

Abstract
Background
Adeno-associated virus (AAV) is an ideal gene therapy vector and is non-immunogenic in many small animal models. The stable gene expression commonly seen in murine models does not necessarily translate to nonhuman primates and higher-order species, highlighting the need for a better understanding of immune activation to these vectors. One capsid variant, AAVrh32.33, demonstrates a unique phenotype in murine muscle, reminiscent of what is often seen in higher-order species. AAVrh32.33 generates a strong CD8+ T-cell response to both capsid and encoded transgene antigens in a manner independent of transgene product or major histocompatability complex haplotype, making it an ideal candidate for studying immune activation to AAV in the mouse.
Methods
To map the H-2b and H-2d dominant epitopes of the AAVrh32.33 capsid, C57BL/6 or Balb/C mice received an intramuscular injection of 1 × 1011 genome copies of AAV2/rh32.33.CB.nLacZ. Three weeks later, splenocytes were harvested and stimulated in vitro with pooled or individual peptides from the AAVrh32.33 capsid peptide library and analysed by an interferon (IFN)-γ enzyme-linked immunosorbent spot assay or intracellular cytokine staining.
Results
The immunodominant epitopes within the AAVrh32.33 capsid responsible for driving CD8+ T-cell responses to the capsid protein in C57BL/6 (SSYELPYVM) and Balb/C (KIPASGGNAL) mice were defined.
Conclusions
Identification of dominant capsid epitopes will make it possible to monitor cellular responses to the AAV capsid in vivo, facilitating mechanistic studies critical to defining how cellular immunity to the AAV capsid arises and, ultimately, how the generation of capsid-specific T cells can be avoided to ensure safety in a gene therapy setting. Copyright © 2009 John Wiley & Sons, Ltd.
Keywords: AAV, capsid epitope, gene therapy, T cells
Introduction
Adeno-associated virus (AAV) is a promising gene delivery vector because of its ability to achieve sustained, high level expression of a packaged gene within target tissues [1,2]. As a result of the stability of expression of foreign transgene products in numerous murine tissues, AAV was generally considered to be minimally immunogenic for many years [1–3]. However, recent studies have demonstrated that it is possible to generate humoral and/or cell-mediated immune responses to vector encoded proteins [4–8]. Furthermore, even in cases where stable transgene expression is achieved in murine models, expression of the identical transgene product in nonhuman primates is often transient, accompanied by a substantial interferon (IFN)-γ -producing T-cell response and a brief elevation in liver transaminases [9]. In a recent clinical trial for hemophilia B, loss of transgene product expression over time and a state of transient transaminitis was suspected to be caused by the presence of AAV2 capsid-specific T cells clearing vector transduced hepatocytes [7]. In addition to the potential of capsid-specific T cells to clear transduced targets in vivo, the AAV capsid can also enhance the generation of transgene specific T cells, which may further contribute to the loss of transgene expression over time [10].
The implication of AAV capsid-driven immunity in the loss of transgene expression after gene transfer has demonstrated a need for further elucidation of the mechanism of T-cell activation to AAV vectors in the host. We recently identified an AAV capsid variant, AAVrh32.33, that elicits qualitatively and quantitatively robust CD8+ T-cell responses to both capsid and transgene antigens in the mouse, allowing mechanistic studies to be conducted within a small animal model [10]. AAVrh32.33, a novel engineered vector isolated from rhesus macaques and phylogenetically closest to AAV4, is evolutionarily and structurally divergent from other AAVs. Importantly, its seroprevalence in human populations is significantly reduced compared to AAV2, 7 and 8, making it attractive as a gene therapy vector were its immunogenicity to be overcome [11]. To allow for quantitative assessment of CD8+ T-cell responses to the AAV capsid, we defined the immunodominant capsid epitopes for AAVrh32.33 in two commonly used mouse strains, C57BL/6 (H-2b) and Balb/C (H-2d). Understanding the T-cell response to the AAV capsid is critical for the safe and efficacious use of recombinant AAV as a vector for gene therapy or genetic vaccines.
Materials and methods
AAVrh32.33 capsid peptide library
An AAVrh32.33 VP1 capsid (NCBI accession no. ACB55318; http://www.ncbi.nlm.nih.gov/) peptide library of 145 peptides was generated as 15-mers overlapping by ten amino acids (Mimotopes, Victoria, Australia) and dissolved in dimethyl sulfoxide (DMSO) at approximately 100 mg/ml. Twelve pools of consecutive peptides were prepared, resulting in 11 pools of 12 peptides each and a final pool containing 13 peptides. Peptides were stored at −20 °C prior to use. Pools and individual peptides were used at 2 μg/ml to avoid the potential toxicity associated with higher peptide/DMSO concentrations.
Animals
Male C57BL/6 and Balb/C mice (6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained in the Animal Facility of Translational Research Laboratories. All experimental procedures involving the use of mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Vector production and purification
Recombinant AAV (rAAV) vector with the AAVrh32.33 viral capsid expressing nLacZ was manufactured as described previously [3] by PennVector at the University of Pennsylvania (Philadelphia, PA, USA). Briefly, an AAV cis-plasmid containing transgene cDNA driven by the chicken β-actin (CB) promoter and flanked by AAV2 inverted terminal repeats was packaged by triple transfection of HEK293 cells using an adenovirus helper plasmid (pAdΔF6), and a chimeric packaging construct containing the AAV2 Rep gene and the AAVrh32.33 Cap gene. Vectors were purified by three rounds of cesium chloride gradient centrifugation. The genome titer [genome copies (GC) per ml] of AAV vectors was determined by a real-time poylmerase chain reaction.
Animal treatments
Mice were anesthetized with ketamine-xylazine (70 and 7 mg/kg of body weight, intraperitoneally). 1 × 1011 GC of recombinant AAV vector was injected into the anterior tibialis muscle in a volume of 50 μl of sterile phosphate-buffered saline (PBS). At various timepoints post-vector injection, mice were sacrificed by CO2 inhalation followed by cervical dislocation to harvest the spleen.
Intracellular cytokine staining
Spleens harvested from treated mice were transferred into Liebowitz’s-15 (L-15; Cellgro, Mediatech, Herndon, VA, USA) at room temperature (RT). The tissue was then homogenized and passed through a 70 μm nylon cell strainer (Fisher Scientific, Pittsburgh, PA, USA) to remove cell clumps and un-dissociated tissue. The cells were centrifuged for 5 min at 1600 r.p.m = 532.5 × g. at RT, resuspended in fresh L-15 media and centrifuged again. The cell pellet was resuspended in T cell assay media [Dulbecco’s modified Eagle’s medium (Cellgro, Mediatech), 10% heat-inactivated fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 1% Pen-Strep (Cellgro, Mediatech), 1% l-glutamine, 10 mm HEPES (Cellgro, Mediatech), 0.1 mm non-essential amino acids (Invitrogen, Carlsbad, CA, USA), sodium pyruvate, and 10−6 m 2-mercaptoethanol (Cellgro, Mediatech)].
After resuspending at a concentration of 107 cells/ml, splenocytes were plated at 106 cells/well in triplicate on 96-well round bottom plates. T cell assay media was supplemented with 1 μg/ml Brefeldin A (GolgiPlug; BD Pharmingen, San Diego, CA, USA) and 20 ng/ml mouse IL-2 (BD Pharmingen). Cells were stimulated with the AAVrh32.33 capsid peptide pool(s) for 5 h at 37 °C, 10% CO2. Control cells were incubated without peptide, or in the presence of PMA/I (PMA: 0.05 μg/ml; I, Ionomycin: 1 μg/ml; Sigma, St Louis, MO, USA). After the stimulation, cells were washed and stained with fluorescein isothiocyanate-conjugated anti-mouse CD8α (Ly-2) antibody and a PE-Cy5-labeled anti-mouse CD4 antibody (BD Pharmingen) for 30 min at 4 °C. Cells were washed with PBS/1% FBS, then permeabilized in Cytofix/Cytoperm solution at 4 °C for 20 min. Cells were washed again thoroughly with 1 × Perm/Wash Buffer and stained with a PE labeled anti-mouse IFN-γ antibody (BD Pharmingen) for 45 min at 4 °C. After washing, cells were examined by flow cytometric analysis. Samples were acquired on an FC500 (Beckman-Coulter, Miami, FL, USA) and analysed using FlowJo software (Tree Star, San Carlos, CA, USA).
IFN-γ enzyme-linked immunosorbent spot (ELISPOT) assay
Splenocytes from treated mice were harvested as described above. After isolation and washing steps splenocytes were overlaid onto a Ficoll-Paque (Amersham Biosciences, Piscataway, NJ, USA) gradient layer and centrifuged for 20 min at 2000 r.p.m = 832 × g at RT to remove red blood cells. The lymphocyte band was then recovered and further washed two times in PBS/1% FBS.
To determine the number of cells secreting IFN-γ in response to antigenic stimulation, an IFN-γ ELISPOT assay was performed according to the manufacturer’s instructions (BD Biosciences, San Diego, CA, USA). Briefly, 96-well plates were coated with capture antibody overnight at 4 °C, and blocked for 2 h at 25 °C with RPMI, 10% FBS, 1% Pen-Strep-l-Glut. Splenocytes were plated at three densities (106, 5 × 105 and 2.5 × 105 cells/well) in T cell assay media supplemented with 2 μg/ml of the AAVrh32.33 capsid peptide library. PMA/Ionomycin (PMA: 0.05 μg/ml; Ionomycin: 1 μg/ml; Sigma) was used to stimulate a separate population of lymphocytes (plated at 2 × 104 cells/well) as a nonspecific, positive control. Cells plated in media alone (no peptide stimulation) served as a negative control. Plates were incubated for 18 h at 37 °C, 5% CO2. After incubation, plates were washed vigorously in deionized water, PBS/0.05% Tween-20 (Sigma), and incubated for 2 h at RT with 2 μg/ml of biotinylated anti-mouse IFN-γ detection antibody. After three washes with PBS/0.05% Tween-20, the plates were incubated with 5 μg/ml of enzyme conjugate (streptavidin-horseradish peroxidase) for 1 h at RT. After a series of washes with PBS/0.05% Tween-20 and PBS, spots were developed using the AEC Substrate Set (BD Pharmingen). Color development was stopped after 8 min by washing with distilled water. Plates were dried overnight at RT and read using the AID ELISPOT reader system (Cell Technology, Columbia, MD, USA). Responses greater than 200 spot-forming units or at least 2 logs over background were considered.
Results and Discussion
AAV mediated gene transfer holds great potential for the treatment of monogenetic disease [12]. However, the safety and efficacy of gene transfer can be negatively effected by host immune responses [13]. Although the impact of transgene specific immunity on the loss of expression has been well established, it is also becoming apparent that T-cell responses to the AAV capsid itself may also threaten the stability of transgene expressing cells after gene transfer [7]. We recently reported that the AAVrh32.33 capsid is highly immunogenic in murine muscle and may be an ideal variant to study the mechanism of immune activation to AAV in a small animal model [10]. In the present study, we sought to identify the immunodominant T cell epitopes of the AAVrh32.33 capsid within both C57BL/6 and Balb/C mice.
Identification of the AAVrh32.33 capsid-specific CD8+ T cell epitope in C57BL/6 mice
To map the H-2b dominant epitope to the AAVrh32.33 capsid, C57BL/6 mice received an intramuscular (i.m.) injection of AAV2/rh32.33.CB.nLacZ at a dose of 1 × 1011 GC. As a negative control, mice were injected with AAV2/8.CB.nLacZ at the same dose. Three weeks later, at the peak of T-cell activation, splenocytes were harvested from the treated mice and stimulated in vitro with the peptide library corresponding to the entire AAVrh32.33 capsid protein. This library was divided into 12 peptide pools, each pool containing 12 or 13 individual 15-mers with a ten amino acid overlapping sequence (Figure 1A). T-cell activation to each peptide pool was determined by IFN-γ ELISpot.
Figure 1.
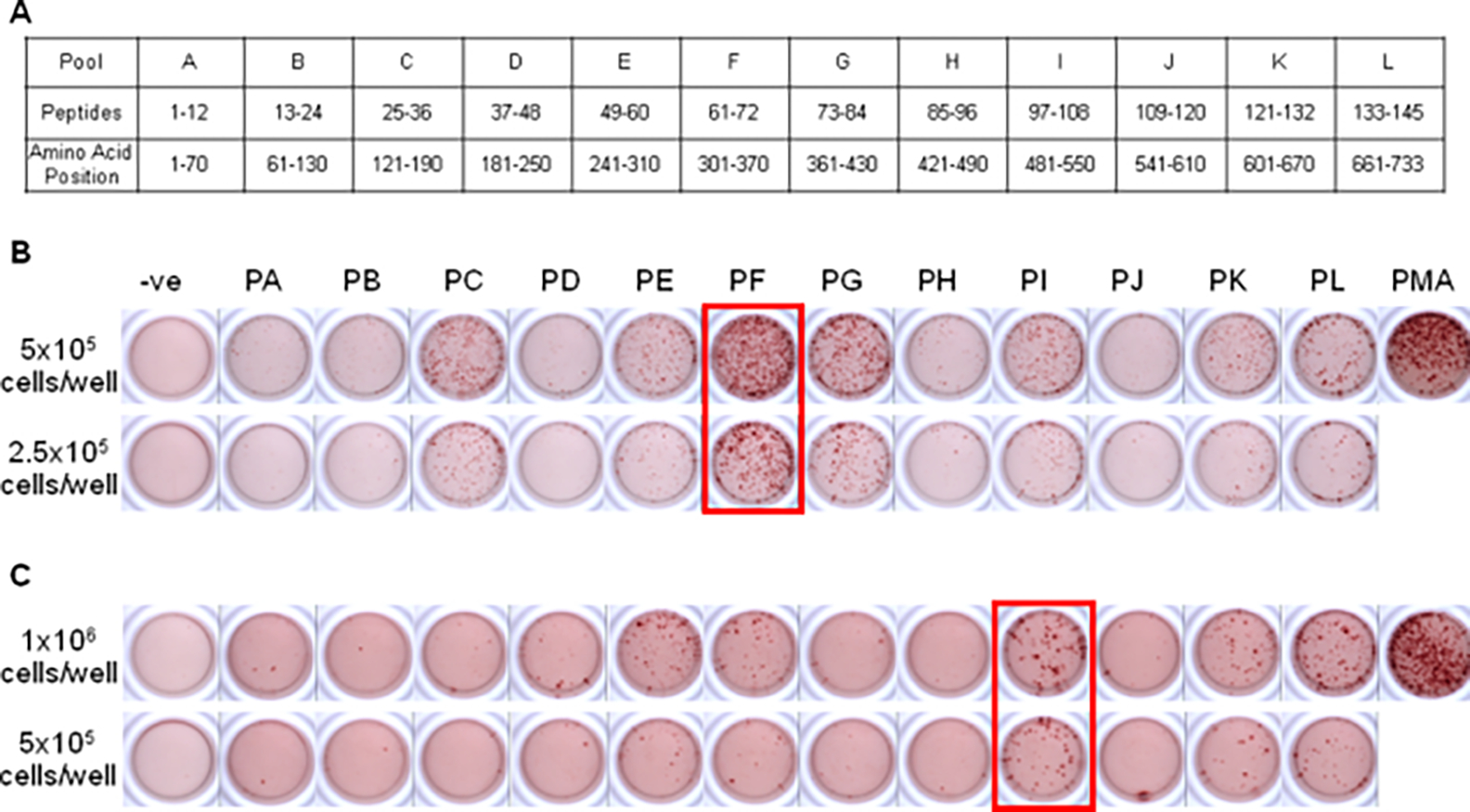
Identification of peptide pools containing H-2b and H-2d T cell epitopes for the AAVrh32.33 capsid. (A) The complete peptide library of the AAVrh32.33 capsid protein was divided into 12 peptide pools (pools A–L), with each pool containing 12 or 13 individual 15-mers, overlapping by ten amino acids. The amino acid positions of each peptide pool have been indicated, according to AAVrh32.33 VP1 numbering (NCBI accession no. ACB55318). Twenty-one days after i.m. injection of 1 × 1011 GC AAV2/rh32.33.CB.nLacZ, splenocytes from (B) C57BL/6 and (C) Balb/C mice were harvested and stimulated in vitro with each of the 12 peptide pools. Cells were seeded at 2.5 × 105 or 5 × 105 cells/well for C57BL/6 mice and at 5 × 105 and 106 cells/well for Balb/C mice. IFN-γ producing T-cell responses were noted for C57BL/6 mice in pools C, E, F, G, I, K and L. For Balb/C mice, positive pools included E, F, I, K and L. A no-peptide negative control and positive PMA/I control were also included. Cells were seeded at 2 × 104 cells/well for PMA/I control wells. Representative data from three independent experiments are shown
Mice injected with the negative control vector generated no IFN-γ -producing T-cell responses to the AAVrh32.33 capsid protein, confirming that T cells induced in response to AAV2/8 could not cross-react with peptide sequences from the AAVrh32.33 capsid (data not shown). This was expected because of the limited degree of primary sequence homology between the two capsid variants; AAVrh32.33 shares only 68% homology with AAV8, compared with AAV2 and AAV8, which are approximately 84% homologous. In C57BL/6 mice receiving AAV2/rh32.33.CB.nLacZ, however, IFN-γ responses to the AAVrh32.33 capsid protein were noted in pools C, E, F, G, I, K and L (Figure 1B). The highest density of spots and the most robust IFN-γ signal was exhibited by peptides within pool F (Figure 1B). For this reason, it was concluded that the immunodominant epitope(s) were located within this pool.
To identify the specific 15-mer peptide sequence(s) responsible for T-cell activation in pool F, all twelve 15-mer peptides comprising this pool were screened individually by IFN-γ ELISpot (data not shown). As a result, two neighboring 15-mer peptides, #67 (VQIFADSSYELPYVM) and #68 (DSSYELPYVMDAGQE), which shared an overlapping ten amino acid sequence, were identified (Figure 2A). To define the T-cell subsets responsible for expressing IFN-γ in response to AAVrh32.33 capsid antigen, splenocytes harvested from AAV2/rh32.33.CB.nLacZ-treated C57BL/6 mice were stimulated in vitro with pool F or peptides 67 and 68 and subjected to intracellular cytokine staining (ICS) followed by flow cytometry. No CD4+ T-cell activation was observed (Figure 2C). It was determined that the CD8+ T cell population was responsible for the production of IFN-γ in these mice (Figure 2B).
Figure 2.
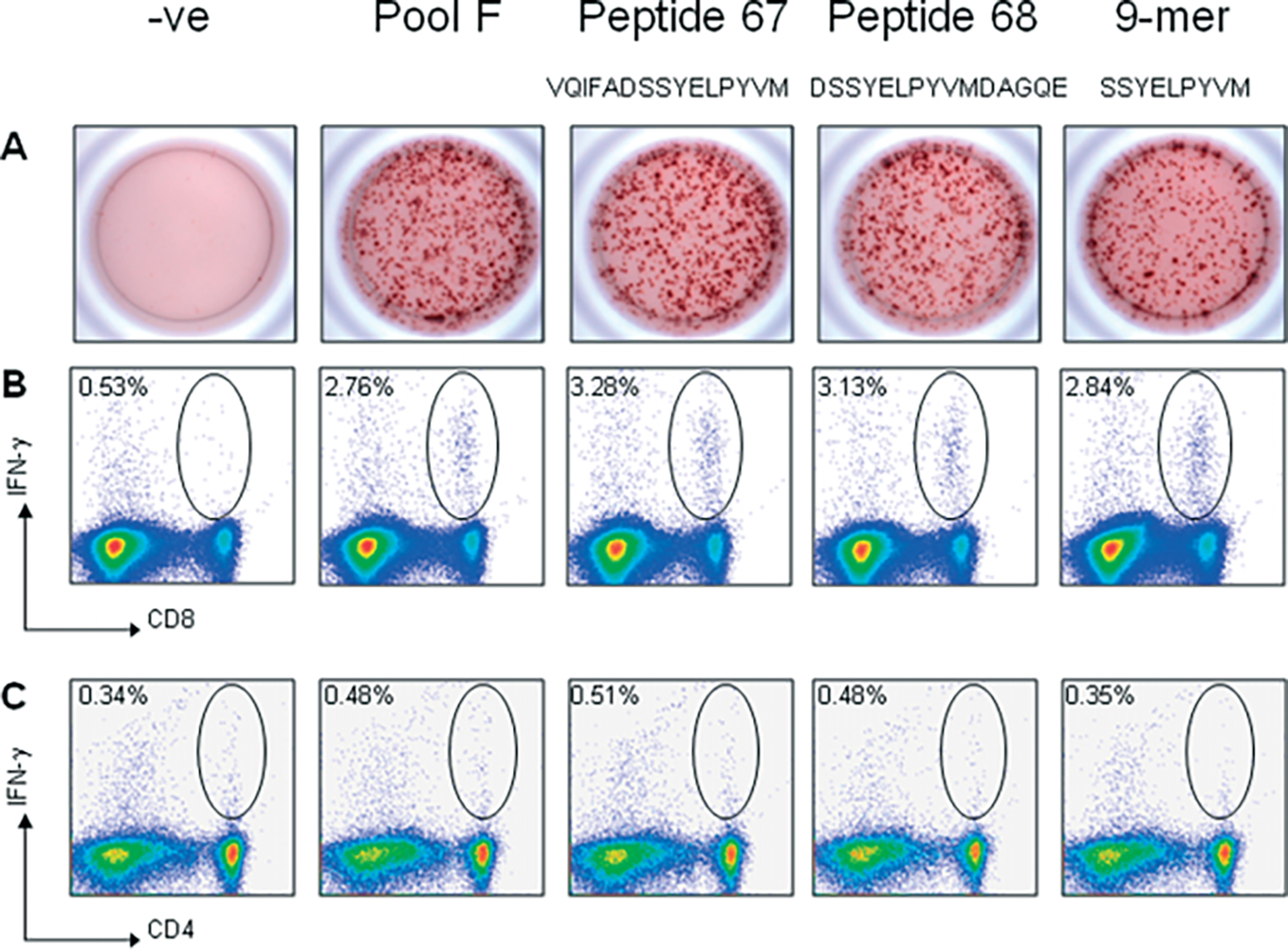
Fine mapping of the H-2b-restricted T cell epitope in the AAVrh32.33 capsid protein. Splenocytes (5 × 105 cells/well) harvested from AAV2/rh32.33.CB.nLacZ-injected C57BL/6 mice at day 21 were stimulated in vitro with 2 μg/ml of either pool F, peptides 67 and 68, or the 9-mer, SSYELPYVM. Samples were analysed by either (A) IFN-γ ELISpot or (B,C) intracellular cytokine staining followed by flow cytometric analysis. Staining was performed using monoclonal antibodies to (B) CD8, (C) CD4, and IFN-γ. Negative control wells were stimulated with no peptide. Representative results from three separate experiments are shown
To identify the potential major histocompatability complex (MHC) class I binders within the contiguous sequence spanning the two overlapping peptides, VQIFADSSYELPYVMDAGQE, two epitope prediction programs were used: HLA-BIND-Bio-Informatics and Molecular Analysis Section (BIMAS) [14] and SYFPEITHI [15]. The highest-ranking candidate epitope sequence predicted by BIMAS for the H2-Kb MHCI molecule was SSYELPYVM (Table 1). No H2-Kb MHCI binders were predicted by SYFPEITHI. SSYELPYVM was also the highest-ranking predicted epitope for the H2-Db MHCI molecule, as predicted by both BIMAS and SYFPEITHI (Table 1). Based on these scores, SSYELPYVM was synthesized for further study.
Table 1.
Predicted MHC class I epitopes
| Strain | Pool | Peptide sequence | MHC class I | Score |
|---|---|---|---|---|
|
| ||||
| C57BL/6 | I | SSYELPYVM | H2-Kb | 6.6 (B); (S)* |
| H2-Db | 24.6 (B); 16 (S) | |||
| Balb/C | F | IPASGGNAL | H2-Kd | 69.1 (B); 16 (S) |
| KIPASGGNAL | H2-Ld | 195 (B); 23 (S) | ||
| H2-Kd | 82.9 (B); 12 (S) | |||
| H2-Dd | 24 (B); (S)* | |||
The highest ranking candidate epitope sequences for C57BL/6 and Balb/C mice as determined by BIMAS and SYFPEITHI are shown. The discrepancy of the scoring system and the inability of SYFPEITHI to identify epitopes scored by BIMAS can be attributed to the general limitations of the epitope binding prediction programs [16,17]. B, BIMAS; S, SYFPEITHI.
Not identified.
To confirm the role of SSYELPYVM as the H-2b immunodominant epitope in the AAVrh32.33 capsid protein, the peptide was tested in C57BL/6 mice, in comparison to pool F and individual 15-mers, peptides 67 and 68, by both IFN-γ ELISpot and ICS (Figure 2). ELISpot results confirm that the 9-mer, SSYELPYVM, is able to stimulate IFN-γ secretion to the same degree as the positive pool F and peptides 67 and 68 (Figure 2A). Flow cytometric staining confirms that SSYELPYVM stimulates IFN-γ production from the CD8+ T cell population, confirming its role as the dominant MHC class I epitope of the AAVrh32.33 capsid in C57BL/6 mice (Figure 2B).
The relative binding affinity of SSYELPYVM for the H2-Kb or H2-Db MHC class I molecules was determined by titrating the amount of peptide necessary to activate T cells in vitro. Splenocytes harvested from AAV2/rh32.33.CB.nLacZ-injected mice were stimulated with SSYELPYVM at concentrations ranging from 5 to 1.6 × 10−10 μg/ml to measure the activation of T cells by IFN-γ ELISpot (Figure 3A). SSYELPYVM had a very strong binding affinity, yielding high IFN-γ-producing T-cell responses even when stimulated with 0.00032 μg/ml of peptide. The binding affinity steadily decreased from 6.4 × 10−5 μg/ml to 5.1 × 10−7 μg/ml, where it remained slightly above background (Figure 3A).
Figure 3.
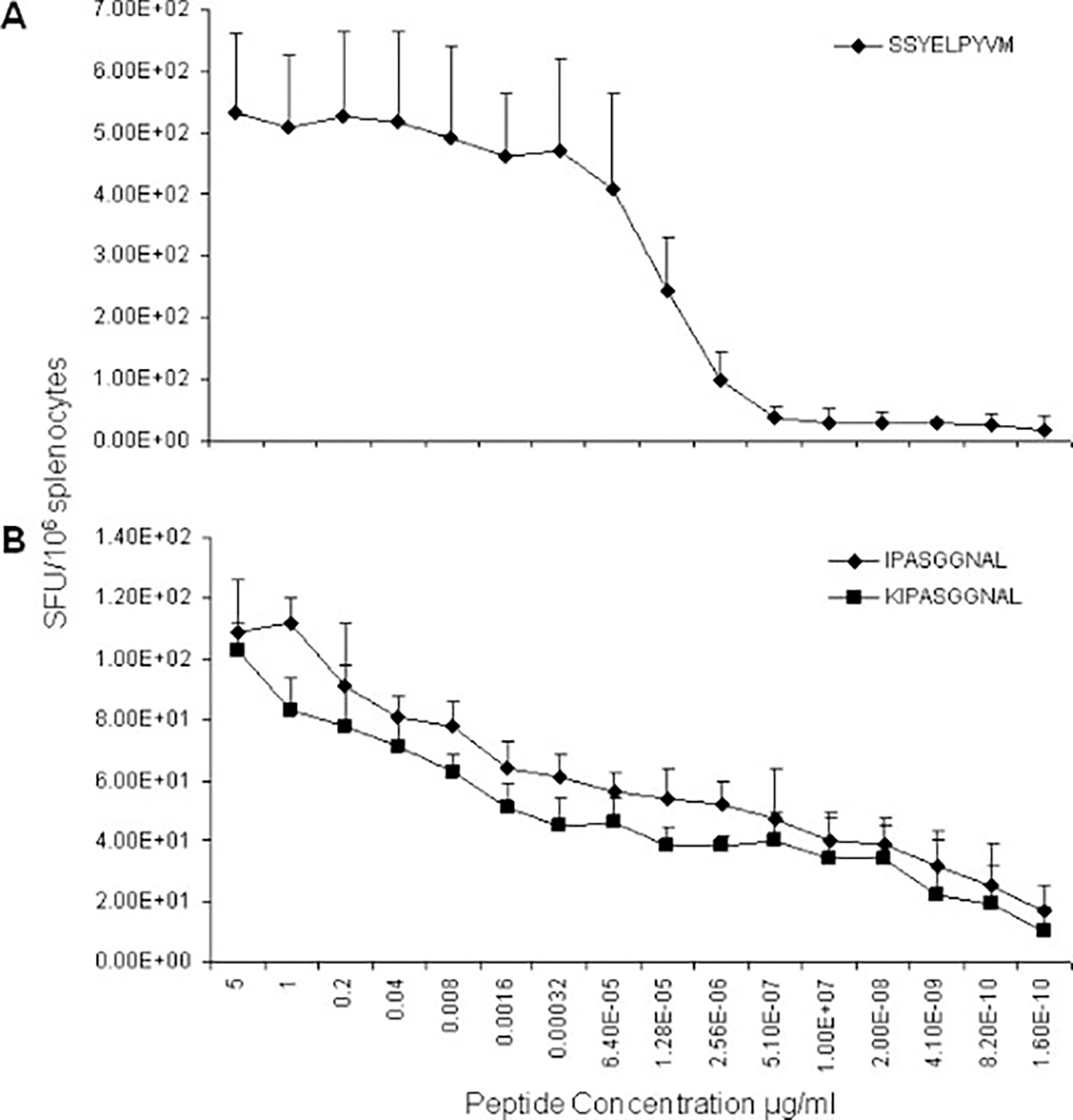
Titration of the concentration of H-2b and H-2d-restricted CD8 T cell epitopes required for T-cell activation in vitro. Three weeks post-injection of 1 × 1011 GC AAV2/rh32.33.CB.nLacZ, splenocytes were harvested from (A) C57BL/6 or (B) Balb/C mice and stimulated with decreasing concentrations of the dominant peptides: SSYELPYVM and (K)IPASGGNAL, respectively. The degree of T-cell activation was measured as spot-forming units/106 splenocytes by IFN-γ ELISpot. Experiments were repeated on two separate occasions; error bars indicate the standard deviation
It is important to note that, although pool F contained the immunodominant epitope, all individual peptides comprising the less positive pools C, E, G, I, K and L were also screened to identify the 15-mer(s) responsible for T-cell activation in C57BL/6 mice. These minor epitopes included LESPQEPDSSSGIGK (peptide 30; pool C), INNNWGLRPKAMRVK (peptide 59; pool E), NFEMAYNFEKVPFHS (peptide 81; pool G), LKYDTHYTLNNRWSN (peptide 101; pool I), HADGHFHPSPLIGGF (peptide 125; pool K) and RWNPEVQFTSNYGNQ (peptide 139; pool L).
Identification of AAVrh32.33 capsid-specific CD8+ T cell epitopes in Balb/C mice
Balb/C mice received an i.m. injection of 1 × 1011 GC of AAV2/rh32.33.CB.nLacZ or AAV2/8.CB.nLacZ as a negative control. Splenocytes harvested at day 21 were stimulated in vitro with the 12 pools corresponding to the entire AAVrh32.33 capsid protein, and analysed for T-cell activation by IFN-γ ELISpot (Figure 1C). Following AAV2/rh32.33.CB.nLacZ, T-cell activation to the AAVrh32.33 capsid was demonstrated in pools E, F, I, K and L (Figure 1C). Pool I resulted in the greatest degree of IFN-γ production and the highest density of spots per well, and was therefore considered to contain the immunodominant T cell epitope (Figure 1C). No IFN-γ -producing T-cell responses to the AAVrh32.33 capsid protein were observed in AAV2/8 injected controls (data not shown).
Fine mapping of the specific peptide sequence(s) from pool I responsible for T-cell activation was performed by stimulating splenocytes with all twelve 15-mer peptides comprising this pool. IFN-γ ELISpot revealed two neighboring 15-mer peptides, peptide #98 (ASQNYKIPASGGNAL) and peptide #99 (KIPASGGNALLKYDT), which shared an overlapping ten amino acid sequence (Figure 4A). ICS was then performed in order to define the T cell population responsible for expressing IFN-γ in response to these peptides. Splenocytes harvested from AAV2/rh32.33.CB.nLacZ-treated Balb/C mice were stimulated in vitro with pool I or with peptides 98 and 99 and stained with monoclonal antibodies to CD4, CD8 and IFN-γ. The IFN-γ -producing T-cell response after stimulation was found to be CD8-mediated (Figure 4B); minimal CD4+ T-cell activation was observed (Figure 4C).
Figure 4.
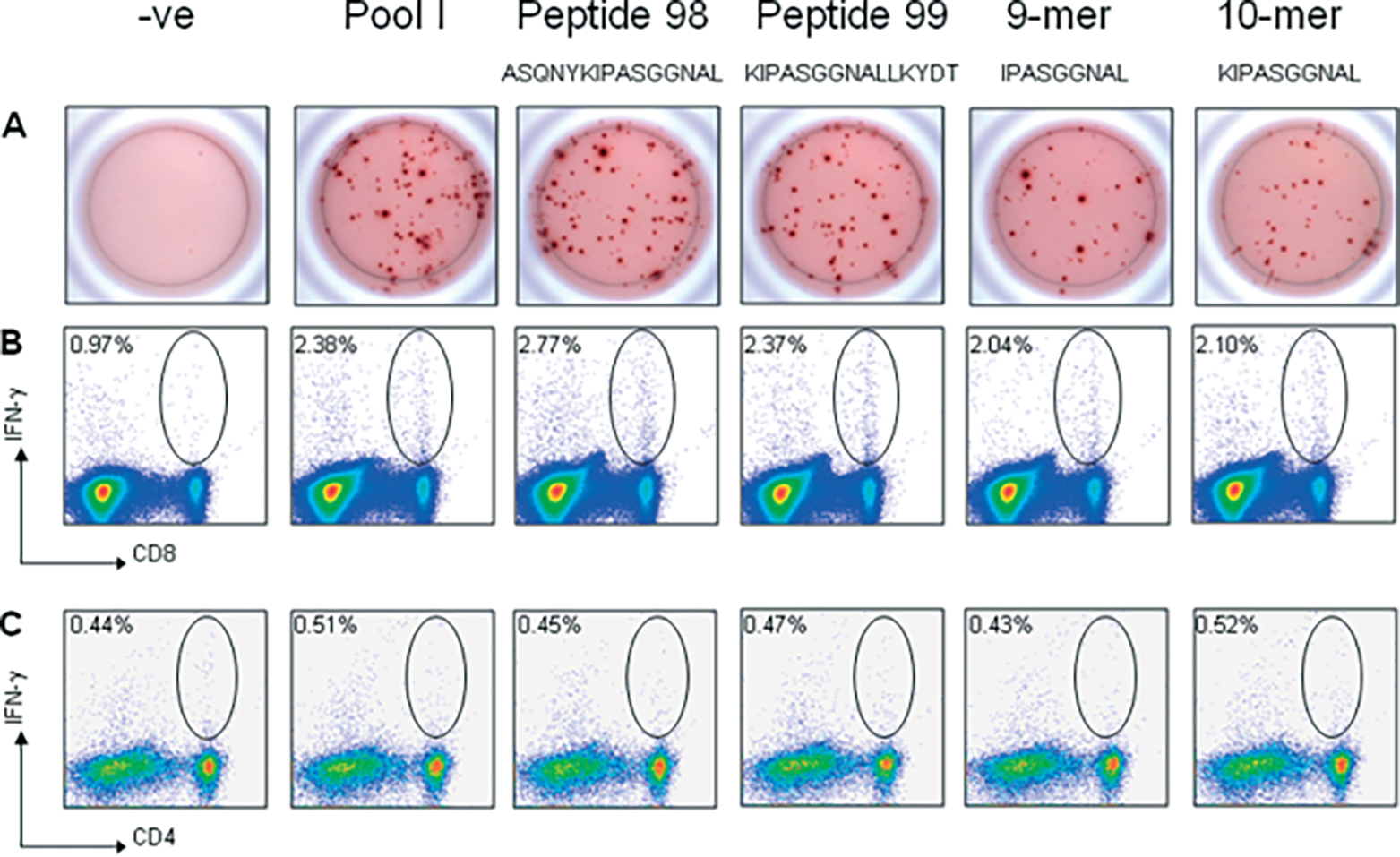
Fine mapping of the H-2d-restricted T cell epitope in the AAVrh32.33 capsid protein. Splenocytes (106 cells/well) harvested from AAV2/rh32.33.CB.nLacZ-injected Balb/C mice at day 21 were stimulated in vitro with 2 μg/ml of either pool I, peptides 98 and 99, the 9-mer, IPASGGNAL, or the 10-mer, KIPASGGNAL. Samples were analysed by either (A) IFN-γ ELISpot or (B,C) intracellular cytokine staining followed by flow cytometric analysis. Staining was performed using monoclonal antibodies to (B) CD8, (C) CD4 and IFN-γ. Negative control wells were stimulated with no peptide. Representative results from three separate experiments are shown
BIMAS and SYFPEITHI were then used to identify the potential MHCI binders within the peptide sequence, ASQNYKIPASGGNALLKYDT (Table 1). For the H2-Kd MHCI molecule, both algorithms gave the highest score to either the 9-mer, IPASGGNAL, or the 10-mer, KIPASGGNAL. (SYFPEITHI also predicted IPASGGNALL as an equivalently likely epitope for H2-Kd. This 10-mer, similar to KIPASGGNAL, is also centred around the 9-mer, IPASGGNAL). BIMAS also predicted the 10-mer KIPASGGNAL as the highest ranking candidate for the H2-Dd MHCI molecule. No H2-Dd MHCI binders were predicted by SYFPEITHI. The 9-mer, IPASGGNAL, was the highest-ranking predicted binder for H2-Ld MHCI by both algorithms (Table 1). Based on these scores, both the 9-mer, IPASGGNAL, and the 10-mer, KIPASGGNAL, were synthesized for further evaluation.
To confirm that (K)IPASGGNAL is the immunodominant H2-d epitope in the AAVrh32.33 capsid protein, both peptides were tested in Balb/C mice by IFN-γ ELISpot and ICS, in comparison to pool I and individual 15-mers, peptide 98 and 99 (Figure 4). Both the 9-mer and the 10-mer stimulated an equivalent degree of IFN-γ secretion, which was comparable to the level of T-cell activation seen with the positive pool I and individual 15-mers 98 and 99 (Figure 4A). Flow cytometric staining confirmed that the T-cell response to these peptides is CD8-mediated (Figure 4B). In conclusion, (K)IPASGGNAL is the dominant MHCI epitope of the AAVrh32.33 capsid in Balb/C mice.
To determine whether the 9-mer or 10-mer is more dominant, the relative binding affinities of IPASGGNAL and KIPASGGNAL for the MHC class I molecule was defined by titrating the amount of peptide necessary for T-cell activation in vitro. Splenocytes harvested from AAV2/rh32.33.CB.nLacZ-injected mice were stimulated with either peptide at concentrations ranging from 5 to 1.6 × 10−10 μg/ml, and the activation of T cells was measured by IFN-γ ELISpot (Figure 3B). Both the 9-mer and 10-mer had a moderate to low binding affinity that appeared to steadily decline as the peptide concentration was lowered. Consistent with our previous findings, the AAVrh32.33 capsid is highly immunogenic in C57BL/6 mice and only moderately so in the Balb/C strain [10]. This explains why the level of T-cell activation achieved by the H-2b dominant epitope is much greater than that seen with the H-2d dominant peptide in their respective mouse strains (Figure 3).
Although pool I contained the H-2d dominant epitope, all individual peptides comprising the less positive pools E, F, K and L were screened to identify the 15-mer(s) responsible for T-cell activation in Balb/C mice. Minor epitopes included: INNNWGLRPKAMRVK (peptide 59; pool E), NLTSTVQIFADSSYE (peptide 66; pool F), GLKHPPPQIFIKNTP (peptide 128; pool K) and YSTGQVAVQIEWEIE (peptide 135; pool L).
An alignment of the AAVrh32.33 capsid epitope sequences with other major AAV serotypes illustrates that the dominant AAVrh32.33 capsid epitopes are poorly conserved across the family (Table 2). Conservation of the H-2b dominant capsid epitope, SSYELPYVM, between AAVrh32.33 and AAV4 is not surprising given the high degree of sequence homology shared between these two closely-related variants. The lack of complete conservation in all other cases was also expected given the structural divergence of AAVrh32.33 and AAV4 from most other serotypes within the AAV family.
Table 2.
Alignment of AAVrh32.33 dominant capsid epitopes with other major AAV serotypes
| C57BL/6 | Balb/C | |
|---|---|---|
|
| ||
| AAVrh32.33 | SSYELPYVM | KIPASGGNAL |
| AAV2 | SEYQLPYVL | - NNSEYS |
| AAV4 | SSYELPYVM | KIPATGSDSL |
| AAV6 | SEYQLPYVL | - NNSNFT |
| AAV7 | SEYQLPYVL | - NNSNFA |
| AAV8 | SEYQLPYVL | - NNSNFA |
| AAV9 | SDYQLPYVL | - NNSEFA |
Sequence alignment of the H-2b and H-2d dominant capsid epitopes of AAVrh32.33 with AAV2, AAV4, AAV6, AAV7, AAV8 and AAV9. Alignment was performed using BioEdit, version 7.0.0 (Ibis Biosciences, Carlsbad, CA, USA). Note that the alignment of the H-2d epitope may vary as a result of amino acid deletions in the sequences of AAV2 and 6–9 relative to AAV4 and AAVrh32.33.
In conclusion, identification of AAV capsid immunodominant epitopes is critical for quantifying T-cell activation to vector capsids. Because of recent concern that capsid-specific T-cell responses may play a role in eliminating vector transduced cells, it is critical to understand the mechanism of immune activation to these vectors. Most AAV serotypes elicit poor capsid T-cell responses in murine models, making the phenomenon observed in higher-order species difficult to study. The strong capsid-specific CD8+ T-cell response generated to AAVrh32.33 in the mouse makes it an ideal candidate to address this issue in a small animal model. Mapping the immunodominant CD8+ T cell epitopes to the AAVrh32.33 capsid in commonly used mouse strains will facilitate mechanistic studies of capsid T-cell activation in C57BL/6 and Balb/C mice.
Acknowledgements
We thank Arbans Sandhu and Julie Johnston at the PennVector Core (University of Pennsylvania) for supplying the rAAV vectors; Deirdre McMenamin and Regina Munden (University of Pennsylvania) for assistance with animal studies. J.M.W. is an inventor on patents licensed to various biopharmaceutical companies. J.M.W. holds equity in, consults for, and receives a grant from ReGenX Biosciences. This work was supported by grants from NIH [P01-HL059407, P30-DK47757 (J.M.W.), T32-AR053461-03 (L.E.M.)] and GSK.
References
- 1.Herzog RW, Yang EY, Couto LB, et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med 1999; 5: 56–63. [DOI] [PubMed] [Google Scholar]
- 2.Snyder RO, Miao C, Meuse L, et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat Med 1999; 5: 64–70. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Calcedo R, Nichols TC, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood 2005;105: 3079–3086. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Chirmule N, Gao G, et al. CD40 ligand-dependent activation of cytotoxic T lymphocytes by adeno-associated virus vectors in vivo: role of immature dendritic cells. J Virol 2000; 74: 8003–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fields PA, Kowalczyk DW, Arruda VR, et al. Role of vector in activation of T cell subsets in immune responses against the secreted transgene product factor IX. Mol Ther 2000; 1: 225–235. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Dobrzynski E, Schlachterman A, et al. Systemic protein delivery by muscle-gene transfer is limited by a local immune response. Blood 2005; 105: 4226–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006; 12: 342–347. [DOI] [PubMed] [Google Scholar]
- 8.Zaiss AK, Muruve DA. Immune responses to adeno-associated virus vectors. Curr Gene Ther 2005; 5: 323–331. [DOI] [PubMed] [Google Scholar]
- 9.Gao G, Wang Q, Calcedo R, et al. AAV-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum Gene Ther 2009; Aug 20 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mays LE, Vandenberghe LH, Xiao R, et al. Adeno-associated virus capsid structure drives CD4-dependent CD8 + T cell response to vector encoded proteins. J Immunol 2009; 182: 6051–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calcedo R, Vandenberghe LH, Gao G, et al. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 2009; 199: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma IM, Weitzman MD. Gene therapy: 20-first century medicine. Annu Rev Biochem 2005; 74: 711–738. [DOI] [PubMed] [Google Scholar]
- 13.Zaiss AK, Muruve DA. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther 2008; 15: 808–816. [DOI] [PubMed] [Google Scholar]
- 14.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 1994; 152: 163–175. [PubMed] [Google Scholar]
- 15.Rammensee H, Bachmann J, Emmerich NP, et al. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999; 50: 213–219. [DOI] [PubMed] [Google Scholar]
- 16.Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes – a framework for modelling, testing, and applications. Methods 2004; 34: 436–443. [DOI] [PubMed] [Google Scholar]
- 17.Pelte C, Cherepnev G, Wang Y, et al. Random screening of proteins for HLA-A*0201-binding nine-amino acid peptides is not sufficient for identifying CD8 T cell epitopes recognized in the context of HLA-A*0201. J Immunol 2004; 172: 6783–6789. [DOI] [PubMed] [Google Scholar]