

Summary
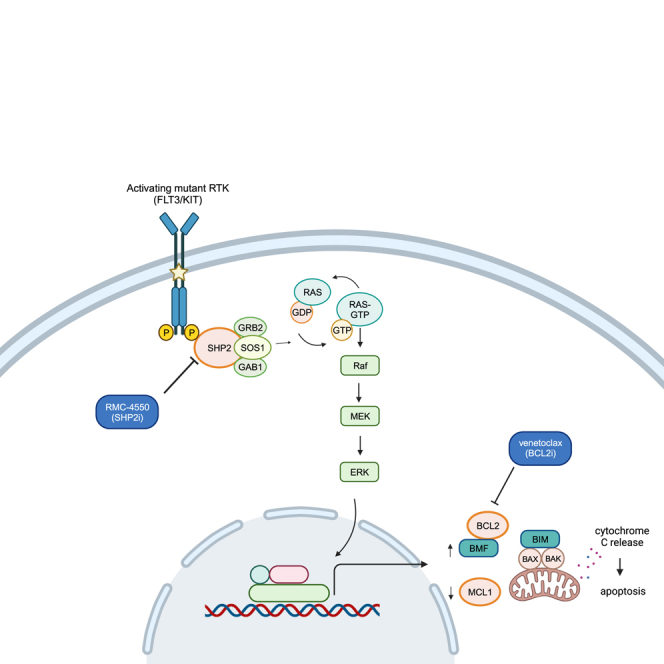
Mutations in the receptor tyrosine kinases (RTKs) FLT3 and KIT are frequent and associated with poor outcomes in acute myeloid leukemia (AML). Although selective FLT3 inhibitors (FLT3i) are clinically effective, remissions are short-lived due to secondary resistance characterized by acquired mutations constitutively activating the RAS/MAPK pathway. Hereby, we report the pre-clinical efficacy of co-targeting SHP2, a critical node in MAPK signaling, and BCL2 in RTK-driven AML. The allosteric SHP2 inhibitor RMC-4550 suppresses proliferation of AML cell lines with FLT3 and KIT mutations, including cell lines with acquired resistance to FLT3i. We demonstrate that pharmacologic SHP2 inhibition unveils an Achilles’ heel of RTK-driven AML, increasing apoptotic dependency on BCL2 via MAPK-dependent mechanisms, including upregulation of BMF and downregulation of MCL1. Consequently, RMC-4550 and venetoclax are synergistically lethal in AML cell lines and in clinically relevant xenograft models. Our results provide mechanistic rationale and pre-clinical evidence for co-targeting SHP2 and BCL2 in RTK-driven AML.
Keywords: acute myeloid leukemia, SHP2, apoptosis, cancer, drug synergy, FLT3, targeted therapies
Graphical abstract
Highlights
-
•
SHP2 inhibition has anti-leukemic activity in AML driven by FLT3 and KIT mutations
-
•
SHP2 inhibition increases AML dependency on BCL2 via RAS/MAPK-dependent mechanisms
-
•
Co-targeting SHP2 and BCL2 has synergistic activity in RTK-driven AML models
Resistance to targeted therapies in AML involves RAS/MAPK signaling hyperactivation. Popescu et al. report that SHP2 inhibition in RTK-driven AML leads to suppression of MAPK pathway signaling and increased apoptotic dependency on BCL2. Simultaneous co-targeting of SHP2 and BCL2 has synergistic anti-leukemic activity in FLT3- and KIT-mutant AML models.
Introduction
Mutations in the receptor tyrosine kinases (RTKs) encoded by FLT3 and KIT are consistently among the most commonly identified molecular lesions in acute myeloid leukemia (AML). These mutations occur in 30%–40% of individuals with AML1,2,3 and are associated with an increased risk of relapse. Individuals with FLT3 internal tandem duplication (ITD) mutations have decreased overall survival, primarily due to high relapse rates.4 Mutations in KIT, though more infrequent, are enriched in individuals with AML driven by CBFB-MYH11 or RUNX1-RUNX1T1 gene fusions and double the risk of relapse in a subtype of AML otherwise associated with good prognosis.5,6 Addition of the KIT inhibitor (KITi) dasatinib to chemotherapy in individuals with newly diagnosed core binding factor (CBF) AML is feasible,7,8 but improvements in relapse-free and overall survival have not been demonstrated in randomized trials. In contrast, FLT3 inhibitors (FLT3i) have proven benefits in individuals with AML with FLT3 mutations in both upfront and relapsed/refractory (R/R) settings. Addition of the FLT3i midostaurin or quizartinib to standard induction chemotherapy demonstrated a survival benefit over chemotherapy alone in newly diagnosed FLT3-mutant AML,9,10 and this strategy is currently the preferred first-line treatment in these affected individuals. The second-generation selective FLT3is quizartinib (AC220) and gilteritinib (ASP2215) showed superior overall survival compared with salvage chemotherapy in R/R FLT3-mutant AML in multinational phase III trials,11,12 establishing a new standard of care. However, remissions induced by these targeted therapies are short-lived, and relapse due to secondary resistance limits clinical benefit.13,14 Given poor outcomes and the lack of consensus-validated standard of care for individuals who relapse on FLT3i, there is a critical unmet need for novel treatment strategies to overcome clinical resistance.
Our group and others recently identified the emergence of polyclonal RAS/MAPK pathway mutations, most commonly in the form of activating NRAS mutations, in individuals relapsing on FLT3i.14,15,16 Functional and biochemical analysis of AML cell lines implicate reactivation of MAPK signaling as a primary mediator of acquired resistance to FLT3i.14 Furthermore, in vitro modeling studies highlight the importance of protective bone marrow microenvironment cytokines in the progression of the MAPK-driven resistance. Specifically, soluble factors such as FLT3 ligand (FLT3-L) and fibroblast growth factor 2 (FGF2) reactivate ERK phosphorylation in residual AML cells and lay the groundwork for overt resistance, characterized by acquisition of intrinsic resistance mutations.17,18
RAS pathway mutations have also been associated with intrinsic and adaptive resistance to other targeted AML therapies, including the IDH2 inhibitor enasidenib19 and the BCL2 inhibitor venetoclax.20 While no studies have investigated the clinical resistance to KITis in AML, RAS mutations have been described in resistance to KITis in individuals with KIT-mutant gastrointestinal stromal tumor (GIST).21 The body of evidence associating activating RAS mutations with clinical resistance to the full spectrum of approved targeted therapeutics in AML identifies a common and critical barrier to improved outcomes in AML.
Tyrosine-protein phosphatase non-receptor 11 (SHP2; encoded by PTPN11) is a protein tyrosine phosphatase (PTP) that functions as a signal-relay molecule for RTK-mediated activation of the RAS/MAPK signaling pathway. Acting downstream of RTKs, SHP2 serves as a scaffold for the assembly of multi-protein complexes at the cell membrane that promote GTP loading of RAS by the guanine nucleotide exchange factor (GEF) SOS1.22 SHP2 also promotes RAS activation by dephosphorylating and inhibiting key RAS-negative regulators such as RAS GTPase-activating protein (RasGAP)23 and Sprouty proteins.24 SHP2 has also been linked to RTK-dependent activation of phosphatidylinositol 3-kinase (PI3K)/AKT25 and JAK/STAT signaling26 and to upregulation of anti-apoptotic gene expression in FLT3-ITD+ cells.27,28 Allosteric SHP2 inhibitors (SHP2i) stabilize the closed, autoinhibited conformation, hampering both the catalytic and the scaffolding functions of SHP2.22,29 Such compounds have demonstrated pre-clinical anti-tumor activity in multiple RTK-driven cancers, including FLT3-mutant AML in vitro and murine models.29,30,31 Moreover, allosteric SHP2i have shown activity in cell lines bearing RAS mutant proteins with substitutions at glycine 12 that remain dependent on GEF-mediated GTP loading for survival signaling.22,32 SHP2i also prevent adaptive resistance to MEK inhibitors in both wild-type and mutant KRAS cancer models.33
Given the pivotal role of SHP2 in RTK signaling, we hypothesize that SHP2 inhibition has activity in multiple subtypes of RTK-driven AML and suppresses key mechanisms of tyrosine kinase inhibitor (TKI) resistance dependent on activated MAPK signaling, particularly those driven by GTP-cycling RAS mutations and cytokine signaling. Although clinical-grade compounds such as TNO-155, RMC-4630, or JAB-3312 are under investigation in phase I/II clinical trials in a myriad of solid malignancies,34 SHP2 inhibition has not yet been clinically explored in AML, and effective targets for potential clinical combinations remain unknown.
Here, we show that the allosteric SHP2i pre-clinical tool compound RMC-455022 potently and selectively suppresses cell proliferation and induces apoptosis in FLT3- and KIT-mutant AML cell lines. Using high-throughput transcriptomics, dynamic BH3 profiling, and biochemical analyses, we demonstrate that SHP2 inhibition exposes an Achilles’ heel of RTK-driven AML cells by increasing apoptotic dependency on BCL2 via multiple MAPK-dependent mechanisms. Exploiting this vulnerability, we found that combining RMC-4550 and the FDA-approved BCL2 inhibitor venetoclax is highly synergistic in AML cell line models with FLT3 or KIT mutations, but also in FLT3-mutant AML cells harboring NRASG12C. Furthermore, the combination is effective in vivo, reducing leukemic burden and improving survival in both cell-line-derived xenograft (CDX) and patient-derived xenograft (PDX) AML models. Altogether, these data provide a mechanistic rationale and pre-clinical evidence for co-targeting of SHP2 and BCL2 as a therapeutic strategy in FLT3- and KIT-mutant AML.
Results
RMC-4550 is active in FLT3- and KIT-mutant AML cell lines
As RTK-driven AML relies upon hyperactive RAS/MAPK signaling for survival, we hypothesized that targeting SHP2 would inhibit proliferation of RTK-driven AML cells, as has been previously reported in solid tumors.22,29,30,32 To test this hypothesis, we assessed the efficacy of RMC-4550 in 10 AML cell lines with RTK or downstream signaling mutations, using a cell viability assay (Figure 1A). As expected, RMC-4550 inhibited cell proliferation in cell lines with RTK mutations, most potently in the FLT3-ITD mutant cell lines Molm-14 and MV4-11 (IC50 = 146 and 120 nM, respectively) and less potently in the KITN822K mutant cell lines Kasumi-1 and SKNO-1 (IC50 = 193 and 480 nM, respectively). To a lesser degree, RMC-4550 also inhibited the growth of the K562 cell line driven by the BCR-ABL1 gene fusion (IC50 = 2,086 nM). In general, pharmacological sensitivity to SHP2 inhibition correlated with the cell lines’ genetic dependency on PTPN11, evaluated in the Cancer Dependency Map portal (DepMap: https://depmap.org) (Figure 1B), with the exception of U937 cells. Although U937 is a cell line highly dependent on PTPN11, it has an activating PTPN11G60R mutation. Substitutions at the G60 residue are shown to bias the open conformation of SHP2 and are associated with resistance to allosteric SHP2 inhibition.35,36 Accordingly, cell lines with activating mutations in PTPN11 (U937, PTPN11G60R) or RAS (OCI-AML3, NRASQ61L; THP-1, NRASG12D; NOMO-1, KRASG13D; HL-60, NRASQ61L) were resistant to RMC-4550 (IC50 > 10 μM, Figure 1A and Table S1).
Figure 1.

Pharmacologic inhibition of SHP2 has anti-proliferative activity in RTK-driven AML cell lines
(A) Dose-response curves representing relative proliferation of nine AML cell lines after 48 h of exposure to serial doses of RMC-4550. Data represent the mean ± SEM of three technical replicates.
(B) Pearson correlation between sensitivity to RMC-4550 represented by IC50 values obtained from (A) and genetic dependency on PTPN11 represented as CRISPR gene effect scores from http://depmap.org. Lower scores denote high genetic dependency. Pearson’s r and two-tailed p shown.
(C) Dose-response curves of Molm-14 cells with doxycycline-inducible expression of PTPN11 variants exposed for 48 h to serial doses of RMC-4550. Data represent the mean ± SEM of three technical replicates.
(D) RAS-GTP levels in four AML cell lines exposed for 90 min to RMC-4550. Data represent the means of three technical replicates ±SD; two-tailed ANOVA with Tukey’s correction for multiple comparisons (∗∗∗∗p ≤ 0.0001, ∗∗∗p ≤ 0.001).
(E) Western blot analysis of four AML cell lines exposed for 90 min to RMC-4550; β-actin was used as loading control. For phospho-ERK expression, band intensities from images were normalized to a total ERK control.
(F) Dose-response curves representing relative proliferation of quizartinib-selected Molm-14 cells with additional mutations in FLT3 and NRAS genes. Data represent the mean ± SEM of three technical replicates.
(G and H) Dose-response curves showing caspase 3/7 expression in Molm-14 cells cultured in regular growth medium or HS-5 stromal cell-conditioned medium after 24 h of exposure to increasing doses of gilteritinib or RMC-4550, normalized to untreated controls. Data represent the means ± SD of three technical replicates.
To determine if the anti-proliferative effect of RMC-4550 is due to on-target SHP2 inhibition, we transduced FLT3-ITD+ Molm-14 cells with doxycycline-inducible constructs to overexpress wild-type PTPN11 or activating PTPN11 variants bearing activating mutations,36 as well as the double PTPN11T253L/Q257M mutation, previously reported to impair the binding of allosteric SHP2.29 The hyperactive PTPN11E76K and the PTPN11T253L/Q257M mutants were profoundly resistant to RMC-4550, while the mildly activating PTPN11S502P and PTPN11G60V mutants exhibited less resistance (Figure 1C). At a biochemical level, in RTK-driven sensitive cell lines, RMC-4550 rapidly impaired RAS GTP loading and repressed downstream phosphorylation of ERK in cell lines with FLT3 and KIT mutations (Figures 1D and 1E), but only minimally in OCIAML-3, a cell line with a constitutive NRASQ61L mutation and low genetic dependency on PTPN11 (Figures S1A and S1B). We observed a comparable level of MAPK signaling inhibition in RTK-driven cell lines after treatment with selective FLT3 and KIT inhibitors (gilteritinib and avapritinib, respectively; Figures S1C and S1D). In addition, RMC-4550 did not suppress RAS-GTP levels in Molm-14 cells with doxycycline-inducible expression of a PTPN11T253L/Q257M mutation, suggesting this is an on-target effect (Figure S1E). In keeping with the idea that the activity of RMC-4550 is mediated primarily through inhibition of MAPK signaling, RMC-4550 exhibited no activity in the JAK2V617F mutant HEL cell line. While RMC-4550 induced modest inhibition of RAS-GTP and ERK phosphorylation in these cells (Figures S1F and S1G), there was no significant inhibition of proliferation (Figure S1H), consistent with these cells’ dependence on JAK2/STAT rather than MAPK signaling.
RMC-4550 is active in FLT3 TKI-resistant cell lines
We next evaluated the activity of RMC-4550 under conditions conferring resistance to FLT3 TKIs. RMC-4550 maintained activity against Molm-14 cells expressing FLT3 tyrosine kinase domain (TKD) mutations associated with secondary resistance to FLT3 TKIs (Figure 1F),13,37 including the “gatekeeper” F691L variant and mutations in the FLT3 activation loop. We also evaluated RMC-4550 efficacy in Molm-14 cells harboring secondary NRASG12C or NRASQ61K mutations that emerged during long-term exposure to the FLT3i quizartinib. Whereas RMC-4550 retained activity in Molm-14 cells with a NRASG12C mutation, the NRASQ61K variant conferred a high degree of resistance (Figure 1F). This finding agrees with the previously reported observation that RASG12C mutants are oncoproteins that retain RAS-GTPase activity and are, therefore, vulnerable to impaired SOS1/2-mediated exchange induced by SHP2 inhibition.32 Moreover, we observed that treatment with increasing doses of RMC-4550 enhanced the anti-proliferative activity of gilteritinib in Molm-14 and Molm-14 NRASG12C cells, but not in Molm-14 NRASQ61K (Figure S1I), highlighting the potential of SHP2 inhibition to overcome resistance to FLT3is in cells with RAS mutations that preserve GTP hydrolysis activity. Cell-extrinsic resistance to FLT3i is associated with ERK reactivation induced by bone marrow microenvironment-protective cytokines.17 Accordingly, we assessed the ability of RMC-4550 and the FLT3i gilteritinib to induce apoptosis in Molm-14 cells cultured in medium conditioned by the immortalized human bone marrow stromal cell line HS5 and in medium enriched with FGF2 and FLT3-L at 10 ng/mL.17,38 When cultured in conditioned and cytokine-enriched media, Molm-14 cells exhibited significantly less apoptosis upon exposure to gilteritinib. In contrast, RMC-4550 was much less vulnerable to resistance to apoptosis induced by conditioned and cytokine-enriched media (Figures 1G, 1H, S1J, and S1K). This finding suggests that SHP2 inhibition has the potential to overcome stromal-mediated resistance to FLT3 TKIs.
SHP2 inhibition alters the transcriptomic profile of RTK-driven AML cell lines
To evaluate the transcriptomic changes induced by SHP2 inhibition and identify potential complementary pathways that could be effectively co-targeted with an SHP2i, we performed RNA sequencing (RNA-seq) in two FLT3-ITD (Molm-14, MV4-11) and one KIT-mutant (SKNO-1) AML cell line treated with RMC-4550 or vehicle (DMSO). Hierarchical clustering revealed significant changes in global gene expression with SHP2i treatment (Figure 2A), and principal-component analysis (PCA) showed distinct gene expression patterns in samples treated with RMC-4550 compared with vehicle (Figures S2A and S2B). Differential gene expression analysis identified 132 overlapping dysregulated genes (fold change >2 compared with untreated control, false discovery rate [FDR] adj p < 0.05) in all three cell lines (Figure 2B). Molm-14 cells exhibited the highest number of differentially expressed genes (3,016) and MV4-11 the lowest (464) (Figures 2B–2E). Following enrichment analysis Gene Ontology (GO) biological process terms, we found the common 132 differentially expressed genes to be involved in processes such as cell-cycle regulation, cell division, interferon response pathways, or cytokine-mediated signaling pathways (Figure S2C).
Figure 2.

SHP2 inhibition alters the gene expression in FLT3- and KIT-mutant AML cell lines
(A) Heatmap showing gene intensity per sample relative to the average level across all samples. Individual genes are shown on the y axis and samples are shown on the x axis. Red and blue cells correspond to higher and lower RNA-seq levels, respectively. One thousand genes selected at random from all differentially expressed genes are shown. Samples are three replicates of Molm-14, MV4-11, and SKNO-1 cells, treated for 24 h with either DMSO or RMC-4550, 500 nM.
(B) Venn diagram showing the number of differentially expressed genes in each cell line and the overlaps between cell lines.
(C–E) Volcano plots displaying differentially expressed genes in each cell line. Highlighted genes above a significance threshold of −log10(pval) ≥ 7 in Molm-14 and MV4-11 and ≥6 in SKNO-1 cells.
(F) Barcode plots representing GSEA results for the indicated gene sets in all cell lines. Normalized enrichment score (NES) and false discovery rate (FDR) were determined by the GSEA computational method.
(G) qRT-PCR data representing gene expression change relative to DMSO-treated cells of IFIT2, OAS1, and OASL genes after 24 h treatment with RMC-4550, 500 nM. Expression of MEKDD was induced with 24 h treatment with doxycycline, 1 μg/mL. Data represent the mean ± SD of three technical replicates, normalized to DMSO-treated controls; t test with Benjamini-Hochberg FDR (Q = 1%) was used for statistical significance (∗∗∗∗Q ≤ 0.0001, ∗∗Q ≤ 0.01; ns, non-significant).
(H) Barcode plot of apoptosis gene set and heatmap representing relative expression of genes. Genes with an average fold change ≥2 or ≤ −2 are shown.
In a cross-cell-line pooled analysis, gene set enrichment analysis (GSEA) for hallmark gene signatures39,40 showed increased expression of genes downregulated by KRAS activation and decreased expression of MYC targets (Figure 2F), consistent with expected transcriptomic changes associated with RAS-MAPK signaling inhibition. Most of the highest enriched gene sets in the pooled analysis were also commonly enriched in each individual cell line (Figures S2D–S2F). Cells treated with RMC-4550 exhibited a marked overexpression of genes implicated in interferon-α (IFN-α) and IFN-γ signaling (Figures 2F and S2G). Interferons are known to be potent inducers of apoptosis in cancer cells via numerous mechanisms.41,42,43 Among IFN-stimulated genes (ISGs) upregulated by RMC-4550 in all cell lines, IFIT2 (or ISG54; fold change compared with untreated control: 3.06 to 18.44) has been shown to induce Bax- and/or Bak-dependent apoptosis via activation of the mitochondrial pathway.43 Similarly, RMC-4550 upregulated the expression of 2′–5′ oligoadenylate synthetase family members OAS1, OAS2, OAS3, and OASL in each cell line (fold change compared with untreated control: 3.5 to 20). These proteins, under IFN stimulation, induce apoptosis by both degrading intracellular RNA via RNase L44 and directly interacting with BCL2 family members.45 Moreover, we observed a significant increase in expression of the IFN-responsive tumor suppressor SAMD9L46 in all cell lines and of the pro-apoptotic X-linked inhibitor of apoptosis-associated factor 1 (XAF1) in the MV4-11 and SKNO-1 cell lines.
Upregulation of IFN gene sets in response to several kinase inhibitors in RTK-driven cancer cell lines has been previously reported.47 We investigated if the overexpression of IFN targets in our dataset is contingent upon SHP2i ability to suppress RAS/MAPK signaling. RMC-4550 significantly upregulated the gene expression of IFIT-2, OAS-1, and OAS-L in Molm-14 cells, but not in cells with doxycycline-induced overexpression of a dominant-active MEK1 allele (MEKS218D/S222D, referred to as MEKDD)48 (Figure 2G), confirming that SHP2i increases transcription of pro-apoptotic IFN targets via suppression of RAS/MAPK signaling.
Consistent with RAS-MAPK inhibition and ISG overexpression, we also noted a significant transcriptional downregulation of genes encoding cell-cycle-related targets of E2F transcription factors and of genes involved in the G2/M checkpoint, suggesting that SHP2i impairs DNA replication and blocks cell-cycle progression (Figure 2F). An intriguing finding was the enrichment of the myogenesis gene set, a sign of phenotypical remodeling of the contractile cytoskeleton (Figure 2F). Genetically and pharmacologically interfering with actomyosin contractility has been shown to induce apoptosis in the HL-60 AML cell line and to modulate oncogenic gene expression through the mechanotransducing system of YAP.49 In EGFR-mutant non-small cell lung cancer (NSCLC), hyperactive YAP is associated with resistance to EGFR and MEK1 inhibition and apoptosis evasion through YAP-mediated repression of pro-apoptotic BMF (Bcl2-modifying-factor).50 In our dataset, the genes mediating apoptosis were not dysregulated to a significant threshold (FDR = 0.28), likely due to differential expression of genes coding for both pro- and anti-apoptotic proteins (Figure 2H). However, we found the pro-apoptotic TRAIL (TNFSF10), BIM (BCL2L11), and BMF to be upregulated across all cell lines. Of these, BMF was upregulated to a greater extent on average and was one of the top 10 most significantly upregulated genes in Molm-14 cells (fold change compared with untreated control: 4.41; FDR 8.1e−8). BMF codes for a BH3-only protein, found at a steady state to be sequestered with actin-associated myosin V motors. Upon anoikis (a type of programmed cell death), it detaches from the cytoskeleton and binds and neutralizes BCL2 family members to allow cytochrome c release from mitochondria.51,52 Anoikis occurs as a consequence of cell detachment from the extracellular matrix (ECM) and cytoskeletal perturbations, preventing ectopic cell growth and readhesion.52
RMC-4550 increases apoptotic dependency on BCL2 in FLT3- and KIT-mutant AML
In light of our RNA-seq findings, we sought to confirm transcriptional upregulation of BMF using qRT-PCR. Treatment with RMC-4550 significantly increased the gene expression levels of BMF in all four cell lines with RTK mutations (Molm-14, MV4-11, Kasumi-1, SKNO-1), but not in OCIAML-3 or HEL cells (Figures 3A and S3A) or in Molm-14 cells with doxycycline-induced expression of MEKDD (Figure 3B), highlighting that SHP2i-mediated suppression of MAPK signaling is required for BMF transcriptional upregulation. At the protein level, BMF was increased upon 24 h exposure to RMC-4550, although the degree of increase varied by cell line. In contrast, consistent changes in other pro- and anti-apoptotic proteins were not observed. Upregulation of the pro-apoptotic protein BIM was noted in MV4-11 cells, but not in the other cell lines tested. We also noticed decreased levels of the MAPK-dependent anti-apoptotic MCL1 in Molm-14 and SKNO-1 cells. No significant change in any of the BH3-family proteins occurred in the OCIAML-3 or HEL cell lines (Figures 3C and S3B). In keeping with our RNA-seq results, Molm14 cells had the greatest degree of SHP2i-induced BMF upregulation on both the transcript and the protein level. Consistent with this, Molm-14 and SKNO1 cells treated for 24 h with RMC-4550 exhibited higher cytoplasmic intracellular levels of BMF compared with untreated cells, as shown by immunofluorescent staining (Figures 3D–3F).
Figure 3.

RMC-4550 increases expression of pro-apoptotic BMF and the dependency of apoptosis on BCL2 in RTK-driven AML cell lines
(A and B) qRT-PCR data representing gene expression change of the BMF gene in (A) Molm-14, MV4-11, Kasumi-1, SKNO-1, and OCIAML-3 cells and in (B) Molm-14 MEKDD cells after 24 h of treatment with RMC-4550. Data represent the mean ± SD of three technical replicates; two-way ANOVA with Sidak’s correction for multiple comparisons was used for statistical significance.
(C) Western blot analysis of pro- and anti-apoptotic proteins in Molm-14, MV4-11, Kasumi-1, and SKNO-1 cells exposed to RMC-4550, 1,000 nM, for 24 h; β-actin was used as the loading control.
(D and E) Representative immunofluorescence images showing cytoplasmic increase in BMF (green) after 24 h of treatment with RMC-4550 in Molm-14 (D) and SKNO-1 (E) cell lines. DAPI (blue) was used as a nuclear stain. Images were acquired at 20× magnification using confocal microscopy. Scale bar, 25 μm.
(F) Plots showing mean fluorescence intensity (MFI) values normalized to cell area of BMF immunofluorescent staining in Molm-14 and SKNO-1 cells untreated and treated with RMC-4450, 1,000 nM. For each condition, images from three independent slides were acquired using confocal microscopy, and regions of interest (ROIs) were drawn around each single cell. Data represent background-subtracted MFI divided by area or each ROI; Wilcoxon signed-rank test was used for statistical significance.
(G) iBH3 profiling data representing normalized cytochrome c release following 16 h of treatment with DMSO or RMC-4550 and exposure to BIM peptide or BCL2, BCLxL, and MCL1 inhibitors in Molm-14, MV4-11, Kasumi-1, and SKNO-1 cells. Data represent the mean ± SD of three technical replicates; two-way ANOVA with Sidak’s correction for multiple comparisons was used for statistical significance (∗∗∗∗p ≤ 0.0001, ∗∗∗p ≤ 0.001, ∗∗p ≤ 0.01, ∗p ≤ 0.05, ns, non-significant).
BMF exerts its pro-death response by binding BCL2 family members. Among its binding partners, BMF has the highest affinity for BCL2.53 To assess the apoptotic dependency on individual BCL2 family proteins, we used a BH3 profiling assay54 to quantify the mitochondrial outer membrane permeabilization (MOMP) as a result of exposure to either BH3 peptides (BIM) or BH3 inhibitors (BCL2, ABT-199/venetoclax; BCLxL, WEHI539; MCL1, S63845) in Molm-14, MV4-11, Kasumi-1, and SKNO-1 cells. The doses of peptides and inhibitors used for this assay were selected based on the baseline apoptotic dependency of each cell line on each BH3 family member. Treatment with RMC-4550 increased global apoptotic priming (measured by increased cytochrome c release after exposure to BIM peptide) and apoptotic dependency on BCL2 (measured by increased cytochrome c release after exposure to ABT-199) in all cell lines. Cytochrome c release in response to exposure to BCLxL or MCL1 inhibitors was inconsistent across cell lines (Figure 3G). These findings suggest that, upon SHP2 inhibition, these AML cells are more primed for apoptosis and their apoptotic dependency shifts toward BCL2. Therefore, co-inhibition of SHP2 and BCL2 may be a mechanistically complementary approach to induce apoptosis. Using a caspase 3/7 assay (Figure S3C), we observed that as monotherapy, RMC-4550 only moderately induced apoptosis in RTK-driven AML cells (Figure S3D) but did so more effectively than the potent MEK inhibitor trametinib (Figure S3E). Taken together, these observations imply that a combination treatment of RMC-4550 and venetoclax can enhance cell death in RTK-driven AML models.
Pharmacologic co-targeting of SHP2 and BCL2 has synergistic anti-proliferative activity in FLT3- and KIT-mutant AML
Having pinpointed BCL2 dependency as a vulnerability of RTK-driven AML cells treated with SHP2i, we hypothesized that RMC-4550 may sensitize these cells to pharmacological inhibition of BCL2 with venetoclax. To test this hypothesis, we treated Molm-14, MV4-11, Kasumi-1, and SKNO-1 cells with both agents and performed formal assessment of drug synergy using a cell viability readout (CellTiterGlo) and the SynergyFinder 3.0 computational tool.55 The combination was highly synergistic in these cell lines. We also observed a high synergy score in Molm-14 NRASG12C, despite reduced sensitivity to RMC-4550 as monotherapy (Figure 4A). In contrast, we did not observe synergistic activity in Molm-14 NRASQ61K cells, other RAS mutant AML cell lines fully insensitive to single-agent SHP2 inhibition, or the JAK2V617 mutant HEL cells (Figure S4A). Interestingly, we observed moderate combinatorial activity at high doses of the two compounds in the BCR-ABL1 mutant cell line K562. Although it is unclear if translationally relevant in the setting of BCR-ABL1 fusions, this effect is in line with previous reports of synergistic co-inhibition of MAPK signaling with gilteritinib and BCL2i in non-FLT3 mutant AML.56 Interestingly, in the FLT3-ITD+ cell lines Molm-14 and MV4-11, the combination of RMC-4550 and venetoclax was more synergistic than gilteritinib and venetoclax (Figure S4B), an active combination that achieved high modified composite complete remission (mCRc) rates in a phase Ib clinical investigation in individuals with R/R FLT3 mutant AML.57 Furthermore, the combination of RMC-4550 and venetoclax induced significantly higher levels of apoptosis in comparison with individual single agents in Molm-14, MV4-11, Kasumi-1, and SKNO-1 cell lines and Molm-14 NRASG12C cells (Figure 4B). In Molm-14 NRASQ61K, OCIAML-3, THP-1, and HEL cell lines, the drug combination was largely ineffective at inducing apoptosis and only minimally increased apoptosis compared with venetoclax alone in K562 and THP1 cells (Figure S4C). We did note that single-agent venetoclax induced higher levels of apoptosis in Molm-14 NRASG12C compared with the parental Molm14 cell line (Figure 4B). To better understand this increased sensitivity to BCL2 inhibition, we performed a comparative baseline BH3 profiling assessment of Molm-14 and Molm-14 NRASG12C cells and observed that the co-mutant cells have a higher mitochondrial permeability to cytochrome c at higher doses of venetoclax, suggesting they are more BCL2 dependent compared with the parental cell line. (Figure S4D). This increased BCL2 dependence is potentially due to the toxic effect of hyperactive RAS signaling previously reported in other RTK-driven cancers.58
Figure 4.

Co-targeting SHP2 and BCL2 is a synergistic therapeutic approach in FLT3- and KIT-mutant AML
(A) Dose-response matrices representing normalized cell viability inhibition following 48 h of treatment with increasing doses of RMC-4550 and venetoclax in the indicated cell lines. Synergy scores were computed using the Bliss method within SynergyFinder v.3.0 software.
(B) Caspase 3/7 expression after 24 h for treatment with RMC-4550 and venetoclax at the indicated concentrations. Results depict caspase 3/7-positive cells normalized to untreated controls. Data represent the mean ± SD of three technical replicates; one-way ANOVA with Dunnett correction for multiple comparisons (∗∗∗∗p ≤ 0.0001).
(C) Western blot analysis of BMF protein after 24 h of treatment with RMC-4550 in Molm-14 and SKNO-1 cells with BMF CRISPR knockout; β-actin was used as loading control.
(D and E) Dose-response curves of apoptosis measured after 24 h of treatment with RMC-4550 and venetoclax in Molm-14 and SKNO-1 BMF KO cells compared with control cells. Data represent the mean ± SD of three technical replicates per condition, normalized to untreated controls.
(F) Western blot analysis showing phospho-ERK and MCL1 dysregulation after treatment for 90 min and 24 h with RMC-4550 and venetoclax; β-actin was used as loading control.
Having demonstrated that SHP2 inhibition upregulates BMF, a potent BCL2 repressor, we aimed to investigate whether BMF is involved in the synergistic activity of RMC-4550 and venetoclax. Using CRISPR-Cas9 ribonucleoprotein transfection, we knocked out BMF in Molm-14 and SKNO-1 cells. Western blotting confirmed the ablation of BMF expression in knockout (KO) cells upon SHP2 inhibition (Figure 4C and Table S2). We treated cells with intact BMF and BMF KO with either increasing doses of RMC-4550 or one dose of RMC-4550 (250 nM for Molm-14, 500 nM for SKNO-1) and then exposed them for 24 h to increasing doses of venetoclax and measured caspase 3/7 levels. The sensitivity to apoptosis in response to either single agent was similar for both the control and the BMF-KO cell lines (Figures S4E and S4F). However, the apoptotic response to venetoclax when co-treated with RMC-4550 was attenuated in BMF-KO cells compared with control BMF-competent cells (Figures 4D and 4E). Although BMF-KO cells still undergo apoptosis, this difference underlines that maximal sensitization to BCL2 inhibition by RMC-4550 does not occur in the absence of BMF, implicating a role for BMF in mediating the drug synergy in both RTK-driven AML models.
Although RMC-4550 initially suppressed ERK phosphorylation in both Molm-14 and SKNO-1 cell lines (Figure 1C), we note rebound activation of pERK after 24 h of treatment (Figure 4F). Notably, the addition of venetoclax successfully reversed the rebound compared with RMC-4550 alone in both cell lines and downregulated MCL1 further in Molm-14 cells (Figure 4F and Table S2). These observations are in line with similar findings reported in Molm-14 cells co-treated with gilteritinib and venetoclax.59 Moreover, we found that, at 24 h, RMC-4550 and venetoclax mildly decreased pERK and downregulated MCL1 even in the presence of the NRASG12C mutation (Figure 4F and Table S2).
To investigate whether the synergic induction of cell death is a consequence of MAPK signaling suppression by SHP2i, we treated doxycycline-inducible MEKDD-overexpressing Molm-14 and MV4-11 cells with RMC-4550 and then exposed them to venetoclax and measured apoptosis. Overexpression of dominant-active MEKDD completely abrogated the apoptotic response to the drug combination. In contrast, doxycycline-inducible overexpression of a dominant-active AKT variant, AKTE17K, only partially attenuated the apoptotic response (Figures S4G and S4H). These findings highlight that effective inhibition of RAS-MAPK signaling is critical for SHP2i-mediated sensitization to venetoclax. In support of this observation, we found that the MEK inhibitor trametinib and venetoclax were also synergistic in inhibiting cell proliferation of the RTK-driven AML cell lines investigated (Figure S4I), in agreement with previous data reporting pre-clinical synergy between the MEK inhibitor cobimetinib and venetoclax in AML.60
The combination of RMC-4550 and venetoclax is active in cell-line-derived xenograft models of AML
We next sought to determine whether the synergistic activity of RMC-4550 and venetoclax observed in vitro could be recapitulated in vivo using CDX models. To emulate multiple distinct clinical scenarios, we evaluated AML cells with FLT3-ITD-Molm-14 cells, Molm-14 cells with acquired NRASG12C mutation, and KIT-mutant-Kasumi-1 cells. Cells were transduced with a firefly luciferase-containing lentiviral construct and then injected into NOD/SCID/IL2Rγnull (NSG) female mice. Upon engraftment, the mice were randomized into four groups receiving treatment with vehicle, RMC-4550 (30 mg/kg, orally, once daily [po, qd]), venetoclax (100 mg/kg, po, qd), or their combination, 5 days per week, for a total of 28 days (Figure 5A). Using in vivo bioluminescence imaging (BLI), we observed that combination treatment reduced in vivo tumor burden (Figures 5B, 5C, and S5A–S5D). In all three CDX models, mice in the combination arm had significantly enhanced survival compared with the control group and with monotherapies (Figures 5D–5F). Collectively, these data demonstrate in vivo combinatorial efficacy of SHP2 and BCL2 inhibition in FLT3- and KIT-mutant AML models, as well as in a model of AML resistant to FLT3i due to secondary NRASG12C mutation.
Figure 5.

Combination therapy with RMC-4550 and venetoclax is effective in vivo in CDX AML models
(A) Schematic representation of the CDX in vivo study design.
(B) Representative images of in vivo BLI assessment of NSG mice engrafted with luciferase-tagged Molm-14 cells during the trial.
(C) Quantification of BLI data from the Molm-14 CDX study, data represent the mean ± SD (n = 5/group).
(D–F) Kaplan-Meier curves representing survival data of subjects from each treatment group in (D) Molm-14, (E) Molm-14 NRASG12C, and (F) Kasumi-1 CDX studies; log rank (Mantel-Cox) test was used for comparison of survival curves and p values are shown.
RMC-4550 and venetoclax combination is effective in patient-derived AML xenografts and primary samples
We next aimed to verify the anti-leukemic activity of RMC-4550 and venetoclax in AML PDX models. To determine a tolerable dosing schedule, we initially treated non-tumor-bearing NSG-SGM3 mice (NSG mice with transgenic expression of human GM-CSF, IL-3, and SCF in an NSG background) with two doses (20 and 30 mg/kg) of RMC-4550 in combination with venetoclax (100 mg/kg). At the end of 28 days of treatment, no significant body weight changes or treatment-related toxicities were reported with either dosing regimen (Figure S6A), and the 30 mg/kg dosing strategy was selected for further PDX studies.
We next tested the combination in two PDX models established from individuals with FLT3-ITD AML (clinical data in Table S3). AML cells were injected into busulfan-conditioned NSG-SGM3 mice, as previously reported.61 After engraftment, mice were randomized into treatment groups: vehicle, RMC-4550 (30 mg/kg, po, qd), venetoclax (100 mg/kg, po, qd), and the combination of the two agents (Figure 6A). Flow-cytometric quantification of human CD45 (hCD45)-expressing cells in murine peripheral blood obtained from intermediate time-point bleedings was used to monitor disease progression (Figure S6B). The combination therapy reduced the percentage of circulating hCD45+ cells compared with both vehicle control and single-agent venetoclax in both PDX models and versus single-agent RMC-4550 in the PDX #1 model (Figures 6B and S6C). After 28 days of treatment or at the pre-determined humane study endpoint, mice were sacrificed and hCD45+ cells were enumerated in murine cardiac blood, spleen, and bone marrow by flow cytometry. Combination therapy led to significant suppression of leukemic burden in comparison with vehicle and venetoclax in all assessed organs (Figures 6C and 6D). The activity of RMC-4550 alone varied by assessed tissue, but the combination demonstrated significant suppression of hCD45+ cells compared with RMC-4550 alone, in spleen for one xenograft model (Figure 6C) and bone marrow for the other (Figure 6D). RMC-4550 alone or in combination also reduced spleen weight and length (Figures 6E, S6D, and S6E). Immunohistochemical staining of murine spleen samples at trial termination additionally showed a marked decreased of hCD45+ cells in mice receiving the combination (Figure 6F).
Figure 6.

Simultaneous SHP2 and BCL2 inhibition is effective in FLT3-mutant AML PDX models and in primary AML samples
(A) Schematic representation of the PDX in vivo study design.
(B) Quantification of hCD45+ cells over the course of treatment in the four treatment groups in the PDX #1 study.
(C and D) Quantification of hCD45+ in target organs at the study termination (PDX #1, n = 6/group; PDX #2, n = 5/group). Data represent the mean ± SD; one-way ANOVA with Tukey correction for multiple comparisons was used for statistical analysis and p values are shown.
(E) Representative images of spleens harvested from subjects in the vehicle or RMC-4550 + venetoclax treatment groups in PDX #1 study.
(F) Representative images of immunohistochemical staining for hCD45 in spleen samples from subjects in the vehicle or RMC-4550 + venetoclax treatment groups in PDX #2 study. Scale bar, 1000 μm.
(G) Violin plots showing differences in CFU-normalized counts in primary samples derived from healthy donors (n = 4) or individuals with AML (n = 4) treated for 14–16 days with RMC-4550, venetoclax, or their combination. Data represent three technical replicates per sample; unpaired t test with Holm-Sidak correction for multiple comparisons was used for statistical analysis and p values are shown.
Finally, in an effort to determine if the combination of RMC-4550 and venetoclax would be expected to suppress normal hematopoiesis, we evaluated the colony-forming unit (CFU) potential of primary bone marrow mononuclear cells (BMMCs) harvested from four healthy donors compared with four individuals with FLT3 mutant AML in the presence of DMSO, RMC-4550 (500 nM), venetoclax (200 nM), or both inhibitors. The combination treatment dramatically inhibited CFU formation in AML samples, but to a lesser extent in healthy donor samples (Figure 6G). Although the observed degree of colony growth inhibition in healthy bone marrow may suggest potential myelosuppression clinically, the efficacy of the combination in AML samples argues in favor of a therapeutic index. Nevertheless, the hematopoietic toxicity of the drug combination should be established clinically.
Discussion
Acquired resistance to FLT3i is a major challenge of the current therapeutic paradigm of FLT3-mutant AML. Single-cell genomic analyses of affected individuals treated with FLT3i have illustrated the heterogeneous nature of on-target and off-target resistance mechanisms.13,14,16 We and others previously reported that clones reactivating the RAS/MAPK pathway under FLT3 inhibition account for clinical resistance in a large proportion of affected individuals.14,16,62 Pre-clinical studies suggest that feedback activation of ERK signaling limits FLT3i efficacy63 and that microenvironmental factors protecting from FLT3i also converge on reactivation of downstream MAPK and PI3K/AKT signaling.17,18 Collectively, these data imply that RAS signaling is a primary mediator of survival signaling in FLT3-mutant AML.
SHP2 inhibition decreases oncogenic RAS-RAF-MEK-ERK signaling through disruption of SOS1-mediated RAS-GTP loading.22 The addition of SHP2i to MEK inhibitors successfully reversed feedback activation of RTK that leads to resistance in various solid malignancies.33,64,65 In FLT3-mutant AML, SHP2is showed promising single-agent activity in cell lines and murine models,29,31 but have not yet been clinically investigated in individuals with AML. In this study, we demonstrate that targeting SHP2 exposes a vulnerability of RTK-driven AML by increasing apoptotic priming and BCL2 dependency through multiple MAPK-dependent mechanisms, resulting in increased sensitivity to venetoclax-induced cell death. Our in vivo and in vitro findings thus provide robust pre-clinical evidence that coupling SHP2 blockade with the clinically available BCL2i venetoclax is a synergistic, effective approach against FLT3- and KIT-mutant AML. This drug combination is amenable to clinical translation and can address several mechanisms of adaptive resistance to FLT3i, including secondary FLT3 TKD mutations, exchange-sensitive RAS mutations, and AML-protective microenvironment crosstalk signals.
We notably show that RMC-4550 retains activity against TKI-resistant Molm-14 cells with NRASG12C, in agreement with published data suggesting that oncogenic RAS G12C variants are sensitive to upstream modulation of SOS1-mediated nucleotide exchange.22,32 Resistance to FLT3i has been previously characterized as a stepwise process, with an early phase of resistance, mediated by bone marrow microenvironmental cytokines that protect and metabolically reprogram residual AML cells, and a late, intrinsic resistance, defined by expansion of NRAS mutant subclones.17,18 Accordingly, we show that RMC-4550 successfully overcomes the paracrine-mediated resistance to FLT3i-induced apoptosis conferred by microenvironmental soluble factors, such as stromal-secreted cytokines, or exogenous FLT3-L and FGF2.
Using RNA-seq, we identified pro-death transcriptomic changes induced by SHP2i, such as upregulation of pro-apoptotic IFN-α- and IFN-γ-stimulated genes. Overexpression of IFN target genes following MAPK inhibition has been previously reported in cancer cell lines driven by oncogenic signaling and was associated with a drug-induced immune reprogramming resulting in a senescence-like state and immune evasion.47 In addition, we observed a significant upregulation of BMF, a pro-apoptotic protein known to transduce mechanical signals to the apoptotic machinery in the context of anoikis. High intracellular amounts of BMF lead to neutralization of anti-apoptotic proteins, particularly BCL2, facilitating BIM/BAX/BAK-mediated apoptosis.51,52 Transcriptional upregulation of both IFN targets and BMF is reversed in the setting of MEK activation, suggesting these changes rely upon effective ERK suppression.
Simultaneous with the overexpression of the BCL2 inactivator BMF, we observed downregulation of MCL1. A recent study reported that activation of RAS/MAPK leads to MCL1 overexpression in AML cell lines resistant to venetoclax, whereas genetic silencing or pharmacologic inhibition of MCL1 restores sensitivity to BCL2 inhibition.66 Using BH3 profiling assays we demonstrate that SHP2 inhibition increases overall sensitivity to BIM-induced apoptosis, but also the dependency of leukemia cells on BCL2. Collectively, these data suggest that SHP2i uncovers an unexpected susceptibility of RTK-driven AML, rendering AML cells dependent on BCL2 for cell death through a complex mechanism contingent on RAS/MAPK suppression.
Indeed, the combination of RMC-4550 and venetoclax synergizes in inhibiting leukemia proliferation, inducing apoptosis, and preventing ERK reactivation. Notably, the combination increases caspase levels even in AML cells, where either agent only modestly induces apoptosis. Recently, it was reported that clinical resistance to venetoclax in AML is associated with monocytic differentiation and a low BCL2/MCL1 gene expression ratio, whereas MEKis could sensitize monocytic AML cells to venetoclax.67,68 We show that RMC-4550 inhibits MAPK signaling and downregulates MCL1, suggesting a mechanism whereby SHP2i can potentially enhance the pharmacologic response to BCL2i in venetoclax-resistant AML.
A synergistic relationship between FLT3i and venetoclax has previously been demonstrated,59,69 and this combination has demonstrated clinical activity in R/R FLT3-mutant AML.57,70 However, this combination would not be expected to have activity in KIT-mutant AML. A recent study attributed the synergy of gilteritinib and venetoclax exclusively to BIM activation.59 The proposed mechanism is that venetoclax displaces BCL2 from BIM, while gilteritinib downregulates MCL1, thereby allowing available, free BIM to initiate apoptosis. In addition to MCL1 downregulation, we observe that BMF is a contributing factor to the synergy between RMC-4550 and venetoclax, as Molm-14 BMF-KO cells show less apoptosis than BMF-competent cells when treated with the combination. We find that BMF overexpression in response to SHP2 inhibition relies entirely on the ability of RMC-4550 to suppress MAPK signaling, considering that expression of BMF is unchanged in SHP2i-resistant MEKDD cells.
Of note, RMC-4550 and venetoclax do not elicit a synergistic induction of apoptosis in the setting of MEKDD overexpression, emphasizing the necessity of efficient MAPK signaling suppression for cytotoxicity. While upregulation of IFN signaling, upregulation of BMF, and downregulation of MCL1 appear to be partially responsible for the induction of apoptosis and BCL2 dependency, the full molecular mechanisms of RMC-4550 and venetoclax synergy remain to be elucidated. Nonetheless, these data suggest that other strategies co-targeting RAS/MAPK signaling and BCL2 would be similarly synergistic. Our findings are in line with those of a previous study reporting anti-leukemic activity of cobimetinib and venetoclax in multiple AML cell lines, exploiting MEKi-mediated MCL1 downregulation.60
Single-agent targeting of the MAPK pathway in AML has been largely ineffective or intolerable clinically.71,72,73 However, the use of MAPK inhibitors to increase sensitivity to BCL2 inhibition has the potential to both enhance clinical activity and facilitate the use of lower, more tolerable doses of drug. In our study, the combination of RMC-4550 and venetoclax was tolerable and effective in inhibiting leukemia progression and improving survival in clinically relevant CDX and PDX AML models. Notably, a small single-center study of azacitidine, venetoclax, and trametinib in heavily pre-treated R/R AML with RAS/MAPK pathway mutations demonstrated modest clinical activity with a response rate of 25%. However, this study did not include affected individuals with RTK mutations. In addition, the combination was poorly tolerated, with 50% of subjects requiring trametinib dose reductions, which potentially limited achievement of effective MAPK inhibition.74 Based on our findings, we hypothesize that the doublet combination of MAPK pathway and BCL2 inhibition may be better tolerated, which would facilitate clinical efficacy in individuals with RTK-driven AML. Moreover, the increased induction of apoptosis observed with SHP2i compared with MEKi (Figures S3D and S3E) suggests that targeting the MAPK pathway upstream may further increase efficacy.
Pre-clinical studies identified MAPK pathway reactivation as a potential mechanism of resistance to SHP2i in AML cell lines.75 Due to the reliance of SHP2i effect and BCL2 synergy on effective suppression of MAPK signaling, we expect that non-cycling RAS mutations will prove resistant to the combination of SHP2 and BCL2 inhibition, as demonstrated by the lack of synergy observed in NRASQ61K mutant cell lines. However, based on evidence of increased BCL2 dependency induced by MAPK pathway inhibition, we expect that upstream MAPK pathway inhibitors with broad activity against multiple RAS isoforms, including those now in clinical development in solid tumors,76 will demonstrate similar synergistic activity with BCL2is and potential for clinical activity in multiple contexts of RAS-mutant/MAPK-driven AML.
Overall, this study supports the idea that by harnessing RAS/MAPK signaling suppression, SHP2 inhibition induces pro-apoptotic changes and an increase in BCL2 dependency that may be opportunistically exploited with concurrent BCL2 inhibition. Our findings provide a strong mechanistic rationale to support clinical investigation of a SHP2 and BCL2 inhibitor combination in RTK-driven AML.
Limitations of the study
Our study faces several limitations. Although we show that co-targeting SHP2 and BCL2 has activity against RAS mutants with preserved GTP hydrolysis activity, such as NRASG12C, we acknowledge that several cycling-independent RAS mutations that can drive resistance to TKIs are insensitive to this combination. Studies investigating broader-spectrum RAS/MAPK inhibitors should be performed to address this limitation. Most of our mechanistic studies were performed in vitro in AML cell line models. Validating our findings in patient-derived primary samples and in the presence of a tumor microenvironment could recapitulate more faithfully the synergy mechanisms in human disease. In this study we demonstrated anti-leukemic activity of the combinatorial inhibition of SHP2 and BCL2 in only two PDX in vivo models. Ultimately, the tolerability and efficacy of the combination treatment in AML need to be determined clinically.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat #3700; RRID:AB_2242334 |
| p44/42 MAPK (Erk1/2) (3A7) Mouse mAb | Cell Signaling Technology | #9107; RRID:AB_10695739 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell Signaling Technology | #9101; RRID:AB_331646 |
| Akt Antibody | Cell Signaling Technology | #9272; RRID:AB_329827 |
| Phospho-Akt (Thr308) Antibody | Cell Signaling Technology | #9275; RRID:AB_329828 |
| Stat5 (D2O6Y) Rabbit mAb | Cell Signaling Technology | #94205; RRID:AB_2737403 |
| Phospho-Stat5 (Tyr694) Antibody | Cell Signaling Technology | #9351; RRID:AB_2315225 |
| Bcl-2 (124) Mouse mAb | Cell Signaling Technology | #15071; RRID:AB_2744528 |
| Bcl-xL Antibody | Cell Signaling Technology | #2762; RRID:AB_10694844 |
| Mcl-1 Antibody | Cell Signaling Technology | #4572; RRID:AB_2281980 |
| Bim (C34C5) Rabbit mAb | Cell Signaling Technology | #2933; RRID:AB_1030947 |
| BMF (E5U2J) Rabbit mAb | Cell Signaling Technology | #50542; RRID:AB_2892182 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat# 925-32211 RRID:AB_2651127 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat# 925-68070, RRID:AB_2651128 |
| Bmf Antibody | Novus Biologicals | Cat# NBP1-76658, RRID:AB_11040482 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647® | Thermo Fisher Scientific | Cat# A-21244, RRID:AB_2535812 |
| Alexa Fluor® 647 anti-Cytochrome c Antibody | BioLegend | Cat# 612310, RRID:AB_2565241 |
| PE anti-mouse CD45 Antibody | BioLegend | Cat# 103105, RRID:AB_312970 |
| APC/Cyanine7 anti-human CD45 Antibody | BioLegend | Cat# 304014, RRID:AB_31440 |
| CD45 (Intracellular Domain) (D9M8I) XP® Rabbit mAb | Cell Signaling Technology | Cat# 13917, RRID:AB_2750898 |
| Bacterial and virus strains | ||
| pCW57.1 | RRID:Addgene_41393 | |
| NEB 10-beta Competent E. coli (High Efficiency) | New England Biolabs | Cat# C3019I |
| Biological samples | ||
| PDX #1 | This paper | HM0007 |
| PDX #2 | Qin et al.61 | CD33-CART-0005 |
| AML patient and healthy donor samples | UCSF Hematologic Malignancies Tissue Bank | |
| Chemicals, peptides, and recombinant proteins | ||
| RMC-4550 | Revolution Medicines | ID: 0694550-018 |
| Venetoclax (ABT-199) | Selleckchem | Cat# S8048; CAS No. 1257044-40-8 |
| Gilteritinib (ASP2215) | Selleckchem | Cat# S7754; CAS No. 1254053-43-4 |
| WEHI-539 HCl | Selleckchem | Cat# S7100 CAS No. 2070018-33-4 |
| Trametinib (GSK1120212) | Selleckchem | Cat# S2673; CAS No. 871700-17-3 |
| S63845 | MedChem Express | Cat# HY-100741 CAS No. : 1799633-27-4 |
| BIM peptide | New England Peptides | Cat# LB4609 |
| Alamethicin | Enzo Life Sciences | Cat BML-A150-0005; CAS No. 27061-78-5 |
| Digitonin | Milipore Sigma | Cat# D5628; Cas No. CAS Number: 11024-24-1 |
| D-Mannitol | Milipore Sigma | Cat# M9647; CAS No. 69-65-8 |
| Succinic acid | Milipore Sigma | Cat# S3674; CAS No. 110-15-6 |
| Ethylenediaminetetraacetic acid | Milipore Sigma | Cat# E6758; CAS No. 60-00-4 |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid | Milipore Sigma | Cat# E3889; CAS No. 67-42-5 |
| HEPES Buffer, N-(2-Hydroxyethyl)piperazineN′-(2-ethanesulfonic acid) | Milipore Sigma | Cat# SRE0065; |
| Glycine | Thermo Fisher Scientific | Cat# BP381-1 CAS No. 56-40-6 |
| Doxycycline Hydrochloride | Milipore Sigma | Cat# D3072 CAS No. 10592-13-9 |
| Recombinant Human GM-CSF | PeproTech | Cat# 300-03; Genpept: P04141 |
| Recombinant Human Flt3-Ligand | PeproTech | Cat# 300-19; Genpept: P49771 |
| Recombinant Human FGF-basic | PeproTech | Cat# 100-18B; Genpept: P09038 |
| Lipofectamine™ 3000 | Thermo Fisher Scientific | Cat# L3000008 |
| Lenti-X™ concentrator | TakaraBio | Cat# 631232 |
| Triton™ X-100 Surfact-Amps™ Detergent Solution | Thermo Fisher Scientific | Cat# 85111 |
| Paraformaldehyde Aqueous Solution | Electron Microscopy Sciences | Cat#15710-S |
| Normal Goat Serum (10%) | Thermo Fisher Scientific | Cat# 50197Z |
| ProLong™ Gold Antifade Mountant with DNA Stain DAPI | Thermo Fisher Scientific | Cat# P36935 |
| D-Luciferin, Potassium Salt | Gold Biotechnology | Cat# LUCK-100; CAS No. 115144-35-9 |
| Hematoxylin Instant | Thermo Fisher Scientific | Cat# 6765015 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7573 |
| Pierce™ BCA Protein Assay Kits | Thermo Fisher Scientific | Cat# 23225 |
| Ras G-LISA Activation Assay Kit (Colorimetric Based) - 96 Assays | Cytoskeleton | Cat# BK131 |
| CellEvent™ Caspase-3/7 Green Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10427 |
| RNeasy Mini Kit (50) | Qiagen | Cat# 74104 |
| Discovery Purple Kit | Roche Diagnostics | Cat# 760229 |
| Deposited data | ||
| CRISPR (DepMap Public, Chronos) | the Cancer Dependency Map | https://depmap.org |
| RNA-seq data | This paper | GEO: GSE217359 |
| Experimental models: Cell lines | ||
| Molm-14 | ATCC | RRID:CVCL_7916 |
| MV4-11 | ATCC | RRID:CVCL_0064 |
| Kasumi-1 | ATCC | RRID:CVCL_0589 |
| SKNO-1 | ATCC | RRID:CVCL_2196 |
| OCIAML-3 | ATCC | RRID:CVCL_1844 |
| NOMO-1 | ATCC | RRID:CVCL_1609 |
| HL-60 | ATCC | RRID:CVCL_0002 |
| THP-1 | ATCC | RRID:CVCL_0006 |
| K562 | ATCC | RRID:CVCL_0004 |
| HEL | ATCC | RRID:CVCL_0001 |
| U-937 | ATCC | RRID:CVCL_0007 |
| HS-5 | ATCC | RRID:CVCL_3720 |
| LentiX 293T | Takara Bio | RRID:CVCL_4401 |
| Molm-14 PTPN11 WT-dox | This paper | N/A |
| Molm-14 PTPN11 E76K-dox | This paper | N/A |
| Molm-14 PTPN11 G60V-dox | This paper | N/A |
| Molm-14 PTPN11 S502P-dox | This paper | N/A |
| Molm-14 PTPN11 T253M/Q257L-dox | This paper | N/A |
| Molm-14 MEK DD-dox | This paper | N/A |
| Molm-14 AKT E17K-dox | This paper | N/A |
| MV4-11 MEK DD-dox | This paper | N/A |
| MV4-11 AKT E17K-dox | This paper | N/A |
| Molm-14 BMF KO | This paper | N/A |
| SKNO-1 BMF KO | This paper | N/A |
| Molm-14 FFLuc | This paper | N/A |
| Molm-14 NRAS G12C FFLuc | This paper | N/A |
| Kasumi-1 FFLuc | This paper | N/A |
| Molm-14 NRAS G12C | McMahon et al.14 | N/A |
| Molm-14 NRAS Q61K | McMahon et al.14 | N/A |
| Molm-14 FLT3 D835H | Smith et al.77 | N/A |
| Molm-14 FLT3 D835V | Smith et al.77 | N/A |
| Molm-14 FLT3 D835Y | Smith et al.77 | N/A |
| Molm-14 FLT3 N841K | Smith et al.77 | N/A |
| Molm-14 FLT3 Y842C | Smith et al.77 | N/A |
| Molm-14 FLT3 F691L | Smith et al.77 | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain #:005557 RRID:IMSR_JAX:005557 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | The Jackson Laboratory | Strain #:013062 RRID:IMSR_JAX:013062 |
| Oligonucleotides | ||
| Human GAPDH Taqman probe | Thermo Fisher Scientific | Hs02758991_g1 |
| Human OASL Taqman probe | Thermo Fisher Scientific | Hs00984387_m1 |
| Human OAS1 Taqman probe | Thermo Fisher Scientific | Hs00973637_m1 |
| Human IFIT2 Taqman probe | Thermo Fisher Scientific | Hs01922738_s1 |
| Human BMF Taqman probe | Thermo Fisher Scientific | Hs00372937_m1 |
| BMF sgRNA guide1: 5’-AAGGCCAGGGCCACAGCAGU-3’ | Synthego | Custom ID |
| BMF sgRNA guide2: 5’- AAGCUCCCGGGUUGGGUCAC-3’ | Synthego | Custom ID |
| BMF sgRNA guide1: 5’- GGAGCCAUCUCAGUGUGUGG-3’ | Synthego | Custom ID |
| Software and algorithms | ||
| GraphPad Prism | http://www.graphpad.com/ | RRID:SCR_002798 |
| FlowJo | https://www.flowjo.com/solutions/flowjo | RRID:SCR_008520 |
| R Project for Statistical Computing | http://www.r-project.org/ | RRID:SCR_001905 |
| STAR | Dobin et al.78 | RRID:SCR_004463 |
| Limma-voom package | Law et al.79 | |
| ShinyGO | http://bioinformatics.sdstate.edu/go/ | RRID:SCR_019213 |
| Gene Set Enrichment Analysis | Subramanian et al.39 | RRID:SCR_003199 |
| NIS Elements | RRID:SCR_014329 | |
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| SynergyFinder 3.0 | Ianevski et al.55 | |
| ICE | Conant et al.80 | |
Resource availablity
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Catherine C. Smith (catherine.smith@ucsf.edu).
Materials availability
Materials generated in this study are available upon request from the lead contact.
Data and code availability
-
•
RNA-sequencing data reported in this study have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus repository under the accession number GEO: GSE217359.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.
Experimental model and study participant details
Cell lines
Molm-14, MV4-11, OCIAML-3, HL-60, NOMO-1, THP-1 were a gift from Dr. Scott Kogan (University of California, San Francisco, San Francisco, CA). U937 were purchased from the Cell and Genome Engineering Core at University of California, San Francisco. Kasumi-1 and SKNO-1 cell lines were purchased from ATCC. K562, HEL and HS5 cells were a gift from Dr. Neil Shah (University of California, San Francisco, San Francisco, CA). Molm-14 NRAS-mutant and FLT3 TKD–mutant cells were generated by culturing Molm-14 parental cells in vitro in media containing escalating doses of quizartinib (0.5–20 nmol/L).77 Quizartinib-resistant cells were subcloned and Sanger sequencing was performed for mutation confirmation. To generate PTPN11-mutant and MEKDD Molm-14 doxycycline-inducible expression cell lines, PTPN11 and MEK variants were cloned into a Gateway tetracycline-inducible destination lentiviral vector, pCW57.1 (Addgene). The cloning was performed by Twist Biosciences. Lenti-X 293T cells (Takara Bio) were infected using Lipofectamine 3000 (Invitrogen) and cultured for 48 hours in DMEM with 10% FBS. The supernatant was harvested, concentrated using Lenti-X concentrator (Takara Bio), then used to infect cell lines. Forty-eight hours following lentiviral infection, cells were selected with puromycin. To generate luciferase-tagged cell lines we transfected parental Molm-14, quizartinib-resistant Molm-14 NRASG12C, and Kasumi-1 cells with the Firefly-luciferase expressing plasmid pLV 416 oFL T2A mCherry, a gift from Neil Shah (University of California, San Francisco, San Francisco, CA).
Cell lines were cultured in RPMI-1640 (Gibco) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin/L-glutamine (Gibco), with the exception of Kasumi-1 and HL-60 cells, cultured with 20% FBS. Culture media for SKNO-1 cell line was supplemented with human GM-CSF (10 ng/mL, PeproTech). Cells tested negative for Mycoplasma by the MycoAlert PLUS Mycoplasma Detection Kit (Lonza). Experiments were performed within three months of cell line thawing. Authentication of all cell lines was performed at the University of California, Berkeley, DNA Sequencing Facility, using short tandem repeat DNA profiling.
Patient samples
Coded primary AML samples were collected, stored, and studies performed after approval of research protocols from the University of California San Francisco Hematologic Malignancies Tissue Bank or the Children’s Hospital of Philadelphia Center for Childhood Cancer Research biorepository by institutional review boards. Informed, written consent according to the Declaration of Helsinki was obtained from patients prior to tissue collection. Clinical information of patient samples are provided in Table S3.
Cell line-derived xenografts
All in vivo xenograft animal studies were performed in full accordance with UCSF Institutional Animal Care and Use Committee (IACUC) at the UCSF Preclinical Therapeutics Core (PTC) or the Children’s Hospital of Philadelphia IACUC and Laboratory Animal Facility. NOD/SCID/IL2Rγnull female mice were injected with cell line-derived xenografts consisting of 5 x 106 Firefly-luciferase-tagged Molm-14, Molm-14 NRASG12C and Kasumi-1 cells suspended in PBS. Tumor volume progression was assessed bi-weekly using a Xenogen IVIS Spectrum imaging system (Perkin Elmer) after injection of 150 mg/kg of D-luciferin (GoldBio). Bioluminescence (BLI) in photons per second per cm2 per steradian (p/s/cm2/sr) was translated to color-coded image to indicate disease burden and quantified using IVIS Living Image (Xenogen) software. ROI were manually drawn around the bodies of the mice to assess signal intensity emitted. Based on bioluminescence-detectable engraftment at dosing start was: mean BLI = 2.72 x 1010 for Molm-14 cell line, 2.84 x 106 for Molm-14 NRASG12C cell line and 1.44 x 106 for Kasumi-1 cell line. Animals were randomized into four groups (n=5 or n=6): a control group receiving vehicle, a group receiving RMC-4550, 30 mg/kg, a group receiving venetoclax 100 mg/kg, and a group receiving the combination of the two agents. Treatments were administered by oral gavage, daily, 5 times a week during weekdays, for 28 days. Animals were sacrificed in compliance to IACUC protocols at the study termination or at humane endpoint, as per UCSF Laboratory Animal Resource Center (LARC) veterinary advice.
Patient-derived xenograft models
Patient derived xenograft models of human FLT3-ITD AML were generated in NSG or triple transgenic NOD.scid.Il2Rγcnull-SGM3 (NSGS) mice (Jackson Laboratory) and serially passaged. Mice were conditioned with busulfan (Sigma) 25mg/kg intraperitoneally on days -2 and -1. Mice were injected with 1 to 1.5 million cells after 1 day of rest. Mice were monitored for weight change weekly during entirety of the trial. Mice were bled by submandibular puncture weekly starting on week 4 from injection and analyzed by FACS for human (hCD45) and mouse CD45 (mCD45). Mice were randomly assigned to a treatment arm once they reached greater than or equal to (0.5% hCD45+ cells / total events) by FACS. Treatment arms were defined as following: vehicle (oral gavage, 5X per week), RMC-4550 (30 mg/kg, oral gavage, 5X per week), venetoclax (100 mg/kg, oral gavage, 5X per week), combination: RMC-4550 (30 mg/kg) and venetoclax (100 mg/kg, oral gavage 5X per week). Mice were treated for 28 days (20 days of dosing), during which they were monitored daily for disease progression and every other week by FACS. Mice were euthanized after 28 days or when they reached humane endpoint, whichever was sooner. Mice were processed upon endpoint and cardiac blood, bone marrow and spleen were analyzed by FACS for hCD45/mCD45. Spleen fragments were processed for immunohistochemical staining.
Method details
Compounds
RMC-4550 was provided by Revolution Medicines. Venetoclax (S8048), gilteritinib (S7754), WEHI-539 (S7100) and trametinib (S2673) were purchased from Selleckchem. S63845 (HY-100741) was purchased from MedChem Express. BIM peptide was purchased from New England Peptide.
Cell viability assays
For cell viability experiments, cells were seeded in three technical replicates in 96-well plates and exposed to increasing concentrations of indicated drug for 48 hours. For doxycycline-inducible cell line experiments, cells were were stimulated for 24 hours with doxycycline 1 ug/mL and maintained in the same concentration of doxycycline for the duration of experiments. Cell viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega), luminescence was measured on a Molecular Devices iD3 multimode plate reader and normalized to untreated controls.
Apoptosis assays
Cells were cultured in appropriate culture media, then seeded in three technical replicates in 96-well clear plates and exposed to increasing concentrations of indicated drug for 24 hours. CellEvent Caspase-3/7 Green Flow Cytometry Kit (Thermo Fisher) was used to measure fraction of apoptotic cells by flow cytometry (BD LSR Fortessa with HTS sampler). Percentage of caspase-3/7 positive cells in each treatment condition was normalized to vehicle-treated control populations (FlowJo software v9, BD Biosciences).
Conditioned media experiments
To generate conditioned media, 106 HS5 cells were plated in 10 mL of complete RPMI in a 10-cm dish and media harvested after 72 hours, cleared by centrifugation and filtration through a 0.22-mm membrane. Molm-14 cells were cultured in conditioned media, complete RPMI and RPMI supplemented with FGF-2 and FLT3-L (10 ng/mL) each, as previously described,17,38 then exposed to indicated doses of inhibitors for 24 hours and assessed for apoptosis.
Active RAS detection assay
For RAS-GTP detection, a Ras GTP-ase immunosorbent assay (G-LISA) Activation Assay Kit (#BK131, Cytoskeleton) was used, as per manufacturer’s protocol. In brief, 1 x 106 cells were incubated with drug treatments for 90 minutes, then cell lysates were harvested. 25 μg of total protein per condition were incubated in triplicate in wells coated with Raf1-RBD. Wells were then washed, and incubated with an anti-Ras antibody (#GL-11) and subsequently with a HRP-conjugated secondary antibody (#GL-02). Absorbance readout at 490 nM was used to assess the colorimetric response.
Immunoblotting
Cells were plated in appropriate media and treated with the indicated concentrations of inhibitors. After 90 minutes or 24-hours incubation, cells were washed in PBS and lysed in buffer (50 mmol/L HEPES, pH 7.4, 10% glycerol, 150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, and 1.5 mmol/L MgCl2) supplemented with protease and phosphatase inhibitors (EMD Millipore). The lysates were clarified by centrifugation, quantitated by BCA assay (Thermo Scientific) and normalized. 20 mg of protein were loaded on 10% Bis-Tris gels then transferred to nitrocellulose membranes. Immunoblotting was performed using the following antibodies from Cell Signaling Technology: anti-β-Actin (8H10D10, #3700), anti-Erk1/2 (3A7, #9107), anti-phoshpo-Erk1/2 (Thr202/Tyr204, #9101), anti-Akt (#9272), anti-phospho Akt (Thr308, #9275), anti-Stat5 (D2O6Y, #94205), anti-phospho-Stat5 (Tyr 694, #9351), anti-Bcl2 (124, #15071), anti-Bcl-xL (#2762), anti-Mcl-1 (#4572), anti-Bim (C34C5, #2933), anti-BMF (E5U2J, #50542). Nitrocellulose membranes were subsequently incubated in a solution of secondary antibodies (IRDye 800CW Goat anti-Rabbit, #935-32211, IRDye 680RD Goat anti-Mouse, #935-68070, LI-COR Biosciences) and scanned on an Odyssey CLx infrared imaging system (LI-COR Biosciences). When needed, band intensities were quantified using the Image Studio software.
RT-qPCR
Cells were incubated for 24 hours with indicated drug concentrations and total RNA was isolated using RNeasy Mini kit (QIAGEN). cDNA was synthesized from 500 ng total RNA using SuperScript III reverse transcriptase (Invitrogen) and qPCR was performed in 384-well plates using Taqman Gene Expression assays (GAPDH: Hs02758991_g1, OASL: Hs00984387_m1, OAS1: Hs00973637_m1, IFIT2: Hs01922738_s1, BMF: Hs00372937_m1,) on a QuantStudio 6 instrument (Thermo Fisher). Differential gene expression was calculated using the 2−ΔΔCt method with GAPDH as a housekeeping control gene in three technical replicates.
RNA-seq
Molm-14, MV4-11 and SKNO-1 cell were exposed to drug treatments, in three biological replicates per condition. RNA samples with RIN scores between 8.4 and 10 were used to generate libraries and sequenced by BGI Genomics using DNBseq platform. mRNA was purified using oligo(dT)-attached magnetic beads and reverse transcribed. The cDNA fragments were end-repaired, 3’-adenylated, and PCR-purified after adaptor ligation. PCR products were quantified on 2100 Analyzer instrument (Agilent), heat-denatured and circularized by a splint oligo sequence to generate circular DNA nanoball (DNB) libraries. DNBs were loaded into a patterned nanoarray and sequenced by Probe-Anchor Synthesis (cPAS), generating 100 bp paired-end reads. Quality metrics for raw sequencing data were evaluated with FastQC. Reads were aligned to the human reference genome GRCh38 and gene counts were quantified using STAR.78 Following raw gene count quantification, the data from each sample were subjected to quality evaluation tests. This was done by manually inspecting the density plot, MA plots, principal component analysis (PCA) plots, correlation heatmap and PCA association plots, as well as using several automated outlier tests, namely sum of Euclidean distance to other samples, Kolmogorov-Smirnov test, mean Pearson correlation with other samples and Hoeffding’s D method. Samples were defined as outliers if they failed two or more of these tests, or if the data were deemed substandard following visual inspection of PCA and MA plots. All samples passed the outlier tests and no issues were identified in MA or PCA plots. Raw counts were normalized using the trimmed mean of M-values (TMM) method. Differential gene expression analysis was carried out using limma-voom79 and the threshold for differential gene expression was an adjusted P < 0.05, corrected for multiple comparisons using the false discovery rate adjustment (FDR) and a fold-change in expression ≥ 2. R version 4.0.4 running on Ubuntu 20.04 LTS was used for data processing and analysis. Gene set enrichment analysis (GSEA v4.2.3)39 was used to identify enriched functional gene sets. Gene ontology (GO enrichment analysis for Biological Process terms was performed using the ShinyGO v0.77 app at https://bioinformatics.sdstate.edu/go with a FDR cutoff of 0.05 and terms sorted by Fold Enrichment.
Immunofluorescence
Molm-14 cells were treated with either vehicle solution or 1 μM of RMC-4550. 5e4 cells were cyto-spun on glass slides using a Cytospin 3 (Shandon) cytocentrifuge, then fixed with 4% paraformaldehyde, then permeabilized and blocked with a solution of 0.1% Triton-X and 10% normal goat serum (#50197Z, Thermo Fisher) in PBS. The slides were incubated overnight with an anti-BMF antibody solution (#NBP1-76658, Novus Biologicals) 10 μg/mL in PBS with 0.01% Tween-20 and 1% BSA, washed, then incubated with a secondary antibody (#A-21244, Invitrogen). Coverslips were mounted using ProLong Gold Antifade Mountant with DAPI (#P36935, Thermo Fisher). Images were acquired with on a Nikon A1R-MP confocal microscope with a 20x magnification objective and processed using Fiji (ImageJ v 2.3.0). ROI were drawn around each individual cell on raw, pre-processed images acquired from three independent fields from each condition in each cell line. Mean fluorescent intensities and surface areas of each ROI were calculated using NIS Elements software.
BH3 profiling
The BH3 profiling of AML cell lines was carried out as previously described.54 In brief, cells were treated for 16 hours with the indicated doses of RMC-4550, then incubated in 96-well plates in a solution containing 0.002% digitonin and either Bim peptide or Bcl2, BclxL or Mcl1 inhibitors, fixed and stained with an anti-cytochrome C (clone 6H2.B4, # 612310, BioLegend). Inhibitors are used in this assay as tool compounds to assess permeabilization of outer mitochondrial membrane to cytochrome C in response to selective inhibition of each BH3-family member protein. The doses of peptides and inhibitors used for this assay were selected based on the baseline apoptotic dependency of each cell line to each BH3 family member. FACS acquisition was performed on a BD Fortessa LSR instrument with HTS sampler. Cytochrome C release was measured by gating cytochrome C negative populations, normalized to vehicle-treated control populations (FlowJo software v9, BD Biosciences).
CRISPR Cas9 KO
BMF knock-out was performed using a Gene Knockout Kit v2 (Synthego), using a multi-guide sgRNA approach. Ribonucleoprotein (RNP) was assembled by combining a 180 pmol mixture of three sgRNA guides (Sequences G1: 5’-AAGGCCAGGGCCACAGCAGU-3’, G2: 5’- AAGCUCCCGGGUUGGGUCAC-3’, G3: 5’- GGAGCCAUCUCAGUGUGUGG-3’) with 30 pmol of Cas9-2NLS nuclease from S. pyogenes in Opti-MEM (Gibco) media and transfected into Molm-14 and SKNO-1 cell lines using a MaxCyte ATX system. RNP-transfected cells and cells transfected with Cas9 only, used as negative controls, were downstream cultured in complete media. Knock-out efficiency was determined by Inference of CRISPR Edits (ICE) analysis80 and functionally assessed by Western Blot.
Immunohistochemistry (IHC) staining
IHC staining was performed at the UCSF Histology and Biomarkers Core on a Ventana BenchMark Ultra instrument using Discovery reagents (Ventana Medical Systems) according to manufacturer’s instructions, except as noted. After deparaffinization and antigen-retrieval, slides were incubated with primary antibody anti-CD45 (1:200, clone: D9M8I, Product ID: 13917 from Cell Signaling). The primary antibody was detected with Discovery Purple kit. Finally, slides were counterstained with hematoxylin (Thermo Scientific Shandon Instant Hematoxylin cat. 6765015). Images were acquired using a Zeiss Axio Scanner.Z1 slide scanner with a 20X Plan-Apochromat objective and processed using Zeiss Zen Lite v3.6.
Colony-forming unit assay
Primary samples from leukapheresis products harvested from either healthy individuals or patients with AML were thawed in DMEM media (Gibco) supplemented with 20% FBS, 2mM EDTA and 500 μg DNase I. 5 x 104 to 2.5 x 105 cells were incubated with DMSO, RMC-4550 (500 nM), venetoclax (200 nM) or the combination of both drugs in methylcellulose media enriched with human cytokines (recombinant human SCF, GM-CSF, G-CSF, IL-3, IL-6, Epo, #HSC005, R&D Systems). The methylcellulose mixture was plated in three replicates to 35 mm culture dishes using regular syringes with 16 gauge non-stick needles and incubated at 37°C, 5% CO2 for 14-16 days. Colony forming units were counted using an Olympus CKX53 light microscope with 4X magnification objective and the counts were normalized to untreated controls.
Quantification and statistical analysis
Details on statistical analysis of experiments can be found in the figure legends. Unpaired t tests were used to compare independent groups. One-way or two-way analysis of variance (ANOVA) tests with were used to compare more than two groups, and correction for multiple comparisons was applied when needed. Statistical significance is denoted as follows: ∗, P ≤ 0.05; ∗∗, P ≤ 0.01; ∗∗∗, P ≤ 0.001; ∗∗∗∗, P ≤ 0.0001, unless stated otherwise. Data are reported as mean ± SD, unless otherwise specified. For cell viability readouts, dose-response curves and absolute IC50 values were calculated using a three parameters nonlinear regression model. Drug synergy assessment was performed using the Bliss method within SynergyFinder v3.0 (https://synergyfinder.fimm.fi/),55 with LL4 curve fitting and cell viability readout data. For in vivo studies, no statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those previously published.30,31 Data collection and analysis were not performed blind to the experimental conditions. GraphPad Prism software (v8.3.1) was used for statistical analysis and data plotting. BioRender.com was used for to draw graphic illustrations.
Acknowledgments
The authors thank the Smith lab members for scientific input and technical support throughout the project, Kevin Shannon for critical reading of the manuscript, Fios Genomics for assistance with bioinformatic analysis of RNA-seq data, Celine Mahieu for assistance with nucleofection protocols, Noura Ismail-Mohammed for technical support with confocal microscopy imaging, and Christopher Chien and Nirali Shah at the NCI for assistance with AML specimens for xenograft studies. This work was supported in part by Revolution Medicines. C.A.C.P. was funded by a Young Investigator Award from the Alex's Lemonade Stand Foundation. C.L. was supported by the NIGMS Predoctoral Training in Biomedical Sciences T32 grant (T32-GM-136547). The Preclinical Therapeutic Core (PTC) at UCSF (P.P., J.A.C.S., V.S.) is supported by Cancer Center Support Grant (CCSG, NCI/NIH) award P30CA082103. S.K.T. is a Scholar of the Leukemia & Lymphoma Society and Joshua Kahan Endowed Chair in Pediatric Leukemia at the Children's Hospital of Philadelphia. C.C.S. is a Leukemia & Lymphoma Society Scholar in Clinical Research and a Damon Runyon-Richard Lumsden Foundation Clinical Investigator supported (in part) by the Damon Runyon Cancer Research Foundation (CI-99-18).
Author contributions
Conceptualization, B.P. and C.C.S.; methodology, B.P., T.C.T., C.A.C.P., P.P., J.M.R., V.S., E.S., and C.C.S.; software, B.P. and C.L.; validation, B.P. and S.W.; formal analysis, B.P., T.C.T., and C.L.; investigation, B.P., T.C.T., S.W., P.P., J.A.C.S., H.H., J.M.R., S.X., and V.S.; resources, C.S., B.J.L., C.A.C.P., J.A.C., V.S., S.K.T., E.S., and C.C.S.; data curation, B.P., H.H., S.X., and C.C.S.; writing – original draft, B.P. and C.C.S.; writing – review & editing, B.P., C.S., C.A.C.P., C.L., S.K.T., E.S., and C.C.S.; visualization, B.P., S.W., and J.A.C.S.; supervision, V.S., E.S., and C.C.S.; project administration, C.S., B.J.L., and C.C.S.; funding acquisition, C.C.S.
Declaration of interests
C.S. is a former employee and stockholder of Revolution Medicines, Inc., and a current employee and stockholder of BridgeBio Pharma, Inc. B.J.L. is a current employee of Revolution Medicines, Inc. J.A.C. is a current employee of Outpace Bio. S.K.T. reports research funding unrelated to this work from Beam Therapeutics; Kura Oncology; Incyte Corporation; scientific advisory boards for Aleta Biotherapeutics, Kura Oncology, and Syndax Pharmaceuticals; consulting for bluebird bio; and travel support from Amgen. C.C.S. reports research funding from AbbVie, Inc., and Revolution Medicines and has served as an advisory board member for AbbVie, Inc., and Genentech.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101290.
Supplemental information
References
- 1.Papaemmanuil E., Gerstung M., Bullinger L., Gaidzik V.I., Paschka P., Roberts N.D., Potter N.E., Heuser M., Thol F., Bolli N., et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A.G., Hoadley K., Triche T.J., Laird P.W., et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolouri H., Farrar J.E., Triche T., Ries R.E., Lim E.L., Alonzo T.A., Ma Y., Moore R., Mungall A.J., Marra M.A., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018;24:103–112. doi: 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abu-Duhier F.M., Goodeve A.C., Wilson G.A., Gari M.A., Peake I.R., Rees D.C., Vandenberghe E.A., Winship P.R., Reilly J.T. FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br. J. Haematol. 2000;111:190–195. doi: 10.1046/j.1365-2141.2000.02317.x. [DOI] [PubMed] [Google Scholar]
- 5.Paschka P., Marcucci G., Ruppert A.S., Mrózek K., Chen H., Kittles R.A., Vukosavljevic T., Perrotti D., Vardiman J.W., Carroll A.J., et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J. Clin. Oncol. 2006;24:3904–3911. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 6.Schnittger S., Kohl T.M., Haferlach T., Kern W., Hiddemann W., Spiekermann K., Schoch C. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107:1791–1799. doi: 10.1182/blood-2005-04-1466. [DOI] [PubMed] [Google Scholar]
- 7.Paschka P., Schlenk R.F., Weber D., Benner A., Bullinger L., Heuser M., Gaidzik V.I., Thol F., Agrawal M., Teleanu V., et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia. 2018;32:1621–1630. doi: 10.1038/s41375-018-0129-6. [DOI] [PubMed] [Google Scholar]
- 8.Marcucci G., Geyer S., Laumann K., Zhao W., Bucci D., Uy G.L., Blum W., Eisfeld A.-K., Pardee T.S., Wang E.S., et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020;4:696–705. doi: 10.1182/bloodadvances.2019000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone R.M., Mandrekar S.J., Sanford B.L., Laumann K., Geyer S., Bloomfield C.D., Thiede C., Prior T.W., Döhner K., Marcucci G., et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erba H.P., Montesinos P., Kim H.-J., Patkowska E., Vrhovac R., Žák P., Wang P.-N., Mitov T., Hanyok J., Kamel Y.M., et al. Quizartinib plus chemotherapy in newly diagnosed patients with FLT3-internal-tandem-duplication-positive acute myeloid leukaemia (QuANTUM-First): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401:1571–1583. doi: 10.1016/S0140-6736(23)00464-6. [DOI] [PubMed] [Google Scholar]
- 11.Cortes J.E., Khaled S., Martinelli G., Perl A.E., Ganguly S., Russell N., Krämer A., Dombret H., Hogge D., Jonas B.A., et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20:984–997. doi: 10.1016/S1470-2045(19)30150-0. [DOI] [PubMed] [Google Scholar]
- 12.Perl A.E., Martinelli G., Cortes J.E., Neubauer A., Berman E., Paolini S., Montesinos P., Baer M.R., Larson R.A., Ustun C., et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 -Mutated AML. N. Engl. J. Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 13.Smith C.C., Paguirigan A., Jeschke G.R., Lin K.C., Massi E., Tarver T., Chin C.-S., Asthana S., Olshen A., Travers K.J., et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130:48–58. doi: 10.1182/blood-2016-04-711820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon C.M., Ferng T., Canaani J., Wang E.S., Morrissette J.J.D., Eastburn D.J., Pellegrino M., Durruthy-Durruthy R., Watt C.D., Asthana S., et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019;9:1050–1063. doi: 10.1158/2159-8290.CD-18-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H., Savage S., Schultz A.R., Bottomly D., White L., Segerdell E., Wilmot B., McWeeney S.K., Eide C.A., Nechiporuk T., et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019;10:244. doi: 10.1038/s41467-018-08263-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peretz C.A.C., McGary L.H.F., Kumar T., Jackson H., Jacob J., Durruthy-Durruthy R., Levis M.J., Perl A., Huang B.J., Smith C.C. Single-cell DNA sequencing reveals complex mechanisms of resistance to quizartinib. Blood Adv. 2021;5:1437–1441. doi: 10.1182/bloodadvances.2020003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traer E., Martinez J., Javidi-Sharifi N., Agarwal A., Dunlap J., English I., Kovacsovics T., Tyner J.W., Wong M., Druker B.J. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016;76:6471–6482. doi: 10.1158/0008-5472.CAN-15-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi S.K., Nechiporuk T., Bottomly D., Piehowski P.D., Reisz J.A., Pittsenbarger J., Kaempf A., Gosline S.J.C., Wang Y.-T., Hansen J.R., et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell. 2021;39:999–1014.e8. doi: 10.1016/j.ccell.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amatangelo M.D., Quek L., Shih A., Stein E.M., Roshal M., David M.D., Marteyn B., Farnoud N.R., de Botton S., Bernard O.A., et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130:732–741. doi: 10.1182/blood-2017-04-779447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiNardo C.D., Tiong I.S., Quaglieri A., MacRaild S., Loghavi S., Brown F.C., Thijssen R., Pomilio G., Ivey A., Salmon J.M., et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135:791–803. doi: 10.1182/blood.2019003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serrano C., Wang Y., Mariño-Enríquez A., Lee J.-C., Ravegnini G., Morgan J.A., Bertagnolli M.M., Beadling C., Demetri G.D., Corless C.L., et al. KRAS and KIT Gatekeeper Mutations Confer Polyclonal Primary Imatinib Resistance in GI Stromal Tumors: Relevance of Concomitant Phosphatidylinositol 3-Kinase/AKT Dysregulation. J. Clin. Oncol. 2015;33:e93–e96. doi: 10.1200/JCO.2013.48.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols R.J., Haderk F., Stahlhut C., Schulze C.J., Hemmati G., Wildes D., Tzitzilonis C., Mordec K., Marquez A., Romero J., et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-NF1- and RAS-driven cancers. Nat. Cell Biol. 2018;20:1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agazie Y.M., Hayman M.J. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell Biol. 2003;23:7875–7886. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanafusa H., Torii S., Yasunaga T., Matsumoto K., Nishida E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J. Biol. Chem. 2004;279:22992–22995. doi: 10.1074/jbc.M312498200. [DOI] [PubMed] [Google Scholar]
- 25.Ivins Zito C., Kontaridis M.I., Fornaro M., Feng G.-S., Bennett A.M. SHP-2 regulates the phosphatidylinositide 3’-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J. Cell. Physiol. 2004;199:227–236. doi: 10.1002/jcp.10446. [DOI] [PubMed] [Google Scholar]
- 26.Ali S., Nouhi Z., Chughtai N., Ali S. SHP-2 regulates SOCS-1-mediated Janus kinase-2 ubiquitination/degradation downstream of the prolactin receptor. J. Biol. Chem. 2003;278:52021–52031. doi: 10.1074/jbc.M306758200. [DOI] [PubMed] [Google Scholar]
- 27.Li L., Modi H., McDonald T., Rossi J., Yee J.-K., Bhatia R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood. 2011;118:1504–1515. doi: 10.1182/blood-2010-06-288910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabinger S.C., Li X.J., Ramdas B., He Y., Zhang X., Zeng L., Richine B., Bowling J.D., Fukuda S., Goenka S., et al. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia. 2013;27:398–408. doi: 10.1038/leu.2012.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y.-N.P., LaMarche M.J., Chan H.M., Fekkes P., Garcia-Fortanet J., Acker M.G., Antonakos B., Chen C.H.-T., Chen Z., Cooke V.G., et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 30.Mainardi S., Mulero-Sánchez A., Prahallad A., Germano G., Bosma A., Krimpenfort P., Lieftink C., Steinberg J.D., de Wit N., Gonçalves-Ribeiro S., et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 31.Pandey R., Ramdas B., Wan C., Sandusky G., Mohseni M., Zhang C., Kapur R. SHP2 inhibition reduces leukemogenesis in models of combined genetic and epigenetic mutations. J. Clin. Invest. 2019;129:5468–5473. doi: 10.1172/JCI130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fedele C., Li S., Teng K.W., Foster C.J.R., Peng D., Ran H., Mita P., Geer M.J., Hattori T., Koide A., et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021;218:e20201414. doi: 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fedele C., Ran H., Diskin B., Wei W., Jen J., Geer M.J., Araki K., Ozerdem U., Simeone D.M., Miller G., et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerr D.L., Haderk F., Bivona T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021;62:1–12. doi: 10.1016/j.cbpa.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Tao Y., Xie J., Zhong Q., Wang Y., Zhang S., Luo F., Wen F., Xie J., Zhao J., Sun X., et al. A novel partially open state of SHP2 points to a “multiple gear” regulation mechanism. J. Biol. Chem. 2021;296:100538. doi: 10.1016/j.jbc.2021.100538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaRochelle J.R., Fodor M., Vemulapalli V., Mohseni M., Wang P., Stams T., LaMarche M.J., Chopra R., Acker M.G., Blacklow S.C. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 2018;9:4508. doi: 10.1038/s41467-018-06823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith C.C., Wang Q., Chin C.-S., Salerno S., Damon L.E., Levis M.J., Perl A.E., Travers K.J., Wang S., Hunt J.P., et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferng T.T., Terada D., Ando M., Tarver T.C., Chaudhary F., Lin K.C., Logan A.C., Smith C.C. The Irreversible FLT3 Inhibitor FF-10101 Is Active Against a Diversity of FLT3 Inhibitor Resistance Mechanisms. Mol. Cancer Therapeut. 2022;21:844–854. doi: 10.1158/1535-7163.MCT-21-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thyrell L., Erickson S., Zhivotovsky B., Pokrovskaja K., Sangfelt O., Castro J., Einhorn S., Grandér D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21:1251–1262. doi: 10.1038/sj.onc.1205179. [DOI] [PubMed] [Google Scholar]
- 42.Thyrell L., Hjortsberg L., Arulampalam V., Panaretakis T., Uhles S., Dagnell M., Zhivotovsky B., Leibiger I., Grandér D., Pokrovskaja K. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2004;279:24152–24162. doi: 10.1074/jbc.M312219200. [DOI] [PubMed] [Google Scholar]
- 43.Stawowczyk M., Van Scoy S., Kumar K.P., Reich N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011;286:7257–7266. doi: 10.1074/jbc.M110.207068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castelli J.C., Hassel B.A., Maran A., Paranjape J., Hewitt J.A., Li X.L., Hsu Y.T., Silverman R.H., Youle R.J. The role of 2’-5’ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh A., Sarkar S.N., Rowe T.M., Sen G.C. A specific isozyme of 2’-5’ oligoadenylate synthetase is a dual function proapoptotic protein of the Bcl-2 family. J. Biol. Chem. 2001;276:25447–25455. doi: 10.1074/jbc.M100496200. [DOI] [PubMed] [Google Scholar]
- 46.Nagamachi A., Matsui H., Asou H., Ozaki Y., Aki D., Kanai A., Takubo K., Suda T., Nakamura T., Wolff L., et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell. 2013;24:305–317. doi: 10.1016/j.ccr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Brägelmann J., Lorenz C., Borchmann S., Nishii K., Wegner J., Meder L., Ostendorp J., Ast D.F., Heimsoeth A., Nakasuka T., et al. MAPK-pathway inhibition mediates inflammatory reprogramming and sensitizes tumors to targeted activation of innate immunity sensor RIG-I. Nat. Commun. 2021;12:5505. doi: 10.1038/s41467-021-25728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunet A., Pagès G., Pouysségur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994;9:3379–3387. [PubMed] [Google Scholar]
- 49.Chang F., Kong S.J., Wang L., Choi B.K., Lee H., Kim C., Kim J.M., Park K. Targeting Actomyosin Contractility Suppresses Malignant Phenotypes of Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2020;21:3460. doi: 10.3390/ijms21103460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurppa K.J., Liu Y., To C., Zhang T., Fan M., Vajdi A., Knelson E.H., Xie Y., Lim K., Cejas P., et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell. 2020;37:104–122.e12. doi: 10.1016/j.ccell.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puthalakath H., Villunger A., O’Reilly L.A., Beaumont J.G., Coultas L., Cheney R.E., Huang D.C., Strasser A. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–1832. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 52.Frisch S.M., Screaton R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 53.Chen L., Willis S.N., Wei A., Smith B.J., Fletcher J.I., Hinds M.G., Colman P.M., Day C.L., Adams J.M., Huang D.C.S. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 54.Ryan J., Montero J., Rocco J., Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol. Chem. 2016;397:671–678. doi: 10.1515/hsz-2016-0107. [DOI] [PubMed] [Google Scholar]
- 55.Ianevski A., Giri A.K., Aittokallio T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022;50:W739–W743. doi: 10.1093/nar/gkac382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janssen M., Schmidt C., Bruch P.-M., Blank M.F., Rohde C., Waclawiczek A., Heid D., Renders S., Göllner S., Vierbaum L., et al. Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood. 2022;140:2594–2610. doi: 10.1182/blood.2021014241. [DOI] [PubMed] [Google Scholar]
- 57.Daver N., Perl A.E., Maly J., Levis M., Ritchie E., Litzow M., McCloskey J., Smith C.C., Schiller G., Bradley T., et al. Venetoclax Plus Gilteritinib for FLT3 -Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022;40:4048–4059. doi: 10.1200/JCO.22.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Unni A.M., Harbourne B., Oh M.H., Wild S., Ferrarone J.R., Lockwood W.W., Varmus H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife. 2018;7:e33718. doi: 10.7554/eLife.33718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu R., Li L., Nguyen B., Seo J., Wu M., Seale T., Levis M., Duffield A., Hu Y., Small D. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct. Targeted Ther. 2021;6:186. doi: 10.1038/s41392-021-00578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han L., Zhang Q., Dail M., Shi C., Cavazos A., Ruvolo V.R., Zhao Y., Kim E., Rahmani M., Mak D.H., et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020;105:697–707. doi: 10.3324/haematol.2018.205534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qin H., Yang L., Chukinas J.A., Shah N., Tarun S., Pouzolles M., Chien C.D., Niswander L.M., Welch A.R., Taylor N., et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J. Immunother. Cancer. 2021;9:e003149. doi: 10.1136/jitc-2021-003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alotaibi A.S., Yilmaz M., Kanagal-Shamanna R., Loghavi S., Kadia T.M., DiNardo C.D., Borthakur G., Konopleva M., Pierce S.A., Wang S.A., et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2021;2:125–134. doi: 10.1158/2643-3230.BCD-20-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bruner J.K., Ma H.S., Li L., Qin A.C.R., Rudek M.A., Jones R.J., Levis M.J., Pratz K.W., Pratilas C.A., Small D. Adaptation to TKI Treatment Reactivates ERK Signaling in Tyrosine Kinase-Driven Leukemias and Other Malignancies. Cancer Res. 2017;77:5554–5563. doi: 10.1158/0008-5472.CAN-16-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmed T.A., Adamopoulos C., Karoulia Z., Wu X., Sachidanandam R., Aaronson S.A., Poulikakos P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019;26:65–78.e5. doi: 10.1016/j.celrep.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sorokin A.V., Kanikarla Marie P., Bitner L., Syed M., Woods M., Manyam G., Kwong L.N., Johnson B., Morris V.K., Jones P., et al. Targeting RAS Mutant Colorectal Cancer with Dual Inhibition of MEK and CDK4/6. Cancer Res. 2022;82:3335–3344. doi: 10.1158/0008-5472.CAN-22-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Q., Riley-Gillis B., Han L., Jia Y., Lodi A., Zhang H., Ganesan S., Pan R., Konoplev S.N., Sweeney S.R., et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Targeted Ther. 2022;7:51. doi: 10.1038/s41392-021-00870-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pei S., Pollyea D.A., Gustafson A., Stevens B.M., Minhajuddin M., Fu R., Riemondy K.A., Gillen A.E., Sheridan R.M., Kim J., et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020;10:536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuusanmäki H., Leppä A.M., Pölönen P., Kontro M., Dufva O., Deb D., Yadav B., Brück O., Kumar A., Everaus H., et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica. 2020;105:708–720. doi: 10.3324/haematol.2018.214882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh Mali R., Zhang Q., DeFilippis R.A., Cavazos A., Kuruvilla V.M., Raman J., Mody V., Choo E.F., Dail M., Shah N.P., et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica. 2021;106:1034–1046. doi: 10.3324/haematol.2019.244020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yilmaz M., Kantarjian H., Short N.J., Reville P., Konopleva M., Kadia T., DiNardo C., Borthakur G., Pemmaraju N., Maiti A., et al. Hypomethylating agent and venetoclax with FLT3 inhibitor “triplet” therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J. 2022;12:77. doi: 10.1038/s41408-022-00670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maiti A., Naqvi K., Kadia T.M., Borthakur G., Takahashi K., Bose P., Daver N.G., Patel A., Alvarado Y., Ohanian M., et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin. Lymphoma, Myeloma & Leukemia. 2019;19:142–148.e1. doi: 10.1016/j.clml.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luger S.M., Wang V.X., Rowe J.M., Litzow M.R., Paietta E., Ketterling R.P., Lazarus H., Rybka W.B., Craig M.D., Karp J., et al. Tipifarnib as maintenance therapy did not improve disease-free survival in patients with acute myelogenous leukemia at high risk of relapse: Results of the phase III randomized E2902 trial. Leuk. Res. 2021;111:106736. doi: 10.1016/j.leukres.2021.106736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jain N., Curran E., Iyengar N.M., Diaz-Flores E., Kunnavakkam R., Popplewell L., Kirschbaum M.H., Karrison T., Erba H.P., Green M., et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin. Cancer Res. 2014;20:490–498. doi: 10.1158/1078-0432.CCR-13-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Desikan S.P., Ravandi F., Pemmaraju N., Konopleva M., Loghavi S., Jabbour E.J., Daver N., Jain N., Chien K.S., Maiti A., et al. A Phase II Study of Azacitidine, Venetoclax, and Trametinib in Relapsed or Refractory Acute Myeloid Leukemia Harboring RAS Pathway-Activating Mutations. Acta Haematol. 2022;145:529–536. doi: 10.1159/000525566. [DOI] [PubMed] [Google Scholar]
- 75.Wei W., Geer M.J., Guo X., Dolgalev I., Sanjana N.E., Neel B.G. Genome-wide CRISPR/Cas9 screens reveal shared and cell-specific mechanisms of resistance to SHP2 inhibition. J. Exp. Med. 2023;220:e20221563. doi: 10.1084/jem.20221563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Punekar S.R., Velcheti V., Neel B.G., Wong K.-K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022;19:637–655. doi: 10.1038/s41571-022-00671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith C.C., Lasater E.A., Zhu X., Lin K.C., Stewart W.K., Damon L.E., Salerno S., Shah N.P. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013;121:3165–3171. doi: 10.1182/blood-2012-07-442871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022;5:123–130. doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-sequencing data reported in this study have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus repository under the accession number GEO: GSE217359.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.