

Summary
The clinical utility of human interleukin-2 (hIL-2) is limited by its short serum half-life, preferential activation of regulatory T (TReg) over immune effector cells, and dose-limiting toxicities. We previously engineered F10 immunocytokine (IC), an intramolecularly assembled cytokine/antibody fusion protein that linked hIL-2 to an anti-IL-2 antibody (denoted F10) that extended IL-2 half-life and augmented the immune effector to TReg ratio. Here, we leveraged molecular engineering to improve the anti-tumor therapeutic efficacy and tolerability of F10 IC by developing an iteration, denoted F10 IC-CBD (collagen binding domain), designed for intratumoral administration and in situ retention based on collagen affinity. F10 IC-CBD retained IL-2 bioactivity exclusively in the tumor and eliminated IL-2-associated toxicities. Furthermore, F10 IC exhibited potent single-agent therapeutic efficacy and synergy with systemic immune checkpoint blockade and elicited an abscopal response in mouse tumors models. This engineered fusion protein presents a prototype for the design of intratumoral therapies.
Keywords: interleukin-2, immunocytokine, cancer, immunotherapy, intratumoral administration, antibody, collagen affinty
Graphical abstract
Highlights
-
•
Engineered an effector-biased interleukin-2 immunocytokine with collagen affinity
-
•
Immunocytokine designed for intratumoral administration and in situ retention
-
•
Immunocytokine retains IL-2 bioactivity in the tumor and eliminates toxicity
-
•
Immunocytokine exhibits potent anti-tumor effects alone and in combination therapy
The anti-cancer utility of interleukin-2 (IL-2) is limited by its short serum half-life, preferential activation of regulatory T cells, and dose-limiting toxicities. Silver et al. engineered an immunocytokine with collagen affinity that retains effector-biased IL-2 bioactivity exclusively in the tumor, eliminates IL-2 toxicity, and augments the therapeutic efficacy of IL-2.
Introduction
Intravenous administration of high-dose (HD) human interleukin-2 (hIL-2) (aldesleukin) was approved by the US Food and Drug Administration for the treatment of metastatic renal cell carcinoma (RCC) in 1992 and metastatic melanoma in 1998. While HD IL-2 elicits clinical responses in 20% of patients with metastatic RCC1 and 16% of patients with metastatic melanoma,2 its clinical use has been limited by its short in vivo half-life3,4 and pleiotropic activation of both immune effector (Eff) and regulatory T (TReg) cells.5,6,7 These limitations necessitate the high dosing of IL-2. However, HD IL-2 is accompanied by dose-limiting toxicity, most prominently vascular leak syndrome (VLS), which results in intravascular fluid accumulation in organs such as the lungs and liver, and ultimately pulmonary edema and liver damage.8,9,10,11
IL-2 engages the IL-2 receptor (IL-2R) by distinct configurations of the IL-2Rα (CD25), IL-2Rβ (CD122), and common γ (γc or CD132) subunits. Signaling occurs with nanomolar affinity (KD ∼1 nM) through the dimeric IL-2Rβγc complex or with picomolar affinity (KD ∼10 pM) through the trimeric IL-2Rαβγc complex; thus, the presence of IL-2Rα increases the affinity of IL-2 for its receptor by 100-fold.5,6,7 The intermediate affinity IL-2Rβγc complex is mainly expressed on natural killer (NK) cells and naive and resting effector CD4+ and CD8+ T cells.5,6,7 Although activated CD4+ and CD8+ T cells can transiently upregulate IL-2Rα, the high-affinity IL-2Rαβγc complex is predominantly expressed on TReg cells, which constitutively express IL-2Rα.5,6,7 TReg cells, which act to dampen anti-tumor responses, are therefore hyper-responsive to IL-2.
To increase the therapeutic potential of hIL-2, we previously engineered an IL-2 cytokine/antibody fusion protein, denoted F10 immunocytokine (IC), that intramolecularly fuses hIL-2 to an engineered anti-IL-2 antibody (F10; derived from MAB602)9,12,13 that sterically occludes the IL-2/IL-2Rα interaction while allosterically enhancing the IL-2/IL-2Rβ interaction.14 This approach extends the circulating half-life of IL-2 (presumably through interaction of the IC fragment crystallizable [Fc] domain with the neonatal Fc receptor [FcRn]),15 and it eliminates the stimulatory advantage conferred to TReg cells by IL-2Rα, thereby enhancing the activation of Effs. Indeed, we demonstrated the immunostimulatory bias of F10 IC compared to the natural IL-2 cytokine in both cellular and animal models.14 However, this molecule is limited in its anti-tumor efficacy and fails to address the dose-limiting toxicity of IL-2, which is thought to be caused by the on-target, off-tumor activity of IL-2 on non-tumor-specific immune cells and pulmonary and hepatic vascular endothelial cells, as well as the indirect effects of IL-2 on vascular permeability exerted through inflammatory cytokines secreted by systemically activated leukocytes.8,9,10,11
In an effort to maximize the anti-tumor efficacy of administered immunotherapies while minimizing their systemic exposure and on-target, off-tumor activity, intratumoral delivery has established clinical relevance.16,17,18 However, unanchored intratumorally administered therapies may still reach systemic circulation via lymphatic draining, diffusion, and/or the leaky tumor vasculature, resulting in the same toxicity as the systemically administered therapy.19,20 Coupling immunotherapies that are designed to engage proteins that are associated with and/or abundant in the tumor microenvironment (TME) with intratumoral administration constitutes a feasible strategy to prevent systemic exposure and retain locally delivered therapies in the tumor. Collagen, the major structural component of extracellular matrix, is of particular interest as a target due to its abundant and ubiquitous expression in tumors.21,22 Indeed, designing cytokines and antibodies with collagen affinity has been previously shown to promote the homing23,24 or anchoring25,26,27,28 of therapies to the tumor microenvironment following intravenous and intratumoral administration, respectively.
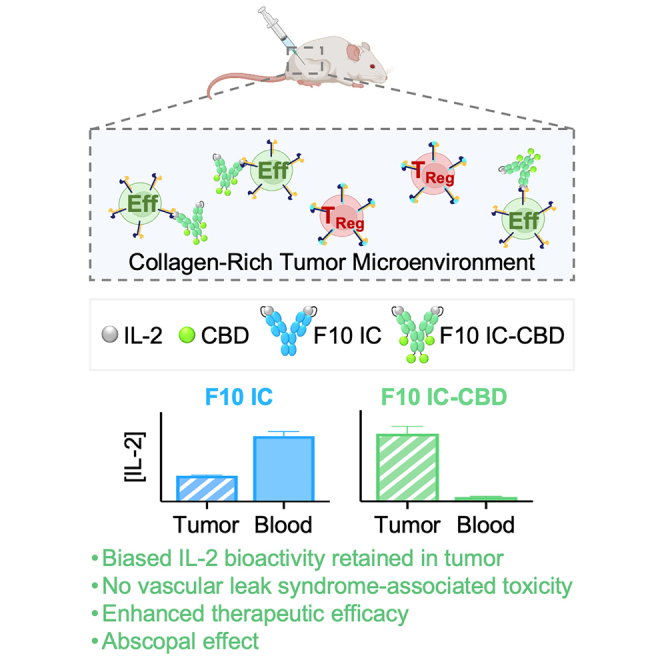
Here, we report the design and characterization of F10 IC-CBD, an intratumorally administered iteration of the Eff-promoting F10 IC that binds to collagen in the TME owing to its genetic fusion to collagen binding domains (CBDs). Upon intratumoral administration, F10 IC-CBD is physically retained in the tumor, thereby increasing its in situ bioavailability. F10 IC minimizes the systemic exposure and toxicity of F10 IC and increases and prolongs the bioactivity of F10 IC exclusively at the tumor, leading to potent single-agent anti-tumor activity. F10 IC-CBD is also capable of eliciting an abscopal response, underscoring the potential for local administration to generate systemic immune responses. Moreover, the anti-tumor activities of F10 IC-CBD safely synergize with immune checkpoint blockade, suggesting potential combination strategies with intratumorally and systemically delivered immunotherapies.
Results
CBD-fused F10 ICs preserve the biophysical activity of the parent F10 IC, occluding the IL-2/IL-2Rα interaction while enhancing the IL-2/IL-2Rβ interaction
The anti-cancer therapeutic efficacy of hIL-2 is hindered by its short half-life3,4 and preferential activation of TReg cells (which constitutively express the high-affinity IL2Rα subunit) at low doses, both of which necessitate high levels of cytokine administration that prompt dose-limiting toxicities. To extend the therapeutic potential of hIL-2, we previously engineered F10 IC, which tethers the C terminus of hIL-2 to the N termini of the light chains of a full-length anti-IL-2 antibody (Figure 1A) that occludes cytokine binding to IL-2Rα while enhancing binding to IL-2Rβ.14 We sought to design an iteration of F10 IC that binds collagen and would therefore be poised for intratumoral administration and retention. The A3 domain of von Willebrand factor (vWF), a plasma protein involved in hemostasis,29 is natively responsible for binding subendothelial collagen and mediating platelet adhesion at sites of vascular injury.29 This domain (hereafter denoted CBD) has been previously employed for genetic fusion to cytokines and covalent chemical conjugation to amine groups in immune checkpoint blockade antibodies to enhance therapeutic localization to tumors following intravenous administration.23,24 To assess the feasibility of extending this strategy to both F10 IC and intratumoral administration, we designed and expressed F10 ICs linked to CBDs in various C-terminal fusion topologies: heavy chain fusion (F10 IC-CBDHC); light chain fusion (F10 IC-CBDLC); or both heavy and light chain fusion (F10 IC-CBDHC/LC) (Figures 1A and S1A–S1C).
Figure 1.

CBD-fused F10 ICs preserve the biophysical and functional activities of parent F10 IC
(A) Schematic of unmodified F10 IC and CBD-fused ICs.
(B–D) Equilibrium biolayer interferometry titrations of hIL-2, F10 IC, CBD-fused F10 ICs, MAB602, and an irrelevant antibody (irrel. Ab, tocilizumab) binding to immobilized hIL-2 (B), hIL-2Rα (C), and hIL-2Rβ (D).
(E and F) STAT5 phosphorylation responses of IL-2Rα− (E) and IL-2Rα+ (F) YT-1 cells stimulated with hIL-2, unmodified F10 IC, or CBD-fused F10 ICs.
(G) IL-2Rα−:IL-2Rα+ YT-1 EC50 ratios for STAT5 phosphorylation assays.
(H–J) STAT5 phosphorylation responses of human CD8+ T (H), Tconv (I), and TReg (J) cells stimulated with hIL-2, unmodified F10 IC, or CBD-fused F10 ICs.
(K) CD8+ T:TReg EC50 ratios.
(L) Tconv:TReg EC50 ratios. Data are depicted as mean ± SD. Full statistical comparisons for (G), (K), and (L) are presented in Table S6. See also Figure S1 and Tables S1–S3.
To assess how these modifications to F10 IC affected the binding of F10 IC to hIL-2 and the IL-2R subunits, biolayer interferometry was performed with immobilized hIL-2, hIL-2Rα, and hIL-2Rβ. Reflecting the intramolecular tethering of hIL-2 to the F10 anti-IL-2 antibody, neither unmodified F10 IC nor any of the CBD-fused F10 ICs bound immobilized hIL-2; binding was only observed for the positive control anti-IL-2 antibody MAB6029,12,13 (Figure 1B; Table S2), which is the parent molecule from which the F10 antibody was evolved through selection for both superior IL-2/IL-2Rα interaction occlusion and enhanced IL-2/antibody binding affinity.14 As expected, neither unmodified F10 IC nor any of the CBD-fused F10 ICs bound immobilized hIL-2Rα (Figure 1C; Table S2), but unmodified F10 IC and all CBD-fused F10 ICs bound immobilized hIL-2Rβ with greater affinity than hIL-2 (Figure 1D; Table S2), due to both bivalent presentation and allosteric enhancement of hIL-2Rβ affinity. Collectively, these binding studies demonstrated that the biophysical properties of the parent F10 IC were preserved upon CBD fusion.
CBD-fused F10 ICs preserve the functional activity of the parent F10 IC, enhancing activation of effector immune cells
To ensure that our engineered CBD-fused F10 ICs recapitulated the signaling behavior of the parent F10 IC, we conducted IL-2R activation studies using IL-2Rα− YT-130 and IL-2Rα+ YT-131 human NK cells as surrogates for immune effector and TReg cells, respectively. Signal transducer and activator of transcription 5 (STAT5) phosphorylation was used as a readout for IL-2-mediated signaling. On IL-2Rα− YT-1 (Eff-like) cells, unmodified and CBD-fused F10 ICs activated signaling with comparable potency to that of hIL-2 (Figure 1E; Table S3). However, on IL-2Rα+ YT-1 (TReg-like) cells, unmodified and CBD-fused F10 ICs induced attenuated activation relative to hIL-2 (Figure 1F; Table S3) due to blockade of IL-2Rα engagement. Comparison of half-maximal signaling activity (EC50) on IL-2Rα− versus IL-2Rα+ YT-1 cells showed that whereas hIL-2 was biased toward TReg-like (IL-2Rα+) cells, unmodified and CBD-fused F10 ICs were biased toward Eff-like (IL-2Rα−) cells (Figure 1G; Table S3). The results of the YT-1 signaling studies were consistent with findings in a mixed population of human peripheral blood mononuclear cells (PBMCs). CD3+CD4+FoxP3– Tconv and CD3+CD4+CD25+FoxP3+ TReg cells were exquisitely sensitive to soluble hIL-2 and substantially less sensitive to unmodified and CBD-fused F10 ICs (Figures 1I and 1J; Table S3). hIL-2 and the unmodified CBD-fused F10 ICs were less potent on CD3+CD8+ T cells compared to CD4+ T cell subsets, and while CD8+ T cells were also more sensitive to hIL-2 than any IC, the magnitude of the sensitivity difference was substantially smaller on CD8+ T cells compared to Tconv or TReg cells (Figure 1H; Table S3). Comparison of EC50 values across Eff and TReg cell subsets revealed that hIL-2 preferentially activated TReg cells over both CD8+ T and Tconv (Figures 1K and 1L; Table S3). In contrast, unmodified and CBD-fused F10 ICs were biased toward the preferential activation of both CD8+ T and Tconv cells over TReg cells (Figures 1K and 1L; Table S3). Taken together, these activation studies confirm that fusion to CBD does not impact the cell signaling profile of the parent F10 IC and that the preferential stimulation of immune effector over TReg cells is maintained.
CBD-fused F10 ICs bind collagen, leading to enhanced tumor retention and reduced systemic IL-2 exposure compared to the parent IC
We next examined the capacity of our engineered CBD-fused F10 ICs to bind collagen. CBD (the A3 domain of vWF factor) is known to bind to collagen types I and III.32,33 Indirect ELISA performed on collagen-coated plates revealed that, as expected, unmodified F10 IC bound to neither collagen type I nor III (Figures 2A and 2B; Table S4). In contrast, all topologies of F10 IC-CBD bound type III collagen, with F10 IC-CBDHC/LC binding with the highest affinity (KD = 35 nM), 11-fold and 4-fold greater than that of F10 IC-CBDHC or F10 IC-CBDLC, respectively (Figure 2A; Table S4). F10 IC-CBDHC/LC bound to collagen type I as well, albeit with lower affinity (KD = 420 nM), whereas binding of F10 IC-CBDHC and F10 IC-CBDLC was undetectable (Figure 2B; Table S4). Importantly, we demonstrate that collagen binding does not interfere with IL-2R signaling, as CBD-fused F10 IC induced equivalent STAT5 activation and immune effector-like cell bias in the presence of collagen type I, collagen type III, or 2% bovine serum albumin (BSA, used as a control) (Figures S2A–S2I). We reasoned that higher affinity collagen interactions would result in superior in situ bioavailability upon intratumoral administration. In support of this hypothesis, recent work has demonstrated that the affinity of locally delivered collagen-binding therapies to collagen is a key determinant of local exposure, with higher matrix affinities yielding superior retention.26 We therefore proceeded with F10 IC-CBDHC/LC (hereafter referred to as F10 IC-CBD) for in vivo characterization.
Figure 2.

CBD-fused F10 ICs bind collagen, leading to enhanced tumor retention and reduced systemic IL-2 exposure compared to the parent IC
(A and B) Binding of unmodified F10 IC and CBD-fused F10 ICs to (A) collagen type I and (B) collagen type III.
(C–E) Near IR-labeled unmodified and F10 IC-CBD were intratumorally injected into CT26 tumor-bearing mice (n = 3/group) and intratumoral fluorescence imaged over time (C). Normalized intratumoral fluorescence over time (D) and area under the curve (AUC) (E) are presented.
(F–G) Unmodified and F10 IC-CBD were intratumorally injected into CT26 tumor-bearing mice (n = 4/group). IL-2 plasma concentrations (F) and AUC (G) are graphed. Data are depicted as mean ± SD. (E) and (G) were analyzed by unpaired Student’s t test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Non-significant comparisons are not depicted. Full statistical comparisons for each panel are presented in Table S6. See also Figure S2 and Tables S4 and S5.
To evaluate whether fusion of F10 IC to CBD would improve intratumoral retention, we utilized in vivo fluorescence imaging to monitor the in situ persistence of fluorescently labeled F10 IC-CBD relative to an equimolar IL-2 dose of unmodified F10 IC following intratumoral injection into CT26 colon carcinoma-bearing mice. Consistent with prior reports that unanchored, intratumorally administered therapies may still reach systemic circulation,19,20 unmodified F10 IC rapidly exited the tumor (Figures 2C and 2D). However, the egress of F10 IC-CBD from the tumor was notably slower (Figures 2C and 2D). 1 day post intratumoral administration, only 28% of administered F10 IC was retained in the tumor, while the majority (70%) of F10 IC-CBD was retained (Figures 2C and 2D). The fitted half-life for F10 IC-CBD (t1/2 = 38.4 h) was longer than that of F10 IC (t1/2 = 11.8 h) (Table S5) resulting in an >2.5-fold higher overall exposure of the tumor to F10 IC-CBD relative to unmodified F10 IC over the 7-day time course, as assessed by the area under the curve (Figure 2E; Table S5).
We conducted a pharmacokinetic study to determine the extent to which the observed intratumoral retention of F10 IC-CBD prevented systemic exposure. Mouse-weight-adjusted equimolar IL-2 doses of fluorescently labeled unmodified F10 IC or F10 IC-CBD were intratumorally administered into CT26 colon carcinoma-bearing mice, and plasma concentration of IL-2 was monitored over 7 days (Figure 2F). F10 IC was detectable in the plasma within 6 h post intratumoral administration and had sustained systemic exposure over time with peak plasma concentration (Cmax = 3.16 μg IL-2/mL) at 24 h (Figure 2F; Table S5). In contrast, F10 IC-CBD remained undetectable in the plasma (Figure 2F) and showed >16-fold lower systemic exposure than F10 IC, as measured by the area under of the curve (Figure 2G; Table S5). While it has been posited that the rapid egress of therapies from the tumor following intratumoral administration may be due to their small size,19,20,26 here we demonstrate that larger biologics such as F10 IC (MW ∼180 kDa) are also rapidly detectable in the serum following intratumoral delivery, further underscoring the utility of anchored intratumoral administration. Collectively, these results demonstrate that CBD-fused F10 ICs bind collagen, and that the lead fusion protein (F10 IC-CBDHC/LC [F10 IC-CBD]) exhibits increased in situ tumor availability, a favorable pharmacokinetic profile, and no systemic exposure following intratumoral administration.
F10 IC-CBD prevents systemic and vascular leak syndrome-associated IL-2 toxicities
VLS is the major dose-limiting toxicity associated with HD IL-2 administration and results in intravascular fluid accumulation in the lungs and liver that leads to pulmonary edema and liver damage.8 VLS is thought to be caused by the on-target off-tumor activity of IL-2 during systemic exposure.8,9,10,11 To evaluate the effects of local administration and retention on systemic and VLS-associated toxicity, we intratumorally administered PBS or a high equimolar IL-2 dose of unmodified F10 IC or F10 IC-CBD into CT26 colon carcinoma-bearing mice and assessed the impact of treatment on mouse weight, pulmonary edema, and liver damage. By 7 days post intratumoral administration, mice treated with unmodified F10 IC had significantly lower body weights, losing an average of 12.4% of their initial body weights (Figure 3A). In contrast, PBS- and F10 IC-CBD-treated mice gained an average of 1.8% and 2.2% of their initial body weights, respectively (Figure 3A). Fluid accumulation in excised lungs 72 h after intratumoral dosing was used as a measure of pulmonary edema. Whereas pulmonary fluid content in PBS- and F10 IC-CBD-treated mice was comparable, F10 IC-treated mice exhibited significantly higher (1.6-fold) pulmonary fluid content (Figure 3B). Serum concentrations of the liver enzymes aspartate aminotransferase (AST) and alanine transaminase (ALT) in circulation were quantified by ELISA as a measurement of liver damage 72 h post intratumoral dosing. F10 IC-treated mice had significantly higher (>1.4-fold) levels of serum ALT compared to both PBS- and F10 IC-CBD-treated mice (Figure 3C). There were no significant differences in serum AST levels across treatment groups (Figure 3D). Overall, these results indicate that increasing retention of F10 IC in the tumor through collagen anchoring prevents the systemic and VLS-associated toxicities associated with IL-2 delivery and demonstrate that F10 IC-CBD has a wider safety margin compared to unmodified F10 IC.
Figure 3.

F10 IC-CBD prevents systemic and vascular leak syndrome-associated IL-2 toxicities
CT26 tumor-bearing mice (n = 5/group) were intratumorally administered PBS or 0.5 mg/kg IL-2 equivalent dose of unmodified F10 IC or F10 IC-CBD. (A) Body weight change, (B) pulmonary fluid content, and serum ALT (C) and ALT (D) concentrations were assessed. Data are depicted as mean ± SD. (A)–(D) were analyzed by ordinary one-way ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Non-significant comparisons are not depicted. Full statistical comparisons for each panel are presented in Table S6.
F10 IC-CBD enhances and prolongs F10 IC bioactivity exclusively in the tumor
The time-dependent effects of intratumoral administration and retention of therapies on immune cell expansion and tumor infiltration have not been previously described. To assess the impact of increasing the intratumoral bioavailability of F10 IC on its bioactivity, we performed a pharmacodynamic immune profiling study. We administered a single dose of PBS or an equimolar IL-2 dose of unmodified F10 IC or F10 IC-CBD into CT26 colon carcinoma-bearing mice and profiled the tumor infiltrating leukocyte (TIL) and splenocyte immune responses at 2, 4, and 7 days post intratumoral administration (Figures 4 and S3).
Figure 4.

F10 IC-CBD enhances and prolongs F10 IC bioactivity in the tumor
CT26 tumor-bearing mice (n = 5/group) were intratumorally administered a single dose of PBS or 2.5 μg IL-2 equivalent of unmodified F10 IC or F10 IC-CBD.
(A) Schematic of experimental design.
(B) Tumor volume, (C) tumor weight, and (D) TILs per mg of tumor tissue at 2, 4, and 7 days post intratumoral injection.
(E) NK, (F) CD8+ T, (G) Tconv, and (H) TReg cell numbers per mg of tumor tissue. Data are depicted as mean ± SD. (B)–(H) were analyzed by two-way ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Non-significant comparisons are not depicted. Full statistical comparisons for each panel are presented in Table S6. See also Figure S3.
While there were no significant differences in tumor volume or weight across treatment groups on days 2 or 4, on day 7, F10 IC-CBD-treated mice had significantly smaller tumors by both volume and weight compared to PBS- or F10 IC-treated mice (Figures 4B and 4C). Moreover, 7 days post intratumoral dosing, F10 IC-CBD-treated mice showed a remarkable increase in the number of CD45+ TILs compared to mice treated with PBS (>5-fold) or F10 IC (>4-fold) (Figure 4D). Similarly, 7 days post intratumoral administration, F10 IC-CBD-treated mice showed >3.5-fold and >5.5-fold average increases in the numbers of CD3−CD49b+ NK, CD3+CD8+ T, and CD3+CD4+FoxP3– Tconv cells relative to PBS- or unmodified F10 IC-treated mice, respectively (Figures 4E–4G). Elevated TReg cell infiltration was also observed in F10 IC-CBD-treated mice compared to PBS- or unmodified F10 IC-treated mice 7 days post intratumoral dosing. However, the enrichment of infiltrating TReg cells 7 days post treatment with F10 IC-CBD was noticeably lower than that of the other immune subsets, reflecting the preferential activation of immune effector cells by F10 IC (Figure 4H). These results were not unexpected based on in vitro signaling data (Figures 1F and 1J; Table S3) showing that although its activity is biased, F10 IC activates both IL-2Rα+ and IL-2Rα− cells. This finding suggests that F10 IC-CBD can engage heterodimeric IL-2R complexes on tumor-infiltrating TReg cells and/or that IL-2 may dissociate from F10 within the immunocytokine, given that the affinities of IL-2 for both IL-2Rα (KD ∼ 3 nM; Table S2) and the high-affinity heterotrimeric IL-2R complex (KD ∼ 10 p.m.)5,6,7 are stronger than that for the F10 single-chain variable fragment (KD ∼ 6 nM).14
Consistent with its rapid egress from the tumor and prolonged systemic exposure, the bioactivity of unmodified F10 IC was largely confined to the spleen, with unmodified F10 IC expanding a markedly higher number of splenocytes compared to both PBS and F10 IC-CBD at both 4 and 7 days post intratumoral dosing (Figure S3D). In addition, significant increases in total numbers of NK, CD8+ T, and TReg cells in the spleen were observed at both 4 and 7 days following unmodified F10 IC treatment (Figures S3E, S3F, and S3H). The largest expansion occurred for NK cells, with unmodified F10 IC-treated mice expanding >6-fold higher numbers of NK cells in the spleen than both PBS- and F10 IC-CBD-treated mice (Figure S3E). No significant differences were observed in the number of Tconv cells across treatment cohorts (Figure S3G). Notably, F10 IC-CBD did not impact the numbers or phenotypes of immune cells in the spleen relative to the PBS control group at any time point, which is likely explained by the retention of IL-2 in the tumor (Figures S3D–S3H).
We previously demonstrated that, in a naive mouse, systemically delivered unmodified F10 IC induces an in vivo bias toward the preferential expansion of immune Eff over TReg cells in the spleen.14 F10 IC-CBD-treated mice showed similar bias toward the preferential expansion of NK (at 2, 4, and 7 days post treatment), CD8+ T (at 7 days post treatment) and Tconv (at 2, 4, and 7 days post treatment) over TReg cells in the tumor (Figures S3A–S3C), with NK and CD8+ T cell biases at days 2 and 7 respectively significantly improved compared to mice treated with unmodified F10 IC or PBS. Consistent with observations regarding systemic exposure, unmodified F10 IC, but not F10 IC-CBD, showed significantly improved bias toward NK cells over TReg cells in the spleen on days 4 and 7 post intratumoral injection (Figures S3I–S3K). Collectively, the results of these studies establish that intratumorally administered F10 IC-CBD increases and prolongs F10 IC bioactivity in the tumor, while the effects of unanchored F10 IC are largely limited to spleen.
Fusion to CBD safely potentiates the single-agent anti-tumor efficacy of F10 IC
While differences in tumor size and TIL numbers were not significantly different across groups at 4 days post intratumoral administration in our pharmacodynamic immune cell profiling study, we observed a trend toward smaller tumor sizes and higher numbers of CD45+ TILs on day 4 for F10 IC-CBD- relative to PBS- or unmodified F10 IC-treated mice (Figures 4B–4D). Based on this observation, we hypothesized that intratumoral dosing at 4-day intervals would maximize the therapeutic potential of F10 IC-CBD. We characterized the single-agent therapeutic performance of F10 IC-CBD in both the highly immunogenic CT26 colon carcinoma model and the poorly immunogenic but highly aggressive B16F10 melanoma model.34 Mice with established tumors were given intratumoral injections of PBS or an equimolar IL-2 dose of unmodified F10 IC or F10 IC-CBD for a total of three doses, each spaced 4 days apart. Mice in all conditions were sacrificed for terminal immune profiling immediately after the last injection (Figures 5, 6, S4, and S5).
Figure 5.

Fusion to CBD safely potentiates the single-agent anti-tumor efficacy of F10 IC in CT26 colon carcinoma
CT26 tumor-bearing mice (n = 6/group) were treated with intratumoral PBS or 2.5 μg IL-2 equivalent of unmodified F10 IC or F10 IC-CBD.
(A) Schematic of study design.
(B) Tumor volume and (C) mouse body weight.
(D) Number of TILs per mg of tumor tissue, (E) NK, (F) CD8+ T, (G) Tconv, (H) TReg, (I) antigen-specific (AH1+) CD8+ T, and (J) PD-1+ CD8+ T cells per mg of tumor tissue at the terminal time point. (B)–(C) were analyzed by two-way ANOVA, and data are depicted as mean ± SD. (D)–(I) were analyzed by ordinary one-way ANOVA, and data are depicted as mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Non-significant comparisons are not depicted. Full statistical comparisons for each panel are presented in Table S6. See also Figures S4 and S6.
Figure 6.

Fusion to CBD safely potentiates the single-agent anti-tumor efficacy of F10 IC in B16F10 melanoma
B16F10 tumor-bearing mice (n = 5/group) were treated with intratumoral PBS or 2.5 μg IL-2 equivalent of unmodified F10 IC or F10 IC-CBD three times, with injections spaced 4 days apart.
(A) Schematic of study design.
(B) Tumor volume and (C) mouse body weight.
(D) Number of TILs per mg of tumor tissue, (E) NK, (F) CD8+ T, (G) Tconv, (H) TReg, (I) antigen-specific (TRP2+) CD8+ T, and (J) PD-1+ CD8+ T cells per mg of tumor tissue at the terminal time point. (B)–(C) were analyzed by two-way ANOVA, and data are depicted as mean ± SD. (D)–(I) were analyzed by ordinary one-way ANOVA, and data are depicted as mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Non-significant comparisons are not depicted. Full statistical comparisons for each panel are presented in Table S6. See also Figure S5.
By both volume and weight, CT26 colon carcinoma-bearing mice treated with F10 IC-CBD had significantly smaller tumors at the terminal time point compared to PBS- and unmodified F10 IC-treated mice (Figures 5B and S4A). In addition, unanchored F10 IC did not show single-agent efficacy and induced significant systemic toxicity, with treated mice losing an average of 4.7% of their initial body weight at the time point of greatest weight change (Figure 5C). In contrast, PBS- and F10 IC-CBD-treated mice gained weight throughout the course of the study and at the terminal time point had gained an average of 3.6% and 4.5% of their initial body weights, respectively (F10 IC-treated mice lost an average of 1.2% of their initial body weights at this time point). These results are consistent with the on-target off-tumor activity and toxicity of unmodified F10 IC, which is further reflected in the significantly increased splenocyte expansion of F10 IC-treated mice relative to PBS- and F10 IC-CBD-treated mice (Figure S4E). At the terminal time point, F10 IC-CBD-treated mice had significantly more TILs compared to both PBS- and F10 IC-treated mice (Figure 5D), representing increases in the numbers of CD3−CD49b+ NK, CD3+CD8+ T, CD3+CD4+FoxP3– Tconv, CD3+CD4+CD45+FoxP3+ TReg, immunodominant antigen (murine leukemia virus envelope [MuLV] gp70)-specific (H-2Ld MuLV gp70+ [AH1+]) CD8+ T, and programmed cell death protein 1 (PD-1)+ CD8+ T cells (Figures 5E–5J). Notably, no significant increases in abundance were observed in any of these populations for unmodified F10 IC-treated mice compared to the PBS-treated cohort. F10 IC-CBD was found to bias toward the expansion of NK and CD8+ T (although not Tconv) cells over TReg cells in the tumor (Figures S4B–S4D).
In contrast to its activity in the CT26 colon carcinoma model, unmodified F10 IC exhibited modest single-agent activity in the B16F10 melanoma model. By both volume and weight, B16F10 tumors of mice treated with F10 IC-CBD were significantly smaller than those treated with PBS or unmodified F10 IC at the terminal time point (Figures 6B and S5A). Interestingly, no significant differences in body weight change were observed across treatment conditions (Figure 6C), despite the presence of on-target off-tumor activity for the unmodified F10 IC, evidenced by the increased number of expanded splenocytes (Figure S5E). The differences observed in body weight changes between the B16F10 and CT26 models were likely due to the genetic background of the mouse strains used, as we previously found that systemically delivered F10 IC also induced significant body weight changes in CT26- but not B16F10-bearing mice.14 At the terminal time point, F10 IC-CBD-treated mice had a significantly higher abundance of TILs compared to both PBS- and F10 IC-treated mice (Figure 6D), with significantly increased numbers of CD3−NK1.1+ NK, CD8+ T, Tconv, TReg, antigen (tyrosinase-related protein-2 [TRP2])-specific (H-2Kb TRP2+) CD8+ T, and PD-1+ CD8+ T cells (Figures 6E–6J). F10 IC-CBD was found to bias toward the expansion of NK and CD8+ T (although not Tconv) cells over TReg cells in the tumor (Figures S5B–S5D).
In both the CT26 and B16F10 models, the numbers of total expanded splenocytes remained similar for PBS- and F10 IC-CBD-treated mice likely due to retention of F10 IC-CBD in the tumor (Figures S4E and S5E). However, unmodified F10 IC-treated mice showed significantly more expanded splenocytes with increases in total numbers of NK, CD8+ T, and TReg cells (Figures S4F, S4G, S4I, S5F, S5G, and S5I). Additionally, in both models, unmodified F10 IC but not F10 IC-CBD induced a greater bias toward NK over TReg cell expansion in the spleen (Figures S4J and S5J). Together, these studies demonstrate the safety and single-agent efficacy of F10 IC-CBD in both highly and poorly immunogenic tumor models, propelled by increased immune cell infiltration exclusively in the tumor and suggest that the relative increase in intratumoral TReg cells observed in F10 IC-CBD-treated mice does not abolish F10 IC-CBD therapeutic efficacy.
Whether or not skewed IL-2 activity provides a therapeutic advantage in the context of intratumoral administration has not been previously assessed. To this end, we sought to understand the relative contributions of both collagen binding and biased IL-2 activity in driving the efficacy of F10 IC-CBD. We generated control immunocytokines consisting of wild-type hIL-2 intramolecularly fused to the irrelevant isotype control monoclonal antibody anti-FITC-E235 both with and without fusion to CBD (denoted Control IC-CBD and Control IC, respectively) (Figures S6A–S6D). These molecules allowed for parity in valency and in vivo half-life, thus enabling direct comparison of biased versus unbiased IL-2. We demonstrated that the unmodified Control IC and CBD-fused Control IC bound identically to the IL-2 cytokine and its receptor subunits, and binding properties were found to be as expected relative to native IL-2 (Figures S6E–S6G). We further showed that Control IC-CBD but not Control IC bound to collagen types I and III (Figures S6H and S6I) and that the control immunocytokines elicited similar STAT5 activation profiles to both one another and to native IL-2 on IL-2Rα− and IL-2Rα+ YT-1 cells in signaling assays performed on plates coated with collagen types I and III (Figures S6J–S6M). We then compared the anti-tumor therapeutic efficacies of unmodified F10 IC and F10 IC-CBD to those of both unmodified and CBD-fused Control IC in the CT26 model of colorectal cancer. We found that F10 IC elicited a stronger anti-tumor effect than Control IC, which had no effect on tumor progression likely due to the potent activation of TReg cells by the bivalent, unmodified IL-2 (Figure S6N). Furthermore, F10 IC-CBD elicited stronger anti-tumor effects than did the unmodified F10 IC, while fusion of CBD did not improve the therapeutic effects of Control IC (Figure S6N). Consistent with our other experiments, F10 IC caused a significant decrease in mouse body weight compared to all other treatments groups (Figure S6O). Collectively, these results indicate that immune effector cell bias is an important feature driving the efficacy of locally administered IL-2 cancer immunotherapies and that this feature is synergistic with the improvements conferred by tumor retention resulting from collagen binding.
F10 IC-CBD safely synergizes with PD-1 blockade and elicits an abscopal response both as a monotherapy as well as in combination with PD-1 blockade
The elevated abundance of PD-1+ CD8+ T cells for mice treated with F10 IC-CBD relative to mice treated with PBS or unmodified F10 IC in both the CT26 and B16F10 models (Figures 5J and 6J) led us to hypothesize that the therapeutic efficacy of F10 IC-CBD could be increased through combination with PD-1-targeted immune checkpoint blockade. While mice treated with F10 IC-CBD harbored the highest number of PD-1-expressing CD8+ T cells, based solely off of PD-1 expression, it is unclear whether this population represents exhausted or activated cells, as PD-1 expression is a marker for both.36 Nevertheless, recent evidence suggests that sustained IL-2-mediated STAT5 signaling induces CD8+ T cell exhaustion.37,38,39,40 In the context of anti-cancer therapy, both PD-1+ activated and exhausted T cells would benefit from PD-1 blockade. Therefore, to assess potential synergy with PD-1 blockade, mice with established CT26 colon carcinomas or B16F10 melanomas were administered a total of three doses, each spaced 4 days apart, of either intratumoral PBS, unmodified F10 IC, or F10 IC-CBD with simultaneous intraperitoneal administration of either PBS or anti-PD-1 (Figures 7 and S7).
Figure 7.

F10 IC-CBD safely synergizes with PD-1 blockade and elicits an abscopal response as a monotherapy and in combination treatments
CT26 and B16F10 tumor-bearing mice (n = 5–6/group) were treated with intratumoral PBS or 2.5 μg IL-2 equivalent of unmodified F10 IC or F10 IC-CBD in combination with 100 μg intraperitoneal PBS or anti-PD-1.
(A) Schematic of CT26 and B16F10 study designs.
(B) CT26 tumor volume and (D) BALB/cJ mouse survival.
(D) B16F10 tumor volume and (E) C57BL/6J mouse survival.
(F) CT26 Tumor volumes in both the primary (injected, top) and abscopal (non-injected, bottom) tumor. (B), (D), and (F) were analyzed by two-way ANOVA, and data are depicted as mean ± SD. (C) and (E) were analyzed by log rank Mantel-Cox test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Selected significance for each panel is shown for ease of visualization. All statistical comparisons for each panel are presented in Table S6. See also Figure S7.
Consistent with single-agent studies, CT26-bearing mice treated with PBS or unmodified F10 IC experienced uncontrolled tumor growth, whereas F10 IC-CBD significantly slowed tumor progression (Figure 7B). Additionally, whereas PBS- and F10 IC-CBD-treated mice gained weight throughout the study, F10 IC-treated mice experienced weight loss and showed a significantly lower net gain in body weight (Figure S7A). Furthermore, while the median survivals of PBS- and F10 IC-treated mice were 20 and 21 days respectively, the survival of F10 IC-CBD-treated mice was significantly extended, with a median survival of 29 days (Figure 7B). Combining systemic PD-1 blockade with intratumoral PBS or unmodified F10 IC still resulted in uncontrolled tumor growth and led to minimal extension of survival, with median survival occurring at 23 and 22.5 days, respectively (Figures 7B and 7C). In contrast, anti-PD-1 treatment markedly potentiated F10 IC-CBD efficacy, significantly inhibiting tumor growth relative to all other treatment conditions (Figure 7B). Additionally, anti-PD-1 in combination with F10 IC-CBD extended median mouse survival to 42 days with 33% of treated mice exhibiting completely halted tumor growth when the experiment was terminated on day 45 (Figure 7C).
In B16F10-bearing mice, unmodified F10 IC resulted in moderate single-agent efficacy compared to PBS treatment but still did not effectively control tumor growth, whereas F10 IC-CBD-treated mice experienced significantly delayed tumor progression (Figure 7D). Whereas the median survival of PBS- and F10 IC-treated mice was 17 and 19.5 days respectively, the median survival of F10 IC-CBD-treated mice was significantly longer (25 days) (Figure 7E). All mice gained weight throughout the study, which was unchanged by the addition of anti-PD-1 (Figure S7B). Additionally, combining systemic PD-1 inhibition with intratumoral PBS or unmodified F10 IC did not effectively control tumor growth and led to minimal extension of survival, with median survival occurring at days 19.5 and 20.5 respectively (Figures 7D and 7E). In contrast, F10 IC-CBD synergized with anti-PD-1 blockade, although to a lesser extent than was observed for CT26. In combination with anti-PD-1, F10 IC-CBD significantly delayed tumor growth relative to all other treatment conditions (Figure 7D), and it extended median overall survival to 29.5 days (Figure 7E).
In both the CT26 and B16F10 models, the greater anti-tumor response in mice that received F10 IC-CBD in combination with anti-PD-1 compared to mice that received anti-PD-1 alone or unmodified F10 IC in combination with anti-PD-1 was consistent with the increased abundance of PD-1+ CD8+ TILs in F10 IC-CBD-treated mice (Figures 5J and 6J). Additionally, the increased synergy of PD-1 blockade with F10 IC-CBD treatment in the CT26 relative to the B16F10 model is consistent with, and likely explained by, the markedly lower numbers of PD-1+ CD8+ T cells in the latter model. The results of these combination treatment studies highlight the therapeutic benefit of combining F10 IC-CBD with PD-1 blockade in both highly and poorly immunogenic tumors.
An outstanding criticism of intratumoral administration concerns whether abscopal responses can be generated from a locally delivered therapy. We assessed whether F10 IC-CBD was capable of eliciting an abscopal response in the context of the CT26 colon carcinoma model, both as a monotherapy and in combination with PD-1 blockade. We found that, distinct from unmodified F10 IC, F10 IC-CBD is capable of inhibiting growth of a non-injected distal tumor upon intratumoral injection both as a monotherapy and in combination with PD-1 checkpoint blockade (Figure 7F). Unsurprisingly, the generated abscopal response for F10 IC-CBD was potentiated when the molecule was used in combination with PD-1 blockade compared to monotherapy treatment (Figure 7F). In the context of our tumor retention data (Figure 2) and immune profiling studies (Figures 5, 6, S4, and S5), these results suggest that local activation of immune cells is essential for the generation of an abscopal response with F10 IC-CBD. These data are consistent with a previous report of a locally delivered tumor-retained IL-2 therapy eliciting an abscopal response,25 and the efficacy of our molecule in both injected and non-injected tumors further demonstrates the promise of F10 IC-CBD as a cancer immunotherapy.
Discussion
While IL-2 has demonstrated clinical responses in 16% and 20% of patients with metastatic melanoma and metastatic RCC respectively,1,2 high doses are required to achieve these effects due to the pleiotropic activities of IL-25,6,7 and the cytokine’s short serum half-life.3,4 However, the clinical use of HD IL-2 is hampered by dose-limiting toxicities,11 most prominently VLS, which ultimately results from systemic IL-2 exposure. Several engineering approaches have been implemented to bias the activity of IL-2 toward immune effector cells (typically by blocking IL-2Rα engagement or fusing to the IL-2Rα subunit) and/or to extend the cytokine’s serum half-life (through recruitment of FcRn engagement).14,41,42,43 However, these strategies are still challenged by systemic exposure of the cytokine, which limits tumor-specific activity and can lead to on-target off-tumor toxicities. As a result, other approaches have been designed to maximize IL-2 anti-tumor efficacy while minimizing toxicity by localizing IL-2 activity to the tumor. These approaches have predominately focused on the systemic delivery of immunocytokines that fuse IL-2 to the C-terminal end of an antibody or antibody fragment that actively targets a tumor-associated antigen or an extracellular matrix component (most commonly alternatively spliced forms of fibronectin). However, the performance of these systemically delivered immunotherapies varies based on expression of the target antigen, which may be heterogeneous across patients and within tumors.44 Furthermore, systemic exposure and on-target off-tumor activity of these intravenously administered therapies remains an inherent issue.
Intratumoral administration of therapeutics has gained clinical relevance due to its perceived ability to achieve high local drug concentrations and reduced systemic exposure. Intratumoral administration is also of clinical relevance due to its ability to achieve anti-tumor effects on distal or metastasized tumors through abscopal effects as such tumors share common tumor antigens, and immune cells at the treated tumor may be locally primed to act systemically.16,17,18,19,20 Various formulations of IL-2 (e.g., wild type,45,46,47,48 PEGylated,49 fused to a tumor-associated antigen targeting antibody,50 fused to a fibronectin targeting diabody51,52) have been administered intratumorally in clinical studies, with varying degrees of efficacy. A shortcoming of this approach is that the intratumoral administration of unanchored cytokines does not prevent their diffusion or leakage into systemic circulation,19,20 with several clinical studies demonstrating that cytokines are rapidly detectable in circulation following intratumoral administration.53,54,55,56 More recent IL-2 formulations have coupled therapies that actively target tumors with local delivery,57 although use of native IL-2 fails to bias expansion of immune effector over TReg cells. Furthermore, intratumorally administered cytokines that target tumor-associated antigens or fibronectin are still challenged by target expression heterogeneity. Collagen, which is the most abundant protein in the body,58 may present a superior target to overcome expression heterogeneity.
The local delivery of therapies that target collagen is a promising strategy for cancer treatment due to the ubiquity of collagen expression across tumor types.21,22 Previous strategies have demonstrated that antibodies and cytokines can be genetically fused or chemically conjugated to collagen binding moieties, such as the A3 domain of vWF,23,24 the extracellular matrix protein lumican,25,26,27 and the CBD of leukocyte associated immunoglobin like receptor 1 (LAIR1)26,28 for systemic (vWF) or intratumoral (lumican and LAIR1) administration.
We present a new approach that combines intratumoral administration and retention (based on collagen affinity) with an intramolecularly assembled IL-2 cytokine/antibody fusion protein that delivers IL-2 with occluded IL-2Rα binding and enhanced IL-2Rβ binding. This strategy addresses all shortcomings of IL-2 therapy by biasing cytokine activity toward immune Eff cells, ablating IL-2 on-target off-tumor activity, and extending IL-2 in vivo half-life through FcRn-mediated recycling.15 With intratumoral administration, we found that our engineered IL-2 therapy, F10 IC-CBD, increased the in situ tumor bioavailability of IL-2, abolished systemic and VLS-associated IL-2 toxicity, and increased, prolonged, and retained the bioactivity of IL-2 in the tumor. Additionally, we demonstrated that F10 IC-CBD significantly inhibited tumor progression in tumor models of disparate immunogenicity and that both immune effector cell bias and tumor localization were essential to therapeutic efficacy. We also found that our engineered immunocytokine safely synergized with systemic PD-1-targeted immune checkpoint blockade and induced abscopal effects in both the absence and presence of immune checkpoint inhibition. Mechanistic studies showed that tumor infiltration of NK, CD8+ T, and Tconv cells was prominent in the response to intratumoral F10 IC-CBD. F10 IC-CBD also led to elevated numbers of infiltrating TReg cells in the tumor, suggesting that therapies targeting TReg inhibition may also potentiate the performance of our engineered IC.
The anti-tumor activity and lack of toxicity observed for F10 IC-CBD demonstrates its therapeutic promise. Future work will be needed to realize the translational potential for this approach. In particular, optimization of the timing and levels of dosing for both the IC and accompanying therapeutics will be required to advance clinical development. Additionally, the combination of F10 IC-CBD with tumor retained and locally delivered and collagen-binding immune checkpoint blockade may also be promising. In addition, it will be important to test the in vivo activity of our molecule in the context of a human immune system to ensure translatability of our approach to the clinic. Overall, we introduce a potentially tumor-agnostic and tumor-retained cytokine/antibody fusion protein that shows promise as a powerful new strategy for the treatment of solid tumors, both as an intratumorally administered monotherapy and in combination with other systemically or locally delivered immunotherapies.
Limitations of the study
An inherent limitation of our approach is that the high affinity of our engineered immunocytokine for collagen makes systemic delivery of the therapy challenging. Owing to the presence of collagen throughout the body, systemic administration of collagen-binding therapies such as F10 IC-CBD may result in off-target accumulation of the molecule, which may limit its efficacy and/or lead to toxicity. Indeed, in preliminary studies we performed assessing different routes of administration, although the majority of delivered protein was localized to the tumor, we observed accumulation of F10 IC-CBD in the collagen-rich tails and vitreous cavities of mice upon intravenous and retro-orbital administration, respectively. While the abscopal effects observed in the present study demonstrate that intratumoral injection can still lead to systemic anti-cancer effects, we note that not all tumors are treatable by this route of administration. Future studies will optimize the systemic delivery of collagen-binding immunocytokines, with a focus on dose optimization and the design of new fusion proteins with lower affinity for collagen.
Finally, to advance translation of our engineered IC, and IL-2 therapies more broadly, it will be necessary to benchmark the activity and therapeutic efficacy of our biased IL-2 therapy against other IL-2-based strategies. Various biased IL-2 therapies are under development, including the following: (1) IL-2 muteins,39,59,60,61,62,63,64,65 mimics,66,67 and receptor masking strategies,68 which disrupt or enhance interfaces of the IL-2 cytokine with its cognate receptor subunits in order to augment the immune effector to TReg ratio; (2) PEGylation69,70,71; (3) biased IL-2-containing immunocytokines that target tumor-associated antigens or immune markers72,73,74,75; (4) IL-2/anti-IL-2 antibody complexes12,13,76,77,78,79; and (5) untargeted biased IL-2 immunocytokines.14,80,81 Each of these IL-2 therapies has a unique set of pharmacokinetic, tumor-targeting, IL-2 receptor subunit engagement, and immune activation properties. It will be particularly important to understand which properties are most important for anti-tumor immunity and to identify the cell types essential for therapeutic response in order to design superior IL-2-based drugs and combination treatments. It is possible that the design of the most effective IL-2 therapy may result from the incorporation of collagen-binding capability into a mutated version of the IL-2 that possesses a collection of the best properties from various molecules. An advantage for our platform is the stabilized unimolecular format, which simplifies manufacturing and clinical advancement. Moreover, the modular design of F10 IC-CBD permits the engineering of both the cytokine and antibody within the immunocytokine to design next-generation biased IL-2 therapies that possess the most important features for anti-tumor efficacy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| ™Antibodies | ||
| Anti-phospho STAT5 Alexa Fluor 647 [PY694] | BD Biosciences | Cat# 562076; RRID: AB_11154412 |
| Tocilizumab | N/A | |
| Anti-human CD3 APC-eFluor780 [UCHT1] | Thermo Fisher Scientific | Cat# 47-0038-42; RRID: AB_1272042 |
| Anti-human CD4 PerCP-Cy5.5 [SK3] | BD Biosciences | Cat# 566923; AB_2739678 |
| Anti-human CD8 BV605 [SK1] | BioLegend | Cat# 344742; RRID: AB_2566513 |
| Anti-human CD25 BV421 [M-A251] | BD Biosciences | Cat# 562442; RRID: AB_11154578 |
| Anti-human FoxP3 PE [236A/E7] | BD Biosciences | Cat# 560852; RRID: AB_10563418 |
| Peroxidase AffiniPure Goat Anti-Human IgG (H + L) | Jackson ImmunoResearch | Cat# 115-035-003; RRID: AB_10015289 |
| Anti-mouse CD16/32 [2.4G2] | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-mouse CD45.2 BV570 [104] | BioLegend | Cat# 109833; RRID: AB_10900987 |
| Anti-mouse CD3 BV785 [145-2C11] | BioLegend | Cat# 100355; RRID: AB_2565969 |
| Anti-mouse CD4 Super Bright 436 [RM4-5] | Thermo Fisher Scientific | Cat# 62-0042-82; RRID: AB_2716977 |
| Anti-mouse CD8a BV650 [53–6.7] | BioLegend | Cat# 100741; RRID: AB_11124344 |
| Anti-mouse CD25 BV510 [PC61] | BioLegend | Cat# 102041; RRID: AB_2562269 |
| Anti-mouse CD49b PerCP-Cy5.5 [DX5] | BioLegend | Cat# 108916; RRID: AB_2129358 |
| Anti-mouse FoxP3 APC [FKJ-16s] | Thermo Fisher Scientific | Cat# 17-5773-82; RRID: AB_469457 |
| Anti-mouse NK1.1 PerCP-Cy5.5 [PK136] | BioLegend | Cat# 108728; RRID: AB_2132705 |
| Anti-mouse PD-1 PE/Dazzle 594 [29F.1A12] | BioLegend | Cat# 135228; RRID: AB_2566006 |
| Anti-mouse FoxP3 PE [FKJ-16s] | Thermo Fisher Scientific | Cat# 12-5773-82; RRID: AB_465936 |
| Anti-mouse PD-1 [RMP1-14] | BioXcell | Cat# BE0146; RRID: AB_10949053 |
| ™Bacterial and virus strains | ||
| One Shot™ MAX Efficiency™ DH5α – T1R Competent Cells | Thermo Fisher Scientific | Cat# 12297016 |
| ™Biological samples | ||
| Human peripheral blood mononuclear cells | Anne Arundel Medical Blood Donor Center | N/A |
| ™Chemicals, peptides, and recombinant proteins | ||
| Poly(beta-amino ester) 4-4-6 transfection reagent | Klapper et al.1 | N/A |
| Magnesium acetate solution | Sigma-Aldrich | Cat# 62052 |
| F10 IC | This study | N/A |
| F10 IC-CBDHC | This study | N/A |
| F10 IC-CBDLC | This study | N/A |
| F10 IC-CBDHC/LC | This study | N/A |
| Control (FITC-E2) IC | Atkins et al.2 | N/A |
| Control (FITC-E2) IC-CBDHC/LC | This study | N/A |
| Human IL-2 | Atkins et al.2 | N/A |
| Biotinylated human IL-2 | Atkins et al.2 | N/A |
| Biotinylated human IL-2Rα | Atkins et al.2 | N/A |
| Biotinylated human IL-2Rβ | Atkins et al.2 | N/A |
| Human Collagen Type I | Sigma-Aldrich | Cat# CC050 |
| Human Collagen Type III | Sigma-Aldrich | Cat# CC054 |
| Ficoll-Paque PLUS density gradient media | Cytiva Life Sciences | Cat# 17144002 |
| ACK Lysing Buffer | Quality Biological | Cat# 118-156-101 |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34957 |
| IRDye® 800CW N-hydroxysuccinimide Ester | LI-COR Biosciences | Cat# 929-70021 |
| NHS-Rhodamine (5/6-carboxy-tetramethyl-rhodamine succinimidyl ester), mixed isomer | Thermo Fisher Scientific | Cat# 46406 |
| eBioscience™ Fixable Viability Dye eFluor™ 780 | Thermo Fisher Scientific | Cat# 65-0865-14 |
| H-2Ld MuLV gp70 Tetramer-SPSYVYHQF-PE | MBL International Corporation | Cat# TB-M521-1 |
| iTAg Tetramer/PE – H-2 Kb TRP2 (SVYDFFVWL) | MBL International Corporation | Cat# TB-5004-1 |
| ™Critical commercial assays | ||
| Plasmid Plus Maxi kit | Qiagen | Cat# 12963 |
| BirA biotin-protein ligase standard reaction kit | Avidity LLC | Cat# BirA500 |
| eBioscience™ FoxP3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| Pharmingen™ Transcription Factor Phospho Buffer Set | BD Biosciences | Cat# 563239 |
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130-096-730 |
| CD45 (TIL) MicroBeads, mouse | Miltenyi Biotec | Cat# 130-110-618 |
| ViaStain™ Acridine Orange/Propidium Iodide Staining Solution | Nexcelom Bioscience | Cat# CS2-0106 |
| ™Experimental models: Cell lines | ||
| Freestyle™ 293-F cells | Thermo Fisher Scientific | Cat# R79007; RRID: CVCL_6642 |
| Human: YT-1 natural killer cell line | Donohue et al.3 | N/A |
| Human: IL-2Ra+ YT-1 natural killer cell line | Konrad et al.4 | N/A |
| Mouse: CT26 cell line | ATCC | Cat# CRL-2638; RRID: CVCL_7254 |
| Mouse: B16-F10 cell line | ATCC | Cat# CRL-6475; RRID: CVCL_0159 |
| ™Experimental models: Organisms/strains | ||
| Mouse: BALB/cJ | The Jackson Laboratory | Strain# 000651; RRID: IMSR_JAX:000651 |
| Mouse: C57BL/6J | The Jackson Laboratory | Straint# 000664; RRID: IMSR_JAX:000664 |
| ™Recombinant DNA | ||
| Plasmid: gWiz_F10_IC_HC | This study | N/A |
| Plasmid: gWiz_F10_IC_LN35_LC | This study | N/A |
| Plasmid: gWiz_gWiz_F10_IC_HC_CBD | This study | N/A |
| Plasmid: gWiz_F10_IC_LN35_LC_CBD | This study | N/A |
| Plasmid: gWiz_Control_IC[FITC-E2]_HC | Atkins et al.2 | N/A |
| Plasmid: gWiz_Control_IC[FITC-E2]_LC | Atkins et al.2 | N/A |
| Plasmid: gWiz_Control_IC[FITC-E2]_HC_CBD | This study | N/A |
| Plasmid: gWiz_Control_IC[FITC-E2]_LC_CBD | This study | N/A |
| Plasmid: gWiz_hIL-2 | Atkins et al.2 | N/A |
| Plasmid: gWiz_hIL-2+BH3 | Atkins et al.2 | N/A |
| Plasmid: gWiz_hIL-2Rα+BH3 | Atkins et al.2 | N/A |
| Plasmid: gWiz_hIL-2Rβ+BH3 | Atkins et al.2 | N/A |
| ™Software and algorithms | ||
| UNICORN™ v7.1 | Cytiva Life Sciences | https://www.cytivalifesciences.com/en/us/shop/chromatography/software/unicorn-7-p-05649 |
| Octet® Data Analysis software v7.1 | FortéBio | N/A |
| Pearl Impulse Software v2.0 | LI-COR Biosciences | N/A |
| Matrix Software | Nexcelom Bioscience | https://www.nexcelom.com/knowledge-base/matrix-software-manual/ |
| GraphPad Prism v9.3.1 | GraphPad | https://www.graphpad.com |
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo |
| ™Other | ||
| Pierce™ Protein G Agarose | Thermo Fisher Scientific | Cat# 20397 |
| Ni-NTA Agarose | Qiagen | Cat# 30210 |
| Octet® Streptavidin (SA) Biosensor | Sartorius | Cat# 18-5019 |
| 1-Step™ Ultra TMB-ELISA Substrate Solution | Thermo Fisher Scientific | Cat# 34028 |
Resource availability
Lead contact
Any further information or requests should be directed to, and will be fulfilled by, Dr. Jamie B. Spangler (jamie.spangler@jhu.edu).
Materials availability
Requests for new materials generated in this paper are to be directed to and will be fulfilled (pending MTA and associated restrictions) by the lead contact.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is availanle from lead contact the upon request.
Experimental model and subject details
Cell lines
Human embryonic kidney (HEK) 293-F cells (Thermo Fisher Scientific) were cultivated in Freestyle 293 Expression Medium (Thermo Fisher Scientific) supplemented with 0.2 U/mL penicillin-streptomycin (Gibco). Unmodified IL-2Rα− YT-130 and IL-2Rα+ YT-131 human NK cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1× minimum non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES, and 100 U/mL penicillin-streptomycin [Gibco]). CT26 and B16F10 cells were purchased from ATCC. CT26 cells were cultured in ATCC modified RPMI 1640 supplemented with 10% FBS and 100 U/mL penicillin-streptomycin (Gibco). B16F10 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 100 U/mL penicillin-streptomycin (Gibco). All cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2.
Human peripheral blood mononuclear cells (PBMCs)
For human peripheral blood mononuclear cell (PBMC) studies, leukopaks were obtained from Anne Arundel Medical Blood Donor Center (Anne Arundel, Maryland, USA).
Mice
6-8-week-old female BALB/cJ (000651) and C57BL/6J (000664) mice purchased from The Jackson Laboratory were used in these studies. Animals were housed in specific pathogen-free conditions and experiments conducted in accordance with the Johns Hopkins University Animal Care and Use Committee (ACUC) under protocol number MO20M285.
Method details
Protein design and purification
The sequences encoding the parent F10 immunocytokine VH and VL regions14 were used in this study to formulate the recombinant F10 IC on the human immunoglobulin (IgG) 1 lambda isotype. We previously generated a human IgG1 lambda isotype Control IC consisting of an anti-fluoresceine-specific (FITC-E2) antibody C-terminally fused to human IL-2 at the light chain.35 The heavy chain (HC) and light chain (LC) of F10 IC, both unmodified and C-terminally fused, via (Gly4Ser)3 linker, to the A3 CBD of von Willebrand factor (vWF) (Table S1), were separately cloned into the gWiz mammalian expression vector (Genlantis) using Gibson Assembly (New England Biolabs). The HC and LC of the Control IC, C-terminally fused via (Gly4Ser)3 linker, to the A3 CBD of vWF, were also separately cloned into the gWiz mammalian expression vector (Genlantis). Unmodified and CBD-fused ICs were recombinantly expressed in HEK 293-F cells via transient co-transfection of plasmids encoding the corresponding heavy chain (HC) and light chain (LC) using the high-yield transfection reagent poly(beta-amino ester) 4-4-6.82 HEK 293-F cells were grown to 1.2×106 cells/mL and diluted to 1.0×106 cells/mL in a fresh culture medium on the day of transfection. A total of 1 mg of maxiprepped (Qiagen), endotoxin-free plasmid DNA (1:2 ratio HC:LC) and 63.15 mg of poly(beta-amino ester), 4-4-6, per L of cells were independently diluted to 0.05 and 3 mg/mL in 25 mM magnesium acetate buffer (Sigma-Aldrich) pH 5.2 respectively, combined by vortexing and incubated at room temperature for 15 min. The resulting DNA/4-4-6 nanoparticles were subsequently added dropwise to the diluted HEK 293-F cells in a shaking flask and the transfected cells incubated, with orbital sharking at 125 rpm, at 37°C with 5% CO2. ICs were purified from cell supernatants 5 days post-transfection via protein G affinity chromatography followed by size-exclusion chromatography (SEC) using a Superdex 200 Increase 10/300 GL column on an ÄKTA fast protein liquid chromatography (FPLC) instrument (Cytiva Life Sciences) equilibrated in HEPES-buffered saline (HBS, 150mM NaCl in 10mM HEPES pH 7.3). Size and purity (>99%) were verified by SDS-PAGE analysis.
The sequences encoding the human IL-2 (hIL-2, residues 1–133), the hIL-2Rα ectodomain (residues 1–217) and the hIL-2Rβ ectodomain (residues 1–214) were previously separately cloned into the gWiz mammalian expression vector with a C-terminal biotin acceptor peptide (BAP; GLNDIFEAQKIEWHE) followed by a hexahistidine tag.35 The sequence of hIL-2 was also previously encoded into the gWiz mammalian expression vector with only a C-terminal hexahistidine tag.35 hIL-2, the hIL-2Rα ectodomain and the hIL-2Rβ were recombinantly expressed in HEK 293-F cells via transient transfection as described above using Ni-NTA agarose (Qiagen) affinity chromatography followed by SEC. For proteins that were biotinylated, prior to SEC, proteins were incubated overnight at 4°C with soluble BirA ligase in 0.5 mM Bicine pH 8.3, 100 mM ATP, 100 mM magnesium acetate, and 500 mM biotin (Avidity). Complete biotinylation was verified by an SDS-PAGE streptavidin shift assay.
Biolayer interferometry binding studies
Binding to IL-2, IL-2Rα and IL-2Rβ was measured on an OctetRED96 system (FortéBio). All proteins were diluted in 1× PBS pH 7.2 containing 0.1% BSA (PBSA) and filtered through a 0.45 μm filter. Biotinylated hIL-2 (50 nM), hIL-2Rα (100 nM), and hIL-2Rβ (50 nM) were immobilized on streptavidin biosensors (Sartorius) for 120 s and baseline measurements generated in PBSA. Binding kinetics were measured by submerging coated biosensors into serially diluted IC for 300 s (association), followed by PBSA for 450 s (dissociation). IL-2, MAB602, and an irrelevant antibody (in house recombinantly expressed Tocilizumab) were used as control analytes. Biosensors were regenerated in 0.1 M glycine pH 2 in between analytes. All measurements were performed at room temperature. Data was processed and kinetic parameters calculated using the Octet Data Analysis software v7.1 assuming a assuming a 1:1 binding model. Equilibrium titration curves were fitted in GraphPad Prism, assuming first order binding interactions. Experiments were reproduced two times with similar results.
YT-1 and PBMC IL-2 induced STAT5 activation studies
For YT-1 studies, approximately 2×105 IL-2Rα− YT-130 and IL-2Rα+ YT-131 human natural killer cells were plated in each well of a 96 well plate and stimulated with serial dilutions of IL-2 or IC in culture medium for 20 min at 37°C, fixed for 10 min at room temperature with paraformaldehyde (Electron Microscopy Sciences) to a final concentration of 1.5%, and permeabilized in ice-cold 100% methanol (Millipore Sigma) overnight at −80°C. Fixed and permeabilized cells were washed with 1× PBS with 0.1% BSA (PBSA), stained for anti-phospho STAT5 Alexa Fluor 647 (PY694, BD Biosciences 562076, 1:50) diluted in 20 μL PBSA for 2 h at RT. Stained cells were then washed with PBSA and acquired on an Attune NxT flow cytometer (Thermo Fisher Scientific). For YT-1 signaling studies on collagen-coated plates, medium binding enzyme-linked immunosorbent assay (ELISA) microplates (Greiner Bio One) were coated with 100 μL of 10 μg/mL purified collagen I or III (Sigma-Aldrich) in 1× PBS or PBS only (control) for 1 h at 37°C. Wells were then washed three times with 200 μL PBS-T. Wells were subsequently blocked with 200 μL of 2% BSA diluted in in PBS-T (0.5% Tween 20 in 1× PBS) at room temperature for 2 h followed by three washes with 100 μL PBS-T and a single 100 μL wash with YT-1 culture media. Following washes, wells were incubated at 37°C for 20 min with titrating amounts of IL-2 or IC and approximately 2×105 IL-2Rα− or and IL-2Rα+ YT-1 cells per well in YT-1 culture media. Following stimulation, YT-1 cells were transferred to a 96 well plate, fixed, permeabilized and stained for anti-phospho STAT5 as described above.
For human peripheral blood mononuclear cell (PBMC) studies, leukopaks were obtained from Anne Arundel Medical Blood Donor Center (Anne Arundel, Maryland, USA). PBMCs were isolated by a Ficoll-Plaque PLUS gradient (Cytiva Life Sciences) followed by ACK red blood cell lysis (Quality Biological). Approximatley 2×106 cells were plated in each well of a 96 well plate and resuspended in 50 μL LIVE/DEAD Aqua (Thermo Fisher Scientific L34957; 1:1000) in 1× PBS and subsequently washed with PBSA. Stained cells were subsequently stimulated with serial dilutions of 40 μL IL-2 or IC in RPMI 1640 for 20 min at 37°C followed by immediate fixation and permeabilization using the Pharmingen Transcription Factor Phospho Buffer Set (BD Biosciences) according to manufacturer protocol. Cells were stained with anti-human CD3 APC-eFluor780 (UCHT1, Thermo Fisher Scientific 47-0038-42; 1:50), anti-human CD4 PerCP-Cy5.5 (SK3, BD Biosciences 566923; 1:100), anti-human CD8 BV605 (SK1, BioLegend 344742; 1:50), anti-human CD25 BV421 (M-A251, BD Biosciences 562442; 1:100), anti-human FoxP3 PE (236A/E7, BD Biosciences 560852, 1:50), and phospho STAT5 Alexa Fluor 647 (PY694, BD Biosciences 562076, 1:50), resuspended in PBSA, and acquired on an Attune NxT flow cytometer. Data was analyzed in FlowJo v10.8.1. Dose-response curves were fitted to a logistic model and half-maximal effective concentrations (EC50) calculated. Experiments were conducted in duplicate and performed three times with similar results.
Collagen-binding enzyme-linked immunosorbent assay (ELISA)
Medium binding ELISA microplates (Greiner Bio One) were coated with 100 μL of 10 μg/mL purified collagen I or III (Sigma-Aldrich) in 1× PBS for 1 h at 37°C. Wells were subsequently blocked with 200 μL of 2% BSA diluted in in PBS-T (0.5% Tween 20 in 1× PBS) at room temperature for 2 h followed by incubation at room temperature for 1 h with 100 μL serial dilutions of IC in PBSA. Wells were then incubated for 1 h at room temperature with Peroxidase AffiniPure Goat Anti-Human IgG (H + L) (Jackson ImmunoResearch 115-035-003; 1:10000) diluted in 100 μL PBSA followed by a 5-10-min room temperature incubation with 100 μL 1-Step Ultra TMB-ELISA Substrate Solution (Thermo Fisher Scientific) in the dark. Reactions were stopped with 100 μL 1 M sulfuric acid and absorbance at 450 nm, corrected with a reference absorbance at 570 nm measured on a BioTek Synergy Mx microplate reader. Three washed were completed with PBST after each step prior to TMB substrate addition. Equilibrium dissociation constants were obtained by nonlinear regression analysis assuming one-site specific binding. Experiments were conducted in duplicate and performed at least three times with similar results.
In vivo live imaging and plasma pharmacokinetic analyses
To assess tumor retention in live imaging studies, ICs were labeled using IRDye 800CW N-hydroxysuccinimide (NHS) Ester (LI-COR Biosciences) according to manufacturer protocol and unreacted dye removed by SEC. 5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) were subcutaneously implanted on the right flank of acclimated, 6-8-week-old female BALB/c mice (n = 3/group). When tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected intratumorally with 2.5 μg IL-2 equivalents of near-IR-labeled IC dilited in 50 μL 1× PBS and imaged on 37°C heated imaging bed under 2.5% isoflurane and 2.5L/min oxygen flow on an LI-COR Pearl Impulse Imager (LI-COR Biosciences) at 0, 6, 12, 24, 36, 48 and 72 h post intratumoral administration. Images were acquired at 800 mm with 85 μm resolution. Intratumoral fluorescence intensity was calculated in the Pearl Impulse Software v2.0 (LI-COR Biosciences) as the sum of pixel intensity values at the tumor minus the product of the background pixel intensity and tumor area. The half-life of the normalized intratumoral fluorescence was calculated assuming one phase decay, and area under the curve (AUC) was determined. Experiments were conducted twice with similar results.
For plasma pharmacokinetic studies, ICs were labeled using NHS-Rhodamine (Thermo Fisher Scientific) according to manufacturer protocol and unreacted dye removed by SEC. 5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) were subcutaneously implanted on the right flank of acclimated, 6-8-week-old female BALB/c mice (n = 4/group). Acclimated, 6-8-week-old, female BALB/c mice bearing tumors of approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), were injected intratumorally with a 0.5 mg/kg body weight IL-2 equivalent of labeled IC diluted in 50 μL 1× PBS. Prior to and at 0, 6, 12, 24, 36, 48, 72, 96, 120, 144, and 168 h post injection, 18 μL blood samples were collected by tail snip and diluted in 90 μL PBS-EDTA (150 mM NaCl, 12 mM Na2HPO4, 5 mM EDTA) pH 8. Samples were centrifuged at 10000×g for 5 min to remove cells and fluorescence (540/590 nm) of the resulting 10-fold diluted plasma samples measured on a BioTek Synergy Mx microplate reader in Nunc microwell 96-well optical-bottom plates (Thermo Fisher Scientific). Standard curves were generated from fluorescence measurements of serially diluted rhodamine-labeled IC in PBS-EDTA and used to determine IC concentration in each plasma sample. IL-2 plasma concentration over time was graphed and AUC determined. Experiments were conducted twice with similar results.
In vivo toxicity studies
5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) were subcutaneously implanted on the right flank of acclimated, 6-8-week-old female BALB/c mice (n = 6/group). When tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected intratumorally with 50 μL 1× PBS or a 0.5 mg/kg body weight IL-2 equivalent of IC in 50 μL 1× PBS. To assess weight change, mice were weighed prior to (baseline) and 7 days after intratumoral administration on an OHAUS Scout SPX123 portable balance and weight change calculated as the difference in body mass between days 7 and baseline. To assess pulmonary and liver toxicity, 72 h post-intratumoral injection, blood samples were collected from the submandibular vein and the lungs harvested and weighed on an OHAUS Scout SPX123 portable balance. Blood samples incubated at room temperature for 2 h and centrifuged at 10000×g for 10 min to remove cells. Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) concentrations measured by single-step ELISA (Abcam). Excised lungs were weighed an OHAUS Scout SPX123 portable balance and lyophilized on a FreeZone 2.5L −50°C Benchtop Freeze Dryer (Labconco). Pulmonary fluid content was calculated as the difference in the weight of the lungs before and after lyophilization. Experiments were conducted twice with similar results.
In vivo pharmacodynamic immune cell profiling study
5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) were subcutaneously implanted on the right flank of acclimated, 6-8-week-old female BALB/c mice (n = 5/group). When tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected intratumorally with a single dose of 50 μL 1× PBS or 2.5 μg IL-2 equivalent dose of IC in 50 μL 1× PBS. At 2, 4, and 7 days post-intratumoral injection, mice were euthanized by cervical dislocation, and the spleens and tumors were harvested for immune cell profiling. Spleens were processed into single-cell suspension by mechanical dissociation through a 70-μm cell strainer and red blood cells removed with ACK lysis buffer. Harvested tumors were measured with a digital caliper and weighed on an OHAUS Scout SPX123 portable balance. Single-cell tumor suspensions were generated using the Mouse Tumor Dissociation Kit with gentleMACS Octo Dissociators with heaters, and the tumor infiltrating leukocytes (TILs) were isolated by magnetic activated cell sorting (MACS) using mouse CD45 MicroBeads (Miltenyi Biotec) according to manufacturer protocol. Splenocytes and TILs were counted on a Cellaca MX high-throughput automated cell counter (Nexcelom Bioscience) with ViaStain Acridine Orange/Propidium Iodide staining solution (Nexcelom Bioscience). Approximately 2×106 splenocytes and TILs in their entirety were plated in each well of a 96-well plate and stained with Fixable Viability Dye eFluor780 (Thermo Fisher Scientific 65-0865-14; 1:2000) in 50 μL 1×PBS followed by surface staining with anti-mouse CD16/32 (2.4G2, BD Biosciences 553142, 1:100) and anti-mouse CD45.2 BV570 (104, BioLegend 109833; 1:50), anti-mouse CD3 BV785 (145-2C11, BioLegend 100355; 1:50), anti-mouse CD4 Super Bright 436 (RM4-5, Thermo Fisher Scientific 62-0042-82; 1:40), anti-mouse CD8a BV650 (53–6.7, BioLegend 102042; 1:50), anti-mouse CD25 BV510 (PC61, BioLegend 102041; 4 μL per sample), and anti-mouse CD49b PerCP-Cy5.5 (DX5, BioLegend 108916; 4 μL per sample) diluted in 50 μL autoMACS Rinsing Solution supplemented with 0.5% BSA (Miltenyi Biotec) per sample. Following surface staining, samples were fixed, permeabilized and stained with anti-mouse FoxP3 APC (FKJ-16s, Thermo Fisher Scientific 17-5773-82; 1:50) using the eBioScience FoxP3/Transcription Factor Fixation Staining Buffer Set (Thermo Fisher Scientific) according to manufacturer protocol. Washes in between steps were performed with autoMACS Rinsing Solution supplemented with 0.5% BSA (Miltenyi Biotec). Samples were resuspended in autoMACS Rinsing Solution supplemented with 0.5% BSA (Miltenyi Biotec) and acquired on an Attune NxT flow cytometer and data analyzed in FlowJo v10.8.1. Experiments were conducted twice with similar results.
In vivo colon carcinoma and melanoma therapeutic models
For single-agent studies, acclimated, 6-8-week-old, female BALB/c and C57BL/6 mice (n = 5–6/group) were injected subcutaneously on the right flank with 5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) or 1 × 106 B16f10 melanoma cells in 50 μL DMEM (Gibco). When tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected intratumorally with 50 μL 1×PBS or a 2.5 μg IL-2 equivalent dose of IC diluted in 50 μL 1×PBS three times with each injection occurring 4 days apart. 1 day after the final dose, mice in all groups were euthanized by cervical dislocation, the spleens and tumors harvested, and single cell suspensions prepared as described above for immune cell profiling. Approximately 2×106 splenocytes and TILs in their entirety were plated in each well of a 96-well plate and stained with Fixable Viability Dye eFluor780 (Thermo Fisher Scientific 65-0865-14; 1:2000) in 50 μL 1×PBS PBS followed by surface staining with anti-mouse CD16/32 (2.4G2, BD Biosciences 553142; 1:100), anti-mouse CD45.2 BV570 (104, BioLegend 109833; 1:50), anti-mouse CD3 BV785 (145-2C11, BioLegend 100355; 1:50), anti-mouse CD4 Super Bright 436 (RM4-5, Thermo Fisher Scientific 62-0042-82; 1:40), anti-mouse CD8a BV650 (53–6.7, BioLegend 102042; 1:50), anti-mouse CD25 BV510 (PC61, BioLegend 102041; 4 μL per sample), anti-mouse CD49b PerCP-Cy5.5 (DX5, BioLegend 108916; 4 μL per sample [BALB/c]) or anti-mouse NK1.1 PerCP-Cy5.5 (PK136, BioLegend 108728; 4 μL per sample [C57BL/6J]), anti-mouse PD-1 PE/Dazzle 594 (29F.1A12, BioLegend 135228; 4 μL per sample), and H-2Ld MuLV gp70 (SPSYVYHQF) PE (MBL International Corp. TB-M521-1; 10 μL per sample [BALB/cJ]) or H-2Kb TRP2 (SVYDFFVWL) APC (MBL International Corp. TB5004-2; 10 μL per sample [C57BL/6]) diluted in 50 μL autoMACS Rinsing Solution supplemented with 0.5% BSA (Miltenyi Biotec) per sample. Following surface staining, samples were fixed, permeabilized and stained with anti-mouse FoxP3 APC (FKJ-16s, Thermo Fisher Scientific 17-5773-82; 1:50 [BALB/c]) or PE (FKJ-16s, Thermo Fisher Scientific 12-5773-82; 1:50 [C57BL/6]) using the eBioScience FoxP3/Transcription Factor Fixation Staining Buffer Set (Thermo Fisher Scientific) according to manufacturer protocol. Washes in between steps were performed with autoMACS Rinsing supplemented with 0.5% BSA (Miltenyi Biotec). Samples were resuspended in autoMACS Rinsing Solution supplemented with 0.5% BSA (Miltenyi Biotec) acquired on an Attune NxT flow cytometer and data analyzed in FlowJo v10.8.1. Tumors were measured with a digital caliper and mouse body weights recorded from an OHAUS Scout SPX123 portable balance until euthanasia criteria (tumor volumes of ≥1500 mm3 in control group) were met. Experiments were conducted twice with similar results.
For immune checkpoint combination studies, acclimated, 6-8-week-old, female BALB/c and C57BL/6 mice (n = 6/group) were injected subcutaneously on the right flank with 5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) or 1 × 106 B16f10 melanoma cells in 50 μL DMEM (Gibco). When tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected both intratumorally with 50 μL 1×PBS or a 2.5 μg IL-2 equivalent dose of IC diluted in 50 μL 1×PBS and intraperitoneally with 100 μL 1×PBS or 100 μg of anti-mouse programmed cell death protein 1 (PD-1) (RMP1.14; BioXcell BE0146) in 100 μL 1×PBS three times with each injection occurring 4 days apart. Tumors were measured with a digital caliper and mouse body weights recorded from an OHAUS Scout SPX123 portable balance until euthanasia criteria (tumor volumes of ≥1500 mm3 in control group) were met. Experiments were conducted twice with similar results.
The assess the presence of an abscopal effect, 6-8-week-old, female BALB/c (n = 5/group) injected subcutaneously on both the right (primary) and left (abscopal) flanks with 5×105 CT26 colon carcinoma cells in 50 μL RPMI 1640 (ATCC modification, Gibco) each. When primary tumors were approximately 25 mm3 (calculated as V = [L×W×W]/2 where L is the length of the longest axis and W is the length of the shortest axis), mice were injected both intratumorally (on the right flank tumor) with 50 μL 1×PBS or a 2.5 μg IL-2 equivalent dose of IC diluted in 50 μL 1×PBS both with and without an intraperitoneally with 100 μL 1×PBS or 100 μg of anti-mouse PD-1 (RMP1.14; BioXcell BE0146) in 100 μL 1×PBS. Mice were treated three times with each injection occurring 4 days apart. Tumors were measured with a digital caliper and mouse body weights recorded from an OHAUS Scout SPX123 portable balance until euthanasia criteria (combined tumor volumes of ≥1500 mm3 in control group) were met.
Quantification and statistical analysis
GraphPad Prism v9.3.1 was used for all graphical representations and statistical analyses. The sample size, number of replicates, mice, definition of center, and dispersion and precision measures, and type of analysis performed are described in the figure legends where applicable. For all studies, significance is defined as p ≤ 0.05 (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001). Significance between all groups is not always depicted in figures. For full analyses see Table S6.
Acknowledgments
This work was supported by funds from National Institutes of Health grants T32AI007417 (A.B.S), R01EB029455 (J.B.S.), R37CA246699 (S.Y.T.), P41EB028239 (J.J.G.), and R01EB029341 (J.B.S.), a Johns Hopkins University Catalyst Award (J.B.S.), a Sanofi iAward (J.B.S.), Melanoma Research Alliance Young Investigator Award 824779 (J.B.S.), V Foundation Scholar Award V2018-005 (J.B.S.), and an Emerson Collective Cancer Research Fund Award (J.B.S.). The authors would like to acknowledge the Johns Hopkins Medicine MRB Molecular Imaging Service and Cancer Functional Imaging Core, Kathryn M. Luly (Johns Hopkins Translational Tissue Engineering Center) for providing the transfection reagent poly(beta-amino ester) 4-4-6 for protein expression, and Jeffrey A. Hubbell (University of Chicago Pritzker School of Molecular Engineering) for his advice on the project. The graphical abstract was created with Biorender.
Author contributions
Conceptualization; A.B.S. and J.B.S. Methodology; AB.S. with input from S.Y.T., J.I., J.G., and J.B.S. Investigation; A.B.S. with contributions from S.Y.T., M.L. and J.W. Formal analysis; A.B.S. and J.B.S. Funding acquisition; J.B.S. and J.J.G. Supervision; J.B.S. Writing; A.B.S. and J.B.S. All authors reviewed this manuscript and approved the final submitted version.
Declaration of interests
Johns Hopkins University has filed intellectual property (WO2020264321A1) entitled “Methods and materials for targeted expansion of immune effector cells” that covers compound F10 IC as well as related compounds, with J.B.S. as a co-inventor. J.I. is a shareholder of ArrowImmune Inc., which is working on immuno-oncology.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101289.
Supplemental information
References
- 1.Klapper J.A., Downey S.G., Smith F.O., Yang J.C., Hughes M.S., Kammula U.S., Sherry R.M., Royal R.E., Steinberg S.M., Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: A retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113:293–301. doi: 10.1002/cncr.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkins M.B., Lotze M.T., Dutcher J.P., Fisher R.I., Weiss G., Margolin K., Abrams J., Sznol M., Parkinson D., Hawkins M., et al. High-Dose Recombinant Interleukin 2 Therapy for Patients With Metastatic Melanoma: Analysis of 270 Patients Treated Between 1985 and 1993. J. Clin. Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 3.Donohue J.H., Rosenberg S.A. The fate of interleukin-2 after in vivo administration. J. Immunol. 1983;130:2203–2208. doi: 10.4049/jimmunol.130.5.2203. [DOI] [PubMed] [Google Scholar]
- 4.Konrad M.W., Hemstreet G., Hersh E.M., Mansell P.W., Mertelsmann R., Kolitz J.E., Bradley E.C. Pharmacokinetics of Recombinant Interleukin 2 in Humans. Cancer Res. 1990;50:2009–2017. [PubMed] [Google Scholar]
- 5.Boyman O., Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012;12:180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 6.Liao W., Lin J.-X., Leonard W.J. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malek T.R., Castro I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity. 2010;33:153–165. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baluna R., Vitetta E.S. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology. 1997;37:117–132. doi: 10.1016/S0162-3109(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 9.Krieg C., Létourneau S., Pantaleo G., Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA. 2010;107:11906–11911. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa K., Miller F.N., Sims D.E., Lentsch A.B., Miyazaki M., Edwards M.J. Mechanisms of Interleukin-2-Induced Hepatic Toxicity. Cancer Res. 1996;56:507–510. [PubMed] [Google Scholar]
- 11.Siegel J.P., Puri R.K. Interleukin-2 toxicity. J. Clin. Oncol. 1991;9:694–704. doi: 10.1200/JCO.1991.9.4.694. [DOI] [PubMed] [Google Scholar]
- 12.Létourneau S., van Leeuwen E.M.M., Krieg C., Martin C., Pantaleo G., Sprent J., Surh C.D., Boyman O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proc. Natl. Acad. Sci. USA. 2010;107:2171–2176. doi: 10.1073/pnas.0909384107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyman O., Kovar M., Rubinstein M.P., Surh C.D., Sprent J. Selective Stimulation of T Cell Subsets with Antibody-Cytokine Immune Complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 14.Leonard E.K., Tomala J., Gould J.R., Leff M.I., Lin J.-X., Li P., Porter M.J., Johansen E.R., Thompson L., Cao S.D., et al. Engineered cytokine/antibody fusion proteins improve delivery of IL-2 to pro-inflammatory cells and promote antitumor activity. bioRxiv. 2023 doi: 10.1101/2023.05.03.539272. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roopenian D.C., Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 16.Champiat S., Tselikas L., Farhane S., Raoult T., Texier M., Lanoy E., Massard C., Robert C., Ammari S., De Baère T., Marabelle A. Intratumoral Immunotherapy: From Trial Design to Clinical Practice. Clin. Cancer Res. 2021;27:665–679. doi: 10.1158/1078-0432.CCR-20-0473. [DOI] [PubMed] [Google Scholar]
- 17.De Lombaerde E., De Wever O., De Geest B.G. Delivery routes matter: Safety and efficacy of intratumoral immunotherapy. Biochim. Biophys. Acta. Rev. Cancer. 2021;1875 doi: 10.1016/j.bbcan.2021.188526. [DOI] [PubMed] [Google Scholar]
- 18.Marabelle A., Tselikas L., de Baere T., Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann. Oncol. 2017;28:xii33–xii43. doi: 10.1093/annonc/mdx683. xii33–xii43. [DOI] [PubMed] [Google Scholar]
- 19.Huang A., Pressnall M.M., Lu R., Huayamares S.G., Griffin J.D., Groer C., DeKosky B.J., Forrest M.L., Berkland C.J. Human intratumoral therapy: Linking drug properties and tumor transport of drugs in clinical trials. J. Control. Release. 2020;326:203–221. doi: 10.1016/j.jconrel.2020.06.029. [DOI] [PubMed] [Google Scholar]
- 20.Milling L., Zhang Y., Irvine D.J. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 2017;114:79–101. doi: 10.1016/j.addr.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riegler J., Labyed Y., Rosenzweig S., Javinal V., Castiglioni A., Dominguez C.X., Long J.E., Li Q., Sandoval W., Junttila M.R., et al. Tumor Elastography and Its Association with Collagen and the Tumor Microenvironment. Clin. Cancer Res. 2018;24:4455–4467. doi: 10.1158/1078-0432.CCR-17-3262. [DOI] [PubMed] [Google Scholar]
- 22.Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A., Hynes R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016;49:10–24. doi: 10.1016/j.matbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishihara J., Ishihara A., Sasaki K., Lee S.S.-Y., Williford J.-M., Yasui M., Abe H., Potin L., Hosseinchi P., Fukunaga K., et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 2019;11:eaau3259. doi: 10.1126/scitranslmed.aau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansurov A., Ishihara J., Hosseinchi P., Potin L., Marchell T.M., Ishihara A., Williford J.-M., Alpar A.T., Raczy M.M., Gray L.T., et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng. 2020;4:531–543. doi: 10.1038/s41551-020-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Momin N., Mehta N.K., Bennett N.R., Ma L., Palmeri J.R., Chinn M.M., Lutz E.A., Kang B., Irvine D.J., Spranger S., Wittrup K.D. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med. 2019;11:eaaw2614. doi: 10.1126/scitranslmed.aaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Momin N., Palmeri J.R., Lutz E.A., Jailkhani N., Mak H., Tabet A., Chinn M.M., Kang B.H., Spanoudaki V., Hynes R.O., Wittrup K.D. Maximizing response to intratumoral immunotherapy in mice by tuning local retention. Nat. Commun. 2022;13:109. doi: 10.1038/s41467-021-27390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Su Z., Zhao W., Zhang X., Momin N., Zhang C., Wittrup K.D., Dong Y., Irvine D.J., Weiss R. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer. 2020;1:882–893. doi: 10.1038/s43018-020-0095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stinson J.A., Sheen A., Momin N., Hampel J., Bernstein R., Kamerer R., Fadl-Alla B., Samuelson J., Fink E., Fan T.M., et al. Collagen-anchored interleukin-2 and interleukin-12 safely reprogram the tumor microenvironment in canine soft tissue sarcomas. Clin. Cancer Res. 2023;29:2110–2122. doi: 10.1158/1078-0432.CCR-23-0006. CCR-23-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shahidi M. Thrombosis and von Willebrand Factor. Adv. Exp. Med. Biol. 2017;906:285–306. doi: 10.1007/5584_2016_122. [DOI] [PubMed] [Google Scholar]
- 30.Yodoi J., Teshigawara K., Nikaido T., Fukui K., Noma T., Honjo T., Takigawa M., Sasaki M., Minato N., Tsudo M. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells) J. Immunol. 1985;134:1623–1630. doi: 10.4049/jimmunol.134.3.1623. [DOI] [PubMed] [Google Scholar]
- 31.Kuziel W.A., Ju G., Grdina T.A., Greene W.C. Unexpected effects of the IL-2 receptor alpha subunit on high affinity IL-2 receptor assembly and function detected with a mutant IL-2 analog. J. Immunol. 1993;150:3357–3365. [PubMed] [Google Scholar]
- 32.Sixma J.J., Schiphorst M.E., Verweij C.L., Pannekoek H. Effect of deletion of the A1 domain of von Willebrand factor on its binding to heparin, collagen and platelets in the presence of ristocetin. Eur. J. Biochem. 1991;196:369–375. doi: 10.1111/j.1432-1033.1991.tb15826.x. [DOI] [PubMed] [Google Scholar]
- 33.Lankhof H., van Hoeij M., Schiphorst M.E., Bracke M., Wu Y.P., Ijsseldijk M.J., Vink T., de Groot P.G., Sixma J.J. A3 domain is essential for interaction of von Willebrand factor with collagen type III. Thromb. Haemost. 1996;75:950–958. [PubMed] [Google Scholar]
- 34.Lechner M.G., Karimi S.S., Barry-Holson K., Angell T.E., Murphy K.A., Church C.H., Ohlfest J.R., Hu P., Epstein A.L. Immunogenicity of Murine Solid Tumor Models as a Defining Feature of In Vivo Behavior and Response to Immunotherapy. J. Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanDyke D., Iglesias M., Tomala J., Young A., Smith J., Perry J.A., Gebara E., Cross A.R., Cheung L.S., Dykema A.G., et al. Engineered human cytokine/antibody fusion proteins expand regulatory T cells and confer autoimmune disease protection. Cell Rep. 2022;41 doi: 10.1016/j.celrep.2022.111478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon S., Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? OncoImmunology. 2017;7 doi: 10.1080/2162402X.2017.1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y., Zhou N., Zhou L., Wang J., Zhou Y., Zhang T., Fang Y., Deng J., Gao Y., Liang X., et al. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021;22:358–369. doi: 10.1038/s41590-020-00850-9. [DOI] [PubMed] [Google Scholar]
- 38.Beltra J.-C., Bourbonnais S., Bédard N., Charpentier T., Boulangé M., Michaud E., Boufaied I., Bruneau J., Shoukry N.H., Lamarre A., Decaluwe H. IL2Rβ-dependent signals drive terminal exhaustion and suppress memory development during chronic viral infection. Proc. Natl. Acad. Sci. USA. 2016;113:E5444–E5453. doi: 10.1073/pnas.1604256113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo F., Yu Z., Li P., Oh J., Spolski R., Zhao L., Glassman C.R., Yamamoto T.N., Chen Y., Golebiowski F.M., et al. An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature. 2021;597:544–548. doi: 10.1038/s41586-021-03861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dammeijer F., Van Gulijk M., Klaase L., Van Nimwegen M., Bouzid R., Hoogenboom R., Joosse M.E., Hendriks R.W., Van Hall T., Aerts J.G. Low-Dose JAK3 Inhibition Improves Antitumor T-Cell Immunity and Immunotherapy Efficacy. Mol. Cancer Ther. 2022;21:1393–1405. doi: 10.1158/1535-7163.MCT-21-0943. [DOI] [PubMed] [Google Scholar]
- 41.Hernandez R., Põder J., LaPorte K.M., Malek T.R. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat. Rev. Immunol. 2022;22:614–628. doi: 10.1038/s41577-022-00680-w. [DOI] [PubMed] [Google Scholar]
- 42.Leonard W.J., Lin J.-X. Strategies to therapeutically modulate cytokine action. Nat. Rev. Drug Discov. 2023;22:827–854. doi: 10.1038/s41573-023-00746-x. [DOI] [PubMed] [Google Scholar]
- 43.Saxton R.A., Glassman C.R., Garcia K.C. Emerging principles of cytokine pharmacology and therapeutics. Nat. Rev. Drug Discov. 2023;22:21–37. doi: 10.1038/s41573-022-00557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frey K., Fiechter M., Schwager K., Belloni B., Barysch M.J., Neri D., Dummer R. Different patterns of fibronectin and tenascin-C splice variants expression in primary and metastatic melanoma lesions: Fibronectin and tenascin-C splice variants expression. Exp. Dermatol. 2011;20:685–688. doi: 10.1111/j.1600-0625.2011.01314.x. [DOI] [PubMed] [Google Scholar]
- 45.Ray A., Williams M.A., Meek S.M., Bowen R.C., Grossmann K.F., Andtbacka R.H.I., Bowles T.L., Hyngstrom J.R., Leachman S.A., Grossman D., et al. A phase I study of intratumoral ipilimumab and interleukin-2 in patients with advanced melanoma. Oncotarget. 2016;7:64390–64399. doi: 10.18632/oncotarget.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weide B., Derhovanessian E., Pflugfelder A., Eigentler T.K., Radny P., Zelba H., Pföhler C., Pawelec G., Garbe C. High response rate after intratumoral treatment with interleukin-2: Results from a phase 2 study in 51 patients with metastasized melanoma. Cancer. 2010;116:4139–4146. doi: 10.1002/cncr.25156. [DOI] [PubMed] [Google Scholar]
- 47.Weide B., Eigentler T.K., Pflugfelder A., Leiter U., Meier F., Bauer J., Schmidt D., Radny P., Pföhler C., Garbe C. Survival after intratumoral interleukin-2 treatment of 72 melanoma patients and response upon the first chemotherapy during follow-up. Cancer Immunol. Immunother. 2011;60:487–493. doi: 10.1007/s00262-010-0957-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langan E.A., Kümpers C., Graetz V., Perner S., Zillikens D., Terheyden P. Intralesional interleukin-2: A novel option to maximize response to systemic immune checkpoint therapy in loco-regional metastatic melanoma. Dermatol. Ther. 2019;32:e12901. doi: 10.1111/dth.12901. [DOI] [PubMed] [Google Scholar]
- 49.Mattijssen V., De Mulder P.H., De Graeff A., Hupperets P.S., Joosten F., Ruiter D.J., Bier H., Palmer P.A., Van den Broek P. Intratumoral PEG-interleukin-2 therapy in patients with locoregionally recurrent head and neck squamous-cell carcinoma. Ann. Oncol. 1994;5:957–960. doi: 10.1093/oxfordjournals.annonc.a058739. [DOI] [PubMed] [Google Scholar]
- 50.Albertini M.R., Hank J.A., Gadbaw B., Kostlevy J., Haldeman J., Schalch H., Gan J., Kim K., Eickhoff J., Gillies S.D., Sondel P.M. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol. Immunother. 2012;61:2261–2271. doi: 10.1007/s00262-012-1286-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weide B., Eigentler T.K., Pflugfelder A., Zelba H., Martens A., Pawelec G., Giovannoni L., Ruffini P.A., Elia G., Neri D., et al. Intralesional Treatment of Stage III Metastatic Melanoma Patients with L19–IL2 Results in Sustained Clinical and Systemic Immunologic Responses. Cancer Immunol. Res. 2014;2:668–678. doi: 10.1158/2326-6066.CIR-13-0206. [DOI] [PubMed] [Google Scholar]
- 52.Danielli R., Patuzzo R., Di Giacomo A.M., Gallino G., Maurichi A., Di Florio A., Cutaia O., Lazzeri A., Fazio C., Miracco C., et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: results of a phase II study. Cancer Immunol. Immunother. 2015;64:999–1009. doi: 10.1007/s00262-015-1704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Herpen C.M., Looman M., Zonneveld M., Scharenborg N., de Wilde P.C., van de Locht L., Merkx M.A.W., Adema G.J., De Mulder P.H. Intratumoral Administration of Recombinant Human Interleukin 12 in Head and Neck Squamous Cell Carcinoma Patients Elicits a T-Helper 1 Profile in the Locoregional Lymph Nodes. Clin. Cancer Res. 2004;10:2626–2635. doi: 10.1158/1078-0432.CCR-03-0304. [DOI] [PubMed] [Google Scholar]
- 54.Chiocca E.A., Yu J.S., Lukas R.V., Solomon I.H., Ligon K.L., Nakashima H., Triggs D.A., Reardon D.A., Wen P., Stopa B.M., et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Herpen C.M., Huijbens R., Looman M., de Vries J., Marres H., van de Ven J., Hermsen R., Adema G.J., De Mulder P.H. Pharmacokinetics and Immunological Aspects of a Phase Ib Study with Intratumoral Administration of Recombinant Human Interleukin-12 in Patients with Head and Neck Squamous Cell Carcinoma: A Decrease of T-bet in Peripheral Blood Mononuclear Cells. Clin. Cancer Res. 2003;9:2950–2956. [PubMed] [Google Scholar]
- 56.Kramer G., Steiner G.E., Sokol P., Handisurya A., Klingler H.C., Maier U., Földy M., Marberger M. Local Intratumoral Tumor Necrosis Factor- α and Systemic IFN- α 2b in Patients with Locally Advanced Prostate Cancer. J. Interferon Cytokine Res. 2001;21:475–484. doi: 10.1089/10799900152434349. [DOI] [PubMed] [Google Scholar]
- 57.Lutz E.A., Jailkhani N., Momin N., Huang Y., Sheen A., Kang B.H., Wittrup K.D., Hynes R.O. Intratumoral nanobody–IL-2 fusions that bind the tumor extracellular matrix suppress solid tumor growth in mice. PNAS Nexus. 2022;1:pgac244. doi: 10.1093/pnasnexus/pgac244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ricard-Blum S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitra S., Ring A.M., Amarnath S., Spangler J.B., Li P., Ju W., Fischer S., Oh J., Spolski R., Weiskopf K., et al. Interleukin-2 Activity Can Be Fine Tuned with Engineered Receptor Signaling Clamps. Immunity. 2015;42:826–838. doi: 10.1016/j.immuni.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carmenate T., Pacios A., Enamorado M., Moreno E., Garcia-Martínez K., Fuente D., León K. Human IL-2 Mutein with Higher Antitumor Efficacy Than Wild Type IL-2. J. Immunol. 2013;190:6230–6238. doi: 10.4049/jimmunol.1201895. [DOI] [PubMed] [Google Scholar]
- 61.Chen X., Ai X., Wu C., Wang H., Zeng G., Yang P., Liu G. A novel human IL-2 mutein with minimal systemic toxicity exerts greater antitumor efficacy than wild-type IL-2. Cell Death Dis. 2018;9:989. doi: 10.1038/s41419-018-1047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levin A.M., Bates D.L., Ring A.M., Krieg C., Lin J.T., Su L., Moraga I., Raeber M.E., Bowman G.R., Novick P., et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine. Nature. 2012;484:529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carmenate T., Ortíz Y., Enamorado M., García-Martínez K., Avellanet J., Moreno E., Graça L., León K. Blocking IL-2 Signal In Vivo with an IL-2 Antagonist Reduces Tumor Growth through the Control of Regulatory T Cells. J. Immunol. 2018;200:3475–3484. doi: 10.4049/jimmunol.1700433. [DOI] [PubMed] [Google Scholar]
- 64.Kobayashi M., Kojima K., Murayama K., Amano Y., Koyama T., Ogama N., Takeshita T., Fukuhara T., Tanaka N. MK-6, a novel not-α IL-2, elicits a potent antitumor activity by improving the effector to regulatory T cell balance. Cancer Sci. 2021;112:4478–4489. doi: 10.1111/cas.15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merchant R., Galligan C., Munegowda M.A., Pearce L.B., Lloyd P., Smith P., Merchant F., To M.D. Fine-tuned long-acting interleukin-2 superkine potentiates durable immune responses in mice and non-human primate. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silva D.-A., Yu S., Ulge U.Y., Spangler J.B., Jude K.M., Labão-Almeida C., Ali L.R., Quijano-Rubio A., Ruterbusch M., Leung I., et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565:186–191. doi: 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quijano-Rubio A., Bhuiyan A.M., Yang H., Leung I., Bello E., Ali L.R., Zhangxu K., Perkins J., Chun J.-H., Wang W., et al. A split, conditionally active mimetic of IL-2 reduces the toxicity of systemic cytokine therapy. Nat. Biotechnol. 2023;41:532–540. doi: 10.1038/s41587-022-01510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lopes J.E., Fisher J.L., Flick H.L., Wang C., Sun L., Ernstoff M.S., Alvarez J.C., Losey H.C. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Charych D.H., Hoch U., Langowski J.L., Lee S.R., Addepalli M.K., Kirk P.B., Sheng D., Liu X., Sims P.W., VanderVeen L.A., et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016;22:680–690. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 70.Ptacin J.L., Caffaro C.E., Ma L., San Jose Gall K.M., Aerni H.R., Acuff N.V., Herman R.W., Pavlova Y., Pena M.J., Chen D.B., et al. An engineered IL-2 reprogrammed for anti-tumor therapy using a semi-synthetic organism. Nat. Commun. 2021;12:4785. doi: 10.1038/s41467-021-24987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rosen D.B., Kvarnhammar A.M., Laufer B., Knappe T., Karlsson J.J., Hong E., Lee Y.-C., Thakar D., Zúñiga L.A., Bang K., et al. TransCon IL-2 β/γ: a novel long-acting prodrug with sustained release of an IL-2Rβ/γ-selective IL-2 variant with improved pharmacokinetics and potent activation of cytotoxic immune cells for the treatment of cancer. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-004991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun Z., Ren Z., Yang K., Liu Z., Cao S., Deng S., Xu L., Liang Y., Guo J., Bian Y., et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat. Commun. 2019;10:3874. doi: 10.1038/s41467-019-11782-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waldhauer I., Gonzalez-Nicolini V., Freimoser-Grundschober A., Nayak T.K., Fahrni L., Hosse R.J., Gerrits D., Geven E.J.W., Sam J., Lang S., et al. Simlukafusp alfa (FAP-IL2v) immunocytokine is a versatile combination partner for cancer immunotherapy. mAbs. 2021;13 doi: 10.1080/19420862.2021.1913791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klein C., Waldhauer I., Nicolini V.G., Freimoser-Grundschober A., Nayak T., Vugts D.J., Dunn C., Bolijn M., Benz J., Stihle M., et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. OncoImmunology. 2017;6 doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Codarri Deak L., Nicolini V., Hashimoto M., Karagianni M., Schwalie P.C., Lauener L., Varypataki E.M., Richard M., Bommer E., Sam J., et al. PD-1-cis IL-2R agonism yields better effectors from stem-like CD8+ T cells. Nature. 2022;610:161–172. doi: 10.1038/s41586-022-05192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee J.-Y., Lee E., Hong S.-W., Kim D., Eunju O., Sprent J., Im S.-H., Lee Y.J., Surh C.D. TCB2, a new anti-human interleukin-2 antibody, facilitates heterodimeric IL-2 receptor signaling and improves anti-tumor immunity. OncoImmunology. 2020;9 doi: 10.1080/2162402X.2019.1681869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamimura D., Sawa Y., Sato M., Agung E., Hirano T., Murakami M. IL-2 In Vivo Activities and Antitumor Efficacy Enhanced by an Anti-IL-2 mAb. J. Immunol. 2006;177:306–314. doi: 10.4049/jimmunol.177.1.306. [DOI] [PubMed] [Google Scholar]
- 78.Spangler J.B., Tomala J., Luca V.C., Jude K.M., Dong S., Ring A.M., Votavova P., Pepper M., Kovar M., Garcia K.C. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity. 2015;42:815–825. doi: 10.1016/j.immuni.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arenas-Ramirez N., Zou C., Popp S., Zingg D., Brannetti B., Wirth E., Calzascia T., Kovarik J., Sommer L., Zenke G., et al. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci. Transl. Med. 2016;8:367ra166. doi: 10.1126/scitranslmed.aag3187. [DOI] [PubMed] [Google Scholar]
- 80.Sahin D., Arenas-Ramirez N., Rath M., Karakus U., Hümbelin M., Van Gogh M., Borsig L., Boyman O. An IL-2-grafted antibody immunotherapy with potent efficacy against metastatic cancer. Nat. Commun. 2020;11:6440. doi: 10.1038/s41467-020-20220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tomala J., Kovarova J., Kabesova M., Votavova P., Chmelova H., Dvorakova B., Rihova B., Kovar M. Chimera of IL-2 Linked to Light Chain of anti-IL-2 mAb Mimics IL-2/anti-IL-2 mAb Complexes Both Structurally and Functionally. ACS Chem. Biol. 2013;8:871–876. doi: 10.1021/cb3007242. [DOI] [PubMed] [Google Scholar]
- 82.Luly K.M., Yang H., Lee S.J., Wang W., Ludwig S.D., Tarbox H.E., Wilson D.R., Green J.J., Spangler J.B. Poly(Beta-Amino Ester)s as High-Yield Transfection Reagents for Recombinant Protein Production. Int. J. Nanomedicine. 2022;17:4469–4479. doi: 10.2147/IJN.S377371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is availanle from lead contact the upon request.