

Abstract

In this study, we report on the green fluorescence exhibited by nitrobenzofurazan-sulfide derivatives (NBD-Si, i = 1–4). The optical responses of these studied compounds in a polar methanol solvent were simulated by the use of time-dependent density functional theory (TD-DFT) employing the Becke-3-Parameter-Lee–Yang–Parr (B3LYP) functional along with the 6-31G(d,p) basis set. The computed energy and oscillator strength (f) results complement the experimental results. The band gap was calculated as the difference between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). Additionally, the density of states (DOS) was computed, providing a comprehensive understanding of the fundamental properties of these materials and further corroborating the experimental data. When the experimental data derived from ultraviolet/visible (UV/visible) and fluorescence spectroscopic techniques and those from simulated spectra are analyzed, the extracted values match up adequately. In addition, the NBD-sulfide compounds exhibit a large Stokes shift up to 85 nm in a polar methanol solvent. They are hypothesized to represent a novel paradigm of excited-state intramolecular charge transfer (ICT). To understand the intrinsic optical properties of NBD-Si materials, an ICT was identified, and its direction within the molecule was evaluated using the ratio of βvect and βtotal, values extracted from the computed nonlinear optical (NLO) properties. Moreover, the reduced density gradient (RDG)-based noncovalent interactions (NCIs) were employed to characterize the strength and type of NBD-Si interactions. Furthermore, noncovalent interactions were identified and categorized using the Quantum Theory of Atoms in Molecules (QTAIM) analysis. Ultimately, the combination of Hirshfeld surface analysis and DFT calculations was utilized to enhance the characterization and rationalization of these NCIs.
1. Introduction
Since the first report on the intramolecular charge transfer (ICT)-based 4-(dimethylamino) benzonitrile (DMABN) molecule, as a groundbreaking example of electron donor/acceptor (D/A), by Lippert et al.,1 there have been great efforts to obtain ICT-based aryl amine compounds. This has attracted considerable attention from the scientific publishing community owing to their potential ability to design new nonlinear optical (NLO) systems,2 electro-optical switches,3 and organic light-emitting diodes.4−6 Furthermore, they find utility solar energy conversion7−11 and as fluorescence sensors. However, quantifying the extent of ICT remains a challenge for the scientific community. First-order hyperpolarizability and therefore the second-order NLO responses are related to an ICT within the molecule.12 Particularly, the investigation of π-conjugated systems with D/A moieties at opposite ends reveals a strong NLO response. Noncentrosymmetric D-π–A dipolar compounds are often used to achieve first- or second-order nonlinear polarizabilities. The electron capacity of the D and the electron-withdrawing capacity of the A groups play a key role in introducing the ICT extent.13−16 The design of NLO materials is a groundbreaking research area for both the theoretical and experimental communities, with numerous applications in optics, optoelectronics, photonics, and nanophotonics.17,18 These NLO materials are greatly desirable due to their facile synthesis and superior flexibility. This offers the opportunity to fine-tune their structural and optoelectronic characteristics by designing chromophores with large first hyperpolarizability (β).19,20
Methods of experimental and quantum chemistry have been widely used to evaluate the properties of NBD appended to morpholine,21 piperidine,22 pyrrolidine,23 and other electrophilic systems involving NBD-Cl, NBD-OCH3, and NBD-OC6H524
The present work reports the predicted optical (ultraviolet–visible (UV–vis) and photoluminescence (PL)) properties of four examples of NBD-sulfides.25 The main objective was to understand the substituent effects on optical responses, validate the calculation method, and examine the extent of the ICT and NLO characteristics based on the findings. By the nucleophilic aromatic substitution (SNAr) method, the electrophile NBD-Cl reacted with mercaptoderivatives 2a–2d acting as nucleophilic molecules (namely, 2a, 2-pyridinethiol; 2b, 4-pyridinethiol; 2c, N-acetylcysteamine; and 2d, N-acetylcysteine), affording the thioether derivatives 3a–3d (see Scheme 1).
Scheme 1. Applying the SNAr Mechanism to Sulfides with an NBD-Cl Moiety to Convert Them into NBD-Si, 3a–3d, Compounds25.

The compounds will hereafter be referred to as NBD-Si (i: 1–4), respectively. The structure of the designed molecules (3a–3d) was supported by 1H and 13C NMR, Fourier-transform infrared (FTIR), and mass spectroscopy, and their optical responses are reported.25 In this study, we employ density functional theory (DFT) and its time-dependent-DFT (TD-DFT) to calculate electronic characteristics, including highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, UV–visible optical absorption and emission spectra, dipole moment variation (Δμ), average polarizability, and NLO properties.
2. Computational Details and Modeling Approach
The Gaussian 09 package26 was employed to optimize the geometry of the NBD-Si (i: 1–4) molecular structures. DFT calculations were performed in polar methanol (CH3OH) using the IEFPCM solvent model27 and the hybrid B3LYP functional28,29 with the 6-31g(d,p) basis set.30
Gauss View 5.0.831 was utilized to visualize and analyze the output files. The stable molecular structure of each molecule was next employed to extract the energy levels of HOMO (εHOMO) and LUMO (εLUMO), dipole moment (μ), and DOS spectrum. Therefore, the TD-DFT method32 was employed to predict the electronic absorption spectra and the obtained results were subsequently compared with other DFT methods, including the Coulomb-attenuated hybrid exchange-correlation functional (CAM-B3LYP),33 the Tamm-Dancoff approximation (TDA),34 and the hybrid B3PW91 exchange-correlation functionals.35 We performed QTAIM analysis to investigate the noncovalent interactions (NCIs). The Multiwfn_3.7 software36 has been utilized to generate the electron localization function (ELF), the localized orbital locator (LOL), and the RDG analyses. We also used the Visual Molecular Dynamics (VMD) program37 to display the iso-surfaces. The Hirshfeld (HS) surface was computed using the Crystal Explorer 17.5 program.38 To explore the NLO NBD-Si characteristics, electric dipole moment, polarizability (α), and static first and second hyperpolarizabilities (β and γ) were computed in methanol solvent.
3. Results and Discussion
3.1. Geometric and Energetic Parameters
First of all, the geometries of the NBD-Si (i: 1–4) compounds in methanol were optimized in both their ground (GS) and excited (ES) states employing the B3LYP and CIS39 approaches with the 6-31g(d,p) basis set, respectively. The geometry-optimized molecular structures are displayed in Figure 1, and the selected geometrical parameters, including bond lengths and dihedral angles, are listed in Table 1.
Figure 1.

Optimized geometric structures of investigated compounds revealing NCIs.
Table 1. Geometric Parameter Obtained in the Ground (S0) Statea.
| bond
length (Å) |
dihedral angle (°) | ||||
|---|---|---|---|---|---|
| structures | L1 | L2 | ϕ | dipole moment (μ, D) | Δμ (D) |
| NBD-S1 | 1.757 (1.695) | 1.807 (1.780) | 2.98 (27.35) | 13.005 (14.779) | 1.774 |
| NBD-S2 | 1.760 (1.699) | 1.799 (1.776) | 1.41 (27.95) | 8.083 (9.191) | 1.108 |
| NBD-S3 | 1.747 (1.695) | 1.838 (1.821) | 0.035 (0.237) | 12.785 (14.905) | 2.120 |
| NBD-S4 | 1.750 (1.697) | 1.840 (1.823) | 4.56 (3.37) | 12.034 (13.805) | 1.771 |
The values in parentheses correspond to excited (S1) state.
According to structural data, the optimized structures are found to mostly contain three forms of noncovalent contacts, namely, N---H, O---H, and N---O bond interactions, which are responsible for their stability. The computed dipole moment of the first excited state is also reported in Table 1. The geometrical parameters associated with these noncovalent contacts in GS and ES states are illustrated in Figure 1. All of these values are found to be shorter than the sum of van der Waals (VdW) radii [2.75 Å (N---H), 2.72 Å (O---H), and 3.07 Å (N--O)]. It is clear that the values of these short contacts decrease upon excitation.
Table 1 highlights that the variation in the dihedral angle ϕ and the electric dipole moment are the main differences between GS and ES. The value of dihedral angle ϕ, calculated between the NBD moiety and the sulfide group, in the ES is larger than that calculated in the GS for NBD-Si (i: 1–3). Ultimately, all of the optimized structures show that the title compounds involve twisted geometry. As shown in Table 1, the dipole moment in each system was found to be higher in the S1 state than that in the S0 one, increasing by 1–3 D. Additionally, we have looked at the bridge length variations (L1 and L2) of the investigated compounds. As seen, the changes in electron delocalization in the molecular structure lead to only a small difference in bond lengths between the GS and ES states.
Using the hybrid functional B3LYP in conjunction with the empirical dispersion correction D3 (B3LYP-D3),40 the structural parameters for all optimized molecular structures remained unchanged (see Table S1, Supporting Information).
3.2. FMOs and Quantum Theoretical Parameters
The frontier molecular orbitals (FMOs), formed mainly by the HOMO/LUMO iso-surfaces, are the significant orbitals in a molecule. The FMOs play an important role in judging optical and electrical performance, in addition to their chemical properties.41−45 The ability to donate electrons is qualified by HOMOs acting as electron donors, and the aptitude to accept electrons is characterized by LUMOs acting as electron acceptors.46Figure 2 depicts the HOMOs and LUMOs contour graphs, with the positive phase colored red and the negative phase colored green.
Figure 2.

HOMO–LUMO contour plots of the studied compounds with gap energy.
It appears that the FMOs distribution in the investigated compounds is almost the same. It is found that the electronic charge density distribution of HOMO is located all over the molecular structures, while the electronic charge density distribution of LUMO is mainly focused on the NBD moiety. This reveals the presence of an ICT inside the studied molecules from sulfide groups to the NBD acceptor moiety. The FMO’s molecular characteristics-based chemical descriptors, including εHOMO, εLUMO, energy gap (Eg), ionization potential (IP), electron affinity (EA), global hardness (η), chemical potential (μ), electronegativity (χ), and electrophilicity (ω), were extracted from calculations, and results are presented in Table 2. The energy gap (Eg) serves as a key indicator of a molecule’s stability and chemical reactivity.47,48 According to our findings, the energy gaps of the investigated molecules ranged from 3.255 to 3.354 eV. The compounds NBD-S2 and NBD-S1 demonstrated the highest and lowest values, respectively. The compound NBD-S2 is characterized by a lower reactivity and greater stability, indicating higher chemical hardness. On the other hand, the compound NBD-S1 is identified as highly reactive, less stable, and soft. Band gap energy is recognized as a significant factor that offers valuable information regarding the photovoltaic efficiency of materials used in photovoltaic applications.49,50
Table 2. Calculated Electronic Parameters and Derived Quantum Chemical Parameters for the Investigated Molecules (EF Represents the Fermi Energy).
| energy
levels |
chemical
reactivity descriptors |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compounds | εHOMO (eV) | εLUMO (eV) | EF (eV) | Eg (eV) | μ (eV) | η (eV) | ω (eV) | χ = –μ (eV) | IP (eV) | EA (eV) |
| NBD-S1 | –6.615 | –3.359 | –4.987 | 3.255 | –4.987 | 1.628 | 7.638 | 4.987 | 6.343 | 3.679 |
| NBD-S2 | –6.742 | –3.388 | –5.065 | 3.354 | –5.065 | 1.677 | 7.648 | 5.065 | 6.574 | 3.745 |
| NBD-S3 | –6.607 | –3.332 | –4.969 | 3.275 | –4.969 | 1.637 | 7.540 | 4.969 | 6.473 | 3.511 |
| NBD-S4 | –6.638 | –3.345 | –4.991 | 3.293 | –4.991 | 1.646 | 7.566 | 4.991 | 6.500 | 3.530 |
Additionally, the neutral, cation, and anion-optimized
structures
are used to compute EA and IP, which elucidate the barrier injection
energies of holes and electrons. The readings of EA and IP are, respectively,
in the ranges of 3.66–3.74 and 6.34–6.56 eV. The best
electron donor is the molecule NBD-S1, which has the lowest
ionization value (IP = 6.342 eV). Although the best electron acceptor
is NBD-S2, which has the highest affinity (EA = 3.744 eV).
Moreover, using εHOMO and εLUMO,
chemical potential (μ), global hardness (η), and electronegativity
(χ) can be determined as follows:  ;
;  ; χ = −μ. Besides this,
the electrophilicity (ω)51,52 has been computed using
both chemical potential and global hardness
; χ = −μ. Besides this,
the electrophilicity (ω)51,52 has been computed using
both chemical potential and global hardness 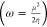 . A molecule with a low electrophilicity
is regarded as a strong nucleophile, whereas a molecule with a high
electrophilicity is regarded as a strong electrophile. The results
indicate that NBD-S1 is a good nucleophile since it has
lower values of (ω), whereas NBD-S4 is a good electrophile.
. A molecule with a low electrophilicity
is regarded as a strong nucleophile, whereas a molecule with a high
electrophilicity is regarded as a strong electrophile. The results
indicate that NBD-S1 is a good nucleophile since it has
lower values of (ω), whereas NBD-S4 is a good electrophile.
Among the electronic properties of the materials under study, εHOMO and εLUMO are particularly crucial, as they facilitate the calculation of both Eg and EF energies. The obtained values are listed in Table 2. The B3LYP-D3/6-31g(d,p) level of theory was also applied to extract electronic parameters for the investigated molecules (see Table S2, Supporting Information). As shown in Table S2, no appreciable changes have been detected.
3.3. Electronic Spectra Analysis of the Investigated NBD-Si Molecules
The optical absorption (UV–vis) and the emission properties of NBD-Si molecules, in methanol, are given in Tables 3 and 4, respectively, while their related electronic spectra are shown in Figure 3. The most intense absorption appears at 409–415 nm. A bathochromic shift of the absorption maxima is evident after functionalization with suitable sulfide groups, such as 2-pyridinethiol. In all cases, the maxima originate from HOMO–LUMO electronic transitions, with significant contributions of 98%. Based on FMO’s distribution analysis, these electronic transitions are associated with the π → π* type.
Table 3. Vertical Wavelength (λ, nm) and the Oscillator Strengths (f) Contributing to S0 → S1 Electronic Transitions for Investigated Molecules, Computed by the TD-B3LYP/6-31g(d,p) Method in Methanol Solvent.
| compounds | λabs,expmax (nm) | fmax | major contribs (%) | minor contribs (%) |
|---|---|---|---|---|
| NBD-S1 | 271 | 0.0095 | H – 5 → L (94%) | H → L + 4 (3%) |
| 306 | 0.1295 | H → L + 1 (96%) | ||
| 426 | 0.3622 | H → L (98%) | ||
| λabs,expmax = 407 nm25 | ||||
| NBD-S2 | 265 | 0.0549 | H – 6 → L (88%) | H → L + 4 (6%) |
| 300 | 0.1246 | H → L + 1 (96%) | ||
| 409 | 0.3611 | H → L (98%) | ||
| λabs,expmax = 400 nm25 | ||||
| NBD-S3 | 265 | 0.0464 | H – 5 → L (81%) | H – 1 → L + 1 (8%), H → L + 2 (7%) |
| 304 | 0.1288 | H → L + 1 (97%) | ||
| 415 | 0.3635 | H → L (98%) | ||
| λabs,expmax = 414 nm25 | ||||
| NBD-S4 | 265 | 0.0474 | H – 6 → L (86%) | H → L + 3 (6%) |
| 305 | 0.128 | H → L + 1 (97%) | ||
| 415 | 0.3733 | H → L (98%) | ||
| λabs,expmax = 411 nm25 | ||||
Table 4. Vertical Wavelength (λ, nm) and the Oscillator Strengths (f) Contributing to S1 → S0 Electronic Transitions of Investigated Molecules, Calculated by the TD-B3LYP/6-31g(d,p) Method in Methanol Solvent. The excitation wavelengths varied from 366 to 380 nm.
| compounds | λPL,expmax (nm) | fmax | major contribs (%) | minor contribs (%) |
|---|---|---|---|---|
| NBD-S1 | 292 | 0.1476 | L → H – 4 (54%), L → H – 2 (23%) | L + 4 → H (8%) |
| 366 | 0.0646 | L + 1 → H (80%) | L + 1 → H – 1 (13%) | |
| 515 | 0.5543 | L → H (88%) | L → H – 1 (8%) | |
| λPL,expmax = 522 nm25 | ||||
| NBD-S2 | 287 | 0.0226 | L + 2 → H – 3 (79%) | L → H – 4 (3%), L + 10 → H – 3 (8%) |
| 366 | 0.0625 | L + 1 → H (90%) | L + 1 → H – 2 (4%) | |
| 513 | 0.5486 | L → H (95%) | ||
| λPL,expmax = 521 nm25 | ||||
| NBD-S3 | 290 | 0.1458 | L → H – 3 (73%) | L + 3 → H (12%) L → H – 4 (8%) |
| 367 | 0.059 | H → L + 1 (95%) | ||
| 507 | 0.5614 | L → H (97%) | ||
| λPL,expmax = 516 nm25 | ||||
| NBD-S4 | 291 | 0.1482 | L → H – 3 (73%), | L + 4 → H (12%), L → H – 5 (5%), L → H – 4 (3%) |
| 365 | 0.0558 | L + 1 → H (95%) | ||
| 503 | 0.5636 | L → H (97%) | ||
| λPL,expmax = 542 nm25 | ||||
Figure 3.

Electronic spectra (UV–vis and PL) achieved at the TD-B3LYP/6-31g(d,p) level of theory of materials.
In fluorescence spectroscopy, the difference in spectral position between the first absorption band’s maximum and the fluorescence emission’s maximum is identified as the Stokes shift (SS), which can be expressed in wavelength: SS = λmaxPL λmaxabs. In some cases, significant reabsorption effects due to the overlap of absorption and emission spectra limit practical applications. To address this, the development of D–A or D-π–A push–pull systems aims to minimize the overlap between absorption and emission spectra, effectively reducing reabsorption losses.53,54 Herein, NBD-sulfide compounds exhibit a large SS up to 85 nm in polar methanol solvent (see Figure 3). The ICT phenomenon is an alternative reason for the large SS. In summary, the extracted values from the experimental data obtained through UV/visible and fluorescence spectroscopic techniques (Tables 3 and 4) align well with the values derived from simulated spectra. Furthermore, the change in dipole moment relaxes the polar solvent around the excited molecule, leading to the lowest energy state.55 This is supported by assignments previously reported by Heberer et al.56 for NBD derivatives, where the lower energy band is attributed to an ICT between the sulfide electron donor and the nitro electron-withdrawing groups of the NBD moiety.
To enhance the result validation, we employed three DFT methods (CAM-B3LYP, TDA, and B3PW91) to predict UV–vis properties for the studied molecules.
The computed results were compared with the available experimental data (see Table S3, Supporting Information). Overall, we consider that the TD-B3LYP/6-31g(d,p) level of theory can be proposed to obtain better results at a reasonable cost.
3.4. DOS Analysis
Nearby orbitals within the boundary area may have quasi-degenerate energy levels, so it may not be practical to use only HOMO and LUMO to define the FMOs.57 Therefore, the DOS spectra for the investigated compounds were generated by the Gauss Sum 3.0 software (the full width at half-maximum (fwhm) is 0.3 eV).58Figure 4 shows the obtained DOS spectra.
Figure 4.

DOS spectra obtained at the B3LYP/6-31g(d,p) level, labeled as (a–d), corresponding to NBD-Si (i = 1–4).
Virtual orbitals, also known as acceptor orbitals, are unoccupied, while filled orbitals are termed donor orbitals. The DOS exhibits a positive value when indicating bonding interactions, a negative value for antibonding interactions, and zero when there are no bonding interactions present.59,60 Furthermore, the DOS spectra directly provide the energy gap.
3.5. RDG Analysis
The molecular stability can be attributed to the presence of relatively weak intra- and intermolecular interactions that can be identified and studied by the NCI methodology. This method uses RDG to provide further evidence for NCIs. The RDG is a fundamental dimensionless parameter based on the relationship between the electron density (ρ) and its derivative,61,62 expressed as
| 1 |
The result of the product of the electron density ρ(r) and the sign of the second-lowest eigenvalues of the electron density Hessian matrix (λ2(r)) is highly significant in anticipating the nature of interaction. The attractive, repulsive, and intermediate interactions correspond to sign(λ2) × ρ < 0 and sign(λ2) × ρ > 0 and sign(λ2) × ρ ≈ 0.63 Two-dimensional (2D) scatter plots and the 3D-RDG isosurface densities of studied molecules in the GS are indicated in Figure 5. Correspondingly, as shown in the RDG plots, there are three kinds of spikes in the zone between −0.05 and 0.05 a.u. The blue regions represent attractive interactions like H-bonds, the red regions signify repulsive interactions, such as steric effects, and the green regions take place for very weak interactions, typically VdW ones.64 Looking at the NCI-RDG graphs, the van der Waals interactions are clearly shown within our NBD molecules. As well, the detection of a blue surface highlights the existence of H-bond interactions. Moreover, red areas are positioned in the center of the rings, demonstrating the effect of steric repulsion.
Figure 5.

RDG scatter plots (left) and NCI plots (right) of the investigated materials in GS.
3.6. ELF and LOL Topological Analyses
ELF and LOL analyses, developed and explained by Silvi and Sain,65 both conduct covalent bonds analysis, revealing areas in the molecular space with high electron pairing potential. ELF and LOL have comparable chemical maps since they depend on the kinetic energy density. Furthermore, ELF is rooted in the analysis of electron pair density, whereas LOL identifies and characterizes the gradients of localized orbitals.66 The ELF and LOL color-filled maps generated by Multiwfn software for the S0 and S1 states of the investigated materials are shown in Figures 6 and 7, respectively. These maps range from 0 to 1. An ELF value greater than 0.5 corresponds to the area in which the bonding and nonbonding electrons are localized, whereas a value under 0.5 indicates a delocalized electronic region.67 LOL values exceed 0.5 in areas where the electron density is controlled by electron localization. Several colors are shown in the ELF and LOL maps. High ELF or LOL values are visually represented by the red color, indicating the presence of localized electrons surrounding hydrogen atoms. The delocalized electron cloud surrounding C, N, S, and O atoms is visually represented by the blue. From the LOL maps, it is evident that the central region of the hydrogen atom appears white, indicating an electron density that surpasses the upper limit of the color scale (0.8). In addition, the red regions between C–C, C–N, C–S, and N–O atoms present covalent interactions with a high LOL value.
Figure 6.

Color-filled map of the ELF (top) and LOL (bottom) of studied molecules in their S0 states.
Figure 7.

Color-filled map of (top) and LOL (bottom) of the studied molecules in their S1 states.
3.7. MEP Analysis
The molecular electrostatic potential (MEP) describes the charge distribution around the molecular structure generated by its nuclei and electrons. The distribution of charge can be used to determine how molecules interact with each other.68,69 It also serves as a tool for identifying reactive sites for nucleophilic and electrophilic reactions.70 The nucleophilic sites assume a high attraction, while the electrophilic sites show a high repulsion. The MEP 3D plots of all structures in both S0 and S1 states were computed by employing the B3LYP/6-31(d,p) level, as illustrated in Figure 8. The figure presents the electrostatic potential values in false colors, progressing in order: red < orange < yellow < green < blue. The positive (blue) areas of the MEP are the preferred nucleophilic attack sites, whereas the negative (red-yellow) areas are the preferred electrophilic attack sites, and green colors are neutral regions. As shown in the MEP three-dimensional (3D) graphs, areas with negative potential are over the electronegative atoms (nitrogen and oxygen atoms) of NBD moieties and the sulfide groups, which support electrophilic attacks.
Figure 8.

MEP 3D plots of studied molecules.
The positive potential zones are over the hydrogen atoms, favoring nucleophilic attack. For all studied molecules, the NBD moieties are very evident in the red-yellow region, confirming the acceptor properties of this group. Consequently, the presence of electrophile and nucleophilic attacks in these molecules supports their push–pull (D–A) nature.
3.8. QTAIM Analysis
The QTAIM analysis, developed by Bader and co-workers,71 identifies the nature and type of intra- and intermolecular H-bonds, including NCIs,72 in a topological manner. According to QTAIM theory, the bond path (BP) is the link between an interacting atom and the saddle point where the gradient of the electron density is zero, also called the critical point (CP). The QTAIM graphs are obtained using Multiwfn software and are illustrated in Figure 9. As shown in the graph, three types of critical points can be identified: a large red spot (+3,–3) represents the nuclear critical point (NCP), a small orange spot (+3,–1) indicates the bond critical point (BCP), and a yellow spot (+3,+1) is the ring critical point (RCP). The BCPs offer an effective measure of the distance between atoms in a molecular system.73−75 Mainly, hydrogen bond interactions can be efficiently characterized and quantified by the use of a variety of topological parameters: electron density ρ (r) and its Laplacian ∇2ρ (r), kinetic energy G (r), potential energy V (r), kinetic energy of Hamiltonian H (r) = G (r) + V (r), and the binding energy BE, calculated by means of the expression provided by Espinosa et al.:76 BE = V (r)/2. Among them, the most significant are the electron density ρ (r) and its Laplacian ∇2ρ (r), which are used to identify the nature of the bond or interaction. A large ρ (r), greater than 0.20 a.u. and negative ∇2ρ (r) values show a covalent bond, whereas small ρ (r), less than 0.10 a.u. and negative ∇2ρ (r) values signify a closed-shell interaction (ionic, H-bond, VdW). In Figure 9, solid lines indicate CBs, while NCIs are shown as dashed lines. Specifically, H-bond interactions can be classified into three categories.77 First, when ∇2ρ (r) > 0 and H (r) > 0, it denotes weak H-bonds. Second, when ∇2ρ (r) > 0 and H (r) < 0, it signifies moderate H-bonds. Lastly, when ∇2ρ (r) < 0 and H (r) < 0, it indicates the strongest H-bonds.
Figure 9.

QTAIM molecular graphics with examples of BCPs for the investigated molecules.
Table 5 presents the topological parameter values at the CBs of various NCI examples. In the QTAIM molecular graphs, NBD-S1 exhibits two BCPs representing N–O and N–H interactions, while NBD-S2 has a single N–O interaction. NBD-S3, in comparison to NBD-S1, features both N–H and N–O interactions. Alternatively, there are four kinds of interactions obtained in the last compound: O–H, N–H, N–O, and C–H interactions. The ρ(r) values of bond critical points vary from 0.0079 au to 0.012 a.u., and their Laplacians range from 0.030 to 0.050 a.u., which are similar to the values offered by Koch and Popelier.78 Also, the positive values of the energy density H (r) justify the weak H-bond. In addition, the band energy appears between −5.382 and −12.602 kJ/mol. The N–H and O–H interactions in NBD-S3 and NBD-S4, respectively, are considered the strongest ones, with BE = 12.602 kJ/mol.
Table 5. Topological Parameters Computed for Investigated Compounds at BCPs.
| compound | interaction | ρ (r) | ∇2ρ (r) | G (r) | V (r) | H (r) | BE (kJ/mol) |
|---|---|---|---|---|---|---|---|
| NBD-S1 | 1 | 0.010 | 0.036 | 0.0075 | –0,0058 | 0.0016 | –7.613 |
| 2 | 0.012 | 0.050 | 0.0110 | –0.0095 | 0.0015 | –12.471 | |
| NBD-S2 | 1 | 0.012 | 0.050 | 0.0110 | –0.0095 | 0.0015 | –12.471 |
| NBD-S3 | 1 | 0.010 | 0.038 | 0.0077 | –0.0059 | 0.0018 | –7.745 |
| 2 | 0.012 | 0.050 | 0.0110 | –0.0096 | 0.0014 | –12.602 | |
| NBD-S4 | 1 | 0.0079 | 0.031 | 0.00603 | –0.0041 | 0.0019 | –5.382 |
| 2 | 0.0083 | 0.030 | 0.0065 | –0.0053 | 0.0012 | –6.957 | |
| 3 | 0.010 | 0.038 | 0.0078 | –0.0059 | 0.0018 | –7.745 | |
| 4 | 0.012 | 0.049 | 0.0110 | –0.0096 | 0.0014 | –12.602 |
3.9. Hirshfeld Surface (HS) Analysis
The analysis of the Hirshfeld surface (HS)79−81 offers insights into the characteristics of molecular surfaces and introduces a novel method to visualize intermolecular interactions. HS analysis is utilized to examine intermolecular interactions involved in the stabilization of crystal packing for the studied compounds.82,83 This visualization is achieved through color coding, which distinguishes short- or long-term contacts.84 HS may be mapped with various properties, such as dnorm, curvedness, and shape index. Figure 10(i–iii) displays the molecular HS (dnorm, curvedness, and shape index) of the studied molecules. The Hirshfeld molecular surfaces are produced using a standard (high) surface resolution. In the case of NBD-S1, the 3D dnorm surfaces are mapped on a fixed color scale ranging from −1.1876 Å (red) to 5.9755 Å (blue). For NBD-S2, the mapping ranges from −1.1858 Å (red) to 6.038 Å (blue). Similarly, for NBD-S3, the range is from −1.2575 Å (red) to 6.4383 Å (blue), and for NBD-S4, it is from −1.2364 Å (red) to 5.8931 Å (blue). Curvedness is mapped within the range of −4.0 to 4.0, while the shape index is mapped in the color range of −1.0 to 1.0. The dnorm attributed is a symmetric function that evaluates the surface distances (di and de) between nuclei inside and outside the HS area, respectively, in relation to their respective van der Waals (vdW) radii. The red and blue regions, corresponding to the negative and positive dnorm values, display shorter and longer contacts, respectively. However, the white color designates the contacts around the VdW radii (dnorm values near zero). On the dnorm surface, the deep red spots indicate intermediate contacts involved in H-bonds.85 The red spots in Figure 10(i) are assigned to N–H···S, N–H···O, and C–H···O hydrogen bonds for compounds NBD-S1 and NBD-S2, C–H···S, C–H···N, N–H···O, and C–H···O H-bonds for compounds NBD-S3 and NBD-S4. Additionally, the presence of red/blue triangles and the expansive green region in the curvedness triangles within the shape index serve as indicators of C–H···π and π···π stacking interactions taking place among the molecules.
Figure 10.


Hirshfeld surfaces mapped over (i) dnorm, (ii) shape index, (iii) curvedness, and (iv) percentage contributions of the various intermolecular contacts and 2-D fingerprint plots of each studied compound.
The 2D depiction of the HS is commonly referred to as a fingerprint,86 as it has the ability to unveil additional insights into the intermolecular contacts. The 2D fingerprint plots can be decomposed to highlight the interactions and the area corresponding to each of these interactions. The HS surfaces and the 2D fingerprint plots have been generated using the Crystal Explorer program.87 The decomposed 2D fingerprint plots of all compounds and all major and minor intermolecular interactions with the percentage of contributions are shown in Figure 10(iv). In this study, the 2D fingerprint plots show various types of interactions that ensure the structure’s cohesion. The overall 2D fingerprint plot showed that the intermolecular interactions of the investigated compounds were dominated by H···O/O···H, H···H, C···N/N···C, C···H/H···C, O···O, and C···O/O···C. The major contribution of Hirschfeld’s total surface in all compounds is attributed to H···O/O···H interactions with 28.9, 26.1, 23.9, and 31.4% for NBD-S1, NBD-S2, NBD-S3, and NBD-S4, respectively. These interactions are characterized by one large spike in the middle of the scattered points. The interactions with the second contribution, however, vary depending on the molecule. NBD-S1 and NBD-S2 exhibit interactions such as H···H, C···N/N···C, C···O/O···C, and C···C. On the other hand, NBD-S3 and NBD-S4 involve interactions such as C···O/O···C, C···H/H···C, O···O, and H···H. The analysis of Hirshfeld surfaces also indicates the existence of additional weak intermolecular interactions that make modest contributions to the surface area (C···S/S···C, N···O/O···N. and H···/S···H).
3.10. NLO Responses for Investigated Compounds
Polarizability and hyperpolarizability measurements can be used to explore the NLO characteristics of the investigated molecules. The significance of these two parameters was crucial for defining ICT properties.88,89 By means of the B3LYP/6-31g(d,p) level of theory, we evaluated the static dipole moment (μ), mean polarizability (α0), polarizability anisotropy (Δα), and static first and second hyperpolarizabilities (β and γ) of the investigated molecules. The relevant expressions used for the calculation are as given below90−93
| 2 |
| 3 |
| 4 |
| 5 |
And,
 |
6 |
where αxx, αyy, and αzz are the polarizability tensor components, and βxxx, βxyy, βxzz, βxxy, βyyy, βyzz, βxxz, βzyy, βzzz and γxxxx, γyyyy, γzzzz, γxxyy, γyyzz, and γzzxx are the first- and second-order hyperpolarizability tensor components along the x, y, and z axes, respectively. The calculated values are given in Table 6. These values have been converted into electrostatic units (for α, 1 a.u. = 0.1482 × 10–24 esu; for β, 1 a.u. = 8.6391 × 10–33 esu and for γ, 1 a.u. = 0.000504 × 10–36 esu).
Table 6. Computed Electric Dipole Moment (μ), Polarizability (α), 1st- and 2nd-Order Hyperpolarizability (β) and (γ), Respectively, for the Studied Compounds.
| polarizability, α × 10–24 esu | 2nd-order hyperpolarizability, γ × 10–36 esu | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| NBD-S1 | NBD-S2 | NBD-S3 | NBD-S4 | NBD-S1 | NBD-S2 | NBD-S3 | NBD-S4 | ||
| αXX | 40.949 | 39.721 | 36.733 | 40.046 | γXXXX | 43.838 | 26.617 | 23.898 | 25.751 |
| αYX | –2.097 | –0.311 | 0.951 | 1.826 | γXXYX | –0.994 | –0.243 | 2.322 | 5.444 |
| αYY | 17.352 | 20.334 | 17.230 | 19.470 | γXXYY | 0.533 | 1. 497 | 1.619 | 3.658 |
| αZX | 7.718 | 6.817 | 40.661 | 3.9125 | γYXYY | –0.627 | –0.118 | 0.316 | 2.089 |
| αZY | 3.148 | 0.532 | 0.438 | –1.159 | γYYYY | 1.725 | 2.974 | 1.530 | 2.927 |
| αZZ | 52.684 | 48.045 | 52.402 | 55.105 | γXXZX | 34.132 | 11.633 | 10.861 | 8.619 |
| α0 = αISO | 36.995 | 36.034 | 35.455 | 38.207 | γXXZY | –1.043 | –0.179 | 1.844 | 2.542 |
| Δα = αANISO | 34.541 | 27.333 | 31.375 | 31.936 | γYXZY | –0.465 | 0.699 | 0.730 | 1.281 |
| 1st-order hyperpolarizability, β × 10–30 esu | γYYZY | 0.806 | 0.177 | 0.098 | 0.368 | ||||
| βXXX | –1.230 | –0.455 | 0.267 | –0.040 | γXXZZ | 42.439 | 8.534 | 7.033 | 5.798 |
| βXXY | –0.152 | –0.035 | 0.206 | 0.462 | γYXZZ | –2.468 | –0.589 | 2.572 | 4.280 |
| βYXY | –0.126 | 0.045 | –0.991 | –0.551 | γYYZZ | –1.362 | –1.004 | –0.617 | 1.866 |
| βYYY | –0.350 | –0.014 | 0.611 | –0.315 | γZXZZ | 111.977 | 49.662 | 42.508 | 42.218 |
| βXXZ | –2.379 | –2.923 | –1.778 | –1.865 | γZYZZ | –5.407 | –2.173 | 5.666 | 5.970 |
| βYXZ | –0.339 | –0.072 | 0.207 | 0.560 | γZZZZ | 520.226 | 382.615 | 365.442 | 391.169 |
| βYYZ | –0.882 | –0.622 | –1.307 | –0.909 | γ0 = γ|| | 129.800 | 86.051 | 81.387 | 88.497 |
| βZXZ | 7.168 | 3.798 | 3.628 | 3.559 | γ⊥ | 43.267 | 28.684 | 27.129 | 29.500 |
| βZYZ | –0.853 | –0.346 | 0.639 | 1.217 | electric dipole moment (debye) | ||||
| βZZZ | 87.016 | 74.188 | 79.671 | 79.848 | μx | 8.396 | –4.646 | –7.934 | –6.592 |
| || (Z) | 50.252 | 42.386 | 45.951 | 46.245 | μy | 1.798 | –0.004 | 4.362 | 2.787 |
| ⊥ (Z) | 16.750 | 14.129 | 15.317 | 15.415 | μz | 9.767 | –6.615 | –9.027 | –9.674 |
| |βvect| | 66.466 | 59.752 | 75.647 | 63.268 | μtot | 13.005 | 8.084 | 12.785 | 12.034 |
| β0 | 83.967 | 70.725 | 76.654 | 77.143 | |||||
| |βvect|/β0 | 0.791 | 0.844 | 0.722 | 0.820 | βvec = ∑iβiμi/μ [i = x,y,z]100 | ||||
| θ (deg) | 37.74 | 32.45 | 43.80 | 34.93 | |||||
The calculated electric dipole moments are found to be 13.005, 8.084, 12.784, and 12.034 D for NBD-S1, NBD-S2, NBD-S3, and NBD-S4, respectively. It is noteworthy that the greatest dipole moment value is observed for compound NBD-S1. The static polarizability (α0), values of the investigated molecules increase in the subsequent order NBD-S3 < NBD-S2 < NBD-S1 < NBD-S4. Similarly, we can see that the anisotropy of polarizability (αaniso) increases in the order: NBD-S2 < NBD-S3 < NBD-S4 < NBD-S1. The hyperpolarizability is a key property of the NLO response, with β0 ranging from 70.725 × 10–30 to 83.967 × 10–30 esu. Additionally, it is noteworthy that the NBD-S1 compound exhibits the highest values for both first- and second-order hyperpolarizabilities, whereas the NBD-S2 compound shows the lowest values in this regard. Notably, the first hyperpolarizability demonstrates a direct correlation with linear polarizability values and an inverse correlation with energy gap values.94−97 In this context, there is a strong correspondence between the βtot values and the observed patterns in Eg values, with the compound having the smallest band gap (3.255 eV) demonstrating the highest βtot value (83.967 × 10–30 esu).
In this section, urea was chosen as a reference because there was no experimental standard for the NLO properties of the molecules of interest (μurea = 1.3732 D, αurea = 5.0477 × 10–24 esu, βurea = 372.8 × 10–33 esu, and γurea = 48 × 10–36 esu).98,99 The first-order hyperpolarizability of all investigated compounds is significantly greater (i.e., ≥70 times) than that of the standard urea, and NBD-S1 exhibits the best NLO characteristics. These results thus confirm that the studied molecules possess good NLO parameters and may be a good candidate for applications in optical devices.
According to Table 6, βtot mainly depends on the βZZZ tensor component, while also showing supplementary contributions from the βZXZ tensor component. Furthermore, based on the collective results presented in Tables 1 and 2, it is evident that compounds with a narrow energy gap and a lower difference in dipole moment between the S0–S1 states (Δμ) will exhibit higher first-order hyperpolarizability.
Conversely, the relationship
between βvect and
βtotal is acknowledged to provide significant information
regarding the direction of the ICT, as the following relationship
holds: 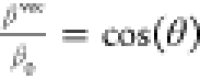 ,100 where θ
characterizes the orientation between the vector formed by the components
of βvec and the dipole moment vector. Note that a
ratio
,100 where θ
characterizes the orientation between the vector formed by the components
of βvec and the dipole moment vector. Note that a
ratio 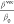 approaching unity means maximum charge
delocalization.101 This phenomenon occurs
due to the dominance of one of the first hyperpolarizability components,
which significantly contributes to the overall first hyperpolarizability.
As a result, a unidirectional alignment parallel to the molecular
dipole moment of ICT takes place. Here, the calculated angle θ
is in the range of 32 to 44°. This confirms the twisted ICT in
all of the investigated NBD-Si molecules.
approaching unity means maximum charge
delocalization.101 This phenomenon occurs
due to the dominance of one of the first hyperpolarizability components,
which significantly contributes to the overall first hyperpolarizability.
As a result, a unidirectional alignment parallel to the molecular
dipole moment of ICT takes place. Here, the calculated angle θ
is in the range of 32 to 44°. This confirms the twisted ICT in
all of the investigated NBD-Si molecules.
4. Conclusions
In this study, we used the TD-B3LYP/6-31g(d,p) level of theory to explain and predict the optical responses of four examples of NBD derivatives in a polar methanol solvent. The studied compounds, labeled NBD-Si (i = 1–4), consist of NBD and sulfide moieties. These molecules absorb within the wavelength range of 250–500 nm and all emit green fluorescence in the 503–515 nm range. They also exhibited large Stokes shifts, up to 500 meV, in polar methanol solvent. We concluded that DFT and its time-dependent extension (TD-DFT) can predict the optical properties of the studied molecules. Additionally, we demonstrate that the optical properties of these investigated systems can be systematically tuned through the choice of substituents; including their nature, extended conjugated systems, and strength.
Findings from QTAIM, ELF, and RDG analyses indicate that the bonding between sulfide and NBD moieties is of electrostatic or hydrogen bond type. The DFT approach is also reliable for determining the NLO properties of the NBD-Si compounds.
In our NLO analysis, we found that small nonplanar NBD-Si molecules with intramolecular charge transfer (ICT) characteristics are excellent candidates for designing NLO materials. Specifically, dipole moment (μ), polarizability (α), and first-order hyperpolarizability (β) were larger in the case of NBD-S1 (μtot = 13.005 D, α0 = 36.995 × 10–24 esu, and β0 = 83.967 × 10–30 esu). The twisted intramolecular charge transfer (TICT) from the sulfide to the respective NBD was clearly elucidated in our NLO studies. Ultimately, we conducted Hirshfeld surface analysis and employed some topological approaches based on DFT, including NCI plots and QTAIM analysis, to further characterize and describe the NCIs.
Acknowledgments
This work was realized in the frame of the Program ERASMUS+ KA107, FSE 2014/2020 P.O.R. Sicilia Project No. 2020-1-IT02-KA107-078488, CUP J49J21003660006. The authors also extend their sincere appreciation to the Ministry of Higher Education and Scientific Research in Tunisia for the technical and financial support provided for this study, based on an agreement between the Ministry of Higher Education and Scientific Research in Tunisia and the American Chemical Society (ACS).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.3c04277.
Geometric parameters at the ground (S0) state for investigated compounds obtained with B3LYP-D3/6-31g(d,p) level of theory (Table S1); computed electronic parameters for investigated molecules at the B3LYP-D3/6-31g(d,p) level of theory, recorded in methanol solvent (Table S2), and selected DFT methods for calculation of vertical wavelength maxima (λ, nm) and their corresponding oscillator strengths (f) contributing to HOMO → LUMO electronic transitions for investigated molecules in methanol solvent (Table S3) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lippert E.; Lüder W.; Moll F.; Nägele W.; Boos H.; Prigge H.; Seibold-Blankenstein I. Umwandlung von Elektronenanregungsenergie. Angew. Chem. 1961, 73, 695–706. 10.1002/ange.19610732103. [DOI] [Google Scholar]
- Grabowski Z. R.; Rotkiewicz K.; Rettig W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4031. 10.1021/cr940745l. [DOI] [PubMed] [Google Scholar]
- Zhang S. Y.; Shu X.; Zeng Y.; Liu Q. Y.; Du Z. Y.; He C. T.; Zhang W. X.; Chen X. M. Molecule-based nonlinear optical switch with highly tunable on-off temperature using a dual solid solution approach. Nat. Commun. 2020, 11, 2752 10.1038/s41467-020-15518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaino T.; Tomaru S. Organic materials for nonlinear optics. Adv. Mater. 1993, 5, 172–178. 10.1002/adma.19930050304. [DOI] [Google Scholar]
- Ong B. S.; Wu Y.; Liu P.; Gardner S. High-performance semiconducting polythiophenes for organic thin-film transistors. J. Am. Chem. Soc. 2004, 126, 3378–3379. 10.1021/ja039772w. [DOI] [PubMed] [Google Scholar]
- Dalton L. R.; Sullivan P. A.; Bale D. H. Electric field poled organic electro-optic materials: state of the art and future prospects. Chem. Rev. 2010, 110, 25–55. 10.1021/cr9000429. [DOI] [PubMed] [Google Scholar]
- Georgieva I.; Aquino A. J.; Plasser F.; Trendafilova N.; Köhn A.; Lischka H. Intramolecular Charge-Transfer Excited-State Processes in 4-(N,N-Dimethylamino)benzonitrile: The Role of Twisting and the πσ* State. J. Phys. Chem. A 2015, 119 (24), 6232–6243. 10.1021/acs.jpca.5b03282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochman M. A.; Durbeej B.; Kubas A. Simulation and Analysis of the Transient Absorption Spectrum of 4-(N,N-Dimethylamino)benzonitrile (DMABN) in Acetonitrile. J. Phys. Chem. A 2021, 125 (39), 8635–8648. 10.1021/acs.jpca.1c06166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissa C.; Calabrese V.; Cavazzini M.; Grisanti L.; Terenziani F.; Quici S.; Painelli A. Tuning the Nature of the Fluorescent State: A Substituted Polycondensed Dye as a Case Study. Chem. - Eur. J. 2013, 19 (3), 924–935. 10.1002/chem.201202154. [DOI] [PubMed] [Google Scholar]
- Henson Z. B.; Müllen K.; Bazan G. C. Design strategies for organic semiconductors beyond the molecular formula. Nat. Chem. 2012, 4 (9), 699–704. 10.1038/nchem.1422. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wei Q.; Cao L.; Fries F.; Cucchi M.; Wu Z.; Scholz R.; Lenk S.; Voit B.; Ge Z.; Reineke S. Organic Light-Emitting Diodes Based on Conjugation-Induced Thermally Activated Delayed Fluorescence Polymers: Interplay Between Intra- and Intermolecular Charge Transfer States. Front. Chem. 2019, 7, 688 10.3389/fchem.2019.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjua M. R. S. A. Nonlinear optical response of a series of small molecules: quantum modification of π-spacer and acceptor. J. Iran. Chem. Soc. 2017, 14 (9), 2041–2054. 10.1007/s13738-017-1141-x. [DOI] [Google Scholar]
- Belyaev A.; Cheng Y. H.; Liu Z. Y.; Karttunen A. J.; Chou P. T.; Koshevoy I. O. Acceptor Phosphonium Fluorophores. Angew. Chem., Int. Ed. 2019, 58 (38), 13456–13465. 10.1002/anie.201906929. [DOI] [PubMed] [Google Scholar]
- Chung H. Y.; Oh J.; Park J.-H.; Cho I.; Yoon W. S.; Kwon J. E.; Kim D.; Park S. Y. Spectroscopic Studies on Intramolecular Charge-Transfer Characteristics in Small-Molecule Organic Solar Cell Donors: A Case Study on ADA and DAD Triad Donors. J. Phys. Chem. C 2020, 124 (34), 18502–18512. 10.1021/acs.jpcc.0c06741. [DOI] [Google Scholar]
- Rout Y.; Montanari C.; Pasciucco E.; Misra R.; Carlotti B. Tuning the Fluorescence and the Intramolecular Charge Transfer of Phenothiazine Dipolar and Quadrupolar Derivatives by Oxygen Functionalization. J. Am. Chem. Soc. 2021, 143 (26), 9933–9943. 10.1021/jacs.1c04173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav I. S.; Jang Y.; Rout Y.; Thomas M. B.; Misra R.; D’Souza F. Near-IR Intramolecular Charge Transfer in Strongly Interacting Diphenothiazene-TCBD and Diphenothiazene-DCNQ Push-Pull Triads. Chem. - Eur. J. 2022, 28 (25), e202200348 10.1002/chem.202200348. [DOI] [PubMed] [Google Scholar]
- Kaur S.; Dhoun S.; Depotter G.; Kaur P.; Clays K.; Singh K. Synthesis, linear and nonlinear optical properties of thermally stable ferrocene-diketopyrrolopyrrole dyads. RSC Adv. 2015, 5, 84643–84656. 10.1039/C5RA17977G. [DOI] [Google Scholar]
- Mori J.; Kaino T. Molecular orbitals of benzoxadiazole compounds with optical nonlinearities. Phys. Lett. A 1988, 127, 259–262. 10.1016/0375-9601(88)90692-5. [DOI] [Google Scholar]
- Kim T. D.; Luo J.; Cheng Z. J.; Shi Z.; Hau S.; Jang S. H.; Zhou X. H.; Tian Y.; Polishak B.; Huang S.; et al. Jen, Binary Chromophore Systems in Nonlinear Optical Dendrimers and Polymers for Large Electrooptic Activities. J. Phys. Chem. C 2008, 112 (21), 8091–9098. 10.1021/jp712037j. [DOI] [Google Scholar]
- Cheng Y. J.; Luo J.; Huang S.; Zhou S.; Shi Z.; Kim T. D.; Bale D. H.; Takahashi S.; Yick A.; Steier W.; et al. Donor-Acceptor Thiolated Polyenic Chromophores Exhibiting Large Optical Nonlinearity and Excellent Photostability. Chem. Mater. 2008, 20, 5047–5054. 10.1021/cm801097k. [DOI] [Google Scholar]
- Chérif I.; Raissi H.; Abieth K.; Gassoumi B.; Caccamo M. T.; Magazu S.; Haj Said A.; Hassen F.; Boubaker T.; Ayachi S. Exploration of intramolecular charge transfer in para-substituted nitrobenzofurazan: Experimental and theoretical analyses. Spectrochim. Acta, Part A 2023, 301, 122939 10.1016/j.saa.2023.12293. [DOI] [PubMed] [Google Scholar]
- Chérif I.; Raissi H.; Abieth K.; Gassoumi B.; Caccamo M. T.; Magazu S.; Haj Said A.; Hassen F.; Boubaker T.; Ayachi S. Computational studies on optoelectronic and nonlinear optical properties of para-substituted nitrobenzofurazan compound. Mater. Today Commun. 2023, 35, 106133 10.1016/j.mtcomm.2023.106133. [DOI] [Google Scholar]
- Chérif I.; Raissi H.; Abieth K.; Gassoumi B.; Caccamo M. T.; Magazu S.; Haj Said A.; Hassen F.; Boubaker T.; Ayachi S. Photophysical and nonlinear optical properties of para-substituted nitrobenzofurazan: A comprehensive DFT investigation. J. Photochem. Photobiol., A 2023, 443, 114850 10.1016/j.jphotochem.2023.114850. [DOI] [Google Scholar]
- Raissi H.; Chérif I.; Aribi I.; Ayachi H.; Haj Said A.; Ayachi S.; Boubaker T. Structure-property relationships in para-substituted nitrobenzofurazans: electrochemical, optical, and theoretical analysis. Chem. Pap. 2022, 76, 4059–4080. 10.1007/s11696-022-02150-y. [DOI] [Google Scholar]
- Bem M.; Radutiu A. C.; Voicescu M.; Caproiu M. T.; Draghici C.; Maganu M.; Enache C.; Constantinescu T.; Balaband A. T. 7-nitrobenzo[c] [1, 2, 5] oxadiazole (nitrobenzofurazan) derivatives with a sulfide group at the 4-position. Synthesis and physical properties. Rev. Roum. Biochim. 2018, 63 (2), 149–155. [Google Scholar]
- Frisch M. J.et al. Gaussian 09, revision A.01; Gaussian Inc.: Wallingford, 2009.
- Tomasi J.; Mennucci B.; Cancès E. The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. 1999, 464, 211–226. 10.1016/S0166-1280(98)00553-3. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Bouazzi D.; Chérif I.; Mehri A.; Touati H.; Caccamo M. T.; Magazù S.; Ayachi S.; Clacens J.-M.; Badraoui B. A joint experimental and theoretical study on Structural, Vibrational and morphological properties of newly synthesized nanocomposites involving Hydroxyapatite-alt-Polyethylene Glycol (HAP/PEG). J. Mol. Liq. 2023, 390, 123192 10.1016/j.molliq.2023.123192. [DOI] [Google Scholar]
- Dennington R.; Keith T.; Millam J.. GaussView, version 5.0.8; Semichem Inc.: Shawnee Mission KS, 2009.
- Petersilka M.; Gossmann U. J.; Gross E. K. U. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212–1215. 10.1103/PhysRevLett.76.1212. [DOI] [PubMed] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Rev. Lett. 2004, 393 (1–3), 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- Hirata S.; Head-Gordon M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. 10.1016/S0009-2614(99)01149-5. [DOI] [Google Scholar]
- Govindarajan M.; Ganasan K.; Periandy S.; Mohan S. DFT (LSDA, B3LYP and B3PW91) Comparative Vibrational Spectroscopic Analysis of α-Acetonaphthone. Spectrochim. Acta, Part A 2010, 76, 12–21. 10.1016/j.saa.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: a multifunctional Wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Spackman P. R.; Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Jayatilaka D.; Spackman M. A. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. 10.1107/S1600576721002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajaji S.; Zaier R.; Ayachi S. Difluorinated cyclopentadithiophene derivatives for green emitters in organic light-emitting diodes: A theoretical investigation. J. Phys. Chem. Solids 2021, 156, 110170 10.1016/j.jpcs.2021.110170. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg J. A Consistent and Accurate ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Durig J. R.; Little T. S.; Gounev T. K.; Gardner J. K.; Sullivan J. F. Infrared and Raman Spectra, Conformational Stability, Vibrational Assignment, and ab Initio Calculations of Chloromethyl Isocyanate. J. Mol. Struct. 1996, 375, 83–94. 10.1016/s0166-1280(96)91235-x. [DOI] [Google Scholar]
- Yoosefian M.; Ansarinik Z.; Etminan N. Density Functional Theory Computational Study on Solvent Effect, Molecular Conformations, Energies and Intramolecular Hydrogen BondStrength in Different Possible Nano-Conformers of Acetaminophen. J. Mol. Liq. 2016, 213, 115–121. 10.1016/j.molliq.2015.10.060. [DOI] [Google Scholar]
- Khalid M.; Ali A.; Jawaria R.; Asghar M. A.; Asim S.; Khan M. U.; Hussain R.; ur Rehman M. F.; Ennis C. H. J.; et al. First principles study of electronic and nonlinear optical properties of A-D-p-A and D-A-D-p-A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 2020, 10 (37), 22273–22283. 10.1039/D0RA02857F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir M. N.; Mirza S. H.; Khalid M.; Ali A.; Khan M. U.; Braga A. A. C. Synthesis, single crystal analysis and DFT based computational studies of 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrim idin-1-ium 3,4,5- trihydroxybenzoate -methanol (DETM). J. Mol. Struct. 2019, 1180, 119–126. 10.1016/j.molstruc.2018.11.089. [DOI] [Google Scholar]
- Ashfaq M.; Tahir M. N.; Kuznetsov A.; Mirza S. H.; Khalid M.; Ali A. DFT and single crystal analysis of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chloro-phenyl)-6-ethylpyrimidin-1- ium:4-hydroxybenzoate:methanol:hydrate (1:1:1:1) (DEHMH). J. Mol. Struct. 2020, 1199, 127041 10.1016/j.molstruc.2019.127041. [DOI] [Google Scholar]
- Shafiq I.; Amanat I.; Khalid M.; Asghar M. A.; Baby R.; Ahmed S.; Alshehri S. M. Influence of azo-based donor modifications on nonlinear optical amplitude of D-π-A based organic chromophores: A DFT/TD-DFT exploration. Synth. Met. 2023, 297, 117410 10.1016/j.synthmet.2023.117410. [DOI] [Google Scholar]
- Lawal M. M.; Govender T.; Maguire G. E.; Honarparvar B.; Kruger H. G. Mechanistic investigation of the uncatalyzed esterifcation reaction of acetic acid and acid halides with methanol: a DFT study. J. Mol. Model. 2016, 22, 235 10.1007/s00894-016-3084-z. [DOI] [PubMed] [Google Scholar]
- Gummidi L.; Kerru N.; Ibeji C. U.; Singh P. Crystal structure and DFT studies of (E)-1-(4-fluorophenyl)-3-(1H-indol-1-yl)-4-styrylazetidin-2-one. J. Mol. Struct. 2019, 1187, 50–58. 10.1016/j.molstruc.2019.03.053. [DOI] [Google Scholar]
- Khan M. U.; Mehboob M. Y.; Hussain R.; Afzal Z.; Khalid M.; Adnan M. Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high-efficiency organic solar cells. Int. J. Quantum Chem. 2020, 120, e26377 10.1002/qua.26377. [DOI] [Google Scholar]
- Khalid M.; Khan M. U.; Razia E.; Shafiq Z.; Alam M. M.; Imran M.; Akram M. S. Exploration of efficient electron acceptors for organic solar cells: Rational design of indacenodithiophene based non-fullerene compounds. Sci. Rep. 2021, 11, 19931 10.1038/s41598-021-99254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr R. G.; Szentpaly L. V.; Liu S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Parr R. G.; Yang W.. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, Oxford, 1989. [Google Scholar]
- Park J. C.; Nam Y. S. Controlling surface defects of non-stoichiometric copper-indium-sulfide quantum dots. J. Colloid Interface Sci. 2015, 460, 173–180. 10.1016/j.jcis.2015.08.037. [DOI] [PubMed] [Google Scholar]
- Meinardi F.; Daniel H. M.; Carulli F.; Colombo A.; Velizhanin K. A.; Makarov N. S.; Simonutti R.; klimov V. I.; Brovelli S. Highly efficient large-area colourless luminescent solar concentrators using heavy-metal-free colloidal quantum dots. Nat. Nanotechnol. 2015, 10, 878–885. 10.1038/nnano.2015.178. [DOI] [PubMed] [Google Scholar]
- Valeur B.Molecular Fluorescence: Principles and Applications; Wiley-VCH: Berlin, Germany, 2001. [Google Scholar]
- Heberer H.; Matschiner H. UV–VIS– und IR-spektroskopische Untersuchungen an N-substituierten 4-Amino-7-nitrobenz-2,1,3-oxadiazolen. J. Prakt. Chem. 1986, 328, 261–274. 10.1002/prac.19863280216. [DOI] [Google Scholar]
- Ans M.; Iqbal J.; Eliasson B.; Saif M. J.; Javed H. M. A.; Ayub K. Designing of non-fullerene 3D star-shaped acceptors for organic solar cells. J. Mol. Model. 2019, 25, 129 10.1007/s00894-019-3992-9. [DOI] [PubMed] [Google Scholar]
- Maddalena F.; Falco C.; Caironi M.; Natali D. Assessing the width of Gaussian density of states in organic semiconductors. Org. Electron. 2015, 17, 304–318. 10.1016/j.orgel.2014.12.001. [DOI] [Google Scholar]
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- Chen M.; Waghmare U. V.; Friend C. M.; Kaxiras F. A density functional study of clean and hydrogen-covered α-MoO3 (010): Electronic structure and surface relaxation. J. Chem. Phys. 1998, 109, 6680–6854. 10.1063/1.477252. [DOI] [Google Scholar]
- Liu S.; Zhao Y.; Zhang C.; Lin L.; Li Y.; Song Y. the novel excited state intramolecular proton transfer broken by intermolecular hydrogen bonds in HOF system. Spectrochim. Acta, Part A 2019, 219, 164–172. 10.1016/j.saa.2019.04.053. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Shahar K.; Paula M. S.; Julia C. G.; Cohen A. J.; Weitao Y. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132 (2010), 6498–6506. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alotaibi M. T. Noncovalent interaction stabilizes the 2, 4-Dinitrophenylhydrazone Derivatives over g-C3N4 surface to enhance optical properties: synthesis, characterization, and DFT investigation. J. Mol. Struct. 2020, 1214, 128192 10.1016/j.molstruc.2020.128192. [DOI] [Google Scholar]
- Boto R. A.; Piquemal J. P.; Contreras-García J. Revealing strong interactions with the reduced density gradient: a benchmark for covalent, ionic and charge-shift bonds. Theor. Chem. Acc. 2017, 136, 139 10.1007/s00214-017-2169-9. [DOI] [Google Scholar]
- Silvi B.; Savin A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. 10.1038/371683a0. [DOI] [Google Scholar]
- Arulaabaranam K.; Mani G.; Muthu S. Computational assessment on wave function (ELF, LOL) analysis, molecular confirmation and molecular docking explores on 2-(5-Amino2- Methylanilino)-4-(3-pyridyl) pyrimidine. Chem. Data Collect. 2020, 29, 100525 10.1016/j.cdc.2020.100525. [DOI] [Google Scholar]
- Prasana J. C.; Muthu S.; Abraham C. S. Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: a potential antiviral drug. Comput. Biol. Chem. 2019, 78, 9–17. 10.1016/j.compbiolchem.2018.11.014. [DOI] [PubMed] [Google Scholar]
- Gadre S. R.; Suresh C. H.; Mohan N. Electrostatic Potential Topology for Probing Molecular Structure, Bonding Reactivity. Molecules 2021, 26, 3289 10.3390/molecules26113289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevvanthi S.; Muthu S.; Raja M. Molecular docking, vibrational spectroscopy studies of (RS)-2-(tert-Butylamino)-1-(3-chlorophenyl)propan-1-one: A potential Adrenaline uptake inhibitor. J. Mol. Struct. 2018, 1173, 251–260. 10.1016/j.molstruc.2018.07.001. [DOI] [Google Scholar]
- Saral A.; Sudha P.; Muthu S.; Sevvanthi S.; Irfan A. Molecular structure spectroscopic Elucidation, IEFPCM solvation (UV– Vis, MEP, FMO, NBO, NLO), molecular docking and biological assessment studies of lepidine (4-Methylquinoline). J. Mol. Liq. 2022, 345, 118249 10.1016/j.molliq.2021.118249. [DOI] [Google Scholar]
- Bader R. F. W.; Austen M. A. Properties of atoms in molecules: atoms under pressure. J. Chem. Phys. 1997, 107, 4271–4285. 10.1063/1.474769. [DOI] [Google Scholar]
- Tariq S.; Khalid M.; Raza A. R.; Rubab S. L.; de AlcântaraMorais S. F.; Khan M. U.; Tahir M. N.; Braga A. A. C. Experimental and computational investigations of new indole derivatives: A combined spectroscopic, SC-XRD, DFT/TD-DFT and QTAIM analysis. J. Mol. Struct. 2020, 1207, 127803 10.1016/j.molstruc.2020.127803. [DOI] [Google Scholar]
- Khalid M.; Ali A.; Abid S.; Tahir M. N.; Khan M. U.; Ashfaq M.; Imran M.; Ahmad A. Facile Ultrasound-Based Synthesis, SC-XRD, DFT Exploration of the Substituted Acyl-Hydrazones: An Experimental and Theoretical Slant towards Supramolecular Chemistry. ChemistrySelect 2020, 5 (47), 14844–14856. 10.1002/slct.202003589. [DOI] [Google Scholar]
- Ali A.; Khalid M.; Abid S.; Tahir M. N.; Iqbal J.; Ashfaq M.; Kanwal F.; Lu C.; Rehman M. F. Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone. Crystals 2020, 10, 778 10.3390/cryst10090778. [DOI] [Google Scholar]
- Ali A.; Khalid M.; Ashfaq M.; Malik A. N.; Tahir M. N.; Assiri M. A.; Imran M.; de AlcântaraMorais S. F.; Braga A. A. C. Preparation, QTAIM and Single-Crystal Exploration of the Pyrimethamine-Based Co-Crystal Salts with Substituted Benzoic Acids. ChemistrySelect 2022, 7 (17), e202200349 10.1002/slct.202200349. [DOI] [Google Scholar]
- Espinosa E.; Molins E.; Lecomte C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. 10.1016/S0009-2614(98)00036-0. [DOI] [Google Scholar]
- Rozas I.; Alkorta I.; Elguero J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. 10.1021/ja0017864. [DOI] [Google Scholar]
- Koch U.; Popelier P. L. A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. A 1995, 99, 9747–9754. 10.1021/j100024a016. [DOI] [Google Scholar]
- Hirshfeld F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta. 1977, 44, 129–138. 10.1007/BF00549096. [DOI] [Google Scholar]
- Spackman M. A.; Jayatilaka D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. 10.1039/B818330A. [DOI] [Google Scholar]
- McKinnon J. J.; Jayatilaka D.; Spackman M. A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 37, 3814–3816. 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]
- Ashfaq M.; Khalid M.; Tahir M. N.; Ali A.; Arshad M. N.; Asiri A. M. Synthesis of Crystalline Fluoro-Functionalized Imines, Single Crystal Investigation, Hirshfeld Surface Analysis, and Theoretical Exploration. ACS Omega 2022, 7 (11), 9867–9878. 10.1021/acsomega.2c00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid M.; Ali A.; Abid S.; Tahir M. N.; Khan M. U.; Ashfaq M.; Imran M.; Ahmad A. Facile Ultrasound-Based Synthesis, SC-XRD, DFT Exploration of the Substituted Acyl-Hydrazones: An Experimental and Theoretical Slant towards Supramolecular Chemistry. ChemistrySelect 2020, 5, 14844–14856. 10.1002/slct.202003589. [DOI] [Google Scholar]
- Ali A.; Khalid M.; Abid S.; Tahir M.; Iqbal J.; Ashfaq M.; Kanwal F.; Lu C.; Rehman M. Green synthesis, SC-XRD, non-covalent interactive potential andelectronic communication via DFT exploration of pyridine-basedhydrazone. Crystals 2020, 10 (9), 778 10.3390/cryst10090778. [DOI] [Google Scholar]
- McKinnon J. J.; Spackman M. A.; Mitchell A. S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr., Sect. B: Struct. Sci. 2004, 60, 627–668. 10.1107/S0108768104020300. [DOI] [PubMed] [Google Scholar]
- Spackman M. A.; Byrom P. G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. 10.1016/S0009-2614(97)00100-0. [DOI] [Google Scholar]
- Jayatilaka D.; Wolff S. K.; Grimwood D. J.; McKinnon J. J.; Spackman M. A. CrystalExplorer: a tool for displaying Hirshfeld surfaces and visualising intermolecular interactions in molecular crystals. Acta Crystallogr., Sect. A: Found. Crystallogr. 2006, 62, s90 10.1107/S0108767306098199. [DOI] [Google Scholar]
- Prasad P. N.; Williams D. J.. Introduction to Nonlinear Optical Effects in Molecules and Polymers; Wiley: New York, 1991. [Google Scholar]
- Geskin V. M.; Lambert C.; Brédas J. L. Origin of High Second- and Third-Order Nonlinear Optical Response in Ammonio/Borato Diphenylpolyene Zwitterions: the Remarkable Role of Polarized Aromatic Groups. J. Am. Chem. Soc. 2003, 125, 15651–15658. 10.1021/ja035862p. [DOI] [PubMed] [Google Scholar]
- Lokhande P. K. M.; Patil D. S.; Kadam M. M.; Sekar N. Theoretical Investigation of Optical and Nonlinear Optical (NLO) Properties of 3-Azabenzanthrone Analogues: DFT and TD-DFT Approach. ChemistrySelect 2019, 4, 10033–10045. 10.1002/slct.201901681. [DOI] [Google Scholar]
- Tillekaratne A. D.; de Silva R. M.; de Silva K. M. N. Push-Pull Porphyrins as Non-Linear Optical Materials: Ab Initio Quantum Chemical Calculations. J. Mol. Struct.: THEOCHEM 2003, 638, 169–176. 10.1016/S0166-1280(03)00566-9. [DOI] [Google Scholar]
- Kleinman D. A. Nonlinear Dielectric Polarization in Optical Media. Phys. Rev. 1962, 126, 1977–1979. 10.1103/PhysRev.126.1977. [DOI] [Google Scholar]
- Janaki A.; Balachandran V.; Lakshmi A. First order molecular hyperpolarizabilities and intramolecular charge transfer from vibrational spectra of NLO material: 2,6-dichloro-4- nitraniline. Ind. J. Pure Appl. Phys. 2013, 51, 601–614. [Google Scholar]
- Khalid M.; Khan M. U.; Shafiq I.; Hussain R.; Mahmood K.; Hussain A.; Jawaria R.; Hussain A.; Imran M.; Assiri M. A. NLO Potential Exploration for D-p-A Heterocyclic Organic Compounds by Incorporation of Various p-Linkers and Acceptor Units. Arab. J. Chem. 2021, 14 (8), 103295 10.1016/j.arabjc.2021.103295. [DOI] [Google Scholar]
- Khalid M.; Khan M. U.; Shafiq I.; Hussain R.; Ali A.; Imran M.; Braga A. A. C.; Ur Rehman M. F.; Akram M. S. Structural modulation of π-conjugated linkers in D−π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties. R. Soc. Open Sci. 2021, 8, 210570 10.1098/rsos.210570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid M.; Shafiq I.; Mahmood K.; Hussain R.; Ur Rehman M. F.; Assiri M. A.; Imran M.; Akram M. S. Effect of different end-capped donor moieties on non-fullerenes based non-covalently fused-ring derivatives for achieving high-performance NLO properties. Sci. Rep. 2023, 13, 1395 10.1038/s41598-023-28118-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid M.; Arshad M. N.; Murtaza S.; Shafiq I.; Haroon M.; Asiri A. M.; de AlcântaraMorais S. F.; Braga A. A. Enriching NLO Efficacy via Designing Non-Fullerene Molecules with the Modification of Acceptor Moieties into ICIF2F: An Emerging Theoretical Approach. RSC Adv. 2022, 12 (21), 13412–13427. 10.1039/D2RA01127A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. M.; Liu Y.; Man L. M.; Zhou J. R.; Liu X. P.; Ni C. L. Crystal structure,vibrational spectra, optical properties and density functional theoretical approach of [Bz4-NH 2 Py] 4 [CdCl 4 ] 2. H 2 O. Vib. Spectrosc. 2017, 93, 23–28. 10.1016/j.vibspec.2017.09.002. [DOI] [Google Scholar]
- Cassidy C.; Halbout J.; Donaldson W.; Tang C. Nonlinear optical properties of urea. Opt. Commun. 1979, 29, 243–246. 10.1016/0030-4018(79)90027-0. [DOI] [Google Scholar]
- Machado D. F. S.; Lopes T. O.; Lima I. T.; da Silva Filho D. A.; de Oliveira H. C. Strong Solvent Effects on the Nonlinear Optical Properties of Z and E Isomers from Azo-Enaminone Derivatives. J. Phys. Chem. C 2016, 120 (31), 17660–17669. 10.1021/acs.jpcc.6b01567. [DOI] [Google Scholar]
- Günter P.Nonlinear Optical Effects and Materials, 4th ed.; Springer: Berlin, 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.