

Summary
During cancer progression, tumorigenic and immune signals are spread through circulating molecules, such as cell-free DNA (cfDNA) and cell-free RNA (cfRNA) in the blood. So far, they have not been comprehensively investigated in gastrointestinal cancers. Here, we profile 4 categories of cell-free omics data from patients with colorectal cancer and patients with stomach adenocarcinoma and then assay 15 types of genomic, epigenomic, and transcriptomic variations. We find that multi-omics data are more appropriate for detection of cancer genes compared with single-omics data. In particular, cfRNAs are more sensitive and informative than cfDNAs in terms of detection rate, enriched functional pathways, etc. Moreover, we identify several peripheral immune signatures that are suppressed in patients with cancer. Specifically, we establish a γδ-T cell score and a cancer-associated-fibroblast (CAF) score, providing insights into clinical statuses like cancer stage and survival. Overall, we reveal a cell-free multi-molecular landscape that is useful for blood monitoring in personalized cancer treatment.
Keywords: multi-omics, cell-free DNA, cell-free RNA, cancer diagnosis, liquid biopsy
Graphical abstract
Highlights
-
•
15 types of variation events are derived from 4 categories of cell-free multi-omics data
-
•
cfRNAs are more sensitive than cfDNAs in terms of detecting cancer-related genes
-
•
Certain immune signatures are suppressed in gastrointestinal cancer patients’ plasma
-
•
cfRNA biomarkers in blood predict clinical statuses like cancer stage and survival
Tao et al. profile 15 types of variation events from cfDNAs and cfRNAs in patients with colorectal or stomach cancer. Integration of genomic, epigenomic, and transcriptomic data in plasma enhances the detection of cancer-related genes, where cfRNA signatures are more sensitive and predictive of cancer status.
Introduction
Extracellular nucleic acid molecules include cell-free DNA (cfDNA) and cell-free RNA (cfRNA). These molecules are typically fragmented but resist full degradation in plasma because of protection from extracellular vesicles or binding proteins (e.g., nucleosomes for cfDNA and ribonucleoproteins for cfRNA). These extracellular molecules have been extensively utilized in assessments of cancer diagnosis and prognosis because cancer-induced modifications in tumor cells are detectable via cfDNAs and cfRNAs in circulating blood.1 Additionally, a patient’s clinical status during cancer treatment is strongly influenced by factors such as the tumor microenvironment and peripheral immune system. For example, a patient’s stromal cell activity and systemic immune response can have substantial impact on treatment outcome (e.g., immunotherapy).2,3 Importantly, cell-free molecules, especially cfRNAs, include signals derived from these non-neoplastic cellular constituents.4
Many cfDNA features (e.g., methylation pattern, mutation, copy number, fragment pattern, and nucleosome footprint) have been utilized for noninvasive assessments of disease diagnosis and prognosis.5,6,7 Many cfRNA features can also serve as biomarkers; such features include the abundance of microRNAs8 and circular RNAs,9 fragment copies, and alternative splicing patterns of mRNAs and long noncoding RNAs.10,11,12 Various RNA-regulatory elements altered in tumor cells, such as RNA editing,13 also have potential applications in the field of liquid biopsy.
Multi-omics data regarding tumor cells and tissues reportedly provide a more holistic understanding of the corresponding diseases compared with single-omics data.14,15 The combined use of multiple cell-free molecules in liquid biopsy can enhance diagnostic efficacy. For example, the utilization of cfRNA and cfDNA facilitates the detection of EGFR mutations in plasma.16 Similarly, a multi-analyte blood assay involving 61 DNA mutations and 8 proteins has demonstrated clinical value in cancer diagnostics.17 However, there has been limited systematic exploration of cell-free multi-omics data involving cfDNAs and cfRNAs in the context of cancer, including prevalent gastrointestinal malignancies such as colorectal cancer (CRC) and stomach adenocarcinoma (STAD). Current methods for diagnosis and monitoring of these two cancers (e.g., endoscopy and tissue biopsy) lack the ability to reveal the heterogeneity of molecular mechanisms among patients.18 Therefore, an in-depth exploration of the common and unique cell-free signatures that characterize these two gastrointestinal cancers could elucidate their extracellular biology and facilitate noninvasive monitoring applications.
Here, we present a systematic evaluation of cell-free multi-omics data, including methylated cfDNA immunoprecipitation sequencing (cfMeDIP-seq) and cfDNA whole genome sequencing (cfWGS) as well as total and small cfRNA sequencing (cfRNA-seq). Each set of matched multi-omics data was derived from a 2- to 3-mL plasma sample. Using CRC and STAD as examples of gastrointestinal cancers, we investigated various alterations within cfDNAs and cfRNAs to delineate a cell-free multi-molecular landscape.
Results
Cell-free multi-omics profiling and data quality control
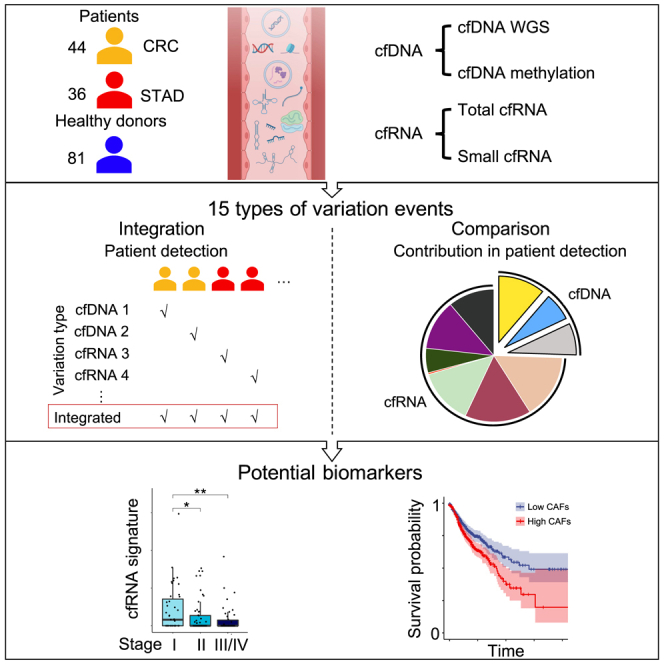
To explore the realm of cell-free multi-omics and conduct comparative analyses in the context of CRC and STAD, we performed sequencing analyses across four types of omics data, involving 161 individuals (Figure 1A, see details in STAR Methods; Figures S1A–S1C; Table S1). We implemented stringent measures to ensure data integrity (see quality control steps in STAR Methods; Table S2). In particular, the intra-omics correlation between samples exceeded 0.75 in each single-omics analysis, inter-omics correlations were near zero (Figure S1D), and the concentrations, read lengths, and read distributions were consistent with previous reports5,12,19 (Figure S2). As expected, there were disparities in the read distributions of sequenced cfDNAs and cfRNAs; cfRNA-seq provided abundant information in exonic regions, whereas cfDNA-seq provided more extensive information in exonic, intronic, and intergenic regions (Figure 1B).
Figure 1.

Cell-free multi-omics data summary and quality control
(A) Cell-free multi-omics data in plasma. Numbers inside and outside of brackets are datasets and samples, respectively, where some samples were mixed for sequencing. The gap in the ring means no paired data.
(B) Multi-omics reads mapped on a housekeeping gene, GAPDH, and a tumor suppressor gene, TP53. The coverage is normalized by total mapped reads. Red dashed line, the average coverage of cfDNA reads mapped on a gene.
(C) Density plots of multi-omics correlation coefficients of genes in tissues (TCGA), cell lines (CCLE), and plasma (this study).
We performed a comprehensive assessment of multi-omics correlations across plasma data (this study), cell line data (Cancer Cell Line Encyclopedia [CCLE]20), and tissue data (The Cancer Genome Atlas [TCGA]21) (Figure 1C). In contrast to cellular data,20,22,23 correlations between cfDNAs and cfRNAs were less pronounced, probably because of the heterogeneous origins of cfDNAs and cfRNAs.4,24 The signals in plasma were derived from various cellular origins, suggesting that altered DNA and RNA sequences in plasma are not derived from similar origins. Thus, the combined insights from diverse cell-free molecular constituents in plasma could potentially enhance the detection of cancer pathologies relative to the use of single-omics data. Accordingly, we utilized combined multi-omics data to facilitate the identification of cancer genes and evaluate their detection capabilities.
Combination of cell-free multi-omics data enhanced the detection of cancer genes in plasma
We comprehensively profiled 15 cell-free molecular variations using multi-omics data (STAR Methods). These variations can be utilized for comprehensive analysis of patients with cancer and healthy donors (HDs), considering multidimensional individual samples (Figure 2A) and gene-based results (Figure 2B). For example, within tumor samples, the TP53 gene is usually depleted at the DNA copy number level and downregulated at the RNA expression level in patients with cancer (Figure 2B). Our investigation revealed that certain cancer patients could not be detected simultaneously through the cfDNA and cfRNA methodologies at 95% specificity, implying that the combination of cfDNA and cfRNA data could substantially enhance the capacity for TP53 detection. The establishment of the 95% specificity benchmark was based upon variation measurements in HDs; an individual with a variation value out of the 95th percentile (e.g., mutation rate or abundance level) in the HD population was considered an outlier.
Figure 2.

Detection of the cancer genes using different types of variations
(A) Overview of the plasma multi-omics variation atlas (including 30 patients with STAD, 23 patients with CRC, and 18 HDs).
(B) cfDNA copy number and cfRNA abundance for TP53. HD, healthy donor; CRC, colorectal cancer; STAD, stomach adenocarcinoma. The dotted lines represent 95% specificity defined by HDs.
(C) Detection capacity for each cancer gene, combing different variations derived from cfDNA (cfWGS and cfMeDIP-seq) and total cfRNA-seq data. Frequently altered genes are defined by a greater than 75% detection ratio.
(D) Distribution of variation types among all genes that are frequently altered.
(E) Altered genes with top detection ratios ranked by cfRNA and cfDNA, respectively.
We quantified the sensitivity of cancer gene detection, defined as the proportion of cancer patients being detected at 95% specificity, and then investigated various types of variations associated with a predetermined ensemble of cancer genes. The cancer gene collection was based on the reference dataset of Catalog of Somatic Mutations in Cancer (COSMIC) hallmark cancer genes,25 which comprises 38 genes with annotated somatic mutations in the colorectal or gastric spectrum. Our data showed that the combination of multiple variations led to a substantial increase in detection capacity compared with the detection rate in cases of single variations (Figure 2C). For 9 cancer genes, including CUX1, SMAD2, QKI, and TP53, a substantial proportion of patients (>75%) exhibited variations at the cfDNA or cfRNA level, implying that variations in these genes frequently occurred among patients with cancer. Specific variations in cfRNA were particularly prevalent, including alternative promoters (average ratio: 17.7%), RNA expression (average ratio: 13.3%), allele-specific expression (average ratio: 12.5%), and RNA splicing (average ratio: 11.4%).
Overall, our results showed that combined cell-free multi-omics data could enhance the detection capacity for each of the pre-defined cancer genes (i.e., sensitivity score with 95% specificity); variations in cfRNA often exhibited a greater effect on sensitivity compared with their cfDNA counterparts among the 38 cancer genes. Next, we expanded the investigation beyond pre-defined cancer genes to include genes that showed variation in patients with cancer (i.e., cancer-related genes).
Compared with variations in cfDNA, variations in cfRNA demonstrated greater sensitivity in cancer-related gene detection
Genes were considered cancer-related when they exhibited outlier behavior (out of 95% in HDs) in more than 75% of patients with cancer. Patient detection rates for all genes across cfDNA and cfRNA are shown in Table S3. Similar to the pre-defined cancer genes, frequently altered genes were mainly identified by analysis of variations in cfRNA (Figure 2D). Generally, variations in cfRNA tended to be more sensitive than variations in cfDNA with respect to cancer detection. This observation in plasma samples is similar to the results of a multi-omics study involving tumor cells, which revealed that variations in RNA constituted 78.23% of all alterations identified across 731 genes with significant recurrent abnormalities.14
We also identified the 30 genes with the highest sensitivity (expressed as the proportion of detected patients) for alterations at either the cfDNA or cfRNA level (Figure 2E). These data emphasize that variations in cfRNA exhibit greater sensitivity than their cfDNA counterparts; the top genes with variations in cfDNA typically exhibited lower sensitivity relative to the top genes with variations in cfRNA. Furthermore, we revealed considerable differences between the top genes recognized by cfDNA and the top genes recognized by cfRNA. These differences indicate that the genetic insights offered by these two modalities are complementary when evaluated within plasma. For example, numerous top genes (e.g., MAP3K7CL, DEK, CLEC1B, and SKAP2) with variations in cfRNA displayed functional associations with oncogenes and immune pathways. Among the top genes with variations in cfDNA, altered mitochondrial DNA sequences were common; these alterations may be associated with the increase metabolic activity present in cancer.26
Identification of differential alterations in cell-free molecules among patients with cancer
In addition to the identification of variations within individual patients, we conducted statistical analyses to detect variations with differential characteristics between two distinct cohorts: patients with cancer vs. HDs and patients with CRC vs. patients with STAD (STAR Methods; Figure 3A). Among these differential alterations, key observations were that cfRNA abundance, cfDNA methylation level, and cfDNA windowed protection score (WPS) primarily exhibited distinct variations within STAD; similarly, cfRNA abundance, cfRNA single-nucleotide variant (SNV), and cfDNA WPS primarily exhibited distinct variations within CRC, with numerical differences (Figure 3A). Many well-known cancer alterations were identified in the differential analysis. For example, increased abundance of cfRNA associated with KRAS was identified in patients with cancer (edgeR exactTest, p = 0.011). Moreover, increased accessibility of the KRAS promoter region was observed among patients with cancer, as indicated by its cfDNA WPS (Figure 3B). As another example, the methylation level of cfDNA associated with PGRMC1, a carbon monoxide-responsive molecular switch associated with EGFR,27 exhibited significant hypomethylation within the promoter region among patients with cancer (edgeR exactTest, p = 0.047) (Figure 3B).
Figure 3.

Differential alterations of various cell-free molecules
(A) Numbers of the differentially altered events in plasma between patients with cancer (n = 80) and HDs (n = 81) and between patients with CRC (n = 44) and patients with STAD (n = 36).
(B) Examples of KRAS cfRNA abundance, KRAS window protection score (WPS), and PGRMC1 cfDNA methylation. The blue blocks, lines, and arrows below each panel are gene models. Black blocks above the promoter region of KRAS, promoter regions; gray blocks, enhancer regions; red arrowhead, open regions in cancer.
(C) PCAs of 3 representative differential alterations among cancer patients and HDs.
(D) Ratio of inter-class distance over intra-class distance for each type of differential alteration. Ratios larger than 1 (dashed line) are colored red.
Next, we evaluated the discriminatory abilities of these diverse alterations in cancer classification (Figures 3C and S3). We quantitatively determined the classification efficacy for each variation using the ratio of inter-class distance to intra-class distance. We found that the variations identified within cfWGS data performed well in cancer detection (i.e., patients with cancer vs. HDs) and the identification of distinct cancer types (i.e., CRC vs. STAD). In contrast, cfMeDIP-seq data exhibited robust identification of cancer types rather than detection of cancer. Notably, microRNA abundance data derived from small cfRNA-seq data displayed suboptimal performance in both scenarios; variations derived from total cfRNA-seq data usually demonstrated superior cancer detection efficacy rather than identification of specific cancer types. Additionally, microbial cfRNA abundance (derived from total cfRNA-seq data) more effectively distinguished the two cancer types compared with features derived from human cfRNAs, consistent with our previous findings12 (Figure 3D).
To compare the performance of combined multi-omics data with single-omics approaches regarding classification, we performed random forest classification and subsequent validation through bootstrapping. Although classification performance regarding STAD vs. HD and STAD vs. CRC was considerably enhanced by using combined multi-omics data, the CRC detection task did not show a similar benefit. However, conclusions based on the current cohort could be affected by the random nature of the bootstrapping procedure and the cancer-type-specific attributes of cell-free nucleic acids. Thus, a broader validation cohort is needed to confirm these findings (Figure S4).
Identification of suppressed immune signatures in plasma using cell-free multi-omics data
We explored the enriched pathways involved in differential alterations (Figure 3A) between patients with cancer and HDs. The analysis showed that cfRNA data had comparatively better information, relative to cfDNA data, regarding the quantity (Figure 4A) and functional attributes (Figure 4B) of the enriched pathways. In terms of upregulated genes, we found that cancer-related pathways were significantly enriched in variations detectable by cfDNA copy number and cfRNA abundance (edgeR exactTest, p < 0.05). Additionally, we identified various immune pathways (e.g., T cell and B cell receptor signaling pathways) that were enriched in genes with downregulated cfRNA abundance among patients (Figures 4B, S5A, and S5B), implying that the plasma of these patients exhibits a state of immunosuppression within these pathways. For example, CD8A, a marker of cytotoxic T cells, and ZAP-70, a gene with a critical activating role in downstream T cell signal transduction pathways,28 were downregulated in the plasma of patients with cancer. CD19, a marker of B cells, was similarly downregulated in these patients. PD-L1, a well-known immune suppressor in cancer, was significantly upregulated in the plasma of patients with CRC (Figure 4C).
Figure 4.

Enriched functional pathways of the differential alterations
(A) Numbers of the enriched pathways for the differentially altered genes defined by different variation events in cancer patients’ plasma.
(B) Kyoto Encyclopedia of Genes and Genomes (KEGG) terms of the top enriched pathways for each differential alteration.
(C) Differential cfRNA abundance for the example genes in the immune pathways. The y axis indicates transcripts per million (TPM).
(D) Example genes in the T cell receptor signaling pathway altered at different omics levels. The p-value represents the significance of the differential alterations. The combined p-value was calculated by Activepathways.
(E) Down-regulated T cell and B cell receptor signaling pathways calculated by the public total and extracellular vesicle-enriched (EV-enriched) cfRNA-seq datasets.
Single-tailed Wilcoxon rank-sum test was used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant. ESCA, esophageal carcinoma; HCC, hepatocellular carcinoma; LUAD, lung adenocarcinoma; PDAC, pancreatic ductal adenocarcinoma.
Multi-omics pathway enrichment analysis (STAR Methods) also confirmed this immunosuppression phenomenon; most instances of downregulation involved cfRNA rather than cfDNA. For example, among the 32 genes with differential variation that were enriched in the T cell receptor signaling pathway, 28 exhibited significant variation in cfRNA abundance, whereas 7 displayed significant variation in cfDNA abundance (Figures 4D, S5C, and S5D).
Next, we validated the immunosuppression phenomenon in published cell-free data for 710 samples, including total cfRNA and extracellular vesicle–enriched cfRNA data for CRC, STAD, esophageal carcinoma (ESCA), hepatocellular carcinoma (HCC), pancreatic ductal adenocarcinoma (PDAC), and lung adenocarcinoma (LUAD) (Table S4). Our validation analysis indicated that the T cell and B cell receptor signaling pathways were significantly downregulated in plasma from patients with these types of cancer (Figure 4E).
In summary, cell-free multi-omics data revealed many cancer-related functional pathways and signatures in plasma. In particular, we identified several downregulated immune signatures in plasma from patients with cancer, which were mostly derived from cfRNAs.
cfRNA features were derived from specific circulating cells and components of the tumor microenvironment
To elucidate the origins of these cfRNA-derived signatures, we performed total RNA-seq of matched samples, including plasma, primary tumor, normal tissue adjacent tumor (NAT), and peripheral blood mononuclear cells (PBMCs) from 16 patients with CRC and 6 HDs (Figure 5A). We used the computational deconvolution tool named "estimating the proportion of immune and cancer cells" (EPIC)29 to assess RNA-seq reads in plasma, tissue, and PBMCs, facilitating the identification of their origins and components. Surprisingly, we found that the cfRNAs in plasma were derived from blood hematopoietic constituents, including lymphocytes (including B cells, CD4 T cells, CD8 T cells, and natural killer [NK] cells) and macrophages, as well as encapsulated elements derived from components of the tumor microenvironment (e.g., endothelial cells and cancer-associated fibroblasts [CAFs]). Notably, cfRNAs in plasma contained CAF-associated signals that were nearly undetectable in RNA from PBMCs (Figure 5B). Overall, the results showed that plasma cfRNAs included peripheral blood cell-associated cues as well as tissue-derived cellular signatures.
Figure 5.

RNA expression signals compared in plasma, PBMCs, and tumors
(A) RNA-seq data in the paired samples of 22 patients with CRC. NAT, normal tissue adjacent tumor.
(B) Inferred signals originating from different cell types for 3 types of RNA-seq data (plasma, PBMCs, and tissue). The y axis indicates the log10-transformed cell type ratio estimated by EPIC. Single-tailed Wilcoxon rank-sum tests were used.
(C) Inferred relative abundance of more cell types.
(D) Correlated pathways of plasma and paired tumor samples. The abundance value of a pathway was averaged from the genes in this pathway. The numbers on the x axis corresponds to the sample identifiers in (A).
To determine which cells were downregulated in patients with cancer, we investigated immune cell abundance via cfRNA-seq data based on LM22 immune cell markers30 (Figure 5C). Plasma from patients with cancer (vs. HDs) and primary tumor samples (vs. adjacent normal tissue) exhibited substantial downregulation of CD8-positive T cells; these phenomena were confirmed by the Wilcoxon rank-sum test (p = 0.006 and p = 0.009, respectively). Similarly, immune cell populations with tumor suppressor potential (i.e., B cells and NK cells) displayed similar downregulation tendencies within plasma and primary tumors. These downregulated immune signatures, present in plasma and tumors, were not prominent in PBMCs (Figure 5C), highlighting the potential for plasma cfRNAs to more effectively identify characteristics of the tumor microenvironment and cancer progression compared with RNA from PBMCs.
We also assessed a broader range of pathways to identify gene expression relationships in paired tumor and plasma samples. This analysis demonstrated significant positive correlations across many pathways, including the Rap1 signaling pathway (Spearman correlation, R = 0.764, p = 0.002), mismatch repair (Spearman correlation, R = 0.698, p = 0.005), cancer-related pathway (Spearman correlation, R = 0.5, p = 0.043), complement and coagulation cascades (Spearman correlation, R = 0.588, p = 0.019), and platelet activation (Spearman correlation, R = 0.533, p = 0.032) (Figure 5D).
In summary, this analysis revealed robust positive correlations between plasma cfRNAs and tumor RNAs with respect to specific cancer- and immune-related signatures. The finding that plasma cfRNAs can detect tumor-derived signals and origins suggests that those cfRNAs can serve as a noninvasive modality for clinical monitoring of cancer progression and patient status.
Plasma cfRNA-derived signatures reveal the clinical status of patients with cancer
To confirm the utility of plasma cfRNA-derived signatures in dynamic monitoring of clinical status among patients with cancer, we performed an investigation of 143 total cfRNA-seq datasets derived from patients with CRC and patients with STAD, using our previously published data (GSE174302).12 We calculated various cell type signature scores and determined their correlations with cancer stage based on the deconvolution of total cfRNA-seq data (STAR Methods; Figures 6A and 6B). We found significant associations between scores corresponding to distinct cell types (e.g., γδ T cells, resting NK cells, M2 tumor-associated macrophages, and CAFs) and overall cancer stage (Figures 6C–6F). Among them, the γδ-T cell score and CAF score showed the greatest positive and negative correlations with cancer stage, respectively (γδ-T cell score: Spearman correlation = −0.2385, p = 0.0022; CAF score: Spearman correlation = 0. 2734, p = 0.0005).
Figure 6.

Clinical status-informative signatures derived from the plasma cfRNA
(A) Correlations between cancer stage and cfRNA-derived cell-type signatures for 77 patients with CRC and 66 patients with STAD. TME, tumor microenvironment; CAF, cancer-associated fibroblast; iCAF, inflammatory CAF; myCAF, myofibroblastic CAF.
(B) Overview of various cfRNA-derived signatures and individual clinical status, ranked by γδ-T cell score and resting NK cell score. The cfRNA-derived scores of (C) γδ T cells, (D) EPIC: CAFs, (E) resting NK T cells, and (F) TIDE: TAM are shown at different cancer stages for CRC and STAD patients. EPIC: CAFs, CAFs score defined by EPIC; TIDE: TAM, TAM score defined by TIDE; TIDE, a computational method named "tumor immune dysfunction and exclusion"; TAM, tumor associated macrophages.
(G) Tumor sizes for 2 subtypes of CRC patients categorized by the γδ-T cell scores (positive, >0; negative, = 0) in plasma. Single-tailed Wilcoxon rank-sum tests were used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(H) Survival with high (top 50%) and low (bottom 50%) scores of EPIC CAFs in the TCGA cohort of 475 patients with colon adenocarcinoma (COAD), 164 patients with rectal adenocarcinoma (READ), and 367 patients with STAD. Log rank test was used for survival time comparison.
Furthermore, we examined the correlations of these two scores with the clinical indexes other than cancer stage. The γδ-T cell score also showed a negative correlation with respect to tumor size in patients with CRC (Figure 6G; Spearman correlation = −0.1700, p = 0.0709), consistent with its anti-tumor function.31,32 Patients with low plasma-derived CAF score in a TCGA cohort (1,006 patients with CRC and patients with STAD) showed significant longer survival time (Figure 6H; Log-rank p = 0.00095). The other signatures correlated with patient survival in the TCGA cohort are described in Figure S6. Correlations of all signatures and clinical indexes are summarized in Table S5.
In summary, this study revealed distinct immune (e.g., γδ T cell) and tumor microenvironment (e.g., CAF) signatures derived from plasma cfRNA-seq data. These signatures have potential as predictive scores to monitor clinical status in terms of cancer stage, tumor size, and survival. However, it is important to emphasize that rigorous validation of these signatures in diverse CRC and STAD cohorts is warranted prior to their use in clinical practice.
Discussion
Conclusion
In this study, we revealed the cell-free nucleic acid landscape for CRC and STAD, using matched data that included genomic, epigenomic, and transcriptomic analyses of plasma. Next, we demonstrated the concept of combined multi-omics in liquid biopsy. Finally, we provided a cfRNA-based utility for dynamic monitoring of cancer status.
Clinical significance of cancer status monitoring via noninvasive biomarkers
Convenient monitoring of patient status, in terms of tumor size, cancer stage, and immune response, is essential during cancer treatment (e.g., immune and neoadjuvant therapies).33 However, methods for evaluating treatment response and effectiveness remain inconvenient and inaccurate.34 Liquid biopsy based on cfDNA/cfRNA biomarkers has emerged as a promising approach because of its noninvasive nature, minimal discomfort, economic viability, and ease of implementation. Quantitative signatures/scores based on noninvasive biomarkers can help clinicians to tailor therapeutic strategies to individual patients. Additionally, the gene signatures and functional pathways inferred from plasma sequencing data can provide targets for investigation of the mechanisms that lead to disparate treatment responses.
Functional targets revealed by the multi-omics data
The present study revealed many enriched pathways related to the tumor microenvironment (Figure 4). For example, the focal adhesion pathway plays an essential role in cellular communication and is strongly correlated with cancer progression.35 Furthermore, there was substantial downregulation of translation pathways in cfRNA data from the plasma of patients with cancer. This phenomenon is consistent with the notion of tumor-educated platelets.36 Additionally, the suppressed immune signatures in plasma match variations in the composition of active immune cell fractions within tumors, as revealed in a previous study.31 Considering the differential immunotherapy responses of patients with cancer,37 these pathways and signatures merit careful investigation across cancer subtypes.
Multi-omics perspectives in liquid biopsy
Similar to recent cfRNA studies,38,39 we did not perform extracellular vesicle enrichment during cfRNA-seq because the full range of cfRNAs in plasma included extracellular vesicle-associated cfRNAs and cfRNAs that originated outside of extracellular vesicles (e.g., ribonucleoproteins). A more thorough exploration of cfRNAs in various extracellular vehicles and ribonucleoproteins is needed. Moreover, the human circulatory system conveys biological signals through vesicular mechanisms and alternative modalities. It is important to recognize that cfDNA and cfRNA constitute a fraction of this substantial biological flux. The interpretation of these complex biological processes in the circulatory system requires greater effort to fully investigate diverse macromolecular components, including proteins and lipids. A full understanding of cell-free molecules also requires a multifaceted approach that incorporates various technological modalities and experimental paradigms, such as chromatin immunoprecipitation sequencing of cell-free nucleosomes carrying active chromatin modifications (cfChIP-seq).40
Limitations of the study
In this study, state-of-the-art technologies enabled simultaneous investigation of cfDNAs, cfDNA methylation patterns, small cfRNAs, and total cfRNAs using a limited amount of plasma (2–3 mL). It is important to acknowledge that some conclusions could have been biased by the specific technology used in this study. For example, cfMeDIP was performed because of its reduced plasma requirement (1–1.5 mL) compared with the bisulfite sequencing method (5–8 mL).41 A bisulfite-based method, such as single-cell whole-genome bisulfite sequencing (scWGBS) would provide more detailed insights regarding DNA methylation compared with cfMeDIP, although it would require considerably greater sequencing depth (>30×).42 The scope of the present study was confined to a subset of patients with gastrointestinal cancer. Consequently, it is important to gain a comprehensive understanding of the cell-free molecule landscape across various cancer types and subtypes. A large cohort study involving multiple clinical centers is also needed to establish a robust predictive model for clinical practice.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Plasma, PBMCs and tissue | Peking University First Hospital | https://www.pkufh.com/ |
| Chemicals, peptides, and recombinant proteins | ||
| Phage lambda DNA | Thermo Fisher Scientific | Cat#SD0011 |
| Recombinant DNase I | TaKaRa | Cat#2270B |
| ERCC RNA Spike-In Control Mixes | Thermo Fisher Scientific | Cat#4456740 |
| ExiSEQ NGS Spike-in | Qiagen | Cat#331535 |
| Critical commercial assays | ||
| Ficoll | TBD | Cat#LTS1077-1 |
| QIAamp MinElute ccfDNA Kit | Qiagen | Cat#55284 |
| Qubit dsDNA HS Assay kit | Thermo Fisher Scientific | Cat#Q32854 |
| Kapa HiFi Hotstart ReadyMix | Roche | Cat# 07958935001 |
| Kapa Hyper Prep Kit | Roche | Cat# 07962363001 |
| NEBNext Multiplex Oligos index | NEB | Cat#E7335L |
| MagMeDIP-seq Package | Diagenode | Cat#C02010040 |
| Plasma/Serum Circulating RNA and Exosomal Purification kit | Norgen | Cat#42800 |
| RNA Clean and Concentrator-5 kit | Zymo | Cat#R1014 |
| SMARTer® Stranded Total RNA-Seq Kit – Pico | TaKaRa | Cat#634413 |
| miRNeasy Serum/Plasma Kit | Qiagen | Cat#217184 |
| QIAseq miRNA Library Kit | Qiagen | Cat#331505 |
| Trizol | Thermo Fisher Scientific | Cat#15596026 |
| NEBNext Ultra™ II RNA Library Prep Kit | NEB | Cat#E7770L |
| Deposited data | ||
| Raw FASTQ data | This paper | GEO: GSE186607 |
| Code | This paper | Zenodo: https://doi.org/10.5281/zenodo.8416584 |
| Software and algorithms | ||
| bwa-mem2 | Vasimuddin et al.43 | https://github.com/bwa-mem2/bwa-mem2 |
| samtools | Li et al.44 | https://github.com/samtools/samtools |
| GATK | DePristo et al.45 | https://github.com/broadinstitute/gatk |
| fastp | Chen et al.46 | https://github.com/OpenGene/fastp |
| bowtie2 | Langmead et al.47 | https://github.com/BenLangmead/bowtie2 |
| featureCounts | Liao et al.48 | https://subread.sourceforge.net/ |
| cutadapt | Martin49 | https://github.com/marcelm/cutadapt |
| STAR | Dobin et al.50 | https://github.com/alexdobin/STAR |
| trim_galore | Krueger et al.51 | https://github.com/FelixKrueger/TrimGalore |
| umi_tools | Smith et al.52 | https://github.com/CGATOxford/UMI-tools |
| bedtools | Quinlan et al.53 | https://github.com/arq5x/bedtools2 |
| MISO | Katz et al.54 | https://github.com/yarden/MISO |
| salmon | Patro et al.55 | https://github.com/COMBINE-lab/salmon |
| rMATs | Shen et al.56 | https://github.com/Xinglab/rmats-turbo |
| kraken2 | Wood et al.57 | https://github.com/DerrickWood/kraken2 |
| MEDIPS | Lienhard et al.58 | https://bioconductor.org/packages/release/bioc/html/MEDIPS.html |
| Hmisc | Frank et al.59 | https://cran.r-project.org/web/packages/Hmisc/index.html |
| edgeR | Robinson et al.60 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| biomaRt | Durinck et al.61 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| clusterProfiler | Yu et al.62 | https://github.com/GuangchuangYu/clusterProfiler/ |
| CIBERSORTx | Newman et al.63 | https://cibersortx.stanford.edu/index.php |
| EPIC | Racle et al.29 | https://github.com/GfellerLab/EPIC |
| TIDE | Jiang et al.64 | https://github.com/foreverdream2/dysfunction_interaction_test/releases |
| ActivePathways | Paczkowska et al.65 | https://cran.r-project.org/web/packages/ActivePathways/index.html |
| randomForest | Breiman66 | https://cran.r-project.org/web/packages/randomForest/index.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhi John Lu (zhilu@tsinghua.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All FASTQ files generated in this study have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All original code has been deposited at Github and is publicly available at https://github.com/tyh-19/Pipeline-for-multiomics. All software being used in this study was summarized with versions and references in Table S6. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
We sequenced 360 cell-free omics datasets (86 cfWGS, 98 cfMeDIP-seq, 127 total cfRNA-seq, and 49 small cfRNA-seq) from 161 individuals (44 colorectal cancer patients, 36 stomach adenocarcinoma patients, and 81 healthy donors). The individuals were recruited from Peking University First Hospital (PKU, Beijing). Informed consent was obtained for all patients. Cell-free genome (cfWGS: individual number = 125), epigenome (cfMeDIP: individual number = 150), and transcriptome (total cfRNA-seq: individual number = 152; small cfRNA-seq: individual number = 73) were profiled. Patients’ age was distributed between 42 and 87 years (median age = 64 years), and most patients (51 out of 80) were diagnosed with stage I/II (Figure S1A). Different subtypes of colorectal cancer and stomach adenocarcinoma were included in the cohort (Figure S1B). More detailed information on age, gender, health status is provided in Table S1. For each person, 2–3 mL plasma sample was divided into 2–4 parts for 2 to 4-omics sequencing. For some samples (mostly from healthy donors), the plasma volumes were limited (less than 2 mL). We mixed these samples from persons with the same gender and similar age, then divided them into 2 to 4 parts (Figure S1C).
Among all participants, 95 were matched in 2-omics data, 84 were matched in 3-omics data, and 42 were matched in 4-omic data (Figure 1A). By requiring enough coverage ratio and total mapped reads (see the detailed analyzing Methods below, Table S2), we kept most datasets (352 out of 360) for the downstream analyses.
This study was approved by the institutional review board of Peking University First Hospital (2018-15). Informed consent was obtained from all participants.
Method details
Sample processing
Peripheral whole blood samples were collected using EDTA-coated vacutainer tubes before any treatment of the patients. Plasma was separated within 2 h after collection. All plasma samples were aliquoted and stored at −80°C before cfDNA and cfRNA extraction. Each sample was divided into 2–4 parts for sequencing different molecular types.
Peripheral blood mononuclear cells (PBMCs) were separated from whole blood by Ficoll. All PBMC samples were stored at −80°C.
Tissue samples were collected during surgery and transferred to liquid nitrogen within 30 min. Normal tissue adjacent tumor (NAT) was collected at least 2 cm away from the primary tumor.
Isolation and sequencing of cfDNA (cfWGS) and cfDNA methylation (cfMeDIP)
cfDNA was extracted from plasma using QIAamp MinElute ccfDNA Kit. DNA concentration was quantified by Qubit dsDNA HS Assay kit. Up to 5 ng plasma cfDNA (∼0.5 mL plasma) was used for cfWGS library with Kapa HiFi Hotstart ReadyMix in 11–13 cycles. Libraries were sequenced on Illumina HiSeq X-ten (∼60.7 million paired-end reads per library) with paired-end read length of 150 bases.
cfDNA methylation (cfMeDIP-seq) library was prepared following a previous protocol.67 Up to 15 ng plasma cfDNA (∼1 mL plasma) were used as input, followed by end repair and A-tailing using Kapa Hyper Prep Kit. Next, adaptors were ligated using NEBNext Multiplex Oligos index. Phage lambda DNA was added to fill the low input to 100 ng. After heat-denature and snap-cool, single-stranded DNA mixture was incubated with 5-mC antibody provided by MagMeDIP-seq Package, followed by 14–16 cycles of library amplification, bead purification, and size selection. Libraries were sequenced on Illumina HiSeq X-ten (∼42.9 million paired-end reads per library) with paired-end read length of 150 bases.
cfWGS data processing and quality control
Raw fastq files were trimmed with trim_galore68 (All software being used in this study were summarized with versions and references in Table S6.), then aligned to hg3869 genome with default parameters using bwa-mem2.43 Reads were further filtered by proper template length (20 bp to 1000 bp) using samtools44 and de-duplicated using GATK MarkDuplicates.45 Base quality was recalibrated using GATK BaseRecalibrator.45
We developed a set of quality control criteria to filter out poor libraries (Table S2). 6 quality control steps were included: 1) relH score (the relative frequency of CpGs) < 1.5; 2) saturation score (300 bp bins correlation) > 0.9; 3) genome depth >0.2; 4) coverage ratio >0.1; 5) mapped ratio >0.9; 6) unique read pairs >2 million. Finally, 2 samples were filtered out.
cfMeDIP-seq data processing and quality control
Methylation data were trimmed by fastp.70 Clean reads were firstly subjected to lambda genome alignment and then hg3869 genome using bowtie271 with “end-to-end” mode. Mapped reads were then de-duplicated by GATK MarkDuplicates.45 For the quality control procedure, we employed MEDIPS58 package to get CpG enrichment metrics and saturation estimation in 300 bp genome-wide bins. featureCounts48 were used to assign reads to each gene.
In data processing, we included 6 quality control steps (Table S2): 1) saturation score >0.9; 2) GoGe score (the observed/expected ratio of CpGs) > 1.2; 3) relH score >1.5; 4) coverage ratio >0.05; 5) mapped ratio> 0.9; 6) unique read pairs >2 million. In total, 3 samples were filtered out.
Isolation and sequencing of cfRNAs (total cfRNA-seq and small cfRNA-seq)
Total cfRNAs were extracted from ∼1 mL of plasma using the Plasma/Serum Circulating RNA and Exosomal Purification kit. Recombinant DNase I was used to digest DNAs. One set of ERCC RNA Spike-In Control Mixes was added. Next, the RNA Clean and Concentrator-5 kit was used to obtain pure total RNA. The total cfRNA library was prepared by SMARTer Stranded Total RNA-Seq Kit – Pico. Libraries were sequenced on Illumina HiSeq X-ten (∼37.5 million paired-end reads per library) with a length of 150 bases.
Small cfRNAs were extracted from ∼1 mL of plasma using the miRNeasy Serum/Plasma Kit. 1 μl ExiSEQ NGS Spike-in was added to the extracted RNA. The small cfRNA library was prepared with the QIAseq miRNA Library Kit. Libraries were sequenced on Illumina HiSeq X-ten (∼40.1 million reads per library), where adaptors linked to the short reads were later removed.
Total cfRNA-seq data processing and quality control
For total cfRNA-seq data, adaptors and low-quality sequences were trimmed using cutadapt.68 Reads shorter than 16 nt were discarded. For template-switch-based RNA-seq data, GC oligos introduced in reverse transcription were trimmed off, after which reads shorter than 30 nt were discarded. The remaining reads were mapped to ERCC’s spike-in sequences, NCBI’s UniVec sequences (vector contamination), and human rRNA sequences sequentially using STAR.50 Then, all reads unmapped in previous steps were mapped to the hg3869 genome index built with the GENCODE72 v27 annotation. Reads unaligned to hg38 were aligned to circRNA junctions.73 For circRNA, only fragments spanning back-splicing junctions were taken into consideration. Duplicates in the aligned reads were removed using GATK MarkDuplicates.45 To avoid the impact of potential DNA contamination, only intron-spanning reads were considered for gene expression quantification.36 Intron-spanning reads were defined as a read pair with a CIGAR string in which at least one mate contains ‘N’ in the BAM files. Reads on exons were counted and aggerated to gene by featureCounts.48
We filtered total cfRNA-seq samples using multiple quality control steps (Table S2): 1) raw read pairs >10 million; 2) clean read pairs (reads remained after trimming low quality and adaptor sequences) > 5 million; 3) aligned read pairs after duplicate removal (aligned to the hg3869 human genome, and circRNA junctions) > 0.5 million; 4) fraction of spike-in read pairs <0.5; 5) ratio of rRNA read pairs <0.55; 6) ratio of mRNA and lncRNA read pairs >0.2; 7) ratio of unclassified read pairs <0.6; 8) number of intron-spanning read pairs >100,000, 9) exonic/intronic reads ratio >1. In total, 3 samples were filtered out.
Small cfRNA-seq data processing and quality control
For small cfRNA-seq data, reads quality lower than 30 or length less than 15 were filtered by trim_galore. The remaining reads were sequentially mapped to ExiSEQ NGS Spike-in (a mix of 52 synthetic 5′ phosphorylated microRNAs), NCBI’s UniVec sequences, and human rRNA sequences, miRNA recorded in miRbase,74 lncRNA, mRNA, piRNA, snoRNA, snRNA, srpRNA, tRNA, transcripts of unknown potential (TUCPs) annotated in MiTranscriptome,75 Y_RNA by bowtie2.71 Mapped reads were sorted and indexed by samtools.44 Duplicates were removed by umi_tools.52
We filtered small cfRNA-seq samples using 2 quality control steps (Table S2): (1) datasets are required to have at least 100,000 reads that overlap with any annotated RNA transcript in the host genome, and (2) over 50% of the reads that map to the host genome also align to any RNA annotation. All small cfRNA-seq samples have enough reads for quantification, and most of the reads are aligned to RNA.
Isolation and sequencing of RNAs in tissue cells and PBMCs
Tissue RNA was extracted by Trizol. The tissue RNA library was prepared with the NEBNext Ultra II RNA Library Prep Kit for Illumina. PBMC was seprated by Ficoll from whole blood. The PBMC RNA library was prepared by SMARTer Stranded Total RNA-Seq Kit – Pico. All libraries were sequenced on Illumina HiSeq X-ten (∼38.8 million per library) with paired-end read length of 150 bases, where adaptors being sequenced were later removed.
Genome annotations
Human gene-centric genome regions and RNA biotypes were extracted from GENCODE v27 gtf file using bedtools.76 Human genome blacklist regions77 were downloaded from ENCODE (https://www.encodeproject.org/). CpG island regions were downloaded from UCSC genome browser (http://genome.ucsc.edu/). CpG shore and shelf were defined as 2 kb and 4 kb flank regions, respectively. Repeated regions were downloaded from RepeatMasker (rmsk) database in UCSC genome browser. Promoter regions were defined as −2000 bp to +500 bp relative to TSS (transcription start site), according to a recent study.78
cfDNA and cfRNA length estimation
The length of cfDNA was summarized using BAM metric “TLEN” (Figure S2B). Insert length of total cfRNA-seq (Figure S2H) was estimated by MISO,54 using long constitutive exons as references.
Correlation calculation among samples and omics
For correlation among samples, experiment reproducibility was checked using high throughput data correlation. Sample-based (i.e., sample A correlated with B by all genes abundances) Pearson correlations and corresponding p-values were calculated by rcorr function in R package Hmisc.79 Inter- or inner-omics correlations among different cancer types were averaged from multiple samples.
For gene correlation among omics, gene-based correlations (e.g., a gene’s DNA copy number and its RNA expression in the matched samples) were calculated. To compare omics correlation in cell lines and tissues, we downloaded RNA expression, DNA copy number, and DNA methylation data from the Cancer Cell Line Encyclopedia (CCLE)20 and the Cancer Genome Atlas (TCGA)21 from UCSC Xena (https://gdc.xenahubs.net/) and the Cancer Dependency Map portal (https://depmap.org/), respectively. Matched 3-omics data (33 stomach and 49 large intestine cell lines; 337 STAD, 307 COAD tissues) were selected for further analysis. For TCGA data, the gene-level copy number data were calculated by taking the segmental mean of the corresponding gene; the DNA methylation data were analyzed by calculating the CpG average beta value in the promoter region (2000 bp upstream and 500 bp downstream of TSS) of each gene; the gene expression data were converted to TPM (transcripts per million) data. Genes with NAs were removed.
Calculation of multiple cfDNA variations
DNA copy number, windowed protection score (WPS), end-motif frequency, and fragment size were calculated based on the cfDNA-seq data. And DNA methylation of the promoter and CpG island was calculated based on cfMeDIP-seq data.
DNA copy number: Copy number was calculated as a gene-centric CPM (counts per million mapped regions) using cfWGS data, where hg38 blacklist regions77 were masked. It was standardized as Z score using HDs’ distribution.
WPS: Windowed protection score (WPS) was calculated as the originally described study with minor modifications to estimate nucleosome occupancy in cfDNA.80 In brief, we used similar parameters as previously described: a minimum fragment size of 120 bp, a maximum fragment size of 180 bp, and a window of 120 bp. To account for variations in sequencing depth between samples, we performed a normalization step by dividing the WPS by the mean depth of randomly selected 1000 background sites in the genome. And then, for each gene, we quantify the nucleosome occupancy in TSS by computing the mean WPS from −150bp to +50bp around TSS.
End-motif frequency: End-motif was calculated following Jiang et al.81 In short, the occurrence of all 5′ end 4-mer sequences (256 in total) of each valid template were counted and normalized as a ratio for each sample. Shannon entropy was calculated from the frequency of motif as motif diversity score (MDS) for each sample (theoretical scale: [0,1]).
Fragment size: The fragment size ratio matrix was calculated following Cristiano et al.82 In short, 100–150 bp and 151–220 bp cfDNA templates were defined as short and long fragments respectively, 504 filtered bins mentioned in the original paper were converted to 469 bins in GRCh38 genome version, the read counts of each fragments type were also adjusted by LOESS-based GC content correction model.
DNA methylation: For each sample, raw counts of cfMeDIP-seq in promoter regions were normalized to CPM for cfDNA methylation level. We also computed counts per 300bp non-overlapping windows, normalized to CPM, and reduced to windows encompassing CpG islands, shores, and shelves.
Calculation of multiple cfRNA variations
All the RNA variations, except for miRNA abundance, were calculated based on the total cfRNA-seq data.
RNA expression/cfRNA abundance: raw counts of miRNAs were normalized to CPM using small cfRNA-seq data; raw counts of the other genes were normalized to TPM using total cfRNA-seq data.
cfRNA alternative promoter: transcript isoform abundance was quantified by salmon55 and normalized to TPM. TPMs of isoforms with transcript start sites within 10 bp (sharing the same promoter) were aggregated as one promoter activity. TPM <1 promoter is defined as an inactive promoter. The promoter with the highest relative promoter activity is defined as the major promoter. The remaining promoters are defined as minor promoters.14
cfRNA single nucleotide variant (SNV): intron-spanning reads were split by GATK SplitNCigarReads45 for confident SNP calling at RNA level. Then, alterations were identified by GATK HaplotypeCaller45 and filtered by GATK VariantFilteration45 with the following 4 criteria: strand bias defined by fisher exact test phred-scaled p-value (FS) < 20, variant confidence (QUAL) divided by the unfiltered depth (QD) > 2, total number of reads at the variant site (DP) > 10, SNP quality (QUAL) > 20. Allele fraction was defined as allele count divided by total count (reference count and allele count).
cfRNA editing: GATK ASEReadCounter45 was used to identify editing sites based on REDIportal.83 The editing ratio was defined as allele count divided by total count.
cfRNA allele specific expression: GATK ASEReadCounter45 were used to identify allele specific expression gene site based on SNP sites. For each individual, Allelic expression (AE, AE = |0.5 − Reference ratio |, Reference ratio = Reference reads/Total reads) was calculated for all sites with ≥16 reads.84
cfRNA splicing: The percent spliced-in (PSI) score of each alternative splicing event was calculated using rMATs-turbo.56
Chimeric cfRNA: Reads unaligned to genome were remapped to chimeric junctions by STAR-fusion50 to identify chimeric RNA. Chimera references were based on GTex85 and ChimerDB-v3.86
Microbial cfRNA abundance: Reads unaligned to genome were classified using kraken257 with its standard database to identify microbial cfRNA at genus level. Potential contaminations were filtered according to previous study.12 Counts at the genus level were also normalized by total genera counts.
Calculation of differential alteration between cancer and healthy control
cfDNA copy number, promoter methylation, and CpG island methylation: exactTest implemented in edgeR60 were used between cancer patients and HDs. |log2FC| > 0.59 and p-value <0.05 was used as the cutoff for defining significant differential alteration.
cfDNA end motif and fragment size: each differentially used motif or differential size fragment were identified by the Wilcoxon rank-sum test for relatively end motif usage or fragment size. p-value <0.05 was used as the cutoff.
cfDNA windowed protection score: each differentially protected gene was identified by the Wilcoxon rank-sum test for windowed protection score. |delta windowed protection score| > 0.5, and p-value <0.05 was used as cutoff.
RNA expression/cfRNA abundance and cf-miRNA abundance: differentially expressed genes were identified using the exactTest method in edgeR.60 |log2FC| > 0.59 and p-value <0.05 was used as cutoff.
cfRNA alternative promoter usage, editing, and SNV: each differentially used promoter or the differentially mutated allelic site or editing site was defined by the Wilcoxon rank-sum test for promoter usage or allele fraction. |delta allele fraction| > 0.2 and p-value <0.05 was used as cutoff.
cfRNA allele specific expression: each differentially expressed allelic site was defined by the Wilcoxon rank-sum test for AE. |delta AE| > 0.1 and p-value <0.05 was used as cutoff.
cfRNA splicing: differential splicing events were identified by the likelihood ratio test implemented in rMATs. |delta PSI| ≥ 0.05 and p-value <0.05 was used as cutoff.
Chimeric cfRNA: differential chimeric RNA events were defined by the fisher exact test between cancer patients and healthy donors. |delta frequency| > 0.1 and p-value <0.05 was used as cutoff.
Microbial cfRNA abundance: each differential genus abundance was defined by the Wilcoxon rank-sum test. |delta AE| > 0.1 and p-value <0.05 was used as cutoff.
Pathway enrichment analysis
For the above differential alterations, up-regulated and down-regulated genes in cancer were annotated by Kyoto Encyclopedia of Genes and Genomes (KEGG).87 For cfRNA SNP, allele specific expression, and editing, the dysregulated sites’ coordinates were assigned to the gene using an R package, biomaRt.61 KEGG enrichment was calculated using clusterProfiler.62
Integrative pathway analysis of multi-omics
Integrative pathway analysis of multi-omics data (i.e., RNA expression, CNA, DNA methylation) was performed using ActivePathways.65 p-values were corrected for multiple testing using the Holm procedure, and 0.05 was set as the cutoff value for significance. And then, the enrichment map was visualized using the plugin enhancedGraphics in Cytoscape.88
Cell type signature score calculation
Cell type signature scores were deconvoluted from the plasma/tissue total RNA-seq data, using CIBERSORTx63 with 1000 permutations. CIBERSORTx uses a reference panel of signature genes of different cell types and implements a support vector regression model to estimate the compositions of a mixture of different cell types’ RNAs. We used panels of tumor microenvironment (TME) cells89 and LM22 panels of immune cells.30 We also used TIDE64 and EPIC29 methods to calculate scores of TME cells. The input to CIBERSORTx, TIDE, and EPIC is the TPM read count matrix of cfRNA abundance. When calculating the score of EPIC:CAFs for the TCGA cohort, the CAF gene list was re-defined using our cfRNA-seq data (significantly correlated with the stage), and the input gene abundance values were derived from the tissue RNA-seq data of TCGA.
Quantification and statistical analysis
The multi-omics integrated p-value was merged by Browns’ Method in the ActivePathways. The Wilcoxon rank-sum test was used for the quantitative data comparisons in Figures 4, 5, and 6. The Kaplan-Meier survival curve was generated to assess the survival rate. The log rank test was used to test for significant differences. All reported p-values were two-sided, unless otherwise specified. Statistical power of differential alteration was calculated through different methods, which has been described in Calculation of differential alteration between cancer and healthy control. The area under the receiver operating characteristic curve was used to assess the ability to distinguish between CRC, STAD and HD in Figure S4.
Acknowledgments
This work is supported by the Tsinghua University Spring Breeze Fund (2021Z99CFY022), the National Natural Science Foundation of China (81972798, 32170671, and 81902384), the Capital’s Funds for Health Improvement and Research (CFH 2022-2-4075), the National Key Research and Development Plan of China (2022ZD0117700), the Tsinghua University Guoqiang Institute grant (2021GQG1020), the Tsinghua University Initiative Scientific Research Program of Precision Medicine (2022ZLA003), and the Bioinformatics Platform of the National Center for Protein Sciences (Beijing) (2021-NCPSB-005). This study was also supported by the Beijing Advanced Innovation Center for Structural Biology, the Bio-Computing Platform of the Tsinghua University Branch of China National Center for Protein Sciences, National High Level Hospital Clinical Research Funding (Interdisciplinary Research Project of Peking University First Hospital).
Author contributions
Y.T., S.X., S.Z., Z.J.L., and P.W. conceived and designed the project. S.X. and P.B. performed the experiments. Y.T., P.B., Y.J., and Y.L. processed the data and completed the bioinformatics analyses. Sample and clinical information were curated by S.Z., S.C., and P.W. All authors contributed to writing the manuscript. All authors have approved the manuscript and agree with publication.
Declaration of interests
The authors declare no competing interests.
Published: November 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101281.
Contributor Information
Qian Lu, Email: lqa01971@btch.edu.cn.
Pengyuan Wang, Email: pengyuan_wang@bjmu.edu.cn.
Zhi John Lu, Email: zhilu@tsinghua.edu.cn.
Supplemental information
References
- 1.Heitzer E., Haque I.S., Roberts C.E.S., Speicher M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019;20:71–88. doi: 10.1038/s41576-018-0071-5. [DOI] [PubMed] [Google Scholar]
- 2.Hiam-Galvez K.J., Allen B.M., Spitzer M.H. Systemic immunity in cancer. Nat. Rev. Cancer. 2021;21:345–359. doi: 10.1038/s41568-021-00347-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett R.L., Puré E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife. 2020;9 doi: 10.7554/eLife.57243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vorperian S.K., Moufarrej M.N., Tabula Sapiens Consortium. Quake S.R., Krasnow M., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., et al. Cell types of origin of the cell-free transcriptome. Nat. Biotechnol. 2022;40:855–861. doi: 10.1038/s41587-021-01188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Pol Y., Mouliere F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell. 2019;36:350–368. doi: 10.1016/j.ccell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Jamshidi A., Liu M.C., Klein E.A., Venn O., Hubbell E., Beausang J.F., Gross S., Melton C., Fields A.P., Liu Q., et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell. 2022;40:1537–1549.e12. doi: 10.1016/j.ccell.2022.10.022. [DOI] [PubMed] [Google Scholar]
- 7.Shen S.Y., Singhania R., Fehringer G., Chakravarthy A., Roehrl M.H.A., Chadwick D., Zuzarte P.C., Borgida A., Wang T.T., Li T., et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–583. doi: 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J., Yu L., Gao X., Hu J., Wang J., Dai Z., Wang J.-F., Zhang Z., Lu S., Huang X., et al. Plasma MicroRNA Panel to Diagnose Hepatitis B Virus–Related Hepatocellular Carcinoma. J. Clin. Oncol. 2011;29:4781–4788. doi: 10.1200/JCO.2011.38.2697. [DOI] [PubMed] [Google Scholar]
- 9.Wang S., Zhang K., Tan S., Xin J., Yuan Q., Xu H., Xu X., Liang Q., Christiani D.C., Wang M., et al. Circular RNAs in body fluids as cancer biomarkers: the new frontier of liquid biopsies. Mol. Cancer. 2021;20:13. doi: 10.1186/s12943-020-01298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y., Wang S., Xi X., Zhang M., Liu X., Tang W., Cai P., Xing S., Bao P., Jin Y., et al. Integrative analysis of long extracellular RNAs reveals a detection panel of noncoding RNAs for liver cancer. Theranostics. 2021;11:181–193. doi: 10.7150/thno.48206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larson M.H., Pan W., Kim H.J., Mauntz R.E., Stuart S.M., Pimentel M., Zhou Y., Knudsgaard P., Demas V., Aravanis A.M., Jamshidi A. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021;12:2357. doi: 10.1038/s41467-021-22444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S., Jin Y., Wang S., Xing S., Wu Y., Tao Y., Ma Y., Zuo S., Liu X., Hu Y., et al. Cancer type classification using plasma cell-free RNAs derived from human and microbes. Elife. 2022;11 doi: 10.7554/eLife.75181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Aroya S., Levanon E.Y. A-to-I RNA Editing: An Overlooked Source of Cancer Mutations. Cancer Cell. 2018;33:789–790. doi: 10.1016/j.ccell.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 14.PCAWG Transcriptome Core Group. Calabrese C., Demircioğlu D., Demircioğlu D., Fonseca N.A., He Y., Kahles A., Lehmann K.V., Liu F., Shiraishi Y., et al. Genomic basis for RNA alterations in cancer. Nature. 2020;578:129–136. doi: 10.1038/s41586-020-1970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sammut S.-J., Crispin-Ortuzar M., Chin S.-F., Provenzano E., Bardwell H.A., Ma W., Cope W., Dariush A., Dawson S.-J., Abraham J.E., et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature. 2022;601:623–629. doi: 10.1038/s41586-021-04278-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krug A.K., Enderle D., Karlovich C., Priewasser T., Bentink S., Spiel A., Brinkmann K., Emenegger J., Grimm D.G., Castellanos-Rizaldos E., et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018;29:700–706. doi: 10.1093/annonc/mdx765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen J.D., Li L., Wang Y., Thoburn C., Afsari B., Danilova L., Douville C., Javed A.A., Wong F., Mattox A., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parikh A.R., Leshchiner I., Elagina L., Goyal L., Levovitz C., Siravegna G., Livitz D., Rhrissorrakrai K., Martin E.E., Van Seventer E.E., et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019;25:1415–1421. doi: 10.1038/s41591-019-0561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao F., Wei A., Hu X., He Y., Zhang J., Xia L., Tu K., Yuan J., Guo Z., Liu H., et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin. Epigenetics. 2020;12:112. doi: 10.1186/s13148-020-00898-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M., et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henrichsen C.N., Vinckenbosch N., Zöllner S., Chaignat E., Pradervand S., Schütz F., Ruedi M., Kaessmann H., Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 23.Shao X., Lv N., Liao J., Long J., Xue R., Ai N., Xu D., Fan X. Copy number variation is highly correlated with differential gene expression: a pan-cancer study. BMC Med. Genet. 2019;20:175. doi: 10.1186/s12881-019-0909-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss J., Magenheim J., Neiman D., Zemmour H., Loyfer N., Korach A., Samet Y., Maoz M., Druid H., Arner P., et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018;9:5068. doi: 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reznik E., Miller M.L., Şenbabaoğlu Y., Riaz N., Sarungbam J., Tickoo S.K., Al-Ahmadie H.A., Lee W., Seshan V.E., Hakimi A.A., Sander C. Mitochondrial DNA copy number variation across human cancers. Elife. 2016;5 doi: 10.7554/eLife.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabe Y., Nakane T., Koike I., Yamamoto T., Sugiura Y., Harada E., Sugase K., Shimamura T., Ohmura M., Muraoka K., et al. Haem-dependent dimerization of PGRMC1/Sigma-2 receptor facilitates cancer proliferation and chemoresistance. Nat. Commun. 2016;7 doi: 10.1038/ncomms11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H., Kadlecek T.A., Au-Yeung B.B., Goodfellow H.E.S., Hsu L.-Y., Freedman T.S., Weiss A. ZAP-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb. Perspect. Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Racle J., de Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6 doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentles A.J., Newman A.M., Liu C.L., Bratman S.V., Feng W., Kim D., Nair V.S., Xu Y., Khuong A., Hoang C.D., et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silva-Santos B., Serre K., Norell H. γδ T cells in cancer. Nat. Rev. Immunol. 2015;15:683–691. doi: 10.1038/nri3904. [DOI] [PubMed] [Google Scholar]
- 33.Byrd D.R., Brierley J.D., Baker T.P., Sullivan D.C., Gress D.M. Current and future cancer staging after neoadjuvant treatment for solid tumors. CA. Cancer J. Clin. 2021;71:140–148. doi: 10.3322/caac.21640. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y., Jiang X., Liu Y., Huang H., Xiong Y., Xiao H., Gong K., Li X., Kuang X., Yang X. Elevated tumor markers for monitoring tumor response to immunotherapy. eClinicalMedicine. 2022;46:101381. doi: 10.1016/j.eclinm.2022.101381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sulzmaier F.J., Jean C., Schlaepfer D.D. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Best M.G., Sol N., Kooi I., Tannous J., Westerman B.A., Rustenburg F., Schellen P., Verschueren H., Post E., Koster J., et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 2015;28:666–676. doi: 10.1016/j.ccell.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.-H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The Immune Landscape of Cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moufarrej M.N., Vorperian S.K., Wong R.J., Campos A.A., Quaintance C.C., Sit R.V., Tan M., Detweiler A.M., Mekonen H., Neff N.F., et al. Early prediction of preeclampsia in pregnancy with cell-free RNA. Nature. 2022;602:689–694. doi: 10.1038/s41586-022-04410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen M., Reddy M., Nolan R., Camunas-Soler J., Khodursky A., Scheller N.M., Cantonwine D.E., Engelbrechtsen L., Mi J.D., Dutta A., et al. RNA profiles reveal signatures of future health and disease in pregnancy. Nature. 2022;601:422–427. doi: 10.1038/s41586-021-04249-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadeh R., Sharkia I., Fialkoff G., Rahat A., Gutin J., Chappleboim A., Nitzan M., Fox-Fisher I., Neiman D., Meler G., et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat. Biotechnol. 2021;39:586–598. doi: 10.1038/s41587-020-00775-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.CCGA Consortium. Oxnard G.R., Klein E.A., Swanton C., Seiden M.V., Liu M.C., Oxnard G.R., Klein E.A., Smith D., Richards D., et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020;31:745–759. doi: 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu M.C., Klein E., Hubbell E., Maddala T., Aravanis A.M., Beausang J.F., Filippova D., Gross S., Jamshidi A., Kurtzman K., et al. Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Ann. Oncol. 2018;29:viii14. doi: 10.1093/annonc/mdy269.048. [DOI] [Google Scholar]
- 43.Vasimuddin M., Misra S., Li H., Aluru S. 2019. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. 20–24 May 2019. pp. 314-324. [Google Scholar]
- 44.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S., Li Y., Chen B., Zhao J., Yu S., Tang Y., Zheng Q., Li Y., Wang P., He X., Huang S. exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018;46:D106–D112. doi: 10.1093/nar/gkx891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 49.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. Next Generation Sequencing Data Analysis. 2011;17 doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 50.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krueger F., Frankie J., Phil E., Afyounian E., Schuster-Boeckler B. FelixKrueger/TrimGalore: v0.6.7 - DOI via Zenodo (0.6.7) Zenodo. 2021 doi: 10.5281/zenodo.5127899. [DOI] [Google Scholar]
- 52.Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen S., Park J.W., Lu Z.-x., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lienhard M., Grimm C., Morkel M., Herwig R., Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;30:284–286. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frank, E., Harrell, J.r., (2020). Charles Dupont. Hmisc: Harrell Miscellaneous. R package version 4.4-1.
- 60.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paczkowska M., Barenboim J., Sintupisut N., Fox N.S., Zhu H., Abd-Rabbo D., Mee M.W., Boutros P.C., PCAWG Drivers and Functional Interpretation Working Group. Reimand J., PCAWG Consortium Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020;11:735. doi: 10.1038/s41467-019-13983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Breiman L. Random Forests. Mach. Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 67.Shen S.Y., Burgener J.M., Bratman S.V., De Carvalho D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019;14:2749–2780. doi: 10.1038/s41596-019-0202-2. [DOI] [PubMed] [Google Scholar]
- 68.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 69.Schneider V.A., Graves-Lindsay T., Howe K., Bouk N., Chen H.-C., Kitts P.A., Murphy T.D., Pruitt K.D., Thibaud-Nissen F., Albracht D., et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27:849–864. doi: 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glažar P., Papavasileiou P., Rajewsky N. 2014. circBase: A Database for Circular RNAs. RNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S., et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu G., Guo Y.A., Ho D., Poon P., Poh Z.W., Wong P.M., Gan A., Chang M.M., Kleftogiannis D., Lau Y.T., et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat. Commun. 2021;12:2229. doi: 10.1038/s41467-021-22463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frank E., Harrell C.D., Jr. 2020. Hmisc: Harrell Miscellaneous. R Package Version 4.4-1. [Google Scholar]
- 80.Snyder M.W., Kircher M., Hill A.J., Daza R.M., Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang P., Sun K., Peng W., Cheng S.H., Ni M., Yeung P.C., Heung M.M.S., Xie T., Shang H., Zhou Z., et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020;10:664–673. doi: 10.1158/2159-8290.CD-19-0622. [DOI] [PubMed] [Google Scholar]
- 82.Cristiano S., Leal A., Phallen J., Fiksel J., Adleff V., Bruhm D.C., Jensen S.Ø., Medina J.E., Hruban C., White J.R., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Picardi E., D'Erchia A.M., Lo Giudice C., Pesole G. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017;45:D750–D757. doi: 10.1093/nar/gkw767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Castel S.E., Levy-Moonshine A., Mohammadi P., Banks E., Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015;16:195. doi: 10.1186/s13059-015-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh S., Qin F., Kumar S., Elfman J., Lin E., Pham L.-P., Yang A., Li H. The landscape of chimeric RNAs in non-diseased tissues and cells. Nucleic Acids Res. 2020;48:1764–1778. doi: 10.1093/nar/gkz1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee M., Lee K., Yu N., Jang I., Choi I., Kim P., Jang Y.E., Kim B., Kim S., Lee B., et al. ChimerDB 3.0: an enhanced database for fusion genes from cancer transcriptome and literature data mining. Nucleic Acids Res. 2017;45:D784–D789. doi: 10.1093/nar/gkw1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Su G., Morris J.H., Demchak B., Bader G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinformatics. 2014;47:8.13.1–8.13.24. doi: 10.1002/0471250953.bi0813s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li B., Pei G., Yao J., Ding Q., Jia P., Zhao Z. Cell-type deconvolution analysis identifies cancer-associated myofibroblast component as a poor prognostic factor in multiple cancer types. Oncogene. 2021;40:4686–4694. doi: 10.1038/s41388-021-01870-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All FASTQ files generated in this study have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All original code has been deposited at Github and is publicly available at https://github.com/tyh-19/Pipeline-for-multiomics. All software being used in this study was summarized with versions and references in Table S6. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.