkpoint inhibitors. Semin. Oncol. 2019;46:380–384. doi: 10.1053/J.SEMINONCOL.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Le X., Paz-Ares L.G., Van Meerbeeck J., Viteri S., Cabrera Galvez C., Vicente Baz D., Kim Y.-C., Kang J.-H., Schumacher K.-M., Karachaliou N., et al. Tepotinib in patients (pts) with advanced non-small cell lung cancer (NSCLC) with MET amplification (METamp) J. Clin. Oncol. 2021;39:9021. doi: 10.1200/JCO.2021.39.15_suppl.9021. [DOI] [Google Scholar]
- 36.Rolfo C., Mack P., Scagliotti G.V., Aggarwal C., Arcila M.E., Barlesi F., Bivona T., Diehn M., Dive C., Dziadziuszko R., et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021;16:1647–1662. doi: 10.1016/J.JTHO.2021.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Paik P.K., Veillon R., Felip E., Cortot A., Sakai H., Mazieres J., Thomas M., Reinmuth N., Raskin J., Conte P.F., et al. METex14 ctDNA dynamics & resistance mechanisms detected in liquid biopsy (LBx) from patients (pts) with METex14 skipping NSCLC treated with tepotinib. J. Clin. Oncol. 2021;39:9012. doi: 10.1200/JCO.2021.39.15_SUPPL.9012. [DOI] [Google Scholar]
- 38.Tsui D.C.C., Drusbosky L.M., Wienke S., Gao D., Bubie A., Barbacioru C., Camidge D.R. Oncogene Overlap Analysis of Circulating Cell-free Tumor DNA to Explore the Appropriate Criteria for Defining MET Copy Number–Driven Lung Cancer. Clin. Lung Cancer. 2022;23:630–638. doi: 10.1016/j.cllc.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimoto A., Matsumoto S., Udagawa H., Itotani R., Usui Y., Umemura S., Nishino K., Nakachi I., Kuyama S., Daga H., et al. A Large-Scale Prospective Concordance Study of Plasma- and Tissue-Based Next-Generation Targeted Sequencing for Advanced Non–Small Cell Lung Cancer (LC-SCRUM-Liquid) Clin. Cancer Res. 2023;29:1506–1514. doi: 10.1158/1078-0432.CCR-22-1749. [DOI] [PubMed] [Google Scholar]
- 40.Hartmaier R.J., Markovets A., Cho B.C., de Langen A.J., Goldberg S.B., Goldman J., Le X., Okamoto I., Riess J.W., Cosaert J., et al. Abstract LB078: Tumor genomics in patients (pts) with advanced epidermal growth factor receptor mutant (EGFRm) non-small cell lung cancer (NSCLC) whose disease has progressed on first-line (1L) osimertinib therapy in the Phase II ORCHARD study. Cancer Res. 2022;82:LB078. doi: 10.1158/1538-7445.AM2022-LB078. [DOI] [Google Scholar]
- 41.Yu H.A., Kerr K., Rolfo C.D., Fang J., Finocchiaro G., Wong K.-H., Veillon R., Kato T., Yang J.C.-H., Nadal E., et al. Detection of MET amplification (METamp) in patients with EGFR mutant (m) NSCLC after first-line (1L) osimertinib. J. Clin. Oncol. 2023;41:9074. doi: 10.1200/JCO.2023.41.16_suppl.9074. [DOI] [Google Scholar]
- 42.Hartmaier R.J., Markovets A., Xu W., Baczynska A., Todd A., Ahn M.-J. Abstract LB294: Baseline and on-treatment plasma-based genomics as a predictor of outcome in SAVANNAH: Savolitinib + osimertinib in EGFRm MET overexpressed/amplified NSCLC post-osimertinib. Cancer Res. 2023;83:LB294. doi: 10.1158/1538-7445.AM2023-LB294. [DOI] [Google Scholar]
- 43.Hartmaier R.J., Markovets A.A., Ahn M.J., Sequist L.V., Han J.Y., Cho B.C., Yu H.A., Kim S.W., Yang J.C.H., Lee J.S., et al. Osimertinib + Savolitinib to Overcome Acquired MET-Mediated Resistance in Epidermal Growth Factor Receptor–Mutated, MET-Amplified Non–Small Cell Lung Cancer: TATTON. Cancer Discov. 2023;13:98–113. doi: 10.1158/2159-8290.CD-22-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun B., Qiu T., Zeng X., Duan J., Bai H., Xu J., Li J., Li J., Hao X., Liu Y., et al. Detection of MET polysomy by next-generation sequencing and its clinical relevance for MET inhibitors. Cancer Res. Commun. 2023;3:532–539. doi: 10.1158/2767-9764.crc-22-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan Y., Sun R., Wang Z., Zhang Y., Xiao X., Liu Y., Xin B., Xiong H., Lu D., Ma J. Detection of MET amplification by droplet digital PCR in peripheral blood samples of non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2023;149:1667–1677. doi: 10.1007/s00432-022-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartzberg L.S., Li G., Tolba K., Bourla A.B., Schulze K., Gadgil R., Fine A., Lofgren K.T., Graf R.P., Oxnard G.R., Daniel D. Complementary Roles for Tissue- and Blood-Based Comprehensive Genomic Profiling for Detection of Actionable Driver Alterations in Advanced NSCLC. JTO Clin. Res. Rep. 2022;3 doi: 10.1016/j.jtocrr.2022.100386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abbosh C., Birkbak N.J., Swanton C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018;15:577–586. doi: 10.1038/S41571-018-0058-3. [DOI] [PubMed] [Google Scholar]
- 48.Chan H.T., Chin Y.M., Low S.K. Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers. 2022;14:3275. doi: 10.3390/CANCERS14133275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanman R.B., Mortimer S.A., Zill O.A., Sebisanovic D., Lopez R., Blau S., Collisson E.A., Divers S.G., Hoon D.S.B., Kopetz E.S., et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10 doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y., Park S., Kim W.S., Lee J.C., Jang S.J., Choi J., Choi C.M. Correlation between progression-free survival, tumor burden, and circulating tumor DNA in the initial diagnosis of advanced-stage EGFR-mutated non-small cell lung cancer. Thorac. cancer. 2018;9:1104–1110. doi: 10.1111/1759-7714.12793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang E.W., Dagogo-Jack I., Kuo A., Rooney M.M., Shaw A.T., Digumarthy S.R. Association between circulating tumor DNA burden and disease burden in patients with ALK-positive lung cancer. Cancer. 2020;126:4473–4484. doi: 10.1002/CNCR.33118. [DOI] [PubMed] [Google Scholar]
- 52.Yu Y., Ren Y., Fang J., Cao L., Liang Z., Guo Q., Han S., Ji Z., Wang Y., Sun Y., et al. Abstract CT158: ctDNA analysis in the savolitinib phase II study in Non-Small Cell Lung Cancer (NSCLC) patients (pts) harboring MET exon 14 skipping alterations (METex14) Cancer Res. 2021;81:CT158. doi: 10.1158/1538-7445.AM2021-CT158. [DOI] [Google Scholar]
- 53.Ahn M.J., Mendoza M.J.L., Pavlakis N., Kato T., Soo R.A., Kim D.W., Liam C.K., Hsia T.C., Lee C.K., Reungwetwattana T., et al. Asian Thoracic Oncology Research Group (ATORG) Expert Consensus Statement on MET Alterations in NSCLC: Diagnostic and Therapeutic Considerations. Clin. Lung Cancer. 2022;23:670–685. doi: 10.1016/J.CLLC.2022.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Esagian S.M., Grigoriadou G.Ι., Nikas I.P., Boikou V., Sadow P.M., Won J.K., Economopoulos K.P. Comparison of liquid-based to tissue-based biopsy analysis by targeted next generation sequencing in advanced non-small cell lung cancer: a comprehensive systematic review. J. Cancer Res. Clin. Oncol. 2020;146:2051–2066. doi: 10.1007/S00432-020-03267-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baldanzi G., Graziani A. Physiological Signaling and Structure of the HGF Receptor. Biomedicines. 2014;3:1–31. doi: 10.3390/BIOMEDICINES3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi W., Jeong K.-C., Park S.-Y., Kim S., Kang E.H., Hwang M., Han J.-Y. MYC amplification-conferred primary resistance to capmatinib in a MET -amplified NSCLC patient: a case report. Transl. Lung Cancer Res. 2022;11:1967–1972. doi: 10.21037/TLCR-22-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henry R.E., Barry E.R., Castriotta L., Ladd B., Markovets A., Beran G., Ren Y., Zhou F., Adam A., Zinda M., et al. Acquired savolitinib resistance in non-small cell lung cancer arises via multiple mechanisms that converge on MET-independent mTOR and MYC activation. Oncotarget. 2016;7:57651–57670. doi: 10.18632/ONCOTARGET.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen A., Wang L., Huang M., Sun J., Chen Y., Shen Y.Y., Yang X., Wang X., Ding J., Geng M. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. 2015;75:4548–4559. doi: 10.1158/0008-5472.CAN-14-2743. [DOI] [PubMed] [Google Scholar]
- 59.Liu S.Y., Bao H., Wang Q., Mao W.M., Chen Y., Tong X., Xu S.T., Wu L., Wei Y.C., Liu Y.Y., et al. Genomic signatures define three subtypes of EGFR-mutant stage II–III non-small-cell lung cancer with distinct adjuvant therapy outcomes. Nat. Commun. 2021;12:6450. doi: 10.1038/s41467-021-26806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niederst M.J., Sequist L.V., Poirier J.T., Mermel C.H., Lockerman E.L., Garcia A.R., Katayama R., Costa C., Ross K.N., Moran T., et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015;6:6377. doi: 10.1038/NCOMMS7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J.K., Lee J., Kim S., Kim S., Youk J., Park S., An Y., Keam B., Kim D.W., Heo D.S., et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 2017;35:3065–3074. doi: 10.1200/JCO.2016.71.9096. [DOI] [PubMed] [Google Scholar]
- 62.Arakawa S., Yoshida T., Shirasawa M., Takayanagi D., Yagishita S., Motoi N., Ohe Y. RB1 loss induced small cell lung cancer transformation as acquired resistance to pembrolizumab in an advanced NSCLC patient. Lung Cancer. 2021;151:101–103. doi: 10.1016/J.LUNGCAN.2020.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Fujino T., Kobayashi Y., Suda K., Koga T., Nishino M., Ohara S., Chiba M., Shimoji M., Tomizawa K., Takemoto T., Mitsudomi T. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J. Thorac. Oncol. 2019;14:1753–1765. doi: 10.1016/J.JTHO.2019.06.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Subject to the healthcare business of Merck KGaA, Darmstadt, Germany, Data Sharing Policy, data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.