

Abstract
Background
We recently reported the isolation and sequencing of 30 novel adenoviruses from chimpanzees, bonobos and gorillas. These adenoviruses are promising candidates for the purpose of expanding the repertoire of adenoviral serotypes that can be used to create vectors for circumventing pre-existing neutralizing antibodies in human populations. We thus aimed to create vectors from 20 of the newly isolated adenoviruses.
Methods
Plasmid molecular clones were created that harbored the complete E1-deleted genomes from 20 of the newly isolated ape adenoviruses belonging to species B, C and E. The plasmids were transfected into human embryonic kidney (HEK) 293 cells to rescue vectors. We also tested normal human sera to determine the extent of pre-existing cross-neutralizing antiadenovirus neutralizing antibodies.
Results
Twelve vectors could be rescued and expanded following transfection into HEK 293 cells with yields (from fifty 150-mm culture dishes) that ranged from 3 × 1011 to 7 × 1013 viral particles. Sera from 50 normal human donors were tested for the presence of neutralizing activity against 21 of the newly isolated ape adenoviruses. Cross-neutralizing activity was generally low, although outliers with high neutralizing activity were frequently detected. Species B ape adenoviruses generally showed the least cross-neutralization with antibodies present in the human sera that were tested.
Conclusions
E1-deleted adenovirus vectors can be created from a wide variety of ape adenoviruses that can be rescued and propagated in HEK 293 cells. The prevalence of pre-existing antibodies that can neutralize these adenoviruses in human populations is low.
Keywords: adenovirus, chimpanzee, gene therapy, vector
Introduction
Adenoviruses are attractive as vectors for human gene therapy and genetic vaccines because of their relative promiscuity in tissue tropism, the high-level expression of the transgene, and the ease of producing high titer virus. The availability of human embryonic kidney (HEK) 293 cells, which express the HAdV-5 E1 region genes and thereby complement the E1 defect in vectors, is also an advantage for creating HAdV-5 vectors. However, there is a high prevalence of pre-existing antibodies to species C human adenoviruses such as HAdV-5 and this has been used in the construction of the great majority of existing vectors [1,2]. Those individuals who initially do not possess neutralizing antibodies quickly develop them and this effectively precludes the possibility of the efficacious re-administration of a vector of the same serotype.
An approach towards administering adenovirus vectors in populations with pre-existing immunity as a result of natural infections or previous vector exposure comprises the development of a series of vectors using virus serotypes to which previous exposure is unlikely. This is especially relevant for vaccine strategies where efficacy in a high percentage of the population is required and multiple administrations of vector will be necessary to effectively boost immune responses. Several groups have pursued this approach by developing vectors based on rare serotypes, such as serotype 35 [1,3] or by using nonhuman adenoviruses such as bovine, canine or ovine adenoviruses [4–6]. We have previously reported the construction of adenoviral vectors based on adenoviruses of species E and B, which were originally isolated from chimpanzees [7–10]. The E1 functions of the species E viruses were efficiently complemented by the HAdV-5 E1 genes expressed in HEK 293 cells, and therefore such vectors could be grown in these cells using standard methods. In the present study, we describe the construction of new ape-derived adenovirus vectors based on newly isolated ape (chimpanzee, gorilla and bonobo) adenoviruses that belong to species B, C and E. We have also explored the likelihood that these vectors may be cross-neutralized by circulating antibodies in humans that have resulted from natural infection with human adenoviruses.
Materials and methods
Viruses and viral DNA
Adenoviruses were isolated from the feces of chimpanzees, bonobos, and gorillas that are in captivity in facilities and zoos throughout the USA as described previously [11]. Adenoviruses were purified and adenoviral DNA was extracted using standard protocols.
Determination of neutralization titers
Individual human serum samples were obtained from the Department of Pathology and Laboratory Medicine at the Hospital of the University of Pennsylvania (Philadelphia, PA, USA). Pooled human immunoglobulins (IVIG) derived from the plasma of normal human donors (manufactured by CSL Behring AG, Bern, Switzerland) were reconstituted in accordance with the manufacturer’s instructions and diluted to a concentration of 10 mg/ml before making serial dilutions. The anti-adenoviral neutralization titers in human serum samples or in IVIG were determined as described previously [12]. Briefly, A549 cells were infected with the purified wild-type adenoviruses (after pre-incubation with serial dilutions of the plasma test article) at a multiplicity of 1000. Infected cells were detected with a fluorescently labeled anti-hexon antibody. The dilution of plasma or serum that reduced infection efficiency by 50% was recorded as the anti-adenoviral neutralization titer. The anti-adenoviral neutralization titers in human serum samples, in IVIG, and in sera obtained from rabbits immunized with HAdV-5 or SAdV-24, were determined using vectors as described previously [8].
Creation of infectious molecular clones of E1-deleted vectors based on novel ape adenovirus isolates
Twenty of the newly discovered adenovirus genomes were converted into E1-deleted molecular plasmid molecular clones by the stepwise incorporation of the left end of the genome, deletion of the E1 functions, and, finally, the incorporation of the remainder of the viral genome using available restriction enzyme sites. Because the particular restriction enzyme sites varied, the exact construction schemes that were followed were different for each adenovirus construct. The construction of the E1-deleted molecular clone of the chimpanzee adenoviruses SAdV-30 and SAdV-36 are shown in Figure 1 and Figure 2 respectively.
Figure 1.
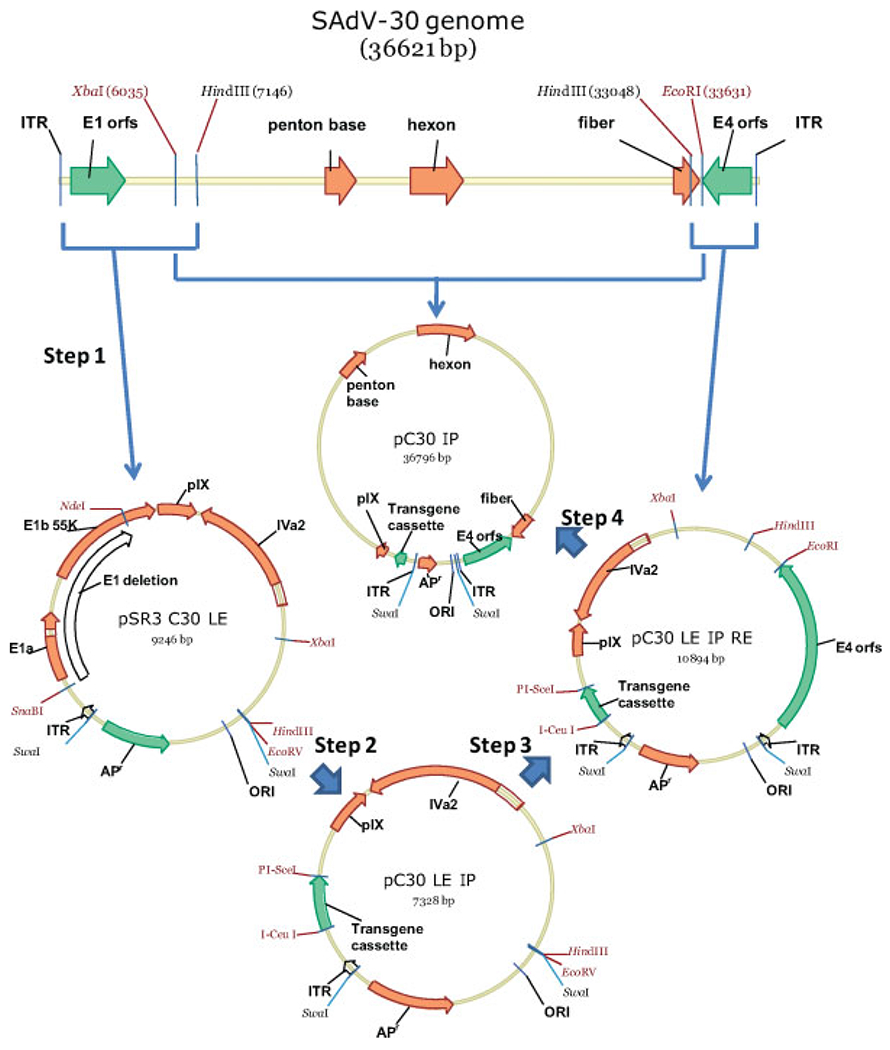
Steps in the construction of the E1-deleted SAdV-30 vector
Figure 2.
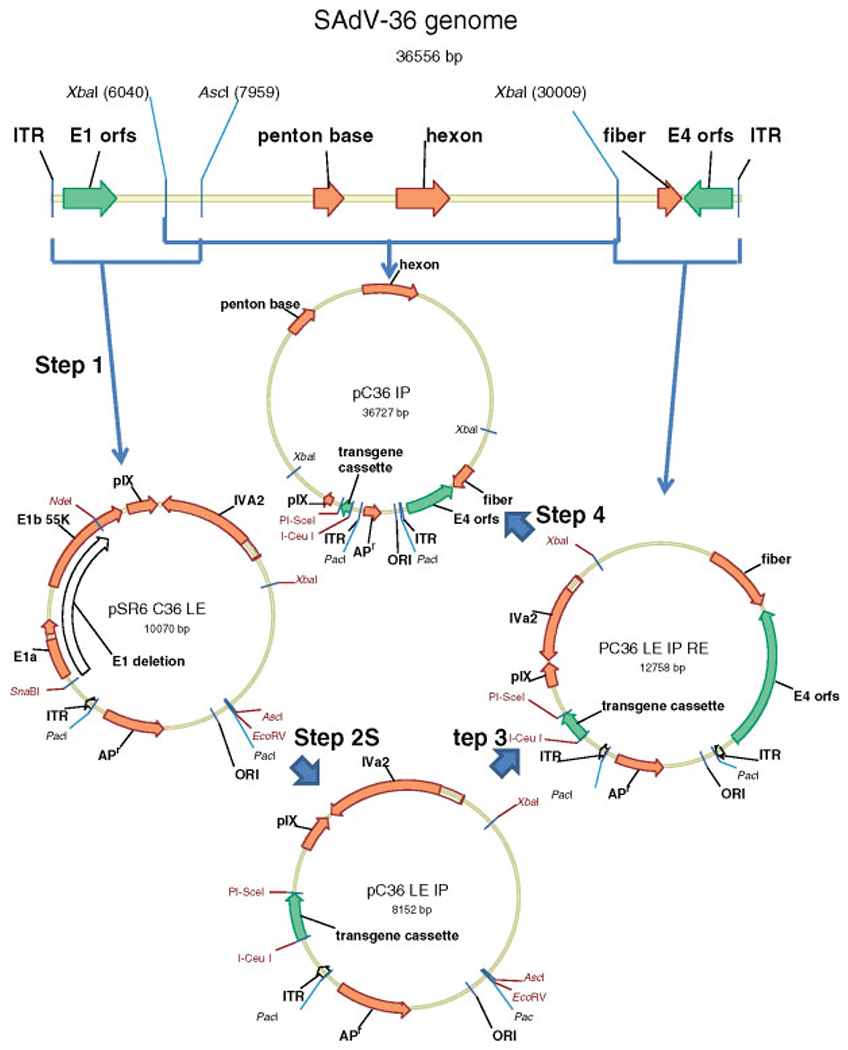
Steps in the construction of the E1-deleted SAdV-36 vector
For SAdV-30 molecular clone construction, the plasmid pSR3 was created by digesting the plasmid pBR322 with the restriction enzymes EcoRI and NdeI and inserting a synthetic DNA fragment containing successive recognition sites for the restriction enzymes SwaI, SmaI, HindIII, EcoRV and SwaI, respectively. To clone the left end of the SAdV-30 viral DNA (Figure 1, step 1), it was digested with the restriction enzyme HindIII and the 7146-bp fragment harboring the left end was inserted into pSR3 digested with SmaI and HindIII to yield the plasmid pSR3C30 LE. The integrity of the left terminus of the cloned DNA was checked by sequencing. To carry out a functional deletion of the E1 region in pSR3 C30 LE, a deletion between the SnaBI (located 455 bp from the left end) and NdeI sites was made (Figure 1, step 2). This removes the entire E1a and E1b 19K coding regions as well as 74% of the E1b 55K coding region at the same time as preserving putative packaging signals and the pIX promoter. A fragment harboring restriction sites for the extremely rare cutter enzymes I-CeuI and PI-SceI was inserted in place of the E1 deletion to yield the plasmid pC30 LE IP. To clone the right end of the SAdV-30 viral DNA, it was digested with the restriction enzyme HindIII and the 3574-bp fragment harboring the right end was inserted into pC30 LE IP digested with EcoRV and HindIII to yield the plasmid pC30 LE.IP RE (Figure 1, step 3). The integrity of the right terminus of the cloned DNA was confirmed as before. The remainder of the SAdV-30 genome was inserted between the unique XbaI and EcoRI sites to produce the plasmid pC30 IP (Figure 1, step 4) containing the complete E1 deleted SAdV-30 genome incorporating an E1 deletion replaced by a fragment harboring I-CeuI and PI-SceI sites that are utilized for the insertion of any desired transgene cassette. Digestion of this plasmid harboring the complete (E1 deleted) SAdV-30 genome with the restriction enzyme SwaI releases the linear infectious DNA for transfection into an E1 complementing cell line such as HEK 293.
For SAdV-36 molecular clone construction, the plasmid pSR6 was created by digesting the plasmid pBR322 with the restriction enzymes EcoRI and NdeI and inserting a synthetic DNA fragment containing successive recognition sites for the restriction enzymes PacI, SmaI, AscI and PacI, respectively. To clone the left end of the SAdV-36 viral DNA (Figure 2, step 1), it was digested with the restriction enzyme AscI and the 7958-bp fragment harboring the left end was inserted into pSR6 digested with SmaI and AscI to yield the plasmid pSR6 C36 LE. The integrity of the left terminus of the cloned DNA was checked by sequencing. To carry out a functional deletion of the E1 region in pSR6 C36 LE (Figure 2, step 2), a deletion between the SnaBI (located 455 bp from the left end) and NdeI sites was made. This removes the entire E1a and E1b 19K coding regions as well as 74% of the E1b 55K coding region at the same time as preserving putative packaging signals and the pIX promoter. A fragment harboring restriction sites for the extremely rare cutter enzymes I-CeuI and PI-SceI was inserted in place of the E1 deletion to yield the plasmid pC36 LE IP. To clone the right end of the SAdV-36 viral DNA (Figure 2, step 3), it was digested with the restriction enzyme XbaI and the 6548 bp fragment harboring the right end was inserted into pC36 LE IP digested with XbaI and EcoRV to yield the plasmid pC36 LE.IP RE. The integrity of the right terminus of the cloned DNA was confirmed as before. The remainder of the SAdV-36 genome between the XbaI sites was inserted into the XbaI site of pC36 LE.IP RE (Figure 2, step 4) to produce the plasmid pC30 IP containing the complete E1 deleted SAdV-36 genome incorporating an E1 deletion replaced by a fragment harboring I-CeuI and PI-SceI sites that are utilized for the insertion of any desired transgene cassette. Digestion of this plasmid harboring the complete (E1 deleted) SAdV-36 genome with the restriction enzyme PacI releases the linear infectious DNA for transfection into an E1 complementing cell line such as HEK 293.
Rescue and propagation of recombinant adenoviruses harboring a transgene cassette expressing the influenza virus nucleoprotein
An expression cassette (approximately 2.5 kb, flanked by restriction sites for I-CeuI and PI-SceI), composed of the human cytomegalovirus immediate early promoter, the codon-optimized influenza A (Puerto Rico/8/34/Mount Sinai) nucleoprotein (NP) coding sequence and the bovine growth hormone polyadenylation signal, was excised from a shuttle plasmid (pShuttle; Clontech Laboratories, Palo Alto, CA, USA) using I-CeuI and PI-SceI and inserted into the adenoviral E1-deleted plasmid molecular clones. The resulting plasmids were then digested by the appropriate enzyme (e.g. SwaI for pC30 FluA NP or PacI for pC36 FluA NP) required to release the infectious viral DNA. HEK 293 cells maintained in DMEM supplemented with 10% fetal bovine serum were transfected with plasmids harboring the complete (E1-deleted) adenoviral genome as described. Cytopathic effects indicating virus rescue was usually observed 2–3 weeks following transfection. Rescued viruses were propagated in HEK 293 cells and purified by cesium chloride gradient centrifugation using standard protocols by Penn Vector core of the University of Pennsylvania.
Inoculation of mice with the adenovirus vectors and assay for T-cell response to the influenza nucleoprotein transgene
Balb/c mice were sedated with intra-peritoneal ketamine/xylazine, and the total adenovirus dose (1011 viral particles per mouse) divided into two 25-μl injections given intramuscularly to the hind leg tibialis anterior muscle. Vaccinated mice were sacrificed 10 days following immunization and their splenic CD8+ T cells were analyzed by stimulating with the immunodominant peptide (TYQRTRALV) of influenza NP and staining for the cytokines interferon (IFN)-γ, interleukin (IL)-2 and tumor necrosis factor (TNF)-α. A peptide of sequence TYQRTRALV corresponding to influenza A NP residues 147–155, which is an immunodominant MHC class I epitope (H-2Kd restricted), was synthesized by Mimotopes (Adelaide, Australia). Splenocytes from control and immunized BALB/c mice (H-2Kd haplotype), were stimulated with the peptide at a concentration of 2 μg/ml for 5 h at 37 °C in 10% CO2 in the presence of 1 μl/ml brefeldin A (GolgiPlug; BD Biosciences, Franklin Lakes, NJ, USA). After washing, cells were stained with fluorescein-labeled anti-mouse CD8 antibody (BD Biosciences). Cells were then washed and permeabilized in Cytofix/Cytoperm (BD Biosciences) for 20 min on ice. Subsequently, the cells were washed again and stained with the anti-cytokine antibodies phycoerythrin (PE)-labeled anti-mouse IFN-γ; (BD Biosciences), PE-Cy7-labeled anti-mouse TNF-α (BD Biosciences) and allophycocyanin-labeled mouse IL-2 antibody (eBioscience, SanDiego, CA, USA). After a final wash, the cells were examined by multi-colour flow cytometry (FC 500; Beckman Coulter, Fullerton, CA, USA) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA) as described previously [13].
Results
Creation of a diverse group of recombinant adenoviruses
To construct and clone the E1 deleted genomes, the strategy originally described for the construction of simian adenoviruses vectors based on SAdV-22, SAdV-23, and SAdV-24 [8] was followed for viruses belonging to species C or E. Briefly, the complete genome was assembled stepwise onto a poly linker fragment containing the appropriate restriction sites. As a first step, the left end of the genome was cloned. Available restriction sites present in the left end fragment (e.g. SnaBI and NdeI in case of SAdV-30) were used to create a functional deletion of E1 and insertion of the I-CeuI and PI-SceI extremely rare cutter sites. This was followed by a stepwise incorporation of the rest of the viral genome. However, because species E1-deleted species B adenoviral vectors are not fully complemented by HEK 293 cells, vectors for the species B adenoviruses were made as chimeric vectors where the left and right ends were derived from SAdV-22 or SAdV-36 (Table 1) as described previously [10].
Table 1.
Twenty E1-deleted molecular clones harboring the influenza nucleoprotein (PR8 strain) transgene were transfected and rescued
| SAdV isolate | Species | Titer | Yield |
|---|---|---|---|
| SAdV-25.2 | E | 5.24 × 1012 | 5.87 × 1013 |
| SAdV-26 | E | – | Poor growth |
| SAdV-27.1* | B | – | Poor growth |
| SAdV-27.2* | B | 1.80 × 1012 | 3.06 × 1012 |
| SAdV-28.1* | B | 5.24 × 1012 | 7.39 × 1013 |
| SAdV-29* | B | 5.24 × 1012 | 3.93 × 1013 |
| SAdV-30 | E | 3.70 × 1012 | 1.18 × 1013 |
| SAdV-31.1 | C | – | Poor growth |
| SAdV-32* | B | 4.51 × 1012 | 2.26 × 1013 |
| SAdV-35.1* | B | – | Poor growth |
| SAdV-36 | E | 2.92 × 1012 | 5.55 × 1012 |
| SAdV-37 | E | 5.52 × 1012 | 2.57 × 1013 |
| SAdV-39 | E | 5.01 × 1012 | 6.14 × 1013 |
| SAdV-40.1 | C | – | Poor growth |
| SAdV-40.2 | C | 2.58 × 1011 | 3.10 × 1011 |
| SAdV-41.1 ** | B | – | Poor growth |
| SAdV-42.2 | C | – | Poor growth |
| SAdV-44 | C | – | Poor growth |
| SAdV-45 | C | 4.73 × 1012 | 3.43 × 1013 |
| SAdV-46** | B | 4.42 × 1012 | 2.48 × 1013 |
| HAdV-5 | C | 5.84 × 1012 | 2.10 × 1013 |
In several cases, the virus showed poor growth in HEK 293 cells and could not be propagated to high titer. The yields from fifty 150-mm culture dish preparations are shown, including the HAdV-5 influenza NP vector preparation in the last row.
Chimeric molecular clones using SAdV-22 right and left ends;
chimeric molecular clones using SAdV-36 right and left ends.
Although all three major capsid proteins (hexon, fiber and penton), harbor neutralization determinants, relatedness among hexon sequences is an accurate predictor of cross-neutralization characteristics. A phylogenetic tree of all published ape adenoviral hexon sequences is shown in Figure 3. Adenoviral isolates to date from apes have belonged to species B, C and E. We have attempted to create adenoviral vectors based on 20 of the 30 isolates that were isolated in our laboratory as shown in Table 1 [11]. Although recombinant adenoviruses could be rescued in all cases, propagation to high titer was successful in 12 instances, as indicated by arrows in Figure 3. Although rescue of recombinant adenovirus vector was visually evident by a cytopathic effect in all cases, we were able to successfully amplify 12 of the 20 constructs that were transfected. The yield obtained in each case, as well as the corresponding titer, is shown in Table 1. Interestingly, there did not appear to be a relationship between the ease of vector rescue and final yield (i.e. vectors that took relatively longer to rescue and propagate did not necessarily have low yields).
Figure 3.
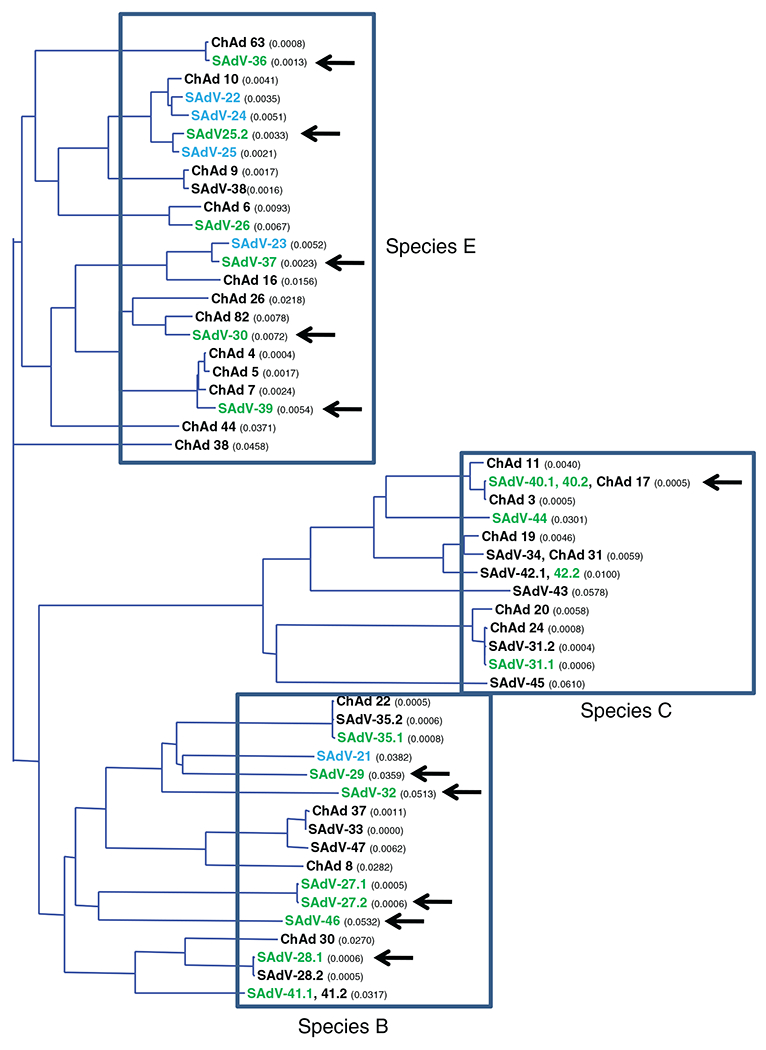
Phylogeny of all available ape adenovirus hexon sequences is shown including isolates from this laboratory (black or green font) (The tree was constructed using the neighbour-joining algorithm as implemented by the AlignX module of Vector NTI software (Invitrogen, Carlsbad, CA, USA). The calculated distance values are shown in parentheses). Isolates from which molecular clones have been constructed are shown in green (present study) or blue [7,8]. The plasmid molecular clones from which rescued vectors expressing the influenza A nucleoprotein were successfully amplified are indicated by arrows
Determination of the prevalence of cross-neutralizing antibodies in humans
Infections with species B, C and E adenoviruses are common in human populations, frequently manifesting with coryza-like symptoms. This results in a fairly high prevalence of detectable neutralizing antibodies against many serotypes such as HAdV-3 (species B), HAdV-4 (species E) and HAdV-5 (species C). It was of interest to determine the degree to which such antibodies may cross-react with the ape adenovirus isolates that are also classified into these three species based on phylogeny [11]. We tested 50 normal blood donors for the presence of cross-neutralizing antibodies against 21 novel ape adenoviruses using the infectivity reduction assay as described in the Materials and methods. The results obtained are shown in Figure 4. It was seen that the prevalence of cross-neutralizing antibodies was highest against the species C adenoviruses and lowest against species B adenoviruses.
Figure 4.
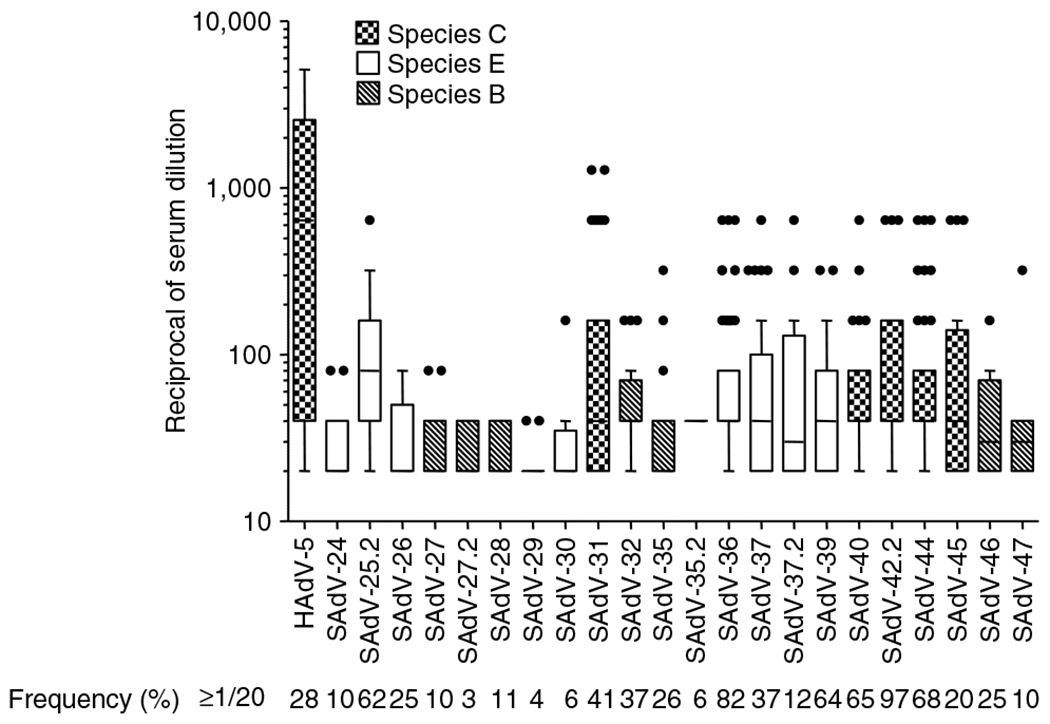
Box and whisker plot of the anti-adenovirus neutralizing activity present in 50 individual plasma samples from normal human blood donors. All 50 samples were tested against each of the indicated adenoviruses in a neutralizing antibody infectivity reduction assay as described in the Materials and methods. Each box encloses 50% of the data, with the upper and lower limits denoting the interquartile range; the horizontal line denoting the median value; the whiskers denoting minimum and maximum values in the data set that fell within an acceptable range; and the circles denoting outliers. The cross-hatching within the boxes is keyed to the species of adenoviruses as indicated. The percentage of the samples that exhibited a neutralization titer that was equal or greater than 1:20 is also indicated
To obtain another picture of the likely prevalence of cross-reacting neutralizing activity against the ape adenoviruses in human populations, we also tested commercially available IVIG (i.e. pooled human immunoglobulins manufactured for human therapeutic use) for neutralizing activity against a panel of ape adenovirus isolates belonging to species B, C, and E (Table 2). In this assay, the neutralization titers against the species B and E isolates were generally low; titers were higher against the two species C serotypes (SAdV-31 and SAdV-40).
Table 2.
Cross-neutralization using rabbit anti-HAdV-5, rabbit anti-SAdV-24 and IVIG
| Neutralization titer using: |
||||
|---|---|---|---|---|
| Adenovirus species | Adenovirus Isolate | Anti-HAdV-5 (rabbit) | Anti-SAdV-24 (rabbit) | IVIG (human) |
| C | HAdV-5 | 1:81920 | <1/20 | 1/640 |
| E | SAdV-24 | <1:20 | 1:655360 | 1:20 |
| E | SAdV-25.2 | <1:20 | 1:655360 | 1:40 |
| SAdV-26 | 1:20 | 1:40960 | 1:20 | |
| SAdV-30 | <1:20 | 1:1280 | 1:20 | |
| SAdV-37 | <1:20 | 1:320 | 1:20 | |
| SAdV-39 | <1:20 | 1:320 | 1:20 | |
| B | SAdV-27 | 1:20 | <1:20 | <1:20 |
| SAdV-28 | <1:20 | <1:20 | 1:40 | |
| SAdV-29 | <1:20 | <1:20 | <1:20 | |
| SAdV-32 | <1:20 | <1:20 | <1:20 | |
| SAdV-35 | <1:20 | <1:20 | 1:20 | |
| C | SAdV-31 | 1:5120 | <1:20 | 1:80 |
| SAdV-40 | <1:20 | <1:20 | 1:40 | |
The anti-adenovirus titers using each of the ape adenovirus isolates shown was determined as described in the Materials and methods. The dilution of the antiserum that reduced infectivity by 50% is shown.
We had previously demonstrated that it was possible to rescue species B E1-deleted vectors in HEK 293 cells by creating chimeric constructs with SAdV-22, a species E adenovirus, as described previously [10]. We therefore used SAdV-22 left and right end fragments for our new species B vectors. However, it was of interest to determine whether this approach of rescuing species B adenovirus vectors by creating chimeric constructs with a species E adenovirus was more generally feasible (and not restricted to SAdV-22). We tested whether SAdV-36 (a species E adenovirus) could be used in place of SAdV-22. As shown in Table 1, species B vectors (SAdV-41.1 and SAdV-46) E1-deleted vectors could be created using left and right end fragments derived from SAdV-36.
Cross-neutralization is an important consideration for re-administration experiments. Very high titer anti-sera that had been raised in rabbits against HAdV-5 and SAdV-24 were also tested for cross-neutralization in this assay (Table 2). We have previously shown that SAdV-24 and SAdV-25 belong to the same neutralization group (i.e. SAdV-25 is neutralized by the anti-SAdV-24 antiserum) [8]. It is interesting to note that SAdV-25.2, which harbors a hexon that is almost identical to SAdV-25, is similarly neutralized efficiently by the anti-SAdV-24 antiserum. (the SAdV-25 and the SAdV-25.2 fiber knob domains are only approximately 43% homologous; data not shown). The cross-neutralization experiment shown in Table 2 was carried out to gauge the extent to which antibodies against HAdV-5 and SAdV-24 (high titer rabbit antisera) would cross-react against some of our ape adenovirus isolates, thereby testing the extent to which cross-neutralization may be observed within, as well as between adenoviral species. We also tested human IVIG against the same panel of adenoviruses. It is interesting to note that anti-serum to SAdV-24 (a species E adenovirus) has a wide range of reactivities against other species E viruses. The panel of species C adenoviruses is much smaller (only SAdV-31 and SAdV-40) and we observe neutralization titers of 1:5120 in the former case and no cross-neutralization in the latter case, respectively. The panel of species E viruses is more extensive (five isolates) and we observe a 2048-fold difference in cross-neutralization titers with the anti-SAdV-24 antiserum.
Elicitation of an anti-influenza nucleoprotein immune response with the SAdV-36 vector
Adenovirus vectors can serve as potent vaccines because of the very strong T-cell response that is elicited against the transgene after inoculation with the vector [2,13]. The SAdV-36 vector expressing influenza NP was one the earliest vectors that we constructed where a reasonably high titer preparation could be made. We compared the activity of the vector created with SAdV-36 expressing the influenza nucleoprotein with that of the HAdV-5-derived vector. The chimpanzee adenovirus vector C36 CMV PI FluA NP and the human HAdV-5 vector AdH5-FluA NP were used to vaccinate BALB/c mice (five mice per group). Vaccinated mice were sacrificed 10 days after immunization and their splenic CD8+ T cells were analyzed by stimulating with the Balb/c immunodominant peptide of influenza NP (TYQRTRALV) and staining for the cytokines IFN-γ, IL-2 and TNF-α (Figure 5). We also determined the subset of the CD8+ T cells that were poly functional (i.e. capable of secreting multiple cytokines), which may be an indication of the quality of the T cell response especially with respect to the formation of a memory population. The overall T cell responses were high with both vectors and were poly functional, although slightly lower with the SAdV-36 vector.
Figure 5.
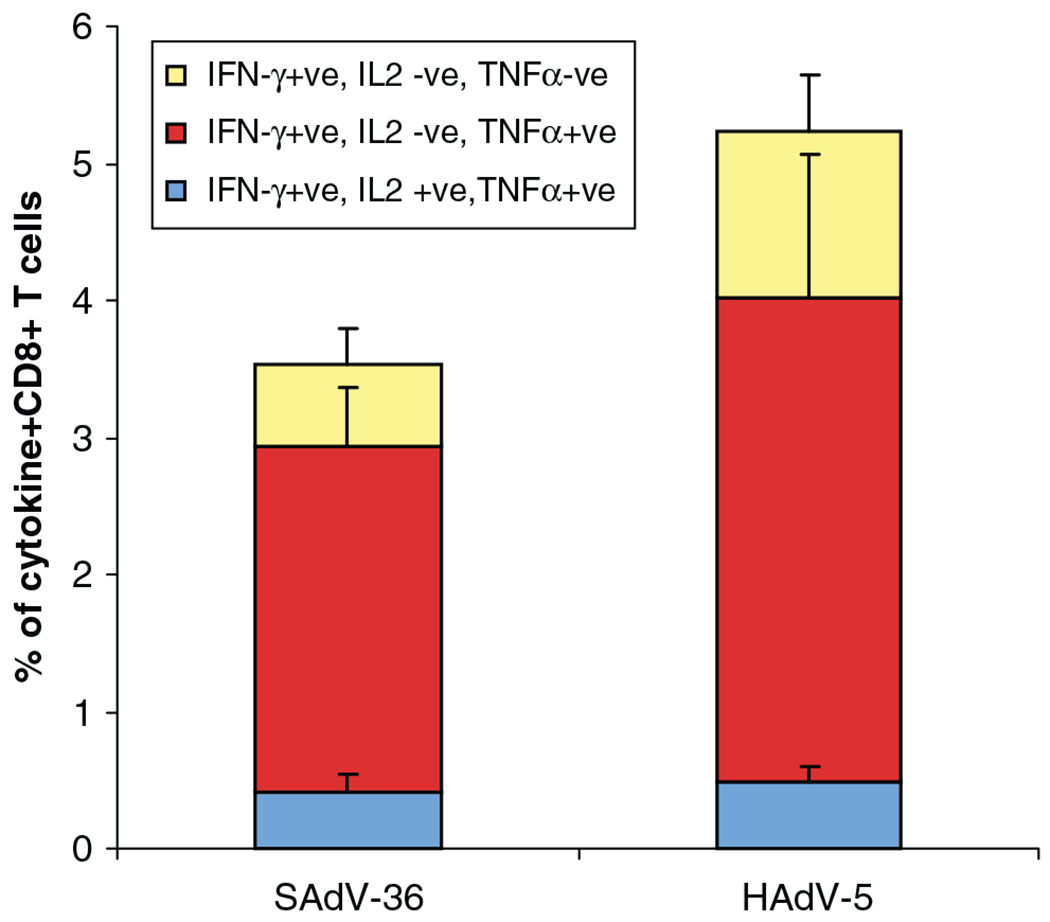
Analysis of CD8+ T lymphocytes from immunized BALB/c mice. Vaccinated mice (five mice per group) were sacrificed 10 days following immunization and their CD8+ T cells were analyzed by stimulating with the immunodominant peptide TYQRTRALV of influenza nucleoprotein and staining for the cytokines IFN-γ, IL-2 and TNF-α. Poly functional CD8+ T cells (i.e. those determined to be secreting multiple cytokines) are indicated
Discussion
We have previously reported on our attempts to expand the repertoire of adenoviral serotypes that are available for the construction of vectors [7–10], with the expectation that this may allow for their widespread use in a setting where neutralizing antibodies in human populations provides the least hindrance. Vectors created from adenoviruses isolated from animals are likely to be not neutralized in humans when first used. However, E1-deleted versions of these vectors cannot be complemented by existing complementing cell lines such as HEK 293 or PER.C6. To create and propagate such vectors, the matching E1-complementing cell lines need to be created in each case. We have previously shown that adenoviruses originally isolated from chimpanzees [14,15] could be engineered to create E1-deleted vectors [8,9] that could be grown in cell lines such as HEK 293, which are commonly used to complement E1 deletions in adenovirus vectors. However, because the available chimpanzee serotypes comprised of only one species B serotype (SAdV-21) and two species E serotypes (SAdV-22/24/25 and SAdV-23) [8], we set out to isolate additional serotypes from great apes: chimpanzees, bonobos, gorillas and orangutans [11]. We were able to purify and sequence over 30 new isolates belonging to 22 new serotypes that could be assigned to species (B, C and E), which are used to categorize human adenoviruses. This proximity in phylogeny between human and chimpanzee bonobo and gorilla adenoviruses (we were unable to amplify the adenoviruses that we could detect in orangutans using human cell lines) likely explains our success in being able to create E1-deleted adenovirus vectors that could be rescued and propagated in HEK 293 cells. However, the titers for eight of the 20 vectors that we rescued were unsuitable for their use in animal experiments. Interestingly, the HEK 293 cell line that expresses the species C (HAdV-5) E1 genes did not complement species C ape adenoviruses better than the species E ape adenoviruses.
The goal of developing a range of vectors with lower neutralization with human sera compared to vectors based on human viruses was realized. Levels of preexisting immunity to the great ape species B adenoviruses were very low; highest neutralizing antibody titers were seen to species C adenoviruses and intermediate levels to species E adenoviruses. However, all the created vectors demonstrated sero-responses improved over that observed with the HAdV-5 vector.
Acknowledgements
We thank the Cooperative Human Tissue Network (CHTN) of the University of Pennsylvania Medical Centre and the National Disease Research Interchange (NDRI) for providing human gut specimens. We also thank the PennVector Core personnel (University of Pennsylvania, Gene Therapy Program) (Hongbo Lu and Jigesha Shah) for their help with purifying adenoviruses and vectors. This work was sponsored by grants from Glaxo-SmithKline and NIDDK (5P30DK047757-15). S.R. and J.M.W. are inventors on patents licensed to various biopharmaceutical companies. J.M.W. is a consultant to ReGenX Holdings, and is a founder, holds equity in, and receives a grant from affiliates of ReGenX Holdings; in addition, he is an inventor on patents licensed to various biopharmaceutical companies, including affiliates of ReGenX Holdings. All animal experiments were conducted following the reviews and recommendations of the Institutional Animal Care and Use Committee of the University of Pennsylvania
References
- 1.Vogels R, Zuijdgeest D, van Rijnsoever R, et al. Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J Virol 2003; 77: 8263–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shenk T Group C adenoviruses as vectors for gene therapy. In Viral Vectors: Gene Therapy and Neuroscience Applications, Kaplitt MG, Lowey AD (eds). Academic Press: San Diego, 1995; 43–54. [Google Scholar]
- 3.Kostense S, Koudstaal W, Sprangers M, et al. Adenovirus types 5 and 35 seroprevalence in AIDS risk groups supports type 35 as a vaccine vector. AIDS 2004; 18: 1213–1216. [DOI] [PubMed] [Google Scholar]
- 4.Kremer EJ, Boutin S, Chillon M, et al. Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. J Virol 2000; 74: 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mittal SK, Middleton DM, Tikoo SK, et al. Pathogenesis and immunogenicity of bovine adenovirus type 3 in cotton rats (Sigmodon hispidus). Virology 1995; 213: 131–139. [DOI] [PubMed] [Google Scholar]
- 6.Xu ZZ, Hyatt A, Boyle DB, et al. Construction of ovine adenovirus recombinants by gene insertion or deletion of related terminal region sequences. Virology 1997; 230: 62–71. [DOI] [PubMed] [Google Scholar]
- 7.Farina SF, Gao GP, Xiang ZQ, et al. Replication-defective vector based on a chimpanzee adenovirus. J Virol 2001; 75: 11603–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy S, Gao G, Lu Y, et al. Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Hum Gene Ther 2004; 15: 519–530. [DOI] [PubMed] [Google Scholar]
- 9.Roy S, Zhi Y, Kobinger GP, et al. Generation of an adenoviral vaccine vector based on simian adenovirus 21. J Gen Virol 2006; 87: 2477–2485. [DOI] [PubMed] [Google Scholar]
- 10.Roy S, Clawson DS, Lavrukhin O, et al. Rescue of chimeric adenoviral vectors to expand the serotype repertoire. J Virol Methods 2007; 141: 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy S, Vandenberghe LH, Kryazhimskiy S, et al. Isolation and characterization of adenoviruses persistently shed from the gastrointestinal tract of nonhuman primates. PLoS Pathog 2009; 5: e1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calcedo R, Vandenberghe LH, Roy S, et al. Host immune responses to chronic adenovirus infections in human and nonhuman primates. J Virol 2009; 83: 2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy S, Kobinger GP, Lin J, et al. Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine 2007; 25: 6845–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowe WP, Hartley JW, Huebner RJ Additional serotypes of the APC virus group. Proc Soc Exp Biol Med 1956; 91: 260–262. [DOI] [PubMed] [Google Scholar]
- 15.Basnight M Jr, Rogers NG, Gibbs CJ Jr, et al. Characterization of four new adenovirus serotypes isolated from chimpanzee tissue explants. Am J Epidemiol 1971; 94: 166–171. [DOI] [PubMed] [Google Scholar]