

Abstract
Background:
CYP2C19 is important in the metabolism of clopidogrel and several antidepressants. This study aimed to characterize the distribution of CYP2C19 star alleles (haplotypes) across diverse African populations compared with global populations.
Methods:
CYP2C19 star alleles and diplotypes were called from high coverage genomes using the StellarPGx pipeline.
Results:
CYP2C19*1 (51%), *2 (17%) and *17 (22%) were the most common star alleles across African populations in this study. It was observed that 3% of African participants had potentially novel CYP2C19 haplotypes.
Conclusion:
This study supports the necessity for CYP2C19 pharmacogenetic testing in African and global clinical settings, as well as the importance of comprehensive star allele characterization in the African context.
Keywords: African populations, CYP2C19, diplotypes, haplotypes, pharmacogenomics, phenotypes, star alleles, StellarPGx
Genetic diversity partly accounts for the variability in drug response within and between populations. This response may range from therapy non-response to severe adverse drug reactions (ADRs) [1–3]. Therefore, consideration of patients' genomic profiles when choosing treatment options has the potential to increase medication safety and efficacy in clinical settings globally [4,5]. Implementation of genotype-guided treatment as part of precision medicine, however, requires comprehensive characterization of variation in genes that encode enzymes involved in drug absorption, distribution, metabolism and excretion (ADME) [6,7].
Here, we present an analysis of CYP2C19 pharmacogenetic variation across African populations, compared with other global populations. CYP2C19 is vital in the metabolism of important antiplatelet medication, such as clopidogrel, and antidepressants (e.g., citalopram, escitalopram and sertraline) [8]. Several CYP2C19 pharmacogenetic variant annotations have been reported in the literature [9]. Given the increase in the burden of depressive disorders, cardiovascular disease and other non-communicable diseases [10], CYP2C19 pharmacogenetic studies are highly relevant in the African context [11,12], and at a global scale [1,13].
CYP2C19 (94 kb long) consists of nine exons and is located in the CYP2C cluster on chromosome 10q23 alongside CYP2C18, CYP2C8 and CYP2C9 (Figure 1) [9]. The Pharmacogene Variation Consortium (PharmVar) has catalogued 39 star (*) alleles (i.e., haplotypes) for CYP2C19 to date (www.pharmvar.org/gene/CYP2C19). These star alleles are defined by various combinations of SNPs, short insertions/deletions (indels) and/or structural variations [9,14]. CYP2C19 phenotype categories range from poor metabolizers (PM) to ultra-rapid metabolizers (UM) largely depending on the distribution of CYP2C19*1 and *38 (normal function), *2 through *4 (non-functional) and *17 (increased function). However, the potential collective functional impact of rare and/or population-specific alleles cannot be overstated.
Figure 1. . Overview of the CYP2C gene locus.
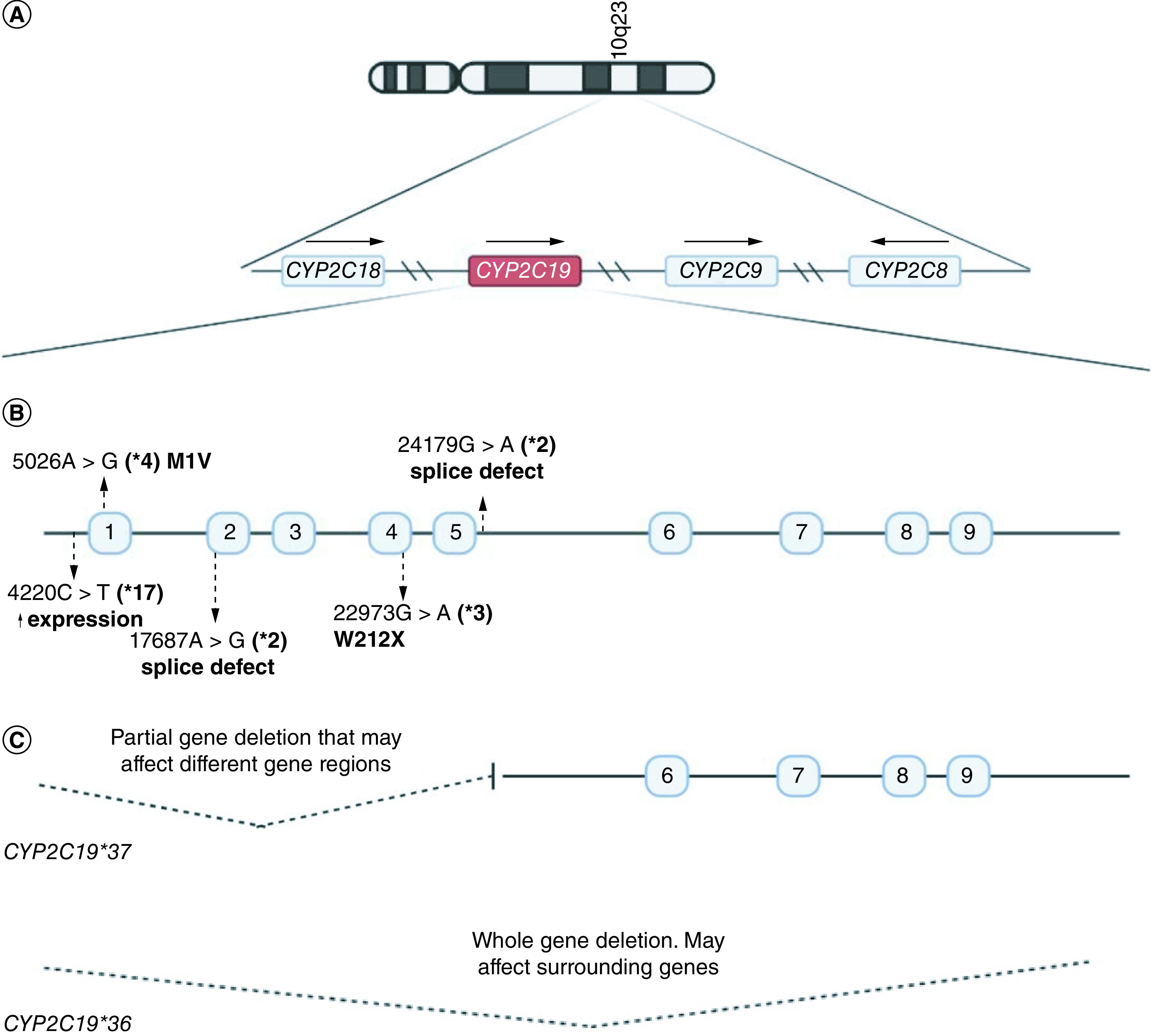
(A) Graphical summary of location of genes on the CYP2C cluster on chromosome 10. This includes CYP2C18, CYP2C19, CYP2C9 and CYP2C8. Arrows indicate whether the genes are on the forward or reverse DNA strands. (B) Core variants which define CYP2C19*2 to *4 and *17. These are previously known common CYP2C19 star alleles across multiple populations. (C) Examples of CYP2C19 structural variations that have been described in literature to date. Numbered boxes represent exons. Variant positions are relative to the RefSeq gene NG_008384.3.
The Clinical Pharmacogenetics Implementation Consortium (CPIC; https://cpicpgx.org/) maintains five up-to-date drug dosing guidelines involving CYP2C19, in particular for clopidogrel and antidepressants. However, these guidelines are largely based on information from patients of European and Asian ancestry. As such, the transferability of these guidelines to African clinical settings may be challenging because frequencies of clinically actionable alleles tend to differ within and between populations [7,15]. Given the higher extent of genetic diversity in African populations, we hypothesize that there may be uncharacterized functionally relevant African-ancestry CYP2C19 variation, especially across understudied ethnolinguistic groups. Furthermore, the recent increase in high-coverage whole genome sequence (WGS) datasets [16,17] and developments in star allele calling [18,19] provide the opportunity to identify rare and/or novel CYP2C19 star alleles.
Therefore, this study aimed to characterize the distribution of CYP2C19 star alleles and predicted phenotypes across diverse African populations, compared with other global populations, based on high coverage WGS data analysis. This study adds to the knowledge on CYP2C19 pharmacogenetic variation, particularly in sub-Saharan African (SSA) populations, which can inform design of genotyping panels, CYP2C19 functional studies, and subsequent clinical pharmacogenetics implementation on the African continent and across the African diaspora. This in turn will help alleviate the burden and costs related to adverse drug reactions to medications such as clopidogrel.
Materials & methods
Ethical conduct of research
Ethics approval for this study was granted by the Human Research Ethics Committee of the University of the Witwatersrand (Protocol no. M220293). This study involved secondary analysis of existing genomic data only, including public data from the 1000 Genomes Project and WGS data from the Africa Wits-INDEPTH Partnership for Genomic Research (AWI-Gen) study. Permission to use data from the AWI-Gen study was granted by the principal investigator, Michèle Ramsay. The AWI-Gen study was approved by the Wits Human Research Ethics Committee Medical under protocols M121029 and M170880.
Population data
High-coverage (30x) WGS data was analyzed in six SSA ethnolinguistic groups represented in the latest 1000 Genomes Project data release [16] and from the AWI-Gen study [20]. The 1000 Genomes Project continental African participants include 108 individuals from the Yoruba in Ibadan (YRI) in Nigeria, 99 individuals from the Esan (ESN) in Nigeria, 99 individuals from the Luhya in Webuye, Kenya (LWK), 85 individuals from the Mende in Sierra Leone (MSL) and 113 individuals from the Mandinka in western divisions of The Gambia (GWD). The AWI-Gen study included South African genomes from 100 southeastern Bantu [SEB]-speaking participants. The total sample size for this study is therefore 604 participants.
WGS datasets (n = 2000) from other global populations represented in the 1000 Genomes Project dataset were incorporated into the study for comparison of CYP2C19 star allele distributions. These include 157 African–American/Afro-Caribbean, 503 European (EUR), 504 east Asian (EAS), 489 south Asian (SAS) and 347 admixed American (AMR) participants.
CYP2C19 star allele calling
CYP2C19 star alleles were called using StellarPGx [18] (Figure 2). StellarPGx is a Nextflow-based pipeline that provides a scalable and reproducible approach to determining allelic variation in CYP genes according to the star allele nomenclature curated by PharmVar.
Figure 2. . Workflow for bioinformatic analysis of the high-coverage input whole genome sequence data represented in the 1000 Genomes and AWI-Gen datasets.

Blue boxes represents data/data points, pink boxes represents tools and green boxes represents key results.
AWI-Gen: Africa Wits-INDEPTH Partnership for Genomic Research; PGx: Pharmacogenomics; VEP: Ensembl variant effect predictor; WGS: Whole genome sequence.
There was no requirement for preselection of variations to be used in star allele assignment in this study because the input participant data was WGS. StellarPGx applied quality by depth >10, and 0.25 < allele balance for heterozygous calls (ABHet) <0.75, among its variant calling filters to improve accuracy in the subsequent CYP2C19 star allele assignment steps. The current StellarPGx workflow version 1.2.6.1 (https://github.com/SBIMB/StellarPGx) uses high-coverage WGS binary alignment map files as inputs (Figure 2). Single nucleotide variations in CYP2C19 were identified using GraphTyper [21]. Genome graph based approaches have been shown to be more reliable in variant discovery due to improved read realignment and reduction in reference allele bias. Following variant detection, StellarPGx assigned candidate star alleles (based on core variants in the sample) and then combined this information with copy number changes (where applicable) detected based on read-depth output from SAMtools [22] to produce final CYP2C19 diplotypes. Core variants in CYP2C19 typically include missense variants, frameshift variants, stop gain/loss, splice site variants and regulatory variants (e.g., for CYP2C19*17). For CYP2C19 structural variations, the known alleles assessed by StellarPGx include *36 (full CYP2C19 deletion) and *37 (partial CYP2C19 deletion). StellarPGx can also detect and phase duplications based on read depth distributions and variant allele depth ratios.
For individuals with novel star alleles, StellarPGx includes a flag in the output indicating the presence of a potential novel allele (as none of the diplotypes in the StellarPGx database would be a match) and recommends extra manual variant inspection (e.g., visualization on the Integrative Genomics Viewer [23]), and experimental validation according to PharmVar criteria. Novel star alleles could be due to occurrence of novel nonsynonymous variants on common haplotype ‘backbones’ (e.g., CYP2C19*1) or novel combinations of existing CYP2C19 core variants defining other haplotypes.
CYP2C19 variant annotation
The Ensembl variant effect predictor (VEP) [24] was used for variant annotation. The VEP plug-ins SIFT [25], Polyphen-2 [26], CADD [27], LRT [28], PROVEAN [29], LOFTEE [30] and VEST4 [31] were used to predict deleteriousness of novel and known allele-defining CYP2C19 variants. The combination of these tools provides a more accurate prediction for variant impacts in pharmacogenes such as CYP2C19, especially with the application of ADME-optimized thresholds [32]. A variant was considered to have a consensus deleterious prediction if at least half of the VEP plugins used to annotate the variant predicted a deleterious impact on CYP2C19.
CYP2C19 phenotype predictions
CYP2C19 metabolizer phenotypes were predicted based on diplotype information from the CPIC guidelines for clopidogrel [1]. Supplementary Table 1 shows how this phenotype prediction was implemented.
Statistical analysis
The summary output from StellarPGx was used to compute star allele and diplotype frequencies. Fisher's exact test was used to determine any significant differences in star allele frequencies between different populations. For correction of multiple testing, we applied the false discovery rate p-value adjustment for dependent observations suggested by Benjamini and Yekutieli [33] for CYP2C19*1, *2 and *17. A p-value < 0.05 indicated a significant difference. Possible deviations from Hardy–Weinberg equilibrium (HWE) were investigated using the Genetics package (v1.3.8.1.3) in R v4.1.0 (www.r-project.org/). A p-value > 0.05 indicated that the allele distributions followed HWE expectations.
Results
In the SSA populations, 3653 unique variants (∼38 variants per kb) were identified in CYP2C19 (± 5-kb flanking regions). Of these variants, 48 (1.3%) were found to be coding variants according to the RefSeq transcript NM_000769.4. In the intronic regions, 3605 (98.6%) variants were identified, with 79 variants being found in the promoter region (NC_000010.11: g.94760741-94762704). The breakdown for all these variants (by type) is shown in Figure 3. In other global populations in this study, 5549 unique variants (∼1000 variants per population) were identified, with an average of ∼11 variants per kb in CYP2C19. Similarly, to the SSA populations, the variants identified in the global populations were largely intronic (∼98%), with fewer being identified as coding variants (∼1.2%).
Figure 3. . Breakdown of CYP2C19 variant annotations obtained from the Ensembl Variant Effect Predictor according to the NM_000769.4 transcript in sub-Saharan African populations in this study.

CYP2C19 star allele frequency distributions
13 previously known CYP2C19 star alleles were identified across all SSA populations in this study. CYP2C19*1, *2 and *17 were the most commonly observed star alleles (Tables 1 & 2). The CYP2C19*2 (loss-of-function allele) frequency was highest in the ESN (20%) and lowest in the GWD (13%) participants, whereas the CYP2C19*17 (increased function allele) frequency was highest in the ESN (26%) and lowest in the SEB (17%) participants. However, there were no significant differences in star allele frequencies between any of the SSA ethnolinguistic groups in this study as determined by the Fisher's exact test. Apart from the commonly observed star alleles, other star alleles observed in SSA had low frequencies and some were present as singletons (Tables 1 & 2). For all star alleles identified there were no significant deviations from HWE.
Table 1. . CYP2C19 star allele distributions across global populations.
| Allele frequencies (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Star allele | CPIC function | Defining variant(s) | Consequence | SSA | African–American/Afro-Caribbean | AMR | SAS | EAS | EUR |
| *1 | Normal function | rs3758581 | Missense (I331V) | 51.4 | 52.7 | 73.3 | 38.5 | 57.2 | 55.8 |
| *2 | No function | rs12769205, rs4244285 | Splice defect, Splice defect | 17.3 | 14.7 | 10.2 | 32.7 | 31 | 14.4 |
| *3 | No function | rs4986893 | Stop-gain (W212X) | <0.1 | 0.3 | 0 | 1.3 | 5.6 | 0 |
| *4 | No function | rs28399504 | Start lost (M1A) | 0 | 0 | 0.3 | 0 | <0.1 | 0.1 |
| *8 | No function | rs41291556 | Missense (W120R) | 0 | 0.3 | 0 | 0.1 | 0 | 0.3 |
| *9 | Decreased function | rs17884712 | Missense (R144H) | 1.4 | 0.6 | 0.1 | 0 | 0 | 0 |
| *10 | Decreased function | rs6413438 | Missense (P227L) | <0.1 | 0 | 0.1 | 0 | 0 | 0 |
| *11 | Normal function | rs58973490 | Missense (R150H) | 0 | 0.3 | 0 | 0 | 0 | 0.2 |
| *12 | Uncertain function | rs55640102 | Stop-lost (X491C) | <0.1 | 0 | 0 | 0 | 0 | 0 |
| *13 | Normal function | rs17879685 | Missense (R410C) | 1.8 | 2.2 | 0.3 | 0 | 0 | 0 |
| *15 | Normal function | rs17882687 | Missense (I19L) | 1.4 | 0.6 | 0 | 0 | 0 | 0 |
| *17 | Increased function | rs12248560 | Regulatory (Expression) | 22.2 | 24.3 | 11.5 | 14.0 | 1.5 | 22.1 |
| *22 | No function | rs140278421 | Missense (R186P) | 0 | 0.3 | 0.1 | 0 | 0 | 0 |
| *24 | No function | rs118203757 | Missense (R335Q) | 0 | 0.3 | 0 | 0 | 0 | 0 |
| *30 | Uncertain function | rs145328984 | Missense (R73C) | <0.1 | 0 | 0 | 0 | 0 | 0 |
| *34 | Uncertain function | rs367543002, rs367543003 | Missense (P3S), Missense (F4L) | 0 | 0 | 0 | 1.2 | 0 | 0 |
| *35 | No function | rs12769205 | Splice defect | 3 | 1.6 | 0.1 | 0 | 0 | 0 |
| *37 | No function | — | Partial gene deletion | 0 | 0 | 0 | 0 | 0 | 0.1 |
| *38 | Normal function | Reference allele | — | 0.4 | 1.3 | 3.8 | 11.8 | 4.5 | 0.7 |
| *39 | Uncertain function | rs17882687, rs17885179 | Missense (I19L), Missense (E122A) | 0.9 | 0.3 | 0 | 0 | 0 | 0 |
AMR: Admixed American; CPIC: Clinical Pharmacogenomics Implementation consortium; EAS: East Asian; EUR: European; SAS: South Asian; SSA: Sub-Saharan Africa.
Table 2. . CYP2C19 haplotype frequencies across six continental African populations in this study.
| Allele frequencies (%) | |||||||
|---|---|---|---|---|---|---|---|
| Haplotype | CPIC function | SEB | YRI | MSL | LWK | GWD | ESN |
| *1 | Normal function | 52 | 49 | 53 | 55 | 52 | 48 |
| *2 | No function | 19 | 17 | 17 | 18 | 13 | 20 |
| *3 | No function | 0 | 0 | 0 | 1 | 0 | 0 |
| *9 | Decreased function | 3 | 0 | 2 | 1 | 1 | 1 |
| *10 | Decreased function | 0 | 0 | 1 | 0 | 0 | 0 |
| *12 | Uncertain function | 1 | 0 | 0 | 0 | 0 | 0 |
| *13 | Normal function | 1 | 2 | 1 | 4 | 3 | 0 |
| *15 | Normal function | 5 | 0 | 0 | 1 | 3 | 1 |
| *17 | Increased function | 17 | 24 | 24 | 18 | 24 | 26 |
| *30 | Uncertain function | 0 | 0 | 0 | 0 | 0 | 1 |
| *35 | No function | 3 | 6 | 2 | 2 | 3 | 2 |
| *38 | Normal function | 1 | 0 | 1 | 0 | 0 | 1 |
| *39 | Uncertain function | 0 | 2 | 0 | 0 | 1 | 2 |
CPIC: Clinical Pharmacogenomics Implementation Consortium; ESN: Esan in Nigeria; GWD: Mandinka in western divisions of The Gambia; LWK: Luhya in Webuye, Kenya; MSL: Mende in Sierra Leone; SEB: Southeastern Bantu; YRI: Yoruba in Ibadan.
In our global comparison, CYP2C19*1 occurred at a significantly higher frequency in the AMR (AF = 73%; p = 3.9 × 10-10) populations and a significantly lower frequency in the south Asian (AF = 39%; p = 8.3 × 10-5) populations compared with SSA (Table 1). Additionally, CYP2C19*2 occurred at significantly lower frequency in the African–American/Afro-Caribbean (AF = 15%; p = 0.006) populations and occurred at significantly higher frequencies in the south Asian (AF = 33%; p = 3.5 × 10-5) and east Asian (AF = 31%; p = 7 × 10-7) populations when compared with SSA (Table 1). Conversely, CYP2C19*17 was present at a significantly higher frequency in SSA (AF = 22%) when compared with AMR (AF = 11%; p = 0.0004), south Asian (AF = 15%; p = 0.001) and east Asian (AF = 2%; p = 4 × 10-15) populations (Table 1). CYP2C19*12 and *30 were observed in the SSA populations but were not called in any of the other global populations, while CYP2C19*4, *8, *11, *22, *24, *34 and *37 were not observed in SSA but were present in various other global populations (Table 1).
CYP2C19 phenotype distributions
CYP2C19 phenotype predictions indicated that only 28% (21–36%) of SSA participants are normal metabolizers (NM) of clopidogrel. Within SSA, the MSL ethnolinguistic group had the lowest number of NMs (21%), and the LWK ethnolinguistic group had the highest number of NMs (36%) (Supplementary Table 3). In our global comparison, the AMR individuals had the highest number of NMs (59%), and the SAS individuals had the lowest number of NMs (22%) (Figure 4 & Supplementary Table 2).
Figure 4. . Distribution of predicted phenotypes for global populations in this study.

Metabolizer status was determined using the Clinical Pharmacogenetics Implementation Consortium's CYP2C19 genotype–phenotype translation guidelines.
AMR: Admixed American participants; EAS: East Asian participants; EUR: European participants; LIM: Likely intermediate metabolizer; LPM: Likely poor metabolizer; SAS: South Asian participants; SSA: Sub-Saharan African participants.
Approximately 5% of the SSA individuals were predicted to be PM of CYP2C19. Within SSA, the SEB and LWK ethnolinguistic groups had the lowest number of PMs (4%), whereas the YRI ethnolinguistic group had the highest number of PMs (6%) (Supplementary Table 3). The PM phenotype was less common (∼1%) across EUR, AMR and African–American/Afro-Caribbean participants.
The PM phenotype was observed at a higher frequency in the east and south Asian populations; however, this was not statistically different (p = 0.08) from the SSA participants (Figure 4 & Supplementary Table 2). The likely PMs were only observed in the SSA participants.
For the rapid metabolizer (RM) phenotype group, there were also no significantly different frequencies across SSA. The frequency across SSA was relatively high (24%) which is mainly accounted for by the *1/*17 diplotype (Supplementary Table 3). The highest observed RM frequency was in MSL (30%), whereas the lowest frequency was observed in LWK (20%) (Supplementary Table 3). Compared with other global populations, the RM phenotype was observed at a higher frequency in the African–American/Afro-Caribbean (39%) and EUR (35%) populations. The frequency of RMs was, however, significantly lower than that observed in the AMR (16%; p = 0.005), SAS (14%; p = 0.0003) and EAS (1%; p = 3 × 10-7) populations when compared with SSA (24%) (Figure 4 & Supplementary Table 2).
We observed the highest frequency of predicted ultrarapid metabolizers (UM) across the SSA populations (5%) when compared with other global populations (UM frequency = 0–4%) in this study (Figure 4 & Supplementary Table 2). The frequency of UMs across SSA ranged from 3 to 7%, with the highest being observed in YRI, MSL and ESN (7%) and the lowest being observed in SEB and LWK (3%) (Supplementary Table 3). None of the differences in UM phenotype distribution were statistically significant.
In SSA, we observed around 6% of participants with indeterminate (IND) CYP2C19 phenotypes. Within SSA this was lowest in YRI (0%) and highest in MSL and LWK (8%). The average number of IND individuals in SSA was lower than the percentage observed in SAS populations (13%) but higher than that in EUR, AMR, African–Amercian/Afro-Caribbean and EAS populations (Figure 4 & Supplementary Table 2). However, these were not statistically significant differences.
Novel CYP2C19 alleles inferred in this study
The CYP2C19 variant positions in this section are based on NG_008384.3, counting from the sequence start, and the protein positions are based on NP_000760.1. The SSA populations had 27 individuals (∼4%) who were predicted to have novel haplotypes. This was greater than the number of individuals with predicted novel haplotypes in AMR (∼3%), EUR (∼1%), EAS (∼0.6%) and African–American/Afro-Caribbean (∼0.6%) participants. However, the SAS populations had a considerably higher proportion of individuals with predicted novel haplotypes (∼11%) compared with SSA (Supplementary Table 4).
Six unique, potentially novel CYP2C19 star alleles were inferred in SSA – at least one in each subpopulation except for YRI (Table 3). Nine participants (∼11%) among the MSL had predicted novel CYP2C19 star alleles. The variants defining the predicted novel haplotypes in this study are already in the SNP database [34]. However, the observed haplotypes in this context have not yet been used to define a star allele by PharmVar and are therefore considered potentially novel, pending final novel star allele or sub-allele assignment by the PharmVar CYP2C19 expert panel.
Table 3. . Inferred novel CYP2C19 haplotypes and unresolved diplotypes in African populations in this study.
| Resolved haplotypes | ||||||
|---|---|---|---|---|---|---|
| # | Background allele | Additional core variant(s) | Consequence(s) | Consensus VEP-based prediction | Count | Country/dataset |
| 1 | *1 | rs146991374∼95056∼A>T | Missense (K432I) | Deleterious | 4 | MSL/1000G |
| 2 | *1 | rs150152656∼ 17767∼C>T | Missense (T130M) | Deleterious | 1 | ESN/1000G |
| 3 | *1 | rs145328984∼17426∼C>T | Missense (R73C) | Deleterious | 1 | ESN/1000G |
| 4 | *1 | rs201306972∼17427∼G>A | Missense (R73H) | Benign | 1 | MSL/1000G |
| 5 | *39 | rs112197069∼17538∼T>A | Missense (F110Y) | Benign | 3 | SEB/AWI-Gen |
| 6 | *2 | rs145119820∼17715∼G>A | Missense (V113I) | Benign | 10 | MSL, LWK, GWD/1000G |
| Unresolved diplotypes with potentially novel star alleles | ||||||
|---|---|---|---|---|---|---|
| # | Background alleles | Additional core variant(s) | Consequence(s) | Consensus VEP-based prediction | Count | Country/dataset |
| 1 | [*1/*2] | rs183701923∼22893∼C>T | Missense (R186C) | Deleterious | 1 | LWK/1000G |
| 2 | [*17/*35] | rs192154563∼95085∼C>T | Missense (R442C) | Deleterious | 1 | LWK/1000G |
| 3 | [*2/*2] | rs201306972∼17427∼G>A+ rs537050749∼24271∼A>G |
Missense (R73H) Missense (N258S) |
Benign | 1 | ESN/1000G |
| 4 | [*1/*17] | rs375760063∼17856∼A>G | Missense (K160E) | Benign | 1 | MSL/1000G |
| 5 | [*1/*17] | rs545642100∼17526∼C>G | Missense (A106G) | Benign | 1 | ESN/1000G |
| 6 | [*2/*17] | rs527499296∼92332∼A>C | Missense (E415D) | Benign | 1 | LWK/1000G |
| 7 | [*1/*3] | rs144056033∼92303∼A>G | Missense (M406V) | Benign | 1 | LWK/1000G |
Note that CYP2C19*1 is defined by rs3758581 (I331V). Variant positions are according to the CYP2C19 RefSeq gene (NG_008384.3).
1000G: 1000 Genomes Project; AWI-Gen: Africa Wits-INDEPTH Partnership for Genomic Research; ESN: Esan in Nigeria; GWD: Mandinka in western divisions of The Gambia; LWK: Luhya in Webuye Kenya; MSL: Mende in Sierra Leone; SEB: Southeastern Bantu; VEP: Ensembl Variant Effect Predictor.
From the variants defining these novel haplotypes, only rs146991374 (defining haplotype 1), rs150152656 (defining haplotype 2) and rs145328984 (defining haplotype 3) had consensus deleterious VEP plugin predictions (Table 3). Notably, the novel haplotype 5 (defined by rs112197069) was only present in the SEB populations. Furthermore, haplotypes 1 (defined by rs146991374) and 6 (defined by rs145119820), which were not present in the SEB participants, were present in more than one individual (Table 3). Haplotype 6 was found in 10 SSA individuals (Table 3).
In addition, there were seven participants for whom StellarPGx predicted presence of potential novel star alleles; however, the backbone allele(s) for the defining nonsynonymous variants could not be determined (Table 3). All these unresolved diplotypes occurred in singletons. Among these, rs183701923 and rs192154563 in unresolved diplotypes 1 and 2, respectively, had a consensus deleterious prediction. Participant IDs relating to the resolved predicted novel haplotypes and unresolved predicted novel diplotypes are provided in Supplementary Table 4. VEP plugin scores for all the CYP2C19 allele-defining variants called in this study are provided in Supplementary Table 5.
Discussion
Assessing variation in CYP2C19 in diverse populations is important because functional variants in this gene might contribute to differences in the efficacy and safety of various important medications such as clopidogrel and antidepressants. Most of the current literature is focused on the distribution of important star alleles in European and Asian populations; far less is known about African populations [3] despite their notable high genetic diversity. Therefore, although precision medicine strategies such as genotype-guided therapy offer much promise in curbing adverse drug reactions and promoting treatment efficacy, there is a risk of poor transferability of Eurocentric guidelines to African populations which may exacerbate existing health disparities [35]. This study provides important information on the distribution of CYP2C19 allelic variation in six diverse SSA populations represented in the 1000 Genomes Project and AWI-Gen datasets. Our findings can inform more comprehensive CYP2C19 pharmacogenetic testing in clinical and research settings. This is pertinent in the African context where there is high genetic heterogeneity and inter-individual response to a number of the relevant aforementioned medications may vary due to CYP2C19 variation.
CYP2C19*2 and *17 are among the actionable star alleles in clinical settings worldwide. CYP2C19*2 (loss of function) is associated with a significant decrease in platelet responsiveness to clopidogrel [1], whereas *17 (increased function) homozygotes are known to be at a greater risk of clopidogrel-related bleeding events [36]. In SSA populations in this study, these two key star alleles occurred at relatively high frequencies similar to those reported in previous studies [36–38] (see also PharmGKB [39]). CYP2C19*2 (no function) and *17 (increased function) frequencies did not differ significantly across the African ethnolinguistic groups in this study despite the large genetic diversity in these populations. In a comparison between SSA and other global populations, CYP2C19*17 was observed at a lower frequency in east Asian (statistically significant), south Asian and AMR populations), whereas CYP2C19*2 and *3 (no function) was more frequent in south and east Asian populations (Table 2). This exemplifies the limitation of using other global populations as a proxy for SSA populations when implementing genotype-guided therapy. In addition to *2 and *17, star alleles such as *9 (decreased function) and *35 (no function), which occur at frequencies >1%, might also be relevant to test for in African settings. However, the estimated frequencies, especially for rarer alleles, should be interpreted with caution given the relatively small sample sizes.
Across the SSA populations in this study, 64–79% of participants had phenotypes (UM, RM, IM, LIM, PM) that would require alterations in clopidogrel and/or antidepressant dosage or medication according to current CPIC guidelines [1]. A similar proportion (∼60%) of these phenotypes combined has been reported in south African Xhosa and Cape Mixed Ancestry participants [37]. This exemplifies the need for CYP2C19 pharmacogenetic testing to minimize adverse drug reactions and promote drug safety across Africa. In our study, phenotype predictions (especially PM, IM and UM) for SSA were significantly different when compared in particular to the SAS and EAS populations. This is in part accounted for by differences in CYP2C19 allele distributions and emphasizes limitations in transferability of actionable star alleles and genotyping platforms based on non-African populations.
We inferred novel CYP2C19 haplotypes that may be used guide new star number assignments by PharmVar. In SSA, 3% of participants carried such haplotypes. Three of the six computationally resolved novel star alleles were found in more than one individual, which provides more evidence for their validation. In general, potential novel star alleles – and alleles with unknown function – contributed to the non-negligible proportion (6%) of SSA participants with indeterminate CYP2C19 phenotypes. Failure to account for this African-ancestry variation may have an impact on the effective implementation of precision medicine across the continent because African populations may have unique CYP2C genetic markers relating to drug response [40]. Furthermore, the calling of novel alleles in this study was facilitated by the use of high coverage WGS data which highlights information that might be missed when using targeted genotyping methods in Africa at present.
This study had some limitations. First, the WGS data used were generated using short read technology. This presents challenges in investigating complex structural variations involving CYP2C19 and other highly similar CYP2C cluster genes (e.g., CYP2C18). For example, StellarPGx initially called HG00268 as having CYP2C19*36 (full gene deletion); however, upon visual inspection on the Integrative Genomics Viewer [23], we revised this to CYP2C19*37 (partial gene deletion) based on the read coverage distribution. Short read data limitations can also account for unresolved singleton novel diplotypes in the study. Secondly, in silico variant effect prediction tools applied in this study do not provide full insight into the relationship between CYP2C19 variants and relevant substrates. This is especially the case for missense, intronic and promoter variants. Therefore, the VEP plugins could not be used to assess the function of novel haplotypes comprehensively. However, molecular functional assays that would address this problem were not within the parameters of this study. Nonetheless, we expect functional effect predictions for stop gain, frameshift and splice defect variants to be consistent with experimental assay validations. Lastly, our phenotype predictions were solely based on the CPIC guidelines outlined for CYP2C19's interaction with clopidogrel. CPIC genotype-phenotype translation for other drugs may have provided different results. However, given the importance of clopidogrel in Africa and globally, it provides a justifiable basis for CYP2C19 phenotype prediction comparisons. It is also important to consider that patient response to clopidogrel and other medications could be influenced by other factors such as age, lifestyle, environment and phenocoversion, whereby genotypic NM patients may present as phenotypic PMs in presence of CYP2C19 inhibitors. We also observed a significant proportion of individuals in SSA with indeterminate CYP2C19 phenotypes (6%). This highlights the shortcomings for the utility of the current CPIC guidelines for genotype–phenotype therapy options in African populations and therefore the need for thorough star allele characterization and phenotypic research across Africa.
Conclusion
This study contributes to the efforts to characterize the distribution of actionable CYP2C19 star alleles and associated phenotypes in SSA and to establish a more comprehensive catalog of potential pharmacogenomically relevant star alleles from African and other global populations. Although no significant CYP2C19 star allele frequency differences were observed across SSA in this study, our results emphasize key differences across global populations. Given our results, CYP2C19*9, *13, *15, *35 and *39 need to be considered, along with *2 and *17, when designing pharmacogenetic testing panels for application in African populations. Furthermore, the relatively large number of predicted novel CYP2C19 star alleles found in SSA emphasizes the need for thorough pharmacogene star allele characterization across Africa, especially in understudied ethnolinguistic groups. This could help guide the development of more comprehensive genotyping panels and phenotype prediction algorithms across Africa and for the African diaspora. In turn, such interventions will be critical in improving the efficacy of medications such as clopidogrel and antidepressants, as well as reducing the risk of adverse drug reactions in patients of African ancestry.
Future perspective
Follow-up studies to this work will entail validation (e.g., using long read resequencing) of novel star alleles inferred in this study and submission to PharmVar for cataloguing because this is recommended before including them in clinical implementation guidelines. Future studies aimed at determining the CYP2C19 functional impact of variants defining these novel alleles, as well as those in unknown function star alleles, are also warranted. Furthermore, it is necessary to evaluate the clinical utility and cost–effectiveness of CYP2C19 genotype-guided therapy in the African context given the extent of pharmacogenetic variation described in this study. Considering that this is a pilot study, the use of more diverse population groups in genetic characterization studies could allow for more informative results and ultimately better the potential for clinical application improvements.
Summary points.
Background
Given the considerable novel African-ancestry variation we found previously in other absorption, distribution, metabolism and excretion (ADME) genes and the extensive genetic diversity in Africa, pharmacogenetic studies are warranted for other key ADME genes.
Pharmacogenetic testing for CYP2C19 is important clinically to optimize patient response to clopidogrel and antidepressants. CYP2C19 is one of the priority pharmacogenes to study in Africa given the increasing burden of cardiovascular and major depressive disorders.
Objective
This study aimed to characterize the distribution of CYP2C19 star alleles (haplotypes) across diverse African populations compared with global populations, with a view toward informing future pharmacogenetic implementations.
Methods
We analyzed 504 high-depth genomes from five African ethnolinguistic groups represented in the 1000 Genomes Project dataset in addition to 100 genomes from South African south eastern Bantu-speaking participants from the Africa Wits-INDEPTH Partnership for Genomic Research study.
CYP2C19 haplotypes and diplotypes were called using the StellarPGx pipeline, and novel allele-defining variants were annotated using Ensembl's variant effect predictor.
Phenotype predictions from diplotypes were made using current guidelines (for clopidogrel) published by the Clinical Pharmacogenomics Implementation Consortium.
Results
CYP2C19*1 (51%), *2 (17%) and *17 (22%) were the most common star alleles across African populations in this study.
3% of African participants had inferred novel haplotypes.
There were no significant differences in star allele frequencies or phenotype proportions between any of the sub-Saharan African (SSA) ethnolinguistic groups.
However, star allele frequencies, as well as phenotype proportions, differed significantly between the African participants and other global participants in our study (most notably south Asian and east Asian participants).
Conclusion
This study contributes to the efforts to characterize the distribution of actionable CYP2C19 star alleles and associated phenotypes in SSA.
Although no significant CYP2C19 star allele frequency differences were observed across SSA in this study, our results emphasize key differences across global populations.
The relatively large number of predicted novel CYP2C19 star alleles found in SSA emphasizes the need for thorough pharmacogene star allele characterization across Africa, especially in understudied ethnolinguistic groups.
This could help guide the development of more comprehensive genotyping panels and phenotype prediction algorithms across Africa and for the African diaspora.
Supplementary Material
Acknowledgments
The authors thank Michèle Ramsay (principal investigator of the AWI-Gen study) for permission and access to the AWI-Gen data (EGAD00001006418) used in this study. We acknowledge the 1000 Genomes Project whose datasets used in this study were generated at the New York Genome Center with funds provided by NHGRI Grant 3UM1HG008901-03S1.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/pgs-2023-0166
Author contributions
RP Booyse, D Twesigomwe and S Hazelhurst designed the study. RP Booyse, D Twesigomwe, S Hazelhurst contributed to acquisition and interpretation of the data. RP Booyse analyzed the data. RP Booyse drafted the manuscript. RP Booyse, D Twesigomwe and S Hazelhurst revised the manuscript for important intellectual content. D Twesigomwe and S Hazelhurst supervised the research. All authors approved the final version of the manuscript.
Financial disclosure
Ross Booyse was supported by the Post Graduate Merit award from the University of the Witwatersrand. David Twesigomwe was supported by funding from the Sydney Brenner Charitable Trust, and his postdoctoral fellowship is hosted at Wits University in partnership with the University of Edinburgh. This study used WGS data from the Africa-Wits INDEPTH Partnership for Genomics Research (AWI-Gen) Collaborative Center which is funded by the NIH/NHGRI (grant no. U54HG006938) as part of the Human, Heredity and
Health in Africa (H3Africa) Consortium. The content is solely the responsibility of the authors and does not necessarily represent the views of the funders. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical disclosure
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
Data sharing statement
The secondary data used in this study is not from a clinical trial. Further details on data availability have been mentioned in the Data Availability Statement in the manuscript.
Data availability statement
The 1000 Genomes Project dataset is publicly available (www.internationalgenome.org/). The 100 high-coverage genomes from the Africa Wits-INDEPTH Partnership for Genomic Research study (EGAD00001006418) are available from the European Genome-Phenome Archive (https://ega-archive.org/).
Code availability statement
The StellarPGx code used in this study can be found at https://github.com/SBIMB/StellarPGx. All custom scripts used in downstream analysis are available at https://github.com/ross-booyse/CYP2C19_PGx.
References
Papers of special note have been highlighted as: •• of considerable interest
- 1.Lee CR, Luzum JA, Sangkuhl K et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin. Pharm. and Ther. 112(5), 959–967 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The Clinical Pharmacogenetics Implementation Consortium guidelines are essential for genotype-guided therapy. This paper in particular details the consensus CYP2C19 genotype–phenotype translation for clopidogrel and implications on drug efficacy and safety.
- 2.Zhao M, Ma J, Li M et al. Cytochrome P450 enzymes and drug metabolism in humans. Int. J. Mol. Sci. 22(23), 12808 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Y, Ingelman-Sundberg M, Lauschke VM. Worldwide distribution of cytochrome p450 alleles: a meta-analysis of population-scale sequencing projects. Clin. Pharmacol. Ther. 102(4), 688–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr DF, Pirmohamed M. Precision medicine in drug safety. Curr. Opin. Toxicol. 23–24, 87–97 (2020). [Google Scholar]
- 5.Desta Z, Gammal RS, Gong L et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2B6 and efavirenz-containing antiretroviral therapy. Clin. Pharmacol. Ther. 106(4), 726–733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arbitrio M, Di Martino MT, Scionti F et al. Pharmacogenomic profiling of ADME gene variants: current challenges and validation perspectives. High-Throughput 7(4), 40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Rocha JEB, Othman H, Botha G et al. The extent and impact of variation in ADME genes in sub-Saharan African populations. Front. Pharmacol. 12, 366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Examines the impact of variation in key absorption, distribution, metabolism and excretion genes in diverse African populations, and how this may impact drug response.
- 8.Whirl-Carrillo M, Huddart R, Gong L et al. An evidence-based framework for evaluating pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. 3(110), 563–572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Botton MR, Whirl-Carrillo M, Del Tredici AL et al. PharmVar GeneFocus: CYP2C19. Clin. Pharm. & Ther. 109(2), 352–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbafati C, Abbas KM, Abbasi-Kangevari M et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396(10258), 1204–1222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alessandrini M, Pepper MS. Priority pharmacogenetics for the African continent: focus on CYP450. Pharmacogenomics 15(3), 385–400 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Sitabule BR, Othman H, Choudhury A et al. Promoting Pharmacogenomics in Africa: Perspectives from Variation in G6PD and Other Pharmacogenes. Clin. Pharmacol. Ther. 113(3), 476–479 (2023). [DOI] [PubMed] [Google Scholar]
- 13.Swen JJ, van der Wouden CH, Manson LE et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: an open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 401(10374), 347–356 (2023). [DOI] [PubMed] [Google Scholar]
- 14.Botton MR, Lu X, Zhao G et al. Structural variation at the CYP2C locus: characterization of deletion and duplication alleles. Hum. Mutat. 40(11), e37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira L, Mutesa L, Tindana P, Ramsay M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genetics 22(5), 284–306 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Byrska-Bishop M, Evani US, Zhao X et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185(18), 3246–3440 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhury A, Aron S, Botigué LR et al. High-depth African genomes inform human migration and health. Nature 586(7831), 741–748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twesigomwe D, Drögemöller BI, Wright GEB et al. StellarPGx: a Nextflow pipeline for calling star alleles in cytochrome P450 genes. Clin. Pharmacol. Ther. 110(3), 741–749 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Hari A, Zhou Q, Gonzaludo N et al. An efficient genotyper and star-allele caller for pharmacogenomics. Genome Res. (1), 61–70 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsay M, Crowther N, Tambo E et al. H3Africa AWI-Gen Collaborative Centre: a resource to study the interplay between genomic and environmental risk factors for cardiometabolic diseases in four sub-Saharan African countries. Glob. Health Epidemiol. Genom. 1, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eggertsson HP, Jonsson H, Kristmundsdottir S et al. Graphtyper enables population-scale genotyping using pangenome graphs. Nat. Genet. 49, 1654–1660 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Li H, Handsaker B, Wysoker A et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16), 2078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson JT, Thorvaldsdóttir H, Winckler W et al. Integrative genomics viewer. Nat. Biotech. 29(1), 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaren W, Gil L, Hunt SE et al. The Ensembl variant effect predictor. Genome Biol. 17(1), 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protocols 4(7), 1073–1082 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. 7(Suppl. 76), (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rentzsch P, Witten D, Cooper GM et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucl. Acids Res. 47(1), 886–894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita A, Kojima K, Patriota AG et al. A fast and robust statistical test based on likelihood ratio with Bartlett correction to identify Granger causality between gene sets. Bioinformatics 26(18), 2349–2351 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16), 2745–2747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karczewski KJ, Francioli LC, Tiao G et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809), 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter H, Douville C, Stenson PD et al. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 14(Suppl. 3), S3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Mkrtchian S, Kumondai M et al. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenomics J. 19(2), 115–126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Inst. Math. Stat. Collect 29(4), 1165–1188 (2001). [Google Scholar]
- 34.Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28(1), 352 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hussein AA, Hamad R, Newport MJ, Ibrahim ME. Individualized medicine in Africa: bringing the practice into the realms of population heterogeneity. Front Genet. 13(1), 873 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biswas M. Global distribution of CYP2C19 risk phenotypes affecting safety and effectiveness of medications. Pharmacogenomics 21(2), 190–199 (2020). [DOI] [PubMed] [Google Scholar]; •• Highlights the impact of key variations in CYP2C19 and the implications in global populations. This offers a crucial comparison with our results.
- 37.Matimba A, Del-Favero J, Van Broeckhoven C, Masimirembwa C. Novel variants of major drug-metabolising enzyme genes in diverse African populations and their predicted functional effects. Hum. Genom. 3(2), 169–190 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drögemöller BI, Wright GE, Niehaus DJ et al. Characterization of the genetic profile of CYP2C19 in two South African populations. Pharmacogenomics 11(8), 1095–1103 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol. Biol. 1015, 311–320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ndadza A, Muyambo S, Mntla P et al. Profiling of warfarin pharmacokinetics-associated genetic variants: Black Africans portray unique genetic markers important for an African specific warfarin pharmacogenetics-dosing algorithm. J. Thromb. Haemost. 19(12), 2957 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.